

Abstract
Antibiotic resistance of bacterial pathogens poses an increasing threat to the wellbeing of our society and urgently calls for new strategies for infection diagnosis and antibiotic discovery. The antibiotic resistance problem to a large extent arises from extensive use of broad-spectrum antibiotics. Ideally, for the treatment of infection, one would like to use a narrow-spectrum antibiotic that specifically targets and kills the disease causing strain. This is particularly important considering the commensal bacterial species that are beneficial and sometimes even critical to the health of a human being. In this contribution, we describe a phage display platform that enables rapid identification of peptide probes for specific bacterial strains. The phage library described herein incorporates 2-acetylphenylboronic acid (APBA) moieties to elicit dynamic covalent binding to the bacterial cell surface. Screening of the library against live bacterial cells yields submicromolar and highly specific binders for clinical strains of Staphylococcus aureus and Acinetobacter baumannii that display antibiotic resistance. We further show that the identified peptide probes can be readily converted to bactericidal agents that deliver generic toxins to kill the targeted bacterial strain with high specificity. The phage display platform described here is applicable to a wide array of bacterial strains, paving the way to facile diagnosis and development of strain-specific antibiotics.
Graphical Abstract
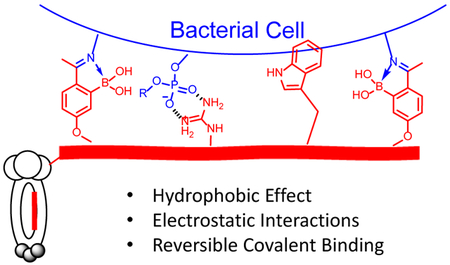
Introduction
Antibiotic resistant bacterial pathogens have become a global threat to public health. Diverse mechanisms of resistance of essentially all current antibiotics have been elucidated in recent years and are continuously being discovered.1 According to the Center for Disease Control, over 2 million antibiotic-resistant infections are reported each year leading to approximately 23,000 deaths in the United States alone. Although resistance occurs naturally, misuse and overuse of broad-spectrum antibiotics in humans and animals accelerates the process. There is thus an urgent need for a change in the way antibiotics are utilized along with the development of novel antimicrobial agents. Ideally, to avoid unintentional elicitation of antibiotic resistance, it would be advantageous to replace widely used broad-spectrum antibiotics with narrow-spectrum antibiotics.2 Furthermore, the use of broad-spectrum antibiotics can cause undesirable disruptions to the microbiota, which plays critical roles in various aspects of human biology.3
The development of narrow-spectrum antibiotics requires novel strategies that enable specific targeting of a bacterial strain of interest. There are only a handful of examples in literature describing targeted antibiotics, including those utilizing antibodies as targeting motifs.4,5 While effective, the development of an antibody drug is nontrivial and can be quite costly. In contrast, screening diverse peptide libraries presents a great opportunity for the discovery of potent and selective targeting motifs for a specific target.6–9 A versatile and high throughput version of such peptide screening strategies makes use of phage display, in which a peptide scaffold of interest is fused to a bacteriophage coat protein. Phage display has been used extensively for studying protein-protein interactions and discovering peptide ligands for various targets including specific biomolecules as well as intact cells.10,11 Until recently, the phage-display technology was limited to presenting peptides only composed of natural, proteinogenic amino acids. Due to technological advances in the field, phage libraries can now be chemically and genetically modified to present unnatural entities, which greatly expand the chemical space of phage displayed molecules.12–14 For example, selective cysteine alkylation has been utilized to create bicyclic peptide libraries on phage.15 Similarly, a glycopeptide library has been developed through oxidative cleavage of an N-terminal serine or threonine that yields a bioorthogonal aldehyde handle for conjugation to carbohydrates.16
Further expanding the chemical space of phage display, we describe herein a novel phage library that incorporates dynamic covalent binding motifs, the advantage of which has been increasingly recognized in medicinal chemistry.17 Specifically, a pair of 2-acetylphenylboronic acid (APBA) moieties was installed onto phage displayed peptides to bind biological amines via dynamic formation of iminoboronates (Figure 1). Screening of this iminoboronate-capable library against live bacterial cells yielded potent and selective binders of Staphylococcus aureus as well as a colistin-resistant strain of Acinetobacter baumannii. Importantly, we show that these peptide binders can be readily converted to targeted antibiotics via conjugation to a bactericidal agent that will be delivered to the intended bacterial strains.
Figure 1.

Modification of Ph.D.-C7C library with APBA warheads. (a) Illustration of a modified phage binding to bacterial cells via iminoboronate formation. (b) Illustration of the cysteine labeling strategy to display 2-APBA on phage. (c) Structure of the chemical modifiers of the C7C phage library. (d) Confirmation of APBA labeling of phage via fluorescent gel imaging after conjugation with Scz-FITC.
Results
Modification of the C7C library to display APBA warheads.
We and others have recently demonstrated the utilization of APBA to bind biological amines via reversible covalent conjugation to give iminoboronates.18–20 In comparison to the corresponding imine, an iminoboronate exhibits much greater thermodynamic stability with typical Kd values in the low millimolar range (1–10 mM). Despite the thermodynamic stabilization, the iminoboronate formation displays accelerated kinetics in both forward and backward directions, leading to quick equilibrium. This dynamic conjugation, analogous to hydrogen bonds, proves to be a powerful mechanism to promote ligand binding to biological targets. Specifically, our recent work shows that incorporating an APBA warhead into cationic peptides can yield selective probes of gram-positive bacteria, which readily label a target bacterium in serum via a combination of reversible covalent and noncovalent interactions.18 We envisioned that introducing such reversible covalent warheads into phage libraries could give a versatile platform to allow discovery of specific probes for diverse bacterial species and strains.
The phage library chosen for modification was the commercially available Ph.D.-C7C library, which displays disulfide-cyclized peptides with seven randomized residues that are fused to the pIII minor coat protein of the M13 phage. The library was modified with APBA moieties via disulfide reduction and selective cysteine alkylation originally described by Derda and coworkers.21 Briefly, the disulfide bond of the C7C peptides was selectively reduced on phage with immobilized TCEP (iTCEP) for 48 hours at 4°C. The reduced cysteines were then alkylated with an APBA derivative, namely APBA-IA, for 2 hours to yield the APBA-dimer library (Figure 1b). The details of APBA-IA synthesis can be found in the Supporting Information (Figure S1–S3). The extent of APBA-IA labeling was monitored by a pulse-chase assay in which biotin-iodoacetamide (Biotin-IA) treatment and streptavidin capture after APBA-IA labeling allowed quantification of phage that APBA-IA failed to label. The minimal streptavidin capture of the APBA-IA treated phage indicates complete labeling of cysteines by APBA-IA (Figure S4). More direct evidence of APBA conjugation was established by treating the modified phage with a fluorophore labeled semicarbazide (Scz-FITC, Figure 1c, d), which we recently reported to conjugate with APBA chemoselectively to form diazaborines.22 The labeled phage was heat denatured and the coat proteins were subjected to fluorescence gel electrophoresis analysis. Reduced phage with and without Biotin-IA labeling were included as negative controls to confirm on the APBA-semicarbazide conjugation. For the APBA labeled phage, a single distinct band was observed that corresponds to the pIII protein (Figure 1d), which is known to run on SDS-PAGE with an apparent molecular weight of 60–65 kDa, larger than its actual molecular weight of 43 kDa.13 Notably, no fluorescent labeling of the pIII protein was observed for the negative controls as expected.
Panning against S. aureus with the APBA-dimer library.
Panning phage libraries directly against live bacterial cells presents an intriguing alterative to panning against target biomolecules. However, earlier efforts along this front have only yielded low affinity (sub to low millimolar) peptide probes.11,23 We postulated that panning the APBA-dimer library against S. aureus has the potential to discover highly potent and selective peptide probes for this bacteria because it is known to overexpress lysine modified phosphoglycerol (Lys-PG) to afford resistance to host defense peptides.24 In fact, Lys-PG synthesis is one of the critical features of S. aureus that makes it a prevalent pathogen.25 To avoid interference of endogenous proteins, which can compete for iminoboronate formation, the APBA-dimer library was screened against S. aureus cells in a suspension containing 10 mg/mL bovine serum albumin (BSA) as an internal competitor (Figure 2a). Three rounds of affinity selection were initiated with an input population of 1010 plaque forming units (pfu) in each round along with extensive washing steps to eliminate non-binders and strong albumin binders by centrifugation. Acid treatment, which is known to disrupt iminoboronate formation,26 was used to release bound phage from S. aureus. The output population typically ranged from 103 to 105 pfu for S. aureus panning. The recovered phage were amplified, labeled with APBA-IA and subjected to the next round of panning. After each round of panning, 10–20 colonies were randomly selected from the output population and subjected to sequencing. Several peptide sequences were observed repeatedly in round 2 and round 3 even within this small set of colonies subjected for sequencing (Table S1). To determine which sequences merited further pursuit, a phage-based microscopy experiment was performed. The individual phage hits were modified with APBA-IA, incubated with S. aureus, and subsequently treated with a FITC-labeled anti-M13 antibody, which binds to the pVIII protein of the M13 phage. The phage-bound S. aureus was imaged via fluorescence microscopy to determine which phage sequences elicited the most potent bacterial labeling (Figure S5). From these results, the five peptide hits that displayed the brightest images were selected and synthesized via solid phase peptide synthesis (KAM1-KAM5, Table S2). Synthesis was performed on a rink amide resin incorporating an orthogonally protected diaminopropionic acid (Dap) residue on the C-terminus, which allowed for on-resin coupling of fluorescein or rhodamine as a fluorescent reporter. A triple glycine linker was installed between Dap and the core C7C peptide to minimize interference of the fluorophore. After peptide synthesis, the pair of cysteines was subsequently modified with APBA-IA. The purity and identity of the final peptides were assessed via LC-MS analysis (Table S3, Figure S6).
Figure 2.

Phage display against S. aureus. (a) Schematic representation of panning against live bacterial cells. Stars denote APBA-IA modification. (b) Flow cytometry analysis of fluorescein labeled KAM5 in for staining S. aureus cells. Data of replicate experiments are given in the Supporting Information (Figure S9), which show consistent results. (c) Flow cytometry comparison of KAM5 to KAM5 Cyclic (no APBA modification) and a naive APBA dimer peptide KAM6. All peptides were labeled with fluorescein and used for cell staining at 1 µM concentration and in the presence of 1 mg/mL BSA. (d) Microscopic images of S. aureus cells stained with 2 µM KAM5. TAMRA labeled KAM5 was used for microscopy due to better photostability of the fluorophore. Scale bar: 10 µm.
Characterization of peptide hits.
To assess S. aureus binding, flow cytometry was used to measure the median fluorescence intensity of the cells that each peptide hit gave. All five peptide hits showed significant binding to S. aureus cells at sub-micromolar concentrations (Figure 2b, Figure S7-S9). Interestingly, all peptides except KAM2 afforded even stronger fluorescence staining of the bacteria in the presence of 1mg/mL BSA than otherwise. Analysis of the bacterial binding curve of KAM5 gave an estimated EC50 of ~1.5 µM for staining S. aureus cells (Figure S8). The bacterial binding potency displayed by KAM5 is orders of magnitude better than the peptides borne out of previous phage display efforts with natural peptide libraries, which only yielded sub to low mM binders.11,23 The S. aureus binding potency of KAM5 is also much greater than that of Hlys-AB1, a rationally designed peptide that incorporates a single APBA motif (Figure S10). Importantly, a negative control peptide KAM6, which was not selected from the screen and contains a random heptapeptide sequence, showed no bacterial staining under the same experimental conditions (Figure 2c). Furthermore, the cyclic precursor of KAM5 (KAM5-Cyclic which has no APBA moieties) elicited little S. aureus staining as well, demonstrating the importance of the APBA warhead (Figure 2c).
The cell staining ability of KAM5 was also evaluated via fluorescence microscopy with a TAMRA labeled peptide. The microscopy studies yielded results consistent with those of flow cytometry: KAM5 at 2 µM concentration gave strong fluorescence staining of S. aureus cells and the addition of BSA up to 10 mg/mL did not inhibit the bacterial staining by KAM5. On the contrary, the BSA addition elicited stronger fluorescence staining of the bacteria, consistent with the flow cytometry results (Figure 2d). The protein-enhanced bacterial binding by KAM5 is not BSA-specific as similar enhancement was observed with human serum albumin (HSA) as well (Figure S11). To better understand this phenomenon, we measured KAM5 binding to these serum proteins via a fluorescence anisotropy experiment (Figure S11). KAM5 did show binding to both BSA and HSA at high protein concentrations, which is perhaps not surprising given these proteins display a large number of surface lysine residues. These results suggest that BSA/HSA may enhance KAM5 binding to S. aureus by forming ternary complexes, although the detailed mechanism requires further investigation.
Further analysis of KAM5 showed equally potent staining of a methicillin-resistant strain of S. aureus (ATCC 43300) in comparison to the strain (ATCC 6358) used in phage display (Figure S12). This is consistent with our hypothesis that KAM5 binds the bacteria via covalent conjugation to Lys-PG, which is present on essentially all S. aureus strains, although its percentage may vary.27 We further analyzed the bacterial selectivity of KAM5 toward different bacterial species. Specifically, KAM5 labeling of Escherichia coli, a model gram-negative bacterium, and Bacillus subtilis, a model gram-positive bacterium, was assessed via flow cytometry (Figure 3a) and fluorescence microscopy (Figure 3b). The data revealed negligible staining of these control bacterial species with up to 10 µM peptide, highlighting the desirable species selectivity towards S. aureus.
Figure 3.

Selective binding of KAM5 to S. aureus over other bacterial species. B. subtilis & E. coli were used as representative gram-positive and gram-negative bacteria species. (a) Results of flow cytometry analysis with fluorescein labeled KAM5 in the presence of 1 mg/mL BSA. Replicate data set is given in the Supporting Information (Figure S9), which shows consistent results. (b) Results of microscopy analysis using TAMRA labeled KAM5 (10 µM). Scale bar: 10 µm.
Comparison to control phage libraries.
To directly assess the advantage of the APBA modified phage library, we performed parallel screening for S. aureus binding using the unmodified C7C library, as well as a C7C-derived library in which the reduced cysteines were alkylated with simple iodoacetamide (C7C-IA library). The S. aureus panning experiments were performed by following the same protocol used for the APBA-dimer library. Three rounds of panning of the unmodified C7C library did yield two repeating sequences (Table S4). However, when synthesized and characterized using flow cytometry, neither of these peptides showed significant bacterial staining up to 10 µM (Figure S13), the highest concentration allowed by our flow cytometry instrument. Even if they did bind S. aureus at higher concentrations, their affinity for the bacteria would be much lower than that of KAM5. It is worth noting that the final output population of the C7C library, in comparison to the APBA-dimer library, contains a proportionally larger number of blank sequences (no peptide displayed), which presumably came from the original, imperfectly created C7C library and did not get selected out during the panning process. The higher percentage of blank sequences suggests that the unmodified C7C library offers few potent binders or “real hits” for S. aureus. Similarly, after three rounds of panning, the C7C-IA library gave a larger number of blank sequences as well, yet only one sequence showing repeats (Table S5). Characterization of this recurring sequence, as well as a randomly picked sequence out of the round 3 output population, failed to show bacterial staining up to 10 µM (Table S6, Figure S13). The failure of these control libraries is consistent with earlier phage display efforts where screening of natural peptide libraries was only able to yield sub-to-low millimolar binders of bacteria at best.11, 23 Collectively, the comparative study presented here clearly showcases the advantage of phage display with dynamic covalent binding motifs.
Generating a targeted antibiotic for S. aureus.
A potent and selective S. aureus binder has the potential to serve as a directing element to develop targeted antibiotics. The preferential binding of KAM5 towards S. aureus over serum proteins as well as other bacterial species makes it an excellent candidate for delivering a nonselective antibiotic to target cells. To test the hypothesis, we conjugated KAM5 to eosin, a phototoxin that upon photoirradiation triggers the production of reactive oxygen species (ROS), killing cells in close proximity (Figure 4a). There has been considerable interest in developing photoinactivation strategies for pathogenic bacteria,28–32 which has been encouraged by the technological advances and therapeutic successes achieved in photodynamic therapy.33 Delivery of a photosensitizer in a species-specific manner would maximize the benefit and minimize the side effect of photodynamic therapy in treating bacterial infections, particularly when the infection site hosts human commensal bacteria. Eosin was conjugated to KAM5 via the Dap residue, similar to the fluorophore labeling protocol (Figure 4b). The conjugate was then assessed for photodynamic inactivation of S. aureus via a titering assay. When S. aureus was mixed with 1 µM and 2 µM KAM5-Eosin and then exposed to blue light for 15 minutes, 74% and 92% cell killing was observed respectively (Figure 4c). Importantly, eosin alone at these concentrations did not elicit cell death nor did KAM5 without the photosensitizer. Extending the bactericidal assay into other strains of S. aureus revealed that KAM5-Eosin worked equally well against the MRSA strain with comparable percentage of cell killing under the same experimental conditions (Figure S14). As expected from the cell binding specificity of KAM5, the cell killing of KAM5-Eosin was specific to S. aureus as no significant cell death was observed for B. subtilis and E. coli treated with 2 µM peptide followed by photoirradiation (Figure 4d). Furthermore, no mammalian cell toxicity by KAM5-Eosin was observed via an MTT assay against HEK 293T cells and Jurkat cells with or without photoirradiation (Figure S15). These results clearly demonstrate the feasibility of developing species-selective antibiotics.
Figure 4.
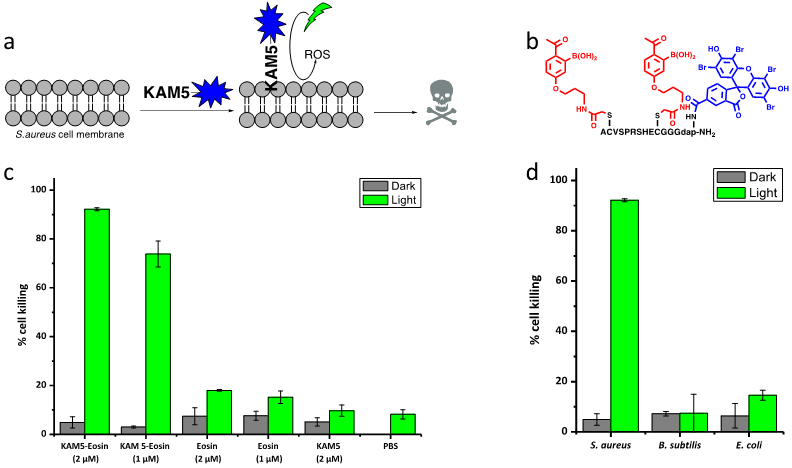
Conjugation of a phototoxin to induce targeted killing of S. aureus. (a) Cartoon representation of photodynamic therapy with KAM5-Eosin. (b) Structure of KAM5-Eosin (c) Percent killing of S. aureus by KAM5-Eosin and controls with and without photoirradiation. (d) Percent killing of several bacterial species with KAM5-Eosin (2 µM) to highlight the S. aureus specificity.
Strain-specific targeting of A. baumannii.
To probe the general applicability of our approach, we panned the APBA-dimer library against a colistin-resistant strain of A. baumannii. A. baumannii has emerged as a major healthcare-associated pathogen,34 which can cause severe infections in lungs and blood. A. baumannii often presents resistance to multiple antibiotics, sometimes even to colistin (polymyxin E), one of the last-resort antibiotics for its treatment. A. baumannii can acquire colistin resistance by modifying its lipooligosaccharide (LOS) with the addition of phosphoethanolamine or 4-aminoarabinose functionalities.35 Some strains even shut down LOS biosynthesis completely and replace the exterior leaflet of the outer membrane with lipoproteins.36,37 The APBA-dimer library on phage was screened against a LOS- mutant of A. baumannii (AB5075, a highly virulent isolate established as a model strain for A. baumannii infection38). Three rounds of panning against the LOS- A. baumannii were executed following the same panning procedure described above for S. aureus except the addition of a negative screen against the wild-type (LOS+) A. baumannii in the second round. After each round of panning, 15 colonies were isolated from the output population and subjected to sequencing, in which convergence was detected starting in round 2 (Table S7). Four different peptide sequences (KAM7–10) were observed repeatedly and synthesized via solid-phase peptide synthesis following the same procedure used for the S. aureus binding peptides (Table S8). Flow cytometry analysis of the peptide hits showed potent binding at sub-µM concentrations (Figure 5a, Figure S16). Similar to what we observed for S. aureus, the presence of BSA did not inhibit the peptides’ binding to the bacteria. Instead, it actually enhanced bacterial staining by KAM8 at sub-µM concentrations to some extent (Figure 5a). Analysis of the concentration profile of the bacterial staining by KAM8 gave an estimated EC50 of ~ 0.3 µM. Also similar to the S. aureus binders, KAM8 binding to A. baumannii requires both the two APBA warheads as well as the specific peptide sequence in between. The negative controls (KAM6 and the cyclic precursor KAM8-Cyclic) showed much reduced binding to the bacteria (Figure S16). The flow cytometry results were further corroborated with fluorescence microscopy studies, which showed bright fluorescence staining of the LOS− A. baumannii with 2 µM KAM8 (Figure 5b). Excitingly, KAM8 stained the bacteria in a strain-specific manner: as seen in the microscopic images, the wild type A. baumannii (LOS+) showed little fluorescence staining under the same conditions that gave strong fluorescence staining of the LOS- strain. Comparative analysis of the strains with flow cytometry gave results consistent with the microscopy studies (Figure 5a). As expected, KAM8 showed no binding to S. aureus or E. coli, which were used as controls to represent gram-positive and gram-negative bacteria respectively (Figure S17).
Figure 5.

Phage display to discover peptide probes for a LOS- strain of A. baumannii (AB5075). (a) Flow cytometry analysis of the representative peptide hit KAM8 in staining A. baumannii (LOS− vs LOS+) in the presence of 1 mg/mL BSA. Replicate data set is presented in the Supporting Information (Figure S9), which shows consistent result. (b) A. baumannii cell staining examined by microscopy with TAMRA labeled KAM8 at 2 µM for LOS− and 10 µM for LOS+. Scale bar: 10 µm. (c) Percent cell killing of A. baumannii (LOS−) with and without photoirradiation. (d) Percent cell killing of the LOS+ versus LOS− strains of A. baumannii with 2 µM KAM8-Eosin. The contrasting outcomes of these strains highlight the high strain specficiy of KAM8.
We further examined the feasibility of converting KAM8 into a targeted antibiotic for the LOS- strain of A. baumannii. Toward this end, we synthesized the KAM8-Eosin conjugate, which upon photoirradiation effectively killed the LOS- A. baumannii cells (2 µM, 15 min light, >90% cell killing, Figure 5c). In contrast, eosin alone at these concentrations did not elicit A. baumannii cell death nor did KAM8 without the photosensitizer. KAM8-Eosin was established as a strain-specific antibiotic of the LOS- A. baumannii as it demonstrated little killing of the wild-type (LOS+) strain under the same conditions (Figure 5d). Collectively, these data showcase that strain-specific bacterial cell killing can be achieved through phage display and selection of the APBA-presenting peptides.
Discussion
This contribution describes the construction and validation of a phage library displaying reversible covalent binding motifs. Specifically, chemical modification of phage displayed peptides yields an APBA dimer library, which allows facile screening against bacterial cells to identify reversible covalent binders of specific bacterial strains. Several earlier reports described phage panning against intact bacterial cells, which however only yielded bacterial binders of sub to low millimolar affinity.11,23 Despite the low affinity binding of the peptide hits, the corresponding intact phage particles showed promise as delivery vehicles of antibiotics, presumably due to the pentavalent display of the peptide binder. For comparison, screening of the APBA dimer library herein yielded bacterial binders of sub-micromolar potency. The contrasting results highlights the promise of chemically modified phage libraries. It is intriguing to speculate that even higher potency binding could be accomplished with better designed phage libraries with reversible covalent warheads. The phage panning against intact cells is remarkably convenient and powerful, allowing facile incorporation of negative screens and internal competitors. Specifically, as we reported in an earlier publication, abundant endogenous protein could compete for iminoboronate conjugation, thereby inhibiting the bacterial binding of an APBA-containing peptide.18 Our data of this study unequivocally show that this protein interference problem can be overcome by including serum albumin in the screening mixture and the reversible covalent binding mechanism can afford highly selective binders in complex biological milieu.
Although further investigation is needed to elucidate the binding mechanisms and targets of the peptide binders identified in this study, we envision the APBA dimer library can be extended to discovering binders of various bacterial pathogens. We believe this is feasible given the iminoboronate chemistry is generally applicable to primary amines, which can be abundant on bacterial cells particularly those showing antibiotic resistance. Amino acid modification of phospholipids, such as Lys-PG synthesis in S. aureus, have been documented for a number of bacterial species as a resistance mechanism to host defense peptides and neutrophil clearance.25,27 Such lipid modification affords a high abundance of amino groups (α- and ε-amine of lysine) on a bacterial surface. Similarly, alanylation of teichoic acids results in abundant alanyl ester structures with α-amines available for iminoboronate conjugation.39,40 Surface modification of gram-negative bacteria is becoming better understood as well. It has been shown that lipopolysaccharide (LPS) or lipooligosaccharide (LOS) modification with phosphoethanolamine and/or 4-aminoarabanose leads to polymyxin resistance for many bacterial species.41 We anticipate these surface modified bacterial strains would be particularly amenable to specific recognition by the iminoboronate-capable peptides. This hypothesis is being tested with ongoing research in our laboratory.
Finally, we demonstrate that a bacterial binder identified from phage display can be readily converted to targeted antibiotics that specifically eradicate the corresponding strain of bacteria. The facile generation of targeted antibiotics is of contemporary importance given the undesirable consequences of broad-spectrum antibiotics, which inevitably cultivate antibiotic resistance and cause damage to human microbiota. Although the work presented here utilizes a phototoxin as the bactericidal agent, it is conceivable that antibiotics of other modes of action, such as those targeting cell membranes including vancomycin and daptomycin, can be utilized to build peptide-antibiotic conjugates, which will expand the scope of our strategy to create targeted antibiotics.
Experimental
Materials and instrumentation.
The Ph.D.™-C7C Phage Display Peptide Library Kit and the E. coli K12 ER2738 strain were purchased from New England Biolabs. All ER2738 cultures were grown in the presence of 20 µg/mL tetracycline. Chemical reagents for small molecule, library synthesis & confirmation and peptide synthesis were purchased from various vendors and used as received. The fluorescent gel was imaged on a BioRad ChemiDoc MP Imaging System. The S. aureus (ATCC 6538) and MRSA (ATCC 43300) were purchased from Microbiologics as a lyophilized pellet. The wild-type A. baumannii (AB5075) is a virulent and multidrug resistant clinical isolate that has been established as a pathogenic model strain (Jacobs et al., 2015; MBio) and the LOS deficient A. baumannii (5075 LOS-) was established from AB5075 through selection for colistin resistance (Boll et al., 2016; PNAS). NMR data of the small molecule was collected on a VNMRS 500 MHz NMR spectrometer. Peptide synthesis was performed on a Tribute peptide synthesizer from Protein Technologies and purified via reverse-phase high performance liquid chromatography (RP-HPLC) on a Waters Prep LC with a Jupiter C18 Column (Phenomenex). Mass spectrometry data were generated using an Agilent 6230 LC TOF mass spectrometer. Fluorescence images were captured on a Zeiss Axio Observer A1 inverted microscope. Flow cytometry analysis was performed on a BD FACSAria cell sorter. Photoinactivation was performed with a X-Cite 120Q (120-Watt lamp) excitation light source accompanied with the Zeiss microscope. Jurkat T lymphocytes were a gift from the Johnson lab at Boston College, HEK293T cells were a gift from the Weerapana lab at Boston College, and mammalian cell cytotoxicity was evaluated on a SpectraMax M5 plate reader along with fluorescence anisotropy.
APBA-IA and peptide synthesis.
Details of the small molecule synthetic route and peptide synthesis are provided in the supplementary information.
APBA-dimer library synthesis.
The Ph.D.™-C7C Phage Display Peptide Library (5 µL, ~1×1013 pfu/mL) was subjected to reduction in the presence of iTCEP (25 µL), in a total volume of 200 µL in TBS (pH 8.5) for 48 hours at 4˚C. APBA-IA (2 mM, 2 µL from a 200 mM DMSO stock) was added to the reduced phage and allowed to conjugate for 2 hours at room temperature. The labeled phage was removed from iTCEP and precipitated to remove excess labeling reagent with 1/6 volume 20% (w/v) PEG-8000, 2.5 M NaCl for 5 hours at 4˚C. Precipitated phage was re-dissolved in PBS (pH 7.4, 100 µL) and the phage titer was calculated according to the M13 Titer Protocol provided by New England Biolabs.
Fluorescent gel analysis.
APBA-IA and Biotin-IA labeled library phage (~1×1010pfu/mL) were subjected to labeling with Scz-FITC (2 mM), synthesized previously13, for 1 hour followed by precipitation. Phage samples were heat denatured at 95˚C for 5 minutes. Samples were subjected to 15% SDS-PAGE for 50 minutes, allowing the lower molecular weight PVIII protein to run off the gel, and imaged.
Panning against whole cells.
S. aureus was grown in LB medium to an OD600~1.0 (~1×109 cfu/mL). The cells (1 mL) were washed with chilled PBS containing 0.05% Tween (PBST) twice and resuspended in PBS (pH 7.4) with 10 mg/mL BSA present. The APBA-labeled phage library (~1×1010 pfu) was added to the cell suspension and allowed to incubate on ice for 1 hour. The cells were washed three times with PBST and three times with PBS to remove unbound phage. Cell-bound phage were incubated with 200 µL elution buffer (Glycine-HCl, pH 2.2, 1 mg/mL) for 15 minutes followed by centrifugation of the cells. The supernatant was removed and neutralized with 150 µL Tris-HCl (pH 9.1). All Eppendorf tubes utilized in the panning procedure were blocked with 10 mg/mL BSA before use. Centrifugation of cells was performed at 5,000 rcf for 5 minutes. The eluted bound phage solution was added to early-log phase ER2738 and amplified for 4.5 hours followed by precipitation to isolate the amplified phage. The amplified phage were labeled with APBA-IA and subjected to the next round of panning. The phage titer was calculated before and after each round of panning to determine the input and output population. Individual phage colonies from each round of panning were amplified in ER2738. Phage DNA was isolated using a Qiagen miniprep kit and sent for sequencing analysis by Eton Bioscience, Inc. The screens against S. aureus with the unmodified C7C library and the IA-alkylated library (C7C-IA) were performed following the same protocol. The C7C-IA library was prepared using the same protocol described for the APBA-dimer library preparation. The screen against A. baumannii (LOS−) was performed following the same protocol; however, a negative screen was introduced against A. baumannii (LOS+) starting in the second round. In the negative screen, the phage library was incubated with A. baumannii (LOS+) for 1 hour, the supernatant was removed and subsequently subjected to the positive screen against A. baumannii (LOS−).
Flow cytometry analysis.
Each bacterial strain was grown to an OD600~0.5, washed and diluted with PBS (pH 7.4). The cells (~1×107 cfu/mL) were incubated with various concentrations of FAM-labeled peptide with or without BSA in PBS. After incubation for 1 hour, samples were subjected to cytometric analysis. Data obtained was analyzed via BD FACSDiva software and median fluorescent values were computed from the generated histograms. All flow cytometry experiments were repeated and generated consistent results (see Figure S9).
Fluorescence microscopy.
Each bacterial strain was grown to an OD600~1.0, washed and diluted with PBS (pH 7.4). The cells (~1×109 cfu/mL) were incubated with various concentrations of TAMRA-labeled peptide with or without BSA in PBS for 1 hour. The same microscopy procedure described in the supplementary information was implemented using filter set 20 HE (excitation: BP 546/12, emission: BP 607/80) suitable for detection of TAMRA fluorescence and images were processed consistently using ImageJ software.
Photoinactivation of bacteria.
Each bacterial strain was grown to an OD600~0.7, washed and diluted with PBS (pH 7.4). The cells (~1×108 cfu/mL) were incubated with eosin-conjugated peptides and various controls for 15 minutes. Half of the bacterial suspension was removed and placed in a 96-well plate (Corning 3595). The well was subjected to photoirradiation on the Zeiss microscope using the 20X objective and fluorescein filter to emit blue light for 15 minutes. Cells were diluted in LB media, spread on LB agar plates and incubated overnight at 37˚C. The A. baumannii (LOS−) strain was spread on LB agar (+ 10 µg/mL polymyxin b) and incubated for 24 hours. The amount of cell killing was calculated by comparing the amount of colonies of treated bacteria to an untreated PBS control. All experiments were repeated and the average cell killing of two trials was plotted.
Supplementary Material
Acknowledgements
We thank Dr. Patrick Autissier for flow cytometry analysis and M. Stephen Trent for providing the A. baumanii strains AB5075 and 5075 LOS-. Financial support for the work is provided by the National Institutes of Health via Grant GM102735 to JG.
Footnotes
Supporting Information. Small molecule synthesis, peptide synthesis and characterization, additional flow cytometry and microscopy results.
References
- (1).Blair JM; Webber MA; Baylay AJ; Ogbolu DO; Piddock LJ Nature rev, 2015, 13, 42. [DOI] [PubMed] [Google Scholar]
- (2).Fischbach MA; Walsh CT Science, 2009, 325, 1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Modi SR; Collins JJ; Relman DA J. Clin. Invest, 2014, 124, 4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Lehar SM; Pillow T; Xu M; Staben L; Kajihara KK; Vandlen R; DePalatis L; Raab H; Hazenbos WL; Morisaki JH; Kim J; Park S; Darwish M; Lee BC; Hernandez H; Loyet KM; Lupardus P; Fong R; Yan D; Chalouni C; Luis E; Khalfin Y; Plise E; Cheong J; Lyssikatos JP; Strandh M; Koefoed K; Andersen PS; Flygare JA; Wah Tan M; Brown EJ; Mariathasan S Nature, 2015, 527, 323. [DOI] [PubMed] [Google Scholar]
- (5).Mariathasan S; Tan MW Trends Mol. Med. 2017, 23, 135. [DOI] [PubMed] [Google Scholar]
- (6).Kumaresan PR; Wang Y; Saunders M; Maeda Y; Liu R; Wang X; Lam KS ACS Comb. Sci, 2011, 13, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Qian Z; Upadhyaya P; Pei D Methods Mol. Biol, 2015, 1248, 39. [DOI] [PubMed] [Google Scholar]
- (8).Jagadish K; Gould A; Borra R; Majumder S; Mushtaq Z; Shekhtman A; Camarero JA Angew. Chem. Int. Ed, 2015, 54, 8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Owens AE; de Paola I; Hansen WA; Liu Y-W; Khare SD; Fasan R J. Am. Chem. Soc, 2017, 139, 12559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hamzeh-Mivehroud M; Alizadeh AA; Morris MB; Church WB; Dastmalchi S Drug. Discov. Today, 2013, 18, 1144. [DOI] [PubMed] [Google Scholar]
- (11).Huang JX; Bishop-Hurley SL; Cooper MA Antimicrob. Agents Chemother, 2012, 56, 4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ng S; Jafari MR; Derda R ACS Chem. Biol, 2012, 7, 123. [DOI] [PubMed] [Google Scholar]
- (13).Mohan K; Weiss GA ACS Chem. Biol, 2016, 11, 1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Day JW; Kim CH; Smider VV; Schultz PG Bioorg. Med. Chem. Lett, 2013, 23, 2598. [DOI] [PubMed] [Google Scholar]
- (15).Heinis C; Rutherford T; Freund S; Winter G Nat. Chem. Biol, 2009, 5, 502. [DOI] [PubMed] [Google Scholar]
- (16).Ng S; Jafari MR; Matochko WL; Derda R ACS Chem. Biol, 2012, 7, 1482. [DOI] [PubMed] [Google Scholar]
- (17).Bandyopadhyay A; Gao J Curr. Opin. Chem. Biol, 2016, 34, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Bandyopadhyay A; McCarthy KA; Kelly MA; Gao J Nat. Commun, 2015, 6, 6561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Cal PM; Frade RF; Cordeiro C; Gois PM Chemistry 2015, 21, 8182. [DOI] [PubMed] [Google Scholar]
- (20).Akcay G; Belmonte MA; Aquila B; Chuaqui C; Hird AW; Lamb ML; Rawlins PB; Su N; Tentarelli S; Grimster NP; Su Q Nat. Chem. Biol, 2016, 12, 931. [DOI] [PubMed] [Google Scholar]
- (21).Jafari MR; Deng L; Kitov PI; Ng S; Matochko WL; Tjhung KF; Zeberoff A; Elias A; Klassen JS; Derda R ACS Chem. Biol, 2014, 9, 443. [DOI] [PubMed] [Google Scholar]
- (22).Bandyopadhyay A; Cambray S; Gao J J. Am. Chem. Soc, 2017, 139, 871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Gasanov U; Koina C; Beagley KW; Aitken RJ; Hansbro PM Infect. Immun, 2006, 74, 566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Andra J; Goldmann T; Ernst CM; Peschel A; Gutsmann T J. Biol. Chem, 2011, 286, 18692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Slavetinsky C; Kuhn S; Peschel A Biochim. Biophys. Acta - Mol. Cell Biol. Lipids, 2017, 1862, 1310. [DOI] [PubMed] [Google Scholar]
- (26).Bandyopadhyay A; Gao J J. Am. Chem. Soc, 2016, 138, 2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Peschel A; Jack RW; Otto M; Collins LV; Staubitz P; Nicholson G; Kalbacher H; Nieuwenhuizen WF; Jung G; Tarkowski A; van Kessel KP; van Strijp JA J. Exp. Med, 2001, 193, 1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Dosselli R; Gobbo M; Bolognini E; Campestrini S; Reddi E ACS Med. Chem. Lett, 2010, 1, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Liu F; Soh Yan Ni A; Lim Y; Mohanram H; Bhattacharjya S; Xing B Bioconjug. Chem, 2012, 23, 1639. [DOI] [PubMed] [Google Scholar]
- (30).Kasimova KR; Sadasivam M; Landi G; Sarna T; Hamblin MR Photochem. Photobiol. Sci, 2014, 13, 1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Johnson GA; Muthukrishnan N; Pellois JP Bioconjug. Chem, 2013, 24, 114. [DOI] [PubMed] [Google Scholar]
- (32).Alves E; Costa L; Carvalho CM; Tome JP; Faustino MA; Neves MG; Tome AC; Cavaleiro JA; Cunha A; Almeida A BMC Microbiol, 2009, 9, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Sperandio FF; Huang YY; Hamblin MR Recent Pat. Antiinfect. Drug Discov, 2013, 8, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Peleg AY; Seifert H; Paterson DL Clin. Microbiol. Rev, 2008, 21, 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Arroyo LA; Herrera CM; Fernandez L; Hankins JV; Trent MS; Hancock RE Antimicrob. Agents Chemother, 2011, 55, 3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Boll JM; Crofts AA; Peters K; Cattoir V; Vollmer W; Davies BW; Trent MS Proc. Natl. Acad. Sci, 2016, 113, E6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Powers MJ; Trent MS Mol. Microbiol, 2018, 107, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Jacobs AC; Thompson MG; Black CC; Kessler JL; Clark LP; McQueary CN; Gancz HY; Corey BW; Moon JK; Si Y; Owen MT; Hallock JD; Kwak YI; Summers A; Li CZ; Rasko DA; Penwell WF; Honnold CL; Wise MC; Waterman PE; Lesho EP; Stewart RL; Actis LA; Palys TJ; Craft DW; Zurawski DV MBio 2014, 5, e01076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kristian SA; Datta V; Weidenmaier C; Kansal R; Fedtke I; Peschel A; Gallo RL; Nizet V J. Bacteriol, 2005, 187, 6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Brown S; Santa Maria JP Jr.; Walker S Annu. Rev. Microbiol, 2013, 67, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Needham BD; Trent MS Nature Rev. Microbiol, 2013, 11, 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.