

Abstract
In diet metabarcoding analyses, insufficient taxonomic coverage of PCR primer sets generates false negatives that may dramatically distort biodiversity estimates. In this paper, we investigated the taxonomic coverage and complementarity of three cytochrome c oxidase subunit I gene (COI) primer sets based on in silico analyses and we conducted an in vivo evaluation using fecal and spider web samples from different invertivores, environments, and geographic locations. Our results underline the lack of predictability of both the coverage and complementarity of individual primer sets: (a) sharp discrepancies exist observed between in silico and in vivo analyses (to the detriment of in silico analyses); (b) both coverage and complementarity depend greatly on the predator and on the taxonomic level at which preys are considered; (c) primer sets’ complementarity is the greatest at fine taxonomic levels (molecular operational taxonomic units [MOTUs] and variants). We then formalized the “one‐locus‐several‐primer‐sets” (OLSP) strategy, that is, the use of several primer sets that target the same locus (here the first part of the COI gene) and the same group of taxa (here invertebrates). The proximal aim of the OLSP strategy is to minimize false negatives by increasing total coverage through multiple primer sets. We illustrate that the OLSP strategy is especially relevant from this perspective since distinct variants within the same MOTUs were not equally detected across all primer sets. Furthermore, the OLSP strategy produces largely overlapping and comparable sequences, which cannot be achieved when targeting different loci. This facilitates the use of haplotypic diversity information contained within metabarcoding datasets, for example, for phylogeography and finer analyses of prey–predator interactions.
Keywords: cytochrome c oxidase subunit I gene, diet analysis, eDNA, false negatives, metabarcoding, PCR primers
1. INTRODUCTION
Diet studies are critical to the understanding of species interactions, trophic structures, and trophic dynamics (Nielsen, Clare, Hayden, Brett, & Kratina, 2018). They have been applied to a vast set of issues in ecology, evolution, and conservation, such as predator/prey interactions and habitat use (Corse et al., 2010; Sánchez‐Hernández, 2014), trophic niche partitioning (Kartzinel et al., 2015; Trevelline et al., 2018), and the delineation of habitats for guiding species conservation (Quéméré et al., 2013), management (Chivers et al., 2013), and habitat restoration (Motte & Libois, 2002). Diet studies have also proved critical in interfacing agriculture and ecology by assessing the effects of agricultural practices or policies on the trophic behaviors of species (Llaneza & López‐Bao, 2015; Mollot et al., 2014) and by evaluating the ecosystem services of wild species, such as in the control of crop pests (Aizpurua et al., 2018; McCracken et al., 2012).
Over the last decade, considerable efforts have been made toward improving diet assessment methods, in particularly those based on high‐throughput sequencing (HTS) and DNA metabarcoding on environmental samples (Taberlet, Bonin, Zinger, & Coissac, 2018; Taberlet, Coissac, Pompanon, Brochmann, & Willerslev, 2012). DNA metabarcoding has been demonstrated as an efficient alternative to traditional methods of diet analysis (e.g., morphological gut content or stable isotope analyses) by improving both the accuracy and taxonomic resolution of prey identifications, as well as the detection of soft‐bodied, small, or rare prey (Clare, 2014; McInnes et al., 2017; Nielsen et al., 2018; Pompanon et al., 2012). While these advances have considerably enlarged our ability to study large‐scale and highly‐resolved trophic networks (Clare, 2014; Evans, Kitson, Lunt, Straw, & Pocock, 2016; Roslin & Majaneva, 2016), they still suffer from a number of methodological issues (reviewed in Alberdi et al., 2019). In particular, false positives and negatives are common in metabarcoding datasets (Alberdi, Aizpurua, Gilbert, & Bohmann, 2018; Corse et al., 2017; Piñol, Mir, Gomez‐Polo, & Agustí, 2014; Taberlet et al., 2018). False positives correspond to experimental/molecular artifacts (e.g., PCR errors, tag switching, or cross‐sample contaminations) leading to the detection of taxa that were not initially present in the sample. Several experimental and bioinformatic procedures based on negative and positive controls and on technical replicates were developed to filter out such artifacts (Alberdi et al., 2018; Corse et al., 2017; Galan et al., 2018). False negatives correspond to taxa that are not detected while being present in the sample. Although false negatives often occur for rare taxa (e.g., Ficetola et al., 2015), they are also produced when the affinity between primer and primer‐binding sites during polymerase chain reaction (PCR) is low (e.g., Elbrecht & Leese, 2017a; Vamos, Elbrecht, & Leese, 2017), which in turn will determine amplification success and hence the taxonomic coverage of a given primer set. Although there is growing use of control samples to assess levels of false positives (De Barba et al., 2014; Beng et al., 2016; Corse et al., 2017; Galan et al., 2018), no post hoc bioinformatic procedures can identify false negatives. Two primary strategies have been adopted to maximize the taxonomic coverage of primer sets used in metabarcoding studies. One strategy consists in designing “universal” primer sets that target DNA of all taxa in the clade of interest (e.g., Leray et al., 2013; Clarke, Soubrier, Weyrich, & Cooper, 2014; Rennstam Rubbmark, Sint, Horngacher, & Traugott, 2018). However, in silico or in vivo (sensu Alberdi et al., 2018; i.e., based on environmental samples) tests demonstrated that many “universal” primer sets were far from having perfect taxonomic coverage (e.g., Deagle, Jarman, Coissac, Pompanon, & Taberlet, 2014; Alberdi et al., 2018; but see Elbrecht & Leese, 2017a). The other strategy consists in using a cocktail of primer sets targeting either the same locus (e.g., Gibson et al., 2014; Corse et al., 2017) or distinct loci (e.g., Kaunisto, Roslin, Sääksjärvi, & Vesterinen, 2017; Olmos‐Pérez, Roura, Pierce, Boyer, & González, 2017; Devloo‐Delva et al., 2018).
Previously, we developed a benchtop‐to‐desktop workflow to analyze the diet of an invertivorous fish. We evaluated the taxonomic coverage of two primer sets targeting the 5′ end of the barcode region of the cytochrome c oxidase subunit I gene (COI) in silico, in vitro (using tissue‐derived invertebrate DNA), and in vivo (using fish feces; Corse et al., 2017). Here, we introduce a new primer set designed to reduce false negatives. We combined it with the previous two primer sets using a “one‐locus‐several‐primer‐sets” (OLSP) strategy to assay environmental samples. We first assessed the taxonomic coverage of the three primer sets with in silico analyses of current available barcodes. We then conducted an in vivo evaluation of the taxonomic and haplotypic diversity coverage and complementarity of the three primer sets on environmental DNAs (eDNA) obtained from a variety of materials as follows: feces from invertivores living in brackish water, freshwater, and terrestrial habitats, and spider webs. Finally, we brought new insights into the diet of the brackish water fish Pomatoschistus microps (Krøyer, 1838) and of the African freshwater fish Epiplatys infrafasciatus (Günther, 1866), and we assessed the biodiversity of invertebrates trapped in spider webs from the Amazon rainforest.
2. MATERIAL AND METHODS
2.1. Primer design
In the workflow we developed previously (see Corse et al., 2017), we used two primers sets (MFZR and ZFZR; for details, see Table 1) for the detection of invertebrate diversity in fish fecal samples. We aimed here to improve our workflow by developing a new COI primer set that covers an additional diversity of prey species and haplotypes. In this perspective, we manually designed a new forward primer (LepLCO) and two degenerate reverse primers (McoiR1 and McoiR2; for details, see Supporting Information Appendix S1). These primers produced ~150 bp amplicons in the 5′ end of the COI barcode region, that largely overlap with the sequences amplified by the primers sets MFZR and ZFZR. Additionally, we evaluated the reverse primer MLepF1‐rev (Brandon‐Mong et al., 2015) when used with LepLCO (see Table 1 for details).
Table 1.
Primers and primer sets evaluated in silico, in vitro, and in vivo in this study
| Primer set | Primer name | Forward (F)/Reverse (R) | Sequence (5′−3′) | Reference | Evaluation of primer sets: | ||
|---|---|---|---|---|---|---|---|
| In silico | In vitro | In vivo | |||||
| MFZR | Uni‐Minibar‐F1 | F | TCCACTAATCACAARGATATTGGTAC | Meusnier et al. (2008) | Yes | Yes | Yes |
| ZBJ‐ArtR2c | R | WACTAATCAATTWCCAAATCCTCC | Zeale, Butlin, Barker, Lees, and Jones (2011) | ||||
| ZFZR | ZBJ‐ArtF1c | F | AGATATTGGAACWTTATATTTTATTTTTGG | Zeale et al. (2011) | Yes | Yes | Yes |
| ZBJ‐ArtR2c | R | WACTAATCAATTWCCAAATCCTCC | Zeale et al. (2011) | ||||
| LFCR | LepLCO | F | RKTCAACMAATCATAAAGATATTGG | This study | Yes | Yes | Yes |
| McoiR2 | R | CCBCCRATTAWAATKGGTATHAC | This study | ||||
| LepLCO/McoiR1 | LepLCO | F | RKTCAACMAATCATAAAGATATTGG | This study | Yes | Yes | No |
| McoiR1 | R | AATCCBCCRATTAWAATKGGTAT | This study | ||||
| LepLCO/MLepF1‐rev | LepLCO | F | RKTCAACMAATCATAAAGATATTGG | This study | Yes | Yes | No |
| MLepF1‐rev | R | CGTGGAAAWGCTATATCWGGTG | Brandon‐Mong et al. (2015) | ||||
| fwhF1/fwhR1 | fwhF1 | F | YTCHACWAAYCAYAARGAYATYGG | Vamos et al. (2017) | Yes | No | No |
| fwhR1 | R | ARTCARTTWCCRAAHCCHCC | Vamos et al. (2017) | ||||
| MG‐LCO1490‐MiSeq/MG‐univR‐MiSeq | MG‐LCO1490‐MiSeq | F | ATTCHACDAAYCAYAARGAYATYGG | Galan et al. (2018) | Yes | No | No |
| MG‐univR‐MiSeq | R | ACTATAAARAARATYATDAYRAADGCRTG | Galan et al. (2018) | ||||
2.2. In silico evaluation of primers
The taxonomic coverage of seven primers sets (of which three include newly designed primers; see Table 1) was estimated for 36 taxa (see Supporting Information Table S1) according to the approach implemented in primerminer‐0.11 (Elbrecht & Leese, 2017b). Briefly, for each taxonomic group, all COI sequences were downloaded from the NCBI nt database using COi, CO1, COXi, COX1 as keywords as well as all COI sequences from the BOLD database (www.boldsystems.org; Ratnasingham & Hebert, 2007) in February 2017. Sequences were clustered with VSEARCH v2.9.0 (Rognes, Flouri, Nichols, Quince, & Mahé, 2016) implemented in primerminer using a 3% dissimilarity threshold to avoid redundancy, and then, the majority consensus sequences of the clusters were aligned. Only sequences that completely covered the primer annealing site were considered. The number of consensus sequences varied among taxa from 1 to 2075 (median 32; considered taxa listed in Supporting Information Table S1). primerminer then provided a penalty score: We used the default value (i.e., 120) to determine whether the consensus sequences should be successfully amplified (score < 120) or not (score > 120) by the primers.
2.3. In vitro selection of primers
The primers sets LepLCO/McoiR1, LepLCO/McoiR2 (LFCR), and LepLCO/MLepF1‐rev were assayed using the DNA from 16 distinct specimens of four invertebrate species (further details in Supporting Information Appendix S1). We selected the primer set that unambiguously amplified all 16 samples: LFCR.
2.4. In vivo evaluation of primers
The taxonomic coverage of MFZR, ZFZR, and LFCR was empirically evaluated through metabarcoding of 107 eDNA samples (Table 2). DNA was extracted from samples following Corse et al. (2017), and the safety measures to prevent cross contamination are described by Monti et al. (2015). Our analysis also included extraction, aerosol, PCR, and tag negative controls (respectively, Text, Tpai, TPCR, and Ttag in Corse et al., 2017) and two different mock community samples (Tpos1 and Tpos2) as positive controls (Table 3). Samples and controls were amplified by PCR in triplicate using tagged primer sets MFZR, ZFZR, and LFCR, resulting in a total of nine separate PCRs per sample/control. The tags used were 11–13 nucleotide long sequences differentiated by at least three different nucleotides (for details see: Corse et al., 2017). These tags were added on the 5′ end of the primers (12 and 8 distinct tags were used to label forward and reverse primers, respectively). Amplicons were then processed and sequenced on an Illumina MiSeq v3 platform with the paired‐end 250‐nucleotide technology. HTS data were then filtered using the variant‐centered (clustering‐free) approach detailed in Corse et al. (2017) (see Supporting Information Figure S1), which minimizes the amount of errors and false positives/negatives. Briefly, reads were merged and assigned to samples based on forward and reverse tag combinations. After trimming off tags and primers, identical reads were pooled into variants (dereplication). Based on positive and negative controls, read counts for each variant in each sample were used to define thresholds for a series of filtering steps to eliminate low‐frequency noise (LFN; sensu De Barba et al., 2014; Supporting Information Figure S1). LFN filtering steps were optimized to keep all variants within mock samples (Tpos1 and Tpos2) and to eliminate unexpected variants, that is, any variants in negative controls, variants in mock samples that are not part of the mock community, variants in eDNA samples that were unexpected given the source habitat, for example, DNA of freshwater organisms in a brackish water sample (see below). Throughout the filtering procedure, the reproducibility of variants was ensured by (a) eliminating PCR replicates that had a high Renkonen distance to other replicates within the same sample and (b) by retaining only variants that were present in at least two different PCR replicates of the same sample (for a similar approach, see Alberdi et al., 2018; Galan et al., 2018). Variants from different primer sets that were identical in their overlapping regions (~130 bp) were combined into contigs. The taxonomic assignment of each variant/contig was conducted (a) automatically using the lowest taxonomic group approach (Corse et al., 2017) and (b) manually using BOLD systems. When assignment levels were insufficient or when the two approaches conflicted, a third assignment method was conducted by building phylogenetic trees and/or considering biogeographic information (Corse et al., 2017). The combined use of these three assignment approaches led to a final taxonomic assignment for each variant/contig. After an initial round of filtering and taxonomic assignment, the variants that were unexpected given their source habitat were identified. Based on the frequency of these unexpected variants, a second round of filtering was run using adjusted LFN thresholds that maximize the elimination of unexpected variants (Supporting Information Figure S1). Finally, to standardize the evaluation of coverage and complementarity between primer sets, all validated variants/contigs were clustered into molecular operational taxonomic units (MOTUs) based on a 3% divergence threshold using complete‐linkage clustering.
Table 2.
Environmental samples analyzed in this study
| Predator | Number of samples | Environmental sample | Region, habitat | Country | Locality | Coordinates | Sampling date |
|---|---|---|---|---|---|---|---|
| Zingel asper (fish) | 46 | Feces | Palearctic, freshwater | France | Durance River | N 44°18′54″, E 5°55′31″ | October‐2014 |
| Pomatoschistus microps (fish) | 15 | Feces | Palearctic, brakish water | France | Vaccarès Lagoon | N 43°33′14″, E 4°30′21″ | May‐2012 |
| Pomatoschistus microps (fish) | 14 | Feces | Palearctic, brakish water | France | Prévost Lagoon | N 43°31′32″, E 3°54′46″ | May‐2012 |
| Unknow bat species | 1 | Feces | Palearctic, terrestrial | France | Rancogne | N 45°41′48″, E 0°24′12″ | August‐2015 |
| Myotis nattereri (bat) | 2 | Feces | Palearctic, terrestrial | France | Vilhonneur | N 45°40′50″, E 0°25′11″ | June‐2015 |
| Pipistrellus pipistrellus (bat) | 1 | Feces | Palearctic, terrestrial | France | Vilhonneur | N 45°40′50″, E 0°25′11″ | July‐2015 |
| Eptesicus serotinus (bat) | 1 | Feces | Palearctic, terrestrial | France | Vilhonneur | N 45°40′50″, E 0°25′11″ | July‐2015 |
| Miniopterus schreibersii (bat) | 4 | Feces | Palearctic, terrestrial | France | Rancogne | N 45°41′48″, E 0°24′12″ | August‐2015 |
| Barbastella barbastellus (bat) | 1 | Feces | Palearctic, terrestrial | France | Rancogne | N 45°41′48″, E 0°24′12″ | August‐2015 |
| Rhinolophus euryale (bat) | 1 | Feces | Palearctic, terrestrial | France | Rancogne | N 45°41′48″, E 0°24′12″ | August‐2015 |
| Rhinolophus ferrumequinum (bat) | 1 | Feces | Palearctic, terrestrial | France | Saint‐Bonnet‐sur‐Gironde | N 45°21′16″, W 0°39′34″ | December‐2015 |
| Myotis emarginatus (bat) | 1 | Feces | Palearctic, terrestrial | France | Saint‐Bonnet‐sur‐Gironde | N 45°21′16″, W 0°39′34″ | December‐2015 |
| Epiplatys infrafasciatus (fish) | 6 | Feces | Equatorial, freshwater | Cameroon | Lokoundje River | N 3°4′41″, E 10°24′02″ | May‐2012 |
| Unknown spider species | 3 | Spider web | Equatorial, terrestrial | French Guiana | Monkey Mountain, SW Kourou | N 5°4′25″, W 52°42′4″ | July‐2016 |
| Araneomorphae (spider) | 5 | Spider web | Equatorial, terrestrial | French Guiana | Monkey Mountain, SW Kourou | N 5°4′25″, W 52°42′4″ | July‐2016 |
| Pholcidae (spider) | 2 | Spider web | Equatorial, terrestrial | French Guiana | Monkey Mountain, SW Kourou | N 5°4′25″, W 52°42′4″ | July‐2016 |
| Micrathena schreibersi (spider) | 3 | Spider web | Equatorial, terrestrial | French Guiana | Monkey Mountain, SW Kourou | N 5°4′25″, W 52°42′4″ | July‐2016 |
Table 3.
Community composition of mock samples used as positive controls (Tpos1 and Tpos2)
| Positive controls | Species | DNA concentration (ng µl−1) | Taxonomic group | Corresponding variant/contig | |
|---|---|---|---|---|---|
| Tpos1 | Tpos2 | Ephemerella ignita | 0.2 | Ephemeroptera | contig_0019 |
| Tpos1 | Tpos2 | Hydropsyche modesta | 0.2 | Trichoptera | contig_0054 |
| Tpos1 | Oligoneuriella rhenana | 0.2 | Ephemeroptera | contig_0077 | |
| Tpos1 | Eisenia andrei | 0.2 | Oligochaeta | contig_0417 | |
| Tpos1 | Tpos2 | Chironomus riparius | 0.2 | Diptera | LFCR_006421 |
| Tpos1 | Dinocras cephalotes | 0.2 | Plecoptera | LFCR_009263 | |
| Tpos1 | Phoxinus cf. phoxinus | 0.2 | Cypriniformes | MFZR_010307 | |
| Tpos2 | Hydropsyche instabilis | 0.2 | Trichoptera | contig_0027 | |
| Tpos2 | Gammarus pulex | 0.2 | Crustacea | contig_0038 | |
| Tpos2 | Planorbarius corneus | 0.2 | Gastropoda | contig_0053 | |
| Tpos2 | Velia saulii | 0.2 | Heteroptera | contig_0055 | |
| Tpos1 | Tpos2 | Zingel asper | 0.8 | Perciformes | LFCR_005960 |
Mock samples were based on pooled DNAs, which were extracted from individual invertebrate specimens.
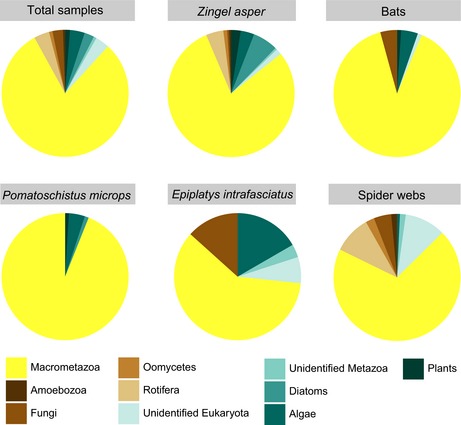
Since all predators in this study were mainly invertivores, we assumed that most macroinvertebrates (and vertebrates) constituted relevant prey and referred to them as “Macrometazoans.” Hence, items that most likely result from passive ingestion or secondary predation such as microinvertebrates (e.g., Amoebozoa, Acari, Tardigrada, Rotifera), diatoms, algae, and plants, as well as potential parasites (e.g., Acanthocephala, Nematoda), were excluded from the analyses (for a similar approach, see Hardy et al., 2017).
To evaluate the coverage and complementarity of the three primer sets, we looked at their performance in detecting different prey groups from the 107 eDNA samples at various taxonomic levels as follows: Phylum, Class, Order, Family, MOTU, and variant. The coverage of each primer set was estimated through the coverage ratio Bc (Ficetola et al., 2010). Here, Bc corresponds to the ratio between the number of taxa detected in samples by a given primer set and the total number of taxa detected by all three primer sets. The complementarity (Com) of the primer sets was assessed by dividing the number of prey items detected by one primer set only by the total number of detected taxa. In addition, we measured samples’ pairwise differences in diet composition between primer sets (Wsd for within‐sample dissimilarity) with pairwise the Bray–Curtis index (Bray & Curtis, 1957). Finally, pairwise Bray–Curtis dissimilarities between samples were also calculated (Bsd for between‐sample dissimilarities).
2.5. Diet analyses
Diet analyses were performed using the Minimal Number of Individuals (MNI; White, 1953) matrix of prey items. The MNI is a semiquantitative statistic that corresponds to the number of distinct variants and/or contigs validated in each sample (see Corse et al., 2017). We further assessed the taxonomic resolution of our diet metabarcoding dataset as a function of predator type, habitat, and geographic location by calculating the mean identification resolution index (IR; see Zarzoso‐Lacoste et al., 2016) calculated as detailed in Corse et al. (2017): A score was attributed for each variant/contig based on the taxonomic level of its final taxonomic assignation (i.e., Species = 6, Genus = 5, Family = 4, Order = 3; Class = 2, Phylum = 1, Kingdom or NA = 0), and then, a mean score among the variants was calculated for each sample.
3. RESULTS
3.1. In silico evaluation of primers
The in silico analysis revealed that theoretical amplification success was generally quite low and varied strongly across primer sets and target taxa (Figure 1). The average in silico amplification success for MFZR, ZFZR, and LFCR was 8%, 34%, and 53%, respectively, and their median primerminer penalty scores were 292,8, 372,8, and 124,7 (Supporting Information Tables S1 and S3). Hence, LFCR is expected to perform better than ZFZR and MFZR in terms of taxonomic coverage.
Figure 1.

In silico evaluation of primer set performance using primerminer. (a) Primer set performance for each taxon in pie charts (green = success; black = failure). On the right, the median number of sequences per taxon used for in silico evaluation of primer sets. (b) Distribution of the median primerminer penalty scores for each primer pair. Mean values are indicated by a triangle within boxplots
3.2. Metabarcoding data
The raw dataset was gathered from 13 distinct MiSeq runs. After preprocessing, the dataset consisted of 15.1 millions (M) of reads that correspond to the three PCR replicates of 107 eDNA samples, 39 negative controls, and 26 mock community samples. Filtering thresholds were determined by run, based on variant occurrence and frequencies (filtering parameters including LFN thresholds are reported in Supporting Information Table S4) and then applied separately for each run. After filtering, 195 variants were validated for MFZR, 237 for ZFZR, and 186 for LFCR. These corresponded to 0.4% of the variants initially identified as COI, but 73% of the reads identified as COI (11.0 M validated reads). After combining the MFZR, ZFZR, and LFCR variants, 168 contigs and 212 variants were obtained. At the end of the filtering process, all the initial eDNA samples contained by at least one variant or contig. Only one negative control (TnegPai1_DNA11; see Supporting Information Table S5) was not eliminated: Two variants assigned to a marine species (Anapagurus hyndmanni; contig_0365 and contig_0745; Supporting Information Table S5) were validated, though not found in any other environmental samples. All seven variants expected in mock samples Tpos1 and Tpos2 were retrieved. In most of the mock samples, however, one or two extra variants were also validated (contig_0238, contig_0124, MFZR_000591; Supporting Information Table S5). As these variants were absent from all other samples/controls, we suggest these arose from organisms ingested by or attached to one of the invertebrate individuals used to build the mock samples. By recovering all of the taxa of the mock communities (Tpos1 and Tpos2) in all the different runs, we assumed that we minimized random fluctuations, making our samples comparable between runs.
A total of 256 distinct Macrometazoan variants/contigs were obtained from eDNA samples (178 were detected by ZFZR, 127 by MFZR, and 163 by LFCR) corresponding to 203 Macrometazoan MOTUs after clustering (143 MOTUs were detected by ZFZR, 99 by MFZR, and 134 by LFCR; Figure 2).
Figure 2.
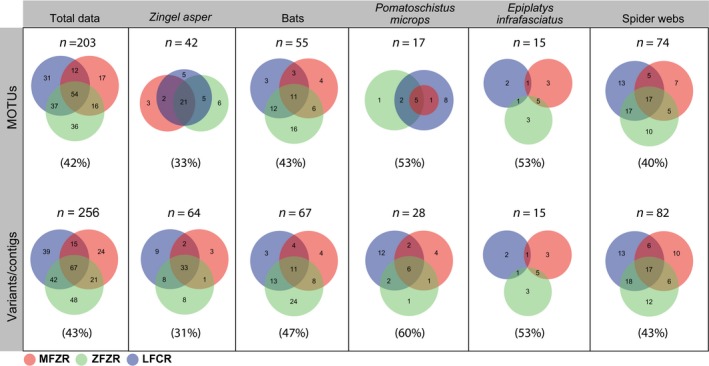
Macrometazoan MOTUs and variants obtained from environmental samples using each primer set. The mean biodiversity complementarity (Com) is in brackets
3.3. In vivo evaluation of the coverage and complementarity of primer sets
Across the whole dataset, the coverage (Bc) differed between primer sets (Kruskal–Wallis test; Χ 2 = 77.23, df = 2, p < 10−15) with LFCR performing 1.1 and 1.2 times better than ZFZR and MFZR, respectively. Mean Bc of primer sets decreased at finer taxonomic level: BcZFZR ranged from 71% to 81%, BcMFZR from 61% to 81%, and BcLFCR from 81% to 93% (Figure 3a). However, for all three primer sets, coverage varied sharply across the different predator categories. For example, ZFZR displayed the highest Bc for bat samples (82% on average) while it displayed the lowest Bc for P. microps samples (55% on average). LFCR exhibited the highest Bc in P. microps samples (98% on average). For the three primer sets, the Bc at variant level was similar to that obtained at the MOTU level. However, we observed that primer set success could vary between variants within MOTUs. Distinct variants within the same MOTU were differentially detected by primer sets for 47% of the 36 Macrometazoan MOTUs for which more than one variant was detected. For instance, in P. microps samples, five different sequences formed the MOTU cluster 119 (Mysidae) of which one variant (contig_0001) was amplified by all primer sets, one by MFZR and ZFZR primer sets only (contig_0468), and three by MFZR only (MFZR_000169, MFZR_004854, MFZR_010569; Supporting Information Table S5; Appendix 2).
Figure 3.

Coverage complementarity and dissimilarity of primer sets. (a) Coverage (Bc) of primer sets, (b) complementarity of primer sets (Com), (c) within‐sample dissimilarity (Wsd), and (d) dissimilarity between environmental samples (Bsd). Mean values are indicated by a triangle within boxplots. Dissimilarities are Bray–Curtis dissimilarities calculated from Minimal Number of Individuals (MNIs). Only Macrometazoans are considered here
The complementarity (Com) of primer sets depended both on the taxonomic level and on the predator type (Figure 3b). The mean Com index differed significantly between the taxonomic levels (Kruskal–Wallis test; Χ 2 = 88.97, df = 5, p < 10−15) and steadily increased from phylum (mean Com = 14.8%) to variant (mean Com = 33.4%; Figure 3b). Although the increase of complementarity from phylum to variant is general, its order of magnitude differed sharply across predators (e.g., Zingel asper vs. E. infrafasciatus; Figure 3b). Furthermore, even at the MOTU and variant levels, the mean Com differed significantly across predator types (Kruskal–Wallis test; Χ 2 = 26.15, df = 4, p = 0.00003 for MOTUs; Χ 2 = 25.02, df = 4, p = 0.00005 for variants).
Similarly to the coverage and to the complementarity, the within‐sample dissimilarity (Wsd) depended on the predator (Kruskal–Wallis test; Χ 2 = 205.11, df = 4, p < 10−15) with the lowest mean values observed for Z. asper (Wsd = 0.12) and P. microps (Wsd = 0.22) and the highest ones observed for E. infrafasciatus (Wsd = 0.65; Figure 3c). Furthermore, for all predators, within‐sample dissimilarity tended to increase with greater taxonomic resolution of prey, especially for spider webs, E. infrafasciatus, and bat samples. Moreover, Wsd values were very close to (or even exceeded in the case of E. infrafasciatus samples) the values of pairwise dissimilarity indexes between samples (Figure 3d).
3.4. Taxonomic identification and resolution
The calculation of IR allowed for a standardized comparison between samples concerning their taxonomic resolution. Across all environmental samples, the mean IR for Macrometazoans was 5.33 (±0.91). IR significantly differed between predator types, habitats, and geographic locations (Figure 4). The mean IR values were close to the maximal value for bats and Z. asper (IR = 6), which corresponded to an average taxonomic assignment of variants to species level. The mean IR was only slightly lower for P. microps samples, while the taxonomic resolution of Macrometazoans in the two types of equatorial samples (E. infrafasciatus feces and spider webs) were close to family level (mean IR = 4.05 ± 0.69).
Figure 4.
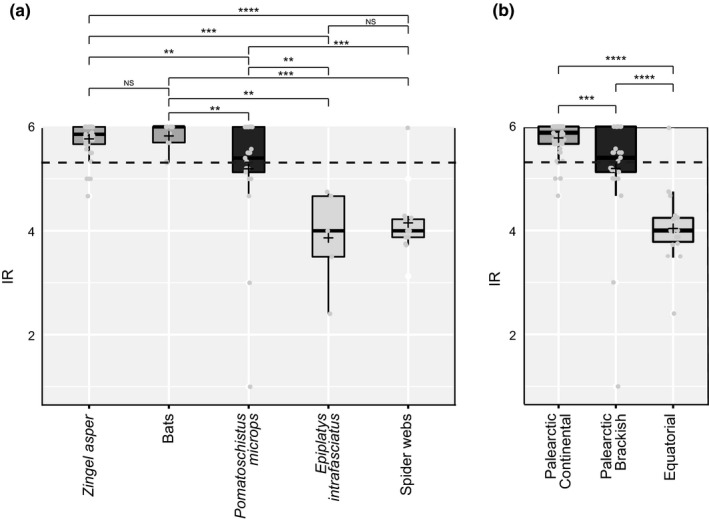
Identification resolution (IR) index of environmental samples. (a) For each predator type, and (b) by habitat/geographic location. Mean values are indicated by “+”. Significance levels of pairwise Kruskal tests are indicated on top: n.s.: nonsignificant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Only Macrometazoans are considered here
3.5. Diet results
The Macrometazoans detected in the 107 environmental samples covered a wide taxonomic array of invertebrates and included some vertebrate prey as well (Figure 5). The proportion of cumulative MNI for non‐Macrometazoans represented <20% of the total dataset and varied from ~10% (P. microps) to ~40% (E. infrafasciatus) of the total (Appendix 1).
Figure 5.
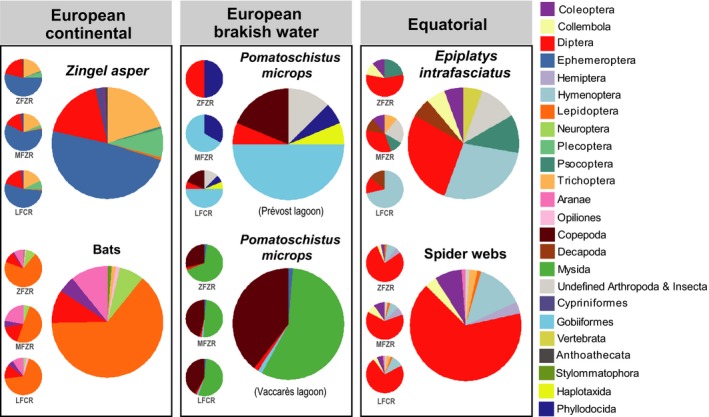
Macrometazoan taxa detected in fecal and spider web samples. The proportions of prey items are based on the cumulative Minimal Number of Individuals (MNIs). Proportions for each primer set are presented to the left of the main pie chart. Only Order‐level taxonomic assignments are presented
Predator DNA was not detected in any of the 46 Z. asper fecal samples. The mean MNI per sample was 3.50 ± 1.97 (Figure 6). Macrometazoan prey of Z. asper was aquatic invertebrates such as Ephemeropera (8 MOTUs), Trichoptera (6 MOTUs), Diptera (13 MOTUs), and Gammaridae (4 MOTUs; Supporting Information Table S5). We also detected DNA from benthic fish species (Barbus barbus and Barbatula sp.) and allochthonous prey (undetermined Nymphalidae). More than one variant was detected for ~30% of Macrometazoan MOTUs. The MOTU assigned to Baetis fuscatus was the most diverse (six distinct variants) and was also the most frequently detected prey (37% of the total MNI).
Figure 6.
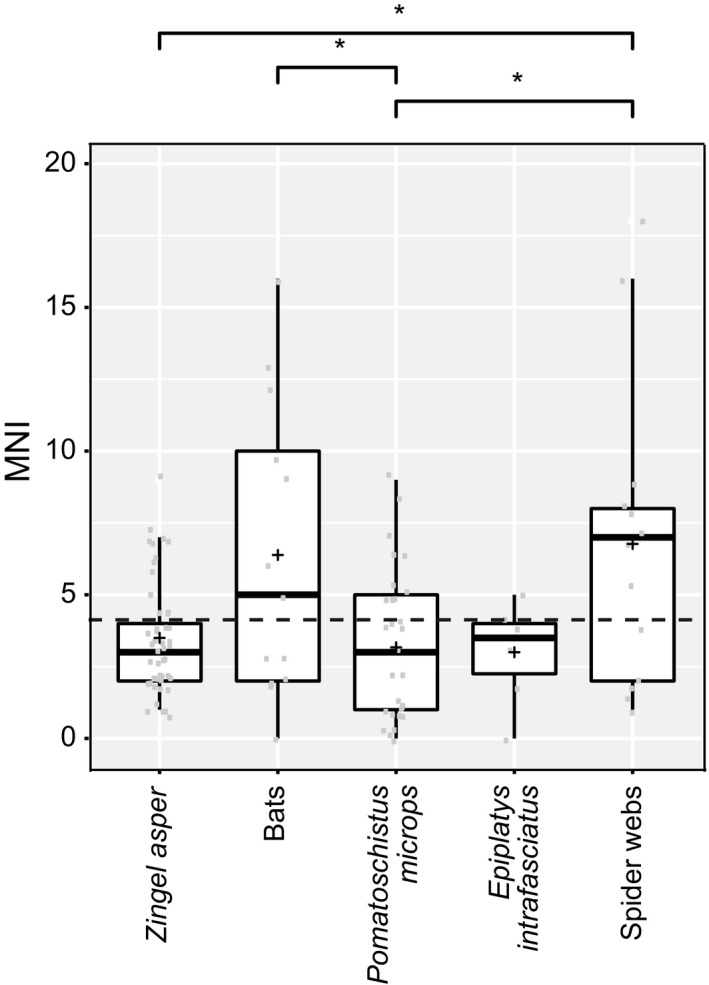
Minimal Number of Individuals (MNI) detected in fecal and spider web samples. Significance levels of pairwise Kruskal tests on top (*p < 0.05). Only Macrometazoans are considered here
Bat DNA was detected in 5 of 13 bat fecal samples. The mean MNI per sample was 6.38 ± 5.09 (Figure 6). Prey DNAs were mainly assigned to flying insects as follows: Lepidoptera (29 MOTUs), Diptera (8 MOTUs) and Neuroptera (3 MOTUs). We also detected ground‐dwelling invertebrates such as Coleoptera (4 MOTUs) and the white‐lipped snail (Cepea hortensis). Lepidoptera were the primary prey while Diptera, Araneae, and Coleoptera constituted secondary prey. This pattern is similar to the prey composition estimated by Galan et al. (2018) for a similar set of bat species.
Predator DNA was detected in all 29 P. microps fecal samples. The mean MNI per sample was 3.17 ± 2.59 (Figure 6). Our data revealed sharp differences among sampling locations in P. microps diet. The P. microps from Prévost Lagoon were mainly piscivorous, predating their syntopic sister species Pomatoschistus minutus (detected in 40% of fecal samples), whereas the P. microps from Vaccarès Lagoon ingested mainly Mysidae (5 MOTUs) and Copepoda (3 MOTUs). Only one fish variant (assigned to Pomatoschistus sp.; one occurrence in fecal sample P16) was detected in Vaccarès.
Predator DNA was detected in all six E. infrafasciatus fecal samples. The mean MNI per sample was 3.00 ± 1.78 (Figure 6). Macrometazoan DNA detected in E. infrafasciatus fecal samples was a mix of aquatic organisms such as crustacean (Atyidae) and terrestrial invertebrates such as ants (Formicidae) and springtails (Collembola), with the ratio of potential allochthonous prey being ~60%. Interestingly, a variant assigned to ray‐finned fishes (Actinopterygii) was detected in one fecal sample.
No DNA of spiders was detected from any spider web samples. The mean MNI per web sample was 6.76 ± 5.34 (Figure 6). Macrometazoans detected in spider webs were mostly flying insects with a high diversity detected for Diptera (52 MOTUs, including 24 Cecidomyiidae MOTUs) and Hymenoptera (9 MOTUs). We also detected variants assigned to Coleoptera, Trichoptera, and Collembola.
4. DISCUSSION
4.1. In vivo tests are essential to determine primer sets
The discrepancies in coverage of primer sets that we observed among in silico, in vitro, or in vivo analyses illustrates the difficulties in predicting the performance of primer sets in complex and degraded eDNA mixtures. In metabarcoding studies, in silico evaluation of PCR performance is often used as a key step for designing and/or selecting primers (e.g., Ficetola et al., 2010; Clarke et al., 2014; Kartzinel & Pringle, 2015; Elbrecht & Leese, 2017a; Elbrecht & Leese, 2017b). However, the actual amplification success of these primers on DNA mixtures or on complex environmental samples often differs substantially from in silico predictions (Alberdi et al., 2018; Corse et al., 2017; Morales & Holben, 2009). Such differences may be explained by two main causes. First, the database used for in silico analyses was built from publicly available sequences, which is likely to miss a lot of the species present in the environmental samples being analyzed. Second, in silico analyses are highly sensitive to the algorithms used for estimating primer performance and overlook other important factors influencing PCR success such as PCR inhibitors in varying amounts and competition among DNA strains for primers, often due to a difference in affinity. Our results and those from Alberdi et al. (2018) emphasize that although in silico and in vitro analyses are important in narrowing down the optimal primer sets (e.g., Elbrecht & Leese, 2017a; Piñol, Senar, & Symondson, 2019), in vivo tests are essential and the ultimate step for selecting primers that maximize biodiversity coverage and minimize PCR biases.
4.2. The use of multiple primers is key to finely describe species diversity
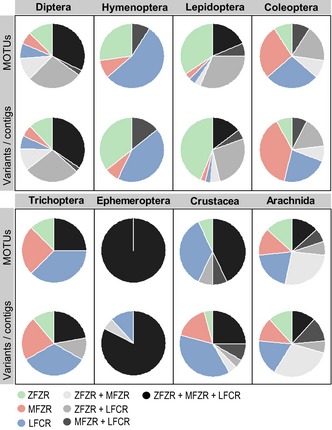
Our three primer sets were shown to perform equally well in detecting higher taxonomic ranks (phyla and classes). On the contrary, there were significant differences in coverage at finer taxonomic levels (e.g., more than 40% of the MOTUs were detected by one primer set only). Interestingly, complementarity slightly increased at the taxonomic level “variant” revealing that even variants belonging to the same MOTUs were not equally detected by all three primer sets. Consequently, the more desirable is a fine taxonomic resolution (MOTUs or variants), the more important is the use of multiple primer sets for revealing greater diversity and decrease false negatives. Furthermore, some primer sets will be more suitable than other depending on the predator. For example, ZFZR has the best coverage for Lepidoptera (Bc = 87%; Appendix 2). When sequencing the diet of bats that eat in majority Lepidoptera (Figure 5), this primer set will be better as it detects more MOTUs than the other primer sets (Figure 2). LFCR, on the other hand, has the best taxonomic coverage for Crustacea (Bc = 86%; Appendix 2) and, consequently, covered 94% of MOTUs and 79% of variants detected in P. microps feces. Overall, our results highlight the need to use multiple primers sets when one wants to tackle finely prey diversity and especially when the prey communities can be highly variable. It should be noted that some authors proposed to use one single highly degenerated primer set as an alternative (Elbrecht & Leese, 2017a, Piñol et al., 2019).
4.3. The “one‐locus‐several‐primer‐sets” strategy
The COI gene is a locus of choice when studying invertebrate diversity and is largely used for this. However, the high variability that makes the COI a powerful marker consequently makes truly universal primer sets hard to design. Yet, several recent attempts, that involved highly degenerated primers, appeared very promising (Elbrecht & Leese, 2017a; Vamos et al., 2017; Galan et al., 2018; see also Figure 1), and comparative in vivo tests (sensu Alberdi et al., 2018) would be desirable to confirm the broad applicability of these highly degenerated primers for exhaustively recovering the biodiversity in environmental samples.
Alternatively, we followed an OLSP strategy (used in Corse et al., 2017; but see also Gibson et al., 2014) targeting the COI gene using three distinct primer sets (MFZR, ZFZR and LFCR). Initially developed for studying the diet of Z. asper, we applied this strategy to other types of invertivores to fully evaluate the applicability of our approach and found that each primer set detected at least some biodiversity that was hidden from the other primer sets (Figure 2). In some cases, we observed critical differences among primer sets: The estimates of the ingested prey communities were sharply different depending on the primer set considered for E. infrafasciatus or for P. microps from Prévost Lagoon (Figure 5). Consequently, whether using one primer set or another is expected to considerably influence the qualitative interpretation of the trophic behavior of these two species. In fact, the differences in prey composition (Wsd) for a same sample obtained with different primers were very close or higher than that observed between samples (Bsd), the former representing technical noise, and the later representing a biological signal (Figure 3c,d), hence confirming the conclusions by Alberdi et al. (2018). The use of several primer sets targeting the same taxa should compensate the high between‐primer set variance in biodiversity detection and expectedly yield a more comprehensive picture of the prey diversity. In addition to maximizing coverage, the OLSP approach produces fully comparable homologous sequences that will facilitate the evaluation of biodiversity at variant level. In our case, all three primer sets target the same locus (the 5′ end of the COI gene) and reads were merged into contigs when overlapping sequences (~130 bp) were identical such that the three different datasets could be either compared together or merged into a single dataset.
The use of amplicon sequence variants instead of MOTUs in metabarcoding analyses was recently suggested to improve and refine measurements of biodiversity (Callahan, McMurdie, & Holmes, 2017; Wares & Pappalardo, 2016). In fact, variant information was used to quantify intraspecific genetic diversity (Elbrecht, Vamos, Steinke, & Leese, 2018; Pedro et al., 2017; Sigsgaard et al., 2016) and was shown to approximate taxa abundance reliably for diet analyses (Corse et al., 2017). However, only robust and reliable datasets free from possible false positives and artifacts can allow such use of variant level information. Consequently, use of variant information in quantitative or semiquantitative ways will require reliable and robust bioinformatics filtering procedures of HTS data (see Corse et al., 2017).
4.4. Public databases and the taxonomic assignment of prey
Both the confidence and the resolution of taxonomic assignment procedures are highly dependent on the completeness of reference sequence databases (e.g., Gibson et al., 2014; Porter et al., 2014; Devloo‐Delva et al., 2018). In this study, we illustrated that the accuracy of taxonomic assignment of prey depended on the environment of the predators (Figure 4b). Palearctic samples (feces from Z. asper, P. microps and bats) displayed a high taxonomic resolution of prey with most variants assigned to species. On the contrary, samples from the tropics (feces of E. infrafasciatus from Africa and spider webs from South America) displayed a much lower taxonomic resolution generally corresponding to family level (Figure 4b). In megadiverse environments and regions, such as equatorial environments, our ability to identify sequences precisely (e.g., at species level) is indeed strongly restricted by relatively incomplete public databases (e.g., Cowart et al., 2015; Beng et al., 2016; Lopes et al., 2017; Stat et al., 2017). Our results therefore illustrate the role of completeness of public databases and the importance in investing into species inventories to better understand food web and species interactions.
4.5. Insights into the diet of Pomatoschistus microps and Epiplatys infrafasciatus
To our knowledge, our OLSP approach using a variant‐based filtering procedure is the first to reveal the trophic ecology of P. microps and E. infrafasciatus with metabarcoding data.
Pomatoschistus species are valuable models for studying adaptation in coastal and estuarine environments (e.g., Pampoulie, Chauvelon, Rosecchi, Bouchereau, & Crivelli, 2001; Larmuseau, Vancampenhout, Raeymaekers, Houdt, & Volckaert, 2010) and historical processes of colonization in the Mediterranean (e.g., Tougard, Folly, & Berrebi, 2014). The trophic interaction of P. microps with other sympatric gobies, such as the sand goby P. minutus, is an important topic of investigation (e.g., Salgado, Cabral, & Costa, 2004; Leitão et al., 2006). Before our study, the knowledge on the diet of P. microps was studied using gut‐content analysis (e.g., Magnhagen & Wiederholm, 1982). The preys we detected were congruent to those previous observations (i.e., mostly zooplankton and benthic organisms) and included Copepoda, Mysidae, Chironomidae, and Annelida (Figure 5; Supporting Information Table S5). In addition, we detected other Pomatoschistus species, which would confirm interspecies predation as already reported in the Pomatoschistus genus (Hamerlynck & Cattrijsse, 1994). Interestingly, important differences were observed between the two sampled localities: P. microps from Vaccarès Lagoon fed mostly on Mysidae whereas P. microps from Prévost fed mostly on P. minutus. The heterogeneous level of interspecific predation may be explained by the scarcity of other Pomatoschistus species in Vaccarès Lagoon (Pampoulie et al., 2001).
Epiplatys species inhabit lentic rivers, usually near the surface under the leaves of plants (e.g., Romand & Morgalet, 1983). Diet of Epiplatys are mainly composed of aquatic insects (Guma'a, 1982; Ndome & Victor, 2002) although terrestrial organisms such as Formicidae may also represent important preys (Romand & Morgalet, 1983). This was the case here, with ~60% of Epiplatys prey being composed of terrestrial organisms such as Formicidae, Collembola, and Psocoptera and only 15% of preys being composed of unambiguously aquatic organisms (Figure 5; Supporting Information Table S5). Collembola may constitute allochthonous prey fallen from riparian vegetation or, given their small size (2–3 mm), secondary prey for E. infrafasciatus (i.e., ingested by their primary prey such as Formicidae). These results are consistent with E. infrafasciatus top‐mouth position that favors capture of prey at the water surface, notably allochthonous ones. We also detected ~25% of algae and fungi in E. infrafasciatus feces, suggesting that this species could also forage aufwuchs communities as reported for E. senegalensis (Ndome & Victor, 2002). Although only based on six fecal samples, our results suggest that E. infrafasciatus exhibits quite versatile foraging behaviors, exploiting both pelagic and benthic habitats and feeding on both allochthonous and aquatic prey.
4.6. The capture of biodiversity by spider webs
Spider webs represent a potential noninvasive source of DNA for conservation, ecology, and management studies (Blake, McKeown, Bushell, & Shaw, 2016; Xu, Yen, Bowman, & Turner, 2015). They are able to trap a part of the local arthropod biodiversity present in natural (even pristine), agricultural, or urban habitats and may serve as “biodiversity capsules” (sensu Boyer, Cruickshank, & Wratten, 2015), especially for flying insects. To date, DNA extracted from spider webs enabled the detection of both the predator and its prey using diagnostic PCR under controlled conditions (Blake et al., 2016; Xu et al., 2015). We here present the first attempt to analyze the biodiversity captured by natural spider webs using metabarcoding. Among our environmental samples, spider webs from the equatorial forest of French Guiana displayed the highest biodiversity by far displaying the highest mean MNI (Figure 6) and the highest MOTU diversity (Supporting Information Tables S6). The main taxa detected were flying insects, especially Diptera (52 MOTUs) and Hymenoptera (9 MOTUs), but also some Coleoptera, Trichoptera, Lepidoptera, and Hemiptera. However, contrary to previous studies using diagnostic PCR (Blake et al., 2016; Xu et al., 2015), we were unable to detect the DNA of the spiders that spun the webs even though our primer sets were able to detect a non‐negligible part of Arachnida in bat fecal samples (Figure 5). Regardless, we illustrate that spider webs do act as natural traps of biodiversity, and we confirmed that they are a promising tool for invertebrate diversity assessments when combined with the power of HTS (as suggested by Xu et al., 2015).
5. CONCLUSIONS
We demonstrated the lack of predictability of both the coverage and complementarity of individual primer sets. Although in silico and in vitro (i.e., based on DNA extracted from isolated organisms) evaluations are useful to narrow down a subset of primer sets for metabarcoding studies (e.g., Elbrecht & Leese, 2017a; Piñol et al., 2019), in vivo evaluation of primers based on a subset of environmental samples is critically informative for minimizing false negatives before conducting larger scale analyses. Furthermore, we formalized the “one‐locus‐several‐primers” (OLSP) strategy that directly addresses primer set coverage biases to minimize false negatives. This strategy produces largely overlapping and comparable sequences, which cannot be achieved when targeting different loci, and facilitates the use of genetic diversity information contained within metabarcoding datasets. However, we emphasize the importance of stringent variant‐based filtering procedures to validate HTS data (Callahan et al., 2016; Corse et al., 2017) before genetic information of metabarcoding datasets can be used to estimate (semi‐)quantitative diversity indices. Our workflow, combining the OLSP strategy and stringent variant‐based filtering, can be easily adapted and extended to other loci and other applications for studying biodiversity through metabarcoding.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
E.C., V.D., C.T., and C.C.Y.X. conceived and designed the study. E.C., V.D., G.A., F.D.M.M., C.F.B.B., J.F.A., R.C., L.Z., D.D., and C.T. collected the samples. G.A. identified invertebrates morphologically. E.C., V.D., and E.M. developed the method. E.C. and C.T. performed the experiments. E.C., E.M., and V.D. analyzed the data. E.C., V.D., and E.M. wrote the paper. All authors drafted the paper.
Supporting information
ACKNOWLEDGMENTS
We warmly thank the staff from the French Agence Française pour la Biodiversité (AFB) and from Aix‐Marseille Université who participated in the fieldwork. We are very grateful to Maxime Galan and Nathalie Charbonnel for kindly sharing bat fecal samples, to Hélène Vignes for her very valuable help in the processing of the MiSeq runs, and to Rémi Grenier for help with the in vitro tests. This work is part of the French Plan National d'Action en faveur de l'apron du Rhône (2012–2016) coordinated by the Direction Régional pour l'Environnement, l'Aménagement et le Logement d'Auvergne‐Rhône‐Alpes and managed by the Conservatoire d'Espaces Naturels Rhône‐Alpes. E.C. was supported by a postdoctoral grant from Electricité de France (EDF) and AFB. This study was funded by the Syndicat Mixte d'Aménagement du Val Durance (SMAVD), the Agence de l'Eau Rhône‐Méditerranée‐Corse (AERMC), and the Conseil Régional de Provence‐Alpes‐Côte d'Azur. D.D. and L.Z. were supported by the LabEx CEBA through the Investissement d'Avenir grant of the Agence Nationale de la Recherche (ANR‐10‐LABX‐25‐01), and by the French Laboratory of Excellence project “TULIP” (ANR‐10‐LABX‐41; ANR‐11‐IDEX‐0002‐02) for data collection. The LabEx CEBA also supported the DNA extractions from spider webs. D.D. was further supported by the People Program (Marie Curie Actions) of the European Union's Seventh Framework Program (FP7/2007‐2013) under REA grant agreement PCOFUND GA‐2013‐609102, through the PRESTIGE program coordinated by Campus France. This work was conducted in accordance with permits from the French Direction Départementale des Territoires des Hautes‐Alpes (DDT 05). Data used in this work were produced through the molecular facilities of LabEx CeMEB (Montpellier), CIRAD (Montpellier), and SCBM (IMBE, Marseille).
APPENDIX 1. Proportion of Macrometazoan and non‐Macrometazoan variants in fecal and spider web samples
The proportion of prey items are based on the cumulative Minimal Number of Individuals (MNIs).
APPENDIX 2. MOTUs and variants coverage of primer sets in some Arthropoda taxa.
Corse E, Tougard C, Archambaud‐Suard G, et al. One‐locus‐several‐primers: A strategy to improve the taxonomic and haplotypic coverage in diet metabarcoding studies. Ecol Evol. 2019;9:4603–4620. 10.1002/ece3.5063
Funding information
Electricité de France (EDF); Agence Française pour la Biodiversité (AFB); Syndicat Mixte d'Aménagement du Val Durance (SMAVD); Agence de l'Eau Rhône‐Méditerranée‐Corse (AERMC); Conseil Régional de Provence‐Alpes‐Côte d'Azur; Agence Nationale de la Recherche (ANR).
Contributor Information
Emmanuel Corse, Email: emmanuel.corse@gmail.com.
Vincent Dubut, Email: vincent.dubut@imbe.fr.
DATA ACCESSIBILITY
Supplementary data deposited in Dryad (https://doi.org/10.5061/dryad.2ck7120) include the unfiltered HTS data from the MiSeq runs used in this paper, as well as the sequence alignment generated with primerminer for in silico evaluation of metabarcoding primer sets.
REFERENCES
- Aizpurua, O. , Budinski, I. , Georgiakakis, P. , Gopalakrishnan, S. , Ibañez, C. , Mata, V. , … Alberdi, A. (2018). Agriculture shapes the trophic niche of a bat preying on multiple pest arthropods across Europe: Evidence from DNA metabarcoding. Molecular Ecology, 27, 815–827. 10.1111/mec.14474 [DOI] [PubMed] [Google Scholar]
- Alberdi, A. , Aizpurua, O. , Bohmann, K. , Gopalakrishnan, S. , Lynggaard, C. , Nielsen, M. , & Gilbert, M. T. P. (2019). Promises and pitfalls of using high‐throughput sequencing for diet analysis. Molecular Ecology Resources, 19(2), 327–348. 10.1111/1755-0998.12960 [DOI] [PubMed] [Google Scholar]
- Alberdi, A. , Aizpurua, O. , Gilbert, M. T. P. , & Bohmann, K. (2018). Scrutinizing key steps for reliable metabarcoding of environmental samples. Methods in Ecology and Evolution, 9, 134–147. 10.1111/2041-210X.12849 [DOI] [Google Scholar]
- Beng, K. C. , Tomlinson, K. W. , Shen, X. H. , Surget‐Groba, Y. , Hughes, A. C. , Corlett, R. T. , & Slik, J. W. F. (2016). The utility of DNA metabarcoding for studying the response of arthropod diversity and composition to land‐use change in the tropics. Scientific Reports, 6, 4603–13. 10.1038/srep24965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake, M. , McKeown, N. J. , Bushell, M. L. , & Shaw, P. W. (2016). DNA extraction from spider webs. Conservation Genetics Resources, 8, 219–221. 10.1007/s12686-016-0537-8 [DOI] [Google Scholar]
- Boyer, S. , Cruickshank, R. H. , & Wratten, S. D. (2015). Faeces of generalist predators as ‘biodiversity capsules’: A new tool for biodiversity assessment in remote and inaccessible habitats. Food Webs, 3, 4603–6. 10.1016/j.fooweb.2015.02.001 [DOI] [Google Scholar]
- Brandon‐Mong, G. J. , Gan, H. M. , Sing, K. W. , Lee, P. S. , Lim, P. E. , & Wilson, J. J. (2015). DNA metabarcoding of insects and allies: An evaluation of primers and pipelines. Bulletin of Entomological Research, 105, 717–727. 10.1017/S0007485315000681 [DOI] [PubMed] [Google Scholar]
- Bray, J. R. , & Curtis, J. T. (1957). An ordination of upland forest communities of southern Wisconsin. Ecological Monographs, 27, 325–349. [Google Scholar]
- Callahan, B. J. , McMurdie, P. J. , & Holmes, S. P. (2017). Exact sequence variants should replace operational taxonomic units in marker‐gene data analysis. The ISME Journal, 11(12), 2639–2643. 10.1038/ismej.2017.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan, B. J. , McMurdie, P. J. , Rosen, M. J. , Han, A. W. , Johnson, A. J. A. , & Holmes, S. P. (2016). DADA2: High‐resolution sample inference from Illumina amplicon data. Nature Methods, 13, 581–583. 10.1038/nmeth.3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chivers, L. S. , Lundy, M. G. , Colhoun, K. , Newton, S. F. , Houghton, J. D. , & Reid, N. (2013). Identifying optimal feeding habitat and proposed Marine Protected Areas (pMPAs) for the black‐legged kittiwake (Rissa tridactyla) suggests a need for complementary management approaches. Biological Conservation, 164, 73–81. 10.1016/j.biocon.2013.04.022 [DOI] [Google Scholar]
- Clare, E. L. (2014). Molecular detection of trophic interactions: Emerging trends, distinct advantages, significant considerations and conservation applications. Evolutionary Applications, 7, 1144–1157. 10.1111/eva.12225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, L. J. , Soubrier, J. , Weyrich, L. S. , & Cooper, A. (2014). Environmental metabarcodes for insects: In silico PCR reveals potential for taxonomic bias. Molecular Ecology Resources, 14, 1160–1170. [DOI] [PubMed] [Google Scholar]
- Corse, E. , Costedoat, C. , Chappaz, R. , Pech, N. , Martin, J.‐F. , & Gilles, A. (2010). A PCR‐based method for diet analysis in freshwater organisms using 18S rDNA barcoding on faeces. Molecular Ecology Resources, 10, 96–108. 10.1111/j.1755-0998.2009.02795.x [DOI] [PubMed] [Google Scholar]
- Corse, E. , Meglécz, E. , Archambaud, G. , Ardisson, M. , Martin, J.‐F. , Tougard, C. , … Dubut, V. (2017). A from‐benchtop‐to‐desktop workflow for validating HTS data and for taxonomic identification in diet metabarcoding studies. Molecular Ecology Resources, 17, e146–e159. 10.1111/1755-0998.12703 [DOI] [PubMed] [Google Scholar]
- Cowart, D. A. , Pinheiro, M. , Mouchel, O. , Maguer, M. , Grall, J. , Miné, J. , & Arnaud‐Haond, S. (2015). Metabarcoding is powerful yet still blind: A comparative analysis of morphological and molecular surveys of seagrass communities. PLoS ONE, 10, e0117562 10.1371/journal.pone.0117562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Barba, M. , Miquel, C. , Boyer, F. , Mercier, C. , Rioux, D. , Coissac, E. , & Taberlet, P. (2014). DNA metabarcoding multiplexing and validation of data accuracy for diet assessment: Application to omnivorous diet. Molecular Ecology Resources, 14, 306–323. 10.1111/1755-0998.12188 [DOI] [PubMed] [Google Scholar]
- Deagle, B. E. , Jarman, S. N. , Coissac, E. , Pompanon, F. , & Taberlet, P. (2014). DNA metabarcoding and the cytochrome c oxidase subunit I marker: Not a perfect match. Biology Letters, 10, 20140562 10.1098/rsbl.2014.0562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devloo‐Delva, F. , Huerlimann, R. , Chua, G. , Matley, J. K. , Heupel, M. R. , Simpfendorfer, C. A. , & Maes, G. E. (2018). How does marker choice affect your diet analysis: Comparing genetic markers and digestion levels for diet metabarcoding of tropical‐reef piscivores. Marine and Freshwater Research, 70, 8–4620. 10.1071/MF17209 [DOI] [Google Scholar]
- Elbrecht, V. , & Leese, F. (2017a). Validation and development of COI metabarcoding primers for freshwater macroinvertebrate bioassessment. Frontiers in Environmental Science, 5, 11. [Google Scholar]
- Elbrecht, V. , & Leese, F. (2017b). primerminer: An R package for development and in silico validation of DNA metabarcoding primers. Methods in Ecology and Evolution, 8, 622–626. [Google Scholar]
- Elbrecht, V. , Vamos, E. E. , Steinke, D. , & Leese, F. (2018). Estimating intraspecific genetic diversity from community DNA metabarcoding data. PeerJ, 6, e4644 10.7717/peerj.4644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, D. M. , Kitson, J. J. , Lunt, D. H. , Straw, N. A. , & Pocock, M. J. (2016). Merging DNA metabarcoding and ecological network analysis to understand and build resilient terrestrial ecosystems. Functional Ecology, 30, 1904–1916. [Google Scholar]
- Ficetola, G. , Coissac, E. , Zundel, S. , Riaz, T. , Shehzad, W. , Bessière, J. , … Pompanon, F. (2010). An In silico approach for the evaluation of DNA barcodes. BMC Genomics, 11, 434 10.1186/1471-2164-11-434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficetola, G. F. , Pansu, J. , Bonin, A. , Coissac, E. , Giguet‐Covex, C. , De Barba, M. , … Taberlet, P. (2015). Replication levels, false presences and the estimation of the presence/absence from eDNA metabarcoding data. Molecular Ecology Resources, 15, 543–556. 10.1111/1755-0998.12338 [DOI] [PubMed] [Google Scholar]
- Galan, M. , Pons, J. B. , Tournayre, O. , Pierre, E. , Leuchtmann, M. , Pontier, D. , & Charbonnel, N. (2018). Metabarcoding for the parallel identification of several hundred predators and their preys: Application to bat species diet analysis. Molecular Ecology Resources, 18, 474–489. [DOI] [PubMed] [Google Scholar]
- Gibson, J. , Shokralla, S. , Porter, T. M. , King, I. , van Konynenburg, S. , Janzen, D. H. , … Hajibabaei, M. (2014). Simultaneous assessment of the macrobiome and microbiome in a bulk sample of tropical arthropods through DNA metasystematics. Proceedings of the National Academy of Sciences of the United States of America, 111(22), 8007–8012. 10.1073/pnas.1406468111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guma'a, S. A. (1982). On the biology of female Epiplatys bifasciatus (Cyprinodontidae) from southern Sudan. Hydrobiologia, 89, 285–300. 10.1007/BF00005716 [DOI] [Google Scholar]
- Hamerlynck, O. , & Cattrijsse, A. (1994). The food of Pomatoschistus minutus (Pisces, Gobiidae) in Belgian coastal waters, and a comparison with the food of its potential competitor P. lozanoi . Journal of Fish Biology, 44, 753–771. 10.1111/j.1095-8649.1994.tb01253.x [DOI] [Google Scholar]
- Hardy, N. , Berry, T. , Kelaher, B. P. , Goldsworthy, S. D. , Bunce, M. , Coleman, M. A. , … Figueira, W. (2017). Assessing the trophic ecology of top predators across a recolonisation frontier using DNA metabarcoding of diets. Marine Ecology Progress Series, 573, 237–254. 10.3354/meps12165 [DOI] [Google Scholar]
- Kartzinel, T. R. , Chen, P. A. , Coverdale, T. C. , Erickson, D. L. , Kress, W. J. , Kuzmina, M. L. , … Pringle, R. M. (2015). DNA metabarcoding illuminates dietary niche partitioning by African large herbivores. Proceedings of the National Academy of Sciences of the United States of America, 112(26), 8019–8024. 10.1073/pnas.1503283112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kartzinel, T. R. , & Pringle, R. M. (2015). Molecular detection of invertebrate prey in vertebrate diets: Trophic ecology of Caribbean island lizards. Molecular Ecology Resources, 15, 903–914. 10.1111/1755-0998.12366 [DOI] [PubMed] [Google Scholar]
- Kaunisto, K. M. , Roslin, T. , Sääksjärvi, I. E. , & Vesterinen, E. J. (2017). Pellets of proof: First glimpse of the dietary composition of adult odonates as revealed by metabarcoding of feces. Ecology and Evolution, 7, 8588–8598. 10.1002/ece3.3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larmuseau, M. H. , Vancampenhout, K. I. M. , Raeymaekers, J. A. , Van houdt, J. K. , & Volckaert, F. A. (2010). Differential modes of selection on the rhodopsin gene in coastal Baltic and North Sea populations of the sand goby, Pomatoschistus minutus . Molecular Ecology, 19, 2256–2268. 10.1111/j.1365-294X.2010.04643.x [DOI] [PubMed] [Google Scholar]
- Leitão, R. , Martinho, F. , Neto, J. M. , Cabral, H. , Marques, J. C. , & Pardal, M. A. (2006). Feeding ecology, population structure and distribution of Pomatoschistus microps (Krøyer, 1838) and Pomatoschistus minutus (Pallas, 1770) in a temperate estuary, Portugal. Estuarine, Coastal and Shelf Science, 66, 231–239. 10.1016/j.ecss.2005.08.012 [DOI] [Google Scholar]
- Leray, M. , Yang, J. Y. , Meyer, C. P. , Mills, S. C. , Agudelo, N. , Ranwez, V. , … Machida, R. J. (2013). A new versatile primer set targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: Application for characterizing coral reef fish gut contents. Frontiers in Zoology, 10, 34 10.1186/1742-9994-10-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llaneza, L. , & López‐Bao, J. V. (2015). Indirect effects of changes in environmental and agricultural policies on the diet of wolves. European Journal of Wildlife Research, 61, 895–902. 10.1007/s10344-015-0966-9 [DOI] [Google Scholar]
- Lopes, C. M. , Sasso, T. , Valentini, A. , Dejean, T. , Martins, M. , Zamudio, K. R. , & Haddad, C. F. (2017). eDNA metabarcoding: A promising method for anuran surveys in highly diverse tropical forests. Molecular Ecology Resources, 17, 904–914. 10.1111/1755-0998.12643 [DOI] [PubMed] [Google Scholar]
- Magnhagen, C. , & Wiederholm, A. M. (1982). Food selectivity versus prey availability: A study using the marine fish Pomatoschistus microps . Oecologia, 55, 311–315. 10.1007/BF00376917 [DOI] [PubMed] [Google Scholar]
- McCracken, G. F. , Westbrook, J. K. , Brown, V. A. , Eldridge, M. , Federico, P. , & Kunz, T. H. (2012). Bats track and exploit changes in insect pest populations. PLoS ONE, 7, e43839 10.1371/journal.pone.0043839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes, J. C. , Alderman, R. , Lea, M.‐A. , Raymond, B. , Deagle, B. E. , Phillips, R. A. , … Jarman, S. N. (2017). High occurrence of jellyfish predation by black‐browed and Campbell albatross identified by DNA metabarcoding. Molecular Ecology, 26, 4831–4845. 10.1111/mec.14245 [DOI] [PubMed] [Google Scholar]
- Meusnier, I. , Singer, G. A. , Landry, J. F. , Hickey, D. A. , Hebert, P. D. , & Hajibabaei, M. (2008). A universal DNA mini‐barcode for biodiversity analysis. BMC Genomics, 9, 214 10.1186/1471-2164-9-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollot, G. , Duyck, P. F. , Lefeuvre, P. , Lescourret, F. , Martin, J.‐F. , Piry, S. , … Tixier, P. (2014). Cover cropping alters the diet of arthropods in a banana plantation: A metabarcoding approach. PLoS ONE, 9, e93740 10.1371/journal.pone.0093740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti, F. , Duriez, O. , Arnal, V. , Dominici, J.‐M. , Sforzi, A. , Fusani, L. , … Montgelard, C. (2015). Being cosmopolitan: Evolutionary history and phylogeography of a specialized raptor, the Osprey Pandion haliaetus . BMC Evolutionary Biology, 15, 255 10.1186/s12862-015-0535-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales, S. E. , & Holben, W. E. (2009). Empirical testing of 16S rRNA gene PCR primer pairs reveals variance in target specificity and efficacy not suggested by in silico analysis. Applied and Environmental Microbiology, 75, 2677–2683. 10.1128/AEM.02166-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motte, G. , & Libois, R. (2002). Conservation of the lesser horseshoe bat (Rhinolophus hipposideros Bechstein, 1800) (Mammalia: Chiroptera) in Belgium. A case study of feeding habitat requirements. Belgian Journal of Zoology, 132, 47–52. [Google Scholar]
- Ndome, C. B. , & Victor, R. (2002). Food and feeding habits of Epiplatys senegalensis (Pisces: Cyprinodontiformes; Cyprinodontidae) in a back water pond in Benin City, Southern Nigeria. West African Journal of Applied Ecology, 3, 105–117. [Google Scholar]
- Nielsen, J. M. , Clare, E. L. , Hayden, B. , Brett, M. T. , & Kratina, P. (2018). Diet tracing in ecology: Method comparison and selection. Methods in Ecology and Evolution, 9, 278–291. 10.1111/2041-210X.12869 [DOI] [Google Scholar]
- Olmos‐Pérez, L. , Roura, Á. , Pierce, G. J. , Boyer, S. , & González, Á. F. (2017). Diet composition and variability of wild Octopus vulgaris and Alloteuthis media (Cephalopoda) paralarvae: A metagenomic approach. Frontiers in Physiology, 8, 321 10.3389/fphys.2017.00321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pampoulie, C. , Chauvelon, P. , Rosecchi, E. , Bouchereau, J. L. , & Crivelli, A. J. (2001). Environmental factors influencing the gobiid assemblage of a Mediterranean Lagoon: Empirical evidence from a long‐term study. Hydrobiologia, 445, 175–181. [Google Scholar]
- Pedro, P. M. , Piper, R. , Bazilli Neto, P. , Cullen, L. , Dropa, M. , Lorencao, R. , … Turati, D. T. (2017). Metabarcoding analyses enable differentiation of both interspecific assemblages and intraspecific divergence in habitats with differing management practices. Environmental Entomology, 46, 1381–1389. 10.1093/ee/nvx166 [DOI] [PubMed] [Google Scholar]
- Piñol, J. , Mir, G. , Gomez‐Polo, P. , & Agustí, N. (2014). Universal and blocking primer mismatches limit the use of high‐throughput DNA sequencing for the quantitative metabarcoding of arthropods. Molecular Ecology Resources, 15, 4603–12. [DOI] [PubMed] [Google Scholar]
- Piñol, J. , Senar, M. A. , & Symondson, W. O. (2019). The choice of universal primers and the characteristics of the species mixture determine when DNA metabarcoding can be quantitative. Molecular Ecology, 28, 407–419. [DOI] [PubMed] [Google Scholar]
- Pompanon, F. , Deagle, B. E. , Symondson, W. O. , Brown, D. S. , Jarman, S. N. , & Taberlet, P. (2012). Who is eating what: Diet assessment using next generation sequencing. Molecular Ecology, 21, 1931–1950. 10.1111/j.1365-294X.2011.05403.x [DOI] [PubMed] [Google Scholar]
- Porter, T. M. , Gibson, J. F. , Shokralla, S. , Baird, D. J. , Golding, G. B. , & Hajibabaei, M. (2014). Rapid and accurate taxonomic classification of insect (class Insecta) cytochrome c oxidase subunit 1 (COI) DNA barcode sequences using a naïve Bayesian classifier. Molecular Ecology Resources, 14, 929–942. [Google Scholar]
- Quéméré, E. , Hibert, F. , Miquel, C. , Lhuillier, E. , Rasolondraibe, E. , Champeau, J. , … Chikhi, L. (2013). A DNA metabarcoding study of a primate dietary diversity and plasticity across its entire fragmented range. PLoS ONE, 8, e58971 10.1371/journal.pone.0058971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnasingham, S. , & Hebert, P. D. N. (2007). BOLD: The barcode of life data system (www.barcodinglife.org). Molecular Ecology Notes, 7, 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennstam Rubbmark, O. , Sint, D. , Horngacher, N. , & Traugott, M. (2018). A broadly applicable COI primer pair and an efficient single‐tube amplicon library preparation protocol for metabarcoding. Ecology and Evolution, 8, 12335–12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rognes, T. , Flouri, T. , Nichols, B. , Quince, C. , & Mahé, F. (2016). VSEARCH: A versatile open source tool for metagenomics. PeerJ, 4, e2584 10.7717/peerj.2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romand, R. , & Morgalet, P. (1983). Ecology and feeding habits of various Cyprinodontidae from Guinea. Revue De Zoologie Africaine, 97, 288–299. [Google Scholar]
- Roslin, T. , & Majaneva, S. (2016). The use of DNA barcodes in food web construction—terrestrial and aquatic ecologists unite!. Genome, 59, 603–628. [DOI] [PubMed] [Google Scholar]
- Salgado, J. P. , Cabral, H. N. , & Costa, M. J. (2004). Feeding ecology of the gobies Pomatoschistus minutus (Pallas, 1770) and Pomatoschistus microps (Krøyer, 1838) in the upper Tagus estuary, Portugal. Scientia Marina, 68, 425–434. [Google Scholar]
- Sánchez‐Hernández, J. (2014). Age‐related differences in prey‐handling efficiency and feeding habitat utilization of Squalius carolitertii (Cyprinidae) according to prey trait analysis. Biologia, 69, 696–704. 10.2478/s11756-014-0347-y [DOI] [Google Scholar]
- Sigsgaard, E. E. , Nielsen, I. B. , Bach, S. S. , Lorenzen, E. D. , Robinson, D. P. , Knudsen, S. W. , … Thomsen, P. F. (2016). Population characteristics of a large whale shark aggregation inferred from seawater environmental DNA. Nature Ecology and Evolution, 1, 0004 10.1038/s41559-016-0004 [DOI] [PubMed] [Google Scholar]
- Stat, M. , Huggett, M. J. , Bernasconi, R. , DiBattista, J. D. , Berry, T. E. , Newman, S. J. , … Bunce, M. (2017). Ecosystem biomonitoring with eDNA: Metabarcoding across the tree of life in a tropical marine environment. Scientific Reports, 7, 12240 10.1038/s41598-017-12501-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taberlet, P. , Bonin, A. , Zinger, L. , & Coissac, E. (2018). Environmental DNA: For biodiversity research and monitoring. Kettering, UK: Oxford University Press. [Google Scholar]
- Taberlet, P. , Coissac, E. , Pompanon, F. , Brochmann, C. , & Willerslev, E. (2012). Towards next‐generation biodiversity assessment using DNA metabarcoding. Molecular Ecology, 21, 2045–2050. 10.1111/j.1365-294X.2012.05470.x [DOI] [PubMed] [Google Scholar]
- Tougard, C. , Folly, J. , & Berrebi, P. (2014). New light on the evolutionary history of the common goby (Pomatoschistus microps) with an emphasis on colonization processes in the Mediterranean Sea. PLoS ONE, 9, e91576 10.1371/journal.pone.0091576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevelline, B. K. , Nuttle, T. , Hoenig, B. D. , Brouwer, N. L. , Porter, B. A. , & Latta, S. C. (2018). DNA metabarcoding of nestling feces reveals provisioning of aquatic prey and resource partitioning among Neotropical migratory songbirds in a riparian habitat. Oecologia, 187, 85–98. 10.1007/s00442-018-4136-0 [DOI] [PubMed] [Google Scholar]
- Vamos, E. E. , Elbrecht, V. , & Leese, F. (2017). Short COI markers for freshwater macroinvertebrate metabarcoding. Metabarcoding and Metagenomics, 1, e14625 10.3897/mbmg.1.14625 [DOI] [Google Scholar]
- Wares, J. P. , & Pappalardo, P. (2016). Can theory improve the scope of quantitative metazoan metabarcoding? Diversity, 8, 4603. [Google Scholar]
- White, T. E. (1953). A method of calculating the dietary percentage of various food animals utilized by Aboriginal peoples. American Antiquity, 18, 396–398. 10.2307/277116 [DOI] [Google Scholar]
- Xu, C. C. Y. , Yen, I. J. , Bowman, D. , & Turner, C. R. (2015). Spider web DNA: A new spin on noninvasive genetics of predator and prey. PLoS ONE, 10, e0142503 10.1371/journal.pone.0142503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarzoso‐Lacoste, D. , Bonnaud, E. , Corse, E. , Gilles, A. , Meglecz, E. , Costedoat, C. , … Vidal, E. (2016). Improving morphological diet studies with molecular ecology: An application for invasive mammal predation on island birds. Biological Conservation, 193, 134–142. 10.1016/j.biocon.2015.11.018 [DOI] [Google Scholar]
- Zeale, M. R. K. , Butlin, R. K. , Barker, G. L. A. , Lees, D. C. , & Jones, G. (2011). Taxon‐specific PCR for DNA barcoding arthropod prey in bat faeces. Molecular Ecology Resources, 11, 236–244. 10.1111/j.1755-0998.2010.02920.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Supplementary data deposited in Dryad (https://doi.org/10.5061/dryad.2ck7120) include the unfiltered HTS data from the MiSeq runs used in this paper, as well as the sequence alignment generated with primerminer for in silico evaluation of metabarcoding primer sets.