

Abstract
Background
Schizophrenia is a chronic, disabling and severe mental disorder, characterised by disturbance in perception, thought, language, affect and motor behaviour. Chlorpromazine and clotiapine are among antipsychotic drugs used for the treatment of people with schizophrenia.
Objectives
To determine the clinical effects, safety and cost‐effectiveness of chlorpromazine compared with clotiapine for adults with schizophrenia.
Search methods
We searched Cochrane Schizophrenia's Trials Register (last update search 16/01/2016), which is based on regular searches of CINAHL, BIOSIS, AMED, Embase, PubMed, MEDLINE, PsycINFO and clinical trials registries. There are no language, date, document type, or publication status limitations for inclusion of records in the Register.
Selection criteria
All randomised clinical trials focusing on chlorpromazine versus clotiapine for schizophrenia. We included trials meeting our selection criteria and reporting useable data.
Data collection and analysis
We extracted data independently. For binary outcomes, we calculated risk ratio (RR) and its 95% confidence interval (CI), on an intention‐to‐treat basis. For continuous data, we estimated the mean difference (MD) between groups and its 95% CI. We employed a random‐effects model for analyses. We assessed risk of bias for included studies and created a 'Summary of findings' table using GRADE.
Main results
We have included four studies, published between 1974 and 2003, randomising 276 people with schizophrenia to receive either chlorpromazine or clotiapine. The studies were poor at concealing allocation of treatment and blinding of outcome assessment. Our main outcomes of interest were clinically important change in global and mental state, specific change in negative symptoms, incidence of movement disorder (dyskinesia), leaving the study early for any reason, and costs. All reported data were short‐term (under six months' follow‐up).
The trials did not report data for the important outcomes of clinically important change in global or mental state, or cost of care. Improvement in mental state was reported using the Positive and Negative Syndrome Scale (PANSS). When chlorpromazine was compared with clotiapine the average improvement scores for mental state using the PANSS total was higher in the clotiapine group (1 RCT, N = 31, MD 11.50 95% CI 9.42 to 13.58, very low‐quality evidence). The average change scores on the PANSS negative sub‐scale were similar between treatment groups (1 RCT, N = 21, MD ‐0.97 95% CI ‐2.76 to 0.82, very low‐quality evidence). There was no clear difference in incidence of dyskinesia (1 RCT, N = 68, RR 3.00 95% CI 0.13 to 71.15, very low‐quality evidence). Similar numbers of participants left the study early from each treatment group (3 RCTs, N = 158, RR 0.68 95% CI 0.24 to 1.88, very low‐quality evidence).
Authors' conclusions
Clinically important changes in global and mental state were not reported. Only one trial reported the average change in overall mental state; results favour clotiapine but these limited data are very difficult to trust due to methodological limitations of the study. The comparative effectiveness of chlorpromazine compared to clotiapine on change in global state remains unanswered. Results in this review suggest chlorpromazine and clotiapine cause similar adverse effects, although again, the quality of evidence for this is poor, making firm conclusions difficult.
Keywords: Humans; Antipsychotic Agents; Antipsychotic Agents/adverse effects; Antipsychotic Agents/therapeutic use; Chlorpromazine; Chlorpromazine/adverse effects; Chlorpromazine/therapeutic use; Dibenzothiazepines; Dibenzothiazepines/adverse effects; Dibenzothiazepines/therapeutic use; Dyskinesia, Drug‐Induced; Dyskinesia, Drug‐Induced/epidemiology; Intention to Treat Analysis; Patient Dropouts; Patient Dropouts/statistics & numerical data; Randomized Controlled Trials as Topic; Risk; Schizophrenia; Schizophrenia/drug therapy
Plain language summary
Direct comparison of two antipsychotics (chlorpromazine versus clotiapine) for treating schizophrenia
Review question
The aim of this review was to find good quality evidence comparing the efficacy of chlorpromazine versus clotiapine for schizophrenia.
Background
Chlorpromazine is one of the first antipsychotics to successfully alleviate the symptoms of psychosis. It was introduced in the 1950s and is still one of the most commonly used antipsychotics. However, chlorpromazine can cause serious side effects, particularly unpleasant movement disorders, causing many people with schizophrenia to stop taking chlorpromazine. During the past 70 years newer medications have been developed and most clinicians now have a wide choice of drugs for managing schizophrenia, however, none cure, and all cause some sort of side effect. The choice of treatment is still challenging. Clotiapine is a newer antipsychotic drug, found to be effective for treating the symptoms of schizophrenia and also known to be effective for treating people with schizophrenia who are resistant to other medications, however, like chlorpromazine it can cause serious movement disorders.
Searching for evidence
Cochrane Schizophrenia's Information Specialist ran an electronic search in January 2016, searching their specialised register for trials that randomised people with schizophrenia to receive either chlorpromazine or clotiapine. The search identified six reports. We inspected these reports and found four trials, published between 1974 and 2003, randomising 276 participants that could be included in the review.
Main results
The four included trials were poorly conducted and did not report data for clinically important change in global or mental state, or cost of care. Improvement in overall mental state was reported and participants receiving clotiapine had better improvement scores than those receiving chlorpromazine. However the trials also reported data for improvement in the negative symptoms, no difference between the two treatments was found. Clotiapine did not cause more movement disorders than chlorpromazine, and similar numbers of participants left the trials early.
Conclusions
There is some very low‐quality evidence that favours clotiapine over chlorpromazine for improving overall mental state. For other outcomes, including adverse effects, there is no evidence of a difference between these two antipsychotics. However these data are very difficult to draw conclusions from, only four small trials provided data and these were poorly conducted. We cannot draw conclusions on the comparative effectiveness of chlorpromazine versus clotiapine from such data.
Summary of findings
Summary of findings for the main comparison. Chlorpromazine compared to Clotiapine for schizophrenia.
| Chlorpromazine compared with Clotiapine for schizophrenia | ||||||
| Patient or population: people with schizophrenia Settings: Intervention: chlorpromazine Comparison: clotiapine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Clotiapine for schizophrenia | Chlorpromazine | |||||
| Global state: clinically important change | Not reported in any study | |||||
| Mental state: 1a. General symptoms ‐ average change (PANSS, short term) | The mean mental state: average improvement scores in the intervention groups was 11.50 higher (9.42 to 13.58 higher) | 31 (1 study) | ⊕⊝⊝⊝ very low1,2,5 | Pre‐stated outcome of clinically important change in mental state not reported | ||
| Mental state: 2c. Specific ‐ average change score for negative symptoms (PANSS ‐ negative short term) | The mean mental state in the intervention groups was 0.97 lower (2.76 lower to 0.82 higher) | 21 (1 study) | ⊕⊝⊝⊝ very low1,2,3 | |||
| Adverse effects: incidence of serious adverse effects. Movement disorders ‐ dyskinesia ‐short‐term | Study population | RR 3.00 (0.13 to 71.15) | 68 (1 study) | ⊕⊝⊝⊝ very low1,2,3 | ||
| 179 per 1000 | 243 per 1000 (168 to 354) | |||||
| Moderate | ||||||
| 49 per 1000 | 67 per 1000 (46 to 97) | |||||
| Adverse effects: clinically significant extrapyramidal symptoms | Not reported in any study | |||||
| Leaving the study early: for any reason | Study population | RR 0.68 (0.24 to 1.88) | 158 (3 studies) | ⊕⊝⊝⊝ very low1,2,4 | ||
| 283 per 1000 | 172 per 1000 (31 to 961) | |||||
| Moderate | ||||||
| 296 per 1000 | 181 per 1000 (33 to 1000) | |||||
| Cost of care | Not reported in any study | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different. Low quality: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low quality: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
1Risk of bias: serious ‐ downgraded by 1: study has an unclear risk of bias for random sequence generation, allocation concealment, or no wash‐out period reported, or source of funding unclear. 2Imprecision: serious ‐ downgraded by 1: there are very few participants, number of events small 3Publication bias: serious ‐ downgraded by 1: the study results only published in a local Japanese journal. 4Inconsistency: serious ‐ downgraded by 1: there was high heterogeneity in the pooled results. 5Indirectness: serious ‐ downgraded by 1: not direct measure of prespecified outcome.
Background
Description of the condition
Schizophrenia can be a chronic, disabling and severe mental disorder that is characterised by disturbance in perception, thought, language, affect and motor behaviour. Common symptoms include hallucinations, delusions, disorganised speech, social withdrawal, flat affect and cognitive impairments. Lifetime prevalence of schizophrenia is around 0.87% (Perala 2007).
Description of the intervention
Chlorpromazine was synthesised in 1951 by Paul Charpentier, and was the first drug that significantly alleviated symptoms of psychosis (Ban 2007). Fifty years after its introduction, chlorpromazine is still one of the most commonly used antipsychotic drugs for management of people with schizophrenia (Adams 2005). Importantly, to evaluate a new drug for treatment of schizophrenia, studies often use chlorpromazine as a 'benchmark' drug (Adams 2014). However, chlorpromazine is associated with adverse effects ranging from inducing seizure, sudden death, neuroleptic malignant syndrome as well as sedation, decreased libido, blurred vision, and orthostatic hypotension (Pakpoor 2014).
Clotiapine has been manufactured since the late 1960s and is prescribed at least in Argentina, Belgium, Israel, Italy, South Africa, Spain, Switzerland and Taiwan for a range of conditions including schizophrenia, bipolar disorder (specifically mania) and other acute psychotic illnesses (Berk 2004). Clotiapine provides rapid treatment for the symptoms of schizophrenia, and its efficiency for people who are not responsive to other typical antipsychotics has been demonstrated (Geller 2005). Like chlorpromazine and other typical antipsychotics, clotiapine is linked to a number of adverse effects including extrapyramidal syndromes and a strong sedative effect (Geller 2005).
How the intervention might work
Chlorpromazine is a phenothiazine neuroleptic, 3‐(2‐chlorophenothiazin‐10‐yl)‐N, N‐dimethylpropan‐1‐amine (Figure 1). Like many antipsychotics, chlorpromazine acts as an antagonist (or blocking agent) for a number of postsynaptic receptors, resulting in the broad range of effects noted previously. Chlorpromazine’s pharmacodynamics varies, but includes antagonist interactions with dopaminergic (especially the D2 subtype), and serotonergic 5‐hydroxytryptamine (5‐HT1 and 5‐HT2) postsynaptic receptors (Amato 2015; Miyamoto 2012).
1.
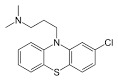
Chlorpromazine structure
Clotiapine is a dibenzothiazepine neuroleptic, named 2‐chloro‐11‐(4‐methyl‐1‐piperazinyl) dibenzo[b,f][1,4]thiazepine (Berk 2004) (Figure 2), and like chlorpromazine acts on multiple serotonergic and dopaminergic postsynaptic receptors. Clotiapine interacts with 5‐HT serotonin receptors in a number of ways, including acting as a blocking agent for 5‐HT3 and down‐regulating 5‐HT2 receptors (clotiapine has also been shown to possess a high affinity for 5HT‐6 receptors) (Geller 2005). There is also evidence that clotiapine demonstrates limited blockage of dopaminergic D2 (Moore 1989) and D4 receptors (Zawilska 1994).
2.
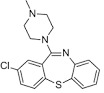
Clotiapine structure
Why it is important to do this review
Chlorpromazine is often considered a 'benchmark' antipsychotic and is used worldwide for treatment of people with schizophrenia, thus, it is important to compare its therapeutic and side effects with other medications. To our knowledge, there is no systematic review comparing the clinical outcomes and side effects of chlorpromazine to clotiapine.
The aim of this review is to compare chlorpromazine to clotiapine, which will then build up a series of Cochrane Reviews to give an overview of chlorpromazine's efficacy compared to other antipsychotics for the treatment of schizophrenia (Table 2).
1. The Cochrane chlorpromazine for people with schizophrenia reviews.
| Review title | Reference |
| Acetophenazine versus chlorpromazine | Bazrafshan 2015 |
| Chlorpromazine dose for people with schizophrenia | Liu 2009 |
| Cessation of medication for people with schizophrenia already stable on chlorpromazine | Almerie 2007 |
| Chlorpromazine versus atypical antipsychotic drugs for schizophrenia | Saha 2013 |
| Chlorpromazine versus clotiapine for schizophrenia | Current review |
| Chlorpromazine versus haloperidol for schizophrenia | Leucht 2008 |
| Chlorpromazine versus metiapine for schizophrenia | Zare 2015 |
| Chlorpromazine versus penfluridol for schizophrenia | Khalili 2015 |
| Chlorpromazine versus piperacetazine for schizophrenia | Eslami 2015 |
| Chlorpromazine versus placebo for schizophrenia | Adams 2014 |
| Chlorpromazine for psychosis induced aggression or agitation | Ahmed 2010 |
Objectives
To determine the clinical effects, safety and cost‐effectiveness of chlorpromazine compared with clotiapine for adults with schizophrenia.
Methods
Criteria for considering studies for this review
Types of studies
We considered all relevant randomised controlled trials (RCTs). If a trial was described as 'double‐blind' but implied randomisation, we included such trials in a sensitivity analysis (see Sensitivity analysis). We excluded quasi‐randomised studies, such as those allocating by alternate days of the week. If people were given additional treatments within chlorpromazine, we planned to only include data if the adjunct treatment was evenly distributed between groups and it was only the chlorpromazine that was randomised.
Types of participants
We included adults, however defined, with schizophrenia or related disorders, including schizophreniform disorder, schizoaffective disorder and delusional disorder, again, by any means of diagnosis. If we found a trial where there was a range of diagnoses, we only included it if the majority of participants had schizophrenia.
We are interested in making sure that information is as relevant to the current care of people with schizophrenia as possible, and so proposed to clearly highlight the current clinical state (acute, early post‐acute, partial remission, remission) as well as the stage (prodromal, first episode, early illness, persistent) and whether the studies primarily focused on people with particular problems (for example, negative symptoms, treatment‐resistant illnesses).
Types of interventions
1. Chlorpromazine
Any dose, any method of administration
2. Clotiapine
Any dose, any method of administration
Types of outcome measures
We divided outcomes into short‐term (less than six months), medium‐term (7 to 12 months), and long‐term (over one year).
Primary outcomes
1. Global state
1.1 Clinically significant improvement in global state, as defined by each study
2. Mental state
2.1 Clinically important change in mental state, as defined by each study
3. Adverse events
3.1 Incidence of clinically important movement disorder, as defined by each study
Secondary outcomes
1. Global state
1.1 Average scores for global state 1.2 Relapse
2. Mental state
2.1 General symptoms ‐ prevalence or average scores 2.2 Specific symptoms ‐ prevalence or average scores 2.2.1 Positive symptoms (delusions, hallucinations, disordered thinking) 2.2.2 Negative symptoms (avolition, poor self care, blunted affect) 2.2.3 Mood ‐ depression
3. Adverse effects
3.1 General ‐ prevalence or average scores 3.2 Specific ‐ prevalence or average scores 3.2.1 Deaths by suicide or natural causes 3.2.2 Movement disorders (extrapyramidal side effects, specifically tardive dyskinesia and neuroleptic malignant syndrome) 3.2.3 Sedation 3.2.4 Dry mouth 3.2.5 Others ‐ categorised by system
4. Leaving the study
5. Behaviour
5.1 General behaviour ‐ prevalence or average scores 5.2 Specific behaviour ‐ prevalence or average scores 5.2.1 Social functioning 5.2.2 Employment status during trial (employed/unemployed) 5.2.3 Occurrence of violent incidents (to self, others, or property)
6. Service use
6.1 Days in hospital 6.2 Readmission due to relapse 6.3 Discharge from hospital (see Differences between protocol and review)
7. Quality of life
7.1 Important or average change in person's quality of life as defined by each study
8. Satisfaction with care
8.1 Important or average change in satisfaction of participant or care provider as defined by each study
9. Cost of care
'Summary of findings' table
We used the GRADE approach to interpret findings (Schünemann 2011) and used GRADEpro GDT to export data from our review to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined, and the sum of available data on all outcomes we rated as important to patient care and decision‐making. We selected the following main outcomes for inclusion in the 'Summary of findings' table.
Global State ‐ clinically important change in global state as defined by each study
Mental State ‐ general ‐ clinically important change in mental state as defined by each study
Mental State ‐ specific ‐ average change in negative symptoms
Adverse effects ‐ incidence of serious adverse events/effects
Adverse effects ‐ clinically important extrapyramidal symptoms
Leaving the study early ‐ for any reason
Cost of care
Search methods for identification of studies
Electronic searches
1. Cochrane Schizophrenia's trials register
The Information Specialist (IS) searched Cochrane Schizophrenia's Study‐Based Register of Controlled Trials using the following search strategy.
(Chlorpromazine AND Clotiapine) in Intervention Field of STUDY
Cochrane Schizophrenia's register of trials is compiled by systematic searches of major resources (including AMED, BIOSIS, CINAHL, EMBASE, MEDLINE, PsycINFO, PubMed, and registries of clinical trials) and their monthly updates, handsearches, grey literature, and conference proceedings (see Group Module). There is no language, date, document type, or publication status limitations for inclusion of records into the register.
Searching other resources
1. Reference searching
We inspected references of all included studies for further relevant studies.
2. Personal contact
For this review, we did not contact the first author of each included study for information regarding unpublished trials.
Data collection and analysis
Selection of studies
ASG and SE independently inspected citations from the searches and identified relevant abstracts. SM and ASE independently re‐inspected a random 20% sample to ensure reliability. ASG and SE obtained and inspected the full reports of the abstracts meeting the review criteria. Again, SM and ASE re‐inspected a random 20% of reports in order to ensure reliable selection. If it had not been possible to resolve disagreement by discussion, we would have attempted to contact the authors of the study for clarification, and if we could not resolve the disagreement we would not have included the trial, but placed it in the table 'Characteristics of studies awaiting classification' until a resolution was made. We included studies that met our inclusion criteria and reported useable data. We would have excluded studies that either did not meet our inclusion criteria or met our inclusion criteria but did not report useable data.
Data extraction and management
1. Extraction
Review authors AB and MZ extracted data from all included studies. In addition, to ensure reliability, SM and ASG independently extracted data from a random sample of these studies, comprising 10% of the total. We extracted data presented only in graphs and figures whenever possible, but only included the data if two reviewers independently had the same result. We attempted to contact study authors through an open‐ended request in order to obtain missing information, or for clarification, whenever necessary. If studies had been multi‐centred, where possible, we would have extracted data relevant to each component centre separately.
2. Management
2.1 Forms
We extracted data onto standard, simple forms provided by Cochrane Schizophrenia.
2.2 Scale‐derived data
We included continuous data from rating scales only if:
the psychometric properties of the measuring instrument had been described in a peer‐reviewed journal (Marshall 2000);
the measuring instrument had not been written or modified by one of the trialists for that particular trial;
the instrument was a global assessment of an area of functioning and not sub‐scores which were not, in themselves, validated or shown to be reliable. However there would be exceptions: we would include sub‐scores from mental state scales measuring positive and negative symptoms of schizophrenia.
Ideally the measuring instrument should either be i. a self‐report, or ii. completed by an independent rater or relative (not the therapist). We realise that this is not often reported clearly; in 'Description of studies' in the Results section we have noted if this is the case or not.
2.3 Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. On the other hand, calculation of change needs two assessments (baseline and endpoint), which can be difficult in unstable and difficult‐to‐measure conditions such as schizophrenia. We intended to primarily use endpoint data, and only use change data if the former were not available. We would have, where necessary, combined endpoint and change data in the analysis as we planned to use mean differences (MDs) rather than standardised mean differences (SMDs) throughout (Deeks 2011).
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we applied the following standards:
For change data
We entered change data, as when continuous data are presented on a scale that included a possibility of negative values (such as change data), it was difficult to tell whether data were skewed or not. We presented and entered change data into statistical analyses.
For endpoint data from studies with fewer than 200 participants:
When a scale started from the finite number 0, we subtracted the lowest possible value from the mean, and divided this by the standard deviation (SD). If this value was lower than 1, it strongly suggested a skew, and we would exclude such data. If this ratio was higher than 1 but below 2, there was a suggestion of skew. We would enter such data and test whether its inclusion or exclusion changed the results substantially. Finally, if the ratio was larger than 2, we would include such data because skew was less likely (Altman 1996; Higgins 2011a).
If a scale started from a positive value (such as the Positive and Negative Syndrome Scale (PANSS), which could have values from 30 to 210) (Kay 1986), we modified the calculation described above to take into account the scale starting point. In such cases skew is present if 2 SD > (S ‐ S min), where S was the mean score and S min was the minimum score.
(Please note, irrespective of the above rules, we would enter endpoint data from studies of at least 200 participants in the analysis because skewed data pose less of a problem in large studies).
2.5 Common measure
To facilitate comparison between trials, we intended to convert variables that could be reported in different metrics, such as days in hospital (mean days per year, per week or per month) to a common metric (e.g. mean days per month).
2.6 Conversion of continuous to binary
Where possible, we made efforts to convert outcome measures to dichotomous data. This can be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It is generally assumed that if there is a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or PANSS) (Kay 1986), this can be considered as a clinically significant response (Leucht 2005). If data based on these thresholds were not available, we used the primary cut‐off presented by the original study authors.
2.7 Direction of graphs
Where possible, we entered data in such a way that the area to the left of the line of no effect indicated a favourable outcome for clotiapine. Where keeping to this made it impossible to avoid outcome titles with clumsy double‐negatives (e.g. 'Not un‐improved') we reported data where the left of the line indicated an unfavourable outcome and noted this in the relevant graphs.
Assessment of risk of bias in included studies
Again review authors MZ and AB worked independently to assess risk of bias by using criteria described in the Cochrane Handbook for Systemic Reviews of Interventions (Higgins 2011b) to assess trial quality. This set of criteria is based on evidence of associations between an overestimate of effect and high risk of bias of the article, such as sequence generation, allocation concealment, blinding, incomplete outcome data and selective reporting.
If the raters disagreed, we made the final rating by consensus, with the involvement of another member of the review group (SM). Where inadequate details of randomisation and other characteristics of trials were provided, we contacted authors of the studies in order to obtain, if possible, further information. We reported non‐concurrence in quality assessment, but if disputes arose as to which category a trial was to be allocated, again, we attempted to resolve by discussion.
Measures of treatment effect
1. Binary data
For binary outcomes we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive (Boissel 1999) than odds ratios (ORs) and that ORs tend to be interpreted as RR by clinicians (Deeks 2000). The number needed to treat for an additional beneficial outcome/harmful outcome (NNTB/NNTH) statistic with its CIs is intuitively attractive to clinicians, but is problematic both in its accurate calculation in meta‐analyses and interpretation (Hutton 2009). For binary data presented in the 'Summary of findings' table, where possible, we calculated illustrative comparative risks.
2. Continuous data
For continuous outcomes we estimated the MD between groups. We preferred not to calculate effect size measures (SMD). However, if scales of very considerable similarity were used, we would presume that there was a small difference in measurement, and we would have calculated effect size and transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster‐randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, study authors often fail to account for intra‐class correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992), whereby P values are spuriously low, CIs unduly narrow, and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
If clustering had not been accounted for in primary studies, we would have presented data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. Where clustering was incorporated into the analysis of primary studies, we would have presented these data as if from a non‐cluster randomised study, but adjusted for the clustering effect.
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the ICC (Design effect = 1 + (m‐1) * ICC) (Donner 2002). If the ICC was not reported we would assume it to be 0.1 (Ukoumunne 1999).
If cluster studies have been appropriately analysed taking into account ICC and relevant data documented in the report, synthesis with other studies would be possible using the generic inverse variance technique.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state, despite a wash‐out phase. For the same reason, cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, we only used data from the first phase of cross‐over studies.
3. Studies with multiple treatment groups
Where a study involved more than two treatment arms, if relevant, we presented additional treatment arms in comparisons. If data were binary we simply added and combined within the two‐by‐two table. If data were continuous we would have combined data following the formula in section 7.7.3.8 (Combining groups) of the Cochrane Handbook for Systemic Reviews of Interventions (Higgins 2011a). Where the additional treatment arms were not relevant, we did not use these data.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss of follow‐up, data must lose credibility (Xia 2009). We chose that, for any particular outcome, should more than 50% of data be unaccounted for, we would not reproduce these data or use them within analyses. If, however, more than 50% of those in one arm of a study were lost, but the total loss was less than 50%, we would address this within the 'Summary of findings' table by downgrading quality. We also downgraded quality within the 'Summary of findings' table, should total loss be 25% to 50%.
2. Binary
Where attrition for a binary outcome was between 0% and 50% and where these data were not clearly described, we presented data on a 'once‐randomised‐always‐analyse' basis (an intention‐to‐treat analysis (ITT)). Where studies did not use an ITT analysis, we presented completer‐only data.
3. Continuous
3.1 Attrition
Where attrition for a continuous outcome was between 0% and 50%, and data only from people who completed the study to that point were reported, we used and presented these data.
3.2 Standard deviations
If SDs were not reported, we first tried to obtain the missing values from the study authors. If not available, where there were missing measures of variance for continuous data, but an exact standard error (SE) and CIs available for group means, and either P value or t value available for differences in mean, we could calculate them according to the rules described in the Cochrane Handbook for Systemic Reviews of Interventions (Higgins 2011a). When only the SE was reported, we calculated SDs by the formula SD = SE * square root (n). Chapters 7.7.3 (Higgins 2011a) and 16.1.3 (Higgins 2011c) of the Cochrane Handbook for Systemic Reviews of Interventions present detailed formulas for estimating SDs from P values, t or F values, CIs, ranges, or other statistics. If these formulae did not apply, we would calculate the SDs according to a validated imputation method based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study's outcome, and thus to lose information. We nevertheless examined the validity of the imputations in a sensitivity analysis, excluding imputed values.
3.3 Assumptions about participants who left the trials early or were lost to follow‐up
Various methods are available to account for participants who leave trials early or are lost to follow‐up. Some trials just present the results of study completers, others use the method of last observation carried forward (LOCF), while more recent methods, such as multiple imputation or mixed‐effects models for repeated measurements (MMRM), have become more of a standard. While the latter methods seem to be somewhat better than LOCF (Leon 2006), we feel that the high percentage of participants leaving the studies early and differences in the reasons for leaving the studies early between groups is often the core problem in randomised schizophrenia trials. We did not, therefore exclude studies based on the statistical approach used but preferred more sophisticated approaches. For example, We preferred MMRM or multiple‐imputation to LOCF, and only presented completer analyses if some kind of ITT data were not available at all. Moreover, we addressed this issue in the 'Incomplete outcome data' domain of the 'Risk of bias' tool.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We simply inspected all studies for clearly outlying people or situations that we had not predicted would arise, and would have fully discussed outliers.
2. Methodological heterogeneity
We considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We simply inspected all studies for clearly outlying methods which we had not predicted would arise and would have discussed such methodological outliers if they arose.
3. Statistical heterogeneity
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I2 statistic
We investigated heterogeneity between studies by considering the I2 method alongside the Chi2 P value. The I2 provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of I2 depends on i. magnitude and direction of effects, and ii. strength of evidence for heterogeneity (e.g. P value from Chi2 test, or a CI for I2). We would have interpreted an I2 estimate greater than or equal to around 50%, accompanied by a statistically significant Chi2 test as statistical evidence of substantial levels of heterogeneity (Deeks 2011). If we had found substantial levels of heterogeneity in the primary outcome, we would have explored reasons for heterogeneity (Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
1. Protocol versus full study
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results. These are described in the Cochrane Handbook for Systemic Reviews of Interventions (Sterne 2011). We tried to locate protocols of included randomised trials. If the protocol was available, we compared outcomes in the protocol with those in the published report. If the protocol was not available, we compared outcomes listed in the methods section of the trial report with actually reported results.
2. Funnel plot
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in the Cochrane Handbook for Systemic Reviews of Interventions (Sterne 2011). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We would not use funnel plots for outcomes where there were 10 or fewer studies, or where all studies were of similar size. In future versions if funnel plots are possible, we will seek statistical advice in their interpretation.
Data synthesis
We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This often seems to be true to us and the random‐effects model takes into account differences between studies even if there is no statistically significant heterogeneity. There is, however, a disadvantage to the random‐effects model; it puts added weight onto small studies which often are the most biased ones. Depending on the direction of effect, these studies can either inflate or deflate the effect size. We chose a random‐effects model for all analyses.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses
1.1 Primary outcomes
We did not anticipate any subgroup analyses.
1.2 Clinical state, stage or problem
We proposed to undertake this review and provide an overview of the effects of chlorpromazine compared with clotiapine for people with schizophrenia in general. In addition, however, if relevant data had been available we would have reported data on subgroups of people in the same clinical state, stage and with similar problems.
2. Investigation of heterogeneity
If inconsistency had been high, we would have reported this. First, we would have investigated whether data had been entered correctly. Second, if data were correct, we would have visually inspected the graph, and successively removed studies outside of the company of the rest to see if homogeneity was restored. For this review we decided that, should this occur with data contributing to the summary finding of no more than around 10% of the total weighting, we would have presented such data. If not, we would not pool data, but would have discussed the issues. We knew of no supporting research for this 10% cut‐off, but were investigating use of prediction intervals as an alternative to this unsatisfactory state.
When unanticipated clinical or methodological heterogeneity was obvious, we simply stated hypotheses regarding these for future reviews or versions of this review. We did not anticipate undertaking analyses relating to clinical or methodological heterogeneity.
Sensitivity analysis
1. Implication of randomisation
We would have included trials in a sensitivity analysis if they had been described in some way as to imply randomisation. For the primary outcomes, if there had been no substantive difference when we added the implied randomised studies to those with better descriptions of randomisation, then we would have employed all data from these studies. If there was a substantive difference then we would have presented these data as 'other data'.
2. Assumptions for lost binary data
Where assumptions had to be made regarding people lost to follow‐up (see Dealing with missing data) we compared the findings of the primary outcomes when we used our assumptions and when we used data only from people who completed the study to that point. If there had been a substantial difference, we would have reported results and discussed them, but continued to employ our assumptions.
If assumptions were needed regarding missing SDs (see Dealing with missing data), we would have compared the findings of the primary outcomes when we used our assumptions and when we used data only from people who completed the study to that point. We would have undertaken a sensitivity analysis, testing how prone results were to change when we only compared completer‐only data to the imputed data using the above assumptions. If there had been a substantial difference, we would have reported results and discussed them, but continued to employ our assumptions.
3. Risk of bias
We would have analysed the effects of excluding trials that were judged to be at high risk of bias across one or more of the domains (see Assessment of risk of bias in included studies) for the meta‐analysis of the primary outcome. If the exclusion of trials at high risk of bias did not substantially alter the direction of effect or the precision of the effect estimates, then we would have included data from these trials in the analysis.
4. Imputed values
We would have undertaken a sensitivity analysis to assess the effects of including data from trials if we had used imputed values for ICC in calculating the design effect in cluster‐randomised trials.
If we noted substantial differences in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we would not pool data from the excluded trials with the other trials contributing to the outcome, but would present them separately.
5. Fixed‐effect and random‐effects
We synthesised all data using a random‐effects model. However, we also synthesised data for the primary outcome using a fixed‐effect model to evaluate whether this altered the significance of the results.
Results
Description of studies
Results of the search
The PRISMA table shows results of our search Figure 3 (Moher 2009).
3.
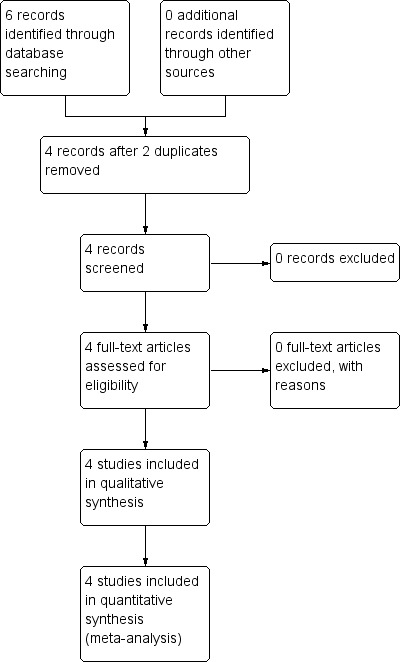
Study flow diagram of trial selection from 2016 electronic search
In the original search we found six reports that were potentially relevant. After removing two duplicates, we inspected full texts of the remaining four reports.
Included studies
All four full‐text articles referred to individual studies and we were able to include all four studies.
1. Methods
One study was a cross‐over trial (Schliefer 2003) and the three remaining studies were parallel trials (Jacobsson 1974; Kaneko 1969; Van Wyk 1971). All included studies were randomised ‐ one, Kaneko 1969, implied randomisation.
2. Length of trials
Trial duration ranged from four weeks (Jacobsson 1974) to 12 weeks (Schliefer 2003, Van Wyk 1971).
3. Participants
Three studies reported that they included people with schizophrenia, one (Jacobsson 1974) included participants with acute psychotic syndromes including "schizophrenic type" illnesses. Only Kaneko 1969 reported the diagnostic criteria ("cases with catatonic excitation, marked irritability, violence and manneristic act; hallucination; delusion; deficiency of initiative and apathy") and described the symptoms required for participants to be included in the study. In total, we included 276 participants in this review.
4. Setting
All included studies were conducted in hospitals.
5. Study size
The average number of participants was 69, ranging from 49 (Jacobsson 1974) to 101 (Van Wyk 1971).
6. Intervention
6.1 Chlorpromazine
All the included studies compared chlorpromazine with clotiapine. The doses of chlorpromazine in the included studies ranged from 40 mg/day to 600 mg/day. The mean dose of chlorpromazine provided by Jacobsson 1974 was 404.5 mg/day ().
6.2 Clotiapine
The doses of clotiapine in the included studies ranged from 40 mg/day (Kaneko 1969) to 240 mg/day (Jacobsson 1974; Kaneko 1969). The mean dose was 125.2 mg/day in Jacobsson 1974. The other studies provided no further details for the prescribed dose of the medication.
6.3 Other treatments
Van Wyk 1971 also included one more treatment arm, thioridazine, in addition to chlorpromazine and clotiapine. We did not include data from this group.
7. Outcomes
Binary and continuous data were available. Outcomes reported by the studies included mental state, leaving the study early and adverse events. None of the included studies reported global state, quality of life, cost of care or behaviour.
7.1 Outcome scales
The following scales provided continuous data for the analyses.
7.1.2 Mental state
i. Positive and Negative Syndrome Scale (PANSS) (Kay 1987)
A brief rating scale used to assess the severity of positive and negative symptoms of schizophrenia, this scale has been reported as an operationalised, drug‐sensitive instrument, providing balanced representation of positive and negative symptoms as well as measuring their relationship to one another. Using PANSS, the patient is rated from 1 to 7 on 30 different symptoms based on interview in addition to reports of family members or clinical staff. PANSS scores can range from 30 to 210, high scores indicating more severity of positive, negative or global psychopathology.
ii. Martens' Symptom Scale (S‐Scale) (Helgason 1983)
This scale is designed for measuring possible improvements in people with schizophrenia. This scale comprises 23 items, measuring severity of signs or symptoms. Two items are bipolar with 9 possible scores, the others are unipolar with 5 possible scores (ranging from absence to high severity of a sign or a symptom).
Excluded studies
We did not exclude any studies. This was partly because the search was very specific.
Awaiting assessment
No studies are awaiting assessment.
Ongoing studies
We did not identify any ongoing studies.
Risk of bias in included studies
Please also see Figure 4 and Figure 5.
4.
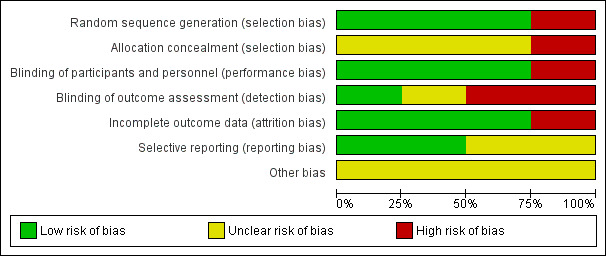
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies
5.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study
Allocation
Three studies (Jacobsson 1974, Schliefer 2003, Van Wyk 1971) were randomised, however only Jacobsson 1974 reported the method used for generating random allocation. In Jacobsson 1974 participants were randomised by pre‐established randomised codes. None of these three studies described how allocation was concealed. Kaneko 1969 did not provide clear information for either randomisation or allocation concealment. Randomisation was implied and we rated this study high risk for selection bias.
Blinding
Jacobsson 1974, Kaneko 1969 and Schliefer 2003 were double‐blind and had low risks of bias for performance bias. Identical capsules and tablets were reported as the most common procedure for blinding both participants and personnel. Van Wyk 1971 had high risk of performance bias as it reported that due to administrative and staff shortage, they couldn't introduce any blind procedures into the trial. Detection bias (blinding of outcome assessment) varied. Schliefer 2003 was low risk, reporting that trained observers were blind to the participants' treatment status. Jacobsson 1974 was high risk, mentioning that a separate envelope for each participant was provided, in case it should be necessary to know what drug an individual participant was receiving, and Kaneko 1969 was at unclear risk of bias, with no clear information regarding blinding of outcome assessment.
Incomplete outcome data
Three included studies (Jacobsson 1974; Kaneko 1969; Schliefer 2003) had low risk of bias for incomplete outcome data. Only Van Wyk 1971 did not provide information about missing data and participants who left the study early.
Selective reporting
Two studies (Jacobsson 1974; Kaneko 1969) reported data for all outcomes. Schliefer 2003 and Van Wyk 1971 did not provide pre‐specified outcomes so it is unclear if they reported all outcomes.
Other potential sources of bias
Schliefer 2003 was financially supported by a research grant from Stanley Medical Research Institute and had no wash‐out period for the study. Jacobsson 1974 and Van Wyk 1971 declared who supplied their drugs but did not report the source of funding. Kaneko 1969 was also unclear about their funding source.
Effects of interventions
See: Table 1
Studies relevant to this review fell into a single comparison. We were able to extract numerical data from four randomised studies.
1. Comparison 1: chlorpromazine versus clotiapine
1.1 Mental state: 1a. General symptoms ‐ average change score (PANSS total, high = good)
For this outcome we found a single study, with a total of 31 people. We found evidence of a clear difference, favouring clotiapine, between chlorpromazine and clotiapine (MD 11.50, 95% CI 9.42 to 13.58, Analysis 1.1).
1.1. Analysis.
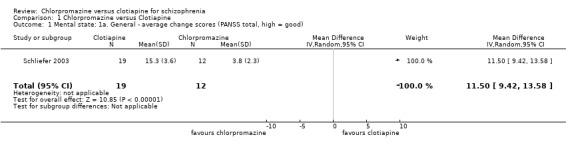
Comparison 1 Chlorpromazine versus Clotiapine, Outcome 1 Mental state: 1a. General ‐ average change scores (PANSS total, high = good).
1.2 Mental state: 1b. General symptoms ‐ average change score (S‐scale, high = poor)
There was a clear difference, favouring chlorpromazine between chlorpromazine and clotiapine (1 RCT, N = 38, MD 1.45, 95% CI 0.38 to 2.52, Analysis 1.2).
1.2. Analysis.
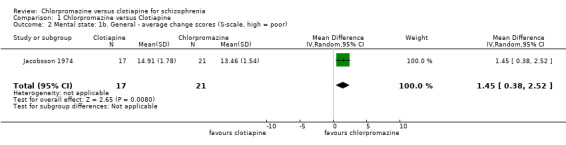
Comparison 1 Chlorpromazine versus Clotiapine, Outcome 2 Mental state: 1b. General ‐ average change scores (S‐scale, high = poor).
1.3 Mental state: 2a. Specific ‐ positive symptoms ‐ frequency
A single study (Kaneko 1969) reported incidence of positive and negative symptoms (Analysis 1.3).
1.3. Analysis.
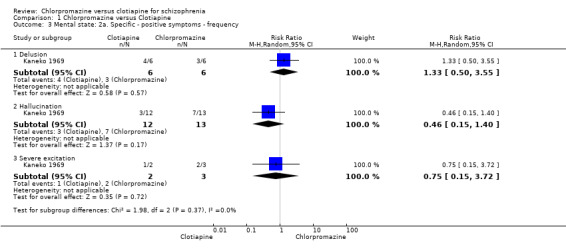
Comparison 1 Chlorpromazine versus Clotiapine, Outcome 3 Mental state: 2a. Specific ‐ positive symptoms ‐ frequency.
We found no evidence of a clear difference between chlorpromazine and clotiapine for several specific symptoms (Analysis 1.3).
1.3.1 Delusions: 1 RCT, N = 12, RR 1.33, 95% CI 0.50 to 3.55
1.3.2 Hallucinations: 1 RCT, N = 25, RR 0.46, 95% CI 0.15 to 1.40
1.3.3 Severe excitation: 1 RCT, N = 5, RR 0.75, 95% CI 0.15 to 3.72
1.4 Mental state: 2a. Specific ‐ positive symptoms ‐ average change score (PANSS‐positive, high = good)
Kaneko 1969 also provided mental state data, reporting the average change scores on the PANSS for both positive and negative symptoms.
For delusions we found evidence of a clear effect, favouring clotiapine (1 RCT, N = 11, MD 7.36, 95% CI 3.54 to 11.18) but no effect for the outcome of 'hallucinations' (1 RCT, N = 16, MD ‐3.00, 95% CI ‐6.75 to 0.75, Analysis 1.4).
1.4. Analysis.
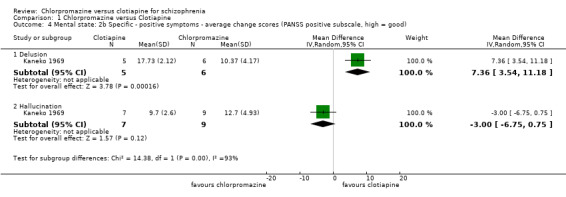
Comparison 1 Chlorpromazine versus Clotiapine, Outcome 4 Mental state: 2b Specific ‐ positive symptoms ‐ average change scores (PANSS positive subscale, high = good).
1.5 Mental state: 2c. Specific ‐ negative symptoms ‐ average change scores (PANSS‐negative, high = good)
For the outcome of 'deficiency of initiative and apathy' we found no evidence of a clear difference between chlorpromazine and clotiapine (MD ‐0.97, 95% CI ‐2.76 to 0.82, Analysis 1.5).
1.5. Analysis.
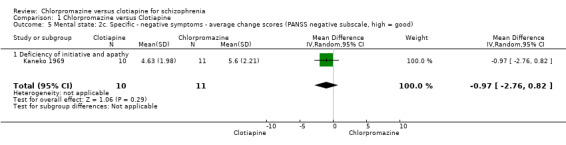
Comparison 1 Chlorpromazine versus Clotiapine, Outcome 5 Mental state: 2c. Specific ‐ negative symptoms ‐ average change scores (PANSS negative subscale, high = good).
1.6 Mental state: 2d. Specific ‐ various symptoms ‐ average change score (S‐Scale, high = poor)
Jacobsson 1974 (N = 49) measured mental state using Martens' Symptom Scale (S‐Scale). Findings were varied. For the outcome of 'delusions' we did not find evidence that chlorpromazine was clearly different in its effects compared with clotiapine (1 RCT, N = 38, MD ‐0.06, 95% CI ‐0.3 to 0.18). For 'hallucinations', however, we did find evidence of an effect, favouring clotiapine (1 RCT, N = 38, MD ‐0.24, 95% CI ‐0.46 to ‐0.02, Analysis 1.6). We also found evidence that chlorpromazine was better in its effects compared with clotiapine for 'influence of thought' (1 RCT, N = 38, MD 0.28, 95% CI 0.17 to 0.39) but no evidence of a clear difference between chlorpromazine and clotiapine for 'splitting' (1 RCT, N = 38, MD 0.01, 95% CI ‐ 0.09 to 0.11, Analysis 1.6).
1.6. Analysis.
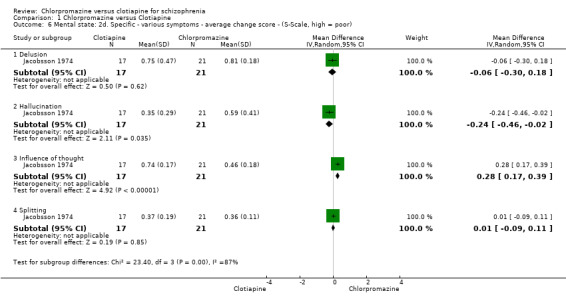
Comparison 1 Chlorpromazine versus Clotiapine, Outcome 6 Mental state: 2d. Specific ‐ various symptoms ‐ average change score ‐ (S‐Scale, high = poor).
1.7 Adverse effects 1. Central nervous system ‐ short term
Kaneko 1969 (N = 68) reported on several general central nervous system effects. The study did not find evidence of a clear difference between chlorpromazine and clotiapine for drowsiness (RR 1.89, 95% CI 0.98 to 3.63), somnipathy (RR 1.38, 95% CI 0.63 to 2.99), unsteadiness (RR 1.71, 95% CI 0.77 to 3.82) nor weakness (RR 0.75, 95% CI 0.42 to 1.34, Analysis 1.7).
1.7. Analysis.
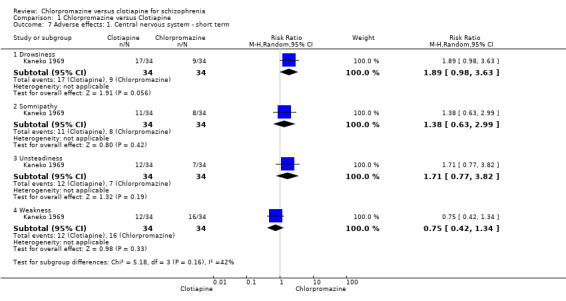
Comparison 1 Chlorpromazine versus Clotiapine, Outcome 7 Adverse effects: 1. Central nervous system ‐ short term.
1.8 Adverse effects: 2. Movement disorders ‐ short term
There was not a clear difference between chlorpromazine and clotiapine for the outcome of akathisia (1 RCT, N = 68, RR 3.00, 95% CI 0.65 to 13.83), dyskinesia (1 RCT, N = 68, RR 3.00, 95% CI 0.13 to 71.15) nor Parkinsonism (2 RCTs, N = 106, RR 1.17, 95% CI 0.87 to 1.57, Analysis 1.8).
1.8. Analysis.
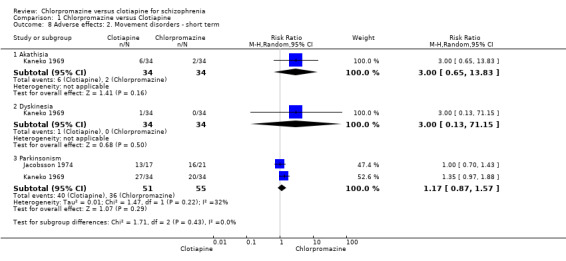
Comparison 1 Chlorpromazine versus Clotiapine, Outcome 8 Adverse effects: 2. Movement disorders ‐ short term.
1.9 Adverse effects: 3. Various other ‐ short term
Kaneko 1969 (N = 68) reported 'weight gain (of not less than 3 kg at the end of study)' and did not find a clear difference between chlorpromazine and clotiapine (RR 1.12, 95% CI 0.71 to 1.75). The same study also reported 'thirst' and, again, found no clear difference between the drugs (RR 1.29, 95% CI 0.54 to 3.06, Analysis 1.9).
1.9. Analysis.
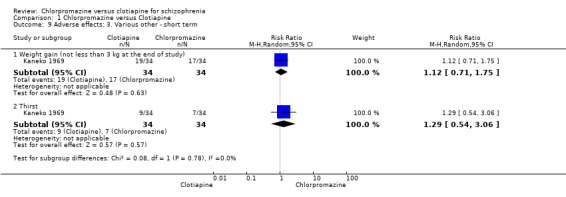
Comparison 1 Chlorpromazine versus Clotiapine, Outcome 9 Adverse effects: 3. Various other ‐ short term.
1.10 Leaving the study early ‐ short term
Around 17% left the studies but there was no clear difference between chlorpromazine and clotiapine (3 RCTs, N = 158, RR leaving for any reason 0.68, 95% CI 0.24 to 1.88), this outcome had moderate levels of heterogeneity (I2 = 47%). Attrition fell to about 14% when the reason of 'due to deterioration or adverse effects' was reported (2 RCTs, N = 117, RR 1.23, 95% CI 0.51 to 2.99) but there was still no difference between groups (Analysis 1.10).
1.10. Analysis.
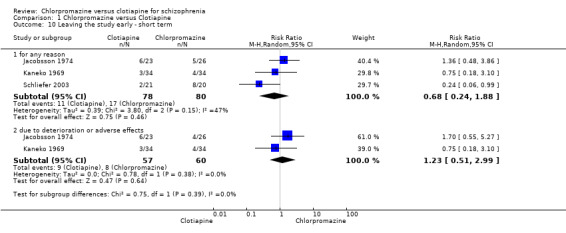
Comparison 1 Chlorpromazine versus Clotiapine, Outcome 10 Leaving the study early ‐ short term.
1.11 Service use: discharge from hospital ‐ short term (12 weeks)
Only Van Wyk 1971 (N = 70), reported data on numbers of participants discharged from hospital by 12 weeks. There is evidence of an effect favouring clotiapine for this outcome (RR 1.49, 95% CI 1.07 to 2.08, Analysis 1.11).
1.11. Analysis.
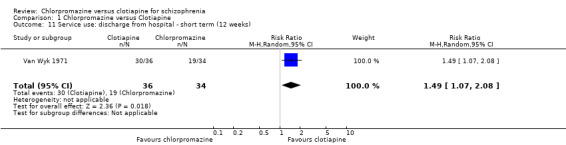
Comparison 1 Chlorpromazine versus Clotiapine, Outcome 11 Service use: discharge from hospital ‐ short term (12 weeks).
We also performed analysis of subclasses of mental state by positive symptoms and the results are reported in Table 3.
2. Mental state: improvement for positive symptoms.
| Outcome or subgroup | Studies | Participants | Statistical method | Effect estimate |
| Mental state: average scores for improvement of positive symptoms (high score = improvement) | 1 | 27 | Mean difference (IV, random, 95% CI) | 2.17 [‐7.98, 12.33] |
| 1. Hallucination | 1 | 16 | Mean difference (IV, random, 95% CI) | ‐3.00 [‐6.75, 0.75] |
| 2. Delusion | 1 | 11 | Mean difference (IV, random, 95% CI) | 7.36 [3.54, 11.18] |
| Mental state: Improved positive symptoms (frequency of marked effects) | 1 | 42 | Risk ratio (M‐H, random, 95% CI) | 0.82 [0.41, 1.62] |
| 1. Hallucination | 1 | 25 | Risk ratio (M‐H, random, 95% CI) | 0.46 [0.15, 1.40] |
| 2. Delusion | 1 | 12 | Risk ratio (M‐H, random, 95% CI) | 1.33 [0.50, 3.55] |
| 3. Severe excitation | 1 | 5 | Risk ratio (M‐H, random, 95% CI) | 0.75 [0.15, 3.72] |
Discussion
Summary of main results
The summary below refers to the outcomes selected for Table 1, and highlights the important findings of this review for evidence‐based decision making.
1. Global state
We could not extract any usable data from the trials comparing chlorpromazine with clotiapine, therefore it is not possible to draw any conclusions about whether clotiapine is more or less effective in improving the global state of people with schizophrenia. This is a real omission of even the few trials we have identified.
2. Mental state: general
Only one trial specifically reported mental state as measured by the PANSS total scores. Results favoured efficacy of chlorpromazine compared with clotiapine but these are short‐term, very low‐quality data, from one tiny trial, reporting a proxy outcome (continuous measure rather than binary), the interpretation of which is problematic in clinical life. A meta‐analysis of three randomised trials that evaluated the global improvements in people with acute psychotic disorders ‐ rather than more narrowly defined and stable schizophrenia ‐ indicated no distinct superiority of clotiapine compared with other standard treatments like chlorpromazine (Berk 2004). At best, this current review suggests no dramatic difference between the two compounds.
3. Mental state: specific
Our findings fail to indicate a clear difference between the efficacy of chlorpromazine versus clotiapine for improving negative symptoms, and both drugs were not effective in improving negative symptoms. This is an important outcome and there is little convincing evidence from any comparison of any treatment for people with schizophrenia that negative symptoms are responsive to drugs. The call for larger, longer, better trials in this area is not specific to the comparison which was the focus of this review.
4. Adverse effects: incidence of movement disorders, and clinically significant extrapyramidal symptoms
For the limited data we had to work with, chlorpromazine and clotiapine seem to produce a similar profile of movement disorders in people with schizophrenia. However, another review suggested that clotiapine may induce fewer movement disorders (Parkinsonism symptoms) (Berk 2004). This is important and merits further investigation of safety and tolerability profiles.
5. Leaving the study early
These studies were short. Around 17% of both groups left early (any reason) and 14% when it came to the specific reason of deterioration or adverse effects. These figures compare favourably to many of the more modern trials where attrition rate is far greater ‐ even over a matter of a few weeks. There seems to be no implication in the studies that attrition was for different reasons in each group. More detailed information would have been helpful and it might have been possible to extract outcome data, even on those who left. Nevertheless, we found no evidence that one or other drug was more unacceptable to either trial participants, or researchers.
7. Service use
At the end of 12 weeks in one study around 80% of people receiving clotiapine were able to be discharged from hospital compared with around 50% receiving chlorpromazine. This was statistically significant. If reproduced this is a most important result. If clotiapine is genuinely faster at getting people better this is very important with enormous implications. This result needs to be reinvestigated.
6. Cost of care
Again, we could not extract any usable data from the trials comparing chlorpromazine with clotiapine, therefore it is not possible to draw any conclusions about whether chlorpromazine is more or less effective than clotiapine in terms of cost. However, as there is little difference in the cost of the drugs, all else being equal, then service use is a good proxy for cost. Should the finding for service use be true and clotiapine really does get people better, faster, then savings by use of clotiapine would be huge.
Overall completeness and applicability of evidence
1. Completeness
There are some relevant limitations for the conclusions of this meta‐analysis, which should be pointed out. Of the four included studies, almost all of them provided some data on the primary outcomes mental state and adverse effects. However, no study reported usable data for the primary outcome 'global state'. Therefore, the evidence on this primary outcome is not complete. In addition, only three studies reported on secondary outcomes. The evidence on prespecified adverse effects is particularly incomplete, as none of the included trials specifically reported important side effects such as death, suicide or cardiac effects. There were also no usable data on behaviour, quality of life, service use, satisfaction with care or cost of care. The total number of participants in the included studies is extremely limited, which can lead to inconclusive results (Davey Smith 1998), regardless of whether the estimations were significant. Therefore, new large trials with better outcome reporting are needed for clear interpretation and precise decisions about the differences between the efficacy of chlorpromazine and clotiapine. These outcomes do not need to depend on sophisticated rating but could be clear, binary and pragmatic.
2. Applicability
2.1 Diagnosis
All of the included studies except Schliefer 2003 were conducted over 40 years ago, which could have led to serious limitations for applicability of this review. Almost all the included studies in this review provided less rigorous criteria for diagnosis of schizophrenia than would, perhaps, be seen in more modern trials. It is likely that some included participants in those studies would reflect other diagnosis than schizophrenia. However, this is rather like normal clinical practice where diagnosis is not an exact science.
2.2 Setting
All included studies were conducted in hospitals and inpatient settings. However, today much schizophrenia is diagnosed and treated in the early stages in the community, which could reduce the applicability of this review for current practice.
Quality of the evidence
The data reported are limited and poor quality; we rated available evidence as very low based on GRADE (Schünemann 2011). Three studies stated that they were randomised and provided information about how this was achieved. One study said it was randomised but provided no information about the randomisation method. Three studies had serious risk of bias for allocation concealment. One study was not blinded. Two studies reported that the assessor was not blinded to which drug the participants were receiving. We also detected inconsistency, imprecision and serious publication bias detected for two studies. None of the included studies reported usable data for most important outcomes as they provided no standard deviation or standard error of mean for estimations.
Potential biases in the review process
The search was based on Cochrane Schizophrenia's Trials Register. There may be some unpublished trials that we are not aware of. At the time this review's trials were published there was pharmaceutical industry interest in the findings and this could have led to publication or reporting bias.
Agreements and disagreements with other studies or reviews
We are not aware of any other systematic reviews on the efficacy of chlorpromazine versus clotiapine for schizophrenia ‐ but would be really interested in finding any.
Authors' conclusions
Implications for practice.
1. For people with schizophrenia
Where clotiapine is available it is unclear of its advantages over chlorpromazine. It may be better at improving mental state compared to chlorpromazine but data are really so limited and of such poor quality that it is impossible to be certain of this. Schizophrenia is such a potentially difficult and damaging illness that it is important to not dismiss older treatments and clotiapine may have its place in treatment of some people who it would 'suit'. People with schizophrenia and their families should expect better data than are available from the few existing trials.
2. For clinicians
This review suggests effects of clotiapine and these effects may be important. We really do not have good data on any outcomes but almost none on global state, and service use. Where clotiapine is used, and there is a dilemma about whether to use it, clinicians could help increase data on the effects of this antipsychotic drug by organising its evaluation in real world settings.
3. For policy makers
Where clotiapine is an available option for use for people with schizophrenia, this review gives no evidence to discourage its inclusion within policy except that the data, when compared with one benchmark antipsychotic ‐ chlorpromazine ‐ is surprisingly and disappointingly thin.
Implications for research.
1. General
Better reporting of trials would have resulted in this review being more informative. We recognise that we are largely using standards of today to judge trials of the past but we anticipate that adherence to the CONSORT statement (Moher 2001) would improve the quality of reporting data for future research.
2. Specific
Where clotiapine is available trials could be undertaken and, as this review shows, such studies are justified if the drug is in use. Long‐term effects are important. We realise that design of such a study takes time and methodical planning but we have given this some thought and outline a draft design (Table 4).
3. Suggestions for design of future study.
| Methods | Allocation: randomised, with sequence generation and concealment of allocation clearly described Blindness: double, tested Duration: 12 months beyond end of intervention at least Raters: independent |
| Participants | Diagnosis: people with schizophrenia ‐ however diagnosed* Age: any Sex: both History: any N = 300** |
| Interventions | 1. Clotiapine: ˜100 mg/day. N = 150 2. Chlorpromazine: ˜400 mg/day. N = 150 |
| Outcomes | Global state ‐ relapse, clinically important change Mental state ‐ general ‐ clinically important change in mental state, average change in negative symptoms Adverse effects ‐ incidence of serious adverse events/effects, clinically important extrapyramidal symptoms Leaving the study early ‐ for any reason Cost of care Service outcomes: admitted, number of admissions, length of hospitalisation, discharge, contacts with psychiatric services Compliance with drugs Economic evaluations: cost‐effectiveness, cost‐benefit |
| Notes | *This could be diagnosed by clinical decision. If funds were permitting all participants could be screened using operational criteria, otherwise a random sample should suffice. **Size of study with sufficient power to highlight about a 10% difference between groups for primary outcome |
Acknowledgements
Cochrane Schizophrenia Editorial Base in Nottingham produces and maintains standard text for use in the Methods section of their reviews. We have used this text as the basis of what appears here and adapted it as required.
The search terms were developed by Cochrane Schizophrenia's Information Specialist and the contact author of this review.
We would like to thank Professor Clive Adams (Co‐ordinating Editor of Cochrane Schizophrenia, Faculty of Medicine and Health Sciences, University of Nottingham) for providing advice, guidance and support throughout the undertaking of this review. We would also like to thank Claire Irving for providing additional support.
We would like to thank Emma Plugge for her peer review and Clare Dooley for copy‐editing the protocol. We would also like to thank Charlott Ziff and Hao Ruiqi for reviewing this version of the review.
Parts of this review were generated using RevMan HAL v 4.0. You can find more information about RevMan here.
Data and analyses
Comparison 1. Chlorpromazine versus Clotiapine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mental state: 1a. General ‐ average change scores (PANSS total, high = good) | 1 | 31 | Mean Difference (IV, Random, 95% CI) | 11.50 [9.42, 13.58] |
| 2 Mental state: 1b. General ‐ average change scores (S‐scale, high = poor) | 1 | 38 | Mean Difference (IV, Random, 95% CI) | 1.45 [0.38, 2.52] |
| 3 Mental state: 2a. Specific ‐ positive symptoms ‐ frequency | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Delusion | 1 | 12 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.50, 3.55] |
| 3.2 Hallucination | 1 | 25 | Risk Ratio (M‐H, Random, 95% CI) | 0.46 [0.15, 1.40] |
| 3.3 Severe excitation | 1 | 5 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.15, 3.72] |
| 4 Mental state: 2b Specific ‐ positive symptoms ‐ average change scores (PANSS positive subscale, high = good) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 Delusion | 1 | 11 | Mean Difference (IV, Random, 95% CI) | 7.36 [3.54, 11.18] |
| 4.2 Hallucination | 1 | 16 | Mean Difference (IV, Random, 95% CI) | ‐3.0 [‐6.75, 0.75] |
| 5 Mental state: 2c. Specific ‐ negative symptoms ‐ average change scores (PANSS negative subscale, high = good) | 1 | 21 | Mean Difference (IV, Random, 95% CI) | ‐0.97 [‐2.76, 0.82] |
| 5.1 Deficiency of initiative and apathy | 1 | 21 | Mean Difference (IV, Random, 95% CI) | ‐0.97 [‐2.76, 0.82] |
| 6 Mental state: 2d. Specific ‐ various symptoms ‐ average change score ‐ (S‐Scale, high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6.1 Delusion | 1 | 38 | Mean Difference (IV, Random, 95% CI) | ‐0.06 [‐0.30, 0.18] |
| 6.2 Hallucination | 1 | 38 | Mean Difference (IV, Random, 95% CI) | ‐0.24 [‐0.46, ‐0.02] |
| 6.3 Influence of thought | 1 | 38 | Mean Difference (IV, Random, 95% CI) | 0.28 [0.17, 0.39] |
| 6.4 Splitting | 1 | 38 | Mean Difference (IV, Random, 95% CI) | 0.01 [‐0.09, 0.11] |
| 7 Adverse effects: 1. Central nervous system ‐ short term | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 Drowsiness | 1 | 68 | Risk Ratio (M‐H, Random, 95% CI) | 1.89 [0.98, 3.63] |
| 7.2 Somnipathy | 1 | 68 | Risk Ratio (M‐H, Random, 95% CI) | 1.38 [0.63, 2.99] |
| 7.3 Unsteadiness | 1 | 68 | Risk Ratio (M‐H, Random, 95% CI) | 1.71 [0.77, 3.82] |
| 7.4 Weakness | 1 | 68 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.42, 1.34] |
| 8 Adverse effects: 2. Movement disorders ‐ short term | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 Akathisia | 1 | 68 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.65, 13.83] |
| 8.2 Dyskinesia | 1 | 68 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 71.15] |
| 8.3 Parkinsonism | 2 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.87, 1.57] |
| 9 Adverse effects: 3. Various other ‐ short term | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 Weight gain (not less than 3 kg at the end of study) | 1 | 68 | Risk Ratio (M‐H, Random, 95% CI) | 1.12 [0.71, 1.75] |
| 9.2 Thirst | 1 | 68 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [0.54, 3.06] |
| 10 Leaving the study early ‐ short term | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 for any reason | 3 | 158 | Risk Ratio (M‐H, Random, 95% CI) | 0.68 [0.24, 1.88] |
| 10.2 due to deterioration or adverse effects | 2 | 117 | Risk Ratio (M‐H, Random, 95% CI) | 1.23 [0.51, 2.99] |
| 11 Service use: discharge from hospital ‐ short term (12 weeks) | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.49 [1.07, 2.08] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Jacobsson 1974.
| Methods | Allocation: randomised by pre‐established randomised code Blindness: double‐blind ‐ tablets of the same appearance and taste Duration: 4 weeks Design: parallel Setting: hospital Country: Sweden |
|
| Participants | Diagnoisis: "acute psychotic syndromes of a schizophrenic type" N = 49 Age: 18‐60 years Sex: 23 M, 26 F History: suffering from acute psychotic syndromes of a schizophrenic type, either during their first episode or on a re‐occurrence of an acute stage Excluded: gross neurological or somatic disorders unrelated to the principal condition, alcoholism or drug addiction, and spontaneous remission |
|
| Interventions | 1. Clotiapine: range: 40‐220 mg/d, maximum dose: 240 mg/d, mean dose: 125.2 mg/d. N = 23 2. Chlorpromazine: range: 200‐600 mg/d, maximum dose: 600 mg/d, mean dose: 404.5 mg/d. N = 26 |
|
| Outcomes | Mental state: general, specific symptoms (Martens' Symptom Scale (S‐Scale)) Adverse effects: incidence of Parkinsonism Leaving the study early Unable to use ‐ Mental state: general ‐ average endpoint score (scale not named ‐ unsure if peer reviewed) Behaviour: Wing rating scale (no SD reported) Adverse effects: haematology, hepatic, ophthalmic (no SD reported), constipation, tiredness ‐ average change score (scale not named ‐ unsure if peer reviewed) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised: quote: "according to a pre‐established randomized code." |
| Allocation concealment (selection bias) | Unclear risk | No details provided for allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "clotiapine or 100 mg chlorpromazine were administered using the double‐blind technique" |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "A separate envelope for each code‐number was also prepared in case it should be necessary to know what drug an individual patient was receiving" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing data were addressed in the study. |
| Selective reporting (reporting bias) | Low risk | All pre‐specified outcomes were reported in the study. |
| Other bias | Unclear risk | Drugs were supplied by AstraZeneca, Mölndal. Source of funding not reported |
Kaneko 1969.
| Methods | Allocation: implied randomisation Blindness: double‐blind ‐ tablets of the same external form Duration: 10 weeks (2 weeks placebo before the study started‐ 8 weeks for intervention) Design: parallel Setting: hospital Country: Japan |
|
| Participants | Diagnosis: schizophrenia N = 68 Age: 18‐60 years Sex: 52 M, 16 F History: including symptoms of severe excited state, stuporous state, hallucination, delusion or apathy. Symptoms were evaluated by a simplified scale for evaluation of psychogenic symptoms (for schizophrenia and for use by doctors) as 12 items, and the intensity of each item was assessed in five grades. Excluded: marked exacerbation or serious side effects, complete remission |
|
| Interventions | 1. Clotiapine: maximum dose: 240 mg/d. N = 34 2. Chlorpromazine: maximum dose: 600 mg/d. N = 34 |
|
| Outcomes | Mental state: average change score (PANSS positive and negative sub‐scale) Adverse effects: movement disorders, central nervous system, weight gain, thirst Leaving the study early Unable to use Mental state: improvement using Keio‐Gijuku University type, simplified scale, (no mean or SD) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | No information was provided for randomisation |
| Allocation concealment (selection bias) | High risk | No information was provided for allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Employed as the experimental method in the present study. was the double‐blind, controlled technique " |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing data and attrition were addressed in the study. |
| Selective reporting (reporting bias) | Low risk | All pre‐specified outcomes were reported in the study. |
| Other bias | Unclear risk | Source of funding not reported |
Schliefer 2003.
| Methods | Allocation: randomised Blindness: double‐blind ‐ identical capsules Duration: 12 weeks Design: cross‐over Setting: both inpatient and outpatients Country: Israel |
|
| Participants | Diagnosis: severe chronic schizophrenia N = 58 Age: 18‐65 years Sex: 44 M, 14 F History: severe chronic active psychotic hospitalised patients with a history of non‐response to at least 3 antipsychotics Excluded: currently in an exacerbation, unstable or serious physical illnesses, suicidality, drug abuse or unstable family situations |
|
| Interventions | 1. Clotiapine: maximum dose: 240 mg/d. N = 19 2. Chlorpromazine: maximum dose: 600 mg/d. N = 12 33 participants received additional trihexyphenidyl for EPS throughout the study and 27 received benzodiazepines for helping to sleep |
|
| Outcomes | Mental state: improvement (average change score PANSS) Leaving the study early Unable to use Mental state: improvement (CGI, NOSIE): no usable data reported for first phase of cross‐over |
|
| Notes | *Only data from first phase of cross‐over used | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomized" |
| Allocation concealment (selection bias) | Unclear risk | No information was provided for allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The design was double‐blind crossover" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "by trained observers blind to the patient’s treatment status" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing data and attrition were addressed in the study. |
| Selective reporting (reporting bias) | Unclear risk | No information was provided for pre‐specified outcomes. |
| Other bias | Unclear risk | There was no wash‐out period in the study. This study was supported by a clinical trial grant from the Stanley Medical Research Institute. |
Van Wyk 1971.
| Methods | Allocation: randomised Blindness: not blind Duration: 12 weeks Design: parallel Setting: hospital Country: South Africa |
|
| Participants | Diagnosis: schizophrenia and "toxic psychoses" N = 101 Age: adults (no further information provided) Sex: male History: acute psychoses, no epilepsy or depressive psychoses |
|
| Interventions | 1. Clotiapine: 40 mg/d orally. N = 36 2. Chlorpromazine: 200 mg/d orally. N = 34 3. Thioridazine: 200 mg/d orally. N = 31 |
|
| Outcomes | Service use: number discharged by 12 weeks Unable to use Mental state: general ‐ average endpoint score (scale not named ‐ unsure if peer reviewed) Adverse effects: adverse effects mentioned in text, but no data reported to support statements |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Allocation to either of the 3 drug groups was at random" |
| Allocation concealment (selection bias) | Unclear risk | No information provided for allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Because of the administrative and staff shortage problems we could not introduce· any 'blind' procedures into the trial" |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "Because of the administrative and staff shortage problems we could not introduce· any 'blind' procedures into the trial" |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No information was provided for attrition and missing data |
| Selective reporting (reporting bias) | Unclear risk | No information was provided for pre‐specified outcomes |
| Other bias | Unclear risk | Clotiapine was supplied by a researcher from Switzerland. No source of funding reported |
EPS: extrapyramidal symptoms F: female M: male N: number SD: standard deviation
Scales
PANSS: Positive and negative syndrome scale S‐Scale: Marten's Symptom scale CGI: Clinical Global Impression NOSIE: Nurses' Observational Scale for Inpatient Evaluation
Differences between protocol and review
We have updated some of the text in the methods to reflect changes made to the template since publication of the protocol. As these changes did not affect our data collection and analyses for these studies we felt we could add to the methods section, so they are published before the next update of this review.
1. Areas of text changed
1.1 Use of scales
We added this sentence to clarify use of sub‐scale data:
the instrument was a global assessment of an area of functioning and not sub‐scores which were not, in themselves, validated or shown to be reliable. However there would be exceptions, we would include sub‐scores from mental state scales measuring positive and negative symptoms of schizophrenia.
1.2 Selection of studies
We added this sentence to clarify our selection of studies for inclusion and exclusion.
We included studies that met our inclusion criteria and reported useable data. We would have excluded studies that either did not meet our inclusion criteria or met our inclusion criteria but did not report useable data.
1.3 Outcome title change
We have, changed 'service utilisation outcome' to 'service use' and added discharge from hospital. Data were reported for this outcome in a trial and we felt it was of interest to reproduce these data for the review. Excluding would mean the trial would have no useable data.
2. Additional authors
Two new authors have joined the team (Asam Bazrafshan and Morteza Zare) since protocol publication.
Contributions of authors
Shahrzad Mazhari (SM): designing the review, development and writing the review, reliability checking for trial selection, reliability checking for data extraction and report writing.
Saeed Esmailian (SE): development and writing of protocol, trial selection and review writing.
Armita Shah‐Esmaili (ASE): provided general advice, reliability checking for trial selection, draft checking.
Ali Shahsavari Goughari (ASG) : development and writing of protocol, trial selection, reliability checking data extraction and review writing
Azam Bazrafshan (AB): data extraction, statistical analysis and review writing.
Morteza Zare (MZ): data extraction, statistical analysis and review writing.
Sources of support
Internal sources
-
Kerman University of Medical Sciences, Kerman, Iran.
all review authors are students at this university
External sources
No sources of support supplied
Declarations of interest
Shahrzad Mazhari: None known Saeed Esmailian: None known Armita Shah‐Esmaili: None known Ali Shahsavari: None known Azam Bazrafshan: None known Morteza Zare: None known
New
References
References to studies included in this review
Jacobsson 1974 {published data only}
- Jacobsson L, Nordn MB. A controlled trial of clotiapine and chlorpromazine in acute schizophrenic syndromes. Acta Psychiatrica Scandinavica 1974;Suppl. 255:55‐71. [DOI] [PubMed] [Google Scholar]
Kaneko 1969 {published data only}
- Kaneko J, Tanimukai H, Kudo Y. A double‐blind, controlled study of the effects of clotiapine and chlorpromazine on schizophrenia. Clinical Psychiatry 1969;11(9):721‐8. [Google Scholar]
Schliefer 2003 {published data only}
- Belmaker R. Clotiapine for schizophrenia. Stanley Foundation Research Programs 2006.
- Geller V, Gorzaltsan I, Shleifer T, Belmaker RH, Bersudsky Y. Clotiapine compared with chlorpromazine in chronic schizophrenia. Schizophrenia Research 2005;80:343‐7. [DOI] [PubMed] [Google Scholar]
- Schliefer T, Bersudsky Y, Geller V, Belmaker RH. Clotiapine in schizophrenia: a controlled study. 16th European College of Neuropsychopharmacology Congress. Prague, Czech Republic, 2003.
Van Wyk 1971 {published data only}
- Wyk AJ, Maris GFT. Chlorpromazine, clotiapine and thioridaz ‐ a comparative clinical trial on Bantu psychotic patients. South African Medical Journal 1971;45:945‐7. [PubMed] [Google Scholar]
Additional references
Adams 2005
- Adams CE, Rathbone J, Thornley B, Clarke M, Borrill J, Wahlbeck K, et al. Chlorpromazine for schizophrenia: a Cochrane systematic review of 50 years of randomised controlled trials. BMC Medicine 2005;3:15. [PUBMED: 16229742] [DOI] [PMC free article] [PubMed] [Google Scholar]
Adams 2014
- Adams CE, Awad GA, Rathbone J, Thornley B, Soares‐Weiser K. Chlorpromazine versus placebo for schizophrenia. Cochrane Database of Systematic Reviews 2014, Issue 1. [DOI: 10.1002/14651858.CD000284.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ahmed 2010
- Ahmed U, Jones H, Adams CE. Chlorpromazine for psychosis induced aggression or agitation. Cochrane Database of Systematic Reviews 2010, Issue 12. [DOI: 10.1002/14651858.CD007445.pub2; PUBMED: 20393959] [DOI] [PubMed] [Google Scholar]
Almerie 2007
- Almerie MQ, Alkhateeb H, Essali A, Matar HE, Rezk E. Cessation of medication for people with schizophrenia already stable on chlorpromazine. Cochrane Database of Systematic Reviews 2007, Issue 1. [DOI: 10.1002/14651858.CD006329; PUBMED: 17253586] [DOI] [PMC free article] [PubMed] [Google Scholar]
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313(7066):1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Amato 2015
- Amato D. Serotonin in antipsychotic drugs action. Behavioural Brain Research 2015;277:125‐35. [DOI] [PubMed] [Google Scholar]
Ban 2007
- Ban TA. Fifty years chlorpromazine: a historical perspective. Neuropsychiatric Disease and Treatment 2007;3(4):495‐500. [PUBMED: 19300578] [PMC free article] [PubMed] [Google Scholar]
Bazrafshan 2015
- Bazrafshan A, Zare M, Okhovati M, Shamsi Meimandi M. Acetophenazine versus chlorpromazine for schizophrenia. Cochrane Database of Systematic Reviews 2015, Issue 4. [DOI: 10.1002/14651858.CD011662] [DOI] [Google Scholar]
Berk 2004
- Berk M, Rathbone J, Mandriota‐Carpenter S. Clotiapine for acute psychotic illnesses. Cochrane Database of Systematic Reviews 2004, Issue 4. [DOI: 10.1002/14651858.CD002304.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bland 1997
- Bland JM. Statistics notes. Trials randomised in clusters. BMJ 1997;315:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use [Apercu sur la problematique des indices d'efficacite therapeutique, 3: comparaison des indices et utilisation. Groupe d'Etude des Indices D'efficacite]. Therapie 1999;54(4):405‐11. [PUBMED: 10667106] [PubMed] [Google Scholar]
Davey Smith 1998
- Davey Smith G, Egger M. Meta‐analysis: unresolved issues and future developments. BMJ 1998;16:221‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. 8th International Cochrane Colloquium; 2000 Oct 25‐28; Cape Town. Cape Town: The Cochrane Collaboration, 2000.
Deeks 2011
- Deeks JJ, Higgins JPT, Altman DG (editors). Chapter 9: Analysing data and undertaking meta‐analyses. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Divine 1992
- Divine GW, Brown JT, Frazier LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7(6):623‐9. [DOI] [PubMed] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21:2971‐80. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne D, Altman DG, Higgins JPT, Curtin F, Worthington HV, Vail A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Eslami 2015
- Eslami Shahrbabaki M, Sharafkhani R, Dehnavieh R, Vali L. Chlorpromazine versus piperacetazine for schizophrenia. Cochrane Database of Systematic Reviews 2015, Issue 6. [DOI: 10.1002/14651858.CD011709] [DOI] [PMC free article] [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(7):7‐10. [DOI] [PubMed] [Google Scholar]
Geller 2005
- Geller V, Gorzaltsan I, Shleifer T, Belmaker RH, Bersudsky Y. Clotiapine compared with chlorpromazine in chronic schizophrenia. Schizophrenia Research 2005;80(2‐3):343‐7. [PUBMED: 16126373] [DOI] [PubMed] [Google Scholar]
Gulliford 1999
- Gulliford MC. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149:876‐83. [DOI] [PubMed] [Google Scholar]
Helgason 1983
- Helgason T. Methodology in Evaluation of Psychiatric Treatment. Cambridge: Cambridge University Press, 1983. [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Green S (editors). Chapter 7: Selecting studies and collecting data. In: Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011b
- Higgins JPT, Altman DG, Sterne JAC (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011c
- Higgins JPT, Deeks JJ, Altman DG (editors). Chapter 16: Special topics in statistics. In: Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Hutton 2009
- Hutton JL. Number needed to treat and number needed to harm are not the best way to report and assess the results of randomised clinical trials. British Journal of Haematology 2009;146(1):27‐30. [DOI] [PubMed] [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and Negative Syndrome Scale (PANSS) Manual. North Tonawanda, NY: Multi‐Health Systems, 1986. [Google Scholar]
Kay 1987
- Kay SR, Fiszbein A, Opfer LA. The Positive and Negative Syndrome Scale (PANSS) for schizophrenia. Schizophrenia Bulletin 1987;13(2):261‐76. [DOI] [PubMed] [Google Scholar]
Khalili 2015
- Khalili N, Vahedian M, Nikvarz N, Piri M. Chlorpromazine versus penfluridol for schizophrenia. Cochrane Database of Systematic Reviews 2015, Issue 8. [DOI: 10.1002/14651858.CD011831] [DOI] [PMC free article] [PubMed] [Google Scholar]
Leon 2006
- Leon AC, Mallinckrodt CH, Chuang‐Stein C, Archibald DG, Archer GE, Chartier K. Attrition in randomized controlled clinical trials: methodological issues in psychopharmacology. Biological Psychiatry 2006;59(11):1001‐5. [PUBMED: 16905632] [DOI] [PubMed] [Google Scholar]
Leucht 2005
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of brief psychiatric rating scale scores. British Journal of Psychiatry 2005;187:366‐71. [PUBMED: 16199797] [DOI] [PubMed] [Google Scholar]
Leucht 2008
- Leucht C, Kitzmantel M, Kane J, Leucht S, Chua WLLC. Haloperidol versus chlorpromazine for schizophrenia. Cochrane Database of Systematic Reviews 2008, Issue 1. [DOI: 10.1002/14651858.CD004278.pub2; PUBMED: 18254045] [DOI] [PubMed] [Google Scholar]
Liu 2009
- Liu X, Haan S. Chlorpromazine dose for people with schizophrenia. Cochrane Database of Systematic Reviews 2009, Issue 2. [DOI: 10.1002/14651858.CD007778; PUBMED: 19370692] [DOI] [PubMed] [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
Miyamoto 2012
- Miyamoto S, Miyake N, Jarskog LF, Fleischhacker WW, Lieberman JA. Pharmacological treatment of schizophrenia: a critical review of the pharmacology and clinical effects of current and future therapeutic agents. Molecular Psychiatry 2012;17:1206‐27. [DOI] [PubMed] [Google Scholar]
Moher 2001
- Moher D, Schulz KF, Altman D. The CONSORT statement: revised recommendations for improving the quality of reports of parallel‐group randomized trials. JAMA 2001;285:1987‐91. [DOI] [PubMed] [Google Scholar]
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG, The PRISMA Group (2009). Preferred Reporting Items for Systematic Reviews and Meta‐Analyses: The PRISMA Statement. PLoS Medicine 2009;6(7):e1000097. [DOI: 10.1371/journal.pmed1000097] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 1989
- Moore NC, Gershon S. Which atypical antipsychotics are identified by screening tests?. Clinical Neuropharmacology 1989;12:167‐84. [DOI] [PubMed] [Google Scholar]
Overall 1962
- Overall JE, Gorham DR. The brief psychiatric rating scale. Psychological Reports 1962;10:799‐812. [Google Scholar]
Pakpoor 2014
- Pakpoor J, Agius M. A review of the adverse side effects associated with antipsychotics as related to their efficacy. Psychiatria Danubina 2014;26 Suppl 1:273‐84. [PUBMED: 25413553] [PubMed] [Google Scholar]
Perala 2007
- Perala J, Suvisaari J, Saarni SI, Kuoppasalmi K, Isometsa E, Pirkola S, et al. Lifetime prevalence of psychotic and bipolar I disorders in a general population. Archives of General Psychiatry 2007;64(1):19‐28. [PUBMED: 17199051] [DOI] [PubMed] [Google Scholar]
Saha 2013
- Saha KB, Sampson S, Zaman RU. Chlorpromazine versus atypical antipsychotic drugs for schizophrenia. Cochrane Database of Systematic Reviews 2013, Issue 7. [DOI: 10.1002/14651858.CD010631] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schünemann 2011
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Sterne 2011
- Sterne JAC, Egger M, Moher D (editors). Chapter 10: Addressing reporting biases. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Intervention. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area‐wide and organisation‐based interventions in health and health care: a systematic review. Health Technology Assessment 1999;3(5):1‐75. [PubMed] [Google Scholar]
Xia 2009
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin 2009;33(7):254‐7. [Google Scholar]
Zare 2015
- Zare M, Bazrafshan A, Okhovati M, Mazhari S, Mousavi R. Chlorpromazine versus metiapine for schizophrenia. Cochrane Database of Systematic Reviews 2015, Issue 5. [DOI: 10.1002/14651858.CD011655] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zawilska 1994
- Zawilska JB, Derbiszewska T, Nowak JZ. Clozapine and other neuroleptic drugs antagonize the light‐evoked suppression of melatonin biosynthesis in chick retina: involvement of the D4‐like dopamine receptor. Neural Transmission 1994;97:107‐17. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Mazhari 2015
- Mazhari S, S. Goughari A, Esmailian S, Shah‐Esmaili A. Chlorpromazine versus clotiapine for schizophrenia. Cochrane Database of Systematic Reviews 2015, Issue 8. [DOI: 10.1002/14651858.CD011810] [DOI] [PMC free article] [PubMed] [Google Scholar]