

Abstract
The DNA repair enzyme ALKBH2 is implicated in both tumorigenesis as well as resistance to chemotherapy in certain cancers. It is currently under study as a potential diagnostic marker and has been proposed as a therapeutic target. To date, however, there exist no direct methods for measuring the repair activity of ALKBH2 in vitro or in biological samples. Herein, we report a highly specific, fluorogenic probe design based on an oligonucleotide scaffold that reports directly on ALKBH2 activity both in vitro and in cell lysates. Importantly, the probe enables the monitoring of cellular regulation of ALKBH2 activity in response to treatment with the chemotherapy drug temozolomide through a simple fluorescence assay, which has only previously been observed through indirect means such as qPCR and western blots. Furthermore, the probe provides a viable high-throughput assay for drug discovery.
Keywords: biosensors, cancer, DNA repair, fluorescent probes, nucleic acids
Deficiencies in alkylative DNA damage repair may give rise to carcinogenic mutations but may also be exploited for the treatment of cancer using alkylating drugs.[1,2] In response to such drugs, cancers can develop resistance through overexpression of DNA repair enzymes.[3] One of the repair enzymes implicated in both tumorigenesis and drug resistance is the demethylase ALKBH2, a human homologue of the bacterial repair enzyme AlkB. ALKBH2 is an FeII/2-oxoglutarate-dependent dioxygenase that repairs the positively charged DNA lesions 1-methyladenine (m1A) and 3-methylcytosine (m3C).[4] Of the eight known human AlkB homologues, ALKBH2 and ALKBH3 are the only ones known to demethylate m1A and m3C in genomic DNA. While ALKBH3 prefers single-stranded substrates, ALKBH2 prefers double-stranded DNA.[5]
The importance of ALKBH2 in DNA repair has been demonstrated in mouse models, in which the knockdown of ALKBH2 results in the accumulation of genomic m1A and m3C,[6] giving rise to transcription stops and error-prone replication bypass.[7] Elevated ALKBH2 activity has also been associated with resistance to chemotherapy. Ina study of brain tumor samples taken from patients treated with alkylating drugs such as temozolomide (TMZ), all samples contained elevated levels of ALKBH2 mRNA over those from patients receiving radiotherapy.[8] TMZ resistance in several glioblastoma cell lines as well as cisplatin resistance in H1299 lung cancer cells was suppressed by siRNA-mediated knockdown of ALKBH2,[9,10] and reduced ALKBH2 activity improves sensitivity to alkylating drugs.[11,12] These findings have stimulated recent efforts to find inhibitors of ALKBH2.[13,14]
In spite of its potential importance to cancer diagnosis, prognosis, and therapy, measuring the repair activity of ALKBH demethylases remains challenging.[5] Current methods for measuring ALKBH2 activity in vitro such as high performance liquid chromatography (LC-MS),[15] gel electrophoresis (PAGE),[11] and most recently melting temperature analysis[16] are limited because they are discontinuous, multistep and often nonspecific for ALKBH2 over the related homologue ALKBH3. While in vitro methods are limited, the situation for measurement of ALKBH2 activity in cells and tissues is even poorer. There is currently no reported method to directly and specifically measure ALKBH2 activity in biological samples such as tissue extracts, cell lysates, or live cells. The most commonly reported methods rely on indirect measurements of ALKBH repair in E. coli expressing ALKBH2 using labeled bacteriophages as reporters.[5,17] Such methods are not viable for human cells.
Recently, fluorescent DNA probes have been developed for the direct analysis of DNA repair.[18] Herein, we describe the first fluorogenic probes for ALKBH2 activity, affording rapid and real-time measurements of repair by ALKBH2 in vitro and in human cell extracts using only a fluorescence microplate reader. The optimized probe 13 is highly specific for ALKBH2 demethylation and is used to directly quantify elevated ALKBH2 activity in a cancer cell line treated with the drug TMZ. Probe 13 is also well suited to conducting high-throughput screens of potential ALKBH2 activators/inhibitors.
Our strategy for developing a light-up fluorescence response employs a 1-methyladenosine quenching (MAQ) mechanism. In this design, the electron-poor m1A base is situated adjacent to the electron-rich fluorescent nucleoside-analogue pyrene, which is quenched by photo-induced charge transfer to the damaged base (Figure 1).[19] Upon demethylation of m1A, the quenching interaction is lost and the probe emits a fluorescence signal. A previously reported MAQ probe for the related enzyme ALKBH3 consisted of a single-stranded 10-mer oligonucleotide with a central m1A lesion and two adjoining pyrene α-nucleosides, which form an excimer[20] (Table 1). While this probe efficiently detected ALKBH3 activity, it was unable to detect ALKBH2 activity. The observed selectivity of the probe was surprising given that both enzymes are reported to have similar catalytic efficiencies with single-stranded DNA substrates.[21] In light of this, we hypothesized that the probe length, its single-stranded character, or the pyrene nucleoside reporter was prohibiting probe repair by ALKBH2.
Figure 1.

The 1-methyladenosine quenching (MAQ) probe design used to detect demethylation by ALKBH2. The pyrene excimer is quenched by photo-induced charge transfer to the damaged base. Following repair of the positively charged lesion, the quenching is lost, resulting in fluorescence emission.
Table 1:
Probe designs for fluorogenic ALKBH2 reporting. Probes contain either the pyrene α-nucleoside (αY) or β-nucleoside (βY) along with 1-methyldeoxyadenosine (m1A). Probes 12, 13, and 13p are designed to form hairpins with a hexaethylene glycol (HEX) linker forming the loop.
| Probe | Probe Sequence | λEx/Em [nm] | F.C.[a] | t1/2[b] |
|---|---|---|---|---|
| MAQ | AAAAm1AαYαYAAA | 345/480 | N.C.[c] | N.C.[c] |
| 1 | AAAAm1AβYAAAA | 345/385 | 12 | 210 |
| 2 | AAAAm1AαYAAAA | 345/385 | N.C.[c] | N.C.[c] |
| 3 | AAAAβYm1AAAAA | 345/385 | N.C.[c] | N.C.[c] |
| 4 | AAAAαYm1AAAAA | 345/385 | N.C.[c] | N.C.[c] |
| 5 | AAAAAAAm1AβYAAAAAA | 345/385 | 25 | 175 |
| 6 | AAAAAAAAAAm1AβYAAAAAAAA | 345/385 | 22 | 200 |
| 7 | AAAAAAAAAAAAAm1AβYAAAAAAAAAA | 345/385 | 18 | 190 |
| 8 | AAAAm1AβYβYAAA | 345/480 | N.C.[c] | N.C.[c] |
| 9 | AAAAAAAm1AβYβYAAAAA | 345/480 | 18 | 165 |
| 10 | AAAAAAAAAAm1AβYβYAAAAAAA | 345/480 | 15 | 200 |
| 11 | AAAAAAAAAAAAAm1AβYβYAAAAAAAAA | 345/480 | 15 | 200 |
| 12 | CGC TTT SST TTT GCG Hex CGC AAA m1AβYβY AAA GCG | 345/480 | 9 | 125 |
| 13 | CGC TTA SSA TTT GCG Hex CGC AAA m1AβYβY AAA GCG | 345/480 | 8 | 80 |
| 13p | COMeGOMeC TTA SSA TTT GCG Hex CGC AAA m1AβYβY AAA GCOMeGOMe | 345/480 | 8 | 80 |
Fold change in fluorescence intensity at emission maxima following repair.
Time of half-maximal repair in seconds based on time to achieve 50% maximum probe signal.
Not calculated.
Given the structural differences between the ALKBH2 and ALKBH3 active sites,[22] we hypothesized that structural changes to the probe might influence activity. Specifically, we considered that the use of the unnatural α-anomer of the pyrene reporter rather than the naturally oriented β-anomer might influence enzymatic recognition of the probe. To test this, probes 1–4 were synthesized, based on A-rich 10-mer oligodeoxynucleotides in which the location and anomer of the pyrene nucleoside were varied. Probes were synthesized on an automated DNA synthesizer using phosphoramidite chemistry. Both anomers of the pyrene nucleoside phosphoramidite were accessed by Normant-cuprate-mediated C-glycosylation of Hoffer’s chlorosugar followed by acidcatalyzed epimerization (see the Supporting Information for full details). A-rich sequences were selected for the flanking regions of the probe because adenine is known to exert little or no fluorescence quenching of pyrene.[23] When tested against recombinant human ALKBH2, probe 1 was the only successfully repaired probe, demonstrating a strong and surprising stereoselective requirement by ALKBH2 for the β-anomer of the pyrene nucleoside (βY) adjacent to m1A (Figure 2a). Furthermore, the location was critical, with a strong preference for location of the fluorescent nucleotide on the 3′ side of m1A (Table 1; compare probes 1 and 3).
Figure 2.

Fold change in fluorescence intensity of a) probes 1–4 or b) probes 9 and 12 (1 µm) incubated with ALKBH2 (0.5 µm) measured at 385 nm (a) or 480 nm (b). c) Selectivity of probe 12 with buffer alone or supplemented with 1.5 mm MgCl2. See Supporting Information for full reaction conditions.
To explore the substrate-length preference of ALKBH2, we prepared probes 5–7, again using a poly(dA) context with total lengths varied from 10–25 nt (Table 1). In probes with a single pyrene, length did not appear to significantly affect the time of half-maximal repair (t1/2); however, the overall fold-change of the probe was affected, with a maximal increased fluorescence of 25-fold at 385 nm achieved using 15-mer probe 5. Probes 8–11 were also synthesized containing two adjacent pyrenes to redshift the probe emission from 385 nm to 480 nm through excimer formation,[24] giving a Stokes shift of 135 nm. A similar trend in length versus fold-change was observed with probe 9 yielding an 18-fold change in fluorescence at 480 nm (Figure 2b); however, the shorter probe 8 was not repaired. Testing probes 8–11 for ALKBH2-versus-ALKBH3 selectivity, we found that all four were good substrates for ALKBH3 (Supporting Information, Figure S1) suggesting that ALKBH3 is indifferent to the identity of the pyrene anomer or length of the substrate over this range.
In order to improve the specificity of probe designs for ALKBH2, we incorporated a short duplex hairpin structure to exploit ALKBH3’s low activity on double-stranded substrates and ALKBH2’s reported preference for duplex DNA.[17,22] Probe 12 incorporates two complementary 15-mer regions connected by a hexaethylene glycol linker. One strand of the probe incorporates an m1A base adjacent to two pyrenes. On the complementary strand, the pyrene excimer is paired opposite two 1′,2′-deoxyribose spacers (Table 1). This pairing has previously been shown to mimic the thermodynamic stability of an A:T base pair and supports normal duplex structure.[25,26] Experiments with purified enzyme revealed that hairpin probe 12 showed a much higher specificity for ALKBH2 than its single-stranded counterpart. Probe 12 yielded a 9-fold increase in fluorescence and a t1/2 of 125 s, almost twice as fast as the single-stranded probes (Figure 2b). Minimal repair of 12 by ALKBH3 was observed, confirming high selectivity of the double-stranded probe for ALKBH2 (Figure 2c). Interestingly, in separate studies we observed non-competitive inhibition of ALKBH2 by duplex DNA when repairing single-stranded probes but not double-stranded probes (Supporting Information, Table S2 and Figures S3 and S4). This effect was not observed with ALKBH3.
Consistent with previous literature reports,[17] we found that the addition of 1.5 mm magnesium to the reaction buffer increased the rate of ALKBH2 demethylation while further suppressing ALKBH3, to yield over 100-fold selectivity for the former based on initial rate measurements (Figure 2c, Table S1). The magnesium-dependent increase in selectivity was not observed with single-stranded substrates, indicating the divalent cation interacts with the duplex probe or its interface with the enzyme, rather than the enzyme alone (Table S1).
In an effort to improve the overall signal of probe 12, we synthesized probe 13, replacing the complementary thymine bases neighboring the pyrene excimer with adenine. While thymine is known to strongly quench pyrene fluorescence, adenine can effectively insulate pyrene against the quenching of nearby pyrimidines.[23] We found that the design change improved the brightness by two-fold while impacting the fold change only minimally (Figure S2).
We validated the ability of probe 13 to assay ALKBH2 inhibitors by measuring the IC50 of pyridine 2,4-dicarboxylic acid (PDCA), a broad-spectrum inhibitor of 2-OG-dependent enzymes[16] (Figure 3). The assay was conducted in a 60 µL reaction volume on a 384-well plate, yielding an IC50 of 2.43 ± 0.12 µm, fully consistent with reported values.[18] To our knowledge, probe 13 enables the first continuous real-time assay of ALKBH2 activity, making it likely the most practical assay available for high-throughput screens.
Figure 3.
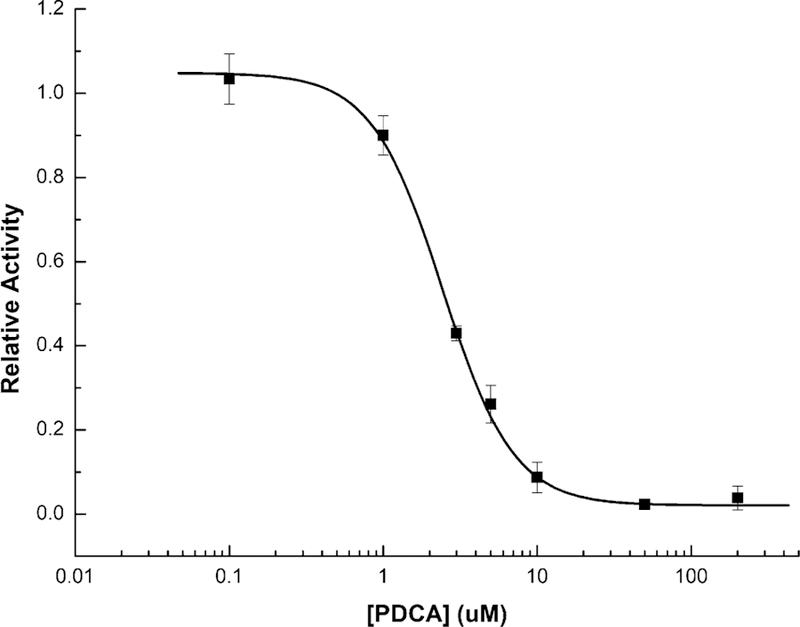
Probe 13 used in high-throughput format for evaluating an inhibitor. Dose-response curve of broad-spectrum demethylase inhibitor pyridine 2,4-dicarboxylic acid (PDCA) with ALKBH2 generated using probe 13. The curve yielded an IC50 measurement of 2.43 ± 0.12 µm.
To test the utility of probe 13 in differentiating activity levels of ALKBH2 in cells, we synthesized a nuclease-protected version by substituting 2′-O-methyl protected nucleotides at the 3′ and 5′ termini of the probe (13p). Tests with purified enzyme confirmed (Supporting Information, Figure S5) that the modified end residues had little or no effect on response. We then tested the probe in lysates from engineered mouse embryonic fibroblasts (MEFs) having varied murine ALKBH2 and ALKBH3 activity.[27] The lysates were tested in a 384-well plate format with 2 µm probe and the necessary cofactors (see the Supporting Information for reaction details). The fluorescence response from each cell line was monitored over an extended period at 37 °C (Figure 4).
Figure 4.
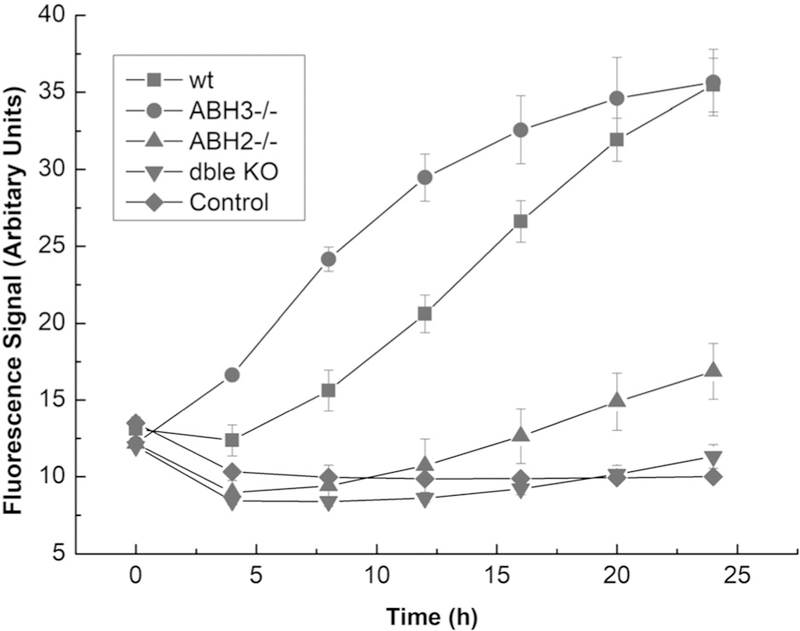
Nuclease-protected probe 13p yields ALKBH2-specific signals in mammaliancell lysates. Shown are fluorescence responses of probe 13p (2 μm)with MEF lysates (1 mgmL−1 total protein). Data collected from 384-well plates over 27 hincubated at 378°C.
The data show that the probe 13p responds to enzymatic activity from cells expressing ALKBH2 (Figure 4). After 24 h, the signals generated from the wild-type and ALKBH3−/− cells were high and equivalent, while ALKBH2−/− cells showed only a small amount of signal over background. The double ALKBH2 knockout and control experiments show almost identically low fluorescence responses as expected. Taken together, these results show that probe 13p is able to measure and differentiate activity levels of ALKBH2 in cell lysates at biologically relevant protein concentrations. Furthermore, it is highly selective for ALKBH2, with only a small amount of signal generated from the ALKBH2 knockout cells. When the lysates were tested in buffer containing EDTA, we observed no signal, consistent with the expectation that the FeII-dependent ALKBH enzymes are responsible for the fluorescence signal (Supporting Information, Figure S6).
Finally, we employed probe 13p to compare ALKBH2 activity in human cell lines. Lysates were generated from U87 and HEK293T cells and their fluorescence response with 13p was compared after 24 h incubation (Figure 5a). The response from the HEK293T cell line, derived from non-cancerous cells, was about twice that of the wild type MEF cells. The human tumor cell line U87 showed significantly reduced ALKBH2 activity, consistent with reports of epigenetic silencing of demethylases in that cell line.[28] We then treated the U87 cell line with the DNA methylating drug TMZ, which is known to induce elevated ALKBH2 expression in certain cancers as a drug resistance mechanism.[9] We found that incubation with TMZ for 24 h resulted in a near doubling of ALKBH2 signal from the U87 lysates (p = 0.007), consistent with qPCR data (Figure 5b).[9] These experiments demonstrate that 13p can be used to directly measure the development of a drug resistance response in cancer cell lines without the need for qPCR or western blots, which are fundamentally indirect measures.
Figure 5.
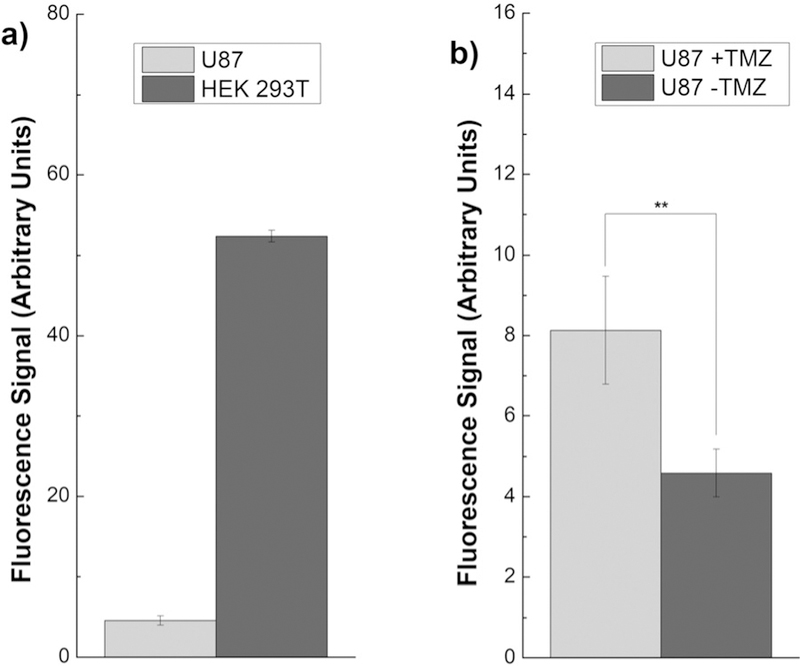
Nuclease-protected probe 13p reports on ALKBH2 activity in lysates derived from human cell lines. Experiments were performed in a384-well plate with 1mgmL−1 total cellular protein, 100 μm TMZ, and fluorescence was measured after 24 h incubation.
Taken together, our data show that hairpin probe 13 and the nuclease-protected variant 13p can be used to measure ALKBH2 activity in vitro and in cell lysates in real time. Importantly, 13p is highly specific and is capable of discriminating between the closely-related enzyme homologues ALKBH2 and ALKBH3, allowing it to quantify ALKBH2 activity levels in cells with minimal background signals. This represents the first probe for the specific measurement of ALKBH2 activity from human cells in real time. The probes reported in this study may also be readily adapted for high-throughput screening of potential small-molecule modulators. While recent efforts have been made to develop both small-molecule and aptamer-based inhibitors of ALKBH2, those efforts have relied on low-throughput CE[14] or DCMS[13] assays that put major constraints on the scope of the screen and design of inhibitors. As a result, the only reported small-molecule inhibitors have relatively low affinity and selectivity, acting as pan-2-OG-dependent dioxygenase inhibitors. Given that assays of specific DNA repair markers are already used in the diagnosis of malignancies,[29] it is possible that probes such as 13p may in the future find relevance in the clinic as well.
Supplementary Material
Acknowledgements
We acknowledge support from the U.S. National Cancer Institute (R01 CA217809 to ETK and R01 CA176611 to TRO), the California Tobacco-Related Disease Research Program (25IP-0020, TRO), and the City of Hope Comprehensive Cancer Center (P30 CA033572, TRO).
Footnotes
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/anie.201807593.
Conflict of interest
The authors declare no conflict of interest.
Contributor Information
David L. Wilson, Department of Chemistry, Stanford University, Stanford, CA 94305 (USA).
Dr. Andrew A. Beharry, Department of Chemical and Physical Sciences, University of Toronto, Mississauga, ON L5L 1C6 (Canada)
Prof. Dr. Eric T. Kool, Department of Chemistry, Stanford University, Stanford, CA 94305 (USA).
References
- [1].Roos WP, Thomas AD, Kaina B, Nat. Rev. Cancer 2016, 16, 20–33. [DOI] [PubMed] [Google Scholar]
- [2].Lord CJ, Ashworth A, Nature 2012, 481, 287–294. [DOI] [PubMed] [Google Scholar]
- [3].Holohan C, Schaeybroeck SV, Longley DB, Johnston PG, Nat. Rev. Cancer 2013, 13, 714–726. [DOI] [PubMed] [Google Scholar]
- [4].Duncan T, Trewick SC, Koivisto P, Bates PA, Lindahl T, Sedgwick B, Proc. Natl. Acad. Sci. USA 2002, 99, 16660–16665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Fedeles BI, Singh V, Delaney JC, Li D, Essigmann JM, J. Biol. Chem 2015, 290, 20734–20742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ringvoll J, et al. , EMBO J 2006, 25, 2189–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Anindya R, DNA Repair 2017, 49, 21–25. [DOI] [PubMed] [Google Scholar]
- [8].Cetica V, Genitori L, Giunti L, Sanzo M, Bernini G, Massimino M, Sardi I, J. Neuro-Oncol 2009, 94, 195–201. [DOI] [PubMed] [Google Scholar]
- [9].Johannessen T-CA, Prestegarden L, Grudic A, Hegi ME, Tysnes BB, Bjerkvig R, Neuro-Oncol 2013, 15, 269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wu S, Xu W, Liu S, Chen B, Wang X, Wang Y, Liu S, Wu J, Acta Pharmacol. Sin 2011, 32, 393–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tran TQ, Gabra MBI, Lowman XH, Yang Y, Reid MA, Pan M, O’Connor TR, Kong M, PLOS Biol 2017, 15, e2002810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Li Q, Huang Y, Liu X, Gan J, Chen H, Yang C-G, J. Biol. Chem 2016, 291, 11083–11093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Woon ECY, et al. , J. Med. Chem 2012, 55, 2173–2184. [DOI] [PubMed] [Google Scholar]
- [14].Yufa R, Krylova SM, Bruce C, Bagg EA, Schofield CJ, Krylov SN, Anal. Chem 2015, 87, 1411–1419. [DOI] [PubMed] [Google Scholar]
- [15].Chen F, Tang Q, Bian K, Humulock ZT, Yang X, Jost M, Drennan CL, Essigmann JM, Li D, Chem. Res. Toxicol 2016, 29, 687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yang T, Cheong A, Mai X, Zou S, Woon ECY, Chem. Commun 2016, 52, 6181–6184. [DOI] [PubMed] [Google Scholar]
- [17].Falnes PØ, Bjørås M, Aas PA, Sundheim O, Seeberg E, Nucleic Acids Res 2004, 32, 3456–3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wilson DL, Kool ET, ACS Chem. Biol 2017, 10.1021/acschembio.7b00919. [DOI] [Google Scholar]
- [19].Manoharan M, Tivel KL, Zhao M, Nafisi K, Netzel TL, J. Phys. Chem 1995, 99, 17461–17472. [Google Scholar]
- [20].Beharry AA, Lacoste S, O’Connor TR, Kool ET, J. Am. Chem. Soc 2016, 138, 3647–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lee D-H, Jin S-G, Cai S, Chen Y, Pfeifer GP, O’Connor TR, J. Biol. Chem 2005, 280, 39448–39459. [DOI] [PubMed] [Google Scholar]
- [22].Yang C-G, Yi C, Duguid EM, Sullivan CT, Jian X, Rice PA, He C, Nature 2008, 452, 961–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wilson JN, Cho Y, Tan S, Cuppoletti A, Kool ET, ChemBioChem 2008, 9, 279–285. [DOI] [PubMed] [Google Scholar]
- [24].Wilson JN, Kool ET, Org. Biomol. Chem 2006, 4, 4265–4274. [DOI] [PubMed] [Google Scholar]
- [25].Smirnov S, Matray TJ, Kool ET, de los Santos C, Nucleic Acids Res 2002, 30, 5561–5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Matray TJ, Kool ET, J. Am. Chem. Soc 1998, 120, 6191–6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Nay SL, Lee D-H, Bates SE, O’Connor TR, DNA Repair 2012, 11, 502–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dunn J, et al. , Br. J. Cancer 2009, 101, 124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Brandwein JM, Kassis J, Leber B, Hogge D, Howson-Jan K, Minden MD, Galarneau A, Pouliot J-F, Br. J. Haematol 2014, 167, 664–670. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.