

Abstract
On-surface synthesis is appearing as an extremely promising research field aimed at creating new organic materials. A large number of chemical reactions have been successfully demonstrated to take place directly on surfaces through unusual reaction mechanisms. In some cases the reaction conditions can be properly tuned to steer the formation of the reaction products. It is thus possible to control the initiation step of the reaction and its degree of advancement (the kinetics, the reaction yield); the nature of the reaction products (selectivity control, particularly in the case of competing processes); as well as the structure, position, and orientation of the covalent compounds, or the quality of the as-formed networks in terms of order and extension. The aim of our review is thus to provide an extensive description of all tools and strategies reported to date and to put them into perspective. We specifically define the different approaches available and group them into a few general categories. In the last part, we demonstrate the effective maturation of the on-surface synthesis field by reporting systems that are getting closer to application-relevant levels thanks to the use of advanced control strategies.
1. Introduction
On-surface synthesis is a newly developing field of research that aims at making use of well-defined solid surfaces as confinement templates to initiate chemical reactions.1−10 The concepts of supramolecular chemistry are applied here to provide well-defined functional surfaces from the “bottom-up” self-assembly of nanometer-sized elementary building-blocks.11 On-surface synthesis represents in a sense an extension of heterogeneous catalysis whereby the initial precursors, the intermediate states, and the reaction products all remain in an adsorbed state, usually in the submonolayer regime. The interest in creating covalent nanoarchitectures directly on surfaces is manifold.12 On-surface synthesis gives access to original reaction mechanisms in mild conditions that would be not easily accessible in standard chemistry conditions,13,14 such as the alkane polymerization,15 the formation of oligophenylene macrocycles,16,17 or the synthesis of polyacenes,18−26 radialene,27 or arynes.28,29 Also, it represents an efficient route to the formation of robust organic networks and 2D polymers.30,31 Finally, the full range of available surface science techniques can deliver exquisite characterization of the different reaction processes with atomic precision.2,32,33
A few examples of surface-supported coupling reactions were reported already in the late 1990s and early 2000s,34−37 but the on-surface synthesis field has really emerged with the seminal work of Grill and co-workers in 2007 demonstrating the covalent coupling of brominated tetraphenyl-porphyrins in ultra-high-vacuum (UHV) environment.38 Rapidly, a wide range of coupling schemes has been proposed, sometimes however with limited success and reproducibility. The quest for effective control and the demonstration of real programing abilities of on-surface synthesis are now crucial to the development of this field and its future applicability in actual devices.39 Most of the processes presented have been reported only for very specific systems, with few case exceptions, and can therefore hardly be considered as general rules. However, thanks to the abundant literature, general tendencies can be drawn that can certainly be described as real strategies and effective tools available to the community. In fact, many elaborate experimental works leading to efficient control of the reaction products have been developed, and they can be classified into a few well-defined categories that are presented in this review. Note that a few review articles presenting particular strategies have been recently proposed, such as the control on terminal alkyne coupling,40 the self-assembly strategy,41 or the use of surface templates,42 thus revealing the need for extracting a general rationale in this topical issue.
The scope of the present review is limited to supported chemical reactions leading to an effective covalent coupling between organic precursors (mainly the formation of C–C or C–N bonds). It is not the aim of this review to describe all existing reactions reported in the literature. For an extensive description of all chemical systems, many reviews are available; see, e.g., recent references (1−10). Also, the reaction mechanisms will in general not be detailed here. In fact they have been scarcely studied, only on a few particular cases. They appeared to be very dependent on the specific molecule–substrate system, and it seems very difficult to extract general tendencies. The reader can refer to published studies for theoretical43−49 or experimental50−55 insights.
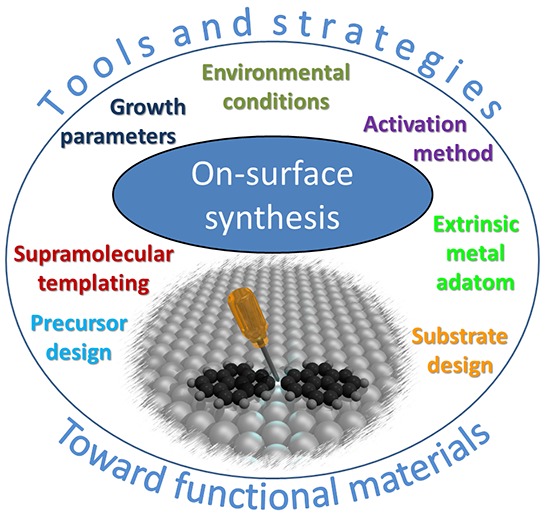
The proposed tools and strategies that are developed in this review consist of control of the reaction initiation, the activation process (e.g., light-induced reactions or the tuning of activation temperature), the reaction yield, or control of the reaction type and the reaction product (selectivity, product configuration). Figure 1 represents the various parameters that are experimentally available to gain such control. A few general categories can be drawn: the precursor and the substrate designs, the use of extrinsic metal adatoms, the supramolecular templating effect, the control on the growth parameters, the environmental control, and the choice of the activation method. We propose here to specifically define the different approaches available and to categorize them. The different experimental studies found in the literature will be listed and classified into each of these categories. Additionally, we demonstrate the effective maturation of the on-surface synthesis field by reporting some systems that are getting closer to application-relevant levels thanks to the use of advanced control strategies. The example of graphene nanoribbons (GNRs) is particularly illustrative due to its wide flexibility and versatility and to the high level of control achieved. Finally, the possibility of measuring properties at the single-molecule level, the development of surface nanopatterning methods, the possibility of growing covalent systems on technology-relevant insulating substrates, and the first attempts to create functional devices based on an on-surface synthesis technique will be detailed altogether in the last section.
Figure 1.

Overview of the diversity of available parameters that can be tuned to gain efficient control on the reaction products during on-surface synthesis. The maturation of the field is demonstrated by the development of efficient methods aimed at creating device-ready functional materials.
Abbreviations for molecular names are used whenever they were found in the original articles. The term Ullmann coupling was used in the general sense of surface-supported dehalogenative C–C coupling. Also, the term Glaser coupling was used in the sense of homocoupling of terminal alkynes.
2. General Concepts for On-Surface Synthesis
On-surface synthesis can be considered as a newly developing type of chemistry that nicely complements our current synthetic capabilities. However, from a methodological point of view, there are much stronger differences with respect to conventional chemistry than a newcomer to the field may imagine. The confinement to two dimensions and the often-used vacuum environment, consequently with an absolute absence of solvents, dramatically change the range of chemical reactions and the experimental techniques that can be applied to characterize the outcomes. In the following we briefly provide some basic notions about these two aspects.
2.1. Reaction Schemes
An important number of review articles present catalog-type descriptions of the state of the art, with regard to the reactions successfully applied to date in on-surface synthesis (see, e.g., recent refs (1−10)). Although not aiming here at the presentation of an exhaustive list of those reactions and their descriptions, we would still like to draw special attention to a few reactions that have been recognized as particularly efficient and robust. In this respect, the unquestionable leader is the Ullmann-type coupling or more generally the dehalogenative coupling of reactants. Indeed, although mechanistically differing from the solution based Ullmann coupling, the halogen activation in surface-supported organic species, ending with bond formation between those activated carbon sites, is often termed as Ullmann-like. However, it should be noted that this broad group of reactions includes important variations. By way of example, such reactions have been successfully demonstrated not only with aryl halides (Figure 2a),1 but also with alkyl (Figure 2b),56 alkenyl (Figure 2c),57 or alkynyl halides (Figure 2d).58 Furthermore, the use of dihalogenated or trihalogenated carbon atoms has been shown to allow for the homocoupling with double (Figure 2e)59 or triple bonds (Figure 2f),60 respectively. As with other reactions, and more explicitly discussed in section 6, also here the supporting surface plays a key role, displaying different mechanisms and reaction evolution (e.g., the presence or absence of metastable organometallic intermediates) depending on the substrate of choice.
Figure 2.

Schematic drawings of selected reactions commonly used in on-surface synthesis, grouped as dehalogenative coupling (a–f), alkyne coupling (g–i), condensation reactions (j, k), direct C–H activation (l, m), and intramolecular dehydrogenation (n).
Among the next most studied reactions we find alkyne coupling, which again embraces a relatively large variety of (often competing) reactions. Thus, although they are generally grouped as Glaser-type reactions, in analogy to the wet-chemistry dehydrogenative alkyne homocoupling (Figure 2g), several other reaction schemes including nondehydrogenative coupling (Figure 2h) and cyclotrimerization (Figure 2i) typically compete.61 Routes to steer and maximize the selectivity toward particular reactions will be discussed in later sections.
Other popular coupling reactions are the condensation of boronic acids (Figure 2j),62,63 and the Schiff base reaction or the formation of imines from the condensation of aldehydes with amines (Figure 2k).64,65 Although again stressing that the list of successfully run reactions on surfaces is much more extensive and treated in greater detail in previous reviews,1−10 we would like to list one more reaction as particularly common, namely, the direct C–H activation of aryls (Figure 2l)66 or alkyls (Figure 2m).15
Note that the above-mentioned reactions are the basis for most of the examples presented in later sections of this review, focused on coupling reactions that result in larger, generally more complex products. Instead, because intramolecular dehydrogenation (Figure 2n) does not lead to molecular coupling, it is hardly addressed in later sections. However, its relevance should be explicitly acknowledged, being the basis for many of the milestones along the development of “on-surface synthesis” in a broader perspective. The following are offered by way of examples: it allowed in one of the early works in 2008 the selective synthesis of fullerenes from aromatic precursor molecules,67 it is a key step in the synthesis of graphene nanoribbons68 or nanographenes,69 and it has allowed the study of highly reactive species like arynes28,29,70 as well as the synthesis of elusive molecules like triangulene71 or higher acenes.19−26
2.2. Characterization and Analysis Techniques
One of the many differences between conventional chemistry and on-surface synthesis is the characterization tools applied within each field. For the latter, the intrinsic presence of surfaces and the often-used vacuum environment place the full battery of typical surface science characterization techniques at the service of this research field. Among them, by far the most popular characterization tool utilized in on-surface chemistry is scanning probe microscopy, which allows characterization molecule by molecule. Thus, this readily sets up an important difference with respect to conventional chemistry, which relies to date solely on ensemble-averaging techniques. Nevertheless, the identification of chemical structures from scanning probe microscopy is not straightforward. Most commonly used is scanning tunneling microscopy, which probes the spatial distribution of low-energy electronic states and is therefore not directly related to the bonding structure. However, several routes have been developed to unmask it. One is the comparison of STM images of the in situ obtained products with those of ex situ prepared products subsequently deposited onto the surface (Figure 3a).72 A similar imaging contrast under equal scanning conditions is evidence of a right assignment. Alternatively, the contrast that a given structure should give in STM can be calculated and compared to the experiment (Figure 3b),73 again supporting a correct assignment if a good agreement is found. In 2009, a breakthrough in scanning probe microscopy allowed the direct visualization of covalent bonding structures by noncontact atomic force microscopy (nc-AFM) with functionalized probes,74 later on becoming an increasingly common approach to probe the chemical structures of reaction intermediates and end products (Figure 3d).33,55,75 The same probe functionalization idea has been applied afterward to STM, with similar success in the resolution of the bonding structures (Figure 3c).25,76,77
Figure 3.

Product structure determination from STM images by comparison of (a) in situ and ex situ synthesized molecules and (b) experimental results with the calculated contrast for a given molecular structure. (c) High-resolution bond-resolving imaging with functionalized probes in STM mode. (d) High-resolution bond-resolving imaging with functionalized probes in nc-AFM mode (further compared to conventional STM imaging and to the chemical structures). (a) Reprinted with permission from ref (72). Copyright 2013 American Chemical Society. (b) Reprinted by permission from ref (73). Copyright 2012 Springer Nature. (c) Reprinted with permission from ref (25). Copyright 2018 Royal Society of Chemistry. (d) Reprinted with permission from ref (33). Copyright 2013 AAAS.
It should be mentioned here that tip manipulation is widely used as proof to confirm the covalent character of the surface-synthesized structures.15,27,38,66,78−94 Covalently coupled compounds or chains are shown to be displaced as a whole without breaking by tip manipulation, while supramolecular architectures would be expected to get destroyed due to lower cohesion interaction. This latter issue remains questionable however because hydrogen-bonded95 or metal–organic91,94,96,97 motifs can also be successfully manipulated and displaced as a whole without destruction. In particular, the organometallic intermediate and the covalent product of terminal alkyne derivatives could be manipulated in a similar way on Ag(111).91
Although in this research field the determination of the chemical bonding structure may be the most relevant use of scanning probe microscopy, it is important to note that it can also be used to probe a variety of physical properties.98 By way of example, scanning tunneling spectroscopy or Kelvin probe microscopy provides information on the energetic and spatial distribution of specific orbitals or on the total electron density, respectively.
However, beyond these single-molecule characterization techniques, ensemble-averaging techniques have provided similarly important information for the development of the field. One such example is core level photoemission (XPS), in which the signal is directly dependent on the chemical bonding environment of the atoms and thus perfectly suited to track chemical changes. By way of example, the graphene nanoribbon synthesis process outlined in Figure 4a has been studied by XPS.50 It is a two-step reaction in which the reactant first polymerizes by Ullmann-type coupling and then, at higher temperatures, transforms into planar GNRs through cyclodehydrogenation. Each of the chemical changes leaves certain fingerprints in the involved atoms, as evidenced in the temperature-dependent C 1s and Br 3d spectra shown in Figure 4b,c, on top of which the dashed lines mark the temperatures associated in that work to each of the chemical processes of the GNR synthesis.50 Similar temperature-dependent data have been subsequently measured with higher resolution, whereby detailed insight into the GNR growth dynamics99 or the influence of the halogen type100 could be obtained.
Figure 4.

(a) Reaction scheme for the synthesis of graphene nanoribbons by an initial polymerization and subsequent cyclodehydrogenation together with the C 1s (b) and Br 3d (c) core level spectra as a function of the sample annealing temperature, evidencing the chemical changes. (d) NEXAFS spectra of the precursor molecule (blue, 0 °C) and GNR (red, 400 °C) under s-polarization (dashed) or p-polarization incidence of the X-rays. The inset includes spectra at intermediate temperatures (0, 120, 270, and 400 °C). (e) Time-resolved mass spectrometry at increasing temperatures monitoring the halogen desorption during the reaction scheme depicted in part a, as well as the associated integrated core level intensity of the carbon and halogen signals (f). (g) Raman breathing mode energy dependence on GNR width. (h) High-resolution electron energy loss spectroscopy of the polymers and GNRs depicted in part a. (i) Angle-resolved photoemission spectroscopy data of dibromo-terphenyl before and after polymerization into poly(paraphenylene). (a–d) Reproduced with permission from ref (50). Copyright 2014 Royal Society of Chemistry. (e, f) Reproduced with permission from ref (99). Copyright 2018 American Chemical Society. (g) Reproduced with permission from ref (107). Copyright 2018 American Chemical Society. (h) Reprinted with permission from ref (105). Copyright 2012 American Physical Society. (i) Reproduced with permission from ref (102). Copyright 2016 American Chemical Society.
Another classical experimental technique in surface science is near edge X-ray absorption spectroscopy (NEXAFS), which thus has also been applied to on-surface synthesis. This technique basically probes electronic transitions between core levels and unoccupied molecular orbitals.101 As discussed above, the former depend on the chemical environment of the atoms, and in a less trivial way so do the latter. Importantly, the signal depends not solely on the energy of the involved orbitals, but also on their alignment with respect to the polarization direction of the exciting light. Consequently, by varying that relative alignment NEXAFS data provide provide key information not only on the electronic properties of the probed samples, but also on their structure and adsorption geometry.101 By way of example, Figure 4d shows NEXAFS data on the same materials discussed above and depicted in Figure 4a. Without getting into the details, the changes in the measured resonances provide evidence for the changes in the electronic properties between the reactant molecule (blue curves) and the GNR (red curves). Instead, the polarization dependence (dashed vs full lines) holds information on the adsorbate’s adsorption geometry. The GNR is planar and lies flat on the substrate surface, which causes the π-resonances to appear intense under p-polarization and is virtually absent under s-polarization. Instead, the nonplanarity of the reactant molecule, brought about by the steric hindrance of the H atoms of neighboring anthracene units, causes the polarization dependence to be much weaker.
Mass spectroscopy has also been applied in on-surface synthesis studies. An example thereof is depicted in Figure 4e,99 in which the desorption of the halogen byproducts resulting from the Ullmann coupling is monitored as a function of the sample temperature. The results show excellent agreement with the simultaneously measured XPS data of the corresponding halogens (Figure 4f).99
Another technique that has been successfully applied to monitor the chemical changes of the molecules is ultraviolet photoemission spectroscopy (UPS), most powerful when measured with angular resolution (ARPES). This can be nicely observed in Figure 4i, displaying a clear change in the photoemission when comparing the as-deposited dibromo-terphenyl reactant molecules with the resultant poly(paraphenylene) polymers after Ullmann coupling on a TiO2 surface. The flat bands associated with the electron confinement within the molecules evolve into a strongly dispersive band as the electrons delocalize along the conjugated wire (Figure 4i).102 Thus, besides providing unambiguous evidence of the chemical changes, ARPES also provides important information on the electronic properties of the materials at each of their stages along a reaction.102−104
Fingerprints on particular chemical structures (and therefore on their changes) can also be obtained from vibrational spectroscopy. One way to address that information is, e.g., high-resolution electron energy loss spectroscopy (HREELS), whereby the energy loss of monochromatic electrons is analyzed as they interact with the probed molecules and excite, among others, their vibrational modes. Focusing again on the same reaction scheme depicted in Figure 4a, the HREELS data after the first polymerization step and after the second cyclodehydrogenation step are compared in Figure 4h, evidencing clear differences and thus the suitability of this technique to track the reactions.105 An alternative technique to assess the material’s vibrations is Raman spectroscopy. In fact, this technique has become particularly important in one of the most booming research directions within on-surface synthesis: the growth of atomically precise graphene nanoribbons. As readily happened with the “parent material” graphene,106 Raman spectroscopy has appeared as an extremely versatile tool to assess the ribbon’s properties, ranging from the edge orientation to the nanoribbon width (Figure 4g),107 or the overall sample quality.106−111 Moreover, because Raman spectroscopy does not require ultra-high-vacuum conditions nor metallic substrates, it represents an ideal noninvasive tool to probe the ribbons (or any other product of interest) after their implementation into device structures or simply after their transfer onto insulating or other functional substrates.
Last but not least, theoretical calculations provide invaluable input of completely disparate kinds. Most typically used are density functional theory (DFT) calculations, although important insight has also been obtained, among others, from tight-binding, molecular dynamics or Monte Carlo simulations. On one hand they are systematically required to support the interpretation of the experimental results, while on the other hand their predictive power allows screening the potentially most interesting problems or systems to address in experiments. In addition, not limited to the properties or systems accessible in experiments, calculations provide deep and thorough insight not obtainable by other means.
Beyond the nature of the reactions and the associated products, another issue of interest is often the quality of the resultant samples. By way of example, covalent networks formed by on-surface synthesis can incorporate a wide range of point defects, due to incomplete reactions or to local distortions in the geometry of the molecular building-block. However, the quality of the networks in terms of crystallographic order is usually barely addressed. Successful network formation would be simply qualified subjectively as being of “good” or “poor” quality. The different defects observed (the modulations in the network topology, the presence of incompletely reacted precursors) can be assigned from the STM images and counted individually to get an estimation of the overall quality. Only the global extension of the networks (the domain size) can be properly quantified. While 1D networks such as GNRs can extend up to several hundreds of nanometers, the domains of 2D networks formed in UHV are usually limited to sizes between 10 and 30 nm. However, in ambient conditions covalent networks with high crystallographic order and extension of several hundreds of nm were obtained (see section 8 and Figure 24a,b). The global morphology of the porous graphene networks has been assessed qualitatively by using Monte Carlo simulations and their comparison with the STM images (see Figure 17a).43 Also, a statistical analysis approach based on the minimal spanning tree (MST) method112 has been proposed to provide a quantitative and comparative estimation of the quality of boronic acid based covalent networks.113
Figure 24.

(a) Reversibility of the dehydration reaction of boronic acids can be achieved by heating in the presence of water pressure. Well-ordered extended networks can be obtained in this way. (b) Sequential STM images showing decomposition/repair cycles consisting of mere decomposition in ambient environment followed by the annealing procedure (each image is 100 × 100 nm2). (c) The homocoupling of DPIA leads to the formation of polymer chains in phenyloctane or to cyclic dimers in trichlorobenzene. (a, b) Adapted with permission from ref (127). Copyright 2012 Royal Society of Chemistry. (c) Reprinted with permission from ref (183). Copyright 2014 Springer Nature.
Figure 17.

Influence of the substrate nature. (a, b) Comparison of the formation of nanoporous graphene polymer from CHP precursor on Cu(111), Au(111), and Ag(111) surfaces. (a) STM images. (b) Corresponding results from Monte Carlo simulations with varying factor P corresponding to the ratio of reactivity to diffusivity. (c) The quality of the nanoporous polymer resulting from the self-condensation reaction of BDBA precursors was estimated by use of a minimal spanning tree (MST) analysis. Results from different deposition conditions are reported (Tc is the evaporation temperature, and Ts is the substrate temperature during deposition). (d–f) Comparative study of the Ullmann coupling reaction of TBB on noble metal surfaces. (d) STM images and corresponding models of the initial state (IS), the organometallic intermediate state (IntS), and the final covalently linked product (FS). Their relative occurrences in the function of the annealing temperature are reported for Cu, Au, and Cu surfaces, together with representative STM images. (e) Representation of the reaction yield for the covalent product as a function of the temperature. (f) Energy diagram representing the relative positions of the intermediate and activated states for the three different surfaces. (a) Adapted with permission from ref (43). Copyright 2012 American Chemical Society. (b) Reprinted with permission from ref (113). Copyright 2011 American Physical Society. (c–e) Adapted with permission from refs (1 and 231). Copyright 2016 Springer Nature.
2.3. Conspectus
A wide range of chemical coupling reactions have been successfully applied to on-surface synthesis. Among the most representative ones are the Ullmann-type coupling or the radical coupling of halogenated species, the Glaser-type coupling or the homocoupling of terminal alkynes, the condensation of boronic acids, the Schiff base reaction or the formation of imines from the condensation of aldehydes with amines, and the direct C–H activation of aryls or alkyls. The reactions performed may be sometimes highly complex, for example, with the formation of stable organometallic intermediates in Ullmann-type reactions or with the formation of various side products in Glaser-type reactions.
The whole set of surface science techniques is available to provide advanced characterization tools to on-surface synthesis, with a strong emphasis on scanning probe microscopy (STM/AFM) that gives direct imaging of the organic structures at the atomic scale. Spectroscopy techniques such as photoelectron (XPS/UPS) or X-ray absorption (XAS/NEXAFS) spectroscopy, and mass (TDS/TPD) or vibrational (HREELS/Raman) spectroscopy, are also widely used. In many cases, strong support from theoretical calculations is required for a correct interpretation of the experimental data.
3. Precursor Design
The rational design of the precursor molecule is naturally of prime importance to control the reaction product. The aromatic backbone can be modulated while preserving the reactive groups. As a result, the lattice size and the symmetry of the resulting network can be finely controlled. In addition, hierarchical coupling strategies have been developed to gain control of the growth process. The reaction pathway can also be tuned by precursor design, for example, by adding side functionality (a substituent that does not contribute directly to the coupling reaction) providing steric hindrance or modulating the whole reactivity. The influence of the halogen type in Ullmann coupling reactions has been extensively studied recently.99,100,114−116 Due to the different strength of the carbon–halogen bond depending on the halogen type, the latter modulates in general the dehalogenation temperature but can also have other indirect effects on the extent, orientation, and structure of the related polymers. Heteroatoms can be introduced into the precursor design for two main reasons. They can act as dopant and adequately tune the electronic properties of the polymer formed. Also, they can be used to create specific functions that can induce the formation of metal–organic complexes with intrinsic or extrinsic metal adatoms.
3.1. Precursor Symmetry
In supramolecular chemistry, the symmetry of the supramolecular networks is directly reflecting the symmetrical arrangement of the molecular binding groups (see Figure 5a).117 This basic principle remains valid for supramolecular chemistry on surfaces. For a given precursor family, 0D,118,119 1D,118,120 or 2D121 structures can form. This strategy can be efficiently applied to on-surface synthesis. This is illustrated in the seminal work of Ullmann coupling between porphyrin units.38 Depending on the number of brominated group attached on the periphery of the precursor, 0D (dimers), 1D (linear chains), or 2D (square domains) structures were obtained (see Figure 5b–e). The comparative formation of individual dimers or extended 1D chains was similarly reported with dibrominated122 or triply halogenated60,123 precursors. More specifically, a variety of different topologies can be realized by varying the number of reactive sites and also the geometry of the precursors. For Schiff base coupling, 1D linear or zigzag polymers and 2D hexagonal or chessboard molecular nanostructures were designed and constructed by these means (see Figure 6a–c).124
Figure 5.
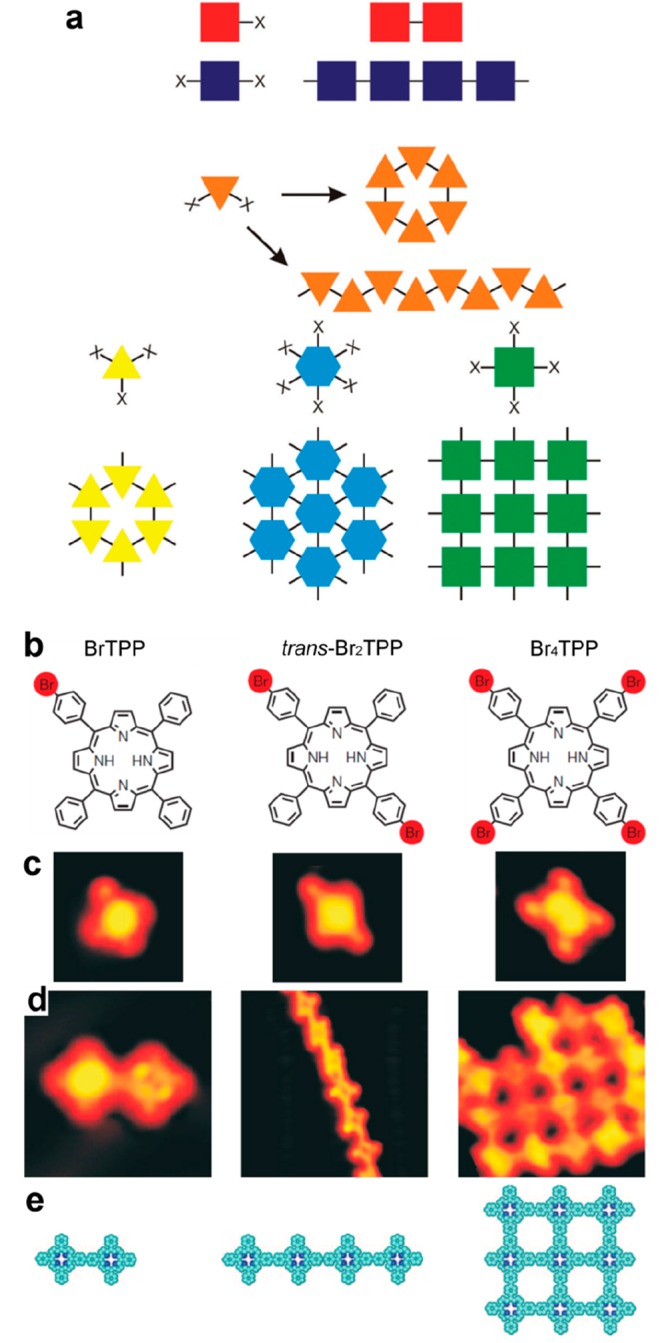
Control of the network dimensionality and topology. (a) Schematic illustration showing the relationship among the precursor functionality, its symmetry, and the topology of the formed networks. (b–e) Mono-, di-, and tetra-bromo functionalized porphyrins (molecular models (b) and corresponding STM images (c)) were used to grow 0D (dimers), 1D (linear chains), or 2D networks, respectively (STM images (d) and corresponding molecular models (e)). (a) Reproduced with permission from ref (6). Copyright 2017 Royal Society of Chemistry. (b–e) Reprinted with permission from ref (38). Copyright 2007 Springer Nature.
Figure 6.
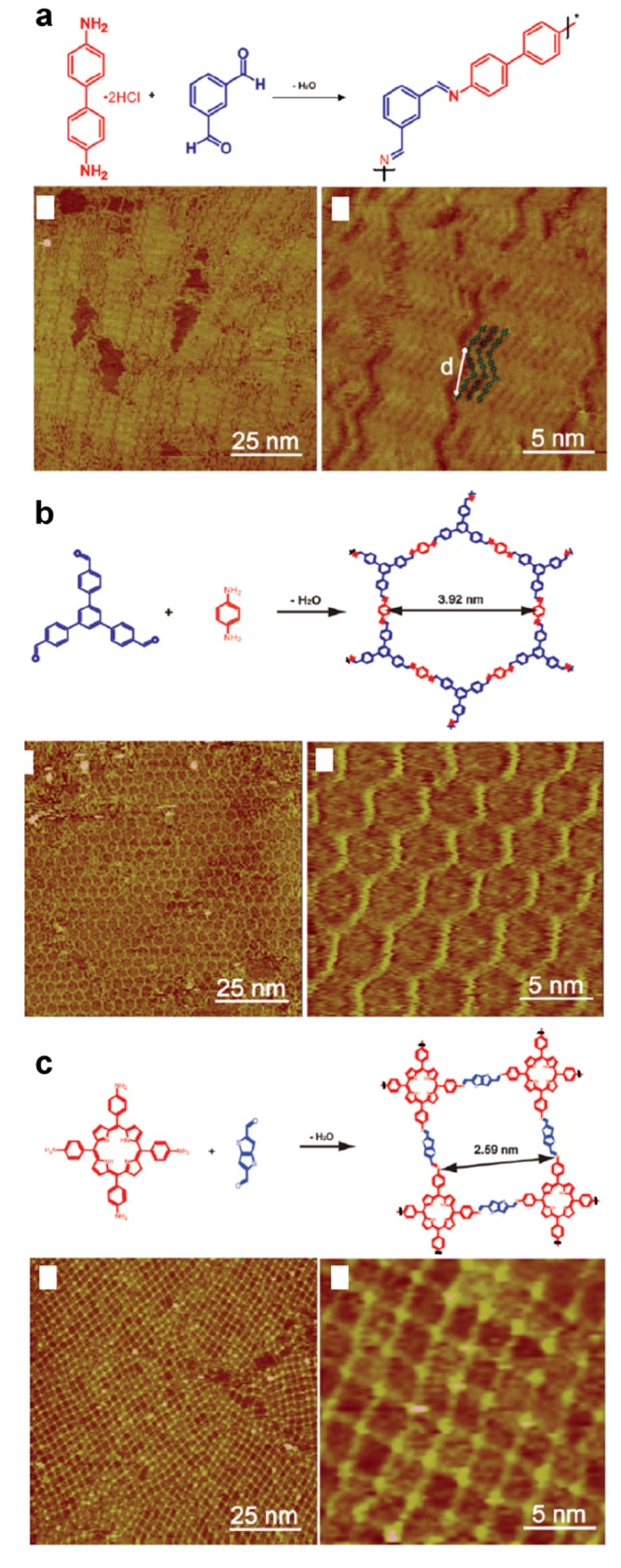
Control of the network dimensionality and topology. Precursor design strategies to build different network dimensionalities and topologies from Schiff base reaction. (a) Zigzag chains. (b) Honeycomb network. (c) Square chessboard network. Reprinted with permission from ref (124). Copyright 2015 AIP Publishing.
3.2. Precursor Size
Similar to what has been proposed for the surface growth of systematic metal–organic frameworks,125,126 the lattice parameter and the pore size of covalently linked networks can be directly modulated by the precursor size. A series of diboronic acid precursors with different backbone sizes and designs were used in ambient conditions on HOPG.127,128 In all cases, the porous networks presented a hexagonal symmetry with pore spacing adequately following the size predicted by DFT. The temperature required for complete polymerization could be related to the precursor size and was reported from 50 °C for the smallest precursor benzene-diboronic (BDBA) to 100 °C for quaterphenyl-diboronic acid.128 Similarly, a series of various polyamines and polyaldehydes has been proposed and successfully used to create Schiff base polymers.64,65,129−135 Here again, the porous networks presented the expected symmetry and pore spacing. The change in network size could directly influence the extension of the conjugation of the aromatic system, thus providing an efficient way to tune the bandgap.134 In some cases, reducing the size of the monomer could have a beneficial effect in terms of reaction selectivity (see section 3.4.1).136 Such an effect was related to a lowering of the steric shielding when reducing the monomer size. It was suggested that the use of nonaromatic precursors resulted in a significant increase of polygonal defects.137 However, the use of different isomeric routes showed no influence on the network quality.137,138
A comparative study of the network quality in UHV for two star-shaped monomers of different size, triply functionalized benzene and triply functionalized trisphenylbenzene, was reported with iodine (TIB, TIPB)139 or ethynyl (TEB, Ext-TEB) reactive groups.140 In principle, similar honeycomb networks are expected after reaction for all these precursors. In the case of the iodinized precursors on the Au(111) surface the smaller monomers formed small patches of ordered networks, and the larger monomers yielded only oligomers. However, the opposite behavior was reported when using the ethynyl functions, which prevents extraction of any generalization. It was suggested that the topological defects and the formation of pores deviating from the ideal hexagonal shape (i.e., pentagons or heptagons) were favored for the larger monomers because the longer pore wall could give more flexibility. Such an effect was also reported for other covalent honeycomb networks.62,141,142
3.3. Sequential Coupling
The confinement of the reactants in the 2D environment of a surface is a clear limitation for the growth of surface-supported covalent networks. Indeed, the multiplication of nucleation centers and the reduced diffusivity of small oligomers represent an important source of defects during the growth process.143 To overcome such effects, one strategy is to introduce a bimodal coupling scheme leading to sequential or hierarchical growth. Here the precursors are functionalized with more than one reactive group leading to different orthogonal reactions with a significant difference in activation temperatures. In this way the formation of intermediates of moderate size is facilitated, and topological defects can possibly be minimized. Eventually, the hierarchical reaction sequence can be manipulated by controlling the reaction parameters.
The coupling of halogenated compounds in an Ullmann-type reaction is based on the formation of a radical upon breaking of the carbon–halogen bond. The strength of the latter bond is directly related to the halogen used. The C–I bond (2.84 eV in iodobenzene) is significantly weaker than the C–Br bond (3.49 eV in bromobenzene),144 and density functional theory (DFT) simulations suggested that on coinage surfaces the energy barriers are around 0.3 eV smaller for deiodination than for debromination.44 This issue has been used to propose a sequential coupling mechanism using two well-separated activation temperatures. With trans-Br2I2-porphyrin (trans-Br2I2TPP), 1D coupling was achieved upon mild temperature annealing and activation of the I sites, and 2D network formation was observed upon higher-temperature annealing and activation of the Br sites (Figure 7a,b).145 A slightly larger domain size was obtained in this way with a reduced number of defects. A similar strategy was used with an asymmetric star-shaped tris-phenylbenzene precursor (BIB).53,146 In a detailed study, it was concluded that the sequential coupling aided in reducing the amount of irregular pores. The same tris-phenylbenzene precursor but bearing two chlorines and one bromine (BCCTP) was also used, with the aid of extrinsic Cu adatoms for the second reaction step.115,147 Higher order was achieved with this strategy. In the case of pyrene derivatives (Br2I2Py), hierarchical dehalogenation was observed but the structures were limited to organometallic chains possibly due to high steric hindrance.148 In another example, the use of a trifunctional precursor bearing two I and one Br (CTPA) could promote the formation of a 1D chain upon mild annealing and of a 2D porous network at higher annealing.149 Finally, this strategy has been successfully used on the surface of bulk calcite.150 In the case of an insulator substrate that is only weakly interacting with the molecular layers, such a hierarchical approach is certainly promising to give access to higher activation temperatures.
Figure 7.

Sequential coupling strategy. (a) Chemical structure of a porphyrin derivative (trans-Br2I2TPP) bearing both Br and I substituents in para positions, respectively. In a first step the I positions react to build 1D chains, and the Br positions react in a second step at a higher annealing temperature to connect the chains and build 2D networks. (b) Corresponding STM images showing the isolated precursors, supramolecular chains, covalent chains, and 2D covalent networks. (c) For a precursor bearing both bromine and boronic acid functions (DBPBA), the condensation of the boronic acid moieties leads in a first step to the formation of boroxine trimers that couple covalently at a higher annealing temperature in a second step. (d) Similar strategy with a para-bromide-boronic acid (BBBA). (a, b) Reprinted by permission from ref (145). Copyright 2012 Springer Nature. (c) Adapted with permission from ref (157). Copyright 2011 Royal Society of Chemistry. (d) Adapted with permission from ref (141). Copyright 2012 American Chemical Society.
The two-step reaction strategy has been demonstrated also with other coupling schemes. It represents actually the basic mechanism used for the formation of graphene nanoribbons (GNRs) on Au(111),151 where the Ullmann coupling between dibromo-bianthryl precursors (DBBA) allows for the efficient formation of 1D covalent structures, that are then locked into robust ribbons thanks to the additional C–C coupling obtained by cyclodehydrogenation (see section 10.1). Similarly, the formation of 5-membered rings inside 1D polymeric chains has been observed.152−155 An additional dehydrogenative coupling step can be initiated laterally between polyphenelyne chains or GNRs to form larger ribbons or even nanoporous graphene (see section 10.2). Ullmann coupling followed by cross-dehydrogenative coupling was also proposed with the precursor bromo-biphenyl to form 1D covalent structures.156 Note that the sequential strategy induced here a remarkable supramolecular templating effect, see section 5.1.
Sequential coupling has been proposed with a combination of Ullmann coupling and boronic acid condensation.141,157 With a dibromobenzene boronic acid precursor (DBPBA), the boronic condensation took place in the crucible prior to evaporation and the second radical coupling led to the formation of domains of only limited size, but the strategy represented a unique way of creating boroxine based networks with the smallest pore size (Figure 7c).157 With a para-bromobenzene diboronic acid precursor (BBBA), the boronic condensation took place directly on the metal surface at room temperature.141 The activation of the second radical coupling produced 2D extended polymeric networks of high quality and with a much higher reaction advancement as compared to boronic acids alone (Figure 7d).113,141 The boronic acid condensation was also proposed in combination with the Schiff base reaction, giving the possibility to create chiral networks.158
With a dihydroxy-dibromo precursor (DN), the dehalogenation reaction was coupled to a dehydration or to a dehydrogenation intramolecular reaction (Figure 8a).159 On Au(111), the precursors were transformed by dehydration or dehydrogenation before the C–Br cleavage occurred (i.e., at lower annealing temperature), and the Ullmann coupling could be activated at higher temperature, leading to the formation of polymer chains. However, on Ag(111) dehalogenation occurred in parallel to dehydration, and the carbon radicals were immediately passivated by the as-formed hydrogen atoms. The formation of oligomers was in this way nicely inhibited.
Figure 8.

Sequential coupling strategy. (a) The DN precursor can experience two distinct reaction pathways associated with different internal reorganizations of the molecule. On Au(111), a dehydrogenation reaction is followed by Ullmann coupling leading to the formation of 1D polymer chains. On Ag(111) a dehydration reaction is accompanied by dehalogenation leading to the formation of individual monomers or dimers. (b) Domino reaction with the ENA precursor. The dehydrogenative coupling of the carboxylic moieties is allowed only after the Glaser coupling of the terminal alkyne has been realized. (a) Adapted with permission from ref (159). Copyright 2017 American Chemical Society. (b) Adapted with permission from ref (161). Copyright 2016 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
A two-step reaction was reported using a biphenyl bis-α-diazo ketone.160 The initial N2 fragmentation produced 1D polymeric alkene chains that underwent further deoxygenation and intramolecular cyclization. Most interestingly, a domino reaction was demonstrated with a monomer combining ethynyl and carboxylic acid moieties (ENA) on the Au(111) surface.161 The dehydrogenative dimerization of the alkyne functionalities (Glaser coupling) was activated upon mild annealing, together with a dehydrogenative coupling of the carboxylic functions leading to the formation of bisacylperoxide connected polymers. This second reaction could be hindered at low coverage through the formation of Au-carboxylate complexes (Figure 8b). Remarkably, the deposition of the ex situ synthesized Glaser coupling product revealed that the activation of the carboxyl functions could occur readily at room temperature. In fact, the annealing was only needed to trigger the Glaser coupling process but not required for the O–O bond formation via dehydrogenative coupling of the carboxylic moieties. It was thus assumed that the structure bearing the butadiyne motif must provide unique properties to the molecule, which seemed to be essential for the peroxide formation. The domino reaction thus represents a unique way to block temporarily a reactive function.
Finally, to achieve control on the selectivity of terminal alkyne reactions, the use of terminal alkynyl halides (BEPB, bBEPB, tBEP) was proposed.58 Polydiynes were obtained in this way with high selectivity against usually encountered side reactions. Note that the formation of alkynyl bonds could also be obtained from triply halogenated precursors.60,123,162 Protection of alkyne groups was proposed using silylation.88 Deprotection and recovering of the pristine alkyne groups was achieved by σ-bond metathesis and corresponding silylation of a coadsorbed carboxylic acid. Homocoupling of the terminal alkynes was then realized.
3.4. Modulating the Reactivity via Specific Functional Groups
The formation of covalent structures can be controlled by providing additional side functional groups (side substituents) to the precursor that do not participate directly in the covalent coupling mechanism but that can alter the reactivity or the growth process.
3.4.1. Steric Hindrance
Alkyl chain substitution in the ortho position of a para-bisarylalkyne resulted in a high selectivity toward Glaser coupling and an almost complete suppression of side products due to steric shielding (see Figure 9a–c).136 High selectivity was similarly obtained by introducing cyano functionality to a linear diethynyl-terphenyl precursor (CN-DETP).90 It was suggested here that also the steric repulsion due to the CN groups was responsible for the selectivity. In addition, the supramolecular interactions between the as-formed nanowires could promote the linear growth mode of the polymer and the formation of a double strand structure (see Figure 9d,e). The introduction of methylidene groups into alkane chains (DMH) favored the formation of the (1 × 3) reconstruction of the Au(110) surface which resulted in a lowering of the activation temperature for alkane polymerization.163 The introduction of two symmetric methyl groups in ortho positions of bromophenyl derivatives was shown to be effective in improving the structural quality of the network formed by the organometallic intermediate.164 The addition of a methyl group in the ortho position of dibromonaphthalene (DBDMN) produced various effects depending on the substrate used.165 On Au(111) only halogen bonding self-assembly was possible. On Ag(111) organometallic chains could form. On Cu(111) the Ullmann coupling occurred in parallel with intermolecular cyclodehydrogenation and ultranarrow GNR motifs with chiral edges formed. Selective C–H bond activation was obtained by making use of the steric hindering effect of phenyl groups.166 The Ullmann coupling was blocked at one (DBMTP precursor) or both (DBOTP precursor) C–Br positions leading to monoselective dehydrogenative C–C coupling (see Figure 10a).
Figure 9.

(a) STM images and corresponding chemical models of the various products that can be formed from terminal alkyne precursors. (b) Distribution of the occurrence of the reaction products obtained from the long version of a diethynyl precursor on Ag(111). (c) By using the shorter precursor version, the reaction selectivity is strongly enhanced toward the formation of the linear homocoupling I. (d) A 2D-like network and a large distribution of linear and 3-fold connections is obtained using DETP precursor, while with CN-DETP precursor (e) only dimer linear chains are formed. (a–c) Adapted with permission from ref (136). Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (d, e) Adapted with permission from ref (90). Copyright 2018 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Figure 10.

(a) Phenyl substituents are used to block the Ullmann coupling at the C–Br positions. Monoselective C–H bond activation and C–C dehydrogenative coupling can be obtained with DBMTP or DBOTP precursors. (b) The selectivity of the cross-coupling reaction between BB and EBB precursors can be efficiently steering by controlling the relative ratio and/or total coverage. (a) Adapted with permission from ref (166). Copyright 2018 American Chemical Society. (b) Adapted with permission from ref (174). Copyright 2017 Royal Society of Chemistry.
3.4.2. Other Modulating Effects
The reactivity of benzene-diboronic acid precursors can in principle be directly tuned through the presence of substituents on the benzene ring: electron withdrawing substituents enhance the reactivity of the boronic acid moiety while electron donating groups lower it.167 However, the changes in reactivity as measured by thermal analysis (thermogravimetry and differential scanning calorimetry) did not follow the changes observed for network formation on a metal surface, eventually exhibiting opposite behaviors.62,168 Regioselectivity was observed between pyridine and pyrazine moieties in dehydrogenative aryl–aryl bond formation.169 The addition of hydroxyl groups in Schiff base networks could notably enhance their stability in acidic and alkali environments.170 Also, it could lead to the formation of cross-junctions stabilizing the multilayer growth of 1D structures.171 However, it was shown that side hydroxyl groups (in DHI precursors) could react with the as-formed imine bonding motifs to form oxazolidine and possibly oxidize with atmospheric O2 to oxazoline.172 As a consequence, distortion of the polymer network structure was observed, but also this additional internal oxidation was suggested to simultaneously act as a means for stabilization by making the network formation irreversible.
The fluorination of tetrabromo-biphenyl (Br4F6BP) was shown to stabilize the organometallic intermediate and therefore inhibit the Ullmann coupling.116 Note that, for this tetrabrominated biphenyl precursor (as well for the nonfluorinated case), site-selective didebromination was found with a marked temperature difference in double vs quadruple activation. The presence of benzene rings with lateral extension (in DAN or AABA precursors) was shown to induce disorder in the networks.134 For olefin metathesis, important changes in the product distribution (oligomer length and formation of cyclic structures) were observed on HOPG by introducing an ester functionality inside the long aliphatic side chains.173
The selectivity toward Ullmann cross-coupling reaction between similar precursors was studied using a mixture of bromobenzene (BB) and ethylbromobenzene (EBB) molecules.174 The addition of an ethyl side group in para position improved the stability of the molecule on Cu(111), especially with regards to the active sites at step edges. Consequently, cross-coupling with bromobenzene was only observed when the ethylbromobenzene was present in large excess (see Figure 10b). Otherwise, the repulsive forces between the two precursors led to a majority of homocoupling products. Going to very low coverages (<0.01 ML) in equimolar ratio was necessary to achieve a majority of cross-coupling reactions.
Two tetrathieno-anthracene isomers were shown to behave very differently on Cu(111), Ni(111), or Pd(111) surfaces. While one of the isomers (2TTA) could promote intramolecular rebonding to produce pentacene, geometrical constraints prevented the other isomer (3TTA) from intramolecular rebonding and promoted oligomerization.175 The efficiency of the σ-bond metathesis of silylated alkynes was shown to be much higher for aromatic precursors (TTEN) than for nonaromatic ones (TPDT).176
3.5. Conspectus
Following the general principles in supramolecular chemistry, the symmetry, the lattice parameter, and the pore size of surface-supported covalently linked networks can be directly modulated by the precursor design. Side effects such as the modification of the reaction efficiency or its selectivity can eventually be observed for smaller precursors. Hierarchical growth can be obtained by introducing two distinct reacting groups leading to different orthogonal reactions with a significant difference in their activation temperatures. It has been proposed with a large variety of combinations of coupling schemes, far beyond simple Ullmann coupling with different halogen types, thus demonstrating that it is a valid approach. The hierarchical strategy can lead to elaborate reaction mechanisms with interesting effects (e.g., domino reaction). However, the benefits are not always straightforward in terms of reaction efficiency and network quality. Finally, the addition of nonreactive functional groups in the precursor design can efficiently increase the reaction selectivity by steric shielding. Other effects such as the improvement of the stability or the reactivity of the precursors, or the introduction of postsynthetic interchain interactions, have been observed.
4. Extrinsic Metal Adatom Strategies
The presence of coadsorbed extrinsic metal adatoms on the surface can be advantageous to promote coupling reactions in two different ways. First, they can act as a catalyst177 and improve the reactivity of the system. Second, they can incorporate into metal–organic networks with a predefined structure acting as a template for the coupling reaction.
4.1. Metal Adatoms as Extrinsic Catalyst
The surface-supported Ullmann coupling reaction is usually catalyzed by the underlying metal substrate, but an alternative mechanism based on the codeposition of additional metal adatoms acting as extrinsic catalyst can take place. This was shown in the case of porphyrin derivatives, for which the presence of Pd or Cu adatoms could efficiently lower the activation temperature on Au(111)178 or on hBN/Ni(111),179 as compared to the same reaction taking place on the same surface without extrinsic adatoms (Figure 11a). Also, a fine kinetics study revealed that different reaction mechanisms are taking place on Au(111). The rate limiting step was found to be the C–C coupling formation in the presence of Pd while it was the initial activation (dehalogenation) in the presence of Cu. It was thus found that Cu is a better catalyst, providing more and longer covalent chains.178 The same strategy has been used to grow polyphenylene on Au(111),147 even achieving long, high-quality chains on Au(100).89 Remarkably, it was shown that codeposition of lanthanide adatoms (Dy) was very efficient in dehalogenating dibromoterphenyl precursors (DBTP) on Ag(111) at room temperature.180 However, in this case the covalent coupling between the molecules was then prohibited upon annealing (see Figure 11b), thus revealing the potential of f-metals as inhibitors of uncontrolled C–C coupling reactions. Also, Ho adatoms were used to lower the dehydrogenation and covalent coupling reaction temperatures of terminal alkynes to as low as 100 and 130 K, respectively.181 High selectivity toward cyclotrimerization was found.
Figure 11.

Metal adatom-directed growth. (a) Evolution of the oligomerization reaction yield of TBB precursors on h-BN/Ni(111) depending on the presence of codeposited Cu or Pd adatoms. R1 and R2 are two different sets of deposition parameters. Pd adatoms catalyze the reaction already at room temperature. (b) The codeposition of Dy adatoms can very efficiently lead to the dehalogenation of DBTP precursors and the formation of organometallic complexes at room temperature, but simultaneously act as an inhibitor for the homocoupling reaction, as compared to the bare Ag(111) surface. (c) Reaction scheme of the multistep mechanism involving the bromo-dichloro precursor BCCTP and codeposited Cu adatoms on Au(111). (d) Corresponding STM images. (a) Adapted with permission from ref (179). Copyright 2016 Royal Society of Chemistry. (b) Adapted with permission from ref (180). Copyright 2017 American Chemical Society. (c, d) Adapted with permission from ref (147). Copyright 2016 Royal Society of Chemistry.
A multistep strategy was proposed with a tris-phenylbenzene precursor bearing two chlorines and one bromine (BCCTP) on Au(111) (Figure 11c,d).147 The bromine was first activated, and then the chlorines with the help of codeposited Cu. Remarkably, this second reaction (dechlorination) could take place already (although only partially) at a slighter lower temperature than the first debromination reaction. This resulted in the formation of a rather high-quality honeycomb network, however of limited extension. The activation temperature of the homocoupling reaction of bromobenzene on Cu(111) was strongly reduced by depositing Co nanoislands.182 At 80 K all deposited molecules diffused to the top of the Co islands where the C–Br bond dissociated. The homocoupling was obtained at 160 K, while 350 K would be required on bare Cu(111).
The catalytic activity of metal adatoms can eventually be preserved on a thin insulating film. On the hexagonal boron nitride layer (hBN) formed on the Ni(111) surface, the radical Ullmann coupling between tris-bromophenylbenzene precursors (TBB) could take place at a remarkably reduced activation temperature upon codeposition with Cu or Pd adatoms (Figure 11a).179 At the liquid–HOPG interface, a pyridine solution of Cu(OAc)2·H2O was added to catalyze the homocoupling reaction of terminal alkynes (DPIA).183
4.2. Hierarchical Growth
Extrinsic adatoms can be used for structural organization as linking elements in predefined metal–organic networks. Heteroatoms should be introduced here in the precursor design to act as a ligand. The covalent coupling reaction then takes place in a second step in a hierarchical manner. This effect was used with a porphyrin precursor incorporating pyridyl groups that easily form metal–organic bonds.184 Here the codeposition of Cu adatoms led to the formation of 1D metal–organic stripes, between which covalent coupling was activated in a second step in a zip-like mechanism (Figure 12a). This metal adatom-directed templating could thus lead to the formation of well-defined dimeric ribbons.
Figure 12.

(a) Sequential strategy combining metal–organic coordination and Ullmann coupling. The linear chains that are formed by coordination with Cu adatoms at room temperature get covalently coupled to form dimeric chains after annealing at 180 °C. (b) Similarly, the metal–organic networks can be used to promote a (Pd adatom-catalyzed) Sonogashira cross-coupling reaction. (a) Adapted with permission from ref (184). Copyright 2013 American Chemical Society. (b) Reproduced with permission from ref (185). Copyright 2017 Royal Society of Chemistry.
Similarly, the preformed metal–organic network could be used as an efficient template to promote a (metal-catalyzed) Sonogashira cross-coupling reaction (Figure 12b).185 It was shown here that this metal adatom-directed hierarchical strategy could efficiently and simultaneously inhibit the homocoupling and enhance the cross-coupling. Internal C–H activation and C–C bond formation were reported inside an organometallic polymer (made from PEPmBr precursor), leading to modification of the C–Ag–C as well as the N–Ag–N coordination angles.186
4.3. Conspectus
The addition of extrinsic metal adatoms can efficiently lower the activation temperature of Ullmann-type coupling reactions. It can even provide a way to catalyze the reactions on nonmetallic substrates. Also, hierarchical growth has been demonstrated with the help of extrinsic metal adatoms: their complexation with the precursors directs the formation of a metal–organic network that acts as a structural template for the subsequent coupling reaction.
5. Supramolecular Templating
The mobility of the precursors is an important factor for surface-supported reactions. Indeed, all reactants are confined in the two-dimensional environment of the interface. At low coverage and/or at high temperature, the formation of a 2D gas phase gives the possibility to explore the full configurational phase space, whereas when approaching the full monolayer coverage and/or at lower temperature, the system condenses in a 2D solid phase that eventually severely reduces the reaction pathway possibilities. In this latter case the precursors are in interaction with each other in a well-defined supramolecular template. The quest for supramolecular templating in surface-supported coupling reactions is motivated by the poor degree of order usually encountered in covalent networks. This effect is related to the irreversible conditions used in UHV that preclude the formation of the thermodynamically most stable structure through defect self-healing. In contrast, supramolecular interactions grant access to a self-healing mechanism that is a prerequisite for the formation of ordered and extended 2D structures.187−189 It is therefore highly desirable to make use of the versatility of supramolecular interactions to steer coupling reactions on a surface. Also, supramolecular interactions can improve the selectivity of the reaction. In a topochemical reaction,190 the precursors are constrained by the lattice forces in a well-ordered phase in which the reactive centers are adequately placed for the reaction to occur. The topochemical or topotactic processes are assumed to maintain the excited molecules in a configuration close to what they adopt in their ground state.
5.1. Control of Structural Order
This issue can be illustrated by the example of the formation of graphene nanoribbons using para-dibromoterphenyl precursors (DBTP) on the Au(111) surface (see Figure 13a).191 The precursors self-assemble through the formation of halogen bonds in an ordered fashion in large domains, with all molecules perfectly aligned. Upon stepwise annealing, the reaction can be sequentially activated: debromination, formation of polyphenylene wires, and finally creation of graphene nanoribbons. All reaction steps take place with minimal perturbation of the previous phase, and the prealignment of the molecules serves thus as an efficient template to steer the alignment of the final nanoribbons. The same reaction mechanism was observed with dibromo-biphenyl (BrPh2Br) or diiodo-biphenyl (IPh2I) on the Ag(111) and Cu(111) surfaces.89 A similar effect has been reported with a terphenyl molecule (BDFPTP) bearing difluoropyridin moieties that could experience two distinct and original reaction pathways on Au(111).192 The self-assembly of the reactants in well-defined molecular domains prior to reaction could greatly enhance the regioselectivity of the dehydrocyclization reaction and suppress the defluorinated coupling, as compared with the reaction obtained from a disordered phase.
Figure 13.

Supramolecular templating effect to steer the growth of covalent networks. (a) The alignment of DBTP precursors in a supramolecular phase is preserved upon annealing and stepwise formation of perfectly aligned polyphenylene chains and fused nanoribbons. (b) The formation of rylene-type graphene nanoribbons from QR precursors at low coverage produces a variety of different bonding configurations. At nearly one monolayer coverage (c), the selectivity toward straight nanoribbon (type I) formation is significantly enhanced due to steric repulsion. (a) Adapted with permission from ref (191). Copyright 2015 American Chemical Society. (b, c) Adapted with permission from ref (193). Copyright 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
Halogen-free formation of graphene nanoribbons was demonstrated using quaterrylene (QR) precursors on Au(111) and Ag(111).193 The regioselectivity between kinked or straight connections could be steered in a high-coverage regime due to the repulsive interactions from adjacent molecules in the dense supramolecular phase (see Figure 13b,c).
A supramolecular template could be realized with a bicomponent system, namely, bis-aminophenyl-diphenylporphyrin (TPP(NH2)2) with C60.194 Here the nanoporous supramolecular phase of TPP(NH2)2 could adequately incorporate fullerene molecules in an ordered fashion, see Figure 14e. The bicomponent structure was in an adequate configuration so that the topochemical coupling of the NH2 moieties to the fullerenes could be realized with a minor structural change upon annealing (Figure 14f). The covalent copolymer consists of 1D strands extended over large distances with perfect order.
Figure 14.

Supramolecular templating effect to steer the growth of covalent networks. (a) Schematic representation of the formation of kinked terphenyl polymer from BBP precursors. The Br adatoms produced from the first dimerization step produces a confinement template (b) that steers the regioselectivity of polymerization reaction leading to well-aligned linear polymers (c), as opposed to the branched polymers formed from nonbrominated 4Ph precursors (d). (e) The nanoporous phase of TPP(NH2)2 can perfectly accommodate C60 molecules that are positioned in an adequate position to covalently bind to the neighboring NH2 moieties upon annealing (f). (a–c) Adapted with permission from ref (156). Copyright 2015 Royal Society of Chemistry. (d) Adapted with permission from ref (66). Copyright 2014 Royal Society of Chemistry. (e, f) Adapted with permission from ref (194). Copyright 2010 American Chemical Society.
The polymerization of diacetylene compounds proceeds in general topochemically as both the monomer and the polymer belong to the same crystal system and have almost identical lattice constants. One-dimensional structures of adequately functionalized diacetylene compounds self-assembled with high order and favorable interchain distance on HOPG or MoS2(0001) surfaces (see Figure 15a). The chain polymerization could be initiated by injecting an electron pulse with the STM tip or by UV light irradiation.35,37,195−200 The supramolecular templating effect was revealed by the high sensitivity of the reaction rate to fine modulations of the interchain distance: the shorter the distance between two carbon atoms which are to be bound by the polymerization reaction is, the higher the reaction rate is.197 Similarly, photochemical homocoupling of terminal alkynes (PEBA) resulted in high yield and with high selectivity under UV light irradiation due to the favorable templating effect of the prior supramolecular organization.201
Figure 15.

Supramolecular templating effect to steer the growth of covalent networks. (a) Topochemical polymerization of diacetylene. The precursors are self-assembled in adequate configuration so that the polymerization takes place with minimum conformational change. (b) The Ullmann coupling of Br4-PTCDA results in a stochastic formation of a graphene-like nanoribbon of various length and orientation. By using in situ polymerized polyphenylene chains (red arrows in part c), linear grooves are created that steer the alignment of the nanoribbons. (a) Adapted with permission from refs (199 and 200). Copyright 2012 and 2008 Royal Society of Chemistry. (b, c) Adapted from ref (86). Creative Commons Attribution 4.0 International License.
Dibromo-terphenyl molecules (DBTP) were used to create a strong covalent 1D template for the growth of a new type of graphene-like nanoribbons on Au(111).86,202 The terphenyl molecules first reacted after annealing to form long polyphenylene linear polymers which served as molecular grooves to perform 1D constraint on the codeposited GNR precursors (Br4-PTCDA). Good alignment and improved structural quality were achieved with this approach (see Figure 15b,c). Instead of using polyphenylene made out of dibromo-terphenyl (DBTP), a similar 1D constraining effect was obtained with dibromo-dianthryl (DBBA) growing into armchair graphene nanoribbons. Introduction of heteroatoms (boron and oxygen) on the edges of a chiral GNR has proven efficient in inducing lateral self-assembly through the formation of inter-ribbon hydrogen bonds.203 Well-aligned GNR arrays with different modes of homochiral and heterochiral inter-ribbon assemblies were observed.
5.2. Control on Reaction Selectivity
With the precursor para-bromo-biphenyl (BBP), Ullmann coupling allowed for the formation of quaterphenyls that subsequently bonded together by cross-dehydrogenative coupling to form 1D covalent structures on Cu(110) (Figure 14a).156 Remarkably here, the first Ullmann reaction created on the surface Br adatoms as byproducts that remained incorporated in the supramolecular structure in a well-ordered fashion (Figure 14b). This templating effect then steered the regioselectivity of the second coupling reaction and allowed for the production of specific 1D polymers (Figure 14c). This is clearly in contrast with the direct cross-dehydrogenative coupling of ex situ synthesized quaterphenyl precursors (4Ph) that enabled the formation of nonregioselective or multiple coupling (Figure 14d).66 Selective synthesis of cis-enediyne was achieved on the Ag(111) surface for ethynyl precursors (BPBE or BEBP) thanks to the concomitant use of Br adatoms.204 Br was added as a substituent on the precursor but was easily released on the surface. The coadsorption of Br had a stabilizing effect on the molecular species through the formation of multiple Br···H hydrogen bonds, which inhibited further hydrogenation reactions potentially leading to unwanted byproducts. The use of larger brominated substituents (2Br-DEBPB) was shown to promote the C–H bond activation of terminal alkynes and to lead to the formation of stable organometallic intermediates on Ag(111) at room temperature.91 Interestingly, the formation of the final covalent product took place at higher temperature than the corresponding nonbrominated precursor. In contrast, in another study hydrogen treatment was used during the thermal activation of brominated precursors to remove residual bromine byproduct from the Au(111) surface (in the form of desorbed HBr).205,206 Such treatment was shown to slightly improve the polymer quality.
The metal–organic intermediate formed using a tris-bromophenylbenzene molecule with Br in meta positions (mTBPB) was responsible for a conformational change of the precursor (from C3h to Cs symmetry) that allowed for the creation of linear organometallic chains.207 These 1D structures were then preserved after thermally activated aryl–aryl coupling. Porous nanoribbons were formed in this way on Ag(111). This was in contrast to the case of Au(111) where stable organometallic intermediates did not form and the system evolved under similar conditions into randomly organized 2D networks. Other cases using the template effect of metal–organic structures have been described in section 4.2 for the formation of dimeric chains184 or to control a Sonogashira cross-coupling reaction.185
The bromobiphenyl (BBP) molecule formed organometallic dimers at room temperature that self-assembled on Ag(111) in dense ordered domains, whereby the (BBP)2-Ag species organized with alternating orientation onto two nonequivalent adsorption sites (with their central Ag adatom sitting on bridge or on hollow sites).208 Upon fine control of the annealing temperature, it was shown that the organometallic dimers corresponding to hollow sites started to react first to form quaterphenyl products, while those corresponding to bridge sites required a slightly higher temperature. The reaction thus took place directly inside the stable self-assembly that acted as a selective template providing two different catalytically active sites of different reactivity. With the same system, it was shown that the reaction pathway could be steered whether a supramolecular phase was formed (at high coverages) or not (at low coverage).209 The formation of an intermediate phase with an open structure was prohibited inside the dense supramolecular domains due to steric hindering. Two reaction pathways with distinct activation energies and attempt rates were thus recognized.
5.3. Other Effects
Stilbene derivatives were incorporated inside a complex three-component supramolecular template.210 The size of the host cavities could finely adapt to accommodate properly the precursors, the isomer intermediates, or the products obtained after [2 + 2] photodimerization. It was shown that the supramolecular templating effect of the host–guest confinement greatly increased the efficiency of the reaction.
Tris(iodophenyl)benzene (TIPB) was deposited inside the pores of a supramolecular nanoporous network made out of PTCDI and melamine on Ag(111).211 The reaction was limited to the dehalogenation step at room temperature, and the confinement inside the nanopores inhibited the formation of organometallic intermediates. This template-assisted study thus provided interesting information about the reaction mechanism.
The use of melamine in Schiff base reaction restricted the advancement of the reaction to the formation of stoichiometric oligomers of finite size.212,213 A similar effect was observed by using a nonplanar tetrahedral (sp3) building-block.214 Here it was suggested that the formation of supramolecular self-assemblies of these oligomers, stabilized by consequent intermolecular interactions, played a role in the limitation of the reaction advancement. The competing formation of an ordered bimolecular supramolecular phase could completely inhibit the reaction or drastically affect the kinetics of its formation.215 For the self-condensation of boronic acids (BDBA), the competing formation of the supramolecular phase acted as a strong kinetic blockade to the irreversible polymerization reaction.62 The reaction could be instantaneously activated by destabilizing the supramolecular phase216 through tip manipulation (Figure 25b) or e-beam irradiation.
Figure 25.

Tip-induced reactions. (a) Individual iodobenzene molecules are manipulated with the STM tip to form biphenyl units. From top to bottom: two iodobenzene molecules adsorbed at the step edge of Cu(111); dissociation of C–I bond in the left molecule; dissociation of C–I bond in the right molecule; removal of the right I atom; displacement of the left radical; association of the two benzene units. (b) The polymerization of BDBA precursor can be instantaneously induced inside the supramolecular phase by local mechanical removal of some molecules with the STM tip. (c) The polymerization of diacetylene can be locally initiated by electron injection with the STM tip at various locations to form poly(diacetylene) (PDA) wires that self-propagate on the surface. (d) “Chemical soldering” is performed when a PDA line reaches a pentamer of phthalocyanine (Pc) molecules that get covalently bonded to the PDA wire, eventually leading to the possibility of creating a cross-junction. (a) Reprinted with permission from ref (36). Copyright 2000 American Physical Society. (b) Reproduced with permission from ref (216) . Copyright 2011 Royal Society of Chemistry. (c) Reproduced with permission from ref (199). Copyright 2012 Royal Society of Chemistry. (d) Adapted with permission from ref (198). Copyright 2011 American Chemical Society.
With the system tris-iodophenyl-benzene (TIPB) on Au(111), covalent networks could be formed as a second layer on top of a supramolecular domain.217 The step edges of the Au(111) surface acted as nucleation sites for the Ullmann coupling reaction, but also as anchoring sites for the as-released iodine. After saturation of the step edges by iodine, the reaction was thus self-limiting. Further deposited monomers remained intact and self-assembled on terraces without reacting. Upon reaching full monolayer coverage, the covalent structures were then propelled on top of the halogen-bonded first layer. An unusual behavior was observed with the simple 1,3-diiiodobenzene (DIB) precursor on Cu(111).218 A large excess of released iodine was produced due to partial desorption of the as-formed radicals, leading to the formation of a close-packed iodine monolayer. On top of it, trimer products only were observed that self-assembled thanks to rather long-range interactions.
Fully reversible inhibition of the Ullmann coupling of dibromodibenzothiophene (DBDBT) was achieved on Au(111) by pre-exposure to H2S gas.219 The capture of the free Au adatoms through the formation of S–Au complexes resulted in the passivation of their catalytic role. The Ullmann coupling could then be activated through the reduction of the S–Au complexes by H2 dosing. The Au adatoms became available through release of as-formed H2S (see Figure 16).
Figure 16.
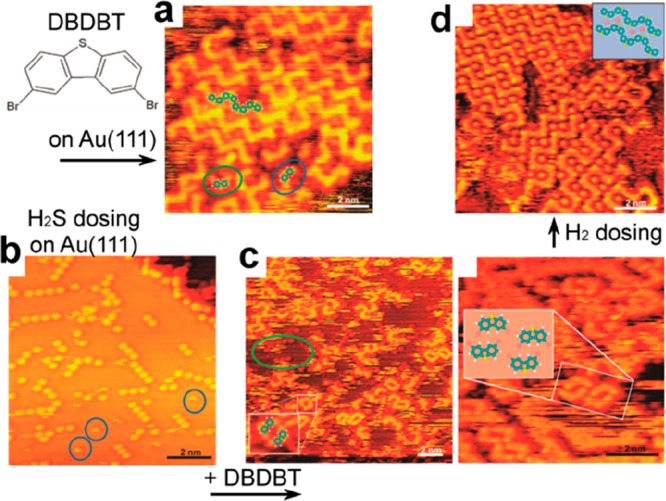
Reversible inhibition of the Ullmann coupling. (a) DBDBT precursors directly deposited on Au(111) form zigzag chains. (b) Predosing of H2S on Au(111) leads to the S-passivation of free Au adatoms and formation of Au–S complexes. (c) Subsequent deposition of DBDT produces intact molecules only. (d) The covalent coupling can be then activated by H2 dosing that releases the Au adatoms by producing H2S. Adapted with permission from ref (219). Copyright 2018 Royal Society of Chemistry.
5.4. Conspectus
Topochemical processes can efficiently lead to high structural order of the reaction products. Supramolecular interactions are used to create a defined template that is preserved along the reaction path. Such an effect is particularly efficient for the growth of aligned para-polyphenylene wires and their lateral fusion into graphene nanoribbons. This strategy can even be extended to the use of additional molecules that do not participate directly in the reaction but that can efficiently provide a specific template. In Ullmann coupling reactions, the halogens released on the surface usually have important roles along the reaction path, by controlling the organization of the reactants or the reaction selectivity. Further effects due to supramolecular interactions have been observed, such as the confinement of the precursors in host–guest cavities or the competing formation of a supramolecular phase that inhibits the reaction.
6. Influence of the Substrate Nature and Symmetry
6.1. Substrate Nature
On-surface reactions are in most cases catalyzed by the supporting metal substrate and are therefore highly dependent on its nature. Up to now, on-surface synthesis has been realized mainly on coinage metal substrates (Cu, Ag, Au) or HOPG (graphite), with few reports on Co,174 Pt,67,220−224 Pd,175,225−227 or insulators (see section 11.3). The substrate nature can affect the reactivity of the precursors (the thermal activation and the formation of stable intermediate states) or the reaction pathway and its selectivity (leading to different product types) and also the growth mode and the overall polymer quality in the case where extended covalent networks are formed. In general, the influence of the substrate nature can be related to the interaction strength with the molecular layer (usually weak physisorption on Au and stronger chemisorption on Ag and Cu228,229), and eventually to the provision of intrinsic adatoms. Note that also the role of the surface thermalization and the energy dissipation have been highlighted and reported to be responsible for the selective stabilization of reaction intermediates.55
The degree of order achieved in polymeric networks is the result of a delicate balance between the diffusivity of the precursors and their reactivity, which are both modulated by the surface nature. The quality of the resulting 2D structures was characterized and quantified using Monte Carlo simulations (Figure 17a,b)43,230 or statistical analyses113 (Figure 17c). In general it was shown that polymerization reactions are governed in a first approximation by the ratio of reactivity to diffusivity and that silver surfaces are more efficient for achieving extended polymeric growth.43,113,136
A comparative study of the catalytic strength for Cu(111), Ni(111), and Pd(111) surfaces has been proposed for tetrathienoanthracene based molecules (2TTA, 3TTA) experiencing intramolecular rebonding or intermolecular coupling and oligomerization.175 The Ullmann-type reaction has been rather extensively studied, and several articles1,94,115,231−237 provide a detailed comparison of the influence of different metal substrates (see, e.g., Figure 17d–f for the case of the tris-bromophenylbenzene precursor (TBB)1,231). Here the reactivity was closely related to the stability of organometallic intermediates. As a general tendency, they were most stable on silver surfaces, somehow stable on copper, and usually nonexistent on gold,1,231 although they could still be observed in some cases.58,86,148,232,238 As a consequence, the final C–C coupling occurred most easily on gold and with the higher activation temperature on silver. However, due to the variety of behaviors encountered, a general rule cannot be drawn and all systems should be investigated on a case-by-case basis.
When several reaction pathways and reaction products can be obtained simultaneously, the substrate nature can eventually achieve partial selectivity and modify the distribution of the reaction products. For example, the cyclotrimerization of alkynes was favored on Au(111) while the homocoupling was preferred on Ag(111) (see Figure 18a,b).61,136,140,239,240 With a dihydroxy-dibromo precursor (DN), the difference in reactivity of the C–Br bond on Au(111) or Ag(111) was used to achieve predominant C–C coupling (on Au(111)) or selective C–H coupling (on Ag(111)), as detailed in section 3.3 and in Figure 8a.159 The monoselective ortho C–H bond activation was demonstrated for two different hydroxyl functionalized precursors (THPB, DHQP) on the Ag(111) surface, while diselective activation was observed on Au(111).241 For a tetrabromo-thiophene derivative (TBTTA), the C–C coupling could be observed only on Ag(111) while on Cu(111) the competing thiophene ring-opening reaction and the degradation of the precursor were observed instead.242 Linear alkanes underwent different C–C bond chemistry (coupling or dissociation) on (110)-oriented metal surfaces.220 Selective dehydrogenation and C–C coupling (polymerization) took place on Au(110) surfaces whereas dehydrogenative pyrolysis resulting in hydrocarbon fragments took place on Pt(110). It was suggested here that the dehydrogenation of alkanes on Au(110) surfaces was an endothermic process, but further C–C coupling between alkyl intermediates was exothermic. On the contrary, due to the much stronger C–Pt bonds, the dehydrogenation on Pt(110) surfaces was energetically favorable, resulting in multiple hydrogen loss followed by C–C bond dissociation. Strict selectivity was demonstrated in the case of competing intra- and intermolecular coupling processes. This has been shown for two different precursors (see Figure 18c) for which Pt(111) inhibited the diffusivity and produced an exclusively intramolecular dehydrogenative reaction, while the Au(111) surface allowed for high diffusivity and consequently intermolecular cross-dehydrogenative coupling.221
Figure 18.

Influence of the substrate nature. (a) The Ext-TEB precursor experiences a homocoupling reaction on Au(111) but a cyclotrimerization reaction on Ag(111) (b), with an advancement controlled by the annealing temperature. (c) The low diffusivity and high reactivity of Pt(111) surfaces induces a cyclodehydrogenation reaction of the individual precursors (reaction path a) while a dehydrogenative polymerization (reaction path b) is obtained for the same two precursors on the poorly reactive and highly diffusing Au(111) surface. (a) Adapted with permission from refs (61 and 140). Copyright 2016 and 2013 Springer Nature. (b) Adapted with permission from refs (61 and 239). Copyright 2016 and 2014 Royal Society of Chemistry. (c) Adapted with permission from ref (221). Copyright 2013 American Chemical Society.
The frequency of photopolymerization occurrence of diacetylene compounds was about 2 orders of magnitude higher on the h-BN(0001) surface243 than on HOPG. This could be explained by the enhanced lifetime of the intermediate excited state, as relaxation was suppressed due to the large bandgap of h-BN.
On the rutile TiO2(110) substrate, it was shown that the concentration of surface hydroxyl groups controlled the C–C coupling reaction of iodinated precursors.244 A moderate amount of hydroxyls was required to allow for the formation of longer chain oligomers. A multistep proton-assisted reaction mechanism was suggested to explain these results.
6.2. Substrate Orientation
Besides its chemical nature, the surface crystallographic orientation is also an important factor governing the reactivity of on-surface synthesis. The effect of the orientation is 2-fold: it modulates the symmetry of the surface ((111)-, (100)-, and (110)-oriented surfaces are hexagonal, square, and rectangular, respectively), and it modulates the coordination number of the surface atoms and their reactivity ((111)-oriented surfaces are the densest and the most inert while (110)-oriented surfaces are the least dense and the most reactive).
As an illustrative example, dibromo-bianthryl precursors (DBBA) can form 1D nanoribbons on a Cu(111) surface, but the reduced diffusivity and enhanced reactivity on Cu(110) completely inhibit intermolecular coupling.245 The 4,4″-dibromo-meta-terphenyl (DMTP) precursor formed linear zigzag oligophenylene chains on Cu(110)246 but cyclic hyperbenzene rings on Cu(111)16 due to a better lattice and symmetry match (see Figure 19a). As compared to Au(111), the Au(100) was shown to promote linear oligomerization for the domino reaction scenario,161 or to foster an oriented 2D growth mode for the sequential Ullmann coupling of porphyrins (trans-Br2I2TPP).145 Also, homocoupling of terminal alkynes (DEBPB) was achieved on Ag(111) while the reaction was limited to the formation of organometallic intermediates on Ag(100) and Ag(110) due to their higher stability on these surfaces.85 It was suggested that the square structure of the Cu(100) surface played a dominant role in directing the molecular assembly of phenylacetylene precursors and served as an active catalyst in the hydrogen tautomerization and C–C coupling to produce tetraphenyl[4]radialene in a cooperative cycloaddition mechanism.27 High selectivity toward the dimerization of dBA though [2 + 2] cycloaddition was observed on Au(100), while on Au(111) the trimerization reaction was equivalently found.247
Figure 19.

Influence of the substrate orientation. (a) The DMTP precursor undergoes two distinct reaction pathways depending on the orientation of the Cu substrate. On Cu(110), 1D oligophenylene zigzag chains are formed while on Cu(111) hyperbenzene macrocycles are formed. (b) Dual reactivity of the INDO4 precursor on Ag substrates. On Ag(111) both coupling types are obtained for the lowest annealing temperature. On Ag(110) the oxidative coupling only is allowed leading to the formation of aligned nanoribbons. On Ag(100) the Knoevenagel coupling alone is obtained at moderate annealing temperature, and both couplings are found at higher temperature. (a) Adapted with permission from ref (246). Copyright 2016 Royal Society of Chemistry. Adapted with permission from ref (16). Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (b) Adapted with permission from ref (248). Copyright 2018 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
A complex orientation-dependent behavior was demonstrated for indacene-tetrone molecules (INDO4) on low-index silver surfaces.13,248 Two competing reaction types were possible for this precursor, the Knoevenagel reaction or an oxidative coupling, and the crystallographic orientation of the supporting surface could control the reactivity (through the activation temperature) and the selectivity of the reaction (see Figure 19b). On Ag(100) the temperature controlled the selectivity: Knoevenagel dimers were formed first, and both coupling types occurred at higher annealing temperatures. The Ag(110) surface was highly selective: Only linear oxidative coupling occurred, and the annealing temperature controlled the reaction advancement (the polymer length). Finally, on Ag(111) both coupling types were observed, but they formed at a sensibly lower annealing temperature. Surprisingly, the densest surface (with highest coordination of the Ag atoms) was the most reactive.
6.3. Substrate Epitaxial Growth
Here we present some systems where the atomic-scale structure of the substrate produces particular epitaxial conditions that play an exceptional role in the surface reaction. In this way, well-identified topochemical reaction mechanisms are reported. This is for example the case on calcite surfaces for which the adsorption strength represents a crucial issue. The use of carboxylic groups allowed for chemisorption of the precursors at specific sites where the intermolecular distances and configuration were favorable for the reaction to take place.47,150 This effect was particularly true for diacetylene polymerization, for which the adsorption sites were preserved from the precursors to the final product.249
Epitaxial effects have been reported for other systems, particularly on (110)-oriented surfaces on which 1D alignment is usually achieved.13,104,250,251 For the aryl–aryl coupling of linear oligophenylene precursors (p-6P, 4Ph) on Cu(110) or Au(110), the particular adsorption sites produced a loss of equivalence of the different carbon atoms and the C–H activation was then achieved selectively in ortho position of the terminal carbons.66,252 An oriented growth guided by registry adaptation was reported for halogenated tetraphenylporphyrins on Au(100).145
The alkane polymerization on Au(110) was realized thanks to an appropriate reconstruction of the surface induced by the adsorption, that produced nanometer-sized 1D channels strictly constraining the orientation of the reactant alkane precursors. As a result, the reaction took place at intermediate temperatures and exclusively at specific terminal or penultimate carbon sites of the chains.15 The increased reactivity of the (1 × 3) reconstruction as compared with the (1 × 2) reconstruction was demonstrated by locally irradiating the sample surface with low-energy electrons (3 to 35 eV). In the irradiated areas, the (1 × 3) reconstruction was induced, and the reaction took place at lower temperature (20 K lower) as compared to the naturally (1 × 2) reconstructed nonirradiated areas.163 Similarly, decarboxylation and polymerization of fatty acids was achieved on Au(110).93
6.4. Substrate Templating
Here we propose some strategies where substrate nanostructuring (at scales larger than the atomic size) was produced and utilized to steer the on-surface reactions. A rather obvious strategy to produce 1D templating is the use of vicinal surfaces. These are surfaces presenting a slight misalignment with respect to a high-symmetry crystal plane that produces a regular distribution of aligned atomic steps. Vicinal surfaces have been successfully used as efficient templates to steer the alignment of various 1D covalent structures such as graphene nanoribbons (see Figure 20a,b).79,103,253−255 In particular, the use of the dibromoterphenyl (DBTP) precursor on the Au(322) surface could produce well-aligned GNRs of well-defined width, the latter selectively emerging from the vicinal terrace width, which provides a fixed number of polyphenylene wires per terrace that can laterally fuse.103 In the case of terminal alkyne reactivity (with DETP), the Ag(877) vicinal surface as compared to Ag(111) could achieve both alignment of the polymer product and selectivity in the coupling scheme (Figure 20c–e).79 It should be noticed however that in some cases the high reactivity of kink sites along the steps edges considerably reduced the length of the covalent wires.255
Figure 20.
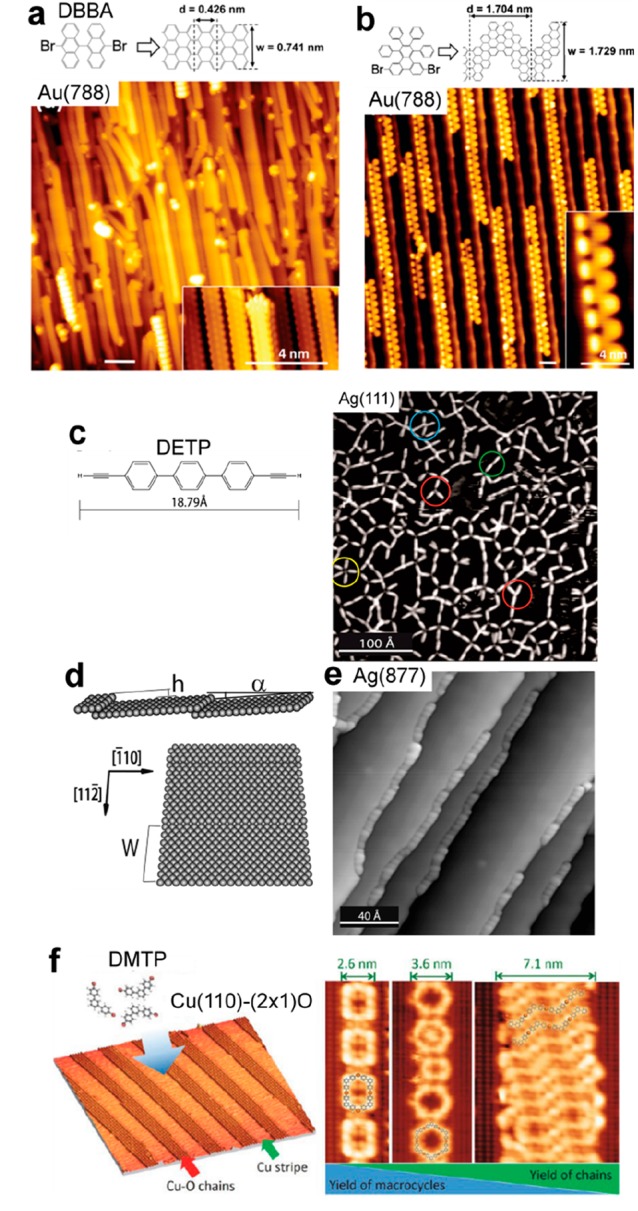
Substrate templating. The regular distribution of aligned atomic steps in vicinal surfaces allows for the formation of perfectly aligned straight (a) or zigzag (b) graphene nanoribbons (GNRs). (c) The reaction of DETP on Ag(111) leads to the formation of various coupling motifs and a 2D-like polymer. Oppositely, on the Ag(877) surface (d), high selectivity toward the linear coupling scheme is obtained, together with the alignment of the linear polymer along the step edges (e). (f) The Cu(110)-(2 × 1)O surface is used as a confinement nanotemplate for the formation of organometallic oligomers from DMTP precursors. Depending on the Cu stripe width, various motifs are selectively formed. (a, b) Reprinted with permission from ref (254). Copyright 2012 American Physical Society. (c–e) Adapted with permission from ref (79). Copyright 2014 American Chemical Society. (f) Adapted with permission from ref (260). Copyright 2016 American Chemical Society.
It was shown that the step edges are active as 1D constrainers for the alkane polymerization reaction on Cu(100) and Cu(111).80 The alkane chains exclusively reacted along (reshaped) step edges at low activation temperatures. These two surfaces, however, share the same preferred step orientation, namely, in both cases aligned along the close-packed [110] (or symmetry related) directions. Different step orientations running along the [112̅] (or symmetry related) direction correspond to the “fully kinked” configuration of the step edge.256 This obviously changes the step reactivity, but also the potential epitaxy with the adsorbate’s structure and its flexibility to reconstruct and accommodate, e.g., zigzag-shaped structures more efficiently. A beautiful example thereof is the alignment of zigzag polyphenylene chains periodically alternating para and meta junctions on top of vicinal silver surfaces.257
As an alternative to vicinal surfaces, an intrinsic surface inhomogeneity can be similarly used for templating. One such case is the uniaxial anisotropy of (110) surfaces of fcc crystal structures. The Cu(110) was used as an atomic-scale guide to produce a collimated motion of CF2 molecules that were manipulated with the STM tip.258 These projectiles were aimed toward a target molecule to induce a coupling reaction. Because the motion of the projectile was perfectly aligned along the atomic rows of the surface, it was possible to precisely control the molecular separation, i.e. to perform the reaction at selected impact parameters. It was shown that head-on collisions favored bimolecular reaction, whereas glancing collisions led only to momentum transfer.
Along similar lines, the native reconstruction of the Au(111) surface and the local uniaxial anisotropy of its zigzag part could also steer the alignment of graphene nanoribbons.259 Very long ribbons (>100 nm) were formed, and their lateral periodicity could vary with coverage at discrete values following a hierarchical templating behavior.
The Cu(110)-(2 × 1)O template represents an interesting approach for 1D nanopatterning. This template contains ordered arrays of alternating stripes of Cu–O chains and bare Cu, the widths of which are controllable. Dibromo-meta-terphenyl precursors (DMTP) were deposited on this surface, and oligomers were formed exclusively on the bare regions of the Cu surface.260 Depending on the width of the bare copper stripes, trapezoidal wave-like chains, arrays of cyclic tetramers or octamers, or zigzag oligomeric chains were obtained (see Figure 20f). It should be noted however that the reaction was limited to the formation of (oligomeric) organometallic species only, due to the limited thermal stability of the oxide template. Nevertheless, interesting effects were reported in relation to this controlled spatial confinement, and this template is probably promising for effective covalent coupling, provided an adequate chemical system is found. A more elaborate process was developed to produce surface templating using NaCl square islands that directed the formation of boronic acid based polymers, see section 11.2.261
6.5. Conspectus
The substrate naturally plays a fundamental role in on-surface synthesis. The nature of the substrate can control the activation temperature or the reaction selectivity. In the case of highly reactive surfaces the degradation of the precursors could be eventually favored. The crystallographic orientation of the surface defines both its symmetry and its reactivity (through the coordination number of the surface atoms). Modulating the surface orientation usually produces various nontrivial effects. Fine epitaxial effects in favor of the reaction have been reported on (110)-oriented surfaces, such as the alkane polymerization on (1 × 3) reconstructed Au(110) or the control on the impact parameter in reaction collisions. Vicinal surfaces usually act as efficient templates for the alignment of polymer chains or graphene nanoribbons.
7. Control of Growth Parameters
In on-surface synthesis, the reaction is confined to the two-dimensional environment of the surface. The reaction parameters available are the substrate (its nature and its structure), the temperature, the concentration of the precursors on the surface (the coverage), and eventually their stoichiometry. The influence of the substrate has been described in a previous section, and the remaining parameters will be discussed in the following. The term “growth parameter” is used here rather than “reaction parameter” because this section is mostly devoted to cases where the coupling reactions lead to the polymeric growth of a given network.
7.1. Kinetics Control
Thermal selectivity was demonstrated in the case of competing intra- and intermolecular coupling processes for tetra-aza-porphyrins bearing ethyl termini (OETAP) on Au(111).262 Room temperature deposition followed by annealing allowed for the precursors to diffuse freely, thus producing intermolecular coupling and formation of phthalocyanine tapes (Figure 21a). However, high-temperature deposition directly activated the intramolecular reaction and produced monomeric phthalocyanine that thus inhibited the intermolecular coupling (Figure 21b). Theoretical modeling (DFT) showed that the decisive step for the dimerization had a significantly lower free energy barrier than for the monomer cyclization. As a result, the monomer ring-closure could occur only if the dimerization was kinetically hindered such as for extremely low coverage, in the absence of nearby nonreacted molecules. Similarly, the Sonogashira coupling was selectively favored at high temperature to form graphyne nanowires from BPBE precursors, as compared to the energetically favored formation of graphdiyne nanowires through hierarchical Glaser and Ullmann coupling.263
Figure 21.
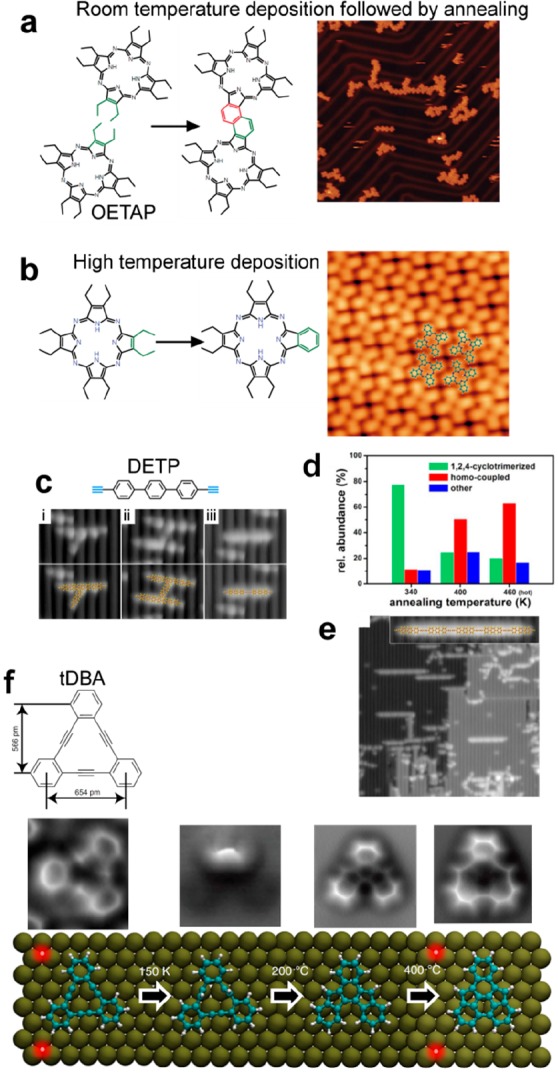
Kinetics control. For OETAP precursors on Au(111), the room temperature deposition followed by annealing resulted in the formation of oligomers (a) while the direct deposition on a hot substrate produced individual phthalocyanines (b) formed from an intramolecular reaction. (c) Different coupling schemes of the DETP precursor lead to different reaction products on Au(110), in particular the cyclotrimerization (i) or the linear homocoupling (iii). The selectivity toward the linear homocoupled product could be sensitively enhanced by increasing the annealing temperature (d), and even more by direct deposition on a hot substrate (e). (f) The precursor tDBA undergoes stepwise internal transformations leading to the formation of stable intermediate species depending on the annealing temperature (the AFM images are presented above the corresponding models). (a, b) Adapted from ref (262). Creative Commons Attribution 4.0 International License. (c–e) Adapted with permission from ref (264). Copyright 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (f) Adapted from ref (265). Creative Commons Attribution 4.0 International License.
The annealing temperature allowed researchers to selectively induce the cyclotrimerization (at moderate temperature) or the homocoupling (at high temperature) reaction of diethynyl-terphenyl (DETP) on Au(110) (Figure 21c–e).264 In fact it was suggested that both reaction pathways have the same intermediate step. Although the trimerization reaction had lower activation energy for the second step, it was also conditioned to the proximity refurbish of hydrogen atoms formed by the first intermediate. In the case of temperatures that are too high, these H atoms were lost and only the high activation pathway leading to homocoupling was thus allowed. Another case of reaction selectivity with temperature was reported for the Knoevenagel reaction vs oxidative coupling on Ag(100) (see section 6.2 and Figure 19b).248
Several examples of molecular internal transformations such as internal cross-coupling and cyclodehydrogenation have been reported for polycyclic aromatic precursors (sometimes partially unsaturated).55,81,265,266 Various kinetically trapped intermediate states were obtained and stabilized by adjusting the annealing temperature. They could be precisely identified by high-resolution AFM imaging, and a realistic scenario for the whole reaction pathway could be drawn in this way (see Figure 21f). For the initial growth of graphene from C2H4 on Re(0001), small C clusters formed that were kinetically trapped.267 They thus appeared as competing and therefore coexisting species for the growth of perfect graphene sheets.
Finally, the temperature can in some cases finely control the reaction advancement of polymerization reactions, by limiting the reaction to the formation of the first reaction steps (dimers or trimers) at moderate annealing or by extending it to the formation of long polymer chains at higher annealing temperatures.13,191,268
7.2. Stoichiometry, Concentration, or Coverage Control
Some studies have reported the effect of precursor concentration and stoichiometry in Schiff base reaction on HOPG.131−133,215 The concentration had important effects on the overall quality of the 2D polymer. Also, a change in the amine to aldehyde ratio could lead to incomplete reaction and a decrease of the dimensionality from 2D networks to 1D linear polymers.131,133 Eventually, the competing formation of an ordered bimolecular supramolecular phase could completely inhibit the reaction.215 It was suggested here that the relative affinities of the building-blocks to the surface, i.e., their adsorption energy, played an important role in the process of polymer formation. The molecule–substrate interaction could be tuned for example by varying the backbone size of the precursors133 or by adding alkyl side chains.215 This effect was utilized together with the dynamic covalent chemistry properties269 of a double imine formation reaction to control in situ the reaction product.130 Alkyl-diamines of varying length were sequentially introduced in an aldehyde solution (see Figure 22). The shorter chains in the imine products were dynamically replaced by the longer chain diamines to optimize the adsorption free energy. The same strategy was used with various precursors producing 1D or 2D polymers.270 The dynamic covalent library could be significantly altered by the addition of coronene as a guest molecule.271 The amplification of specific products was observed in this way.
Figure 22.

Reaction of aldehyde A with diamine B2, B6, or B12 is dynamically controlled by adequate molecular stoichiometry. The shorter chains in the imine products can be replaced directly on the HOPG surface by the longer chain diamines to optimize the adsorption free energy. Adapted with permission from ref (130). Copyright 2014 Springer Nature.
In another study using a tetrakis(formylphenyl)pyrene and various ditopic linear diamines, three different polymorphic covalent networks were obtained: rhombus, parallelogram, and Kagome.135 A clear tuning of the polymorph distribution was obtained by adjusting the monomer concentration while keeping the precursor ratio constant (see Figure 23a–c). Because all polymorphs had the same number of covalent bonds and a similar local configuration, and thus roughly the same free energy, it was suggested that the polymorph distribution was directly related to the differences in their densities, following the general trend demonstrated in concentration-dependent supramolecular assemblies. The molecular ratio of diboronic acid and hexahydroxy-triphenylene could tune the formation of covalent networks based on boronate esters, boroxine, or intermediate mixed products.272 In another work, it was shown that the total concentration of the precursors, at fixed stoichiometry, could have a similar effect.273 The esterification of carboxylic acid and alcohol on HOPG could be activated by increasing the precursor concentration either by ultrasonication or by extended stirring.274
Figure 23.

(a) Reaction of the tetra-aldehyde 1 with the aromatic diamines 2, 3, and 4 of various lengths leads to the polymorphic imine based networks with rhombus, parallelogram, and Kagome structure. (b) Corresponding STM images and chemical models. (c) Distribution of the occurrences of the different polymorphs for different network sizes. The Kagome structure preferentially forms at low concentration while the parallelogram (and to certain amount the rhombus) structure is preferred at high concentration. (d) On Ag(111) the DMTP precursor preferentially forms hyperbenzene macrocycles in low-concentration conditions (low deposition flux), but 1D oligomers in high-concentration conditions (high deposition flux). (a–c) Adapted with permission from ref (135). Copyright 2017 American Chemical Society. (d) Adapted with permission from ref (276). Copyright 2017 American Chemical Society.
The effect of stoichiometry in imine formation has been also studied in UHV.275 When using two similar star-shaped precursors, tris-formylphenyl-benzene (TFPB) and tris-aminophenyl-benzene (TAPB), the highest reaction advancement and network quality were obtained for a 1:1 stoichiometry. When using the same trialdehyde (TFPB) with the linear diamino-terphenyl precursor (DATP), the tuning of the stoichiometry allowed exploring rationally various oligomeric products with well-controlled structure.
In UHV conditions, the term concentration refers to surface concentration or surface coverage. Phase equilibrium is usually observed between self-assembled domains (in a dense phase) and highly mobile molecules in a dilute 2D gas phase. The deposition flux also plays an important role as it will allow the growth process to take place in high-concentration conditions (for high flux) or low-concentration conditions (for low flux).
For the domino reaction on Au(111) (ENA precursor, see section 3.3 and Figure 8b), low-coverage conditions allowed the formation of metal–organic compounds that inhibited the second reaction step.161 Only at high coverages was the full reaction possible. For the same precursor on Ag(111), high selectivity toward the alkyne trimerization was obtained at low coverage while the alkyne dimerization was preferentially obtained at high coverage. It was suggested that the trimerization reaction required more free space to reach the reactive relative orientation of the alkynes.277 For the halogen-free formation of graphene nanoribbons (GNRs) from quaterrylene (QR), regioselectivity was achieved by coverage control (see section 5.1 and Figure 13b,c).193 Two different orientations of the poly(paraphenylene) (PPP) chains formed from dibromobenzene (dBB) on Cu(110) were obtained depending on the surface coverage.104 The polymer chains were incommensurate and oriented along the ⟨11̅0⟩ direction at lower coverage while they were commensurate and oriented along both ⟨11̅2⟩ and ⟨11̅2̅⟩ equivalent directions at saturation coverage (1 ML). The cross-coupling reaction of bromobenzene (BB) and ethylbromobenzene (EBB) on Cu(111) was only made possible at very low coverages (<0.01 ML) to avoid repulsive forces between the two precursors (see Figure 10b).174 At large coverages (0.5–0.7 ML) the homocoupling was observed as the majority. Finally, the high-dilution principle was applied to Ullmann coupling on Ag(111) to control the formation of macrocycles based on the meta-terphenyl precursor (DMTP).276 The use of very low-deposition flux (<0.1 ML/h) allowed for efficient formation (>80% yield) of hyperbenzene macrocyles vs oligomeric open chains, as compared to standard deposition flux (>5 ML/h) for which the macrocycle yield was limited to ∼6% (see Figure 23b). The high probability of polymer cyclization in a low-concentration environment was related to the good reversibility of the C–Ag bond in the intermediate organometallic state and an efficient defect self-healing mechanism. The lateral periodicity of aligned GNRs on the reconstructed Au(111) surface could be varied into discrete values by controlling the coverage, thanks to the hierarchical templating behavior of the surface reconstruction.259
7.3. Conspectus
The reaction temperature controls the mobility of the precursors and their collision probability, and thermal selectivity is sometimes achieved in the case of competing reactions. The concentration of the precursors plays an important role in the overall quality of the networks formed at the liquid/solid interface. Also, the nature of the products can be tuned by modulating the concentration and the stoichiometry of the precursors in a dynamic covalent chemistry approach. The observed products are the result of a fine balance between their intrinsic stability and their affinity for surface adsorption. In general, the influence of supramolecular interactions is much more important at high coverages when dense phases are formed. Good selectivity can be thus achieved in UHV conditions by varying the surface coverage or the deposition flux.
8. Environmental Control
In ultra-high-vacuum (UHV) conditions, the system molecule-on-substrate represents a highly diluted environment and the chemical coupling reaction was shown to be completely irreversible. Growth in conditions allowing the reversibility of the coupling reaction could be obtained in an atmospheric environment, at the solid/liquid or solid/gas interface. In such a case, covalent networks with high crystallographic order and spatial extension up to several hundreds of nanometers can form. This issue was nicely illustrated in the case of diboronic acids that could grow in perfect and extended covalent networks on a HOPG surface upon annealing in a closed reactor in the presence of water vapor (Figure 24a). The latter was brought in the form of a droplet278 or released from hydrated copper sulfate.127 The network can decompose slowly in an ambient environment due to the residual humidity, but it can be reversibly recovered by repeating the same heating procedure (Figure 24b).
Remarkably, in reversible conditions the covalent coupling is a dynamic process269 that can be used to modify in situ the structures produced. The Schiff base formed from the reaction between aldehydes and diamines could thus be tuned in real time by adding sequentially longer chain diamines that replaced the previous ones in the final reaction product (dynamic combinatorial libraries, see also section 7.2 and Figure 22).130,270,271
The solvent nature usually has a strong influence on supramolecular self-assemblies, and it was shown that it could also govern the reaction pathway in a Glaser coupling reaction (DPIA precursor), leading to the formation of a linear polymer in phenyloctane or to cyclic dimers in trichlorobenzene (see Figure 24c).183 The pH can be easily tuned at the aqueous liquid/solid interface, which represents an efficient way to drive the reaction products or the reaction advancement, for example, by stabilizing intermediate states.279 Electropolymerization could be initiated at the liquid/solid interface by application of controlled electrochemical potential or potential pulses, the number of which directly influenced the polymer chain length.280−283
Finally, it was shown that the solvent-free reaction at the gas/solid interface in atmospheric conditions on HOPG had beneficial effects as compared to the liquid/solid interface. The nucleation was reduced, and the polymer extension was increased. Also, the sample homogeneity and its contamination were improved.158,284
8.1. Conspectus
While in vacuum (UHV) the coupling reaction is irreversible, conditions achieving reaction reversibility can be efficiently achieved in an atmospheric environment. As a result, highly ordered extended networks have been obtained on the basis of the imine or the boronic acid chemistries. Also, the networks can be dynamically and reversibly modified or repaired. Finally, at the liquid/solid interface the reaction is directly influenced by the solvent type or the pH, and electrochemical reactions can be initiated.
9. Alternative Activation Methods
Covalent coupling reactions usually require thermal activation, but alternative activation methods have been developed, by means of manipulation with the tip of a scanning probe microscope or external light or electron beam irradiation. By avoiding thermal annealing, it is in particular possible to gain better control on the diffusivity of the precursors and therefore on their self-assembled structure prior to and after reaction. In addition, it is possible to control finely the localization of the reaction and the polymer growth.
9.1. Tip-Induced Reaction
Manipulation of surfaces with a high level of control can be achieved at the single atom level with the tip of a scanning tunneling microscope (STM).285,286 The STM tip has also been identified as an efficient manipulation tool to control covalent coupling reactions on surfaces. In a pioneering work, individual iodobenzene molecules were dissociated, displaced, and reassembled to form biphenyl units in a kind of “hand-made” Ullmann coupling reaction (see Figure 25a).36,287 Polyyne formation was achieved by tip manipulation from the corresponding dibromoolefin on NaCl/Cu(111). Long polyynes, up to the octayne Ph-(C≡C)8-Ph, were prepared in this way.288 Tip manipulation was used on a Cu(110) surface to induce reaction between two CF2 units with a controlled collision geometry, i.e., at selected impact parameters.258
The self-condensation reaction of boronic acid (BDBA) could be initiated locally inside a dense hydrogen-bonded layer by reducing the tip–surface distance and removing some of the molecules, thus providing to the system the free space required for the reaction to take place (see Figure 25b).216 Oligomerization could be induced by patterning a surface area with high tip bias (>3 V).289 Here the reaction from halogenated precursors (2TTA) was however limited to the formation of organometallic oligomers.
The topochemical polymerization of diacetylene compounds on HOPG could be initiated locally inside a self-assembled phase of diacetylene compounds by injecting an electron pulse with the STM tip (see Figure 25c and the description of diacetylene polymerization in section 5.1 and in Figure 15a).37,196−199 Spatial control on the order of 1 nm could be achieved. Remarkably, when phthalocyanine molecules were codeposited on top of the self-assembled diacetylene precursor layer, so-called chemical soldering could be realized (see Figure 25d).198 The self-propagating reaction terminated by creating a covalent bond between the polymer wire and the phthalocyanine in the case where the latter was adequately positioned with regards to the reactive species at the end of the wire. A covalent junction could be realized at both sides of the phthalocyanine, opening up the possibility to create complex organic circuitry on a surface.
Nanocorrals of defined shape were created on an HOPG surface covalently modified with aryl molecules by removing part of the grafted monolayer (nanografting technique,290 see Figure 26a).291 The newly exposed areas immediately accommodated diacetylene compounds with an alignment directed by both the scanning direction during nanografting and the graphite orientation.292 The diacetylene topochemical polymerization was then initiated inside the nanocorrals, and the polymer chain length was limited by the nanocorral host extension (Figure 26a). High control on both chain length and orientation was therefore obtained. The same strategy has been applied to the formation of Schiff base 2D polymers (nanoshaving).293
Figure 26.

Tip-induced reactions. (a) Nanocorrals are created at the surface of a covalently modified HOPG surface by mechanical nanoshaving. PCDA precursors preferentially adsorb inside the nanocorrals, and diacetylene polymerization can be locally initiated with the STM tip. The length of the as-formed PDA wires is limited by the nanocorral host extension. (b) Schematics of the cSPL technique: the AFM tip coated with an organometallic catalyst is brought into contact with an alkene-terminated SAM to locally epoxidize it. In a second step, selective grafting of a nucleophile is achieved. (a) Reproduced with permission from ref (291). Copyright 2017 Royal Society of Chemistry. (b) Adapted with permission from ref (294). Copyright 2016 American Chemical Society.
Various dendronized polymer wires were manipulated with the tip of an AFM and positioned next to each other at defined connecting points (mechanical connection). Chemical soldering was then initiated by UV irradiation providing covalent connections between the polymer chains at the predefined positions.82−84
In catalytic scanning probe lithography (cSPL),295,296 the tip of an AFM was modified to accommodate a catalyst, usually a metal deposited as a thin film or in nanoparticule form. Part of the reactants was provided on a surface in the form of a self-assembled monolayer (SAM) bearing reactive end groups, and the remaining reactants were solvated in the surrounding liquid. A chemical coupling reaction was initiated locally by mechanical application of the catalytic tip, and various chemical compounds could be grafted selectively, with a spatial resolution in the order of a few tens of nanometers.297−304 The technique could be extended to the use of a homogeneous catalyst immobilized onto the tip upon modification with an appropriate ligand (see Figure 26b).294,305−307 The use of a homogeneous catalyst in principle opens the way to a large variety of chemical reactions. It was shown that the catalytic activity of the tip-supported catalyst was barely affected during the reaction process, and large areas could be covalently modified without significant loss of the reactivity.
9.2. Light-Induced and Electron-Beam-Induced Reactions
Light irradiation is a powerful approach for initiating on-surface reactions. It is fast and reliable and can be used in various environments. Light irradiation can be efficiently used to initiate topochemical reactions, i.e., for such reactions producing very small byproducts and when the reaction is expected to induce minor conformational rearrangements. In general, the use of light as an external activation source is advantageous because the reaction initiation can be decoupled from the precursor diffusion that is thermally activated, thus allowing, e.g., an efficient supramolecular templating effect. In particular, the competitive thermal desorption that is usually encountered can thus be avoided. Also, the use of excited-state chemistry can in principle open additional reaction pathways. A part of the results presented here on surface-supported photochemical reactions was recently summarized in some review articles.308−310
The topochemical reaction and the polymerization of diacetylene compounds could be initiated from a well-ordered supramolecular assembly by UV light irradiation. This has been realized on various metallic or insulating substrates: HOPG,195,196,200,311−315 MOS2(0001),313 graphene on SiC,316 Au(111),311 h-BN,243 and calcite.249
Stilbene derivatives first underwent a trans to cis isomerization upon short UV irradiation that positioned the C=C bonds in adequate configuration inside the supramolecular template. Upon further irradiation, a [2 + 2] photodimerization occurred.210,317−319 Note that a precursor with both azobenzene and diacetylene groups could successfully photopolymerize and/or undergo cis–trans isomerization in a fully controlled way depending on irradiation wavelength,314 although the diacetylene polymerization did not depend on the azobenzene isomer form. This combined approach could be promising to gain a high level of control on the reaction. Light irradiation was used to cause degradation of a diboronic acid (ABBA) based nanoporous covalent network containing azobenzene groups.320 The high stress induced by the photoisomerization of the azobenzene moieties resulted in nearly complete destruction of the covalent structure. In this way, phthalocyanine guests incorporated into the network could be controllably released.
As mentioned in section 9.1, light irradiation can be used as chemical soldering to initiate covalent connections between polymer chains that were first manipulated into favorable predefined positions.82−84 Photodimerization of an anthracene derivative was performed at the HOPG–liquid interface.321 The precursor first self-assembled in an ordered phase in flat-lying configuration, and the apparent height increased after irradiation, revealing the realization of an out-of-plane dimerization. It was suggested here that the dimerization occurred in the liquid phase and that the dimer products were gradually replacing the unreacted monomers in the self-assembled monolayer.
Photopolymerization of C60 has been realized on a calcite (10.4) substrate, leading to oriented covalently linked domains.322 The orientation of these domains and the direction of the coupling reaction were dictated by the anisotropic lattice mismatch between the pristine C60 monolayer film and the calcite substrate. UV light irradiation revealed particularly efficient in increasing the reaction yield for the formation of polymeric chains on KCl(001) (see section 11.3 and Figure 34).92
Figure 34.
Formation of polymerized chains on a bulk alkali-halide substrate (a) without or (b) with the help of UV light irradiation. Adapted with permission from ref (92). Copyright 2018 Springer Nature.
Laser light irradiation was shown to induce simultaneous bond breaking of C–Br and C–OH bonds in the bromo-hydroxyphenyl precursor on Ag(111).323 The bond scission happened at low temperature (80 K), and the polymerization between the as-formed biradicals occurred at significantly reduced temperature (room temperature, RT). Using a sterically hindered precursor (DBT) that inhibited direct C–C coupling, it was demonstrated further that light irradiation induced C–Br bond breaking at room temperature,324 which represented the rate-determining step for the complete Ullmann coupling process.44 Because light irradiation enabled the use of lower temperature, better control on the growth mode of polymeric chains was achieved using aminophenyl prophyrins (trans-TPP(NH2)2) on Ag(110).325 Also, here a stereoselective photopolymerization was demonstrated due to the strong interaction with the substrate.
For photochemical Glaser coupling on Ag(111), light irradiation at room temperature allowed researchers to limit the coupling reaction to the formation of dimers, as compared to thermal activation that produced longer oligomers.326 Photochemical Glaser coupling was also demonstrated at the liquid–HOPG interface, and a high selectivity was observed due to topochemical reaction.201
Finally, low-energy electron irradiation has been used to indirectly initiate covalent reactions. The polymerization reaction of boronic acid (BDBA) was activated by global illumination with a low-energy electron beam (15 eV).216 Partial desorption of the precursors was thus induced, that provided to the remaining molecules enough free space to react readily. In the case of alkane (DMH) polymerization on Au(110) (see section 6.3), the increased reactivity of the (1 × 3) reconstruction as compared with the (1 × 2) reconstruction was demonstrated by locally irradiating the sample surface with low-energy electrons.163 In the irradiated areas, the (1 × 3) reconstruction was induced and the reaction took place at lower temperature (20 K lower) as compared to the naturally (1 × 2) reconstructed nonirradiated areas.
9.3. Conspectus
The tip of the scanning probe microscope (STM/AFM) is an efficient tool to manipulate molecules on a surface and initiate chemical reactions. Beyond a fundamental interest to control and observe in parallel the different reaction steps, it is possible to create elaborate structures at the nanometer scale and beyond. Although limited to single-molecule investigations, large-area patterning could be still envisioned using high-density probe arrays.295
Light irradiation has been efficiently used for a wide range of surface-supported reactions such as diacetylene polymerization, C–H activation, Glaser and Ullmann coupling, or maleimide polymerization. In principle, the use of light irradiation as activation means facilitates the in situ imaging of the reaction by limiting the diffusivity of the precursors that is normally induced by thermal activation. We expect light irradiation to be advantageously used in the future in advanced hierarchical coupling reaction strategies. Indeed, in this way two completely orthogonal activation types could be used, and a higher level of control should be gained.
10. Graphene Nanoribbons (GNRs) as Representative Examples of Tailored and Controlled On-Surface Synthesis
Among the different kinds of molecular structures that have been synthesized by means of on-surface synthesis, graphene nanoribbons (GNRs) have definitely attracted the most attention.9,68 The reason for it is found in two interrelated aspects. On the one hand, this family of materials holds enormous interest for many different applications, ranging from thermoelectric devices,327,328 batteries,329 capacitors,330,331 photodetectors,332 or gas sensors,333 to transistors334 or directly integrated circuits.335 One of the sources for such great versatility is the enormous variability they display in their optoelectronic properties as a function of their precise atomic structure.9,68,336 On the other hand, the marked dependence between structure and functionality underlines the strict requirement of atomic precision in the synthesis of the GNRs. To date, this has only been achieved from the bottom-up approach. Although in this respect there have also been important advances in wet-chemistry,337−341 the first successful proof of atomic precision in GNRs was obtained by on-surface synthesis, which still remains the approach whereby the largest pool of GNRs have been grown.9,68,151 Thus, it is the great potential for applications, coupled with on-surface synthesis as a viable route to fully exploit it, which has acted as the main driving force behind the fascinating developments in GNR synthesis.
Sharing many of the superlative properties of its parent material graphene, GNRs owe the additional tunability of their electronic properties to the boundary conditions imposed by the ribbon’s edges.9,68,336 As a result, with control of the GNR width, the crystallographic orientation of its edges, and the detailed edge structure, the electronic properties of GNRs can be changed, e.g., from a semiconducting band structure with a widely tunable gap to a metallic band structure further characterized by correlated low-dimensional magnetism.9,68,336 In combination with additional controllable parameters like the presence of heteroatoms, the formation of heterostructures, or tailored strain, the possibilities to shape the electronic properties of GNRs according to our needs are extraordinary.9,68 Given such a promising scenario, several approaches explained heretofore for on-surface synthesis in general have been applied also to the controlled synthesis of atomically precise GNRs. In the following, we provide some representative examples.
10.1. Precursor Design for Tunable GNR Structures
The first pioneering work reporting the synthesis of atomically precise graphene nanoribbons made use of 10,10′-dibromo-9,9′-bianthracene (DBBA, Figure 27a) and proved its adequate reactivity on Au(111) and Ag(111).151 In a first step, Ullmann coupling is activated, giving rise to highly nonplanar polymeric chains through radical step growth.151,342,343 At higher temperature a second reaction is activated, namely, the cyclodehydrogenation in between the interconnected anthracene units of the polymer, affording atomically precise graphene nanoribbons (Figure 27a).151,342,343 These first GNRs show edges oriented along the armchair direction (30° deviated from a graphene lattice vector) and 7 atoms across its width. They are thus termed 7-aGNRs. In that same article and following a similar two-step process, another GNR with chevron edge structure was synthesized as well (Figure 27i), evidencing the generality of this approach and thereby its promise for further developments.151
Figure 27.

(a) Schematic representation of the reactant, intermediate polymer, and nanoribbon end product during synthesis of 7-aGNRs. Associated STM images with the overlaid models are displayed below the polymer and GNR. Below, some representative reactants and associated microscopy images of the resulting GNRs (with superimposed wireframe structures) are displayed for varying GNRs. Different edge orientations are displayed (b, armchair; c, chiral; d, zigzag). Different widths of armchair ribbons are displayed (e, 5-aGNR; f, 9-aGNR; g, 13-aGNR). Different edge structures are shown (h, cove edge; i, chevron; j, fluoranthene functionalized zigzag edge). Differently doped structures are shown (k, boron heteroatoms; l, nitrile functional groups; m, N heteroatoms. (a, i) Adapted with permission from ref (151). Copyright 2010 Springer Nature. (b) Adapted with permission from ref (371). Copyright 2013 Springer Nature. (d, j) Adapted with permission from ref (152). Copyright 2016 Springer Nature. (e) Adapted from ref (350). Creative Commons Attribution 4.0 International License. (f) Reproduced with permission from ref (107). Copyright 2018 American Chemical Society. (g) Adapted with permission from ref (348). Copyright 2013 American Chemical Society. (h) Adapted with permission from ref (352). Copyright 2015 American Chemical Society. (k) Adapted from ref (357). Creative Commons Attribution 4.0 International License. (l) Adapted with permission from ref (361). Copyright 2017 American Chemical Society. (m) Adapted with permission from ref (356). Copyright 2014 Springer Nature.
Indeed, ever since this seminal work was published in 2010,151 the research on atomically precise GNRs boomed, with most efforts focused on the development of new reactants that lead to different GNR structures. A first important point to consider in the reactant design was found to be a minimization of steric shielding, which may otherwise limit the initial polymerization step. By way of example, the coupling of 10,10′-dibromo-9,9′-bianthracene (Figure 27a) and the coupling of its monoanthracene equivalent have been compared. The intramolecular steric hindrance of the former makes the precursor nonplanar, with an alternating tilt of each anthracene unit with respect to the substrate. As a result, after the halogen activation, two molecules can approach each other with alternatingly tilted anthracene units. This lowers the intermolecular steric hindrance and thus allows the radicals to get close enough to form a covalent C–C bond.9 Instead, the monoanthracene reactant lies flat on the surface. After the halogen activation, the steric repulsion between the hydrogen atoms next to the C radical of either molecule hinders the radical coupling.9 The same steric shielding is found if cyclodehydrogenation of a bianthracene precursor (and thereby its planarization) sets in before the dehalogenation.344 This scenario was observed for chlorinated reactants, whose C–Cl bond displays a higher activation temperature threshold than the brominated counterpart and, most importantly, than the cyclodehydrogenation step.344
Besides granting a successful GNR synthesis, the reactants have been designed so as to provide ribbons with different edge orientations (Figure 27b–d),151,152,345−347 widths (Figure 27e–g),107,151,348−351 edge structure (Figure 27h–j),151,152,352−354 as well as different heteroatoms or functional group doping (Figure 27k–m).347,355−364 As mentioned above, each of these variations brings about important changes in their electronic properties,9,68 which can be further refined by combining the GNRs into heterostructures.354,356,365−368 In fact, the careful design of precursors even allows building periodic heterostructures into the GNRs, whereby topological band engineering has appeared as an additional and powerful handle on the GNR properties.369,370
10.2. Atomically Precise GNRs through Hierarchic Coupling Schemes
To date, the reaction scheme utilized in the seminal GNR synthesis work,151 consisting of Ullmann coupling followed by intramolecular cyclodehydrogenation (Figure 27a), has been the most used approach by far.9,68 However, in a few instances a hierarchic coupling going beyond that common scheme has been applied as well. Although the intramolecular cyclodehydrogenation shows a lower activation temperature threshold, the intermolecular cyclodehydrogenation can also be activated at higher temperatures. That is, following the initial GNR synthesis, those readily formed ribbons can be laterally fused into wider ones at higher temperatures. This concept was first demonstrated on Ag(111) with 7-aGNRs that fused into 14-aGNRs and 21-aGNRs, depending on the number of 7-aGNRs involved.73 Later on, the same reaction was performed on Au(111), starting not only from 7-aGNRs (Figure 28a–f),372−374 but also from 9-aGNRs,374 5-aGNRs,375 or poly(paraphenylene) (PPP),191,376 which can be considered as the narrowest possible armchair-oriented GNR (3-aGNR). As a result, GNRs with a great variety of widths have been obtained, including GNR heterojunctions between well-defined segments of different widths. These systems have been extremely useful for learning about the structure–property relations linking those varying widths or the heterojunction structures with their corresponding electronic properties. However, the downside to this approach is that it relies on random fusion events, lacking the selectivity toward particular GNR structures that is ideally required for potential applications. In this respect, one way to get around this limitation is the use of substrate templating,103 as will be discussed later.
Figure 28.

(a) Schematics of the hierarchic reaction process that, starting from DBBA, first forms polymers and then 7-aGNRs, which in turn serve as reactants for the next step forming GNRs of different widths (and the associated heterostructures) through the lateral fusion of the ribbons. Representative images of 7-aGNRs (b), 14-aGNRs (c), and 21-aGNRs (d), as well as of the associated 7-14-aGNR (e) and 7-14-21-aGNRs heterojunctions, are shown below. (g) STM image of a sample with hierarchically grown GNR heterojunctions through combination of the reactants displayed in part h. (i) Magnified STM image of such a GNR heterojunction. (a–f) Reproduced with permission from ref (373). Copyright 2017 American Chemical Society. (g–i) Reproduced with permission from ref (368). Copyright 2018 American Chemical Society.
It is worth mentioning that the same approach has also been used to laterally fuse GNRs not holding straight edges. One such example made use of width-modulated GNRs alternating 7- and 13-atom-wide segments.353 Their lateral fusion results in the formation of regular and nanometer-scaled pores in between the GNRs, and the process can be extended to culminate in the formation of two-dimensional nanoporous graphene patches with several tens of nanometers in length and width.353 The use of different precursors has later on demonstrated the tunability in size and structure of the pores.377
Hierarchic coupling has also been applied to the synthesis of GNR heterostructures combining, as described in section 3.3, reactant molecules functionalized with Br and I, two halogens with different activation temperatures.368 If two different precursor molecules that share the same halogen functionalization are combined, they will randomly couple to one type or another. As a result, an uncontrolled sequence and position of heterojunctions is obtained along the length of a ribbon that, although useful for fundamental studies, is difficult to incorporate into functional nanodevices. However, by using precursors with different halogens, the growth sequence can be influenced. Thereby, although still maintaining a stochastic contribution, the resulting GNR structures can be made to exhibit preferentially a single heterojunction interface instead of a random sequence of them (Figure 28g–i).368
10.3. Controlling the GNR Growth through Appropriate Substrates
Most of the chemical processes involved in the GNR synthesis and their associated energy barriers depend notably on the substrate material and surface structure. While on one hand this limits the range of potential substrates to be used, on the other hand their appropriate choice can be used as an additional parameter to control the reaction and thus the resulting products,378,379 their distribution, or their alignment.253,254,346,360,380−382
A beautiful example is the comparative reactivity of 10,10′-dibromo-9,9′-bianthracene (DBBA) on (111) surfaces of three coinage metals, Au, Ag, and Cu (Figure 29). On Au(111) the precursor reacts following the scheme readily described in Figure 27a. As expected, upon annealing first the polymerization through Ullmann coupling sets in, followed by a subsequent cyclodehydrogenation step at a higher temperature that affords 7-aGNRs (Figure 27a).151,342,343 Instead, on Cu(111) the strong interaction between reactant and substrate drives a completely different reaction process in which the key factor is the catalytic activation (dehydrogenation) of the C2 and C2′ atoms, rather than the halogenated C10,10′ positions.378,379,383 Further proof of it is that the same end products are obtained independently of the reactant halogenation (or its absence).379,383 As a result, chiral graphene nanoribbons are obtained instead of armchair ones (Figure 29b),108,378,379,383 consequently with utterly different electronic properties.346 On Ag(111), with an intermediate reactivity between Au(111) and Cu(111), the reaction proceeds as on Au(111), following the Ullmann coupling and cyclodehydrogenation sequence for which the reactant was initially designed and affording 7-aGNRs (Figure 29c).73,151,235 One minor difference is that, as occurs with most other reactants undergoing Ullmann coupling, the presence of a metal–organic intermediate is observed on Ag(111) but not on Au(111).235 In addition, the increased reactivity of Ag(111) with respect to Au(111) still makes the synthesis more challenging. A planarization through cyclodehydrogenation of the reactants has been occasionally observed before formation of the polymers through Ullmann coupling,73,235 thereby preventing the ultimate GNR formation (Figure 29d).235
Figure 29.

Representative STM images and wireframe chemical structures of the different end products obtained from 10,10′-dibromo-9,9′-bianthracene on (a) Au(111), (b) Cu(111), and (c, d) Ag(111). On the latter, both 7-aGNRs (c) and layers of cyclodehydrogenated monomers (d) have been observed alike. (a) Adapted with permission from ref (151). Copyright 2010 Springer Nature. (b) Reproduced with permission from ref (384). Copyright 2015 American Chemical Society. (c, d) Adapted from ref (235). Creative Commons Attribution 4.0 International License.
It is worth noting that not only is the particular substrate material critical in the GNR synthesis, but also its surface structure. By way of example, the same precursor discussed above reacts differently on two disparate surface planes of Cu, namely, Cu(111) and Cu(110).245 On Cu(111) it polymerizes first and then forms GNRs through cyclodehydrogenation. In contrast, on the less compact (and therefore presumably more reactive) Cu(110) surface, the intramolecular cyclodehydrogenation occurs first. As a result, the polymerization is quenched, and the end products are 0D nanographene molecules instead of 1D GNRs.
Alternatively, periodically stepped surfaces have been used, not to modify the reaction schemes and the product structures, but to align them. This approach was initially used to obtain a uniaxial azimuthal alignment of 7-aGNRs on Au(788),253,254 although later on its use was extended to different GNRs (9-aGNRs,380 chiral (3,1)-GNRs17) and different stepped surfaces like Au(322).346 In doing so, ensemble-averaging techniques like angle-resolved photoemission spectroscopy253,254,346,380,382,385 or Raman380−382 spectroscopy could be applied along well-defined orientations, obtaining valuable information on the anisotropic optoelectronic properties of the GNRs.
Beyond the alignment, such periodically spaced steps have also demonstrated their applicability as templates to control hierarchical coupling schemes like the one described above, in which poly(paraphenylene) (PPP) chains were synthesized first, and subsequently fused at higher temperature into wider aGNRs through intermolecular cyclodehydrogenation. When performed on flat Au(111), this process leads to a disordered mixture of aGNRs of varying widths, in addition to a few remnant PPP wire segments (Figure 30a). Instead, on Au(322), with regular terraces whose width can fit two side-by-side PPP chains at most, only homogeneous and uniaxially aligned GNRs are formed apart from the remnant unfused PPP wires. These ribbons are identified as selectively synthesized 6-aGNR, formed by the substrate-limited two-by-two fusion of PPP chains (Figure 30b).103
Figure 30.
Representation of the different end products obtained from the high-temperature lateral fusion of PPP on Au(111) (a) and on Au(322) (b). While on the former different widths of GNRs are obtained, on the latter, apart from some PPP remaining unreacted, only 6-aGNRs are obtained. Adapted with permission from ref (103). Copyright 2018 American Chemical Society.
The catalytic properties of the substrates can be controlled not only by its material or surface structure. As readily discussed in section 4, also the addition of extrinsic catalytic adatoms can be used, without the need for changing substrates, to tune and guide the reactivity of the systems. This has also been shown for the synthesis of GNRs: By adding Ni atoms onto a Au(111) surface, 10,10′-dibromo-9,9′-bianthracene (DBBA, the reactant used to synthesize 7-aGNRs) displays a notably different reactivity.386 Readily at RT it is dehalogenated and forms a polymeric organometallic intermediate. While the subsequent C–C coupling into polyanthrylene is observed at similar temperatures of ∼200 °C, in the following cyclodehydrogenation step at ∼400 °C the Ni atoms catalyze the intermolecular fusion, leading to a substantially larger amount of laterally fused GNRs of 14, 21, or 28 carbon atoms in width than without the presence of Ni adatoms.386
10.4. Reaction Kinetics as a Source of Control over GNR Structures
Another important source of control over the GNR structures is the reaction kinetics, which can be manipulated via appropriate reactant design, by the choice of substrate, or by the growth parameters like temperature, deposition rate, or coverage. Regarding the reactant design, one can, e.g., compare 10,10′-dibromo-9,9′-bianthracene (Figure 27b), designed to render 7-aGNRs, with 2,2′-dibromo-9,9′-bianthracene (Figure 27c), designed to render chiral (3,1)-GNRs. Unexpectedly, as readily mentioned above, on Cu(111) both precursors form chiral ribbons through an activation of the same C2 and C2′ atoms. However, with the former the activation involves C–H bonds, while with the latter it involves more labile C–Br bonds. The latter thus facilitates the initial polymerization, ending up with much longer chiral GNRs by placing the halogen functionalization at the right carbon atom.345
A comparison of the reaction kinetics associated with the same two precursors on Au(111), on which armchair and chiral GNRs are formed, respectively, is also very interesting. At this point it is important to remember the nonplanarity of these reactants, in which the two anthracene units display alternate tilts with respect to the surface. For the “chiral precursor” (Figure 27c), the reactant adsorbs with the anthracene ends that hold the Br atoms facing toward the surface, while for the “armchair precursor” (Figure 27b) the Br atoms are at midheight along the tilted anthracenes. For the Br atoms thus lying closer to the catalytic surface for the “chiral precursor”, their dehalogenation temperature threshold is lowered. However, even more notable is the change in the activation temperature of the polymer cyclodehydrogenation. In this case it rather relates to the completely different strain between the two resulting polymers. The anthracene units within the “armchair polymer” are linked covalently along their short axis by a single bond that allows free rotational movement with respect to their neighbors. This freedom allows the alternating tilt of the anthracene units along the polymer backbone, driven by a minimization of the steric hindrance between opposing H atoms in neighboring anthracenes. The “chiral polymer” faces a similar steric hindrance. However, in this case the anthracene units are covalently linked along both their long and short axes. As a result, their rotational freedom is greatly reduced and a substantial strain accumulated. Sterically induced strain has been shown to weaken the involved C–H bonds and thereby the cyclodehydrogenation barriers,69,342,343 in turn explaining the lower threshold temperature displayed by the “chiral polymers”.
Effects like these have important implications. On one hand, lowering the required temperatures in GNR synthesis may allow the use of new substrates that could not withstand higher temperatures. On the other hand, the varying kinetics of the associated reactions can have a substantial impact on the resulting products. In this respect it is worth noting that the radical step growth during the Ullmann polymerization can be easily quenched by available H atoms that saturate the radicals. Because cyclodehydrogenation liberates H atoms, it may compete with the polymerization and greatly lower the average length of resultant GNRs if both processes take place simultaneously. This was initially demonstrated with the growth of chiral (3,1)-GNRs, which display a particularly low cyclodehydrogenation temperature.345 As the substrate temperature during the precursor deposition was increased, the average GNR length decreased due to the quenching of the radical step growth by H atoms liberated in early cyclodehydrogenation events. Instead, the longest GNRs were obtained at the lowest substrate temperatures allowing for polymerization, only to cyclodehydrogenate the structures in a subsequent annealing step.345
Closely related findings were reported later on for the growth of 9-aGNRs on Au(111).99 Two different precursor molecules were used, one functionalized with bromine (Figure 27f) and the iodinated analogue. The polymeric structure obtained from both precursors is the same, therefore sharing the cyclodehydrogenation energy barrier and consequently the activation temperature threshold. However, with the dehalogenation temperature being lower for iodine than for bromine, the iodinated precursor displays a larger separation in temperatures between the polymerization and cyclodehydrogenation steps. As a result, the average GNR length could be enhanced by a factor of 3 changing from Br to I.99
Manipulation of the reaction kinetics through other parameters like the temperature or coverage has also been demonstrated as a useful route to control the resulting GNR structures. Focusing first on the temperature, the different activation thresholds of sequential reactions allow selective synthesis of particular structures that could be seen as intermediates along the reaction path toward a different end product. An example hereof could be the previously described examples of hierarchic synthesis of GNRs through their lateral fusion. That is, on Au(322) the end product for DBTP is 6-aGNRs.103 However, by staying below the cyclodehydrogenation temperature, poly(paraphenylene) can be synthesized exclusively.103 Similar scenarios can occur also at the intramolecular level. This is for example the case with sulfur-doped precursors of chevron GNRs: beyond the initial polymerization and cyclodehydrogenation that readily forms the doped GNRs, another subsequent reaction can be triggered at higher temperatures that cleaves the C–S bonds and leaves a pure-hydrocarbon GNR structure behind (Figure 31).364
Figure 31.

Chemical structure and STM and nc-AFM images of the different polymers and GNRs obtained from the precursor displayed at the left as a function of the annealing temperature. A sulfur-doped GNR is obtained at 720 K annealing temperature. At 840 K, the S atoms are cleaved and the final product is a pure-hydrocarbon GNR. Adapted with permission from ref (364). Copyright 2018 Springer Nature.
Regarding the influence of reactant coverage, an explanatory example is that shown in Figure 13b,c, in which quaterrylene (QR) molecules are used as reactants that fuse through cyclodehydrogenation and give rise to 5-aGNRs.193 The lowest reaction barriers are observed for fusion at an angle (most commonly 150°), rather than with a parallel alignment. As a result, kinked structures of short 5-aGNR segments are preferentially obtained at low precursor coverages. However, as the initial coverage of reactants is enhanced, that scenario changes substantially. The compact self-assembled arrangement of reactants on Au(111) favors the linear fusion into straight 5-aGNRs, while hindering the kinked products through steric shielding.193 Notably, the steric shielding does not stem from the molecular structure itself, but from the surrounding molecules, making the surface coverage an important parameter for control of the reactions.
10.5. Conspectus
The growth of atomically precise graphene nanoribbons is an intensively sought challenge. The great interest that this type of materials has generated and the viability of on-surface synthesis to create them has thus been an important driving force for the development of this whole research field. Besides representing a direct link to potential applications, there have been countless efforts in their synthesis that applied most of the approaches described in earlier sections. A convenient reactant design, the use of hierarchic coupling schemes, and the use of appropriate substrates to guide the reactivity or to align the resulting ribbons, as well as a rational use of the reaction kinetics, have all been proven to be suitable to control the ultimate GNR products.
11. Toward Functional Materials
Besides the unquestionable interest of on-surface synthesis merely as a complement to conventional wet-chemistry, this chemical approach also holds great promise for the development of applications and the synthesis of molecular materials with tailored functionalities of many kinds (e.g., electrical, optical, catalytic, or magnetic). In this regard, among many noteworthy carbon-only materials that have been obtained by on-surface synthesis with particular functionalities in mind, we find for example porous graphene,353,387 graphyne based nanostructures,79,162 or the graphene nanoribbons described above.68 However, not only are the materials themselves required to develop real applications. Often they require patterning into well-defined mesoscopic structures or transfer or synthesis directly on insulating or semiconducting surfaces. Some of these functionality-related issues are addressed in the sections below, starting with in situ functionality tests and followed by other aspects like nanopatterning, growth on insulating substrates, transfer methods, and ultimate device fabrication.
11.1. Testing Functionality at the Single-Molecule Level
Beyond conventional devices, molecular electronics has long been addressing the potential functionality of single- or few-molecule based device structures.388 The field of on-surface synthesis is in excellent synergy with molecular electronics, allowing on one hand the creation of new, previously inaccessible molecular structures, while on the other hand it may help overcome one of molecular electronics’ main problems: the ill-defined and/or unknown structure of the contacts between electrodes and functional molecules. In addition, with the most common technique used within the field of on-surface synthesis being scanning probe microscopies, important insight into the molecule’s functionality at the single-molecule level can be readily obtained by using, e.g., the scanning probe and a conducting substrate as the leads of a two-terminal device.
The simplest experiments of this kind have addressed the evolution of the current between conductive tip and substrate as a function of their distance while lifting a molecular wire contacted on one side by the tip and with the other side adsorbed on the substrate (Figure 32a). The general procedure in such experiments is, after the synthesis of the material to be studied, its approach with a scanning probe while measuring the tunneling current. At some point, a direct contact between probe and sample is obtained (Figure 32b). Thereafter, the tip is continuously retracted, and if the tip-to-molecule bonding is strong enough, the molecular wire can be lifted along. The evolution of the current vs distance holds key information on the charge transport mechanism. It should be noted that the contact between the molecular wire and the STM tip is of critical importance for the charge transport. Because the lifting procedure implies an associated strain, the contact structure may vary during the tip retractions (whether due to a molecule or tip atom rearrangement), typically causing notable jumps in the current signal. Nevertheless, the overall trend is generally as follows. During the approach, the current corresponds to the standard current measured in scanning tunneling microscopy, and the resulting slope when plotting the current on a logarithmic scale (generally termed the β constant) relates to the tunneling barrier, that is, to an effective work function of tip and sample. Instead, during retraction the tunneling occurs through the molecular wire, yielding a new β slope that depends on the charge carrier effective mass, on the molecular bandgap, and on the energy level alignment, becoming virtually independent of distance (thus with a strongly reduced β) if tunneling resonantly through a molecular orbital that is delocalized along the wire.389
Figure 32.

(a) Artistic view of a 7-aGNR being lifted with a scanning probe. (b) Conductance as a function of tip–sample distance during a lifting experiment performed on 7-aGNRs at U = 0.1 V, displaying a steeper approach curve and a reduced slope during retraction. (c) Light emission spectrum from a 7-aGNR in a suspended geometry and U = 1.8 V. (d) dI/dV conductance spectra of chiral (3,1)-GNRs previously manipulated onto NaCl as a function of the tip–sample distance during a lifting experiment. (e) Tunneling decay constant β of 7-aGNRs obtained from lifting experiments as a function of bias. (f) Schematic view of a lifting experiment with porphyrin chains and STM images of the chain before and after various pulling events. (g) STM image of a magnetic Fe-porphyrin coupled to four low-bandgap, chiral (3,1)-GNRs. (a–c) Adapted with permission from ref (395). Copyright 2012 American Chemical Society. (d) Adapted with permission from ref (394). Copyright 2012 American Chemical Society. (e) Adapted with permission from ref (393). Copyright 2012 Springer Nature. (f) Adapted with permission from ref (391). Copyright 2016 American Chemical Society. (g) Adapted from ref (398). Creative Commons Attribution 4.0 International License.
The pioneering experiment performing such measurements on polymers obtained by on-surface synthesis was the lifting of polyfluorene wires from a Au(111) surface. The wires could be lifted up more than 20 nm, and the conductance as a function of tip–sample distance showed not only the expected exponential decay, but also characteristic oscillations as one molecular unit after another was detached from the surface.78 Thereafter, similar experiments have been applied to different polymers like those formed by alternating donor–acceptor units,390 porphyrin chains (Figure 32f),391 and polythiophene392 or graphene nanoribbons (Figure 32a) without393−396 and also with structural defects along its backbone.396 In many of those cases, however, the measurement’s complexity was increased to obtain additional information. For example, bias-dependent experiments have been performed to quantify the tunneling decay constant β not only at low bias, but also as a function of the energy level alignment (Figure 32e).391,393,394 In doing so, the quasiballistic transport could indeed be measured when tunneling resonantly through delocalized orbitals.391,393,394 Also, the differential conductance spectra as a function of the tip–sample distance (Figure 32d)391,394 provide valuable information on the voltage drop distribution across the tip–molecule–substrate junctions. Alternatively, additional channels were acquired and studied in the lifting geometry, as is the light emission from such a junction (Figure 32a,c),392 or the friction at the molecule–substrate interface.397
Very interesting experiments have also been performed on increasingly complex molecular structures obtained by mixing different precursors. The functionality of the so-created heterojunctions can again be tested with the scanning probe microscope. An example thereof is displayed in Figure 32g, which shows a magnetic porphyrin coupled to four low-bandgap graphene nanoribbons.398 The magnetic functionality of the porphyrin is maintained in spite of its coupling to the GNRs, while the ribbons, acting as the leads of a potential molecular device structure, also show unchanged electronic properties.398 All in all, this experiment thus shows that molecular device structures with atomically precise and predetermined contact geometries can be created by on-surface synthesis, further keeping the functionality of their different components uncompromised. Other interesting experiments probing the functionality of heterostructures are for example those that mixed porphyrins as light-emitters with polythiophene chains as linkers, measuring the electroluminescence of single molecules as current was passed through the tip–linker–chromophore–linker–substrate circuit.154,399 In terms of conductance, interesting heterostructures through which the current transport was studied are GNR heterojunctions mixing semiconducting 7-aGNRs and metallic 5-aGNRs. The multiexponential current dependence on tip–sample distance was measured through different kinds of heterojunctions (single or double heterojunctions, as well as different GNR segment lengths) as the ribbons were lifted, obtaining important information on the response of model heterostructures that may be used as tunnel barriers or diodes.367 A conceptually very beautiful experiment was also the conductance measurement through polyfluorene chains interlinked with three-terminal single-molecule nodes. After contacting one of the chains with the scanning probe and lifting the heterostructure, a circuit is formed in which, from the node to the substrate, two chains sustain the current flow in parallel. Furthermore, the nature of the linker was varied so as to provide conduction paths displaying conjugation and/or cross-conjugation, and the associated effect on the current flow was characterized.400
Note here that not only can the intrinsic functionality of molecular architectures be probed, but also that of the interfaces between the molecular structures and their substrates, which may be equally interesting for potential applications. By way of example, a strong hybridization between molecular π states and the metal bands of ferromagnetic substrates can be exploited to tailor the spin-injection into organic materials and to control the magnetism of the interface.401 In this regard, the polymerization of dibromoterphenyl (DBTP) into polyphenylene chains has been reported on a ferromagnetic GdAu2 thin film on Au(111), whereby it was shown that the polymer growth preserves the ferromagnetic ordering of the substrate.401 Another example is the formation of a covalent honeycomb network by Ullmann coupling from TBPB precursors on a Au-rich bimetallic surface on Pd(111).226 Subsequent thermal processing induced a Pd-rich surface covered by the nanoporous network, and the accessibility of Pd active sites was demonstrated through CO adsorption.
Altogether, on-surface synthesis provides the chance to create complex, functional molecular structures and interfaces with atomic precision, while the associated experimental tools typically available for their characterization allow measuring their properties with single-molecule resolution. Not being merely limited to the flow of current, the overall functionality of such promising molecular structures can thus be tested in great detail and at the single-molecule level.
11.2. Nanopatterning and Host–Guest Accommodation
A few strategies have been proposed using on-surface synthesis to create surface patterns or templates in the mesoscopic range. It was proposed to take advantage of the high resistance against annealing of diboronic acid (BDBA) based polymers to use them as a mask to selectively deposit host material in a nanolithography-like process.261 A nanostructured low-binding-energy NaCl ultrathin film was used as a sacrificial layer in a series of hierarchical self-assembly steps (see Figure 33a). In the submonolayer regime, NaCl formed square-shaped islands on the Ag(100) surface. After deposition of BDBA on such a patterned surface, the molecular precursors did not bind on top of the NaCl islands but rather on the free parts of the metal substrate. There the polymerization reaction could be selectively activated. After annealing, the NaCl islands evaporated while the 2D polymer remained unaffected. This resulted in an effective nanostructuring of the surface and the formation of large square areas of uncovered Ag embedded inside the polymer layer. In this way a negative mask was created that could be used to selectively accommodate Fe that formed nanostructured islands inserted into the polymer mask. Also, with increasing Fe coverage, Fe islands could grow and coalesce under the polymer film and were thus able to lift up the polymer layer.
Figure 33.

(a) Schematics of the lithography process in UHV. On a Ag(100) surface, a mask is formed by depositing square-shaped NaCl islands, and the 2D-polymer from BDBA precursors is grown around. Bottom: STM images of the BDBA polymer surrounding NaCl islands (left) and of the square-shape patterned BDBA polymer after annealing and NaCl evaporation (right). (b) The template formed by the BDBA polymer network on HOPG induces structural modifications of the growth mode of C60 multilayers; a quasi-close-packing structure is obtained in contrast to the standard close packing structure obtained without the polymer template. (a) Adapted with permission from ref (261). Copyright 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (b) Adapted with permission from ref (402). Copyright 2017 American Chemical Society.
The Cu(110)-(2 × 1)O nanotemplate described in section 6.4 (Figure 20f) can be regarded as an approach for 1D patterning.260 Interesting effects were reported thanks to the effective control of the oxide interstripe distance.
Nanopatterning was also proposed by manipulation with the STM tip.291,403 The pristine surface of grafted HOPG was recovered at the liquid/solid interface by local scratching of an area of controlled shaped and then used to selectively accommodate alternative materials present in the solution (nanografting, see Figure 26b, section 9.1). Diacetylene (PCDA) polymerization was initiated inside nanocorrals of defined shape on HOPG. Depending on the relative orientation of the nanocorral and the HOPG surface, high control on the polymer size and direction could be achieved.291 Formation of organometallic oligomers of tetrathienoanthracene (2TTA) on a defined patterned area was achieved by applying high bias voltage (>3 V) with the STM tip.289 In catalytic scanning probe lithography (cSPL) (see Figure 26b), a chemical coupling reaction was initiated locally by mechanical application of a catalyst-modified tip. Various chemical compounds could be grafted selectively, with a spatial resolution on the order of a few tens of nanometers.294,297−306
Nanoporous covalent networks could be used as host–guest systems to accommodate molecular species of adequate size. In particular, fullerene (C60) molecules have appropriate size and are fitted exactly inside the pores of the mesh formed by the benzene diboronic acid (BDBA) polymer (see Figure 33b).402−404 The predefined template of the polymer network could even be transferred to multilayer growth of fullerenes.402,403 The distribution in pore size in the nanoporous network formed from tris-bromophenylbenzene (TBPB) allowed selective accommodation of one, two, or three C60 guests inside the pentagonal, hexagonal, or heptagonal pores, respectively.142 A similar polymer template with a larger pore size was used to accommodate Cu-phthalocyanine (CuPc) guests320 in the azobenzene-containing network (ABBA precursor). Remarkably, in this case the photoisomerization of the azobenzene pore walls and the resulting degradation of the framework could be used as an effective and controllable release mechanism of the guest molecules.
11.3. Growth on Insulating Substrates and Decoupling Strategies
The realization of covalent systems on technologically relevant substrates represents an important challenge for the development of future applications. Indeed, the adsorption on a metal surface results in severe quenching of the optoelectronic properties, whereas the latter can be preserved on an inert insulating substrate. For example, the frequency of the photopolymerization occurrence of diacetylene compounds was about 2 orders of magnitude higher on the h-BN(0001) surface243 than on HOPG. This could be explained by the enhanced lifetime of the intermediate excited state, as relaxation was suppressed due to the large bandgap of h-BN. However, the catalytic activity of a metal surface is often required to initiate the coupling reaction, and strong adsorption energy is required to avoid desorption during thermal activation. Nevertheless, some strategies combining these contradictory specifications have been proposed, and a few promising results have been obtained toward gaining an effective decoupling from the substrate. Several examples have been reported concerning covalent coupling on calcite substrate,47,150,249,322,405 for which the strong chemisorption through carboxylic acid groups to the substrate represents a critical step. First attempts on semiconductor substrates (TiO2(110)87,102,244,406 or passivated Ge(001)407,408) are limited to the formation of oligomeric fragments or 1D chains of limited length. The formation of hexacene by surface-assisted reduction on a H-passivated Si(001) surface was reported.24 An interesting alternative is the use of an ultrathin decoupling layer on top of the metal substrate to electronically and spatially separate the organic film from the metal. A limited number of attempts are reported, making use of graphene or hexagonal boron-nitride (h-BN) layers, but also of NaCl or MoSx islands, or even through halogen or silicon intercalation or multilayer formation. A very promising result has been recently reported on a bulk KCl(001) surface.92 Oriented 1D chains were observed using a dimaleimide precursor either spontaneously or upon light irradiation (see Figure 34). The radical polymerization was favored by adequate epitaxial conditions.
The precursor (I6-CHP) used for the growth of the so-called nanoporous graphene43,409 has been studied on h-BN on Rh(111).410 It was shown that the six peripheral iodine atoms were inequivalent in the adsorbed configuration and that the different C–I bonds would therefore dissociate at different annealing temperatures, leaving the possibility to create partially halogenated dimeric species. At the highest annealing temperature (850 K), fully dehalogenated oligomeric structures were found; however, they were of much more limited extension compared to the polymeric networks grown from the same parent molecule on metal surfaces. Tris(bromophenyl)-benzene (TBB) was comparatively studied on both h-BN and on graphene on Ni(111).411 Similarly, only oligomeric species of limited extension were obtained after annealing in both cases. DFT calculations suggested a large (positive) influence of the metal substrate underneath in the reaction process. The activation temperature of the reaction could be substantially reduced on h-BN by using codeposition of Cu or Pd adatoms.179
A substoichiometric molybdenum sulfide phase (s-MoSx) was grown in island form on Cu(111) and used as a catalytically active substrate.412 There a good selectivity toward precursor adsorption followed by the formation of cyclic tetramers through Ullmann coupling was found as compared to bare Cu(111).
Partial adsorption of linear terfluorene oligomers on NaCl islands on Au(111) has been reported.413 Here the Ullmann coupling between DBTF precursors was first realized on the bare metal surface, and NaCl was then deposited at high coverage, leading to NaCl island growth under the polymer wires and lifting up of the latter. Polymeric connections between bare metal and insulating layers were thus realized. Polyyne formation was achieved on a NaCl thin film on Cu(111) by tip manipulation.288
Postsynthetic decoupling was achieved in some cases through in situ formation of an iodine414 or silicon374,415 intercalation layer. The covalent networks formed on Ag(111)414 or Au(111)374,414,415 were exposed in UHV to iodine pressure or Si evaporation that grew in a monolayer fashion below the organic layer. This process led to an effective decoupling of the polymer, both electronically and structurally. An octanethiol SAM was intercalated postsynthetically between graphene nanoribbons and the Au(788) substrate, thus allowing effective etching-free transfer on a silicon substrate.416 For the system tris-iodophenyl-benzene (TIPB) on Au(111), the covalent networks could be formed as a second layer on top of a supramolecular domain, thus potentially providing an effective decoupling.217 Fabrication of a true bilayer covalent network in an eclipsed stacking configuration was demonstrated by imine reaction thanks to strong π–π interactions between tetrathiafulvalene moieties.417
11.4. Transfer Methods and Devices
For many or even most of the devices in which surface-synthesized materials may be used, the functional molecules need to be electronically decoupled from the supporting substrate. Because most on-surface synthesis reactions studied to date take place on metal substrates, the potential implementation of the reaction products into devices would normally require their transfer onto a different substrate from that on which it has been synthesized. While this applies generally, to date only GNRs have been implemented into real devices. Thus, although conceptually similar procedures can be foreseen to be applied to other types of materials in the future, in the following we describe some of the methods that have readily proved successful in the transfer of on-surface-synthesized GNRs onto a variety of different surfaces, as well as some of the subsequent processing associated with the ultimate device creation.
Readily from its very beginning, in the seminal work reporting the first atomically precise synthesis of GNRs, the importance of their transfer onto insulating surfaces was not only acknowledged but also addressed experimentally.151 Ribbons synthesized on Au films on mica were transferred onto SiO2/Si by simple repeated mechanical pressing of the two samples against each other. However, although the process was proved to be successful with Raman spectroscopy,151 its application to the fabrication of devices failed.418 Instead, alternative methods were developed. The first device made with on-surface-synthesized GNRs again made use of ribbons grown on Au films on mica.418 The transfer (Figure 35a) consisted of spinning a poly(methyl methacrylate) (PMMA) film onto the GNRs, which is subsequently baked to form a PMMA/GNR/Au/mica stack. The mica is then delaminated from the Au by means of HF. After rinsing with water, the PMMA/GNR/Au film is transferred into Au etchant. Thereafter, the remaining PMMA/GNR film is rinsed again and drawn onto the target substrate, followed by a bake to remove residual water. The sample is finally stripped of PMMA with acetone, leaving exposed GNRs on an insulating substrate.418 Similar transfer procedures have been adopted by other groups thereafter,419 or also varied to, e.g., avoid the initial PMMA coating of the ribbons. This is done by first detaching the mica substrate from the Au film with HCl. The floating gold film is then picked up with the target substrate, with the GNRs facing the dielectric surface. Subsequent annealing removes residual water and improves the adhesion of the GNR/Au film to the substrate surface. In a last step, gold etching yields exposed GNRs on top of the substrate of choice.353,420,421
Figure 35.

Schematic representation of a GNR transfer process sacrificing (a) or not (b) the gold substrate. (c) Raman spectra of 9-aGNRs as grown on Au and after the transfer and device fabrication. (d) Schematic view (top) and scanning electron micrograph (bottom) of a GNR based device. (e) Transport characteristics of a 9-aGNR based field-effect transistor as sketched in the inset. (a) Reprinted with permission from ref (418). Copyright 2013 AIP Publishing. (b) Reprinted with permission from ref (416). Copyright 2018 AIP Publishing. (c–e) Adapted from ref (420). Creative Commons Attribution 4.0 International License.
The transfer methods described above, although successfully used in the fabrication of devices, have the drawback of sacrificing the Au substrate used for the GNR synthesis. Sacrificing the precious metal substrates implies a substantial added cost, and for that same reason limits their use to thin films. Thus, the development of alternative methods not sacrificing the underlying substrate not only lowers the costs, but also allows the use of a greater variety of substrates, including unlimited reuse of single crystals. As opposed to polycrystalline gold films, single crystals can be used to synthesize uniaxially aligned GNRs (see section 10.3). Preserving such GNR alignment and controlling it with respect to the device structure is thus desirable, since it allows optimizing the density of GNRs bridging across the contacts. An example of such a nonsacrificial transfer process is depicted in Figure 35b,416 in which the GNRs grown on Au are covered with a hydrogen silsesquioxane (HSQ) thin film. The sample is then immersed in a solution of alkanethiols, which intercalate between GNRs and Au forming a self-assembled monolayer. This allows the delamination of the HSQ/GNR from the Au, and its subsequent transfer onto a substrate of choice, only followed by the final removal of the HSQ film by tetramethylammonium (Figure 35b).416 An alternative nonsacrificial technique that has similarly allowed the transfer of uniaxially aligned GNRs grown on appropriate single crystals is the bubbling method.381,422 Inspired in its previous application to two-dimensional graphene,423 the method consists of spinning a poly(methyl methacrylate) (PMMA) film on the GNRs and placing the sample in an electrochemical cell with a NaOH aqueous solution. Hydrogen gas bubbles emerge at the negatively biased cathode, in particular at the GNR/Au interface, due to the reduction of water. Those gas bubbles detach the PMMA/GNR film from the Au single crystal, after which the film is cleaned in distilled water and fished out with the substrate of choice.381
Several other methods have also been applied to bring GNRs onto arbitrary surfaces. However, they start out from GNRs readily in solution or powder. Thus, for standard materials described within this review, synthesized on substrates and supported by substrates, an additional step would be required to get the materials into a substrate-free solution or powder before applying the transfer procedure. Some examples are listed below.
One such transfer method is that based on the exceptional solubility of graphitic materials in chlorosulfonic acid (CSA),333,424 which to date has only been applied to GNRs previously synthesized in solution. After preparation of a GNR solution in CSA, its transfer onto another solvent leads to reaction or mixing of CSA with the solvent (depending on its nature, whether deionized water or other organic solvents), while the insoluble and hydrophobic GNRs remain at the liquid–air interface. In doing that, they self-assemble into uniform thin films whose thickness can be varied with the solution concentration. Single GNR thick layers can be easily obtained, within which the GNRs adopt an edge-on geometry to optimize their π–π stacking. A substrate of choice can be subsequently used to fish out the film.333,424
Just as it also works as an alternative deposition method for smaller molecules like GNR reactants,425 direct contact transfer can also be applied for GNR deposition onto arbitrary substrates once they are available as a powder. This has been proven with solution-synthesized chevron GNRs,337 whose powder was subsequently applied to a fiberglass applicator. Annealing the applicator to remove contaminants and then pressing it onto a silicon surface under ultra-high-vacuum conditions led to GNR exfoliation onto the surface,337 providing cleaner samples than if deposited, e.g., by drop-casting of a GNR dispersion in conventional solvents.338
Unless a bottom-contact geometry is used,419 the ultimate device fabrication additionally requires the nanofabrication of contacts on top of the GNRs, as schematically shown and further displayed with a scanning electron microscope micrograph in Figure 35d.420 This process may involve different lithographic methods that also need to be compatible with the GNR integrity. The technique most commonly used for GNR characterization after transfer onto insulating substrates is Raman spectroscopy. It provides invaluable information on the detailed GNR structure and allows the quantification of potential GNR degradation during the transfer procedures. An example thereof is given in Figure 35c where the Raman spectra of 9-aGNRs are shown as grown on Au(111) films and after transfer and device fabrication.420 The fact that the spectra look identical, including the presence of the radial breathing-like mode (RBLM) peak at 311.5 cm–1, is evidence that the GNR width and edge structure remains intact after the whole processing, yielding device response curves like that displayed in Figure 35e.420 To end with, we would like to remind the reader again that GNRs are interesting for a number of different applications in which also their optical properties play key roles. Their optical response is thus an important parameter to be studied and optimized. With this in mind, it has been shown that the coupling of GNRs to plasmonic nanontenna arrays can enhance the photoluminescence and Raman scattering intensity of the ribbons by more than an order of magnitude if averaged over large areas, and by 3 orders of magnitude in the hot spots of plasmonic antennas.426
11.5. Conspectus
This section illustrates the advancement achieved in the on-surface synthesis field toward the elaboration and the advanced characterization of materials with a direct interest for applications. Various functionalities of on-surface-synthesized compounds, such as their conductivity and their optoelectronic and mechanical properties have been tested at the single-molecule level thanks to the powerful manipulation abilities of scanning probe microscopy. Elaborate strategies have been developed to create and take advantage of patterned structures made of covalent networks. Important efforts have been furnished toward the formation of covalent structures directly on insulating substrates or at least with an efficient decoupling from the metallic surface. Last but not least, real electronic devices have been produced and characterized, although until now they have been limited to the use of graphene nanoribbons only.
12. Conclusions
A wide set of tools and strategies are nowadays available to provide efficient control of on-surface reactions. Various approaches have been successfully demonstrated, such as the tuning of the symmetry, the size and the reactivity of the precursors, the implementation of sequential reactions, the supramolecular or substrate templating, the use of adatom catalyst, the control on the reaction and growth parameters, the use of environmental conditions, and the light irradiation or the direct tip manipulation. In all examples mentioned in this review, scanning probe microscopy appears as a particularly efficient tool for the characterization of the reaction product at the single-molecule level.
On-surface synthesis is certainly positioned as a basic science research field. In this regard, the progress made in terms of understanding of surface chemistry processes at the single-molecule level is already very impressive. Nonetheless, thanks to the high level of control achieved, there still exists a marked tendency toward the elaboration and the investigation of more and more complex systems exhibiting a large diversity of functions. Examples thereof would be the control of a molecular rotor,427 the reversible tuning of the doping level in graphene nanoribbons,428 the rational control of the electronic properties,429 the control of the impact parameter in reaction collisions,258 the use of nonplanar precursors,214 or the challenge of chiral control430−432 or halogen removal.205,206 A critical issue that definitely remains is the control of the extension of 2D networks. Despite various efforts to grow covalent patterns at the micrometer scale or more, the domain sizes are usually still limited to the range of a few tens of nanometers. Also, the importance of entropic effects is yet poorly studied and certainly underestimated.433 Importantly, the time scale has not yet been explored, but some aspects of the dynamics of the reactions may in the future be accessible through appropriate experimental developments.434
The expected application fields for on-surface synthesis are broad and numerous. Examples thereof would be molecular (opto-)electronics, nanoplasmonics, magnetism, green chemistry, sensors, or membranes for gas separation or water purification. Future developments of on-surface synthesis will go through the realization of functional devices. In this regard, significant progress has been made and a real effort in this direction is provided by the community. The important challenges consist of realizing an up-scaling of the fabrication processes. Remarkably, the CVD growth of graphene nanoribbons has already been developed and implemented in a few laboratories.375,416,435−438 The issue of transferring the molecular systems from the metal surface to different substrates has been also thoroughly addressed, as detailed in section 11.4. In addition, the recent demonstration of the growth of polymer chains directly on a bulk insulator is certainly highly attractive and promising.92 Finally, a critical issue for molecular electronics consists of achieving electrical contacts and bridging the nano- to mesoscale gap. There, the on-surface synthesis approach is revealed to be particularly efficient because it can control the organic–inorganic contact at the atomic scale, as demonstrated by the lift-up experiments detailed in section 11.1.
For all these reasons, on-surface synthesis appears now as a mature field of research that possesses the rare ability to bring together basic and applied research interests. It is expected to attract an even wider community and to provide more astonishing results in the near future. An important generic limitation to on-surface synthesis is that it is restricted to the use of single crystal surfaces. Also, the formation of the products is constrained to quantities corresponding to a maximum of a single monolayer. A fundamental breakthrough in the field will be made by achieving an up-scaling of the processes, opening the way to mass production. For this, it will be necessary to find a way to regenerate the surface to allow for the realization of cyclic processes of the reactions, as is the case in traditional catalysis. Inspiration could be searched for in the notorious efforts readily put into graphene-related research, for which recyclable synthesis and large scale transfer processes have been successfully developed.439−442
Acknowledgments
The OMNT team and in particular Nicolas Brefuel, Xavier Bouju, Frédéric Chérioux, Fabien Silly, and André-Jean Attias are gratefully acknowledged for initiating and motivating this work. Financial support from Centre National de la Recherche Scientifique (CNRS), Aix-Marseille University, and Agence Nationale de la Recherche (ANR grant ANR-17-CE08-0010 “DUALITY”, ANR grant ANR-16-JTIC-0002 “PHOTONET”), as well as from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement 635919) and from the Spanish Ministry of Economy, Industry and Competitiveness (MINECO, grant MAT2016-78293-C6), is acknowledged.
Glossary
Glossary
- 0D/1D/2D/3D
zero-/one-/two-/three-dimensional
- 1,4-PDI
1,4-phenylene diisocyanate
- 2,3-DHTA
2,3-dihydroxyterephthalaldehyde
- 2,5-DHTA
2,5-dihydroxyterephthalaldehyde
- 2Br-DEBPB
2,5-diethynyl-1,4-bis(4-bromophenylethynyl)benzene
- 2TTA
trithieno[2′,3′:5,6:3′,2’:3,4:3′,2’:7,8]-anthra[1,2-b]thiophene
- 3TTA
trithieno-[2′,3′:3,4:2′,3′:7,8:3′,2’:5,6]anthra-[2,1-b]thiophene
- 4ATTF
tetrathiafulvalene tetraaldehyde
- 4Ph
para-quaterphenyl
- AABA
4-(2-(9-(2-(4-aminophenyl)ethynyl)anthracen-10-yl)ethynyl)benzenamine
- ABBA
4,4′-phenylazobenzoyl diboronic acid
- AFM
atomic force microscopy
- ASB
4,4′-diaminostilbene dihydrochloride
- BB
bromobenzene
- BBBA
p-bromobenzene boronic acid
- bBEBP
4,4′-di(bromoethynyl)-1,1′-biphenyl
- BBP
4-bromobiphenyl
- BCCTP
1,3-bis(p-chlorophenyl)-5-(p-bromophenyl)benzene
- BDBA
1,4-benzenediboronic acid
- BDFPTP
4,4′-bis(2,6-difluoropyridin-4-yl)-1,1’:4′,1″-terphenyl
- BEBP
4-(bromoethynyl)-1,1′-biphenyl
- BIB
1,3-bis(p-bromophenyl)-5-(p-iodophenyl)benzene
- BPBE
1,1′-biphenyl,4-bromo-4′-ethynyl
- BPCPPCA
2-(4-bromophenyl)-6-(4-chlorophenyl)pyridine-4-carboxylic acid
- BPDA
4,4′-biphenyldiboronic acid
- BrPh2Br
4,4″-dibromo-p-biphenyl
- BrTPP
5-(4-bromophenyl)-10,15,20-tri(phenyl)porphyrin
- Br2-DNPH2
5,15-(dinaphthalene)-10,20-(dibromo)-porphyrin
- Br2-DPPH2
5,15-(diphenyl)-10,20-(dibromo)-porphyrin
- Br2I2Py
1,8-dibromo-3,6-diiodopyrene
- Br2-MPPH2
5-(phenyl)-10,20-(dibromo)-porphyrin
- Br4F6BP
3,3′,5,5′-tetrabromo-2,2′,4,4′,6,6′-hexafluorobiphenyl
- Br4-PTCDA
1,6,7,12-tetrabromo-3,4,9,10-perylene-tetracarboxylic-dianhydride
- Br4Py
1,3,6,8-tetrabromopyrene
- Br4TPP
tetra(4-bromophenyl)porphyrin
- BTA
benzene-1,3,5-tricarbaldehyde
- BTCMB
1,4-bis(trichloromethyl)benzene
- BuOMT
3-butoxy-4-methylthiophene
- C8MT
3-octyl-4-methylthiophene
- C8OMT
3-octyloxy-4-methylthiophene
- CHP
hexaiodo-cyclohexa-m-phenylene
- CN-DETP
4,4″-diethynyl-[1,1’:4′,1″-terphenyl]-3,3″-dicarbonitrile
- COR
coronene
- CTPA
halogenated carbonyl-bridged triphenylamine
- DAAQ
2,6-diaminoanthraquinone
- DAN
1,4-diaminonaphthalene
- DAPh
diazuleno[1,2-c:2′,1′-g]phenanthrene
- DATP
4,4″-diamino-p-terphenyl
- dBA
2,3-dibromoanthracene
- dBB
1,4-dibromobenzene
- DBBA
10,10′-dibromo-9,9′-bianthryl
- DBDBT
2,8-dibromodibenzothiophene
- DBDMN
1,5-dibromo-2,6-dimethylnaphthalene
- DBH
benzo[2,1-g:3,4-g’]dichrysene (dibenzo[i,o]heptahelicene
- DBMBP
4-(dibromomethyl)-1,1′-biphenyl
- DBMTP
4′,6′-dibromo-meta-terphenyl
- DBOTP
3′,5′-dibromo-ortho-terphenyl
- DBPBA
3,5-dibromophenylboronic acid
- DBT
5,11-dibromo-tetracene
- DBTF
α,ω-dibromoterfluorene
- DBTP
4,4″-dibromo-p-terphenyl
- DCTP
4,4″-dichloro-1,1’:4′,1″-terphenyl
- DEB
1,4-di(eicosyl)benzene
- DEBPB
2,5-diethynyl-1,4-bis(phenylethynyl)benzene
- DETP
4,4″-diethynyl-1,1’:4′,1″-terphenyl
- DFT
density functional theory
- DHI
3,3′-dihydroxybenzidine
- DHQP
4,4″-dihydroxy-p-quaterphenyl
- DIB
1,3-diiodobenzene
- DIBA
10,10′-diiodo-9,9′-bianthryl
- DIEDOT
2,5-diiodo-3,4-ethylenedioxythiophene
- DITF
diiodoterfluorene
- DMH
18,19-dimethylidene-hexatriacontane
- DMTP
4,4″-dibromo-meta-terphenyl
- DN
(R)-(−)-6,6′-dibromo-1,1′-bi-2-naphthol
- DP-DBBA
10,10′-dibromo-2,2′-diphenyl-9,9′-bianthracene
- DPIA
5-(dodecyloxy)-N1,N3-di(prop-2-ynyl)isophthalamide
- EBB
4-ethylbromobenzene
- ENA
6-ethynyl-2-naphthoic acid
- Ext-TEB
1,3,5-tris(4-ethynylphenyl)benzene
- GNR
graphene nanoribbon
- HHTP
2,3,6,7,10,11-hexahydroxytriphenylene
- Hn3O
triepoxyhexacene
- I6-CHP
5,5′,5″,5′″,5″″,5′″″-hexaiodocyclohexa-m-phenylene
- INDO4
s-indacene-1,3,5,7(2H,6H)-tetrone
- IPh2I
4,4″-diiodo-p-biphenyl
- NDCA
2,6-naphthalenedicarboxylic acid
- MA
1,3,5-triazine-2,4,6-triamine (melamine)
- mTBPB
1,3,5-tris(3-bromophenyl)benzene
- OETAP
2,3,7,8,12,13,17,18-octaethyl-5,10,15,20-tetraazaporphyrin
- OTPA
2,5-dioctyloxy-terephthalaldehyde
- p-6P
para-sexiphenyl
- pap
pyrazino[2,3-f][4,7]phenanthroline
- Pc
phthalocyanine
- PCDA
10,12-pentacosadiynoic acid
- PDA
poly(diacetylene)
- PEBA
4-ethynylbenzoic
- PEPmBr
5-(2-(4-bromophenyl)ethynyl)pyrimidine
- PPDA
p-phenylenediamine
- PPP
poly(paraphenylene)
- PTCDI
3,4,9,10-perylene tetracarboxylic acid diimide
- QR
quaterrylene
- STM
scanning tunneling microscopy
- TAPB
1,3,5-tris(4-aminophenyl)benzene
- TBB
1,3,5-tris(4-bromophenyl)benzene
- TBD
3,5-bis-tert-butylbenzenediazonium
- tBEP
1,3,5-tris(bromoethynyl)benzene
- TBPB
1,3,5-tris(4-bromophenyl)benzene
- TBr4PP
5,10,15,20-tetrakis(4-bromophenyl)porphyrin
- TBTTA
tetrabromo-tetrathienoanthracene
- TCMB
trichloromethylbenzene
- tDBA
dehydrobenzo[12]annulene
- TEB
1,3,5-triethynyl-benzene
- TFPB
1,3,5-tris(4-formylphenyl)benzene
- THPB
1,3,5-tris(4-hydroxyphenyl)benzene
- TIB
1,3,5-triiodobenzene
- TIPB
1,3,5-tris(4-iodophenyl)benzene
- TMA
trimesic acid
- TPA
terephthaldicarboxaldehyde
- TPDT
3-(1,5-bis(trimethylsilyl)penta-1,4-diyn-3-ylidene)penta-1,4-diyne-1,5-diyl)bis(trimethylsilane
- trans-Br2I2TPP
5,15-bis(4′-bromophenyl)-10,20-bis(4′-iodophenyl)porphyrin
- trans-Br2TPP
5,15-bis(4-bromophenyl)-10,20-bisphenylporphyrin
- trans-TPP(NH2)2
5,15-bis(4-aminophenyl)-10,20-diphenylporphyrin
- TTEN
2,3,6,7-tetrakis((trimethylsilyl)ethynyl) naphthalene
- UHV
ultra-high-vacuum
- UV
ultraviolet
- XPS
X-ray photoelectron spectroscopy
Biographies
Sylvain Clair is a physicist who graduated from Ecole Centrale Paris (France) and Technische Universität Wien (Austria). He first worked as an engineer at the CERN laboratory in Switzerland and then obtained his Ph.D. in 2004 from EPFL (Switzerland) under the supervision of J. V. Barth. He has been a postdoctoral fellow in the group of M. Kawai at RIKEN in Japan and in the group of F. Rosei at INRS in Canada. Since 2008 he has been working as a CNRS research scientist at IM2NP laboratory in Marseille (France). His research interests include the investigation of supramolecular self-assemblies at surfaces using scanning probe microscopy techniques combined with spectroscopic measurements. He has conducted pioneering work on the formation of boronic acid based covalent networks, and he is now developing on-surface synthesis methods to create original functional surfaces.
Dimas G. de Oteyza performed his Ph.D. studies at the Max-Planck Institute for Metals Research and obtained the degree from the Universidad Autónoma de Madrid in 2007. Thereafter he has worked at the National Institute for Materials Science, Donostia International Physics Center (DIPC), at the Molecular Foundry of the Lawrence Berkeley National Laboratory, at the University of California at Berkeley, and at the Centro de Física de Materiales. He is currently an Ikerbasque Research Professor at DIPC. The research throughout his career has been mainly devoted to the investigation of physicochemical phenomena in organic materials and organic–inorganic interfaces, including thin film growth, self-assembly, interface electronics, and chemical reactions. To date he has received honors like the Fonda Fasella Award, the Friedrich Wilhelm Bessel Award, and an ERC Starting Grant with the objective to study and advance the field of on-surface synthesis.
The authors declare no competing financial interest.
References
- Dong L.; Liu P. N.; Lin N. Surface-Activated Coupling Reactions Confined on a Surface. Acc. Chem. Res. 2015, 48, 2765–2774. 10.1021/acs.accounts.5b00160. [DOI] [PubMed] [Google Scholar]
- Björk J.; Hanke F. Towards Design Rules for Covalent Nanostructures on Metal Surfaces. Chem. - Eur. J. 2014, 20, 928–934. 10.1002/chem.201303559. [DOI] [PubMed] [Google Scholar]
- Sun Q.; Zhang R. Y.; Qiu J.; Liu R.; Xu W. On-Surface Synthesis of Carbon Nanostructures. Adv. Mater. 2018, 30, 1705630. 10.1002/adma.201705630. [DOI] [PubMed] [Google Scholar]
- Janica I.; Patroniak V.; Samori P.; Ciesielski A. Imine-Based Architectures at Surfaces and Interfaces: From Self-Assembly to Dynamic Covalent Chemistry in 2D. Chem. - Asian J. 2018, 13, 465–481. 10.1002/asia.201701629. [DOI] [PubMed] [Google Scholar]
- Shen Q.; Gao H. Y.; Fuchs H. Frontiers of On-Surface Synthesis: From Principles to Applications. Nano Today 2017, 13, 77–96. 10.1016/j.nantod.2017.02.007. [DOI] [Google Scholar]
- Lackinger M. Surface-Assisted Ullmann Coupling. Chem. Commun. 2017, 53, 7872–7885. 10.1039/C7CC03402D. [DOI] [PubMed] [Google Scholar]
- Di Giovannantonio M.; Contini G. Reversibility and Intermediate Steps as Key Tools for the Growth of Extended Ordered Polymers via On-Surface Synthesis. J. Phys.: Condens. Matter 2018, 30, 093001. 10.1088/1361-648X/aaa8cb. [DOI] [PubMed] [Google Scholar]
- Hu J. B.; Liang Z. F.; Shen K. C.; Sun H. L.; Jiang Z.; Song F. Recent Progress in the Fabrication of Low Dimensional Nanostructures via Surface-Assisted Transforming and Coupling. J. Nanomater. 2017, 2017, 4796538. 10.1155/2017/4796538. [DOI] [Google Scholar]
- Talirz L.; Ruffieux P.; Fasel R. On-Surface Synthesis of Atomically Precise Graphene Nanoribbons. Adv. Mater. 2016, 28, 6222–6231. 10.1002/adma.201505738. [DOI] [PubMed] [Google Scholar]
- Goronzy D. P.; Ebrahimi M.; Rosei F.; Arramel; Fang Y.; De Feyter S.; Tait S. L.; Wang C.; Beton P. H.; Wee A. T. S.; et al. Supramolecular Assemblies on Surfaces: Nanopatterning, Functionality, and Reactivity. ACS Nano 2018, 12, 7445–7481. 10.1021/acsnano.8b03513. [DOI] [PubMed] [Google Scholar]
- Barth J. V.; Costantini G.; Kern K. Engineering Atomic and Molecular Nanostructures at Surfaces. Nature 2005, 437, 671. 10.1038/nature04166. [DOI] [PubMed] [Google Scholar]
- Dong R. H.; Zhang T.; Feng X. L. Interface-Assisted Synthesis of 2D Materials: Trend and Challenges. Chem. Rev. 2018, 118, 6189–6235. 10.1021/acs.chemrev.8b00056. [DOI] [PubMed] [Google Scholar]
- Kalashnyk N.; Mouhat K.; Oh J.; Jung J.; Xie Y.; Salomon E.; Angot T.; Dumur F.; Gigmes D.; Clair S. On-Surface Synthesis of Aligned Functional Nanoribbons Monitored by Scanning Tunneling Microscopy and Vibrational Spectroscopy. Nat. Commun. 2017, 8, 14735. 10.1038/ncomms14735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Marchi F.; Galeotti G.; Simenas M.; Tornau E. E.; Pezzella A.; MacLeod J.; Ebrahimi M.; Rosei F. Room-Temperature Surface-Assisted Reactivity of a Melanin Precursor: Silver Metal–Organic Coordination versus Covalent Dimerization on Gold. Nanoscale 2018, 10, 16721–16729. 10.1039/C8NR04002H. [DOI] [PubMed] [Google Scholar]
- Zhong D. Y.; Franke J. H.; Podiyanachari S. K.; Blomker T.; Zhang H. M.; Kehr G.; Erker G.; Fuchs H.; Chi L. F. Linear Alkane Polymerization on a Gold Surface. Science 2011, 334, 213–216. 10.1126/science.1211836. [DOI] [PubMed] [Google Scholar]
- Fan Q. T.; Wang C. C.; Han Y.; Zhu J. F.; Hieringer W.; Kuttner J.; Hilt G.; Gottfried J. M. Surface-Assisted Organic Synthesis of Hyperbenzene Nanotroughs. Angew. Chem., Int. Ed. 2013, 52, 4668–4672. 10.1002/anie.201300610. [DOI] [PubMed] [Google Scholar]
- Chen M.; Shang J.; Wang Y. F.; Wu K.; Kuttner J.; Hilt G.; Hieringer W.; Gottfried J. M. On-Surface Synthesis and Characterization of Honeycombene Oligophenylene Macrocycles. ACS Nano 2017, 11, 134–143. 10.1021/acsnano.6b05709. [DOI] [PubMed] [Google Scholar]
- Sanchez-Sanchez C.; Nicolai A.; Rossel F.; Cai J. M.; Liu J. Z.; Feng X. L.; Mullen K.; Ruffieux P.; Fasel R.; Meunier V. On-Surface Cyclization of ortho-Dihalotetracenes to Four- and Six-Membered Rings. J. Am. Chem. Soc. 2017, 139, 17617–17623. 10.1021/jacs.7b10026. [DOI] [PubMed] [Google Scholar]
- Zuzak R.; Dorel R.; Krawiec M.; Such B.; Kolmer M.; Szymonski M.; Echavarren A. M.; Godlewski S. Nonacene Generated by On-Surface Dehydrogenation. ACS Nano 2017, 11, 9321–9329. 10.1021/acsnano.7b04728. [DOI] [PubMed] [Google Scholar]
- Kruger J.; Garcia F.; Eisenhut F.; Skidin D.; Alonso J. M.; Guitian E.; Perez D.; Cuniberti G.; Moresco F.; Pena D. Decacene: On-Surface Generation. Angew. Chem., Int. Ed. 2017, 56, 11945–11948. 10.1002/anie.201706156. [DOI] [PubMed] [Google Scholar]
- Urgel J. I.; Hayashi H.; Di Giovannantonio M.; Pignedoli C. A.; Mishra S.; Deniz O.; Yamashita M.; Dienel T.; Ruffieux P.; Yamada H.; et al. On-Surface Synthesis of Heptacene Organometallic Complexes. J. Am. Chem. Soc. 2017, 139, 11658–11661. 10.1021/jacs.7b05192. [DOI] [PubMed] [Google Scholar]
- Kruger J.; Eisenhut F.; Alonso J. M.; Lehmann T.; Guitian E.; Perez D.; Skidin D.; Gamaleja F.; Ryndyk D. A.; Joachim C.; et al. Imaging the Electronic Structure of On-Surface Generated Hexacene. Chem. Commun. 2017, 53, 1583–1586. 10.1039/C6CC09327B. [DOI] [PubMed] [Google Scholar]
- Zuzak R.; Dorel R.; Kolmer M.; Szymonski M.; Godlewski S.; Echavarren A. M. Higher Acenes by On-Surface Dehydrogenation: From Heptacene to Undecacene. Angew. Chem., Int. Ed. 2018, 57, 10500–10505. 10.1002/anie.201802040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhut F.; Kruger J.; Skidin D.; Nikipar S.; Alonso J. M.; Guitian E.; Perez D.; Ryndyk D. A.; Pena D.; Moresco F.; et al. Hexacene Generated on Passivated Silicon. Nanoscale 2018, 10, 12582–12587. 10.1039/C8NR03422B. [DOI] [PubMed] [Google Scholar]
- Colazzo L.; Mohammed M. S. G.; Dorel R.; Nita P.; García Fernández C.; Abufager P.; Lorente N.; Echavarren A. M.; de Oteyza D. G. On-Surface Synthesis of Heptacene on Ag(001) from Brominated and Non-Brominated Tetrahydroheptacene Precursors. Chem. Commun. 2018, 54, 10260–10263. 10.1039/C8CC04402C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger J.; Eisenhut F.; Skidin D.; Lehmann T.; Ryndyk D. A.; Cuniberti G.; Garcia F.; Alonso J. M.; Guitian E.; Perez D.; et al. Electronic Resonances and Gap Stabilization of Higher Acenes on a Gold Surface. ACS Nano 2018, 12, 8506–8511. 10.1021/acsnano.8b04046. [DOI] [PubMed] [Google Scholar]
- Li Q.; Gao J. Z.; Li Y. Y.; Fuentes-Cabrera M.; Liu M. X.; Qiu X. H.; Lin H. P.; Chi L. F.; Pan M. H. Self-Assembly Directed One-Step Synthesis of [4] Radialene on Cu(100) Surfaces. Nat. Commun. 2018, 9, 3113. 10.1038/s41467-018-05472-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlicek N.; Schuler B.; Collazos S.; Moll N.; Perez D.; Guitian E.; Meyer G.; Pena D.; Gross L. On-Surface Generation and Imaging of Arynes by Atomic Force Microscopy. Nat. Chem. 2015, 7, 623–628. 10.1038/nchem.2300. [DOI] [PubMed] [Google Scholar]
- Pavlicek N.; Majzik Z.; Collazos S.; Meyer G.; Perez D.; Guitian E.; Pena D.; Gross L. Generation and Characterization of a meta-Aryne on Cu and NaCl Surfaces. ACS Nano 2017, 11, 10768–10773. 10.1021/acsnano.7b06137. [DOI] [PubMed] [Google Scholar]
- Sakamoto J.; van Heijst J.; Lukin O.; Schluter A. D. Two-Dimensional Polymers: Just a Dream of Synthetic Chemists?. Angew. Chem., Int. Ed. 2009, 48, 1030–1069. 10.1002/anie.200801863. [DOI] [PubMed] [Google Scholar]
- Lackinger M. On-Surface Polymerization - a Versatile Synthetic Route to Two-Dimensional Polymers. Polym. Int. 2015, 64, 1073–1078. 10.1002/pi.4943. [DOI] [Google Scholar]
- Klappenberger F. Two-Dimensional Functional Molecular Nanoarchitectures – Complementary Investigations with Scanning Tunneling Microscopy and X-Ray Spectroscopy. Prog. Surf. Sci. 2014, 89, 1–55. 10.1016/j.progsurf.2013.10.002. [DOI] [Google Scholar]
- de Oteyza D. G.; Gorman P.; Chen Y. C.; Wickenburg S.; Riss A.; Mowbray D. J.; Etkin G.; Pedramrazi Z.; Tsai H. Z.; Rubio A.; et al. Direct Imaging of Covalent Bond Structure in Single-Molecule Chemical Reactions. Science 2013, 340, 1434–1437. 10.1126/science.1238187. [DOI] [PubMed] [Google Scholar]
- Xi M.; Bent B. E. Mechanisms of the Ullmann coupling reaction in adsorbed monolayers. J. Am. Chem. Soc. 1993, 115, 7426–7433. 10.1021/ja00069a048. [DOI] [Google Scholar]
- Grim P. C. M.; De Feyter S.; Gesquiere A.; Vanoppen P.; Ruker M.; Valiyaveettil S.; Moessner G.; Mullen K.; De Schryver F. C. Submolecularly Resolved Polymerization of Diacetylene Molecules on the Graphite Surface Observed with Scanning Tunneling Microscopy. Angew. Chem., Int. Ed. Engl. 1997, 36, 2601–2603. 10.1002/anie.199726011. [DOI] [Google Scholar]
- Hla S.-W.; Bartels L.; Meyer G.; Rieder K.-H. Inducing All Steps of a Chemical Reaction with the Scanning Tunneling Microscope Tip: Towards Single Molecule Engineering. Phys. Rev. Lett. 2000, 85, 2777–2780. 10.1103/PhysRevLett.85.2777. [DOI] [PubMed] [Google Scholar]
- Okawa Y.; Aono M. Materials science - Nanoscale control of chain polymerization. Nature 2001, 409, 683–684. 10.1038/35055625. [DOI] [PubMed] [Google Scholar]
- Grill L.; Dyer M.; Lafferentz L.; Persson M.; Peters M. V.; Hecht S. Nano-Architectures by Covalent Assembly of Molecular Building Blocks. Nat. Nanotechnol. 2007, 2, 687–691. 10.1038/nnano.2007.346. [DOI] [PubMed] [Google Scholar]
- Avinash M. B.; Govindaraju T. Architectonics: Design of Molecular Architecture for Functional Applications. Acc. Chem. Res. 2018, 51, 414–426. 10.1021/acs.accounts.7b00434. [DOI] [PubMed] [Google Scholar]
- Liu J.; Chen Q. W.; Wu K. On-Surface Construction of Low-Dimensional Nanostructures with Terminal Alkynes: Linking Strategies and Controlling Methodologies. Chin. Chem. Lett. 2017, 28, 1631–1639. 10.1016/j.cclet.2017.04.022. [DOI] [Google Scholar]
- Zhou X.; Dai J. X.; Wu K. Steering On-Surface Reactions with Self-Assembly Strategy. Phys. Chem. Chem. Phys. 2017, 19, 31531–31539. 10.1039/C7CP06177C. [DOI] [PubMed] [Google Scholar]
- Fan Q. T.; Zhu J. F. Controlling the Topology of Low-Dimensional Organic Nanostructures with Surface Templates. Acta Phys. Chim. Sin. 2017, 33, 1288–1296. [Google Scholar]
- Bieri M.; Nguyen M. T.; Gröning O.; Cai J. M.; Treier M.; Ait-Mansour K.; Ruffieux P.; Pignedoli C. A.; Passerone D.; Kastler M.; et al. Two-Dimensional Polymer Formation on Surfaces: Insight into the Roles of Precursor Mobility and Reactivity. J. Am. Chem. Soc. 2010, 132, 16669–16676. 10.1021/ja107947z. [DOI] [PubMed] [Google Scholar]
- Björk J.; Hanke F.; Stafstrom S. Mechanisms of Halogen-Based Covalent Self-Assembly on Metal Surfaces. J. Am. Chem. Soc. 2013, 135, 5768–5775. 10.1021/ja400304b. [DOI] [PubMed] [Google Scholar]
- Björk J.; Zhang Y. Q.; Klappenberger F.; Barth J. V.; Stafstrom S. Unraveling the Mechanism of the Covalent Coupling Between Terminal Alkynes on a Noble Metal. J. Phys. Chem. C 2014, 118, 3181–3187. 10.1021/jp5002918. [DOI] [Google Scholar]
- Björk J. Reaction Mechanisms for On-Surface Synthesis of Covalent Nanostructures. J. Phys.: Condens. Matter 2016, 28, 083002. 10.1088/0953-8984/28/8/083002. [DOI] [PubMed] [Google Scholar]
- Guo C. Y.; Wang Y.; Kittelmann M.; Kantorovitch L.; Kuhnle A.; Floris A. Mechanisms of Covalent Dimerization on a Bulk Insulating Surface. J. Phys. Chem. C 2017, 121, 10053–10062. 10.1021/acs.jpcc.7b02687. [DOI] [Google Scholar]
- Nagoya A.; Tetsuka H.; Ohba N. Mechanisms of Covalent Coupling Reaction of Dibromofluoranthene on Au(111). J. Phys. Chem. C 2018, 122, 17756–17763. 10.1021/acs.jpcc.8b03730. [DOI] [Google Scholar]
- Björk J.Kinetic and Thermodynamic Considerations in On-Surface Synthesis. In On-Surface Synthesis II; Springer International Publishing, 2018; pp 19–34. [Google Scholar]
- Batra A.; Cvetko D.; Kladnik G.; Adak O.; Cardoso C.; Ferretti A.; Prezzi D.; Molinari E.; Morgante A.; Venkataraman L. Probing the Mechanism for Graphene Nanoribbon Formation on Gold Surfaces through X-Ray Spectroscopy. Chem. Sci. 2014, 5, 4419–4423. 10.1039/C4SC01584C. [DOI] [Google Scholar]
- Rieger A.; Schnidrig S.; Probst B.; Ernst K. H.; Wackerlin C. Identification of On-Surface Reaction Mechanism by Targeted Metalation. J. Phys. Chem. C 2017, 121, 27521–27527. 10.1021/acs.jpcc.7b10019. [DOI] [Google Scholar]
- Riss A.Mechanistic Insights into Surface-Supported Chemical Reactions. In On-Surface Synthesis II; Springer International Publishing, 2018; pp 1.– . [Google Scholar]
- Eichhorn J.; Nieckarz D.; Ochs O.; Samanta D.; Schmittel M.; Szabelski P. J.; Lackinger M. On-Surface Ullmann Coupling: The Influence of Kinetic Reaction Parameters on the Morphology and Quality of Covalent Networks. ACS Nano 2014, 8, 7880–7889. 10.1021/nn501567p. [DOI] [PubMed] [Google Scholar]
- Di Giovannantonio M.; Tomellini M.; Lipton-Duffin J.; Galeotti G.; Ebrahimi M.; Cossaro A.; Verdini A.; Kharche N.; Meunier V.; Vasseur G.; et al. Mechanistic Picture and Kinetic Analysis of Surface-Confined Ullmann Polymerization. J. Am. Chem. Soc. 2016, 138, 16696–16702. 10.1021/jacs.6b09728. [DOI] [PubMed] [Google Scholar]
- Riss A.; Paz A. P.; Wickenburg S.; Tsai H. Z.; De Oteyza D. G.; Bradley A. J.; Ugeda M.; Gorman P.; Jung H. S.; Crommie M. F.; et al. Imaging Single-Molecule Reaction Intermediates Stabilized by Surface Dissipation and Entropy. Nat. Chem. 2016, 8, 678–683. 10.1038/nchem.2506. [DOI] [PubMed] [Google Scholar]
- Sun Q.; Cai L. L.; Ding Y. Q.; Ma H. H.; Yuan C. X.; Xu W. Single-Molecule Insight into Wurtz Reactions on Metal Surfaces. Phys. Chem. Chem. Phys. 2016, 18, 2730–2735. 10.1039/C5CP06459G. [DOI] [PubMed] [Google Scholar]
- Sun Q.; Cai L. L.; Ma H. H.; Yuan C. X.; Xu W. The Stereoselective Synthesis of Dienes through Dehalogenative Homocoupling of Terminal Alkenyl Bromides on Cu(110). Chem. Commun. 2016, 52, 6009–6012. 10.1039/C6CC01059H. [DOI] [PubMed] [Google Scholar]
- Sun Q.; Cai L. L.; Ma H. H.; Yuan C. X.; Xu W. Dehalogenative Homocoupling of Terminal Alkynyl Bromides on Au(111): Incorporation of Acetylenic Scaffolding into Surface Nanostructures. ACS Nano 2016, 10, 7023–7030. 10.1021/acsnano.6b03048. [DOI] [PubMed] [Google Scholar]
- Sun Q.; Tran B. V.; Cai L. L.; Ma H. H.; Yu X.; Yuan C. X.; Stohr M.; Xu W. On-Surface Formation of Cumulene by Dehalogenative Homocoupling of Alkenyl gem-Dibromides. Angew. Chem., Int. Ed. 2017, 56, 12165–12169. 10.1002/anie.201706104. [DOI] [PubMed] [Google Scholar]
- Sun Q.; Yu X.; Bao M.; Liu M.; Pan J.; Zha Z.; Cai L.; Ma H.; Yuan C.; Qiu X.; et al. Direct Formation of C-C Triple Bonded Structural Motifs by On-Surface Dehalogenative Homocoupling of Tribromomethyl Molecules. Angew. Chem., Int. Ed. 2018, 57, 4035–4038. 10.1002/anie.201801056. [DOI] [PubMed] [Google Scholar]
- Klappenberger F.; Zhang Y.-Q.; Björk J.; Klyatskaya S.; Ruben M.; Barth J. V. On-Surface Synthesis of Carbon-Based Scaffolds and Nanomaterials Using Terminal Alkynes. Acc. Chem. Res. 2015, 48, 2140–2150. 10.1021/acs.accounts.5b00174. [DOI] [PubMed] [Google Scholar]
- Clair S.; Abel M.; Porte L. Growth of Boronic Acid Based Two-Dimensional Covalent Networks on a Metal Surface under Ultrahigh Vacuum. Chem. Commun. 2014, 50, 9627–9635. 10.1039/C4CC02678K. [DOI] [PubMed] [Google Scholar]
- Zwaneveld N. A. A.; Pawlak R.; Abel M.; Catalin D.; Gigmes D.; Bertin D.; Porte L. Organized Formation of 2D Extended Covalent Organic Frameworks at Surfaces. J. Am. Chem. Soc. 2008, 130, 6678–6679. 10.1021/ja800906f. [DOI] [PubMed] [Google Scholar]
- Tanoue R.; Higuchi R.; Enoki N.; Miyasato Y.; Uemura S.; Kimizuka N.; Stieg A. Z.; Gimzewski J. K.; Kunitake M. Thermodynamically Controlled Self-Assembly of Covalent Nanoarchitectures in Aqueous Solution. ACS Nano 2011, 5, 3923–3929. 10.1021/nn200393q. [DOI] [PubMed] [Google Scholar]
- Xu L. R.; Zhou X.; Yu Y. X.; Tian W. Q.; Ma J.; Lei S. B. Surface-Confined Crystalline Two-Dimensional Covalent Organic Frameworks via on-Surface Schiff-Base Coupling. ACS Nano 2013, 7, 8066–8073. 10.1021/nn403328h. [DOI] [PubMed] [Google Scholar]
- Sun Q.; Zhang C.; Kong H. H.; Tan Q. G.; Xu W. On-Surface Aryl-Aryl Coupling via Selective C-H Activation. Chem. Commun. 2014, 50, 11825–11828. 10.1039/C4CC05482B. [DOI] [PubMed] [Google Scholar]
- Otero G.; Biddau G.; Sanchez-Sanchez C.; Caillard R.; Lopez M. F.; Rogero C.; Palomares F. J.; Cabello N.; Basanta M. A.; Ortega J.; et al. Fullerenes from Aromatic Precursors by Surface-Catalysed Cyclodehydrogenation. Nature 2008, 454, 865–868. 10.1038/nature07193. [DOI] [PubMed] [Google Scholar]
- Corso M.; Carbonell-Sanromà E.; de Oteyza D. G.. Bottom-Up Fabrication of Atomically Precise Graphene Nanoribbons. In On-Surface Synthesis II; Springer International Publishing, 2018; pp 113–152. [Google Scholar]
- Treier M.; Pignedoli C. A.; Laino T.; Rieger R.; Mullen K.; Passerone D.; Fasel R. Surface-Assisted Cyclodehydrogenation Provides a Synthetic Route Towards Easily Processable and Chemically Tailored Nanographenes. Nat. Chem. 2011, 3, 61–67. 10.1038/nchem.891. [DOI] [PubMed] [Google Scholar]
- Schuler B.; Fatayer S.; Mohn F.; Moll N.; Pavlicek N.; Meyer G.; Pena D.; Gross L. Reversible Bergman Cyclization by Atomic Manipulation. Nat. Chem. 2016, 8, 220–224. 10.1038/nchem.2438. [DOI] [PubMed] [Google Scholar]
- Pavlicek N.; Mistry A.; Majzik Z.; Moll N.; Meyer G.; Fox D. J.; Gross L. Synthesis and Characterization of Triangulene. Nat. Nanotechnol. 2017, 12, 308–311. 10.1038/nnano.2016.305. [DOI] [PubMed] [Google Scholar]
- Bebensee F.; Bombis C.; Vadapoo S. R.; Cramer J. R.; Besenbacher F.; Gothelf K. V.; Linderoth T. R. On-Surface Azide-Alkyne Cycloaddition on Cu(111): Does It ″Click″ in Ultrahigh Vacuum?. J. Am. Chem. Soc. 2013, 135, 2136–2139. 10.1021/ja312303a. [DOI] [PubMed] [Google Scholar]
- Huang H.; Wei D. C.; Sun J. T.; Wong S. L.; Feng Y. P.; Neto A. H. C.; Wee A. T. S. Spatially Resolved Electronic Structures of Atomically Precise Armchair Graphene Nanoribbons. Sci. Rep. 2012, 2, 983. 10.1038/srep00983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross L.; Mohn F.; Moll N.; Liljeroth P.; Meyer G. The Chemical Structure of a Molecule Resolved by Atomic Force Microscopy. Science 2009, 325, 1110–1114. 10.1126/science.1176210. [DOI] [PubMed] [Google Scholar]
- Monig H. Copper-Oxide Tip Functionalization for Submolecular Atomic Force Microscopy. Chem. Commun. 2018, 54, 9874–9888. 10.1039/C8CC05332D. [DOI] [PubMed] [Google Scholar]
- Nguyen G. D.; Tsai H. Z.; Omrani A. A.; Marangoni T.; Wu M.; Rizzo D. J.; Rodgers G. F.; Cloke R. R.; Durr R. A.; Sakai Y.; et al. Atomically Precise Graphene Nanoribbon Heterojunctions from a Single Molecular Precursor. Nat. Nanotechnol. 2017, 12, 1077–1082. 10.1038/nnano.2017.155. [DOI] [PubMed] [Google Scholar]
- Hieulle J.; Carbonell-Sanroma E.; Vilas-Varela M.; Garcia-Lekue A.; Guitian E.; Pena D.; Pascual J. I. On-Surface Route for Producing Planar Nanographenes with Azulene Moieties. Nano Lett. 2018, 18, 418–423. 10.1021/acs.nanolett.7b04309. [DOI] [PubMed] [Google Scholar]
- Lafferentz L.; Ample F.; Yu H.; Hecht S.; Joachim C.; Grill L. Conductance of a Single Conjugated Polymer as a Continuous Function of Its Length. Science 2009, 323, 1193–1197. 10.1126/science.1168255. [DOI] [PubMed] [Google Scholar]
- Cirera B.; Zhang Y. Q.; Bjork J.; Klyatskaya S.; Chen Z.; Ruben M.; Barth J. V.; Klappenberger F. Synthesis of Extended Graphdiyne Wires by Vicinal Surface Templating. Nano Lett. 2014, 14, 1891–1897. 10.1021/nl4046747. [DOI] [PubMed] [Google Scholar]
- Zhang J. J.; Chang C. R.; Yang B.; Cao N.; Peng C. C.; Zhang H. M.; Tang D. T. D.; Glorius F.; Erker G.; Fuchs H.; et al. Step-Edge Assisted Direct Linear Alkane Coupling. Chem. - Eur. J. 2017, 23, 6185–6189. 10.1002/chem.201605744. [DOI] [PubMed] [Google Scholar]
- Stetsovych O.; Svec M.; Vacek J.; Chocholousova J. V.; Jancarik A.; Rybacek J.; Kosmider K.; Stara I. G.; Jelinek P.; Stary I. From Helical to Planar Chirality by On-Surface Chemistry. Nat. Chem. 2017, 9, 213–218. 10.1038/nchem.2662. [DOI] [PubMed] [Google Scholar]
- Barner J.; Al-Hellani R.; Schluter A. D.; Rabe J. P. Synthesis with Single Macromolecules: Covalent Connection between a Neutral Dendronized Polymer and Polyelectrolyte Chains as well as Graphene Edges. Macromol. Rapid Commun. 2010, 31, 362–367. 10.1002/marc.200900438. [DOI] [PubMed] [Google Scholar]
- Barner J.; Mallwitz F.; Shu L. J.; Schluter A. D.; Rabe J. P. Covalent Connection of Two Individual Polymer Chains on a Surface: An Elementary Step Towards Molecular Nanoconstructions. Angew. Chem., Int. Ed. 2003, 42, 1932–1935. 10.1002/anie.200250059. [DOI] [PubMed] [Google Scholar]
- Al-Hellani R.; Barner J.; Rabe J. P.; Schluter A. D. Covalent Connection of Individualized, Neutral, Dendronized Polymers on a Solid Substrate using a Scanning Force Microscope. Chem. - Eur. J. 2006, 12, 6542–6551. 10.1002/chem.200600171. [DOI] [PubMed] [Google Scholar]
- Liu J.; Chen Q. W.; Xiao L. H.; Shang J.; Zhou X.; Zhang Y. J.; Wang Y. F.; Shao X.; Li J. L.; Chen W.; et al. Lattice-Directed Formation of Covalent and Organometallic Molecular Wires by Terminal Alkynes on Ag Surfaces. ACS Nano 2015, 9, 6305–6314. 10.1021/acsnano.5b01803. [DOI] [PubMed] [Google Scholar]
- Liu M. Z.; Liu M. X.; She L. M.; Zha Z. Q.; Pan J. L.; Li S. C.; Li T.; He Y. Y.; Cai Z. Y.; Wang J. B.; et al. Graphene-Like Nanoribbons Periodically Embedded with Four- and Eight-Membered Rings. Nat. Commun. 2017, 8, 14924. 10.1038/ncomms14924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolmer M.; Zebari A. A. A.; Prauzner-Bechcicki J. S.; Piskorz W.; Zasada F.; Godlewski S.; Such B.; Sojka Z.; Szymonski M. Polymerization of Polyanthrylene on a Titanium Dioxide (011)-(2 × 1) Surface. Angew. Chem., Int. Ed. 2013, 52, 10300–10303. 10.1002/anie.201303657. [DOI] [PubMed] [Google Scholar]
- Gao H. Y.; Held P. A.; Amirjalayer S.; Liu L. C.; Timmer A.; Schirmer B.; Arado O. D.; Monig H.; Muck-Lichtenfeld C.; Neugebauer J.; et al. Intermolecular On-Surface sigma-Bond Metathesis. J. Am. Chem. Soc. 2017, 139, 7012–7019. 10.1021/jacs.7b02430. [DOI] [PubMed] [Google Scholar]
- Zhou X.; Bebensee F.; Shen Q.; Bebensee R.; Cheng F.; He Y.; Su H.; Chen W.; Xu G. Q.; Besenbacher F.; et al. On-Surface Synthesis Approach to Preparing One-Dimensional Organometallic and Poly-p-Phenylene Chains. Mater. Chem. Front. 2017, 1, 119–127. 10.1039/C6QM00142D. [DOI] [Google Scholar]
- Klappenberger F.; Hellwig R.; Du P.; Paintner T.; Uphoff M.; Zhang L.; Lin T.; Moghanaki B. A.; Paszkiewicz M.; Vobornik I.; et al. Functionalized Graphdiyne Nanowires: On-Surface Synthesis and Assessment of Band Structure, Flexibility, and Information Storage Potential. Small 2018, 14, 1704321. 10.1002/smll.201704321. [DOI] [PubMed] [Google Scholar]
- Liu J.; Chen Q.; He Q.; Zhang Y.; Fu X.; Wang Y.; Zhao D.; Chen W.; Xu G. Q.; Wu K. Bromine Adatom Promoted C-H Bond Activation in Terminal Alkynes at Room Temperature on Ag(111). Phys. Chem. Chem. Phys. 2018, 20, 11081–11088. 10.1039/C7CP07972A. [DOI] [PubMed] [Google Scholar]
- Para F.; Bocquet F.; Nony L.; Loppacher C.; Féron M.; Cherioux F.; Gao D. Z.; Canova F. F.; Watkins M. B. Micrometre-Long Covalent Organic Fibres by Photoinitiated Chain-Growth Radical Polymerization on an Alkali-Halide Surface. Nat. Chem. 2018, 10, 1112–1117. 10.1038/s41557-018-0120-x. [DOI] [PubMed] [Google Scholar]
- Wu L. Q.; Cai Z. Y.; Liu M. Z.; Ye W.; Ju H. X.; Zhu J. F.; Zhong D. Y. Decarboxylation of Fatty Acids on Anisotropic Au(110) Surfaces. J. Phys. Chem. C 2018, 122, 9075–9080. 10.1021/acs.jpcc.8b01847. [DOI] [Google Scholar]
- Koch M.; Gille M.; Viertel A.; Hecht S.; Grill L. Substrate-Controlled Linking of Molecular Building Blocks: Au(111) vs. Cu(111). Surf. Sci. 2014, 627, 70–74. 10.1016/j.susc.2014.04.011. [DOI] [Google Scholar]
- Selvanathan S.; Peters M. V.; Schwarz J.; Hecht S.; Grill L. Formation and Manipulation of Discrete Supramolecular Azobenzene Assemblies. Appl. Phys. A: Mater. Sci. Process. 2008, 93, 247–252. 10.1007/s00339-008-4827-1. [DOI] [Google Scholar]
- Urgel J. I.; Ecija D.; Auwarter W.; Barth J. V. Controlled Manipulation of Gadolinium-Coordinated Supramolecules by Low-Temperature Scanning Tunneling Microscopy. Nano Lett. 2014, 14, 1369–1373. 10.1021/nl4044339. [DOI] [PubMed] [Google Scholar]
- Kong H. H.; Wang L. K.; Sun Q.; Zhang C.; Tan Q. G.; Xu W. Controllable Scission and Seamless Stitching of Metal-Organic Clusters by STM Manipulation. Angew. Chem., Int. Ed. 2015, 54, 6526–6530. 10.1002/anie.201501701. [DOI] [PubMed] [Google Scholar]
- Gross L. Recent Advances in Submolecular Resolution with Scanning Probe Microscopy. Nat. Chem. 2011, 3, 273–278. 10.1038/nchem.1008. [DOI] [PubMed] [Google Scholar]
- Di Giovannantonio M.; Deniz O.; Urgel J. I.; Widmer R.; Dienel T.; Stolz S.; Sanchez-Sanchez C.; Muntwiler M.; Dumslaff T.; Berger R.; et al. On-Surface Growth Dynamics of Graphene Nanoribbons: The Role of Halogen Functionalization. ACS Nano 2018, 12, 74–81. 10.1021/acsnano.7b07077. [DOI] [PubMed] [Google Scholar]
- Galeotti G.; Di Giovannantonio M.; Lipton-Duffin J.; Ebrahimi M.; Tebi S.; Verdini A.; Floreano L.; Fagot-Revurat Y.; Perepichka D. F.; Rosei F.; et al. The Role of Halogens in On-Surface Ullmann Polymerization. Faraday Discuss. 2017, 204, 453–469. 10.1039/C7FD00099E. [DOI] [PubMed] [Google Scholar]
- Stöhr J. In NEXAFS Spectroscopy; Ertl G., Lüth H., Car R., Rocca M. A., Freund H.-J., Eds.; Springer Series in Surface Sciences; Springer-Verlag: Berlin, 1992; Vol. XV, p 404. [Google Scholar]
- Vasseur G.; Abadia M.; Miccio L. A.; Brede J.; Garcia-Lekue A.; de Oteyza D. G.; Rogero C.; Lobo-Checa J.; Ortega J. E. Pi Band Dispersion along Conjugated Organic Nanowires Synthesized on a Metal Oxide Semiconductor. J. Am. Chem. Soc. 2016, 138, 5685–5692. 10.1021/jacs.6b02151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino-Diez N.; Lobo-Checa J.; Nita P.; Garcia-Lekue A.; Basagni A.; Vasseur G.; Tiso F.; Sedona F.; Das P. K.; Fujii J.; et al. Switching from Reactant to Substrate Engineering in the Selective Synthesis of Graphene Nanoribbons. J. Phys. Chem. Lett. 2018, 9, 2510–2517. 10.1021/acs.jpclett.8b00796. [DOI] [PubMed] [Google Scholar]
- Vasseur G.; Fagot-Revurat Y.; Sicot M.; Kierren B.; Moreau L.; Malterre D.; Cardenas L.; Galeotti G.; Lipton-Duffin J.; Rosei F.; et al. Quasi One-Dimensional Band Dispersion and Surface Metallization in Long-Range Ordered Polymeric Wires. Nat. Commun. 2016, 7, 10235. 10.1038/ncomms10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner C.; Leyssner F.; Stremlau S.; Utecht M.; Saalfrank P.; Klamroth T.; Tegeder P. Electronic Structure of a Subnanometer Wide Bottom-Up Fabricated Graphene Nanoribbon: End States, Band Gap, and Dispersion. Phys. Rev. B: Condens. Matter Mater. Phys. 2012, 86, 085444. 10.1103/PhysRevB.86.085444. [DOI] [Google Scholar]
- Malard L. M.; Pimenta M. A.; Dresselhaus G.; Dresselhaus M. S. Raman Spectroscopy in Graphene. Phys. Rep. 2009, 473, 51–87. 10.1016/j.physrep.2009.02.003. [DOI] [Google Scholar]
- Talirz L.; Sode H.; Dumslaff T.; Wang S. Y.; Sanchez-Valencia J. R.; Liu J.; Shinde P.; Pignedoli C. A.; Liang L. B.; Meunier V.; et al. On-Surface Synthesis and Characterization of 9-Atom Wide Armchair Graphene Nanoribbons. ACS Nano 2017, 11, 1380–1388. 10.1021/acsnano.6b06405. [DOI] [PubMed] [Google Scholar]
- Han P.; Akagi K.; Canova F. F.; Shimizu R.; Oguchi H.; Shiraki S.; Weiss P. S.; Asao N.; Hitosugi T. Self-Assembly Strategy for Fabricating Connected Graphene Nanoribbons. ACS Nano 2015, 9, 12035–12044. 10.1021/acsnano.5b04879. [DOI] [PubMed] [Google Scholar]
- Saito R.; Furukawa M.; Dresselhaus G.; Dresselhaus M. S. Raman Spectra of Graphene Ribbons. J. Phys.: Condens. Matter 2010, 22, 334203. 10.1088/0953-8984/22/33/334203. [DOI] [PubMed] [Google Scholar]
- Sasaki K.; Yamamoto M.; Murakami S.; Saito R.; Dresselhaus M. S.; Takai K.; Mori T.; Enoki T.; Wakabayashi K. Kohn Anomalies in Graphene Nanoribbons. Phys. Rev. B: Condens. Matter Mater. Phys. 2009, 80, 155450. 10.1103/PhysRevB.80.155450. [DOI] [Google Scholar]
- Yang R.; Shi Z. W.; Zhang L. C.; Shi D. X.; Zhang G. Y. Observation of Raman G-Peak Split for Graphene Nanoribbons with Hydrogen-Terminated Zigzag Edges. Nano Lett. 2011, 11, 4083–4088. 10.1021/nl201387x. [DOI] [PubMed] [Google Scholar]
- Dussert C.; Rasigni G.; Rasigni M.; Palmari J.; Llebaria A. Minimal Spanning Tree: A New Approach for Studying Order and Disorder. Phys. Rev. B: Condens. Matter Mater. Phys. 1986, 34, 3528–3531. 10.1103/PhysRevB.34.3528. [DOI] [PubMed] [Google Scholar]
- Ourdjini O.; Pawlak R.; Abel M.; Clair S.; Chen L.; Bergeon N.; Sassi M.; Oison V.; Debierre J.-M.; Coratger R.; et al. Substrate-Mediated Ordering and Defect Analysis of a Surface Covalent Organic Framework. Phys. Rev. B: Condens. Matter Mater. Phys. 2011, 84, 125421. 10.1103/PhysRevB.84.125421. [DOI] [Google Scholar]
- Di Giovannantonio M.; El Garah M.; Lipton-Duffin J.; Meunier V.; Cardenas L.; Fagot-Revurat Y.; Cossaro A.; Verdini A.; Perepichka D. F.; Rosei F.; et al. Insight into Organometallic Intermediate and Its Evolution to Covalent Bonding in Surface-Confined Ullmann Polymerization. ACS Nano 2013, 7, 8190–8198. 10.1021/nn4035684. [DOI] [PubMed] [Google Scholar]
- Shi K. J.; Yuan D. W.; Wang C. X.; Shu C. H.; Li D. Y.; Shi Z. L.; Wu X. Y.; Liu P. N. Ullmann Reaction of Aryl Chlorides on Various Surfaces and the Application in Stepwise Growth of 2D Covalent Organic Frameworks. Org. Lett. 2016, 18, 1282–1285. 10.1021/acs.orglett.6b00172. [DOI] [PubMed] [Google Scholar]
- Lischka M.; Michelitsch G. S.; Martsinovich N.; Eichhorn J.; Rastgoo-Lahrood A.; Strunskus T.; Breuer R.; Reuter K.; Schmittel M.; Lackinger M. Remote Functionalization in Surface-Assisted Dehalogenation by Conformational Mechanics: Organometallic Self-Assembly of 3,3 ’,5,5 ’-Tetrabromo-2,2 ’,4,4 ’,6,6 ’-Hexafluorobiphenyl on Ag(111). Nanoscale 2018, 10, 12035–12044. 10.1039/C8NR01987H. [DOI] [PubMed] [Google Scholar]
- Desiraju G. R. Chemistry Beyond the Molecule. Nature 2001, 412, 397–400. 10.1038/35086640. [DOI] [PubMed] [Google Scholar]
- Yokoyama T.; Yokoyama S.; Kamikado T.; Okuno Y.; Mashiko S. Selective Assembly on a Surface of Supramolecular Aggregates of Controlled Size and Shape. Nature 2001, 413, 619–621. 10.1038/35098059. [DOI] [PubMed] [Google Scholar]
- Rabot C.; Clair S.; Kim Y.; Kawai M. Scanning Tunneling Microscopy Observations of Benzoic Acid Molecules Coadsorbed with Single-Walled Carbon Nanotubes on Au(111) Surface. Jpn. J. Appl. Phys. Part 1 2007, 46, 5572–5576. 10.1143/JJAP.46.5572. [DOI] [Google Scholar]
- Clair S.; Pons S.; Seitsonen A. P.; Brune H.; Kern K.; Barth J. V. STM Study of Terephthalic Acid Self-Assembly on Au(111): Hydrogen-Bonded Sheets on an Inhomogeneous Substrate. J. Phys. Chem. B 2004, 108, 14585–14590. 10.1021/jp049501n. [DOI] [Google Scholar]
- Payer D.; Comisso A.; Dmitriev A.; Strunskus T.; Lin N.; Woll C.; DeVita A.; Barth J. V.; Kern K. Ionic Hydrogen Bonds Controlling Two-Dimensional Supramolecular Systems at a Metal Surface. Chem. - Eur. J. 2007, 13, 3900–3906. 10.1002/chem.200601325. [DOI] [PubMed] [Google Scholar]
- Cai L.; Yu X.; Liu M.; Sun Q.; Bao M.; Zha Z.; Pan J.; Ma H.; Ju H.; Hu S.; et al. Direct Formation of C–C Double-Bonded Structural Motifs by On-Surface Dehalogenative Homocoupling of gem-Dibromomethyl Molecules. ACS Nano 2018, 12, 7959–7966. 10.1021/acsnano.8b02459. [DOI] [PubMed] [Google Scholar]
- Shu C. H.; Liu M. X.; Zha Z. Q.; Pan J. L.; Zhang S. Z.; Xie Y. L.; Chen J. L.; Yuan D. W.; Qiu X. H.; Liu P. N. On-Surface Synthesis of Poly(p-Phenylene Ethynylene) Molecular Wires via In Situ Formation of Carbon-Carbon Triple Bbond. Nat. Commun. 2018, 9, 2322. 10.1038/s41467-018-04681-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. H.; Guan C. Z.; Zheng Q. N.; Wang D.; Wan L. J. Molecular Engineering of Schiff-Base Linked Covalent Polymers with Diverse Topologies by Gas-Solid Interface Reaction. J. Chem. Phys. 2015, 142, 101905. 10.1063/1.4906271. [DOI] [PubMed] [Google Scholar]
- Stepanow S.; Lingenfelder M.; Dmitriev A.; Spillmann H.; Delvigne E.; Lin N.; Deng X.; Cai C.; Barth J. V.; Kern K. Steering Molecular Organization and Host-Guest Interactions using Tailor-Made Two-Dimensional Nanoporous Coordination Systems. Nat. Mater. 2004, 3, 229. 10.1038/nmat1088. [DOI] [PubMed] [Google Scholar]
- Schlickum U.; Decker R.; Klappenberger F.; Zoppellaro G.; Klyatskaya S.; Ruben M.; Silanes I.; Arnau A.; Kern K.; Brune H.; et al. Metal-Organic Honeycomb Nanomeshes with Tunable Cavity Size. Nano Lett. 2007, 7, 3813–3817. 10.1021/nl072466m. [DOI] [PubMed] [Google Scholar]
- Guan C. Z.; Wang D.; Wan L. J. Construction and Repair of Highly Ordered 2D Covalent Networks by Chemical Equilibrium Regulation. Chem. Commun. 2012, 48, 2943–2945. 10.1039/c2cc16892h. [DOI] [PubMed] [Google Scholar]
- Dienstmaier J. F.; Medina D. D.; Dogru M.; Knochel P.; Bein T.; Heckl W. M.; Lackinger M. Isoreticular Two-Dimensional Covalent Organic Frameworks Synthesized by On-Surface Condensation of Diboronic Acids. ACS Nano 2012, 6, 7234–7242. 10.1021/nn302363d. [DOI] [PubMed] [Google Scholar]
- Tanoue R.; Higuchi R.; Ikebe K.; Uemura S.; Kimizuka N.; Stieg A. Z.; Gimzewski J. K.; Kunitake M. In Situ STM Investigation of Aromatic Poly(azomethine) Arrays Constructed by ″On-Site″ Equilibrium Polymerization. Langmuir 2012, 28, 13844–13851. 10.1021/la302863h. [DOI] [PubMed] [Google Scholar]
- Ciesielski A.; El Garah M.; Haar S.; Kovaricek P.; Lehn J. M.; Samori P. Dynamic Covalent Chemistry of Bisimines at the Solid/Liquid Interface Monitored by Scanning Tunnelling Microscopy. Nat. Chem. 2014, 6, 1017–1023. 10.1038/nchem.2057. [DOI] [PubMed] [Google Scholar]
- Sun X. L.; Fan L. X.; Zhou X.; Tian W. Q.; Guo Z. X.; Li Z. B.; Li X. K.; Lei S. B. Surface Confined Synthesis of Porphyrin Containing Two-Dimensional Polymers: the Effect of Rigidity and Preferential Adsorption of Building Blocks. Chem. Commun. 2015, 51, 5864–5867. 10.1039/C5CC00659G. [DOI] [PubMed] [Google Scholar]
- Yu Y. X.; Sun J.; Lei S. B. Surface-Confined Synthesis of One-Dimensional Schiff Base Polymers Investigated by Scanning Tunneling Microscopy. J. Phys. Chem. C 2015, 119, 16777–16784. 10.1021/acs.jpcc.5b04938. [DOI] [Google Scholar]
- Yu Y. X.; Lin J. B.; Wang Y.; Zeng Q. D.; Lei S. B. Room Temperature On-Surface Synthesis of Two-Dimensional Imine Polymers at the Solid/Liquid Interface: Concentration Takes Control. Chem. Commun. 2016, 52, 6609–6612. 10.1039/C6CC02005D. [DOI] [PubMed] [Google Scholar]
- Xu L. R.; Yu Y. X.; Lin J. B.; Zhou X.; Tian W. Q.; Nieckarz D.; Szabelski P.; Lei S. B. On-Surface Synthesis of Two-Dimensional Imine Polymers with a Tunable Band Gap: a Combined STM, DFT and Monte Carlo Investigation. Nanoscale 2016, 8, 8568–8574. 10.1039/C5NR07663C. [DOI] [PubMed] [Google Scholar]
- Mo Y. P.; Liu X. H.; Wang D. Concentration-Directed Polymorphic Surface Covalent Organic Frameworks: Rhombus, Parallelogram, and Kagome. ACS Nano 2017, 11, 11694–11700. 10.1021/acsnano.7b06871. [DOI] [PubMed] [Google Scholar]
- Gao H. Y.; Wagner H.; Zhong D. Y.; Franke J. H.; Studer A.; Fuchs H. Glaser Coupling at Metal Surfaces. Angew. Chem., Int. Ed. 2013, 52, 4024–4028. 10.1002/anie.201208597. [DOI] [PubMed] [Google Scholar]
- Yue J. Y.; Liu X. H.; Sun B.; Wang D. The On-Surface Synthesis of Imine-Based Covalent Organic Frameworks with Non-Aromatic Linkage. Chem. Commun. 2015, 51, 14318–14321. 10.1039/C5CC05689F. [DOI] [PubMed] [Google Scholar]
- Liu X. H.; Mo Y. P.; Yue J. Y.; Zheng Q. N.; Yan H. J.; Wang D.; Wan L. J. Isomeric Routes to Schiff-Base Single-layered Covalent Organic Frameworks. Small 2014, 10, 4934–4939. 10.1002/smll.201400899. [DOI] [PubMed] [Google Scholar]
- Schlögl S.; Heckl W. M.; Lackinger M. On-Surface Radical Addition of Triply Iodinated Monomers on Au(111) - The Influence of Monomer Size and Thermal Post-Processing. Surf. Sci. 2012, 606, 999–1004. 10.1016/j.susc.2012.02.011. [DOI] [Google Scholar]
- Zhang Y. Q.; Kepcija N.; Kleinschrodt M.; Diller K.; Fischer S.; Papageorgiou A. C.; Allegretti F.; Bjork J.; Klyatskaya S.; Klappenberger F.; et al. Homo-Coupling of Terminal Alkynes on a Noble Metal Surface. Nat. Commun. 2012, 3, 1286. 10.1038/ncomms2291. [DOI] [PubMed] [Google Scholar]
- Faury T.; Clair S.; Abel M.; Dumur F.; Gigmes D.; Porte L. Sequential Linking to Control the Growth of a Surface Covalent Organic Framework. J. Phys. Chem. C 2012, 116, 4819. 10.1021/jp300417g. [DOI] [Google Scholar]
- Blunt M. O.; Russell J. C.; Champness N. R.; Beton P. H. Templating Molecular Adsorption Using a Covalent Organic Framework. Chem. Commun. 2010, 46, 7157–7159. 10.1039/c0cc01810d. [DOI] [PubMed] [Google Scholar]
- Chen J. J.; Zhu E. B.; Liu J.; Zhang S.; Lin Z. Y.; Duan X. F.; Heinz H.; Huang Y.; De Yoreo J. J. Building Two-Dimensional Materials One Row at a Time: Avoiding the Nucleation Barrier. Science 2018, 362, 1135–1139. 10.1126/science.aau4146. [DOI] [PubMed] [Google Scholar]
- McMillen D. F.; Golden D. M. Hydrocarbon Bond Dissociation Energies. Annu. Rev. Phys. Chem. 1982, 33, 493–532. 10.1146/annurev.pc.33.100182.002425. [DOI] [Google Scholar]
- Lafferentz L.; Eberhardt V.; Dri C.; Africh C.; Comelli G.; Esch F.; Hecht S.; Grill L. Controlling On-Surface Polymerization by Hierarchical and Substrate-Directed Growth. Nat. Chem. 2012, 4, 215–220. 10.1038/nchem.1242. [DOI] [PubMed] [Google Scholar]
- Eichhorn J.; Strunskus T.; Rastgoo-Lahrood A.; Samanta D.; Schmittel M.; Lackinger M. On-Surface Ullmann Polymerization via Intermediate Organometallic Networks on Ag(111). Chem. Commun. 2014, 50, 7680–7682. 10.1039/C4CC02757D. [DOI] [PubMed] [Google Scholar]
- Shi K. J.; Zhang X.; Shu C. H.; Li D. Y.; Wu X. Y.; Liu P. N. Ullmann Coupling Reaction of Aryl Chlorides on Au(111) using Dosed Cu as a Catalyst and the Programmed Growth of 2D Covalent Organic Frameworks. Chem. Commun. 2016, 52, 8726–8729. 10.1039/C6CC03137D. [DOI] [PubMed] [Google Scholar]
- Lischka M.; Fritton M.; Eichhorn J.; Vyas V. S.; Strunskus T.; Lotsch B. V.; Björk J.; Heckl W. M.; Lackinger M. On-Surface Polymerization of 1,8-Dibromo-3,6-Diiodopyrene – a Comparative Study on Au(111) vs. Ag(111) by STM, XPS, and NEXAFS. J. Phys. Chem. C 2018, 122, 5967–5977. 10.1021/acs.jpcc.7b10403. [DOI] [Google Scholar]
- Steiner C.; Gebhardt J.; Ammon M.; Yang Z. C.; Heidenreich A.; Hammer N.; Gorling A.; Kivala M.; Maier S. Hierarchical On-Surface Synthesis and Electronic Structure of Carbonyl-Functionalized One- and Two-Dimensional Covalent Nanoarchitectures. Nat. Commun. 2017, 8, 14765. 10.1038/ncomms14765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittelmann M.; Nimmrich M.; Lindner R.; Gourdon A.; Kuhnle A. Sequential and Site-Specific On-Surface Synthesis on a Bulk Insulator. ACS Nano 2013, 7, 5614–5620. 10.1021/nn402018w. [DOI] [PubMed] [Google Scholar]
- Cai J. M.; Ruffieux P.; Jaafar R.; Bieri M.; Braun T.; Blankenburg S.; Muoth M.; Seitsonen A. P.; Saleh M.; Feng X.; et al. Atomically Precise Bottom-Up Fabrication of Graphene Nanoribbons. Nature 2010, 466, 470–473. 10.1038/nature09211. [DOI] [PubMed] [Google Scholar]
- Ruffieux P.; Wang S. Y.; Yang B.; Sanchez-Sanchez C.; Liu J.; Dienel T.; Talirz L.; Shinde P.; Pignedoli C. A.; Passerone D.; et al. On-Surface Synthesis of Graphene Nanoribbons with Zigzag Edge Topology. Nature 2016, 531, 489. 10.1038/nature17151. [DOI] [PubMed] [Google Scholar]
- Liu J.; Dienel T.; Liu J. Z.; Groening O.; Cai J. M.; Feng X. L.; Mullen K.; Ruffieux P.; Fasel R. Building Pentagons into Graphenic Structures by On-Surface Polymerization and Aromatic Cyclodehydrogenation of Phenyl-Substituted Polycyclic Aromatic Hydrocarbons. J. Phys. Chem. C 2016, 120, 17588–17593. 10.1021/acs.jpcc.6b05495. [DOI] [Google Scholar]
- Chong M. C.; Sosa-Vargas L.; Bulou H.; Boeglin A.; Scheurer F.; Mathevet F.; Schull G. Ordinary and Hot Electroluminescence from Single-Molecule Devices: Controlling the Emission Color by Chemical Engineering. Nano Lett. 2016, 16, 6480–6484. 10.1021/acs.nanolett.6b02997. [DOI] [PubMed] [Google Scholar]
- Di Giovannantonio M.; Urgel J. I.; Beser U.; Yakutovich A.; Wilhelm J.; Pignedoli C. A.; Ruffieux P.; Narita A.; Müllen K.; Fasel R. On-Surface Synthesis of Indenofluorene Polymers by Oxidative Five-Membered Ring Formation. J. Am. Chem. Soc. 2018, 140, 3532–3536. 10.1021/jacs.8b00587. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Sun Q.; Chen H.; Tan Q. G.; Xu W. Formation of Polyphenyl Chains through Hierarchical Reactions: Ullmann Coupling Followed by Cross-Dehydrogenative Coupling. Chem. Commun. 2015, 51, 495–498. 10.1039/C4CC07953A. [DOI] [PubMed] [Google Scholar]
- Schlögl S.; Sirtl T.; Eichhorn J.; Heckl W. M.; Lackinger M. Synthesis of Two-Dimensional Phenylene-Boroxine Networks Through In Vacuo Condensation and On-Surface Radical Addition. Chem. Commun. 2011, 47, 12355–12357. 10.1039/c1cc13896k. [DOI] [PubMed] [Google Scholar]
- Yue J. Y.; Mo Y. P.; Li S. Y.; Dong W. L.; Chen T.; Wang D. Simultaneous Construction of Two Linkages for the On-Surface Synthesis of Imine-Boroxine Hybrid Covalent Organic Frameworks. Chem. Sci. 2017, 8, 2169–2174. 10.1039/C6SC03590F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong H. H.; Yang S.; Gao H. Y.; Timmer A.; Hill J. P.; Arado O. D.; Monig H.; Huang X. Y.; Tang Q.; Ji Q. M.; et al. Substrate-Mediated C-C and C-H Coupling after Dehalogenation. J. Am. Chem. Soc. 2017, 139, 3669–3675. 10.1021/jacs.6b10936. [DOI] [PubMed] [Google Scholar]
- Liu L. C.; Klaasen H.; Timmer A.; Gao H. Y.; Barton D.; Monig H.; Neugebauer J.; Fuchs H.; Studer A. alpha-Diazo Ketones in On-Surface Chemistry. J. Am. Chem. Soc. 2018, 140, 6000–6005. 10.1021/jacs.8b02599. [DOI] [PubMed] [Google Scholar]
- Held P. A.; Gao H.-Y.; Liu L.; Mück-Lichtenfeld C.; Timmer A.; Mönig H.; Barton D.; Neugebauer J.; Fuchs H.; Studer A. On-Surface Domino Reactions: Glaser Coupling and Dehydrogenative Coupling of a Biscarboxylic Acid To Form Polymeric Bisacylperoxides. Angew. Chem., Int. Ed. 2016, 55, 9777–9782. 10.1002/anie.201602859. [DOI] [PubMed] [Google Scholar]
- Li X.; Zhang H.; Chi L.. On-Surface Synthesis of Graphyne-Based Nanostructures. Adv. Mater. 2018, 1804087. 10.1002/adma.201804087. [DOI] [PubMed] [Google Scholar]
- Sun K.; Chen A.; Liu M.; Zhang H.; Duan R.; Ji P.; Li L.; Li Q.; Li C.; Zhong D.; et al. Surface-Assisted Alkane Polymerization: Investigation on Structure–Reactivity Relationship. J. Am. Chem. Soc. 2018, 140, 4820–4825. 10.1021/jacs.7b09097. [DOI] [PubMed] [Google Scholar]
- Fritton M.; Otte K.; Bjork J.; Biswas P. K.; Heckl W. M.; Schmittel M.; Lackinger M. The Influence of Ortho-Methyl Substitution in Organometallic Self-Assembly - a Comparative Study on Cu(111) vs. Ag(111). Chem. Commun. 2018, 54, 9745–9748. 10.1039/C8CC04854A. [DOI] [PubMed] [Google Scholar]
- Liu J.; Xia B. W.; Xu H.; Lin N. A. Controlling the Reaction Steps of Bifunctional Molecules 1,5-Dibromo-2,6-dimethylnaphthalene on Different Substrates. J. Phys. Chem. C 2018, 122, 13001–13008. 10.1021/acs.jpcc.8b04651. [DOI] [Google Scholar]
- Fan Q. T.; Werner S.; Tschakert J.; Ebeling D.; Schirmeisen A.; Hilt G.; Hieringer W.; Gottfried J. M. Precise Monoselective Aromatic C-H Bond Activation by Chemisorption of Meta-Aryne on a Metal Surface. J. Am. Chem. Soc. 2018, 140, 7526–7532. 10.1021/jacs.8b01658. [DOI] [PubMed] [Google Scholar]
- Tokunaga Y.; Ueno H.; Shimomura Y.; Seo T. Formation of Boroxine: Its Stability and Thermodynamic Parameters in Solution. Heterocycles 2002, 57, 787–790. 10.3987/COM-02-9464. [DOI] [Google Scholar]
- Faury T.; Dumur F.; Clair S.; Abel M.; Porte L.; Gigmes D. Side Functionalization of Diboronic Acid Precursors for Covalent Organic Frameworks. CrystEngComm 2013, 15, 2067–2075. 10.1039/c3ce26494g. [DOI] [Google Scholar]
- Kocic N.; Liu X. S.; Chen S. J.; Decurtins S.; Krejci O.; Jelinek P.; Repp J.; Liu S. X. Control of Reactivity and Regioselectivity for On-Surface Dehydrogenative Aryl-Aryl Bond Formation. J. Am. Chem. Soc. 2016, 138, 5585–5593. 10.1021/jacs.5b13461. [DOI] [PubMed] [Google Scholar]
- Mo Y. P.; Liu X. H.; Sun B.; Yan H. J.; Wang D.; Wan L. J. The Intramolecular H-Bonding Effect on the Growth and Stability of Schiff-Base Surface Covalent Organic Frameworks. Phys. Chem. Chem. Phys. 2017, 19, 539–543. 10.1039/C6CP06894D. [DOI] [PubMed] [Google Scholar]
- Yu Y. X.; Yang L.; Liu C. H.; Tian W. Q.; Wang Y.; Lei S. B. The Hierarchical Construction of Cross-Junctions of Molecular Wires with Covalent and Noncovalent Interactions at the Liquid/Solid Interface. Chem. Commun. 2016, 52, 8317–8320. 10.1039/C6CC02975B. [DOI] [PubMed] [Google Scholar]
- Xu L. R.; Cao L. L.; Guo Z. X.; Zha Z. Q.; Lei S. B. Side-Functionalized Two-Dimensional Polymers Synthesized via On-Surface Schiff-Base Coupling. Chem. Commun. 2015, 51, 8664–8667. 10.1039/C5CC02232K. [DOI] [PubMed] [Google Scholar]
- Liu C. H.; Park E.; Jin Y. H.; Liu J.; Yu Y. X.; Zhang W.; Lei S. B.; Hu W. P. Surface-Confined Dynamic Covalent System Driven by Olefin Metathesis. Angew. Chem., Int. Ed. 2018, 57, 1869–1873. 10.1002/anie.201711040. [DOI] [PubMed] [Google Scholar]
- Lewis E. A.; Marcinkowski M. D.; Murphy C. J.; Liriano M. L.; Therrien A. J.; Pronschinske A.; Sykes E. C. H. Controlling selectivity in the Ullmann reaction on Cu(111). Chem. Commun. 2017, 53, 7816–7819. 10.1039/C7CC02901B. [DOI] [PubMed] [Google Scholar]
- Dinca L. E.; MacLeod J. M.; Lipton-Duffin J.; Fu C. Y.; Ma D. L.; Perepichka D. F.; Rosei F. Tailoring the Reaction Path in the On-Surface Chemistry of Thienoacenes. J. Phys. Chem. C 2015, 119, 22432–22438. 10.1021/acs.jpcc.5b05418. [DOI] [Google Scholar]
- Meng X.; Liu L.; García F.; Álvarez B.; Pérez D.; Gao H.-Y.; Peña D.; Fuchs H. Effect of Central π-System in Silylated-Tetraynes on σ-Bond Metathesis on Surfaces. J. Phys. Chem. C 2018, 122, 6230–6235. 10.1021/acs.jpcc.8b00519. [DOI] [Google Scholar]
- Giannakakis G.; Flytzani-Stephanopoulos M.; Sykes E. C. H. Single-Atom Alloys as a Reductionist Approach to the Rational Design of Heterogeneous Catalysts. Acc. Chem. Res. 2019, 52, 237. 10.1021/acs.accounts.8b00490. [DOI] [PubMed] [Google Scholar]
- Adisoejoso J.; Lin T.; Shang X. S.; Shi K. J.; Gupta A.; Liu P. N.; Lin N. A Single-Molecule-Level Mechanistic Study of Pd-Catalyzed and Cu-Catalyzed Homocoupling of Aryl Bromide on an Au(111) Surface. Chem. - Eur. J. 2014, 20, 4111–4116. 10.1002/chem.201304443. [DOI] [PubMed] [Google Scholar]
- Zhao W.; Dong L.; Huang C.; Win Z. M.; Lin N. Cu- and Pd-Catalyzed Ullmann Reaction on a Hexagonal Boron Nitride Layer. Chem. Commun. 2016, 52, 13225–13228. 10.1039/C6CC05029H. [DOI] [PubMed] [Google Scholar]
- Cirera B.; Bjork J.; Otero R.; Gallego J. M.; Miranda R.; Ecija D. Efficient Lanthanide Catalyzed Debromination and Oligomeric Length-Controlled Ullmann Coupling of Aryl Halides. J. Phys. Chem. C 2017, 121, 8033–8041. 10.1021/acs.jpcc.7b02172. [DOI] [Google Scholar]
- Hellwig R.; Uphoff M.; Paintner T.; Björk J.; Ruben M.; Klappenberger F.; Barth J. V. Ho-Mediated Alkyne Reactions at Low Temperatures on Ag(111). Chem. - Eur. J. 2018, 24, 16126–16135. 10.1002/chem.201803102. [DOI] [PubMed] [Google Scholar]
- Lewis E. A.; Murphy C. J.; Pronschinske A.; Liriano M. L.; Sykes E. C. H. Nanoscale Insight into C-C Coupling on Cobalt Nanoparticles. Chem. Commun. 2014, 50, 10035–10037. 10.1039/C4CC03678F. [DOI] [PubMed] [Google Scholar]
- Zhang X. M.; Liao L. Y.; Wang S.; Hu F. Y.; Wang C.; Zeng Q. D. Polymerization or Cyclic Dimerization: Solvent Dependent Homo-Coupling of Terminal Alkynes at HOPG Surface. Sci. Rep. 2015, 4, 3899. 10.1038/srep03899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T.; Shang X. S.; Adisoejoso J.; Liu P. N.; Lin N. Steering On-Surface Polymerization with Metal-Directed Template. J. Am. Chem. Soc. 2013, 135, 3576–3582. 10.1021/ja311890n. [DOI] [PubMed] [Google Scholar]
- Zhang R.; Lyu G. Q.; Li D. Y.; Liu P. N.; Lin N. A. Template-Controlled Sonogashira Cross-Coupling Reactions on a Au(111) Surface. Chem. Commun. 2017, 53, 1731–1734. 10.1039/C6CC10091K. [DOI] [PubMed] [Google Scholar]
- Shen Q.; Larkin E. J.; Delaney C.; Cheng Y. C.; Miao C. Y.; Zhou X.; Liu L. C.; Huang W.; Gao H. Y.; Draper S. M.; et al. Coordination Reactions of 5-(2-(4-Bromophenyl)ethynyl)pyrimidine in On-Surface Synthesis. J. Phys. Chem. C 2018, 122, 8954–8959. 10.1021/acs.jpcc.8b00696. [DOI] [Google Scholar]
- Palma C. A.; Samori P.; Cecchini M. Atomistic Simulations of 2D Bicomponent Self-Assembly: From Molecular Recognition to Self-Healing. J. Am. Chem. Soc. 2010, 132, 17880–17885. 10.1021/ja107882e. [DOI] [PubMed] [Google Scholar]
- Barth J. V. Molecular Architectonic on Metal Surfaces. Annu. Rev. Phys. Chem. 2007, 58, 375–407. 10.1146/annurev.physchem.56.092503.141259. [DOI] [PubMed] [Google Scholar]
- Barth J. V. Fresh Perspectives for Surface Coordination Chemistry. Surf. Sci. 2009, 603, 1533–1541. 10.1016/j.susc.2008.09.049. [DOI] [Google Scholar]
- Schmidt G. M. J. Photodimerization in the Solid State. Pure Appl. Chem. 1971, 27, 647. 10.1351/pac197127040647. [DOI] [Google Scholar]
- Basagni A.; Sedona F.; Pignedoli C. A.; Cattelan M.; Nicolas L.; Casarin M.; Sambi M. Molecules-Oligomers-Nanowires-Graphene Nanoribbons: A Bottom-Up Stepwise On-Surface Covalent Synthesis Preserving Long-Range Order. J. Am. Chem. Soc. 2015, 137, 1802–1808. 10.1021/ja510292b. [DOI] [PubMed] [Google Scholar]
- Chen Q. W.; Cramer J. R.; Liu J.; Jin X.; Liao P. L.; Shao X.; Gothelf K. V.; Wu K. Steering On-Surface Reactions by a Self-Assembly Approach. Angew. Chem., Int. Ed. 2017, 56, 5026–5030. 10.1002/anie.201700745. [DOI] [PubMed] [Google Scholar]
- Cai Z. Y.; She L. M.; He Y. Y.; Wu L. Q.; Cai L.; Zhong D. Y. Halogen-Free On-Surface Synthesis of Rylene-Type Graphene Nanoribbons. Macromol. Chem. Phys. 2017, 218, 1700155. 10.1002/macp.201700155. [DOI] [Google Scholar]
- Sedona F.; Di Marino M.; Sambi M.; Carofiglio T.; Lubian E.; Casarin M.; Tondello E. Fullerene/Porphyrin Multicomponent Nanostructures on Ag(110): From Supramolecular Self-Assembly to Extended Copolymers. ACS Nano 2010, 4, 5147–5154. 10.1021/nn101161a. [DOI] [PubMed] [Google Scholar]
- Takami T.; Ozaki H.; Kasuga M.; Tsuchiya T.; Mazaki Y.; Fukushi D.; Ogawa A.; Uda M.; Aono M. Periodic Structure of a Single Sheet of a Clothlike Macromolecule (Atomic Cloth) Studied by Scanning Tunneling Microscopy. Angew. Chem., Int. Ed. Engl. 1997, 36, 2755–2757. 10.1002/anie.199727551. [DOI] [Google Scholar]
- Miura A.; De Feyter S.; Abdel-Mottaleb M. M. S.; Gesquiere A.; Grim P. C. M.; Moessner G.; Sieffert M.; Klapper M.; Mullen K.; De Schryver F. C. Light- and STM-Tip-Induced Formation of One-Dimensional and Two-Dimensional Organic Nanostructures. Langmuir 2003, 19, 6474–6482. 10.1021/la027051p. [DOI] [Google Scholar]
- Mandal S. K.; Okawa Y.; Hasegawa T.; Aono M. Rate-Determining Factors in the Chain Polymerization of Molecules Initiated by Local Single-Molecule Excitation. ACS Nano 2011, 5, 2779–2786. 10.1021/nn103231j. [DOI] [PubMed] [Google Scholar]
- Okawa Y.; Mandal S. K.; Hu C. P.; Tateyama Y.; Goedecker S.; Tsukamoto S.; Hasegawa T.; Gimzewski J. K.; Aono M. Chemical Wiring and Soldering toward All-Molecule Electronic Circuitry. J. Am. Chem. Soc. 2011, 133, 8227–8233. 10.1021/ja111673x. [DOI] [PubMed] [Google Scholar]
- Okawa Y.; Akai-Kasaya M.; Kuwahara Y.; Mandal S. K.; Aono M. Controlled Chain Polymerisation and Chemical Soldering for Single-Molecule Electronics. Nanoscale 2012, 4, 3013–3028. 10.1039/c2nr30245d. [DOI] [PubMed] [Google Scholar]
- Okawa Y.; Takajo D.; Tsukamoto S.; Hasegawa T.; Aono M. Atomic Force Microscopy and Theoretical Investigation of the Lifted-Up Conformation of Polydiacetylene on a Graphite Substrate. Soft Matter 2008, 4, 1041–1047. 10.1039/b718402f. [DOI] [PubMed] [Google Scholar]
- Colazzo L.; Sedona F.; Moretto A.; Casarin M.; Sambi M. Metal-Free on-Surface Photochemical Homocoupling of Terminal Alkynes. J. Am. Chem. Soc. 2016, 138, 10151–10156. 10.1021/jacs.6b03589. [DOI] [PubMed] [Google Scholar]
- Liu M. Z.; Liu M. X.; Zha Z. Q.; Pan J. L.; Qiu X. H.; Li T.; Wang J. B.; Zheng Y.; Zhong D. Y. Thermally Induced Transformation of Nonhexagonal Carbon Rings in Graphene-like Nanoribbons. J. Phys. Chem. C 2018, 122, 9586–9592. 10.1021/acs.jpcc.8b02565. [DOI] [Google Scholar]
- Wang X. Y.; Urgel J. I.; Barin G. B.; Eimre K.; Di Giovannantonio M.; Milani A.; Tommasini M.; Pignedoli C. A.; Ruffieux P.; Feng X. L.; et al. Bottom-Up Synthesis of Heteroatom-Doped Chiral Graphene Nanoribbons. J. Am. Chem. Soc. 2018, 140, 9104–9107. 10.1021/jacs.8b06210. [DOI] [PubMed] [Google Scholar]
- Wang T.; Lv H.; Fan Q.; Feng L.; Wu X.; Zhu J. Highly Selective Synthesis of cis-Enediynes on a Ag(111) Surface. Angew. Chem., Int. Ed. 2017, 56, 4762–4766. 10.1002/anie.201701142. [DOI] [PubMed] [Google Scholar]
- Tran B. V.; Pham T. A.; Grunst M.; Kivala M.; Stohr M. Surface-Confined 2 + 2 Cycloaddition Towards One-Dimensional Polymers Featuring Cyclobutadiene Units. Nanoscale 2017, 9, 18305–18310. 10.1039/C7NR06187K. [DOI] [PubMed] [Google Scholar]
- Bronner C.; Bjork J.; Tegeder P. Tracking and Removing Br during the On-Surface Synthesis of a Graphene Nanoribbon. J. Phys. Chem. C 2015, 119, 486–493. 10.1021/jp5106218. [DOI] [Google Scholar]
- Ammon M.; Sander T.; Maier S. On-Surface Synthesis of Porous Carbon Nanoribbons from Polymer Chains. J. Am. Chem. Soc. 2017, 139, 12976–12984. 10.1021/jacs.7b04783. [DOI] [PubMed] [Google Scholar]
- Zhou X.; Bebensee F.; Yang M.; Bebensee R.; Cheng F.; He Y.; Shen Q.; Shang J.; Liu Z.; Besenbacher F.; et al. Steering Surface Reaction at Specific Sites with Self-Assembly Strategy. ACS Nano 2017, 11, 9397–9404. 10.1021/acsnano.7b04900. [DOI] [PubMed] [Google Scholar]
- Zhou X.; Wang C. G.; Zhang Y. J.; Cheng F.; He Y.; Shen Q.; Shang J.; Shao X.; Ji W.; Chen W.; et al. Steering Surface Reaction Dynamics with a Self-Assembly Strategy: Ullmann Coupling on Metal Surfaces. Angew. Chem., Int. Ed. 2017, 56, 12852–12856. 10.1002/anie.201705018. [DOI] [PubMed] [Google Scholar]
- Xue J. D.; Xu J.; Hu F. Y.; Liao L. Y.; Li M.; Duan W. B.; Zeng Q. D.; Wang C. Highly Efficient Photodimerization of Olefins in a Nanotemplate on HOPG by Scanning Tunneling Microscopy. Phys. Chem. Chem. Phys. 2014, 16, 25765–25769. 10.1039/C4CP04154B. [DOI] [PubMed] [Google Scholar]
- Judd C. J.; Champness N. R.; Saywell A. An On-Surface Reaction Confined within a Porous Molecular Template. Chem. - Eur. J. 2018, 24, 56–61. 10.1002/chem.201704693. [DOI] [PubMed] [Google Scholar]
- Liu X. H.; Wang D.; Wan L. J. Surface Tectonics of Nanoporous Networks of Melamine-Capped Molecular Building Blocks formed through Interface Schiff-Base Reactions. Chem. - Asian J. 2013, 8, 2466–2470. 10.1002/asia.201300546. [DOI] [PubMed] [Google Scholar]
- Jensen S.; Fruchtl H.; Baddeley C. J. Coupling of Triamines with Diisocyanates on Au(111) Leads to the Formation of Polyurea Networks. J. Am. Chem. Soc. 2009, 131, 16706–16713. 10.1021/ja9043032. [DOI] [PubMed] [Google Scholar]
- Yue J. Y.; Markoulides M.; Regan A. C.; Li S. Y.; Chronakis N.; Gourdon A.; Chen T.; Yan H. J.; Wang D. Construction of 2D Nanoporous Networks by Coupling On-Surface Dynamic Imine Chemistry and Dipole-Stabilized Self-Assembly. Chem. Commun. 2017, 53, 428–431. 10.1039/C6CC08482F. [DOI] [PubMed] [Google Scholar]
- Yu Y. X.; Zheng Y. L.; Lei S. B. From a Two-Dimensional Supramolecular Network to One-Dimensional Covalent Polymer at the Liquid/Solid Interface: Insight into the Role of the Stoichiometric Ratio of the Precursors. J. Phys. Chem. C 2017, 121, 593–599. 10.1021/acs.jpcc.6b11553. [DOI] [Google Scholar]
- Clair S.; Ourdjini O.; Abel M.; Porte L. Tip- or Electron Beam-Induced Surface Polymerization. Chem. Commun. 2011, 47, 8028. 10.1039/c1cc12065d. [DOI] [PubMed] [Google Scholar]
- Peyrot D.; Silly F. On-Surface Synthesis of Two-Dimensional Covalent Organic Structures versus Halogen-Bonded Self-Assembly: Competing Formation of Organic Nanoarchitectures. ACS Nano 2016, 10, 5490–5498. 10.1021/acsnano.6b01938. [DOI] [PubMed] [Google Scholar]
- Rastgoo-Lahrood A.; Bjork J.; Heckl W. M.; Lackinger M. 1,3-Diiodobenzene on Cu(111) - an Exceptional Case of On-Surface Ullmann Coupling. Chem. Commun. 2015, 51, 13301–13304. 10.1039/C5CC04453G. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Fernandez J.; Schmidt S. B.; Lauritsen J. V. Sulfur-Driven Switching of the Ullmann Coupling on Au(111). Chem. Commun. 2018, 54, 3621–3624. 10.1039/C8CC01007B. [DOI] [PubMed] [Google Scholar]
- Cai Z. Y.; Liu M. Z.; She L. M.; Li X. L.; Lee J.; Yao D. X.; Zhang H. M.; Chi L. F.; Fuchs H.; Zhong D. Y. Linear Alkane CC Bond Chemistry Mediated by Metal Surfaces. ChemPhysChem 2015, 16, 1356–1360. 10.1002/cphc.201500097. [DOI] [PubMed] [Google Scholar]
- Pinardi A. L.; Otero-Irurueta G.; Palacio I.; Martinez J. I.; Sanchez-Sanchez C.; Tello M.; Rogero C.; Cossaro A.; Preobrajenski A.; Gomez-Lor B.; et al. Tailored Formation of N-Doped Nanoarchitectures by Diffusion-Controlled on-Surface (Cyclo)-Dehydrogenation of Heteroaromatics. ACS Nano 2013, 7, 3676–3684. 10.1021/nn400690e. [DOI] [PubMed] [Google Scholar]
- Sanchez-Valencia J. R.; Dienel T.; Groning O.; Shorubalko I.; Mueller A.; Jansen M.; Amsharov K.; Ruffieux P.; Fasel R. Controlled Synthesis of Single-Chirality Carbon Nanotubes. Nature 2014, 512, 61–64. 10.1038/nature13607. [DOI] [PubMed] [Google Scholar]
- Pinardi A. L.; Martinez J. I.; Jancarik A.; Stara I. G.; Stary I.; Lopez M. F.; Mendez J.; Martin-Gago J. A. Sequential Formation of N-Doped Nanohelicenes, Nanographenes and Nanodomes by Surface-Assisted Chemical (Cyclo)Dehydrogenation of Heteroaromatics. Chem. Commun. 2014, 50, 1555–1557. 10.1039/c3cc47399f. [DOI] [PubMed] [Google Scholar]
- Otero-Irurueta G.; Martinez J. I.; Bueno R. A.; Palomares F. J.; Salavagione H. J.; Singh M. K.; Mendez J.; Ellis G. J.; Lopez M. F.; Martin-Gago J. A. Adsorption and Coupling of 4-Aminophenol on Pt(111) Surfaces. Surf. Sci. 2016, 646, 5–12. 10.1016/j.susc.2015.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. X.; Jin Q.; Shu C. H.; Hua X.; Long Y. T.; Liu P. N. Dehydrogenative Homocoupling of Tetrafluoro-Benzene on Pd(111) via Para-Selective C-H Activation. Chem. Commun. 2017, 53, 6347–6350. 10.1039/C7CC01476G. [DOI] [PubMed] [Google Scholar]
- Larrea C. R.; Baddeley C. J. Fabrication of a High-Quality, Porous, Surface-Confined Covalent Organic Framework on a Reactive Metal Surface. ChemPhysChem 2016, 17, 971–975. 10.1002/cphc.201600027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens T. V. W.; Volkening S.; Zambelli T.; Wintterlin J. Direct Observation of Surface Reactions of Acetylene on Pd(111) with Scanning Tunneling Microscopy. J. Phys. Chem. B 1998, 102, 6521–6528. 10.1021/jp981383e. [DOI] [Google Scholar]
- Tautz F. S. Structure and Bonding of Large Aromatic Molecules on Noble Metal Surfaces: The Example of PTCDA. Prog. Surf. Sci. 2007, 82, 479–520. 10.1016/j.progsurf.2007.09.001. [DOI] [Google Scholar]
- Hammer B.; Norskov J. K. Why Gold is the Noblest of all the Metals. Nature 1995, 376, 238–240. 10.1038/376238a0. [DOI] [Google Scholar]
- Whitelam S.; Tamblyn I.; Haxton T. K.; Wieland M. B.; Champness N. R.; Garrahan J. P.; Beton P. H. Common Physical Framework Explains Phase Behavior and Dynamics of Atomic, Molecular, and Polymeric Network Formers. Phys. Rev. X 2014, 4, 011044. 10.1103/PhysRevX.4.011044. [DOI] [Google Scholar]
- Dong L.; Wang S.; Wang W.; Chen C.; Lin T.; Adisoejoso J.; Lin N.. Transition Metals Trigger On-Surface Ullmann Coupling Reaction: Intermediate, Catalyst and Template. In On-Surface Synthesis; Advances in Atom and Single Molecule Machines; Gourdon S., Ed.; Springer-Verlag: Berlin, 2016; pp 23–42. 10.1007/978-3-319-26600-8_2 [DOI] [Google Scholar]
- Pham T. A.; Song F.; Nguyen M. T.; Li Z. S.; Studener F.; Stohr M. Comparing Ullmann Coupling on Noble Metal Surfaces: On-Surface Polymerization of 1,3,6,8-Tetrabromopyrene on Cu(111) and Au(111). Chem. - Eur. J. 2016, 22, 5937–5944. 10.1002/chem.201504946. [DOI] [PubMed] [Google Scholar]
- Walch H.; Gutzler R.; Sirtl T.; Eder G.; Lackinger M. Material- and Orientation-Dependent Reactivity for Heterogeneously Catalyzed Carbon-Bromine Bond Homolysis. J. Phys. Chem. C 2010, 114, 12604–12609. 10.1021/jp102704q. [DOI] [Google Scholar]
- Gutzler R.; Walch H.; Eder G.; Kloft S.; Heckl W. M.; Lackinger M. Surface Mediated Synthesis of 2D Covalent Organic Frameworks: 1,3,5-Tris(4-Bromophenyl)Benzene on Graphite(001), Cu(111), and Ag(110). Chem. Commun. 2009, 45, 4456–4458. 10.1039/b906836h. [DOI] [PubMed] [Google Scholar]
- Simonov K. A.; Generalov A. V.; Vinogradov A. S.; Svirskiy G. I.; Cafolla A. A.; McGuinness C.; Taketsugu T.; Lyalin A.; Mårtensson N.; Preobrajenski A. B. Synthesis of Armchair Graphene Nanoribbons from the 10,10’-Dibromo-9,9′-Bianthracene Molecules on Ag(111): the Role of Organometallic Intermediates. Sci. Rep. 2018, 8, 3506. 10.1038/s41598-018-21704-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Bernardo I.; Hines P.; Abyazisani M.; Motta N.; MacLeod J.; Lipton-Duffin J. On-Surface Synthesis of Polyethylenedioxythiophene. Chem. Commun. 2018, 54, 3723–3726. 10.1039/C8CC01465E. [DOI] [PubMed] [Google Scholar]
- Ren J.; Larkin E.; Delaney C.; Song Y.; Jin X.; Amirjalayer S.; Bakker A.; Du S.; Gao H.; Zhang Y.-Y.; et al. Chemistry of 4-[(4-Bromophenyl)Ethynyl]Pyridine at Metal Surfaces Studied by STM. Chem. Commun. 2018, 54, 9305–9308. 10.1039/C8CC03986K. [DOI] [PubMed] [Google Scholar]
- Saywell A.; Gren W.; Franc G.; Gourdon A.; Bouju X.; Grill L. Manipulating the Conformation of Single Organometallic Chains on Au(111). J. Phys. Chem. C 2014, 118, 1719–1728. 10.1021/jp409323g. [DOI] [Google Scholar]
- Liu J.; Ruffieux P.; Feng X. L.; Mullen K.; Fasel R. Cyclotrimerization of Arylalkynes on Au(111). Chem. Commun. 2014, 50, 11200–11203. 10.1039/C4CC02859G. [DOI] [PubMed] [Google Scholar]
- Gao H. Y.; Franke J. H.; Wagner H.; Zhong D. Y.; Held P. A.; Studer A.; Fuchs H. Effect of Metal Surfaces in On-Surface Glaser Coupling. J. Phys. Chem. C 2013, 117, 18595–18602. 10.1021/jp406858p. [DOI] [Google Scholar]
- Li Q.; Yang B.; Lin H. P.; Aghdassi N.; Miao K. J.; Zhang J. J.; Zhang H. M.; Li Y. Y.; Duhm S.; Fan J.; et al. Surface-Controlled Mono/Diselective ortho C-H Bond Activation. J. Am. Chem. Soc. 2016, 138, 2809–2814. 10.1021/jacs.5b13286. [DOI] [PubMed] [Google Scholar]
- Gutzler R.; Cardenas L.; Lipton-Duffin J.; El Garah M.; Dinca L. E.; Szakacs C. E.; Fu C. Y.; Gallagher M.; Vondracek M.; Rybachuk M.; et al. Ullmann-Type Coupling of Brominated Tetrathienoanthracene on Copper and Silver. Nanoscale 2014, 6, 2660–2668. 10.1039/C3NR05710K. [DOI] [PubMed] [Google Scholar]
- Makarova M. V.; Okawa Y.; Verveniotis E.; Watanabe K.; Taniguchi T.; Joachim C.; Aono M. Self-Assembled Diacetylene Molecular Wire Polymerization on an Insulating Hexagonal Boron Nitride (0001) surface. Nanotechnology 2016, 27, 395303. 10.1088/0957-4484/27/39/395303. [DOI] [PubMed] [Google Scholar]
- Kolmer M.; Zuzak R.; Zebari A. A. A.; Godlewski S.; Prauzner-Bechcicki J. S.; Piskorz W.; Zasada F.; Sojka Z.; Bleger D.; Hecht S.; et al. On-Surface Polymerization on a Semiconducting Oxide: Aryl Halide Coupling Controlled by Surface Hydroxyl Groups on Rutile TiO2(011). Chem. Commun. 2015, 51, 11276–11279. 10.1039/C5CC02989A. [DOI] [PubMed] [Google Scholar]
- Simonov K. A.; Vinogradov N. A.; Vinogradov A. S.; Generalov A. V.; Zagrebina E. M.; Svirskiy G. I.; Cafolla A. A.; Carpy T.; Cunniffe J. P.; Taketsugu T.; et al. From Graphene Nanoribbons on Cu(111) to Nanographene on Cu(110): Critical Role of Substrate Structure in the Bottom-Up Fabrication Strategy. ACS Nano 2015, 9, 8997–9011. 10.1021/acsnano.5b03280. [DOI] [PubMed] [Google Scholar]
- Dai J. Y.; Fan Q. T.; Wang T.; Kuttner J.; Hilt G.; Gottfried J. M.; Zhu J. F. The Role of the Substrate Structure in the On-Surface Synthesis of Organometallic and Covalent Oligophenylene Chains. Phys. Chem. Chem. Phys. 2016, 18, 20627–20634. 10.1039/C6CP03551E. [DOI] [PubMed] [Google Scholar]
- Koch M.; Gille M.; Hecht S.; Grill L. Steering a Cycloaddition Reaction via the Surface Structure. Surf. Sci. 2018, 678, 194–200. 10.1016/j.susc.2018.05.014. [DOI] [Google Scholar]
- Kalashnyk N.; Salomon E.; Mun S. H.; Jung J.; Giovanelli L.; Angot T.; Dumur F.; Gigmes D.; Clair S. The Orientation of Silver Surfaces Drives the Reactivity and the Selectivity in Homo-Coupling Reactions. ChemPhysChem 2018, 19, 1802–1808. 10.1002/cphc.201800406. [DOI] [PubMed] [Google Scholar]
- Richter A.; Haapasilta V.; Venturini C.; Bechstein R.; Gourdon A.; Foster A. S.; Kuhnle A. Diacetylene Ppolymerization on a Bulk Insulator Surface. Phys. Chem. Chem. Phys. 2017, 19, 15172–15176. 10.1039/C7CP01526G. [DOI] [PubMed] [Google Scholar]
- Lipton-Duffin J. A.; Miwa J. A.; Kondratenko M.; Cicoira F.; Sumpter B. G.; Meunier V.; Perepichka D. F.; Rosei F. Step-by-Step Growth of Epitaxially Aligned Polythiophene by Surface-Confined Reaction. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 11200–11204. 10.1073/pnas.1000726107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton-Duffin J. A.; Ivasenko O.; Perepichka D. F.; Rosei F. Synthesis of Polyphenylene Molecular Wires by Surface-Confined Polymerization. Small 2009, 5, 592–597. 10.1002/smll.200801943. [DOI] [PubMed] [Google Scholar]
- Cai Z. Y.; She L. M.; Wu L. Q.; Zhong D. Y. On-Surface Synthesis of Linear Polyphenyl Wires Guided by Surface Steric Effect. J. Phys. Chem. C 2016, 120, 6619–6624. 10.1021/acs.jpcc.6b00284. [DOI] [Google Scholar]
- Ruffieux P.; Cai J. M.; Plumb N. C.; Patthey L.; Prezzi D.; Ferretti A.; Molinari E.; Feng X. L.; Mullen K.; Pignedoli C. A.; et al. Electronic Structure of Atomically Precise Graphene Nanoribbons. ACS Nano 2012, 6, 6930–6935. 10.1021/nn3021376. [DOI] [PubMed] [Google Scholar]
- Linden S.; Zhong D.; Timmer A.; Aghdassi N.; Franke J. H.; Zhang H.; Feng X.; Mullen K.; Fuchs H.; Chi L.; et al. Electronic Structure of Spatially Aligned Graphene Nanoribbons on Au(788). Phys. Rev. Lett. 2012, 108, 216801. 10.1103/PhysRevLett.108.216801. [DOI] [PubMed] [Google Scholar]
- Saywell A.; Schwarz J.; Hecht S.; Grill L. Polymerization on Stepped Surfaces: Alignment of Polymers and Identification of Catalytic Sites. Angew. Chem., Int. Ed. 2012, 51, 5096–5100. 10.1002/anie.201200543. [DOI] [PubMed] [Google Scholar]
- Ortega J. E.; Vasseur G.; Piquero-Zulaica I.; Matencio S.; Valbuena M. A.; Rault J. E.; Schiller F.; Corso M.; Mugarza A.; Lobo-Checa J. Structure and Electronic States of Vicinal Ag(111) Surfaces with Densely Kinked Steps. New J. Phys. 2018, 20, 073010. 10.1088/1367-2630/aacbb7. [DOI] [Google Scholar]
- Piquero-Zulaica I.; Garcia-Lekue A.; Colazzo L.; Krug C. K.; Mohammed M. S. G.; Abd El-Fattah Z. M.; Gottfried J. M.; de Oteyza D. G.; Ortega J. E.; Lobo-Checa J. Electronic Structure Tunability by Periodic meta-Ligand Spacing in One-Dimensional Organic Semiconductors. ACS Nano 2018, 12, 10537–10544. 10.1021/acsnano.8b06536. [DOI] [PubMed] [Google Scholar]
- Anggara K.; Leung L.; Timm M. J.; Hu Z. X.; Polanyi J. C. Approaching the Forbidden Fruit of Reaction Dynamics: Aiming Reagent at Selected Impact Parameters. Sci. Adv. 2018, 4, eaau2821 10.1126/sciadv.aau2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno C.; Paradinas M.; Vilas-Varela M.; Panighel M.; Ceballos G.; Pena D.; Mugarza A. On-Surface Synthesis of Superlattice Arrays of Ultra-Long Graphene Nanoribbons. Chem. Commun. 2018, 54, 9402. 10.1039/C8CC04830D. [DOI] [PubMed] [Google Scholar]
- Fan Q. T.; Dai J. Y.; Wang T.; Kuttner J.; Hilt G.; Gottfried J. M.; Zhu J. F. Confined Synthesis of Organometallic Chains and Macrocycles by Cu-O Surface Templating. ACS Nano 2016, 10, 3747–3754. 10.1021/acsnano.6b00366. [DOI] [PubMed] [Google Scholar]
- Clair S.; Ourdjini O.; Abel M.; Porte L. Two-Dimensional Polymer as a Mask for Surface Nanopatterning. Adv. Mater. 2012, 24, 1252. 10.1002/adma.201200063. [DOI] [PubMed] [Google Scholar]
- Cirera B.; Gimenez-Agullo N.; Bjork J.; Martinez-Pena F.; Martin-Jimenez A.; Rodriguez-Fernandez J.; Pizarro A. M.; Otero R.; Gallego J. M.; Ballester P.; et al. Thermal Selectivity of Intermolecular versus Intramolecular Reactions on Surfaces. Nat. Commun. 2016, 7, 11002. 10.1038/ncomms11002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T.; Huang J. M.; Lv H. F.; Fan Q. T.; Feng L.; Tao Z. J.; Ju H. X.; Wu X. J.; Tait S. L.; Zhu J. F. Kinetic Strategies for the Formation of Graphyne Nanowires via Sonogashira Coupling on Ag(111). J. Am. Chem. Soc. 2018, 140, 13421–13428. 10.1021/jacs.8b08477. [DOI] [PubMed] [Google Scholar]
- Lin T.; Zhang L. D.; Bjork J.; Chen Z.; Ruben M.; Barth J. V.; Klappenberger F. Terminal Alkyne Coupling on a Corrugated Noble Metal Surface: From Controlled Precursor Alignment to Selective Reactions. Chem. - Eur. J. 2017, 23, 15588–15593. 10.1002/chem.201701735. [DOI] [PubMed] [Google Scholar]
- Kawai S.; Haapasilta V.; Lindner B. D.; Tahara K.; Spijker P.; Buitendijk J. A.; Pawlak R.; Meier T.; Tobe Y.; Foster A. S.; et al. Thermal Control of Sequential On-Surface Transformation of a Hydrocarbon Molecule on a Copper Surface. Nat. Commun. 2016, 7, 12711. 10.1038/ncomms12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiotari A.; Nakae T.; Iwata K.; Mori S.; Okujima T.; Uno H.; Sakaguchi H.; Sugimoto Y. Strain-Induced Skeletal Rearrangement of a Polycyclic Aromatic Hydrocarbon on a Copper Surface. Nat. Commun. 2017, 8, 16089. 10.1038/ncomms16089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artaud A.; Magaud L.; Ratter K.; Gilles B.; Guisset V.; David P.; Martinez J. I.; Martin-Gago J. A.; Chapelier C.; Coraux J. Size-Selective Carbon Clusters as Obstacles to Graphene Growth on a Metal. Nano Lett. 2018, 18, 4812–4820. 10.1021/acs.nanolett.8b01379. [DOI] [PubMed] [Google Scholar]
- Peyrot D.; Silly M. G.; Silly F. Temperature-Triggered Sequential On-Surface Synthesis of One and Two Covalently Bonded Porous Organic Nanoarchitectures on Au(111). J. Phys. Chem. C 2017, 121, 26815–26821. 10.1021/acs.jpcc.7b08296. [DOI] [Google Scholar]
- Qian C.; Qi Q. Y.; Jiang G. F.; Cui F. Z.; Tian Y.; Zhao X. Toward Covalent Organic Frameworks Bearing Three Different Kinds of Pores: The Strategy for Construction and COF-to-COF Transformation via Heterogeneous Linker Exchange. J. Am. Chem. Soc. 2017, 139, 6736–6743. 10.1021/jacs.7b02303. [DOI] [PubMed] [Google Scholar]
- Yu Y. X.; Lin J. B.; Lei S. B. Dynamic Covalent Chemistry of Imine Polymers at the Liquid/Solid Interface Investigated by Scanning Tunneling Microscopy. RSC Adv. 2017, 7, 11496–11502. 10.1039/C6RA27395E. [DOI] [Google Scholar]
- Sun J.; Lei S. B. Guest-Induced Structural Transformation and Its Effect on the Redistribution of the Surface Confined Combinatorial Libraries. J. Phys. Chem. C 2018, 122, 12315–12321. 10.1021/acs.jpcc.8b03215. [DOI] [Google Scholar]
- Yu L.; Li Z. B.; Wang D. Construction of Boronate Ester Based Single-Layered Covalent Organic Frameworks. Chem. Commun. 2016, 52, 13771–13774. 10.1039/C6CC07399A. [DOI] [PubMed] [Google Scholar]
- Liu C. H.; Yu Y. X.; Zhang W.; Zeng Q. D.; Lei S. B. Room-Temperature Synthesis of Covalent Organic Frameworks with a Boronic Ester Linkage at the Liquid/Solid Interface. Chem. - Eur. J. 2016, 22, 18412–18418. 10.1002/chem.201603547. [DOI] [PubMed] [Google Scholar]
- Ha N. T. N.; Gopakumar T. G.; Yen N. D. C.; Mende C.; Smykalla L.; Schlesinger M.; Buschbeck R.; Ruffer T.; Lang H.; Mehring M.; et al. Ester Formation at the Liquid-Solid Interface. Beilstein J. Nanotechnol. 2017, 8, 2139–2150. 10.3762/bjnano.8.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Z. M.; Yang B. A.; Lin H. P.; Tang Y. Y.; Tang Z. Y.; Zhang J. J.; Zhang H. M.; Li Y. Y.; Xie Y. S.; Li Q.; et al. Structural Variation in Surface-Supported Synthesis by Adjusting the Stoichiometric Ratio of the Reactants. ACS Nano 2016, 10, 4228–4235. 10.1021/acsnano.5b07601. [DOI] [PubMed] [Google Scholar]
- Fan O. T.; Wang T.; Dai J. Y.; Kuttner J.; Hilt G.; Gottfried J. M.; Zhu J. F. On-Surface Pseudo-High-Dilution Synthesis of Macrocycles: Principle and Mechanism. ACS Nano 2017, 11, 5070–5079. 10.1021/acsnano.7b01870. [DOI] [PubMed] [Google Scholar]
- Klaasen H.; Liu L.; Meng X.; Held P. A.; Gao H.-Y.; Barton D.; Mück-Lichtenfeld C.; Neugebauer J.; Fuchs H.; Studer A. Reaction Selectivity in On-Surface Chemistry by Surface Coverage Control - Alkyne Dimerization versus Alkyne Trimerization. Chem. - Eur. J. 2018, 24, 15303–15308. 10.1002/chem.201802848. [DOI] [PubMed] [Google Scholar]
- Dienstmaier J. F.; Gigler A. M.; Goetz A. J.; Knochel P.; Bein T.; Lyapin A.; Reichlmaier S.; Heckl W. M.; Lackinger M. Synthesis of Well-Ordered COF Monolayers: Surface Growth of Nanocrystalline Precursors versus Direct On-Surface Polycondensation. ACS Nano 2011, 5, 9737–9745. 10.1021/nn2032616. [DOI] [PubMed] [Google Scholar]
- Di Giovannantonio M.; Kosmala T.; Bonanni B.; Serrano G.; Zema N.; Turchini S.; Catone D.; Wandelt K.; Pasini D.; Contini G.; et al. Surface-Enhanced Polymerization via Schiff-Base Coupling at the Solid-Water Interface under pH Control. J. Phys. Chem. C 2015, 119, 19228–19235. 10.1021/acs.jpcc.5b05547. [DOI] [Google Scholar]
- Sakaguchi H.; Matsumura H.; Gong H. Electrochemical Epitaxial Polymerization of Single-Molecular Wires. Nat. Mater. 2004, 3, 551–557. 10.1038/nmat1176. [DOI] [PubMed] [Google Scholar]
- Sakaguchi H.; Matsumura H.; Gong H.; Abouelwafa A. M. Direct Visualization of the Formation of Single-Molecule Conjugated Copolymers. Science 2005, 310, 1002–1006. 10.1126/science.1117990. [DOI] [PubMed] [Google Scholar]
- Yang L. Y. O.; Chang C.; Liu S.; Wu C.; Yau S. L. Direct Visualization of an Aniline Admolecule and its Electropolymerization on Au(111) with In Situ Scanning Tunneling Microscope. J. Am. Chem. Soc. 2007, 129, 8076. 10.1021/ja072201+. [DOI] [PubMed] [Google Scholar]
- Chen S.; Wu C.; Yau S. In Situ Scanning Tunneling Microscopy of the Adsorption and Polymerization of Aniline on Au(111) Electrode in Nitric Acid. J. Electroanal. Chem. 2014, 729, 121–127. 10.1016/j.jelechem.2014.07.003. [DOI] [Google Scholar]
- Spitzer S.; Rastgoo-Lahrood A.; Macknapp K.; Ritter V.; Sotier S.; Heckl W. M.; Lackinger M. Solvent-Free On-Surface Synthesis of Boroxine COF Monolayers. Chem. Commun. 2017, 53, 5147–5150. 10.1039/C7CC01131H. [DOI] [PubMed] [Google Scholar]
- Eigler D. M.; Schweizer E. K. Positioning Single Atoms with a Scanning Tunnelling Microscope. Nature 1990, 344, 524–526. 10.1038/344524a0. [DOI] [Google Scholar]
- Meyer G.; Repp J.; Zöphel S.; Braun K.-F.; Hla S. W.; Fölsch S.; Bartels L.; Moresco F.; Rieder K. H. Controlled Manipulation of Atoms and Small Molecules with a Low Temperature Scanning Tunneling Microscope. Single Mol. 2000, 1, 79. 10.1002/(SICI)1438-5171(200004)1:1<79::AID-SIMO79>3.0.CO;2-R. [DOI] [Google Scholar]
- Hla S.-W.; Rieder K.-H. STM Control of Chemical Reactions: Single-Molecule Synthesis. Annu. Rev. Phys. Chem. 2003, 54, 307–330. 10.1146/annurev.physchem.54.011002.103852. [DOI] [PubMed] [Google Scholar]
- Pavlicek N.; Gawel P.; Kohn D. R.; Majzik Z.; Xiong Y. Y.; Meyer G.; Anderson H. L.; Gross L. Polyyne Formation via Skeletal Rearrangement Induced by Atomic Manipulation. Nat. Chem. 2018, 10, 853–858. 10.1038/s41557-018-0067-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinca L. E.; MacLeod J. M.; Lipton-Duffin J.; Fu C.; Ma D.; Perepichka D. F.; Rosei F. Tip-Induced C-H Activation and Oligomerization of Thienoanthracenes. Chem. Commun. 2014, 50, 8791–8793. 10.1039/C4CC03719G. [DOI] [PubMed] [Google Scholar]
- Rosa L. G.; Liang J. Atomic Force Microscope Nanolithography: Dip-pen, Nanoshaving, Nanografting, Tapping Mode, Electrochemical and Thermal Nanolithography. J. Phys.: Condens. Matter 2009, 21, 483001. 10.1088/0953-8984/21/48/483001. [DOI] [PubMed] [Google Scholar]
- Verstraete L.; Hirsch B. E.; Greenwood J.; De Feyter S. Confined Polydiacetylene Polymerization Reactions for Programmed Length Control. Chem. Commun. 2017, 53, 4207–4210. 10.1039/C7CC00885F. [DOI] [PubMed] [Google Scholar]
- Verstraete L.; Smart J.; Hirsch B. E.; De Feyter S. Unidirectional Supramolecular Self-Assembly inside Nanocorrals via In Situ STM Nanoshaving. Phys. Chem. Chem. Phys. 2018, 20, 27482–27489. 10.1039/C8CP05316B. [DOI] [PubMed] [Google Scholar]
- Bilbao N.; Yu Y. X.; Verstraete L.; Lin J. B.; Lei S. B.; De Feyter S. The Impact of Grafted Surface Defects on the On-Surface Schiff-Base Chemistry at the Solid-Liquid Interface. Chem. Commun. 2018, 54, 9905–9908. 10.1039/C8CC03867H. [DOI] [PubMed] [Google Scholar]
- Mesquita V.; Botton J.; Valyaev D. A.; François C.; Patrone L.; Balaban T. S.; Abel M.; Parrain J. L.; Chuzel O.; Clair S. Catalytic Scanning Probe Nanolithography (cSPL): Control of the AFM Parameters in Order to Achieve Sub-100-nm Spatially Resolved Epoxidation of Alkenes Grafted onto a Surface. Langmuir 2016, 32, 4034–4042. 10.1021/acs.langmuir.6b00543. [DOI] [PubMed] [Google Scholar]
- Garcia R.; Knoll A. W.; Riedo E. Advanced Scanning Probe Lithography. Nat. Nanotechnol. 2014, 9, 577–587. 10.1038/nnano.2014.157. [DOI] [PubMed] [Google Scholar]
- Carnally S. A. M.; Wong L. S. Harnessing Catalysis to Enhance Scanning Probe Nanolithography. Nanoscale 2014, 6, 4998–5007. 10.1039/C4NR00618F. [DOI] [PubMed] [Google Scholar]
- Muller W. T.; Klein D. L.; Lee T.; Clarke J.; McEuen P. L.; Schultz P. G. A Strategy for the Chemical Synthesis of Nanostructures. Science 1995, 268, 272–273. 10.1126/science.268.5208.272. [DOI] [PubMed] [Google Scholar]
- Blasdel L. K.; Banerjee S.; Wong S. S. Selective Borohydride Reduction using Functionalized Atomic Force Microscopy Tips. Langmuir 2002, 18, 5055–5057. 10.1021/la0258695. [DOI] [Google Scholar]
- Peter M.; Li X. M.; Huskens J.; Reinhoudt D. N. Catalytic Probe Lithography: Catalyst-Functionalized Scanning Probes as Nanopens for Nanofabrication on Self-Assembled Monolayers. J. Am. Chem. Soc. 2004, 126, 11684–11690. 10.1021/ja048027w. [DOI] [PubMed] [Google Scholar]
- Blackledge C.; Engebretson D. A.; McDonald J. D. Nanoscale Site-Selective Catalysis of Surface Assemblies by Palladium-Coated Atomic Force Microscopy Tips: Chemical Lithography without Electrical Current. Langmuir 2000, 16, 8317–8323. 10.1021/la000674n. [DOI] [Google Scholar]
- Davis J. J.; Coleman K. S.; Busuttil K. L.; Bagshaw C. B. Spatially Resolved Suzuki Coupling Reaction Initiated and Controlled Using a Catalytic AFM Probe. J. Am. Chem. Soc. 2005, 127, 13082–13083. 10.1021/ja043235+. [DOI] [PubMed] [Google Scholar]
- Davis J. J.; Bagshaw C. B.; Busuttil K. L.; Hanyu Y.; Coleman K. S. Spatially Controlled Suzuki and Heck Catalytic Molecular Coupling. J. Am. Chem. Soc. 2006, 128, 14135–14141. 10.1021/ja064840a. [DOI] [PubMed] [Google Scholar]
- Paxton W. F.; Spruell J. M.; Stoddart J. F. Heterogeneous Catalysis of a Copper-Coated Atomic Force Microscopy Tip for Direct-Write Click Chemistry. J. Am. Chem. Soc. 2009, 131, 6692–6694. 10.1021/ja9015974. [DOI] [PubMed] [Google Scholar]
- Zhang K.; Fu Q.; Pan N.; Yu X. X.; Liu J. Y.; Luo Y.; Wang X. P.; Yang J. L.; Hou J. G. Direct Writing of Electronic Devices on Graphene Oxide by Catalytic Scanning Probe Lithography. Nat. Commun. 2012, 3, 1194. 10.1038/ncomms2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valyaev D. A.; Clair S.; Patrone L.; Abel M.; Porte L.; Chuzel O.; Parrain J. L. Grafting a Homogeneous Transition Metal Catalyst onto a Silicon AFM Probe: a Promising Strategy for Chemically Constructive Nanolithography. Chem. Sci. 2013, 4, 2815–2821. 10.1039/c3sc50979f. [DOI] [Google Scholar]
- Botton J.; Gratzer K.; François C.; Mesquita V.; Patrone L.; Balaban T. S.; Clair S.; Parrain J. L.; Chuzel O. Spatially Resolved Acyl Transfer on Surface by Organo-Catalytic Scanning Probe Nanolithography (o-cSPL). Chem. Sci. 2018, 9, 4280–4284. 10.1039/C8SC00294K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosford J.; Valles M.; Krainer F. W.; Glieder A.; Wong L. S. Parallelized Biocatalytic Scanning Probe Lithography for the Additive Fabrication of Conjugated Polymer Structures. Nanoscale 2018, 10, 7185–7193. 10.1039/C8NR01283K. [DOI] [PubMed] [Google Scholar]
- Dai H. L.; Geng Y. F.; Zeng Q. D.; Wang C. Photo-Regulation of 2D Supramolecular Self-Assembly: On-Surface Photochemistry Studied by STM. Chin. Chem. Lett. 2017, 28, 729–737. 10.1016/j.cclet.2016.09.018. [DOI] [Google Scholar]
- Guo C.; Li M.; Kang S. Z. Photochemical Reactions in Self-Assembled Organic Monolayers Characterized by using Scanning Tunneling Microscopy. ChemPhysChem 2016, 17, 802–811. 10.1002/cphc.201501018. [DOI] [PubMed] [Google Scholar]
- Frath D.; Yokoyama S.; Hirose T.; Matsuda K. Photoresponsive Supramolecular Self-Assemblies at the Liquid/Solid Interface. J. Photochem. Photobiol., C 2018, 34, 29–40. 10.1016/j.jphotochemrev.2017.12.005. [DOI] [Google Scholar]
- Endo O.; Furuta T.; Ozaki H.; Sonoyama M.; Mazaki Y. Structures of 17,19-Hexatriacontadiyne Monolayers on Au(111) Studied by Infrared Reflection Absorption Spectroscopy and Scanning Tunneling Microscopy. J. Phys. Chem. B 2006, 110, 13100–13106. 10.1021/jp055161+. [DOI] [PubMed] [Google Scholar]
- Takajo D.; Inaba A.; Sudoh K. Two-Dimensional Solid-State Topochemical Reactions of 10,12-Pentacosadiyn-1-ol Adsorbed on Graphite. Langmuir 2014, 30, 2738–2744. 10.1021/la4049314. [DOI] [PubMed] [Google Scholar]
- Giridharagopal R.; Kelly K. F. Substrate-Dependent Properties of Polydiacetylene Nanowires on Graphite and MoS2. ACS Nano 2008, 2, 1571–1580. 10.1021/nn800287x. [DOI] [PubMed] [Google Scholar]
- Zhang X. M.; Xu S. D.; Li M.; Shen Y. T.; Wei Z. Q.; Wang S.; Zeng Q. D.; Wang C. Photo-Induced Polymerization and Isomerization on the Surface Observed by Scanning Tunneling Microscopy. J. Phys. Chem. C 2012, 116, 8950–8955. 10.1021/jp2115884. [DOI] [Google Scholar]
- Qiao Y. H.; Zeng Q. D.; Tan Z. Y.; Xu S. D.; Wang D.; Wang C.; Wan L. J.; Bai C. L. Photoinduced Organic Nanowires from Self-Assembled Monolayers. J. Vac. Sci. Technol., B: Microelectron. Process. Phenom. 2002, 20, 2466–2469. 10.1116/1.1526601. [DOI] [Google Scholar]
- Deshpande A.; Sham C. H.; Alaboson J. M. P.; Mullin J. M.; Schatz G. C.; Hersam M. C. Self-Assembly and Photopolymerization of Sub-2 nm One-Dimensional Organic Nanostructures on Graphene. J. Am. Chem. Soc. 2012, 134, 16759–16764. 10.1021/ja307061e. [DOI] [PubMed] [Google Scholar]
- Liao L. Y.; Li Y. B.; Zhang X. M.; Geng Y. F.; Zhang J. Y.; Xie J. L.; Zeng Q. D.; Wang C. STM Investigation of the Photoisomerization and Photodimerization of Stilbene Derivatives on HOPG Surface. J. Phys. Chem. C 2014, 118, 15963–15969. 10.1021/jp505511e. [DOI] [Google Scholar]
- Yang G. Z.; Wan L. J.; Zeng Q. D.; Bai C. L. Photodimerization of P2VB on Au(111) in Solution Studied with Scanning Tunneling Microscopy. J. Phys. Chem. B 2003, 107, 5116–5119. 10.1021/jp026270g. [DOI] [Google Scholar]
- Abdel-Mottaleb M. M. S.; De Feyter S.; Gesquiere A.; Sieffert M.; Klapper M.; Mullen K.; De Schryver F. C. Photodimerization of Cinnamate Derivatives Studied by STM. Nano Lett. 2001, 1, 353–359. 10.1021/nl010034k. [DOI] [Google Scholar]
- Liu C. H.; Zhang W.; Zeng Q. D.; Lei S. B. A Photoresponsive Surface Covalent Organic Framework: Surface-Confined Synthesis, Isomerization, and Controlled Guest Capture and Release. Chem. - Eur. J. 2016, 22, 6768–6773. 10.1002/chem.201601199. [DOI] [PubMed] [Google Scholar]
- Kikkawa Y.; Kihara H.; Takahashi M.; Kanesato M.; Balaban T. S.; Lehn J. M. Two-Dimensional Structures of Anthracene Derivatives: Photodimerization and Host-Guest Chemistry. J. Phys. Chem. B 2010, 114, 16718–16722. 10.1021/jp108069a. [DOI] [PubMed] [Google Scholar]
- Lindner R.; Rahe P.; Kittelmann M.; Gourdon A.; Bechstein R.; Kuhnle A. Substrate Templating Guides the Photoinduced Reaction of C-60 on Calcite. Angew. Chem., Int. Ed. 2014, 53, 7952–7955. 10.1002/anie.201309128. [DOI] [PubMed] [Google Scholar]
- Shen Q.; He J. H.; Zhang J. L.; Wu K.; Xu G. Q.; Wee A. T. S.; Chen W. Self-Assembled Two-Dimensional Nanoporous Molecular Arrays and Photoinduced Polymerization of 4-Bromo-4 ’-Hydroxybiphenyl on Ag(111). J. Chem. Phys. 2015, 142, 101902. 10.1063/1.4906116. [DOI] [PubMed] [Google Scholar]
- Basagni A.; Ferrighi L.; Cattelan M.; Nicolas L.; Handrup K.; Vaghi L.; Papagni A.; Sedona F.; Di Valentin C.; Agnoli S.; et al. On-Surface Photo-Dissociation of C-Br Bonds: Towards Room Temperature Ullmann Coupling. Chem. Commun. 2015, 51, 12593–12596. 10.1039/C5CC04317D. [DOI] [PubMed] [Google Scholar]
- Basagni A.; Colazzo L.; Sedona F.; Di Marino M.; Carofiglio T.; Lubian E.; Forrer D.; Vittadini A.; Casarin M.; Verdini A.; et al. Stereoselective Photopolymerization of Tetraphenylporphyrin Derivatives on Ag(110) at the Sub-Monolayer Level. Chem. - Eur. J. 2014, 20, 14296–14304. 10.1002/chem.201403208. [DOI] [PubMed] [Google Scholar]
- Gao H. Y.; Zhong D. Y.; Moenig H.; Wagner H.; Held P. A.; Timmer A.; Studer A.; Fuchs H. Photochemical Glaser Coupling at Metal Surfaces. J. Phys. Chem. C 2014, 118, 6272–6277. 10.1021/jp411889e. [DOI] [Google Scholar]
- Mazzamuto F.; Nguyen V. H.; Apertet Y.; Caer C.; Chassat C.; Saint-Martin J.; Dollfus P. Enhanced Thermoelectric Properties in Graphene Nanoribbons by Resonant Tunneling of Electrons. Phys. Rev. B: Condens. Matter Mater. Phys. 2011, 83, 235426. 10.1103/PhysRevB.83.235426. [DOI] [Google Scholar]
- Sevincli H.; Sevik C.; Cagin T.; Cuniberti G. A Bottom-Up Route to Enhance Thermoelectric Figures of Merit in Graphene Nanoribbons. Sci. Rep. 2013, 3, 1228. 10.1038/srep01228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Raji A. R. O.; Tour J. M. Graphene-Wrapped MnO2-Graphene Nanoribbons as Anode Materials for High-Performance Lithium Ion Batteries. Adv. Mater. 2013, 25, 6298–6302. 10.1002/adma.201302915. [DOI] [PubMed] [Google Scholar]
- Liu M. K.; Tjiu W. W.; Pan J. S.; Zhang C.; Gao W.; Liu T. X. One-Step Synthesis of Graphene Nanoribbon-MnO2 Hybrids and their All-Solid-State Asymmetric Supercapacitors. Nanoscale 2014, 6, 4233–4242. 10.1039/c3nr06650a. [DOI] [PubMed] [Google Scholar]
- Li L.; Raji A. R. O.; Fei H. L.; Yang Y.; Samuel E. L. G.; Tour J. M. Nanocomposite of Polyaniline Nanorods Grown on Graphene Nanoribbons for Highly Capacitive Pseudocapacitors. ACS Appl. Mater. Interfaces 2013, 5, 6622–6627. 10.1021/am4013165. [DOI] [PubMed] [Google Scholar]
- Freitag M.; Low T.; Zhu W. J.; Yan H. G.; Xia F. N.; Avouris P. Photocurrent in Graphene Harnessed by Tunable Intrinsic Plasmons. Nat. Commun. 2013, 4, 1951. 10.1038/ncomms2951. [DOI] [PubMed] [Google Scholar]
- Pour M. M.; Lashkov A.; Radocea A.; Liu X. M.; Sun T.; Lipatov A.; Korlacki R. A.; Shekhirev M.; Aluru N. R.; Lyding J. W.; et al. Laterally Extended Atomically Precise Graphene Nanoribbons with Improved Electrical Conductivity for Efficient Gas Sensing. Nat. Commun. 2017, 8, 820. 10.1038/s41467-017-00692-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwierz F. Graphene Transistors. Nat. Nanotechnol. 2010, 5, 487–496. 10.1038/nnano.2010.89. [DOI] [PubMed] [Google Scholar]
- Kang J. H.; Sarkar D.; Khatami Y.; Banerjee K. Proposal for All-Graphene Monolithic Logic Circuits. Appl. Phys. Lett. 2013, 103, 083113. 10.1063/1.4818462. [DOI] [Google Scholar]
- Wakabayashi K.; Sasaki K.; Nakanishi T.; Enoki T. Electronic States of Graphene Nanoribbons and Analytical Solutions. Sci. Technol. Adv. Mater. 2010, 11, 054504. 10.1088/1468-6996/11/5/054504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radocea A.; Sun T.; Vo T. H.; Sinitskii A.; Aluru N. R.; Lyding J. W. Solution-Synthesized Chevron Graphene Nanoribbons Exfoliated onto H:Si(100). Nano Lett. 2017, 17, 170–178. 10.1021/acs.nanolett.6b03709. [DOI] [PubMed] [Google Scholar]
- Vo T. H.; Shekhirev M.; Kunkel D. A.; Morton M. D.; Berglund E.; Kong L. M.; Wilson P. M.; Dowben P. A.; Enders A.; Sinitskii A. Large-Scale Solution Synthesis of Narrow Graphene Nanoribbons. Nat. Commun. 2014, 5, 3189. 10.1038/ncomms4189. [DOI] [PubMed] [Google Scholar]
- Schwab M. G.; Narita A.; Hernandez Y.; Balandina T.; Mali K. S.; De Feyter S.; Feng X. L.; Mullen K. Structurally Defined Graphene Nanoribbons with High Lateral Extension. J. Am. Chem. Soc. 2012, 134, 18169–18172. 10.1021/ja307697j. [DOI] [PubMed] [Google Scholar]
- Abbas A. N.; Liu G.; Narita A.; Orosco M.; Feng X. L.; Mullen K.; Zhou C. W. Deposition, Characterization, and Thin-Film-Based Chemical Sensing of Ultra-long Chemically Synthesized Graphene Nanoribbons. J. Am. Chem. Soc. 2014, 136, 7555–7558. 10.1021/ja502764d. [DOI] [PubMed] [Google Scholar]
- Joshi D.; Hauser M.; Veber G.; Berl A.; Xu K.; Fischer F. R. Super-Resolution Imaging of Clickable Graphene Nanoribbons Decorated with Fluorescent Dyes. J. Am. Chem. Soc. 2018, 140, 9574–9580. 10.1021/jacs.8b04679. [DOI] [PubMed] [Google Scholar]
- Björk J.; Stafstrom S.; Hanke F. Zipping Up: Cooperativity Drives the Synthesis of Graphene Nanoribbons. J. Am. Chem. Soc. 2011, 133, 14884–14887. 10.1021/ja205857a. [DOI] [PubMed] [Google Scholar]
- Blankenburg S.; Cai J. M.; Ruffieux P.; Jaafar R.; Passerone D.; Feng X. L.; Mullen K.; Fasel R.; Pignedoli C. A. Intraribbon Heterojunction Formation in Ultranarrow Graphene Nanoribbons. ACS Nano 2012, 6, 2020–2025. 10.1021/nn203129a. [DOI] [PubMed] [Google Scholar]
- Jacobse P. H.; van den Hoogenband A.; Moret M. E.; Gebbink R. J. M. K.; Swart I. Aryl Radical Geometry Determines Nanographene Formation on Au(111). Angew. Chem., Int. Ed. 2016, 55, 13052–13055. 10.1002/anie.201606440. [DOI] [PubMed] [Google Scholar]
- de Oteyza D. G.; Garcia-Lekue A.; Vilas-Varela M.; Merino-Diez N.; Carbonell-Sanroma E.; Corso M.; Vasseur G.; Rogero C.; Guitian E.; Pascual J. I.; et al. Substrate-Independent Growth of Atomically Precise Chiral Graphene Nanoribbons. ACS Nano 2016, 10, 9000–9008. 10.1021/acsnano.6b05269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino-Diez N.; Li J. C.; Garcia-Lekue A.; Vasseur G.; Vilas-Varela M.; Carbonell-Sanroma E.; Corso M.; Ortega J. E.; Pena D.; Pascual J. I.; et al. Unraveling the Electronic Structure of Narrow Atomically Precise Chiral Graphene Nanoribbons. J. Phys. Chem. Lett. 2018, 9, 25–30. 10.1021/acs.jpclett.7b02767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.-Y.; Urgel J. I.; Barin G. B.; Eimre K.; Di Giovannantonio M.; Milani A.; Tommasini M.; Pignedoli C. A.; Ruffieux P.; Feng X.; et al. Bottom-Up Synthesis of Heteroatom-Doped Chiral Graphene Nanoribbons. J. Am. Chem. Soc. 2018, 140, 9104–9107. 10.1021/jacs.8b06210. [DOI] [PubMed] [Google Scholar]
- Chen Y. C.; de Oteyza D. G.; Pedramrazi Z.; Chen C.; Fischer F. R.; Crommie M. F. Tuning the Band Gap of Graphene Nanoribbons Synthesized from Molecular Precursors. ACS Nano 2013, 7, 6123–6128. 10.1021/nn401948e. [DOI] [PubMed] [Google Scholar]
- Zhang H. M.; Lin H. P.; Sun K. W.; Chen L.; Zagranyarski Y.; Aghdassi N.; Duhm S.; Li Q.; Zhong D. Y.; Li Y. Y.; et al. On-Surface Synthesis of Rylene-Type Graphene Nanoribbons. J. Am. Chem. Soc. 2015, 137, 4022–4025. 10.1021/ja511995r. [DOI] [PubMed] [Google Scholar]
- Kimouche A.; Ervasti M. M.; Drost R.; Halonen S.; Harju A.; Joensuu P. M.; Sainio J.; Liljeroth P. Ultra-Narrow Metallic Armchair Graphene Nanoribbons. Nat. Commun. 2015, 6, 10177. 10.1038/ncomms10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdurakhmanova N.; Amsharov N.; Stepanow S.; Jansen M.; Kern K.; Amsharov K. Synthesis of Wide Atomically Precise Graphene Nanoribbons from Para-Oligophenylene Based Molecular Precursor. Carbon 2014, 77, 1187–1190. 10.1016/j.carbon.2014.06.010. [DOI] [Google Scholar]
- Liu J. Z.; Li B. W.; Tan Y. Z.; Giannakopoulos A.; Sanchez-Sanchez C.; Beljonne D.; Ruffieux P.; Fasel R.; Feng X. L.; Mullen K. Toward Cove-Edged Low Band Gap Graphene Nanoribbons. J. Am. Chem. Soc. 2015, 137, 6097–6103. 10.1021/jacs.5b03017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno C.; Vilas-Varela M.; Kretz B.; Garcia-Lekue A.; Costache M. V.; Paradinas M.; Panighel M.; Ceballos G.; Valenzuela S. O.; Pena D.; et al. Bottom-Up Synthesis of Multifunctional Nanoporous Graphene. Science 2018, 360, 199–203. 10.1126/science.aar2009. [DOI] [PubMed] [Google Scholar]
- Costa P. S.; Teeter J. D.; Enders A.; Sinitskii A. Chevron-Based Graphene Nanoribbon Heterojunctions: Localized Effects of Lateral Extension and Structural Defects on Electronic Properties. Carbon 2018, 134, 310–315. 10.1016/j.carbon.2018.03.054. [DOI] [Google Scholar]
- Bronner C.; Stremlau S.; Gille M.; Brausse F.; Haase A.; Hecht S.; Tegeder P. Aligning the Band Gap of Graphene Nanoribbons by Monomer Doping. Angew. Chem., Int. Ed. 2013, 52, 4422–4425. 10.1002/anie.201209735. [DOI] [PubMed] [Google Scholar]
- Cai J. M.; Pignedoli C. A.; Talirz L.; Ruffieux P.; Sode H.; Liang L. B.; Meunier V.; Berger R.; Li R. J.; Feng X. L.; et al. Graphene Nanoribbon Heterojunctions. Nat. Nanotechnol. 2014, 9, 896–900. 10.1038/nnano.2014.184. [DOI] [PubMed] [Google Scholar]
- Kawai S.; Saito S.; Osumi S.; Yamaguchi S.; Foster A. S.; Spijker P.; Meyer E. Atomically Controlled Substitutional Boron-Doping of Graphene Nanoribbons. Nat. Commun. 2015, 6, 8098. 10.1038/ncomms9098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloke R. R.; Marangoni T.; Nguyen G. D.; Joshi T.; Rizzo D. J.; Bronner C.; Cao T.; Louie S. G.; Crommie M. F.; Fischer F. R. Site-Specific Substitutional Boron Doping of Semiconducting Armchair Graphene Nanoribbons. J. Am. Chem. Soc. 2015, 137, 8872–8875. 10.1021/jacs.5b02523. [DOI] [PubMed] [Google Scholar]
- Nguyen G. D.; Toma F. M.; Cao T.; Pedramrazi Z.; Chen C.; Rizzo D. J.; Joshi T.; Bronner C.; Chen Y. C.; Favaro M.; et al. Bottom-Up Synthesis of N = 13 Sulfur-Doped Graphene Nanoribbons. J. Phys. Chem. C 2016, 120, 2684–2687. 10.1021/acs.jpcc.5b09986. [DOI] [Google Scholar]
- Basagni A.; Vasseur G.; Pignedoli C. A.; Vilas-Varela M.; Pena D.; Nicolas L.; Vitali L.; Lobo-Checa J.; de Oteyza D. G.; Sedona F.; et al. Tunable Band Alignment with Unperturbed Carrier Mobility of On-Surface Synthesized Organic Semiconducting Wires. ACS Nano 2016, 10, 2644–2651. 10.1021/acsnano.5b07683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonell-Sanroma E.; Hieulle J.; Vilas-Varela M.; Brandimarte P.; Iraola M.; Barragan A.; Li J. C.; Abadia M.; Corso M.; Sanchez-Portal D.; et al. Doping of Graphene Nanoribbons via Functional Group Edge Modification. ACS Nano 2017, 11, 7355–7361. 10.1021/acsnano.7b03522. [DOI] [PubMed] [Google Scholar]
- Durr R. A.; Haberer D.; Lee Y. L.; Blackwell R.; Kalayjian A. M.; Marangoni T.; Ihm J.; Louie S. G.; Fischer F. R. Orbitally Matched Edge-Doping in Graphene Nanoribbons. J. Am. Chem. Soc. 2018, 140, 807–813. 10.1021/jacs.7b11886. [DOI] [PubMed] [Google Scholar]
- Kawai S.; Nakatsuka S.; Hatakeyama T.; Pawlak R.; Meier T.; Tracey J.; Meyer E.; Foster A. S. Multiple Heteroatom Substitution to Graphene Nanoribbon. Sci. Adv. 2018, 4, eaar7181 10.1126/sciadv.aar7181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y.; Qi J.; Zhang Y.-F.; Huang L.; Zheng Q.; Lin X.; Cheng Z.; Zhang Y.-Y.; Feng X.; Du S.; et al. Tuning the Morphology of Chevron-Type Graphene Nanoribbons by Choice of Annealing Temperature. Nano Res. 2018, 11, 6190–6196. 10.1007/s12274-018-2136-3. [DOI] [Google Scholar]
- Chen Y. C.; Cao T.; Chen C.; Pedramrazi Z.; Haberer D.; de Oteyza D. G.; Fischer F. R.; Louie S. G.; Crommie M. F. Molecular Bandgap Engineering of Bottom-Up Synthesized Graphene Nanoribbon Heterojunctions. Nat. Nanotechnol. 2015, 10, 156–160. 10.1038/nnano.2014.307. [DOI] [PubMed] [Google Scholar]
- Carbonell-Sanroma E.; Brandimarte P.; Balog R.; Corso M.; Kawai S.; Garcia-Lekue A.; Saito S.; Yamaguchi S.; Meyer E.; Sanchez-Portal D.; et al. Quantum Dots Embedded in Graphene Nanoribbons by Chemical Substitution. Nano Lett. 2017, 17, 50–56. 10.1021/acs.nanolett.6b03148. [DOI] [PubMed] [Google Scholar]
- Jacobse P. H.; Kimouche A.; Gebraad T.; Ervasti M. M.; Thijssen M.; Liljeroth P.; Swart I. Electronic Components Embedded in a Single Graphene Nanoribbon. Nat. Commun. 2017, 8, 119. 10.1038/s41467-017-00195-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner C.; Durr R. A.; Rizzo D. J.; Lee Y.-L.; Marangoni T.; Kalayjian A. M.; Rodriguez H.; Zhao W.; Louie S. G.; Fischer F. R.; et al. Hierarchical On-Surface Synthesis of Graphene Nanoribbon Heterojunctions. ACS Nano 2018, 12, 2193–2200. 10.1021/acsnano.7b08658. [DOI] [PubMed] [Google Scholar]
- Groning O.; Wang S. Y.; Yao X. L.; Pignedoli C. A.; Barin G. B.; Daniels C.; Cupo A.; Meunier V.; Feng X. L.; Narita A.; et al. Engineering of Robust Topological Quantum Phases in Graphene Nanoribbons. Nature 2018, 560, 209–213. 10.1038/s41586-018-0375-9. [DOI] [PubMed] [Google Scholar]
- Rizzo D. J.; Veber G.; Cao T.; Bronner C.; Chen T.; Zhao F. Z.; Rodriguez H.; Louie S. G.; Crommie M. F.; Fischer F. R. Topological Band Engineering of Graphene Nanoribbons. Nature 2018, 560, 204–208. 10.1038/s41586-018-0376-8. [DOI] [PubMed] [Google Scholar]
- van der Lit J.; Boneschanscher M. P.; Vanmaekelbergh D.; Ijas M.; Uppstu A.; Ervasti M.; Harju A.; Liljeroth P.; Swart I. Suppression of Electron-Vibron Coupling in Graphene Nanoribbons Contacted via a Single Atom. Nat. Commun. 2013, 4, 2023. 10.1038/ncomms3023. [DOI] [PubMed] [Google Scholar]
- Wang S. Y.; Kharche N.; Girao E. C.; Feng X. L.; Mullen K.; Meunier V.; Fasel R.; Ruffieux P. Quantum Dots in Graphene Nanoribbons. Nano Lett. 2017, 17, 4277–4283. 10.1021/acs.nanolett.7b01244. [DOI] [PubMed] [Google Scholar]
- Ma C. X.; Liang L. B.; Xiao Z. C.; Puretzky A. A.; Hong K. L.; Lu W. C.; Meunier V.; Bernholc J.; Li A. P. Seamless Staircase Electrical Contact to Semiconducting Graphene Nanoribbons. Nano Lett. 2017, 17, 6241–6247. 10.1021/acs.nanolett.7b02938. [DOI] [PubMed] [Google Scholar]
- Deniz O.; Sanchez-Sanchez C.; Dumslaff T.; Feng X. L.; Narita A.; Mullen K.; Kharche N.; Meunier V.; Fasel R.; Ruffieux P. Revealing the Electronic Structure of Silicon Intercalated Armchair Graphene Nanoribbons by Scanning Tunneling Spectroscopy. Nano Lett. 2017, 17, 2197–2203. 10.1021/acs.nanolett.6b04727. [DOI] [PubMed] [Google Scholar]
- Chen Z. P.; Wang H. I.; Bilbao N.; Teyssandier J.; Prechtl T.; Cavani N.; Tries A.; Biagi R.; De Renzi V.; Feng X. L.; et al. Lateral Fusion of Chemical Vapor Deposited N = 5 Armchair Graphene Nanoribbons. J. Am. Chem. Soc. 2017, 139, 9483–9486. 10.1021/jacs.7b05055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino-Diez N.; Garcia-Lekue A.; Carbonell-Sanroma E.; Li J. C.; Corso M.; Colazzo L.; Sedona F.; Sanchez-Portal D.; Pascual J. I.; de Oteyza D. G. Width-Dependent Band Gap in Armchair Graphene Nanoribbons Reveals Fermi Level Pinning on Au(111). ACS Nano 2017, 11, 11661–11668. 10.1021/acsnano.7b06765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhirev M.; Zahl P.; Sinitskii A. Phenyl Functionalization of Atomically Precise Graphene Nanoribbons for Engineering Inter-ribbon Interactions and Graphene Nanopores. ACS Nano 2018, 12, 8662–8669. 10.1021/acsnano.8b04489. [DOI] [PubMed] [Google Scholar]
- Han P.; Akagi K.; Canova F. F.; Mutoh H.; Shiraki S.; Iwaya K.; Weiss P. S.; Asao N.; Hitosugi T. Bottom-Up Graphene-Nanoribbon Fabrication Reveals Chiral Edges and Enantioselectivity. ACS Nano 2014, 8, 9181–9187. 10.1021/nn5028642. [DOI] [PubMed] [Google Scholar]
- Schulz F.; Jacobse P. H.; Canova F. F.; van der Lit J.; Gao D. Z.; van den Hoogenband A.; Han P.; Gebbink R. J. M. K.; Moret M. E.; Joensuu P. M.; et al. Precursor Geometry Determines the Growth Mechanism in Graphene Nanoribbons. J. Phys. Chem. C 2017, 121, 2896–2904. 10.1021/acs.jpcc.6b12428. [DOI] [Google Scholar]
- Senkovskiy B. V.; Haberer D.; Usachov D. Y.; Fedorov A. V.; Ehlen N.; Hell M.; Petaccia L.; Di Santo G.; Durr R. A.; Fischer F. R.; et al. Spectroscopic Characterization of N = 9 Armchair Graphene Nanoribbons. Phys. Status Solidi RRL 2017, 11, 1700157. 10.1002/pssr.201700157. [DOI] [Google Scholar]
- Senkovskiy B. V.; Pfeiffer M.; Alavi S. K.; Bliesener A.; Zhu J.; Michel S.; Fedorov A. V.; German R.; Hertel D.; Haberer D.; et al. Making Graphene Nanoribbons Photoluminescent. Nano Lett. 2017, 17, 4029–4037. 10.1021/acs.nanolett.7b00147. [DOI] [PubMed] [Google Scholar]
- Senkovskiy B. V.; Fedorov A. V.; Haberer D.; Farjam M.; Simonov K. A.; Preobrajenski A. B.; Martensson N.; Atodiresei N.; Caciuc V.; Blugel S.; et al. Semiconductor-to-Metal Transition and Quasiparticle Renormalization in Doped Graphene Nanoribbons. Adv. Elec. Mater. 2017, 3, 1600490. 10.1002/aelm.201600490. [DOI] [Google Scholar]
- Sanchez-Sanchez C.; Dienel T.; Deniz O.; Ruffieux P.; Berger R.; Feng X. L.; Mullen K.; Fasel R. Purely Armchair or Partially Chiral: Noncontact Atomic Force Microscopy Characterization of Dibromo-Bianthryl-Based Graphene Nanoribbons Grown on Cu(111). ACS Nano 2016, 10, 8006–8011. 10.1021/acsnano.6b04025. [DOI] [PubMed] [Google Scholar]
- Han P.; Akagi K.; Canova F. F.; Mutoh H.; Shiraki S.; Iwaya K.; Weiss P. S.; Asao N.; Hitosugi T. Reply to ″Comment on ’Bottom-Up Graphene-Nanoribbon Fabrication Reveals Chiral Edges and Enantio selectivity’″. ACS Nano 2015, 9, 3404–3405. 10.1021/acsnano.5b01687. [DOI] [PubMed] [Google Scholar]
- Senkovskiy B. V.; Usachov D. Y.; Fedorov A. V.; Haberer D.; Ehlen N.; Fischer F. R.; Gruneis A. Finding the Hidden Valence Band of N = 7 Armchair Graphene Nanoribbons with Angle-Resolved Photoemission Spectroscopy. 2D Mater. 2018, 5, 035007. 10.1088/2053-1583/aabb70. [DOI] [Google Scholar]
- Xing S.; Liu B.; Wang W.; Guo J.; Wang W. On-Surface Synthesis of Graphene Nanoribbons Catalyzed by Ni Atoms. Chem. - Asian J. 2018, 13, 2023–2026. 10.1002/asia.201800610. [DOI] [PubMed] [Google Scholar]
- Blankenburg S.; Bieri M.; Fasel R.; Mullen K.; Pignedoli C. A.; Passerone D. Porous Graphene as an Atmospheric Nanofilter. Small 2010, 6, 2266–2271. 10.1002/smll.201001126. [DOI] [PubMed] [Google Scholar]
- Aradhya S. V.; Venkataraman L. Single-Molecule Junctions beyond Electronic Transport. Nat. Nanotechnol. 2013, 8, 399–410. 10.1038/nnano.2013.91. [DOI] [PubMed] [Google Scholar]
- Joachim C.; Magoga M. The Effective Mass of an Electron when Tunneling through a Molecular Wire. Chem. Phys. 2002, 281, 347–352. 10.1016/S0301-0104(02)00372-5. [DOI] [Google Scholar]
- Nacci C.; Ample F.; Bleger D.; Hecht S.; Joachim C.; Grill L. Conductance of a Single Flexible Molecular Wire Composed of Alternating Donor and Acceptor Units. Nat. Commun. 2015, 6, 7397. 10.1038/ncomms8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang G. W.; Chen S. Z.; Wang W. H.; Lin T.; Chen K. Q.; Shang X. S.; Liu P. N.; Lin N. Resonant Charge Transport in Conjugated Molecular Wires beyond 10 nm Range. J. Am. Chem. Soc. 2016, 138, 11140–11143. 10.1021/jacs.6b07416. [DOI] [PubMed] [Google Scholar]
- Reecht G.; Scheurer F.; Speisser V.; Dappe Y. J.; Mathevet F.; Schull G. Electroluminescence of a Polythiophene Molecular Wire Suspended between a Metallic Surface and the Tip of a Scanning Tunneling Microscope. Phys. Rev. Lett. 2014, 112, 047403. 10.1103/PhysRevLett.112.047403. [DOI] [PubMed] [Google Scholar]
- Koch M.; Ample F.; Joachim C.; Grill L. Voltage-Dependent Conductance of a Single Graphene Nanoribbon. Nat. Nanotechnol. 2012, 7, 713–717. 10.1038/nnano.2012.169. [DOI] [PubMed] [Google Scholar]
- Jacobse P. H.; Mangnus M. J. J.; Zevenhuizen S. J. M.; Swart I. Mapping the Conductance of Electronically Decoupled Graphene Nanoribbons. ACS Nano 2018, 12, 7048–7056. 10.1021/acsnano.8b02770. [DOI] [PubMed] [Google Scholar]
- Chong M. C.; Afshar-Imani N.; Scheurer F.; Cardoso C.; Ferretti A.; Prezzi D.; Schull G. Bright Electroluminescence from Single Graphene Nanoribbon Junctions. Nano Lett. 2018, 18, 175–181. 10.1021/acs.nanolett.7b03797. [DOI] [PubMed] [Google Scholar]
- Koch M.; Li Z.; Nacci C.; Kumagai T.; Franco I.; Grill L. How Structural Defects Affect the Mechanical and Electrical Properties of Single Molecular Wires. Phys. Rev. Lett. 2018, 121, 047701. 10.1103/PhysRevLett.121.047701. [DOI] [PubMed] [Google Scholar]
- Kawai S.; Benassi A.; Gnecco E.; Sode H.; Pawlak R.; Feng X. L.; Mullen K.; Passerone D.; Pignedoli C. A.; Ruffieux P.; et al. Superlubricity of Graphene Nanoribbons on Gold Surfaces. Science 2016, 351, 957–961. 10.1126/science.aad3569. [DOI] [PubMed] [Google Scholar]
- Li J. C.; Merino-Diez N.; Carbonell-Sanroma E.; Vilas-Varela M.; de Oteyza D. G.; Pena D.; Corso M.; Pascual J. I. Survival of Spin State in Magnetic Porphyrins Contacted by Graphene Nanoribbons. Sci. Adv. 2018, 4, eaaq0582 10.1126/sciadv.aaq0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong M. C.; Reecht G.; Bulou H.; Boeglin A.; Scheurer F.; Mathevet F.; Schull G. Narrow-Line Single-Molecule Transducer between Electronic Circuits and Surface Plasmons. Phys. Rev. Lett. 2016, 116, 036802. 10.1103/PhysRevLett.116.036802. [DOI] [PubMed] [Google Scholar]
- Nacci C.; Viertel A.; Hecht S.; Grill L. Covalent Assembly and Characterization of Nonsymmetrical Single-Molecule Nodes. Angew. Chem., Int. Ed. 2016, 55, 13724–13728. 10.1002/anie.201605421. [DOI] [PubMed] [Google Scholar]
- Abadia M.; Ilyn M.; Piquero-Zulaica I.; Gargiani P.; Rogero C.; Ortega J. E.; Brede J. Polymerization of Well-Aligned Organic Nanowires on a Ferromagnetic Rare-Earth Surface Alloy. ACS Nano 2017, 11, 12392–12401. 10.1021/acsnano.7b06374. [DOI] [PubMed] [Google Scholar]
- Cui D. L.; Ebrahimi M.; Rosei F.; Macleod J. M. Control of Fullerene Crystallization from 2D to 3D through Combined Solvent and Template Effects. J. Am. Chem. Soc. 2017, 139, 16732–16740. 10.1021/jacs.7b08642. [DOI] [PubMed] [Google Scholar]
- Plas J.; Ivasenko O.; Martsinovich N.; Lackinger M.; De Feyter S. Nanopatterning of a Covalent Organic Framework Host-Guest System. Chem. Commun. 2016, 52, 68–71. 10.1039/C5CC07557B. [DOI] [PubMed] [Google Scholar]
- Cui D.; MacLeod J. M.; Ebrahimi M.; Perepichka D. F.; Rosei F. Solution and Air Stable Host/Guest Architectures from a Single Layer Covalent Organic Framework. Chem. Commun. 2015, 51, 16510–16513. 10.1039/C5CC07059G. [DOI] [PubMed] [Google Scholar]
- Richter A.; Vilas-Varela M.; Peña D.; Bechstein R.; Kühnle A. Homocoupling of Terminal Alkynes on Calcite (10.4). Surf. Sci. 2018, 678, 106–111. 10.1016/j.susc.2017.12.012. [DOI] [Google Scholar]
- Kolmer M.; Zuzak R.; Steiner A. K.; Zajac L.; Engelund M.; Godlewski S.; Szymonski M.; Amsharov K. Fluorine-Programmed Nanozipping to Tailored Nanographenes on Rutile TiO2 Surfaces. Science 2019, 363, 57. 10.1126/science.aav4954. [DOI] [PubMed] [Google Scholar]
- Olszowski P.; Zapotoczny B.; Prauzner-Bechcicki J. S.; Vilas-Varela M.; Perez D.; Guitian E.; Pena D.; Szymonski M. Aryl Halide C-C Coupling on Ge(001):H Surfaces. J. Phys. Chem. C 2015, 119, 27478–27482. 10.1021/acs.jpcc.5b08883. [DOI] [Google Scholar]
- Berner N. C.; Sergeeva Y. N.; Sergeeva N. N.; Senge M. O.; Cafolla A. A.; McGovern I. T.. Intermolecular Dehalogenation Reactions on Passivated Germanium(001). arXiv 2015, arXiv:1505.06031. [Google Scholar]
- Bieri M.; Treier M.; Cai J. M.; Ait-Mansour K.; Ruffieux P.; Gröning O.; Gröning P.; Kastler M.; Rieger R.; Feng X. L.; et al. Porous Graphenes: Two-Dimensional Polymer Synthesis with Atomic Precision. Chem. Commun. 2009, 45, 6919–6921. 10.1039/b915190g. [DOI] [PubMed] [Google Scholar]
- Dienel T.; Gomez-Diaz J.; Seitsonen A. P.; Widmer R.; Iannuzzi M.; Radican K.; Sachdev H.; Mullen K.; Hutter J.; Groning O. Dehalogenation and Coupling of a Polycyclic Hydrocarbon on an Atomically Thin Insulator. ACS Nano 2014, 8, 6571–6579. 10.1021/nn501906w. [DOI] [PubMed] [Google Scholar]
- Morchutt C.; Bjork J.; Krotzky S.; Gutzler R.; Kern K. Covalent Coupling via Dehalogenation on Ni(111) Supported Boron Nitride and Graphene. Chem. Commun. 2015, 51, 2440–2443. 10.1039/C4CC07107G. [DOI] [PubMed] [Google Scholar]
- Bao Y.; Yang M.; Tan S. J. R.; Liu Y. P.; Xu H.; Liu W.; Nai C. T.; Feng Y. P.; Lu J.; Loh K. P. Substoichiometric Molybdenum Sulfide Phases with Catalytically Active Basal Planes. J. Am. Chem. Soc. 2016, 138, 14121–14128. 10.1021/jacs.6b09042. [DOI] [PubMed] [Google Scholar]
- Bombis C.; Ample F.; Lafferentz L.; Yu H.; Hecht S.; Joachim C.; Grill L. Single Molecular Wires Connecting Metallic and Insulating Surface Areas. Angew. Chem., Int. Ed. 2009, 48, 9966–9970. 10.1002/anie.200904645. [DOI] [PubMed] [Google Scholar]
- Rastgoo-Lahrood A.; Björk J.; Lischka M.; Eichhorn J.; Kloft S.; Fritton M.; Strunskus T.; Samanta D.; Schmittel M.; Heckl W. M.; et al. Post-Synthetic Decoupling of On-Surface-Synthesized Covalent Nanostructures from Ag(111). Angew. Chem., Int. Ed. 2016, 55, 7650–7654. 10.1002/anie.201600684. [DOI] [PubMed] [Google Scholar]
- Deniz O.; Sanchez-Sanchez C.; Jaafar R.; Kharche N.; Liang L.; Meunier V.; Feng X.; Mullen K.; Fasel R.; Ruffieux P. Electronic Characterization of Silicon Intercalated Chevron Graphene Nanoribbons on Au(111). Chem. Commun. 2018, 54, 1619–1622. 10.1039/C7CC08353J. [DOI] [PubMed] [Google Scholar]
- Ohtomo M.; Sekine Y.; Hibino H.; Yamamoto H. Graphene Nanoribbon Field-Effect Transistors Fabricated by Etchant-Free Transfer from Au(788). Appl. Phys. Lett. 2018, 112, 021602. 10.1063/1.5006984. [DOI] [Google Scholar]
- Dong W. L.; Li S. Y.; Yue J. Y.; Wang C.; Wang D.; Wan L. J. Fabrication of Bilayer Tetrathiafulvalene Integrated Surface Covalent Organic Frameworks. Phys. Chem. Chem. Phys. 2016, 18, 17356–17359. 10.1039/C6CP01804A. [DOI] [PubMed] [Google Scholar]
- Bennett P. B.; Pedramrazi Z.; Madani A.; Chen Y. C.; de Oteyza D. G.; Chen C.; Fischer F. R.; Crommie M. F.; Bokor J. Bottom-Up Graphene Nanoribbon Field-Effect Transistors. Appl. Phys. Lett. 2013, 103, 253114. 10.1063/1.4855116. [DOI] [Google Scholar]
- Candini A.; Martini L.; Chen Z. P.; Mishra N.; Convertino D.; Coletti C.; Narita A.; Feng X. L.; Mullen K.; Affronte M. High Photoresponsivity in Graphene Nanoribbon Field-Effect Transistor Devices Contacted with Graphene Electrodes. J. Phys. Chem. C 2017, 121, 10620–10625. 10.1021/acs.jpcc.7b03401. [DOI] [Google Scholar]
- Llinas J. P.; Fairbrother A.; Barin G. B.; Shi W.; Lee K.; Wu S.; Choi B. Y.; Braganza R.; Lear J.; Kau N.; et al. Short-Channel Field-Effect Transistors with 9-Atom and 13-Atom Wide Graphene Nanoribbons. Nat. Commun. 2017, 8, 633. 10.1038/s41467-017-00734-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S. T.; Kojima T.; Nakae T.; Sakaguchi H. Wide Graphene Nanoribbons Produced by Interchain Fusion of Poly(p-Phenylene) via Two-Zone Chemical Vapor Deposition. Chem. Commun. 2017, 53, 7034–7036. 10.1039/C7CC02849K. [DOI] [PubMed] [Google Scholar]
- Passi V.; Gahoi A.; Senkovskiy B. V.; Haberer D.; Fischer F. R.; Gruneis A.; Lemme M. C. Field-Effect Transistors Based on Networks of Highly Aligned, Chemically Synthesized N = 7 Armchair Graphene Nanoribbons. ACS Appl. Mater. Interfaces 2018, 10, 9900–9903. 10.1021/acsami.8b01116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Zheng Y.; Xu X. F.; Dubuisson E.; Bao Q. L.; Lu J.; Loh K. P. Electrochemical Delamination of CVD-Grown Graphene Film: Toward the Recyclable Use of Copper Catalyst. ACS Nano 2011, 5, 9927–9933. 10.1021/nn203700w. [DOI] [PubMed] [Google Scholar]
- Shekhirev M.; Vo T. H.; Pour M. M.; Lipatov A.; Munukutla S.; Lyding J. W.; Sinitskii A. Interfacial Self-Assembly of Atomically Precise Graphene Nanoribbons into Uniform Thin Films for Electronics Applications. ACS Appl. Mater. Interfaces 2017, 9, 693–700. 10.1021/acsami.6b12508. [DOI] [PubMed] [Google Scholar]
- Teeter J. D.; Costa P. S.; Zahl P.; Vo T. H.; Shekhirev M.; Xu W. W.; Zeng X. C.; Enders A.; Sinitskii A. Dense Monolayer Films of Atomically Precise Graphene Nanoribbons on Metallic Substrates Enabled by Direct Contact Transfer of Molecular Precursors. Nanoscale 2017, 9, 18835–18844. 10.1039/C7NR06027K. [DOI] [PubMed] [Google Scholar]
- Pfeiffer M.; Senkovskiy B. V.; Haberer D.; Fischer F. R.; Yang F.; Meerholz K.; Ando Y.; Gruneis A.; Lindfors K. Enhanced Light-Matter Interaction of Aligned Armchair Graphene Nanoribbons using Arrays of Plasmonic Nanoantennas. 2D Mater. 2018, 5, 045006. 10.1088/2053-1583/aacf2b. [DOI] [Google Scholar]
- Murphy C. J.; Smith Z. C.; Pronschinski A.; Lewis E. A.; Liriano M. L.; Wong C.; Ivimey C. J.; Duffy M.; Musial W.; Therrien A. J.; et al. Ullmann Coupling Mediated Assembly of an Electrically Driven Altitudinal Molecular Rotor. Phys. Chem. Chem. Phys. 2015, 17, 31931–31937. 10.1039/C5CP05294G. [DOI] [PubMed] [Google Scholar]
- Simonov K. A.; Vinogradov N. A.; Vinogradov A. S.; Generalov A. V.; Svirskiy G. I.; Cafolla A. A.; Martensson N.; Preobrajenski A. B. Effect of Electron Injection in Copper-Contacted Graphene Nanoribbons. Nano Res. 2016, 9, 2735–2746. 10.1007/s12274-016-1162-2. [DOI] [Google Scholar]
- Skidin D.; Erdmann T.; Nikipar S.; Eisenhut F.; Kruger J.; Gunther F.; Gemming S.; Kiriy A.; Voit B.; Ryndyk D. A.; et al. Tuning the Conductance of a Molecular Wire by the Interplay of Donor and Acceptor Units. Nanoscale 2018, 10, 17131–17139. 10.1039/C8NR05031G. [DOI] [PubMed] [Google Scholar]
- Mairena A.; Wackerlin C.; Wienke M.; Grenader K.; Terfort A.; Ernst K. H. Diastereoselective Ullmann Coupling to Bishelicenes by Surface Topochemistry. J. Am. Chem. Soc. 2018, 140, 15186–15189. 10.1021/jacs.8b10059. [DOI] [PubMed] [Google Scholar]
- Li J. Y.; Martin K.; Avarvari N.; Wackerlin C.; Ernst K. H. Spontaneous Separation of On-Surface Synthesized Tris-Helicenes into Two-Dimensional Homochiral Domains. Chem. Commun. 2018, 54, 7948–7951. 10.1039/C8CC04235G. [DOI] [PubMed] [Google Scholar]
- Yang B.; Cao N.; Ju H. X.; Lin H. P.; Li Y. Y.; Ding H. H.; Ding J. Q.; Zhang J. J.; Peng C. C.; Zhang H. M.; et al. Intermediate States Directed Chiral Transfer on a Silver Surface. J. Am. Chem. Soc. 2019, 141, 168–174. 10.1021/jacs.8b05699. [DOI] [PubMed] [Google Scholar]
- Björk J. Thermodynamics of an Electrocyclic Ring-Closure Reaction on Au(111). J. Phys. Chem. C 2016, 120, 21716–21721. 10.1021/acs.jpcc.6b08755. [DOI] [Google Scholar]
- Patera L. L.; Bianchini F.; Africh C.; Dri C.; Soldano G.; Mariscal M. M.; Peressi M.; Comelli G. Real-Time Imaging of Adatom-Promoted Graphene Growth on Nickel. Science 2018, 359, 1243–1246. 10.1126/science.aan8782. [DOI] [PubMed] [Google Scholar]
- Saito M.; Suda Y.; Furukawa S.; Nakae T.; Kojima T.; Sakaguchi H. Formation of Dibenzopentalene-linking Polymers under the Two-zone CVD and Wet Conditions. Chem. Lett. 2017, 46, 1099–1101. 10.1246/cl.170396. [DOI] [Google Scholar]
- Chen Z. P.; Zhang W.; Palma C. A.; Rizzini A. L.; Liu B. L.; Abbas A.; Richter N.; Martini L.; Wang X. Y.; Cavani N.; et al. Synthesis of Graphene Nanoribbons by Ambient-Pressure Chemical Vapor Deposition and Device Integration. J. Am. Chem. Soc. 2016, 138, 15488–15496. 10.1021/jacs.6b10374. [DOI] [PubMed] [Google Scholar]
- Sakaguchi H.; Kawagoe Y.; Hirano Y.; Iruka T.; Yano M.; Nakae T. Width-Controlled Sub-Nanometer Graphene Nanoribbon Films Synthesized by Radical-Polymerized Chemical Vapor Deposition. Adv. Mater. 2014, 26, 4134–4138. 10.1002/adma.201305034. [DOI] [PubMed] [Google Scholar]
- Sakaguchi H.; Song S. T.; Kojima T.; Nakae T. Homochiral Polymerization-Driven Selective Growth of Graphene Nanoribbons. Nat. Chem. 2017, 9, 57–63. 10.1038/nchem.2614. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zheng Y.; Xu X. F.; Dubuisson E.; Bao Q. L.; Lu J.; Loh K. P. Electrochemical Delamination of CVD-Grown Graphene Film: Toward the Recyclable Use of Copper Catalyst. ACS Nano 2011, 5, 9927–9933. 10.1021/nn203700w. [DOI] [PubMed] [Google Scholar]
- Pizzocchero F.; Jessen B. S.; Whelan P. R.; Kostesha N.; Lee S.; Buron J. D.; Petrushina I.; Larsen M. B.; Greenwood P.; Cha W. J.; et al. Non-Destructive Electrochemical Graphene Transfer from Reusable Thin-Film Catalysts. Carbon 2015, 85, 397–405. 10.1016/j.carbon.2014.12.061. [DOI] [Google Scholar]
- Pham V. P.; Jang H. S.; Whang D.; Choi J. Y. Direct Growth of Graphene on Rigid and Flexible Substrates: Progress, Applications, and Challenges. Chem. Soc. Rev. 2017, 46, 6276–6300. 10.1039/C7CS00224F. [DOI] [PubMed] [Google Scholar]
- Xin H.; Li W. A Review on High Throughput Roll-To-Roll Manufacturing of Chemical Vapor Deposition Graphene. Appl. Phys. Rev. 2018, 5, 031105. 10.1063/1.5035295. [DOI] [Google Scholar]