

Abstract
Motivation
Facilitated by technological improvements, pharmacologic and genetic perturbational datasets have grown in recent years to include millions of experiments. Sharing and publicly distributing these diverse data creates many opportunities for discovery, but in recent years the unprecedented size of data generated and its complex associated metadata have also created data storage and integration challenges.
Results
We present the GCTx file format and a suite of open-source packages for the efficient storage, serialization and analysis of dense two-dimensional matrices. We have extensively used the format in the Connectivity Map to assemble and share massive datasets currently comprising 1.3 million experiments, and we anticipate that the format’s generalizability, paired with code libraries that we provide, will lower barriers for integrated cross-assay analysis and algorithm development.
Availability and implementation
Software packages (available in Python, R, Matlab and Java) are freely available at https://github.com/cmap. Additional instructions, tutorials and datasets are available at clue.io/code.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Computational analysis of datasets generated by treating diverse cell types with pharmacological and genetic perturbagens has proven useful for functional relationship discovery (Hughes et al., 2000; Lamb et al., 2006; Weinstein et al., 1997). To enable such discovery, the NIH Common Fund’s Library of Network-Based Cellular Signatures (LINCS) has brought together several high-dimensional assays to systematically characterize the effects of perturbagens on human cells (Keenan, 2018).
Additionally, the scale of the perturbation-based datasets produced has multiplied in recent years to encompass millions of samples. Such large-scale, holistic representations of perturbation provide an incredible opportunity for systems-based research on health and disease, but also introduce data management challenges unique to perturbation-based functional compendia. In particular, two challenges that are crucial to address in order to facilitate analysis of these initially heterogeneous data are standardized formatting of assay output and ease of access to arbitrary ranges of output datasets.
While the more mature field of DNA sequencing has largely converged on a standard set of file formats and data types, raw forms of perturbational data are more diverse and can range from flow cytometry readouts for mRNA (Subramanian et al., 2017) to mass spectrometry traces for protein phosphorylation (Abelin et al., 2016) to quantitative data extracted from microscopy images for morphological profiling (Bray et al, 2016). Each of these diverse data types has associated metadata, and so additional relevant metadata annotations on literature pathways (Liberzon et al., 2011), drug targets and mechanisms of action (Corsello et al., 2017), are also key to interpretation of analysis results.
Although the LINCS consortium thoughtfully considered the challenges in integrating heterogeneous data in its establishment of standards for metadata prior to public data deposition (Vempati et al., 2014), adoption of a standard for data deposition in itself does not necessarily ease access for computation during exploratory data analysis. To address this, we present the GCTx file format along with open-source software packages that we have developed. GCTx relies on robust HDF5 technology to make large, dense matrices of data and metadata annotations easy to store and explore. Importantly, the format’s utility is not just theoretical: to date, we have aggregated, analyzed, and publicly distributed millions of profiles‘ worth of data from LINCS and other large compendia (Supplementary Material S1).
2 The GCTx format and code libraries
Choice of a data representation technology. Text-based formats like GCT (Supplementary Material S2) have long been used in gene expression analysis (Eisen et al., 1998). However, the dramatically increasing size of datasets in more recent years has made storage as plain text impractical (Fig. 1B). In addition, without a governing data model, text formats cannot efficiently represent relationships between rows, columns and metadata. Furthermore, text formats make it cumbersome to retrace the provenance of an element in the data matrix as it passes through multiple stages of a data processing pipeline, which is an important requirement for reproducibility.
Fig. 1.
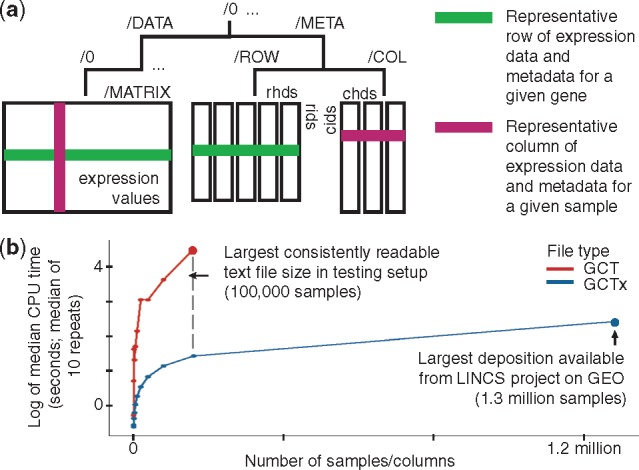
(a) Schematic of a GCTx file. (b) Parse times are faster for GCTx files compared with text-based files; more details in Supplementary Material S3
The GCTx format. The format we developed that addresses these issues is a schema built on HDF5 that we term GCTx. HDF5 supports a platform-independent file format capable of unlimited size, rapid read/write capabilities and selective parsing of a subset of a dataset without loading the entire file into memory first (The HDF Group, 1997–2018). In addition, HDF5 has a vibrant developer community that supports multiple programming languages and operating systems. GCTx adopts a lightweight, shallow hierarchy optimized for representing matrices and associated annotations while retaining several of the key benefits of HDF5’s infrastructure. This shallow hierarchy of component nodes decreases random access time when compared to deeply nested alternatives and also enables simple and efficient extension in the data matrix (appending it to the /MATRIX node) or dataset (incrementing the numerically indexed group name to ‘/1’, ‘/2’, etc. and then appending data and metadata) dimensions (Fig. 1A). More broadly, the standardization of data and metadata representation that GCTx provides frees developers from having to repeatedly customize their analytical code and to think about how to optimally use or represent data in HDF5. This is important because the HDF5 format only provides a generic data model; standardizing the representation of data and metadata consequently encourages reproducibility of data analyses.
Open source software packages integrated with GCTx. To facilitate adoption of the GCTx format with existing bioinformatics and data science tools, we also developed four open source software packages in Python (‘cmapPy’), R (‘cmapR’), Matlab (‘cmapM’) and Java (‘cmapJ’), which simplify input, output, conversion and analysis of GCTx files by representing these file inputs as native data structures readily compatible with powerful data analysis tools.
3 Conclusions
We present GCTx, an HDF5-based file format designed for efficient storage and rapid access of dense data matrices paired with metadata annotations. The format’s ability to store multiple distinct datasets and annotations enable a single file to contain an entire workflow’s worth of content, which aids reproducibility in analyses and collaboration. Importantly, the format's utility is not just theoretical: to date, we have compiled ∼1.3 million samples and made them freely available in the GCTx format (Supplementary Material S3).
Worth noting is that HDF5-based formats have previously been used in genomics (Millard et al., 2011; Sommer et al., 2013); however, these prior formats differ from GCTx in that most of them involve using deep hierarchies to store a variety of experimental design and modeling data with assay output. While this can be a useful structure, our primary needs deviated sufficiently from the features of other HDF5-based formats to merit the development of our own format. Additionally, although relational databases and cloud-based object stores are also capable of storing and efficiently serving massive datasets, we have found that--even as cloud-based object stores become more commonplace--users still request downloadable file-based representations of data for use on their personal computers or traditional login servers. Although this may change over time, we consequently decided that a file-based format would best address the majority of current user needs. To ease adoption of GCTx, we also present four open-source packages that make GCTx straightforward to incorporate with existing tools.
Supplementary Material
Acknowledgements
The authors thank Jodi Hirschman for technical editing of the manuscript and helpful conversations, Andrew Tubelli for graphic design assistance, and Wen Niuw for thorough feedback on software packages and the text. They are grateful to all our users and contributors for using, testing and providing valuable feedback and additions to both the data and software. They also thank the LINCS consortium for data and the LINCS Data Coordination and Integration Center for user testing and input on metadata stored.
Funding
This work was supported in part by the grants from NIH Common Fund's LIbrary of Integrated Network-Based Cellular Signatures (LINCS) program (U54HL127366) and from the NIH Big Data to Knowledge (BD2K) program (5U01HG008699).
Conflict of Interest: none declared.
References
- Abelin J.G. et al. (2016) Reduced-representation phosphosignatures measured by quantitative targeted MS capture cellular states and enable large-scale comparison of drug-induced phenotypes. Mol. Cell. Proteomics, 15, 1622–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray M.-A. et al. (2016) Cell Painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes. Nat. Protoc., 11, 1757–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsello S.M. et al. (2017) The Drug Repurposing Hub: a next-generation drug library and information resource. Nat. Med., 23, 405–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen M.B. et al. (1998) Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA, 95, 14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes T.R. et al. (2000) Functional discovery via a compendium of expression profiles. Cell, 102, 109–126. [DOI] [PubMed] [Google Scholar]
- Keenan A.B. (2018) The library of integrated network-based cellular signatures NIH program: system-level cataloging of human cells response to perturbations. Cell Syst., 6, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb J. et al. (2006) The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science, 313, 1929–1935. [DOI] [PubMed] [Google Scholar]
- Liberzon A. et al. (2011) Molecular signatures database (MSigDB) 3.0. Bioinformatics, 27, 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millard B.L. et al. (2011) Adaptive informatics for multifactorial and high-content biological data. Nat. Methods, 8, 487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer C. et al. (2013) CellH5: a format for data exchange in high-content screening. Bioinformatics, 29, 1580–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A. et al. (2017) A next generation connectivity map: l 1000 platform and the first 1, 000, 000 profiles. Cell, 171, 1437–1452.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The HDF Group. (1997-2018) Hierarchical Data Format, version 5. http://www.hdfgroup.org/HDF5/.
- Vempati U.D. et al. (2014) Metadata standard and data exchange specifications to describe, model, and integrate complex and diverse high-throughput screening data from the library of integrated network-based cellular signatures (LINCS). J. Biomol. Screen, 19, 803–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein J.N. et al. (1997) An information-intensive approach to the molecular pharmacology of cancer. Science, 275, 343–349. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.