

Abstract
The conjugation of antibodies to drugs and drug carriers improves delivery to target tissues. Widespread implementation and effective translation of this pharmacologic strategy awaits the development of affinity ligands capable of a defined degree of modification and highly efficient bioconjugation without loss of affinity. To date, such ligands are lacking for the targeting of therapeutics to vascular endothelial cells. To enable site-specific, click-chemistry conjugation to therapeutic cargo, we used the bacterial transpeptidase, sortase A, to attach short azidolysine containing peptides to three endothelial-specific single chain antibody fragments (scFv). While direct fusion of a recognition motif (sortag) to the scFv C-terminus generally resulted in low levels of sortase-mediated modification, improved reaction efficiency was observed for one protein, in which two amino acids had been introduced during cloning. This prompted insertion of a short, semi-rigid linker between scFv and sortag. The linker significantly enhanced modification of all three proteins, to the extent that unmodified scFv could no longer be detected. As proof of principle, purified, azide-modified scFv was conjugated to the antioxidant enzyme, catalase, resulting in robust endothelial targeting of functional cargo in vitro and in vivo.
Graphical Abstract
INTRODUCTION
Endothelial cells (ECs) have a critical role in the regulation of regional blood flow, coagulation, host defense, and extravascular transport and constitute an important therapeutic site in a variety of thrombotic, ischemic, and inflammatory conditions.1–6 The ability to alter endothelial biology by local therapeutic delivery represents a significant medical need - not only because of the potential to directly improve disease outcomes, but also to modulate passage of other macromolecules or drug carriers out of the vascular space and into target organs.7,8 Vascular immunotargeting, a pharmacologic strategy in which ligands with affinity for endothelial cell surface determinants are attached to therapeutic cargo, has been repeatedly shown to enhance drug delivery to ECs.9–12 Full realization of the benefits of this approach requires both detailed understanding of the biology of endothelial surface molecules and precise, translational implementation of the targeting system.
To date, the vast majority of endothelial drug delivery systems have relied on monoclonal antibodies to achieve targeting.13 For the initial design and testing of these systems, hybridoma-derived, full-length antibodies have a number of advantages: they are easily produced, have prolonged circulation, and typically tolerate nonselective chemical modification and bioconjugation without appreciable loss of binding ability.14,15 Several disadvantages, however, may limit their utility in the translation of vascular immunotargeting to clinical medicine. These include relatively large size, Fc fragment-mediated activation of the immune system, and cross-linking of endothelial target antigens, leading to changes in vascular function or phenotype.16–18 An alternative strategy involves recombinant affinity ligands, like single-chain antibody fragments (scFv), single-domain antibodies, or other engineered protein domains.19,20 These molecules allow production in well-defined prokaryotic or eukaryotic systems, can be genetically manipulated (e.g., for affinity maturation), and lack the constant domains of antibody heavy and light chains responsible for some mechanisms of clearance and immune system activation.17,21
From the standpoint of conjugation chemistry, the transition from full-length antibodies to smaller recombinant affinity ligands like scFv entails both significant challenge and potential opportunity. The relative sensitivity of the latter class of molecules to amine- and thiol-based conjugation has motivated development of techniques for site-specific modification.22–24 By the elimination of the heterogeneous degree of modification and regioisomerism inherent in less specific chemistries, these technologies stand to improve drug targeting by all classes of affinity ligands.25
We recently reported C-terminal biotinylation of a high-affinity, species cross-reactive scFv specific for the platelet-endothelial cell adhesion molecule-1 (PECAM-1 or CD31) and showed robust endothelial targeting of streptavidin-labeled nanoparticles following oriented attachment of the modified affinity ligand.26 These results demonstrated the promise of this approach and motivated development of an alternate means of site-specific modification of endothelial-specific scFv that would allow covalent attachment of targeting ligand to cargo and avoid the immunogenicity of the biotin–streptavidin complex. Here, we report use of a particularly versatile technique: transpeptidation by the bacterial enzyme, sortase A.27 Sortase covalently modifies recombinant proteins tagged with a short recognition motif (sortag) and can be used to functionalize the N- or C-termini of recombinant proteins through attachment of short, appropriately modified peptides.28 While this approach has been used to modify a variety of different types of affinity ligands,29–31 few studies have investigated the reaction efficiency across multiple targeting ligands of the same class, and its application to vascular immunotargeting has yet to be reported. For example, the sortase reaction has been optimized for individual proteins32 and the enzyme mutated to improve catalytic efficiency,33,34 but each new protein typically requires trial-and-error design of sortagged constructs and empiric testing of their modification. In an effort to identify common determinants of scFv reaction efficiency, we studied sortasemediated C-terminal modification of three distinct, endothelial-targeting ligands and found that insertion of a short, semi-rigid amino-acid linker between the C-terminus of the protein and the sortag enhanced the reaction efficiency of all three proteins, both increasing yield and simplifying purification. We demonstrated the utility of this approach through oriented bioconjugation to the antioxidant enzyme, catalase, creating an intercellular adhesion molecule-1 (ICAM-1 or CD54) targeted therapeutic capable of delivery of functional antioxidant enzyme to the endothelium in vitro and in vivo.
RESULTS
Production, Characterization, and Sortase Modification of scFv–LPETGG Proteins.
To enable site-specific modification by sortase, we fused a six amino acid sortag, LPETGG, and triple FLAG tag directly to the C-terminus of three previously described scFv, specific for mouse ICAM-1 (mICAM-1),35 mouse PECAM-1 (mPECAM-1),36 and human ICAM-1 (hICAM-1).37 The molecular design of the scFv–LPETGG construct is shown in Figure S1. Following purification, size-exclusion high-performance liquid chromatography (HPLC) demonstrated a single major peak for each scFv–LPETGG protein (Figure S2). Cell-based ELISAs demonstrated specific binding to each target antigen, with affinities similar to those previously reported for nonsortagged scFv (Table S1).35–37
Figure 1A shows the schema for the sortase-mediated reaction, in which the FLAG tag is replaced with a short peptide containing –GGG at the N-terminus, added in 5-fold excess to drive formation of the desired end-product. Each peptide (Table S2) contains a C-terminal azidolysine residue, to enable oriented click-chemistry conjugation, and a fluorophore-modified lysine residue, to facilitate measurement of reaction progression and efficiency. Fluorescence scans of sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis of the reaction mixtures for each scFv–LPETGG protein (Figure 1B) show low reaction efficiency overall based on the percentage incorporation of TAMRA–azide–GGG peptide. Significantly greater C-terminal modification is seen for one clone, α-mPECAM-1 scFv, compared to α-mICAM-1 and α-hICAM-1 scFv (Table S3). Consistent with these results, Coomassie staining (Figure S3A) shows a band at the expected size for C-terminally modified α-mPECAM-1 scFv–LPETGG (slightly smaller than the unmodified protein due to replacement of the triple FLAG tag by peptide) but not for the other two scFv-LPETGG proteins.
Figure 1.

Sortase modification of scFv-LPETGG proteins. (a) Schematic of the reversible sortase (SrtA) reaction, in which the transpeptidase enzyme removes the C-terminal FLAGx3 tag and appends a fluorescently labeled, azide-modified peptide. (b) SDS-PAGE analysis of reaction mixtures shows clear difference in the reaction efficiency of the three clones. Solid arrowhead indicates size of modified scFv–LPETGG, whereas open arrowhead indicates free TAMRA–azide–GGG peptide (runs as two bands on the gel). (c) HPLC analysis of scFv–LPETGG reaction mixtures confirms difference in α-mPECAM-1 scFv reaction efficiency vs the other two clones. Note that free FAM–azide–GGG peptide elutes in two peaks on SEC. (d) Primary structure of each protein at the scFv C-terminus, showing distinct positioning of amino acids VD (encoded by the sequence of restriction enzyme site SalI). (e) Predictive 3-D modeling of each scFv–LPETGG clone, showing the putative positions of VH (blue) and VL (orange), with the C-terminal amino acids of framework region 4 (FR4) shown in red and amino acids VD shown in green.
A second method of analysis, size-exclusion HPLC, was used to confirm these results. In these experiments, each scFv–LPETGG was reacted with green fluorescent FAM–azide–GGG peptide, which eluted from the column in two peaks at approximately 6.2 and 6.8 min. The use of a second peptide helped to confirm that the observed reaction characteristics were not dependent on one specific peptide structure. HPLC results were in close agreement with SDS-PAGE analysis for all three proteins (Figure 1C and Table S3). Likewise, absorbance measurements at 280 nm matched results of Coomassie staining of gels, showing a second peak corresponding to modified α-mPECAM-1 scFv–LPETGG protein (Figure S3C).
Sequence Analysis and 3D Modeling of scFv–LPETGG Proteins.
The significantly higher reaction efficiency of α-mPECAM-1 scFv–LPETGG prompted re-examination of its C-terminal structure. To enable cloning into the pMT/LPETGG vector, a restriction enzyme site had been appended to the 3′ end of the α-mPECAM-1 variable light chain (VL), inserting the amino acids Val–Asp (VD) between the scFv C-terminus and the sortag. In contrast, the same restriction enzyme site was introduced into the α-mICAM-1 and α-hICAM-1 clones by making a single nucleotide change in the fourth framework region of VL. This modification replaced the native amino acids Val–Glu (VE) with the similar sequence VD and truncated the final two amino acids of framework region 4 (Figure 1D).
To assess the importance of these differences in primary structure, the Rosetta antibody protocol was used to predict the 3D structure of each protein.38 While the algorithm was unable to directly model the C-terminal sortag, the predicted structures (Figure 1E) suggest that the difference in C-terminal sequence is critical in extending the enzyme recognition site beyond the surface of the α-mPECAM-1 scFv, increasing its accessibility.
Sortase Modification of scFv–linker–LPETGG proteins.
To directly test the role of sortag accessibility in the efficiency of C-terminal scFv modification, each protein was synthesized with a 13-amino acid linker, (SSSSG)2SAA, between the end of VL and the enzyme recognition motif. This semi-rigid linker was chosen because of previous success incorporating it into the design of scFv-based fusion protein therapeutics as a means of separating affinity ligand and cargo protein domains.35,36,39–41 The molecular design of the scFv–linker–LPETGG construct is shown in Figure S1. As expected, scFv–linker–LPETGG proteins were slightly larger than their scFv–LPETGG counterparts, with similar purity seen on sizeexclusion HPLC (Figure S2). Cell-based ELISAs revealed nearly identical affinities for their respective target ligands (Table S1).
Figure 2A shows SDS-PAGE analysis of sortase-mediated reactions for each scFv–linker–LPETGG protein. Fluorescence scans demonstrate significantly greater C-terminal modification of each linker-containing protein, as compared to scFv–LPETGG (Table S3, p < 0.01). Moreover, Coomassie staining demonstrated near complete consumption of each scFv–linker–LPETGG, with bands corresponding to unmodified protein replaced by bands at the expected size of modified scFv (Figure S3B). Interestingly, even the α-hICAM-1 scFv-linker-LPETGG reaction mixture, which has lower reaction efficiency based on fluorescent measurements, shows complete consumption of unmodified protein.
Figure 2.

Sortase modification of scFv–linker–LPETGG proteins. (a) SDS-PAGE analysis of sortase reaction mixtures for each of the three scFv clones shows marked increase in reaction efficiency. Solid arrowhead indicates the size of modified scFv–linker–LPETGG, whereas the open arrowhead indicates free TAMRA peptide. (b) HPLC analysis of scFv–LPETGG reaction mixtures shows similar increase in reaction efficiency. (c) Comparison of reaction efficiencies of scFv–LPETGG and scFv–linker–LPETGG for each clone: *, p < 0.02 for α-mPECAM-1 scFv–LPETGG vs α-mICAM-1 and α-hICAM-1 scFv–LPETGG; **, p < 0.01 for each scFv–linker–LPETGG vs scFv–LPETGG. (d) HPLC of purified scFv–linker–FAM–azide for each clone showing normalized fluorescence (note the ~0.2 min delay in position of fluorescence vs absorbance peaks due to positioning of respective detectors).
Nearly identical reaction efficiencies were found on HPLC analysis of scFv–linker–LPETGG reaction mixtures (Figure 2B,C and Table S3). Absorbance measurements at 280 nm again matched the results of Coomassie staining, showing complete elimination of peaks corresponding to unmodified scFv–linker–LPETGG (Figure S3D–F). The elimination of unmodified protein simplified purification of peptide-modified scFv, with a single protein species seen on HPLC (Figure 2D) following the removal of sortase and excess peptide.
Time, Calcium, and Sortase Dependence of scFv Modification.
We next tested a variety of aspects of the sortase-mediated reaction, including its kinetics and dependence on calcium and sortase enzyme concentration. α-mPECAM-1 scFv–LPETGG and scFv–linker–LPETGG were compared to show the effect of the semi-rigid linker (Figure 3). Additional results are shown in Figure S4 and Tables S3–S5. Reactions progressed similarly at RT, reaching equilibrium at approximately 8 h. Reactions at 37C reached equilibrium faster but generally resulted in less C-terminal scFv modification (Table S4). Varying calcium concentrations were tested because of the known role of the divalent cation in modulating the catalytic function of sortase.29 C-terminal modification of both scFv–linker–LPETGG and scFv-LPETGG were detectable over a wide range of concentrations, although concentrations of 100 μM and above were needed for maximal modification of linker containing proteins (Table S5). Figure 3B shows that a calcium concentration of 1 mM or greater was required for maximal modification of α-mPECAM-1 scFv–LPETGG and that the efficiency of this reaction remained lower than that of mPECAM-1 scFv–linker–LPETGG across all concentrations (p < 0.01 for each comparison).
Figure 3.

Comparison of sortase reactions of α-mPECAM-1 scFv–LPETGG and scFv–linker–LPETGG (a) Kinetics, (b) calcium dependence, and (c) effect of ratio of sortase to scFv concentration on C-terminal modification of α-mPECAM-1 scFv–LPETGG and scFv–linker–LPETGG. The left panels show quantitation, and the right panels show SDS-PAGE analysis: *, p < 0.01 for α-mPECAM-1 scFv–linker–LPETGG vs scFv–LPETGG at each calcium concentration; **, p < 0.01 for α-mPECAM-1 scFv–linker–LPETGG vs scFv–LPETGG at 1:1 and 2:1 ratios of [SrtA]/[scFv]; #, p is not significant (0.03 for 4:1 and 0.41 for 8:1 ratios), with Bonferroni correction for multiple t tests.
Finally, various concentrations of sortase enzyme were tested, with the idea that diminished sortag accessibility might be overcome by increasing the ratio of [sortase] to [scFv]. Consistent with this hypothesis, reaction efficiency increased for α-mPECAM-1 scFv–LPETGG at higher ratios (Figure 3). In contrast, increasing the ratio of [sortase] to [scFv] actually decreased C-terminal modification of scFv–linker–LPETGG proteins (Figures 3 and S4 and Table S6). Interestingly, coomassie staining of these gels consistently showed complete elimination of unmodified protein, even as the amount of fluorescent product declined (Figure S4).
C-Terminal Modification Preservation of scFv Affinity and Selectivity.
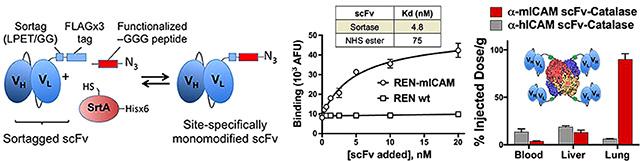
Having successfully purified site-specifically modified scFv–linker–LPETGG, we next sought to determine the effect of C-terminal modification on binding to target antigen. In contrast to affinities calculated from cell-based enzyme-linked immunosorbent assays (ELISAs; Table S1), which are influenced by the secondary anti-FLAG antibody, fluorescent labeling of the scFv allowed direct measurement of equilibrium binding parameters. Site-specifically modified scFv were compared to both their parental antibodies and scFv modified with nonselective amine-based chemistry. Figure 4A,B shows the results of flow cytometry based binding assays. In each case, the affinity of C-terminal modified scFv–linker–LPETGG was nearly an order of magnitude higher than NHS-ester modified scFv–linker–LPETGG, but similar to that of the NHS-ester modified fluorescent parental antibody (Figure S5).
Figure 4.

Affinity and specificity of C-terminal modified scFv–linker–LPETGG. Left panels show flow cytometry based binding curves of sitespecifically modified (a) α-mICAM-1 scFv and (a) α-hICAM-1 scFv to mouse and human ICAM-1 expressing cells, respectively. Binding is specific, with essentially no fluorescent signal seen on wild-type cells. The right panels show NHS-ester modified scFv–linker–LPETGG, which, despite low-level modification (degree of labeling of 1–2 fluorophores per scFv), demonstrates compromised binding affinities. (c) Immunofluorescence of cytokine-activated MS1 mouse endothelial cells shows a similar pattern of staining for site-specifically modified α-mICAM-1 scFv, as compared to fluorescent parental antibody, although the latter is somewhat brighter due to use of a different fluorophore (AlexaFluor 488 vs FITC) and a higher degree of modification. Staining is specific for mouse ICAM-1, with no signal seen for the non-species cross-reactive α-hICAM-1 scFv.
Binding of modified scFv–linker–LPETGG was also tested on MS1 mouse endothelial cells, which express low levels of ICAM-1 at baseline and significantly higher levels following activation with the inflammatory cytokine, TNF-α. Fluorescent staining by C-terminal modified α-mICAM-1 scFv–linker–LPETGG closely matched the pattern of staining seen with fluorescent parental antibody.
scFv Targeting of Catalase to Endothelial Cells in vitro and in vivo.
Having established the improved affinity of site-specifically modified scFv compared to traditional amine reactive modification, we took advantage of the C-terminal azide to achieve oriented conjugation to the antioxidant enzyme catalase, which had been modified to introduce dibenzocyclooctyne (DBCO) groups. Figure 5A shows the Cu-free click chemistry conjugation strategy.
Figure 5.

scFv-targeting of catalase to ICAM-1 in vitro and in vivo. (a) Schematic of bioconjugation reaction of site-specifically modified α-mICAM-1 scFv–linker–LPETGG (i.e., scFv–linker–FAM–azide) and DBCO-modified catalase to give α-mICAM-1 scFv-catalase. (b) Biodistribution (1 h) of α-mICAM-1 scFv-(125I–catalase) shows significant accumulation in lung, kidney, and spleen and decrease in the percent of injected dose per gram in the blood as compared to untargeted α-hICAM-1 scFv control (*, p < 0.005; **, p < 0.001; #, p = not significant or >0.0083, with Bonferroni correction). (c) Binding of α-mICAM-1 scFv–(125I–catalase) to cytokine activated MS1 cells (*, p < 0.01 for α-mICAM-1 scFv–catalase vs α-hICAM-1 scFv–catalase). (d) Protection of cytokine-activated MS1 cells from H2O2-mediated cell death. α-mICAM-1 scFv–catalase is more potent than untargeted controls, significantly reducing 51Cr release even at low doses (*, p < 0.05 for α-mICAM-1 scFv–catalase vs α-hICAM-1 scFv–catalase and DBCO–catalase).
Figure 5B shows the biodistribution of α-mICAM-1 scFv-catalase conjugates 1 h after intravenous injection, with accumulation seen in mouse lung, kidney, heart, and spleen. This pattern of distribution is typical of ICAM-1 targeted proteins and nanoparticles.42–44 The ability to bind activated endothelial cells was confirmed in vitro with mTNF-activated murine endothelial cells (Figure 5C), which bind α-mICAM-1 scFv-catalase conjugates but not α-hICAM-1 scFv targeted controls. As shown in Figure 5D, α-mICAM-1 scFv-catalase conjugates also protected MS1 cells from H2O2-mediated cell death, whereas α-hICAM-1 scFv-catalase conjugates were no different than untargeted catalase.
DISCUSSION
The clinical successes of monoclonal and bispecific antibodies and their drug conjugates45,46 have spurred interest in next generation targeting ligands, including engineered fragments and site-specific conjugation techniques. Apart from tumor cells, the vascular endothelium is perhaps the most logical target for therapeutic delivery, given its poor baseline uptake of drugs and involvement in a wide array of physiologic and pathophysiologic processes. Adhesion molecules expressed on the surface of endothelial cells are a natural choice for targeting due to their role in recruitment of leukocytes and other blood cells to areas of pathologic or inflamed vasculature.13 Indeed, drug carriers and macromolecules directed to these determinants accumulate within blood vessels and have shown the potential to alter coagulation, the inflammatory response, edema formation, and organ function.11,12,37,39,47
While promising as a theoretical pharmacologic strategy, vascular immunotargeting faces a number of practical hurdles to effective translation. Chief among these are frequent reliance on monoclonal antibodies as targeting moieties and the nearuniversal use of nonoriented, nonselective techniques for attachment of affinity ligands to therapeutic payload. These issues are interrelated because full-length antibodies are notably tolerant to amine- and thiol-based modification and bioconjugation. Unfortunately, antibodies are also uniquely designed to elicit immune responses, clearance upon opsonization of foreign material, and cross-linking of antigens, which, in the case of endothelial membrane proteins, may lead to endocytic uptake, cell activation, and barrier disruption.18,48–50 Over the past several years, a number of recombinant, high-affinity protein ligands have been developed for targeting constitutively expressed endothelial determinants, like PECAM-1 and ICAM-1, without the aforementioned disadvantages of full-length antibodies.35,36 These ligands maintain their structure and function when fused genetically to protein cargo and have shown the capacity for effective fusion protein targeting. Nonetheless, their function is readily compromised by nonspecific chemical modification, limiting their capacity for targeting other forms of therapeutics.
We report here the first steps toward integration of recombinant protein affinity ligands into a wide array of endothelial drug delivery systems: efficient, site-specific modification and covalent, oriented bioconjugation. Our initial efforts have focused on scFv, a natural choice for drug-delivery systems, given their direct relationship to monoclonal antibodies. The predictable protein fold and 3-D structure of scFv are advantageous for sortase modification as well, in that key determinants of reaction efficiency can theoretically be established for the entire class of proteins, cutting down on trial-and-error design and empiric testing of each new targeting ligand. With this in mind, we investigated sortase-mediated C-terminal modification of a series of three, distinct, endothelial-targeting scFv, identifying common themes and ultimately creating a platform for potential standardization of this process.
First, our data suggest that direct fusion of the sortag to the scFv C-terminus results in relatively inefficient modification and even small modifications of the native sequence ablate reactivity. The cause is presumably poor accessibility to the sortase enzyme, an explanation previously cited in the literature28 and supported by both our 3-D modeling and the increase in reaction efficiency seen with insertion of amino acids between VL and the sortag. A second conclusion comes from comparison of α-mPECAM-1 scFv–LPETGG and scFv–linker–LPETGG proteins, which differ in both the length and identity of the amino acids separating scFv and sortag. While altering calcium and sortase concentrations compensated for some of the gap in reaction efficiency, the longer, semi-rigid linker was superior across all conditions tested. Indeed, the uniform improvement in reaction efficiency following insertion of each of the scFv into the scFv–linker–LPETGG construct suggests its potential as a platform for high-efficiency C-terminal modification of all scFv. Consistent with this idea, certain conditions were found to be optimal across all three proteins tested: reaction at room temperature, calcium concentration greater than 100 μM, and a [sortase]-to-[scFv] ratio of no more than 1:1. Moreover, the modified scFv demonstrated preserved affinity for target antigen and remained functional, even after conjugation to a large therapeutic enzyme like catalase.
A platform for the standardization of C-terminal modification of scFv should allow rapid expansion of the repertoire of site-specifically modified targeting ligands and integration into a variety of preclinical drug delivery systems. A similar effort to standardize the C-terminal modification of human IgG1 heavy and κ light chains has recently been published and supports the notion of a platform approach for high-efficiency sortase modification of an entire class of structurally related proteins.51 Interestingly, the authors report that direct fusion of a sortag to the C-terminus of the heavy chain constant region is sufficient, whereas a short glycine–serine linker is needed to achieve similar modification of the kappa light chain. Having defined these parameters, the authors demonstrated an ~80% modification of 10 different monoclonal antibodies with a handful of pentaglycine-conjugated small-molecule drugs.
While clearly capable of producing high-affinity, sitespecifically modified endothelial-targeting scFv, some aspects of the sortase reaction with scFv–linker–LPETGG proteins require further study. For example, we did not investigate the effect of varying the [peptide]-to-[scFv] ratio on the efficiency or yield of the sortase reaction. Likewise, it is unknown why one of the proteins, α-hICAM-1 scFv–linker–LPETGG, shows a consistently lower yield of fluorescently modified protein (in comparison to α-mICAM-1 and α-mPECAM-1 scFv-linker–LPETGG) despite the apparent consumption of all starting material. The loss of yield is exacerbated by higher ratios of [sortase] to [scFv], suggesting hydrolysis of the sorting motif, a well-described side reaction in which the enzyme truncates the protein without ligating a –GGG peptide.52 This side reaction is likely also responsible for another perplexing result, the fact that reactions run at 37 °C produce less C-terminal modification (especially at later time points). Because the sortase reaction is reversible and the hydrolytic side reaction is not, the increase in temperature could skew the reaction toward truncation, rather than transpeptidation, of scFv–linker–LPETGG proteins. Previous reports have described a number of methods for reducing the impact of the side reaction, including increasing the concentration of –GGG peptide, altering the sequence of the sortag or surrounding amino acids, and the screening of sortase variants or mutants.53 These techniques or others that have been shown to increase ligation efficiency may need to be explored if hydrolytic side reactions are found to be a recurring limitation of scFv–linker–LPETGG proteins.54
EXPERIMENTAL PROCEDURES
Ethics.
Animal studies were carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted by the NIH, under protocol nos. 805837 and 805708 approved by University of Pennsylvania IACUC.
Cell Lines.
Stably transfected Drosophila S2 cells were maintained in Schneider’s complete medium (Thermo Fisher Scientific, Philadelphia, PA) with 25 μg/mL blasticidin (Invitrogen, Carlsbad, CA) and transitioned to serum free Insect-Xpress (Lonza, Walkersville, MD) supplemented with Glutamax and 0.5 mM CuSO4 for recombinant protein expression. Mouse and human ICAM-1 specific hybridomas (clones YN1/1.7.4 and R6.5) and the mouse endothelial cell line, MS1, were purchased from ATCC (Manassas, VA). Hybridomas were maintained in RPMI1640 with 25 mM HEPES, 10% FBS, and 1× antibiotic–antimycotic (Life technologies, Grand Island, NY). MS1 cells were grown in Dulbecco’s modified Eagle medium with 10% FBS and 1× antibiotic–antimycotic. Human umbilical vein endothelial cells (HUVECs) were purchased from Lonza (Walkersville, MD) and maintained using EGM BulletKit (Lonza, Walkersville, MD).
Antibodies and Other Reagents.
Monoclonal antibodies were purified from hybridoma supernatant using a protein G affinity column (Thermo Fisher Scientific). Recombinant mouse TNF-α was purchased from Corning (Corning, NY). Synthetic peptides with fluorophore- and azide-modified amino acids (Table S2) were purchased from Thermo Fisher Scientific. AlexaFluor488-NHS ester was purchased from Thermo Fisher Scientific. Bovine liver catalase was purchased from Sigma (Saint Louis, MO).
Cloning, Expression, and Purification of Bacterial sortase A.
The 59 amino acid truncated mutant of Staphylococcus aureus sortase A (Sa-SrtAΔ59)29 was polymerasechain reaction (PCR) amplified from pET28a (Addgene plasmid no. 51138) to append a N-terminal His6 tag and NdeI and EcoRI sites for ligation into the pRSET-A vector (Invitrogen). This eliminated the thrombin cleavage site separating the His6 tag from the SrtA protein in the original vector. The construct was transformed into competent Escherichia coli Shuffle cells (NEB). A fresh bacterial stab was inoculated into 500 mL of ZYP-5052 autoinduction media (Amresco, Solon, OH) containing ampicillin (100 μg/mL) and antifoam 204 (Sigma-Aldrich, Saint Louis, MO) and grown for 3–4 days at room temperature (RT). Following centrifugation, bacteria were lysed using a solution of B-PER (Thermo Fisher Scientific), 0.4 mg/mL lysozyme (Amresco), 0.4 μg/mL DNase (Sigma), and Complete Mini-EDTA-free protease inhibitor tablets (Sigma). Lysates were freeze–thawed, and the supernatant was purified using Ni-NTA agarose (Qiagen, Germantown, MD) per manufacturer protocol.
Cloning, Expression, Purification, and Modeling of Sortagged scFv.
The cDNA of mICAM-1, mPECAM-1, and hICAM-1 specific scFv35–37 were modified by PCR to add 5′ BglII and/or 3′ SalI sites and allow insertion into the vector pMT/LPETGG. This plasmid was derived from the S2 expression vector, pMT/BiP/V5-His, by addition of a sequence encoding a C-terminal SrtA recognition motif (LPETGG), triple FLAG tag, and stop codon to prevent the transcription of V5 and 6xHis tags. scFv cDNAs were also ligated into a second vector, pMT/linker–LPETGG, in which sequence encoding a 13 amino acid linker, (SSSSG)2SAA, had been added between the SalI site and the sortag (Figure S1).
Each scFv–LPETGG and scFv–linker–LPETGG plasmid was co-transfected with pCoBLAST in S2 cells and selected with blasticidin to generate stable cell lines. Expression and purification were performed as previously described.35 Briefly, proteins were harvested from S2 cell supernatant using an anti-FLAG (M2) affinity column (Sigma-Aldrich), and assessed for purity via SDS–PAGE and HPLC. Yields were approximately 1–2 mg protein/L supernatant. Predicted 3D scFv structures were created using the Rosie server. The .pdb files were analyzed using MacPyMol software.
SrtA Modification of Sortagged scFv Proteins and Analysis of Reaction Mixtures.
Conjugation of –GGG peptides to the C-terminus of sortagged scFv–LPETGG and scFv–linker–LPETGG proteins was performed as follows: 10 μM sortagged protein was mixed with an equimolar concentration of SrtA (i.e., 10 μM, unless otherwise specified), a 5-fold excess of –GGG peptide (50 μM), and 1 mM CaCl2 (unless otherwise specified) in Tris-buffered saline at pH 7.5. For convenience, reactions were left overnight (~16 h) at RT unless otherwise specified. Reaction mixtures were purified via incubation with Ni–NTA resin for 30 min at RT and centrifugation to remove sortase, followed by desalting on a 10DG column (BioRad, Hercules, CA), per manufacturer protocol, to remove excess –GGG peptide.
For analysis of reaction mixtures, SDS-PAGE gels were scanned using a Typhoon 9400 molecular imager (Amersham, Piscataway, NJ) and analyzed using ImageJ software. Reaction efficiency was calculated as:
where “fluorescence scFv” was the integrated intensity of the fluorescent band seen at the appropriate size for modified protein (~30 kDa for scFv–LPETGG and ~35 kDa for scFv–linker–LPETGG), and “fluorescence (scFv plus peptide)” was the sum of all fluorescent bands.
Reaction mixtures were also analyzed by HPLC using a Yarra 1.8 μm SEC-X150 LC column 150 × 4.6 mm with 0.1 M potassium phosphate buffer (pH 7.0) as a mobile phase and a flow rate of 0.35 mL/min. For each run, absorbance values at 495 nm were normalized using Prism 7.0 software (GraphPad, San Diego, CA), with 0 defined as no absorbance and 1.0 defined as the maximum absorbance within the data set. Reaction efficiency was calculated as:
where “AUC Abs495(scFv)” is the area under the curve (AUC) of the normalized absorbance peak at the appropriate elution time for modified scFv and “AUC Abs495(scFv plus peptide)” is the sum of AUCs for peaks corresponding to modified scFv and residual peptide. AUCs were calculated using Prism 7.0 software (GraphPad).
NHS Ester-Mediated Fluorescent Modification of Proteins.
Proteins were reacted for 1 h at RT in phosphate-buffered saline (PBS) adjusted to pH 8.3 with 1 M NaHCO3 buffer. A ratio of 1:4 mAb/NHS ester AlexaFluor488 and a ratio of 1:8 scFv/NHS ester AlexaFluor488 were found to produce an average degree of labeling of 1–2 fluorophores per protein, respectively.
Cell-Binding Assays and Microscopy.
ELISAs using M2 anti-FLAG antibody were performed on live cells as previously described.35 Flow-based binding assays were done using an Accuri C6 flow cytometer (BD Biosciences, San Jose, CA). For cell staining, MS1 or HUVEC cell monolayers were grown on 8-chamber glass μ-slides (Ibidi) coated with 50 μg/mL fibronectin. Cells were stimulated for 8 h with mouse human TNF (mTNF, 10 ng/mL), respectively, and stained with 20 nM fluorescently labeled scFv or antibody for 30 min at 37 °C. Cells were washed with media, fixed for 10 min with 2% paraformaldehyde, and then washed with PBS. Cells were permeabilized with 0.1% Triton X-100 and stained with DAPI (Life Technologies, Grand Island, NY).
Bioconjugation of Modified scFv to Catalase and Protection from H2O2 Injury.
Catalase at 20 mg/mL was modified with dibenzyocyclooctyne–(polyethylene glycol)4–N-hydroxysuccinimidyl ester (DBCO–PEG4–NHS ester) (Sigma-Aldrich) at a ratio of 1:20 for 1 h at RT and desalted using a 10-DG column. DBCO-modified catalase was incubated overnight at RT at a 1:4 ratio with GGGK[FAM]GGSK[N3]-modified α-mICAM-1 or α-hICAM-1 scFv or left unconjugated as a control.
Endothelial protection from H2O2 mediated injury was measured as previously described.55 Briefly, confluent monolayers of MS1 cells mouse ECs were stimulated with mTNF and loaded with 51Cr (~100000 counts per minute (cpm) per well) for 8 h at 37 °C. Cells were rinsed to remove free isotope and then treated with targeted scFv-catalase versus untargeted scFv–catalase versus unconjugated DBCO–catalase for 30 min, followed by 3 washes with serum-free media to remove nonspecifically bound protein. Cells were then incubated with 2 mM H2O2 in serum-free medium for 5 h at 37 °C and lysed with 1N NaOH and 0.1% Triton-X-100. 51Cr Radioactivity in cell supernatant versus lysate was measured using a WIZARD Automatic γ Counter (PerkinElmer, Waltham, MA), and the percent leakage was calculated as (cpm supernatant)/(cpm lysate plus supernatant).
Radiolabeling, Cell Binding, and Biodistribution of scFv–Catalase Conjugates.
Catalase was directly radioiodinated with [125I]NaI (PerkinElmer, Waltham, MA) using iodination beads and purified using Zeba desalting spin columns (Thermo Fisher Scientific). Radiolabeling efficiency was >95% and free iodine was <5% post-purification. DBCO-modified 125I–catalase was conjugated to azide-modified scFv at a 1:4 ratio overnight at RT.
For cell-binding experiments, MS1 cells were stimulated with mTNF for 8 h prior to incubation with scFv–catalase conjugates for 30 min at 37 °C. Cells were washed three times with media prior to lysis with 1N NaOH and 0.5% Triton-X-100. Radioactivity was measured in cell lysate (“bound”) versus supernatant and washes (“unbound”) using a WIZARD Automatic γ Counter (PerkinElmer, Waltham, MA). In all cases, >95% of the input activity was recovered.
For biodistribution, C57BL/6 mice weighing 20–30 g were anesthetized with 2–2.5% isoflurane. 125I–catalase–scFv conjugates were injected via jugular vein. Post-injection (1 h), blood was withdrawn from the inferior vena cava and animals were euthanized. Organs, including lung, liver, kidney, spleen, heart, and thyroid tissues, were harvested and weighed and radioactivity was measured by γ-counting.
Data Analysis and Statistics.
Significant differences between means were determined using Prism 6.0 software (GraphPad, La Jolla, CA). Either t tests (for comparison of two groups) or one-way analysis of variance (ANOVAs, for the comparison of multiple groups) was performed, followed by an appropriate multiple comparison test and calculation of multiplicity adjusted p values. For ANOVAs, Tukey’s multiple comparison test was used, whereas a Bonferroni correction was used in the case of multiple t tests.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a Center for Targeted Therapeutics and Translational Nanomedicine (CT3N) Pilot Grant Award (ITMAT) at the University of Pennsylvania (C.F.G.). The work is also supported by the National Institute of Health grant nos. T32 HL007439 (C.F.G.), K08-HL130430 (C.F.G.), R01-HL-128398 (V.R.M.), R01-HL-126874 (V.R.M.), R21-CA187657 (A.T.), and R21-EB018863 (A.T.).
ABBREVIATIONS
- ECs
endothelial cells
- scFv
single-chain antibody fragments
- PECAM-1
platelet–endothelial cell adhesion molecule 1
- ICAM-1
intercellular adhesion molecule 1
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bioconjchem.7b00592.
Figures showing molecular design of sortagged scFv proteins; SEC–LC, SDS-PAGE, and HPLC analysis; and flow-binding assays. Tables showing antigenic specificity and affinities, peptide characteristics, and reaction efficiencies of C-terminal scFv modification. (PDF)
REFERENCES
- (1).Aird WC (2003) The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 101, 3765–3777. [DOI] [PubMed] [Google Scholar]
- (2).Maniatis NA, and Orfanos SE (2008) The endothelium in acute lung injury/acute respiratory distress syndrome. Curr. Opin Crit Care 14, 22–30. [DOI] [PubMed] [Google Scholar]
- (3).Sánchez-Aranguren LC, Prada CE, Riaño-Medina CE, and Lopez M (2014) Endothelial dysfunction and preeclampsia: role of oxidative stress. Front. Physiol 5 (372), 1 DOI: 10.3389/fphys.2014.00372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Al-Lamki RS, Bradley JR, and Pober JS (2008) Endothelial cells in allograft rejection. Transplantation 86, 1340–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Callaway CW (2012) Endothelial damage after cardiac arrest—”Endotheliitis. Resuscitation 83, 667–668. [DOI] [PubMed] [Google Scholar]
- (6).Brenner JS, Greineder C, Shuvaev V, and Muzykantov V (2015) Endothelial nanomedicine for the treatment of pulmonary disease. Expert Opin. Drug Delivery 12, 239–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Cantelmo AR, Conradi L-C, Brajic A, Goveia J, Kalucka J, Pircher A, Chaturvedi P, Hol J, Thienpont B, Teuwen L-A, et al. (2016) Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 30, 968–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).McIntosh DP, Tan X-Y, Oh P, and Schnitzer JE (2002) Targeting endothelium and its dynamic caveolae for tissue-specific transcytosis in vivo: a pathway to overcome cell barriers to drug and gene delivery. Proc. Natl. Acad. Sci. U. S. A 99, 1996–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Muzykantov VR, Christofidou-Solomidou M, Balyasnikova I, Harshaw DW, Schultz L, Fisher AB, and Albelda SM (1999) Streptavidin facilitates internalization and pulmonary targeting of an anti-endothelial cell antibody (platelet-endothelial cell adhesion molecule 1): A strategy for vascular immunotargeting of drugs. Proc. Natl. Acad. Sci. U. S. A 96, 2379–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Muro S, Dziubla T, Qiu W, Leferovich J, Cui X, Berk E, and Muzykantov VR (2006) Endothelial Targeting of High-Affinity Multivalent Polymer Nanocarriers Directed to Intercellular Adhesion Molecule 1. J. Pharmacol. Exp. Ther 317, 1161–1169. [DOI] [PubMed] [Google Scholar]
- (11).Shuvaev VV, Christofidou-Solomidou M, Bhora F, Laude K, Cai H, Dikalov S, Arguiri E, Solomides CC, Albelda SM, Harrison DG, et al. (2009) Targeted detoxification of selected reactive oxygen species in the vascular endothelium. J. Pharmacol. Exp. Ther 331, 404–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Hood ED, Greineder CF, Dodia C, Han J, Mesaros C, Shuvaev VV, Blair IA, Fisher AB, and Muzykantov VR (2012) Antioxidant protection by PECAM-targeted delivery of a novel NADPH-oxidase inhibitor to the endothelium in vitro and in vivo. J. Controlled Release 163, 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Muzykantov VR (2013) Targeted Drug Delivery to Endothelial Adhesion Molecules. ISRN Vasc. Med 2013, 1–27. [Google Scholar]
- (14).Lindmo T, Boven E, Cuttitta F, Fedorko J, and Bunn PA (1984) Determination of the immunoreactive function of radiolabeled monoclonal antibodies by linear extrapolation to binding at infinite antigen excess. J. Immunol. Methods 72, 77–89. [DOI] [PubMed] [Google Scholar]
- (15).Stephanopoulos N, and Francis MB (2011) Choosing an effective protein bioconjugation strategy. Nat. Chem. Biol 7, 876–884. [DOI] [PubMed] [Google Scholar]
- (16).Wu AM, and Senter PD (2005) Arming antibodies: prospects and challenges for immunoconjugates. Nat. Biotechnol 23, 1137–1146. [DOI] [PubMed] [Google Scholar]
- (17).Ricklin D, Hajishengallis G, Yang K, and Lambris JD (2010) Complement: a key system for immune surveillance and homeostasis. Nat. Immunol 11, 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Mei H, Campbell JM, Paddock CM, Lertkiatmongkol P, Mosesson MW, Albrecht R, and Newman PJ (2014) Regulation of Endothelial Cell Barrier Function by Antibody-driven Affinity Modulation of Platelet Endothelial Cell Adhesion Molecule-1 (PECAM-1). J. Biol. Chem 289, 20836–20844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Park S, Kang S, Veach AJ, Vedvyas Y, Zarnegar R, Kim J-Y, and Jin MM (2010) Self-assembled nanoplatform for targeted delivery of chemotherapy agents via affinity-regulated molecular interactions. Biomaterials 31, 7766–7775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Goldberg SD, Cardoso RMF, Lin T, Spinka-Doms T, Klein D, Jacobs SA, Dudkin V, Gilliland G, and O’Neil KT (2016) Engineering a targeted delivery platform using Centyrins. Protein Eng., Des. Sel 29, 563–572. [DOI] [PubMed] [Google Scholar]
- (21).Aragnol D, and Leserman LD (1986) Immune clearance of liposomes inhibited by an anti-Fc receptor antibody in vivo. Proc. Natl. Acad. Sci. U. S. A. 83, 2699–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Albrecht H, Burke PA, Natarajan A, Xiong C-Y, Kalicinsky M, DeNardo GL, and DeNardo SJ (2004) Production of Soluble ScFvs with C-Terminal-Free Thiol for Site-Specific Conjugation or Stable Dimeric ScFvs on Demand. Bioconjugate Chem. 15, 16–26. [DOI] [PubMed] [Google Scholar]
- (23).Constantinou A, Epenetos AA, Hreczuk-Hirst D, Jain S, Wright M, Chester KA, and Deonarain MP (2009) Site-Specific Polysialylation of an Antitumor Single-Chain Fv Fragment. Bioconjugate Chem. 20, 924–931. [DOI] [PubMed] [Google Scholar]
- (24).Messerschmidt SKE, Kolbe A, Müller D, Knoll M, Pleiss J, and Kontermann RE (2008) Novel Single-Chain Fv′ Formats for the Generation of Immunoliposomes by Site-Directed Coupling. Bioconjugate Chem. 19, 362–369. [DOI] [PubMed] [Google Scholar]
- (25).Agarwal P, and Bertozzi CR (2015) Site-Specific Antibody–Drug Conjugates: The Nexus of Bioorthogonal Chemistry, Protein Engineering, and Drug Development. Bioconjugate Chem. 26, 176–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Greineder CF, Hood ED, Yao A, Khoshnejad M, Brenner JS, Johnston IH, Poncz M, Gottstein C, and Muzykantov VR (2016) Molecular engineering of high affinity single-chain antibody fragment for endothelial targeting of proteins and nanocarriers in rodents and humans. J. Controlled Release 226, 229–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Mao H, Hart SA, Schink A, and Pollok BA (2004) Sortase-Mediated Protein Ligation: A New Method for Protein Engineering. J. Am. Chem. Soc 126, 2670–2671. [DOI] [PubMed] [Google Scholar]
- (28).Guimaraes CP, Witte MD, Theile CS, Bozkurt G, Kundrat L, Blom AEM, and Ploegh HL (2013) Site-specific C-terminal and internal loop labeling of proteins using sortase-mediated reactions. Nat. Protoc 8, 1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Warden-Rothman R, Caturegli I, Popik V, and Tsourkas A (2013) Sortase-tag expressed protein ligation: combining protein purification and site-specific bioconjugation into a single step. Anal. Chem. 85, 11090–11097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Swee LK, Guimaraes CP, Sehrawat S, Spooner E, Barrasa MI, and Ploegh HL (2013) Sortase-mediated modification of αDEC205 affords optimization of antigen presentation and immunization against a set of viral epitopes. Proc. Natl. Acad. Sci. U. S. A 110, 1428–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Massa S, Vikani N, Betti C, Ballet S, Vanderhaegen S, Steyaert J, Descamps B, Vanhove C, Bunschoten A, van Leeuwen FWB, et al. (2016) Sortase A-mediated site-specific labeling of camelid single-domain antibody-fragments: a versatile strategy for multiple molecular imaging modalities. Contrast Media Mol. Imaging 11, 328–339. [DOI] [PubMed] [Google Scholar]
- (32).Hagemeyer CE, Alt K, Johnston APR, Such GK, Ta HT, Leung MKM, Prabhu S, Wang X, Caruso F, and Peter K (2014) Particle generation, functionalization and sortase A–mediated modification with targeting of single-chain antibodies for diagnostic and therapeutic use. Nat. Protoc 10, 90–105. [DOI] [PubMed] [Google Scholar]
- (33).Chen I, Dorr BM, and Liu DR (2011) A general strategy for the evolution of bond-forming enzymes using yeast display. Proc. Natl. Acad. Sci. U. S. A 108, 11399–11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Chen L, Cohen J, Song X, Zhao A, Ye Z, Feulner CJ, Doonan P, Somers W, Lin L, and Chen PR (2016) Improved variants of SrtA for site-specific conjugation on antibodies and proteins with high efficiency. Sci. Rep 6, 1 DOI: 10.1038/srep31899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Greineder CF, Chacko A-M, Zaytsev S, Zern BJ, Carnemolla R, Hood ED, Han J, Ding B-S, Esmon CT, and Muzykantov VR (2013) Vascular Immunotargeting to Endothelial Determinant ICAM-1 Enables Optimal Partnering of Recombinant scFv-Thrombomodulin Fusion with Endogenous Cofactor. PLoS One 8, e80110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Greineder CF, Brenza JB, Carnemolla R, Zaitsev S, Hood ED, Pan DC, Ding B-S, Esmon CT, Chacko AM, and Muzykantov VR (2015) Dual targeting of therapeutics to endothelial cells: collaborative enhancement of delivery and effect. FASEB J. 29, 3483–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Greineder CF, Johnston IH, Villa CH, Gollomp K, Esmon CT, Cines DB, Poncz M, and Muzykantov VR (2017) ICAM-1–targeted thrombomodulin mitigates tissue factor–driven inflammatory thrombosis in a human endothelialized microfluidic model. Blood Advances 1, 1452–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Lyskov S, Chou F-C, Conchúir SÓ, Der BS, Drew K, Kuroda D, Xu J, Weitzner BD, Renfrew PD, Sripakdeevong P, et al. (2013) Serverification of Molecular Modeling Applications: The Rosetta Online Server That Includes Everyone (ROSIE). PLoS One 8, e63906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Ding B-S, Hong N, Christofidou-Solomidou M, Gottstein C, Albelda SM, Cines DB, Fisher AB, and Muzykantov VR (2009) Anchoring fusion thrombomodulin to the endothelial lumen protects against injury-induced lung thrombosis and inflammation. Am. J. Respir. Crit. Care Med 180, 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Zaitsev S, Spitzer D, Murciano J-C, Ding B-S, Tliba S, Kowalska MA, Marcos-Contreras OA, Kuo A, Stepanova V, Atkinson JP, et al. (2010) Sustained thromboprophylaxis mediated by an RBC-targeted pro-urokinase zymogen activated at the site of clot formation. Blood 115, 5241–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Zaitsev S, Kowalska MA, Neyman M, Carnemolla R, Tliba S, Ding B-S, Stonestrom A, Spitzer D, Atkinson JP, Poncz M, et al. (2012) Targeting recombinant thrombomodulin fusion protein to red blood cells provides multifaceted thromboprophylaxis. Blood 119, 4779–4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Murciano J-C, Muro S, Koniaris L, Christofidou-Solomidou M, Harshaw DW, Albelda SM, Granger DN, Cines DB, and Muzykantov VR (2003) ICAM-directed vascular immunotargeting of antithrombotic agents to the endothelial luminal surface. Blood 101, 3977–3984. [DOI] [PubMed] [Google Scholar]
- (43).Rossin R, Muro S, Welch MJ, Muzykantov VR, and Schuster DP (2007) In vivo Imaging of 64Cu-Labeled Polymer Nanoparticles Targeted to the Lung Endothelium. J. Nucl. Med 49, 103–111. [DOI] [PubMed] [Google Scholar]
- (44).Zern BJ, Chacko A-M, Liu J, Greineder CF, Blankemeyer ER, Radhakrishnan R, and Muzykantov V (2013) Reduction of Nanoparticle Avidity Enhances the Selectivity of Vascular Targeting and PET Detection of Pulmonary Inflammation. ACS Nano 7, 2461–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Alley SC, Okeley NM, and Senter PD (2010) Antibodydrug conjugates: targeted drug delivery for cancer. Curr. Opin. Chem. Biol 14, 529–537. [DOI] [PubMed] [Google Scholar]
- (46).Kontermann R (2012) Dual targeting strategies with bispecific antibodies. mAbs 4, 182–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Howard MD, Greineder CF, Hood ED, and Muzykantov VR (2014) Endothelial targeting of liposomes encapsulating SOD/catalase mimetic EUK-134 alleviates acute pulmonary inflammation. J. Controlled Release 177, 34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Chiba R, Nakagawa N, Kurasawa K, Tanaka Y, Saito Y, and Iwamoto I (1999) Ligation of CD31 (PECAM-1) on Endothelial Cells Increases Adhesive Function of vβ3 Integrin and Enhances β1 Integrin-Mediated Adhesion of Eosinophils to Endothelial Cells. Blood 94, 1319–1329. [PubMed] [Google Scholar]
- (49).Van Buul JD, van Rijssel J, van Alphen FPJ, van Stalborch A-M, Mul EPJ, and Hordijk PL (2010) ICAM-1 Clustering on Endothelial Cells Recruits VCAM-1. J. Biomed. Biotechnol 2010, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Muro S, Wiewrodt R, Thomas A, Koniaris L, Albelda SM, Muzykantov VR, and Koval M (2003) A novel endocytic pathway induced by clustering endothelial ICAM-1 or PECAM-1. J. Cell Sci 116, 1599–1609. [DOI] [PubMed] [Google Scholar]
- (51).Beerli RR, Hell T, Merkel AS, and Grawunder U (2015) Sortase Enzyme-Mediated Generation of Site-Specifically Conjugated Antibody Drug Conjugates with High In vitro and In vivo Potency. PLoS One 10, e0131177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Möhlmann S, Mahlert C, Greven S, Scholz P, and Harrenga A (2011) In vitro Sortagging of an Antibody Fab Fragment: Overcoming Unproductive Reactions of Sortase with Water and Lysine Side Chains. ChemBioChem 12, 1774–1780. [DOI] [PubMed] [Google Scholar]
- (53).Heck T, Pham P-H, Yerlikaya A, Thöny-Meyer L, and Richter M (2014) Sortase A catalyzed reaction pathways: a comparative study with six SrtA variants. Catal. Sci. Technol 4, 2946–2956. [Google Scholar]
- (54).Wang HH, Altun B, Nwe K, and Tsourkas A (2017) Proximity-Based Sortase-Mediated Ligation. Angew. Chem., Int. Ed 56, 5349–5352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Hood ED, Chorny M, Greineder CF, Alferiev IS, Levy RJ, and Muzykantov VR (2014) Endothelial targeting of nanocarriers loaded with antioxidant enzymes for protection against vascular oxidative stress and inflammation. Biomaterials 35, 3708–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.