

Abstract
Background
Multiple analgesic strategies for pain relief during labour are available. Recently remifentanil, a short‐acting opioid, has recently been used as an alternative analgesic due to its unique pharmacological properties.
Objectives
To systematically assess the effectiveness of remifentanil intravenous patient‐controlled analgesia (PCA) for labour pain, along with any potential harms to the mother and the newborn.
Search methods
We searched the Cochrane Pregnancy and Childbirth Group's Trials Register (9 December 2015), ClinicalTrials.gov, the WHO International Clinical Trials Registry Platform (ICTRP), handsearched congress abstracts (November 2015), and reference lists of retrieved studies.
Selection criteria
Randomised controlled trials (RCTs) and cluster‐randomised trials comparing remifentanil (PCA) with another opioid (intravenous (IV)/intramuscular (IM)), or with another opioid (PCA), or with epidural analgesia, or with remifentanil (continuous IV), or with remifentanil (PCA, different regimen), or with inhalational analgesia, or with placebo/no treatment in all women in labour including high‐risk groups with planned vaginal delivery.
Data collection and analysis
Two review authors independently assessed trials for inclusion, extracted data, and appraised study quality.
We contacted study authors for additional information other than incomplete outcome data. We performed random‐effects meta‐analysis.
To reduce the risk of random error in meta‐analysis we performed trial sequential analysis. We included total zero event trials and used a constant continuity correction of 0.01 (ccc 0.01) for meta‐analysis. We applied the Grades of Recommendation, Assessment, Development, and Evaluation (GRADE) approach to assess the quality of evidence.
Main results
Twenty RCTs with 3569 women were included. Of those, 10 trials (2983 participants) compared remifentanil (PCA) to an epidural, four trials (216 participants) to another opioid (IV/IM), three trials (215 participants) to another opioid (PCA), two trials (135 participants) to remifentanil (continuous IV), and one trial (20 participants) to remifentanil (PCA, different regimen). No trials were identified for the remaining comparisons.
Methodological quality of studies was moderate to poor. We assessed risk of bias as high for blinding issues and incomplete outcome data in 65% and 45% of the included studies, respectively.
There is evidence of effect that women in the remifentanil (PCA) group were more satisfied with pain relief than women in the other opioids (IV/IM) group (standardised mean difference (SMD) 2.11, 95% confidence interval (CI) 0.72 to 3.49, four trials, very low‐quality evidence), and that women were less satisfied compared to women in the epidural group (SMD ‐0.22, 95% CI ‐0.40 to ‐0.04, seven trials, very low‐quality evidence).
There is evidence of effect that remifentanil (PCA) provided stronger pain relief at one hour than other opioids administered IV/IM (SMD ‐1.58, 95% CI ‐2.69 to ‐0.48, three trials, very low‐quality evidence) or via PCA (SMD ‐0.51, 95% CI ‐1.01 to ‐0.00, three trials, very low‐quality evidence). Pain intensity was higher in the remifentanil (PCA) group compared to the epidural group (SMD 0.57, 95% CI 0.31 to 0.84, six trials, low‐quality evidence).
Data were limited on safety aspects for both the women and the newborns. Only one study analysed maternal apnoea in a comparison of remifentanil (PCA) versus epidural and reported that half of the women in the remifentanil and none in the epidural group had an apnoea (very low‐quality evidence). There is no evidence of effect that remifentanil (PCA) was associated with an increased risk for maternal respiratory depression when compared to epidural analgesia (RR 0.91, 95% CI 0.51 to 1.62, ccc 0.01, three trials, low‐quality evidence) and no reliable conclusion might be reached compared to remifentanil (continuous IV) (all study arms included zero events, two trials, low‐quality evidence). In one trial of remifentanil (PCA) versus another opioid (IM) three out of 18 women in the remifentanil and none out of 18 in the control group had a respiratory depression (very low‐quality evidence).
There is no evidence of effect that remifentanil (PCA) was associated with an increased risk for newborns with Apgar scores less than seven at five minutes compared to epidural analgesia (RR 1.26, 95% CI 0.62 to 2.57, ccc 0.01, five trials, low‐quality evidence) and no reliable conclusion might be reached compared to another opioid (IV) and compared to remifentanil (PCA, different regimen) both with zero events in all study arms (one trial, very‐low quality evidence). In one trial of remifentanil (PCA) versus another opioid (PCA) none out of nine newborns in the remifentanil and three out of eight in the opioid (PCA) group had Apgar scores less than seven (very‐low quality evidence).
There is evidence that remifentanil (PCA) was associated with a lower risk for the requirement of additional analgesia when compared to other opioids (IV/IM) (RR 0.57, 95% CI 0.40 to 0.81, three trials, moderate‐quality evidence) and that it was associated with a higher risk compared to epidural analgesia (RR 9.27, 95% CI 3.73 to 23.03, ccc 0.01, six trials, moderate‐quality evidence). There is no evidence of effect that remifentanil (PCA) reduced the requirement for additional analgesia compared to other opioids (PCA) (RR 0.76, 95% CI 0.45 to 1.28, three trials, low‐quality evidence).
There is evidence that there was no difference in the risk for caesarean delivery between remifentanil (PCA) and other opioids (IV/IM) (RR 0.63, 95% CI 0.30 to 1.32, ccc 0.01, four trials, low‐quality evidence) and epidural analgesia (RR 1.0, 95% CI 0.82 to 1.22, ccc 0.01, nine trials, moderate‐quality evidence), respectively. Pooled meta‐analysis revealed an increased risk for caesarean section under remifentanil (PCA) compared to other opioids (PCA) (RR 2.78, 95% CI 0.99 to 7.82, two trials, very low‐quality evidence). However, a wide range of clinically relevant and non‐relevant treatment effects is compatible with this result.
Authors' conclusions
Based on the current systematic review, there is mostly low‐quality evidence to inform practice and future research may significantly alter the current situation. The quality of evidence is mainly limited by poor quality of the studies, inconsistency, and imprecision. More research is needed on maternal and neonatal safety outcomes (maternal apnoea and respiratory depression, Apgar score) and on the optimal mode and regimen of remifentanil administration to provide highest efficacy with reasonable adverse effects for mothers and their newborns.
Plain language summary
Patient‐controlled analgesia with remifentanil versus alternative analgesic methods for pain relief in labour
What is the issue
Pain relief during labour can be provided in a number of different ways. These include epidural analgesia, by injection of anaesthetic medication around the nerve roots in the spine, intramuscular or continuous intravenous opioids, and inhalational analgesia such as with nitrous oxide. Remifentanil is a relatively recently introduced potent, short‐acting opioid, which gives control over pain relief.
Why is this important
Labour pain may be associated with adverse effects for the mother and her baby and can result in prolonged labour.
This review aimed to compare remifentanil given via a patient‐controlled analgesia (PCA) device with other opioids given via the same way or via an intramuscular or intravenous injection, with epidural analgesia, with different regimens of remifentanil (PCA) or with remifentanil as a continuous intravenous infusion, with inhalational analgesia, or with no treatment for women during normal vaginal birth. Our main outcomes of interest were satisfaction with pain relief, pain scores, side effects for the women and their babies, need for additional analgesia and the risk for a caesarean section.
What evidence did we find
A search of the literature was performed in November/December 2015 and updated in December 2016. We found 20 randomised controlled trials with 3569 women. The methodological quality of studies was moderate to poor.
Women who received PCA with remifentanil were more satisfied with their pain relief than women receiving other opioids either by intravenous or intramuscular injection (four trials, 216 women, very low‐quality evidence). Remifentanil (PCA) provided stronger pain relief at one hour than the other opioids by intravenous or intramuscular injection (three trials, 180 women) and using PCA (three trials, 215 women), both very low‐quality evidence but with moderate‐quality evidence that remifentanil (PCA) was associated with a reduced need for additional analgesia compared to other intravenous or intramuscular opioids (three trials, 190 women). The number of women with need for additional analgesia was not different with remifentanil (PCA) or opioids (PCA) (three trials, 215 women, low‐quality evidence). Remifentanil (PCA) increased the risk for a maternal respiratory depression compared to other intramuscular opioids (one trial, 36 women, very low‐quality evidence). The newborn babies were not more likely to have low Apgar scores at five minutes after birth under remifentanil (PCA) compared to other intravenous or intramuscular opioids (one trial, 88 newborns, very low‐quality evidence), but newborns have a lower risk under remifentanil (PCA) compared to other opioids (PCA) (one trial, 17 newborns, very low‐quality evidence). Remifentanil (PCA) was not associated with an increased risk for caesarean section when compared with intravenous or intramuscular opioids (four trials, 215 women, low‐quality evidence), but compared to other opioids (PCA) (two trials, 143 women, very low‐quality evidence).
Women were slightly less satisfied with remifentanil (PCA) compared to an epidural for pain relief (seven trials, 2135 women, very low‐quality evidence). Pain intensity was higher in the remifentanil (PCA) group compared to the epidural group (six trials, 235 women, low‐quality evidence), with a higher need for additional analgesia (six studies, 1037 women, moderate‐quality evidence). Remifentanil (PCA) increased the risk for a maternal respiratory arrest compared to an epidural (one trial, 38 women, very low‐quality evidence). Remifentanil (PCA) was not associated with an increased risk of respiratory depression in mothers compared to an epidural (three trials, 687 women, low‐quality evidence). The newborn babies were not more likely to have low Apgar scores at five minutes after birth (five trials, 1322 newborns, low‐quality evidence). The number of women requiring caesarean section was not different with remifentanil (PCA) or epidural analgesia (moderate‐quality evidence).
What does this mean
Our confidence in the results of the current review is limited since the quality of evidence is mostly low. No definite conclusion can be drawn with respect to side effects for women and newborns as well as for the comparators remifentanil given via a continuous infusion or via PCA with a different regimen since there are too few studies with few participants that reported on these. No eligible study examined remifentanil (PCA) versus inhalational analgesia or no treatment. More research is needed, especially on side effects of remifentanil (PCA) for women and newborns.
Summary of findings
Background
Nowadays, multiple strategies are available to provide pain relief during labour, such as central neuraxial analgesia (e.g. epidural analgesia), parenteral opioids, and inhalational analgesia. According to the guidelines of the American Society of Anaesthesiologists (ASA) and the College of Obstetricians and Gynaecologists (ACOG), epidural analgesia is recommended as the most flexible, effective and least depressing to the central nervous system analgesic modality in obstetrics (Goetzl 2002). However, obstetric anaesthesiologists are occasionally faced with women who cannot receive this type of labour analgesia due to absolute or relative contraindications, e.g. woman receiving prophylactic anticoagulants (Moghbeli 2008), or women with significant coagulation disorders. Pregnant women may also ask for alternatives to central neuraxial analgesia for personal reasons. Moreover, central neuraxial analgesia may also technically not be possible to perform in women requesting pain relief for labour. Finally, there are many places in the world which do not offer epidural pain relief either at all, or only on a very limited basis (Saravanakumar 2007).
A common method for pain relief in labour is the use of opioids (e.g. pethidine) administered either via the intravenous (IV) or intramuscular (IM) route. In 2008, a survey in the United Kingdom on the prescription of IM opioids (e.g. pethidine) for labour analgesia concluded that pethidine lacks efficacy as an analgesic and has adverse effects on both the mother and the neonate (Tuckey 2008). Nevertheless, pethidine, morphine or diamorphine, and other long‐acting opioids are still frequently used (Tuckey 2008); a situation that does not differ markedly when compared with other European countries (Schnabel 2011).
These findings are in notable contrast to German and other European countries' guidelines on acute pain relief. Concerning the use of pethidine, the German guidelines on the management of acute pain relief in labour recommend that pethidine is not suited due to neurotoxic effects. Especially for the IM application route of pethidine, a negative recommendation (“Grade of Recommendation: A”) was stated (AWMF guidelines 2009, AWMF‐Register Nr. 001 ‐ 025, download on 29 November 2011).
Another alternative for labour analgesia is achieved by inhalational analgesia using, e.g. nitrous oxide. In principle, this method ensures that the mother stays awake and laryngeal reflexes remain intact. The fact that inhaled interventions for pain relief are usually easy to administer with limited preparation time and fast onset account for their popularity in some countries (Irestedt 1994; Kranke 2013). However, the existing body of evidence with respect to nitrous oxide and other inhaled molecules has been the subject of two systematic reviews with controversial results concerning the effectiveness as a labour analgesic (Klomp 2012; Rosen 2002).
The described discrepancy between scientific evidence and recommendations on the one hand, and the current clinical practice on the other hand, demands a closer look at the current body of evidence to discover alternative techniques that might be promising in view of efficacy (pain relief) and safety for both the mother and the neonate. For several reasons described above, there is a need for an effective and safe systemic analgesic for labour pain, which can be used as an alternative to central neuraxial analgesia in obstetrics. Due to its unique pharmacodynamic and pharmacokinetic profile (fast on‐ and offset), remifentanil might be an alternative opioid for labour analgesia (Egan 1993). Several surveys and narrative reviews focusing on opioids in obstetrics showed that remifentanil is gaining popularity (Lavand'homme 2009).
Proponents of the use of remifentanil for labour analgesia claim that it should be routinely available as an alternative for labour analgesia in those women who either do not want, can not have, or do not need, epidural analgesia (Hill 2008). However, opponents argue that not only does remifentanil produce negative respiratory effects for both the mother and the neonate, but also that the available evidence supporting the use of remifentanil is limited (Van de Velde 2008).
Therefore, it is essential to develop an evidence‐based decision basis for labour pain management and to promote a shared decision‐making process with parturients. In case of superiority of newer, more efficient and safer techniques, these techniques should be implemented when possible and safe to avoid unnecessary suffering and decrease potential negative impact on parental as well as neonatal outcomes.
Description of the condition
Pain during labour can be very intense and many pregnant women are anxious about the pain they will experience. This holds true also for women who have received prepared childbirth training (Melzack 1984). The anatomic and neurophysiologic basis underlying the pain of childbirth along with different pain‐management strategies are described in detail in an overview of systematic reviews dealing with pain management for women in labour (Jones 2012). The choice and demands of pain relief differ between countries and cultures and likewise the willingness to face and endure labour pain (Callister 2003; Callister 2010; Kartchner 2003; Semenic 2004; Weber 1996; Wilkinson 2010). Labour pain may be associated with adverse effects on both the mother and the fetus, mainly by elevated plasma catecholamine levels, respiratory changes and associated shifts in pCO2 and pH. Furthermore, intense pain may also result in prolonged labour (Reynolds 2011). Therefore, it is important to provide women with various options for pain control during labour.
Description of the intervention
Remifentanil, first described in 1991 (James 1991), is a very short‐acting opioid with an analgesic potency that is about 200 times higher compared to morphine (Westmoreland 1993). It acts as a specific agonist on the μ‐opioid‐receptor. The metabolisation of remifentanil through nonspecific tissue and plasma esterases decreases its half‐life to only a few minutes, leading to a rapid decline of action in the patient. The fast on‐ and offset of the drug action facilitates its controllability. Especially, when applied in a patient‐controlled manner, remifentanil analgesia allows enhanced flexibility and controllability for obstetrics. The action of remifentanil, as well as safety concerns are not affected by impaired liver or kidney function of the recipients (Bosilkovska 2012; Hohne 2004). Known side effects of remifentanil include respiratory depression, nausea, pruritus, and decreased heart rate and blood pressure. It is mostly used in anaesthesiology, e.g. as a component of total intravenous anaesthesia (TIVA) combined with propofol due to its predictable pharmacokinetics irrespective of organ function and the lack of accumulation. Owing to the unique pharmacodynamic and pharmacokinetic characteristics of remifentanil, it is increasingly used for labour pain relief. The comparable rapid metabolisation of IV‐administered remifentanil in adults and neonates suggests only a limited risk to cause prolonged side effects for the newborn.
How the intervention might work
Remifentanil has been used for anaesthesia for many years, providing effective and controllable analgesia for different kinds of surgical procedures by acting as a μ‐agonist. Due to its characteristics (fast onset, short half‐life), it can be administered in a patient‐controlled mode, giving the parturient the opportunity of pain relief when required. Therefore, remifentanil might be an alternative to other opioids and to epidural analgesia.
Why it is important to do this review
Remifentanil patient‐controlled analgesia (PCA) for labour analgesia is becoming increasingly popular in some countries, while in other countries there is a remaining reluctance towards its use due to the fear of possible adverse effects based on a few reported severe outcomes secondary to remifentanil administration for labour pain (Bonner 2012; Pruefer 2012). Previously, some of the published trials have been partially summarised in systematic reviews, which either deal with the comparison of remifentanil PCA versus epidural analgesia (Liu 2014), or remifentanil versus pethidine (Leong 2011), or both of those comparisons in addition to fentanyl and nitrous oxide as comparators (Schnabel 2011) in the obstetrics setting. However, none of those reviews, in contrast to the current review, defined adverse events associated with this intervention as their primary outcome. Moreover, an up‐to‐date systematic review with the comprehensive reporting and high‐quality standard of a Cochrane review, including the commitment for a subsequent update process, is still lacking.
Objectives
To systematically assess the effectiveness of remifentanil patient‐controlled analgesia (PCA) for labour analgesia, along with any potential harms to the mother and the baby.
Methods
Criteria for considering studies for this review
Types of studies
We included individually‐randomised controlled trials (RCTs) and planned to include cluster‐randomised trials. Cross‐over trials and quasi‐RCTs were not included. We planned to include trials which were only published in abstract form, if sufficient information in the abstract was available to allow an assured decision on inclusion.
Types of participants
All women in labour with planned vaginal delivery, including high‐risk groups, e.g. preterm labour or following induction of labour were eligible.
We did not include trials involving women scheduled for caesarean delivery.
Types of interventions
We compared remifentanil administered via a patient‐controlled analgesia (PCA) device versus:
another opioid using a different mode (nurse‐/midwife‐controlled intravenous infusion (IV)) or route (intramuscular (IM)/subcutaneous (SC)) of administration;
another opioid using the same mode of administration (PCA);
epidural analgesia or other central neuraxial blocks (e.g. combined spinal‐epidural analgesia (CSE));
remifentanil using a different mode (continuous IV administration) of administration;
remifentanil using the same mode (PCA), but different regimen (e.g. increasing bolus versus constant bolus);
nitrous oxide (or other forms of inhalational analgesia);
placebo or no treatment.
We included trials describing all modes of IV pain control with remifentanil using a PCA pump at any stage during labour. There were no restrictions regarding the lockout interval, the amount of remifentanil delivered with each bolus dose, whether adjusted doses due to the patient’s body weight, e.g. 0.5 μg/kg of actual/ideal body weight, or a dosing scheme, e.g. with increasing doses depending on the efficacy in order to find an appropriate dose. Further, we included trials investigating a regimen with only bolus doses as well as trials investigating regimen that combined a defined amount of continuous administration of remifentanil with additional bolus doses of remifentanil upon request.
Both the bolus doses as well as the basal rates could be steady or variable over the course of time. In the intervention group, no other analgesics were allowed for simultaneous administration. However, this did not exclude the prior use of other parenteral (opioid) analgesics or other methods of pain relief administered to the parturients during the conduct of the study (i.e. escape analgesia, e.g. Entonox).
Types of outcome measures
Primary outcomes
Satisfaction with pain relief (as defined by trialists).
-
Adverse events for women:
apnoea (≥ 20 s of zero respiratory rate);
respiratory depression (less than nine breaths/minute);
oxygen desaturation (SpO2 ≤ 95%, ≤ 92%);
hypotension;
bradycardia;
nausea;
vomiting;
pruritus;
postpartum haemorrhage (≥ 1000 mL);
sedation at one hour after onset of analgesia.
-
Adverse events for newborns:
Apgar score less than seven at five minutes;
Apgar score at five minutes;
need for naloxone;
depressed baby;
fetal heart rate (FHR)/cardiotocography (CTG) abnormalities or non‐reassuring fetal status;
neonatal neurologic and adaptive capacity score (NACS).
Secondary outcomes
Pain intensity (as defined by trialists) at 30 minutes to one hour ('early') and at two hours ('late')
Additional analgesia required (escape analgesia)
Rate of unscheduled caesarean delivery
Rate of assisted vaginal birth
Augmented labour (e.g. use of oxytocin)
Satisfaction with childbirth experience (as defined by trialists)
Sense of control in labour
Effect (negative) on mother/baby interaction
Breastfeeding initiation (as defined by trialists)
Umbilical cord base excess (arterial and venous)
Umbilical cord pH (arterial and venous)
Need for neonatal resuscitation (e.g. CPAP (continuous positive airway pressure), bag or mask ventilation, intubation)
Long‐term childhood development (as defined by trialists)
Cost (as defined by trialists
Search methods for identification of studies
The following methods section of this review is based on a standard template used by Cochrane Pregnancy and Childbirth with review‐specific modifications.
Electronic searches
We searched Cochrane Pregnancy and Childbirth’s Trials Register by contacting their Information Specialist (9 December 2015). We updated this search on 10 December 2016 and added the results to Studies awaiting classification.
The Register is a database containing over 23,000 reports of controlled trials in the field of pregnancy and childbirth. For full search methods used to populate Pregnancy and Childbirth’s Trials Register including the detailed search strategies for CENTRAL, MEDLINE, Embase and CINAHL; the list of handsearched journals and conference proceedings, and the list of journals reviewed via the current awareness service, please follow this link to the editorial information about the Cochrane Pregnancy and Childbirth in the Cochrane Library and select the ‘Specialized Register ’ section from the options on the left side of the screen.
Briefly, the Cochrane Pregnancy and Childbirth’s Trials Register is maintained by their Information Specialist and contains trials identified from:
monthly searches of the Cochrane Central Register of Controlled Trials (CENTRAL);
weekly searches of MEDLINE (Ovid);
weekly searches of Embase (Ovid);
monthly searches of CINAHL (EBSCO);
handsearches of 30 journals and the proceedings of major conferences;
weekly current awareness alerts for a further 44 journals plus monthly BioMed Central email alerts.
Search results are screened by two people and the full text of all relevant trial reports identified through the searching activities described above is reviewed. Based on the intervention described, each trial report is assigned a number that corresponds to a specific Pregnancy and Childbirth review topic (or topics), and is then added to the Register. The Information Specialist searches the Register for each review using this topic number rather than keywords. This results in a more specific search set which has been fully accounted for in the relevant review sections (Included studies; Excluded studies; Studies awaiting classification; Ongoing studies).
Search results were screened by two people (SW, YJ) and the full texts of all relevant trial reports identified through the searching activities described above were reviewed.
In addition, we searched ClinicalTrials.gov (26 November 2015) and the WHO International Clinical Trials Registry Platform (ICTRP) (27 November 2015) for unpublished, planned and ongoing trial reports. Our search terms were detailed in Appendix 1. We updated this search in December 2016 and added the results to Studies awaiting classification.
Searching other resources
We handsearched the congress abstracts of the American Society of Anesthesiologists (ASA), from 2000 to 18 November 2015, the International Anesthesia Research Society (IARS), from 2003 to 26 November 2015, and the European Society of Anaesthesiology (ESA), from 2004 to 26 November 2015. We updated this search in December 2016
We also searched the reference lists of retrieved studies. We did not apply any language or date restrictions.
Data collection and analysis
Selection of studies
Two review authors (SW, YJ) independently assessed for inclusion all the potential studies that were identified as a result of the search strategy (Appendix 2). We resolved any disagreement through discussion or, if required, we consulted a third review author (PK).
We created a study flow diagram to map the number of records identified, included and excluded.
Data extraction and management
We used a form to extract data (Appendix 3). For eligible studies, two review authors (SW, YJ) extracted the data using the agreed form. We resolved discrepancies through discussion or, if required, we consulted a third review author (PK). When information regarding any of the above was unclear, we attempted to contact authors of the original reports to provide further details. We entered data into Review Manager 5 software (RevMan 2014) and checked for accuracy. A detailed description of the included studies is provided under the section Characteristics of included studies.
Assessment of risk of bias in included studies
Two review authors (SW, YJ) independently assessed risk of bias (RoB) for each study using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) (Appendix 4). We resolved any disagreement by discussion or by involving further review authors (PK, AA).
(1) Random sequence generation (checking for possible selection bias)
We described for each included study the method used to generate the allocation sequence in sufficient detail to allow an assessment of whether it should produce comparable groups.
We assessed the method as:
low risk of bias (any truly random process, e.g. random number table; computer random number generator);
high risk of bias (any non‐random process, e.g. odd or even date of birth; hospital or clinic record number);
unclear risk of bias.
(2) Allocation concealment (checking for possible selection bias)
We described for each included study the method used to conceal allocation to interventions prior to assignment and assessed whether the intervention allocation could have been foreseen in advance of, or during recruitment, or changed after assignment.
We assessed the methods as:
low risk of bias (e.g. telephone or central randomisation; sequentially numbered opaque sealed envelopes);
high risk of bias (open random allocation; unsealed or non‐opaque envelopes, alternation; date of birth);
unclear risk of bias.
(3.1) Blinding of participants and personnel (checking for possible performance bias)
We described for each included study the methods used, if any, to blind study participants and personnel from knowledge of which intervention a participant received. We considered that studies were at low risk of bias if they were efficiently blinded (methods used for blinding were plausible), or if we judged that the lack of blinding would be unlikely to affect results. We assessed blinding separately for subjective and objective outcomes. Most of the outcomes being assessed were defined as subjective outcomes with the exception of umbilical cord base excess/pH, vomiting and postpartum haemorrhage which were defined as objective outcomes. All GRADE‐relevant outcomes were subjective outcomes.
We assessed the methods as:
low, high or unclear risk of bias for participants;
low, high or unclear risk of bias for personnel.
(3.2) Blinding of outcome assessment (checking for possible detection bias)
We described for each included study the methods used, if any, to blind outcome assessors from knowledge of which intervention a participant received. We assessed blinding separately for subjective and objective outcomes.
We assessed methods used to blind outcome assessment as:
low, high or unclear risk of bias.
(4) Incomplete outcome data (checking for possible attrition bias due to the amount, nature and handling of incomplete outcome data)
We described for each included study, and for each outcome or class of outcomes (adverse events for mothers and newborns), the completeness of data including attrition and exclusions from the analysis. We stated whether attrition and exclusions were reported and the numbers included in the analysis at each stage (compared with the total randomised participants), reasons for attrition or exclusion where reported, and whether missing data were balanced across groups or were related to outcomes (see Characteristics of included studies, 'Risk of bias' table). We further assessed for each included study the cross‐over rates, escape rates (rescue analgesia), type of data analysis (full‐intention‐to‐treat (F‐ITT), partial‐ITT, per‐protocol‐analysis, as‐treated analysis), and the methods used for imputation of missing data. We assessed attrition bias separately for each outcome or class of outcome (Table 6).
1. Attrition bias: Outcome level (GRADE‐relevant outcomes).
| Study |
No. randomised (Remifentanil/ control) |
No. analysed (Remifentanil/ control) |
Overall assessment for risk of attrition bias | Outcome level_Risk of bias | |||||
| Satisfaction with pain relief | AE for women | AE for newborns | Pain intensity | Additional analgesia | Rate of CS | ||||
| Balki 2007 | 10/ 10 |
10/ 10 |
Low | Low | Low | Low | Low | Low | Low |
| Blair 2005 | 20/ 20 |
20/ 19 |
High | High | High | High | Unclear | Unclear | |
| Calderon 2006 | 12/ 12 |
12/ 12 |
Low | Low | Low | Low | Low | Low | |
| Douma 2010 | 60/ 60/ 60 |
52/ 53/ 54 |
High | High | High | High | Low | Low | High |
| Douma 2011 | 14/ 12 |
10/ 10 |
High | High | Low | High | High | Low | Low |
| Douma 2015 | 57/ 59 |
49/ 49 |
High | High | High | High | Unclear | Unclear | High |
| El‐Kerdawy 2010 | 15/ 15 |
15/ 15 |
Low | Low | Low | Low | Low | Low | |
| Evron 2005 | 43/ 45 |
43/ 45 |
Unclear | Low | High | Low | Low | Low | Low |
| Evron 2008 | 213 NA/ NA/ NA/ NA |
192 44/ 50/ 49/ 49 |
Low | Low | Low | Low | |||
| Freeman 2015 | 709/ 705 |
687/ 671 |
High | High | High | High | High | High | High |
| Ismail 2012 | 380/ 380/ 380 |
380/ 380/ 380 |
Low | Low | Low | Low | Low | Low | |
| Khooshideh 2015 | 41/ 41 |
41/ 41 |
Low | Low | Low | Low | Low | ||
| Ng 2011 | 34/ 34 |
34/ 34 |
Low | Low | Low | Low | Low | Low | Low |
| Shen 2013 | 30/ 30 |
27/ 26 |
High | High | High | High | High | High | |
| Stocki 2014 | 20/ 20 |
19/ 20 |
Low | Low | Low | Low | Low | Low | Low |
| Stourac 2014 | 13/ 15 |
12/ 12 |
High | High | High | Low | High | Low | |
| Thurlow 2002 | 18/ 18 |
18/ 18 |
Unclear | Low | Low | Low | High | High | |
| Tveit 2012 | 19/ 20 |
17/ 20 |
High | High | High | High | High | Low | High |
| Volikas 2001 | 9/ 8 |
9/ 8 |
Low | Low | Low | Low | Low | Low | |
| Volmanen 2008 | 27/ 25 |
24/ 21 |
High | High | High | High | High | High | High |
Abbreviations:
AE: adverse events, CS: caesarean section
We assessed methods as:
low risk of bias (e.g. no missing outcome data after randomisation; missing outcome data less than 15%, and reported, and balanced across groups, and unrelated to true outcome; full‐ and partial‐ITT);
high risk of bias (e.g. missing outcome data greater than 15% or numbers or reasons for missing data not reported or imbalanced across groups; ‘as‐treated’ analysis done with substantial departure of intervention received from that assigned at randomisation);
unclear risk of bias.
(5) Selective reporting (checking for reporting bias)
We described for each included study how we investigated the possibility of selective outcome reporting bias and what we found.
We assessed the methods as:
low risk of bias (if a study protocol was available and all of the study’s pre‐specified primary and secondary outcomes have been reported in the final study report);
high risk of bias (where not all pre‐specified primary and secondary outcomes have been reported; one or more reported primary outcomes were not pre‐specified; outcomes of interest were reported incompletely and so cannot be used; study failed to include results of a key outcome that would have been expected to have been reported);
unclear risk of bias (if no published study protocol was available).
(6) Other bias (checking for bias due to problems not covered by (1) to (5) above)
We described for each included study any important concerns we had about other possible sources of bias (e.g. early stopping of the trial without pre‐defined stopping rules).
We assessed whether each study was free of other problems that could put it at risk of bias:
low risk of other bias;
high risk of other bias;
unclear whether there is risk of other bias.
(7) Overall risk of bias
We made explicit judgements about whether studies were at high risk of bias, according to the criteria given in the Handbook (Higgins 2011). With reference to the potential biases stated above (1 to 6), we assessed the likely magnitude and direction of the bias and whether we considered it was likely to have an impact on the findings. We explored the impact of the level of bias through undertaking sensitivity analyses ‐ seeTable 7; Table 8; Table 9
2. Sensitivity analysis: Selection bias (random sequence generation, allocation concealment).
|
Sensitivity analysis: Selection bias |
Statistical method | All studies | 'high risk of bias'‐studies excluded | Impact on robustness (95% CI) | ||
| n | Effect estimate | n | Effect estimate | |||
| 1. Remifentanil (PCA) versus another opioid (IV/IM) | ||||||
| 1.1 Satisfaction with pain relief | SMD (IV, Random), 95% CI | 4, all at low risk of bias | ||||
| 1.3 Oxygen desaturation (SpO2 < 95%) | RR (MH, Random), 95% CI | 2, all at low risk of bias | ||||
| 1.4 Nausea (and vomiting) | RR (MH, Random), 95% CI | 4, all at low risk of bias | ||||
| 1.6 Pruritus | RR (IV, Random), 95% CI, 0/0 cell counts | 2, all at low risk of bias | ||||
| 1.10 FHR/CTG abnormalities, non‐reassuring fetal status | RR (MH, Random), 95% CI | 2, all at low risk of bias | ||||
| 1.11 Pain intensity 'early' (30 min/1 h) | SMD (IV, Random), 95% CI | 3, all at low risk of bias | ||||
| 1.13 Additional analgesia required (escape analgesia) | RR (MH, Random), 95% CI | 3, all at low risk of bias | ||||
| 1.14 Rate of caesarean delivery | RR (MH, Random), 95% CI | 4, all at low risk of bias | ||||
| 2. Remifentanil (PCA) versus another opioid (PCA) | ||||||
| 2.2 Oxygen desaturation (SpO2 < 95%) | RR (MH, Random), 95% CI | 2, all at low risk of bias | ||||
| 2.10 Need for naloxone | RR (IV, Random), 95% CI, 0/0 cell counts | 2, all at low risk of bias | ||||
| 2.12 NACS at 15/30 min | MD (IV, Random), 95% CI | 2, all at low risk of bias | ||||
| 2.13 Pain intensity 'early' (30 min/1 h) | SMD (IV, Random), 95% CI | 3, all at low risk of bias | ||||
| 2.15 Additional analgesia required (escape analgesia) | RR (MH, Random), 95% CI | 3, all at low risk of bias | ||||
| 2.16 Rate of caesarean delivery | RR (MH, Random), 95% CI | 2, all at low risk of bias | ||||
| 3. Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE) | ||||||
| 3.1 Satisfaction with pain relief | SMD (IV, Random), 95% CI | 7 | ‐0.22 [‐0.40, ‐0.04] | 6 | ‐0.20 [‐0.46, 0.07] | Yes (CI includes 0) |
| 3.3 Respiratory depression (< 9, < 8 breaths/min) | RR (IV, Random), 95% CI, 0/0 cell counts | 3 | 0.91 [0.51, 1.62] | 2 | 0.91 [0.52, 1.61] | No |
| 3.4 Oxygen desaturation (SpO2 < 92%) | RR (MH, Random), 95% CI | 3 | 3.24 [1.66, 6.32] | 2 | 5.83 [0.40, 84.06] | Yes (CI includes 1) |
| 3.5 Oxygen desaturation (SpO2 < 95%) | RR (MH, Random), 95% CI | 3 | 3.27 [2.32, 4.61] | 2 | 5.44 [2.11, 14.02] | Yes (effect and CI increased) |
| 3.6 Hypotension | RR (IV, Random), 95% CI, 0/0 cell counts | 4 | 0.59 [0.37, 0.94] | 3 | 0.57 [0.00, 2.4E7] | Yes (CI includes 1) |
| 3.7 Bradycardia | RR (IV, Random), 95% CI, 0/0 cell counts | 2, all at low risk of bias | ||||
| 3.8 Nausea | RR (MH, Random), 95% CI | 8 | 1.49 [1.19, 1.86] | 7 | 1.41 [1.09, 1.83] | No |
| 3.9 Vomiting | RR (MH, Random), 95% CI | 6 | 1.63 [1.25, 2.13] | 5 | 1.82 [1.29, 2.57] | No |
| 3.10 Pruritus | RR (MH, Random), 95% CI | 7 | 0.75 [0.48, 1.18] | 6 | 0.81 [0.45, 1.45] | No |
| 3.11 Sedation (1 h) | MD (IV, Random), 95% CI | 3, all at low risk of bias | ||||
| 3.12 Apgarscore ≤ 7 (< 7) at 5 min | RR (IV, Random), 95% CI, 0/0 cell counts | 5, all at low risk of bias | ||||
| 3.13 Apgarscore at 5 min | MD (IV,), 95% CI | 3, all at low risk of bias | ||||
| 3.14 Need for naloxone | RR (IV, Random), 95% CI, 0/0 cell counts | 2, all at low risk of bias | ||||
| 3.15 FHR/CTG abnormalities, non‐reassuring fetal status | RR (MH, Random), 95% CI | 5, all at low risk of bias | ||||
| 3.16 Pain intensity 'early' (1 h) | SMD (IV, Random), 95% CI | 6, all at low risk of bias | ||||
| 3.18 Additional analgesia required | RR (IV, Random), 95% CI, 0/0 cell counts | 6 | 9.27 [3.73, 23.03] | 5 | 5.29 [1.2, 23.3] | No |
| 3.19 Rate of caesarean delivery | RR (MH, Random), 95% CI | 9, all at low risk of bias | ||||
| 4. Remifentanil (PCA) versus remifentanil (continuous IV) | ||||||
| 4.1 Respiratory depression (< 8 breaths/min) | RR (IV, Random), 95% CI, 0/0 cell counts | 2, all at low risk of bias | ||||
| 4.3 Hypotension | RR (IV, Random), 95% CI, 0/0 cell counts | 2, all at low risk of bias | ||||
| 4.4 Bradycardia | RR (IV, Random), 95% CI, 0/0 cell counts | 2, all at low risk of bias | ||||
| 4.5 Nausea (and vomiting) | RR (MH, Random), 95% CI | 2, all at low risk of bias | ||||
| 4.8 Need for naloxone | RR (IV, Random), 95% CI, 0/0 cell counts | 2, all at low risk of bias | ||||
All RR for outcomes including 0/0 cell counts (zero/zero event trials) were calculated using TSA (constant continuity correction, 0.01). Review Manager 5 produces computational errors when both the intervention and control group have zero events. By using TSA there is no possibility to choose the MH method (only IV) which may cause small deviations within results.
Abbreviations:
[95% CI]: 95% confidence interval; IV: Inverse Variance; MD: mean difference; MH: Mantel‐Haenszel; n: number of participants; RPCA: Remifentanil PCA; RR: risk ratio; SMD: standardised mean difference
3. Sensitivity analysis: Blinding (performance and detection bias).
|
Sensitivity analysis: Blinding (performance and detection bias) |
Statistical method | All studies | 'high risk of bias'‐studies excluded | Impact on robustness (95% CI) | ||
| n | Effect estimate | n | Effect estimate | |||
| 1. Remifentanil (PCA) versus another opioid (IV/IM) | ||||||
| 1.1 Satisfaction with pain relief | SMD (IV, Random), 95% CI | 4 | 2.11 [0.72, 3.49] | 2 | 2.46 [‐0.34, 5.26] | Yes (CI includes 0) |
| 1.3 Oxygen desaturation (SpO2 < 95%) | RR (MH, Random), 95% CI | 2 | 0.48 [0.00, 47.37] | 1 | 0.05 [0.00, 0.82] | Yes (CI < 1: favours RPCA) |
| 1.4 Nausea (and vomiting) | RR (MH, Random), 95% CI | 4 | 0.54 [0.29, 0.99] | 2 | 0.36 [0.06, 2.29] | Yes (CI includes 1) |
| 1.6 Pruritus | RR (IV, Random), 95% CI, 0/0 cell counts | 2, all at low risk of bias | ||||
| 1.10 FHR/CTG abnormalities, non‐reassuring fetal status | RR (MH, Random), 95% CI | 2, all at low risk of bias | ||||
| 1.11 Pain intensity 'early' (30 min/1 h) | SMD (IV, Random), 95% CI | 3 | ‐1.58 [‐2.69, ‐0.48] | 2 | ‐1.28 [‐2.62, 0.07] | Yes (CI includes 0) |
| 1.13 Additional analgesia required (escape analgesia) | RR (MH, Random), 95% CI | 3 | 0.57 [0.40, 0.81] | 2 | 0.48 [0.25, 0.91] | No |
| 1.14 Rate of caesarean delivery | RR (MH, Random), 95% CI | 4 | 0.70 [0.34, 1.41] | 2 | 0.63 [0.30, 1.31] | No |
| 2. Remifentanil (PCA) versus another opioid (PCA) | ||||||
| 2.2 Oxygen desaturation (SpO2 < 95%) | RR (MH, Random), 95% CI | 2 | 1.28 [0.49, 3.30] | 1 | 1.64 [1.25, 2.15] | Yes (CI > 1: favours opioid) |
| 2.10 Need for naloxone | RR (IV, Random), 95% CI, 0/0 cell counts | 2 | 0.03 [0.00, 1.8E8] | 1 | 0.00 [0.00, 0.06] | Yes (CI < 1: favours RPCA) |
| 2.12 NACS at 15/30 min | MD (IV, Random), 95% CI | 2 | 1.11 [‐0.65, 2.87] | 1 | 0.20 [‐0.93, 1.33] | Yes (direction of effect changed, CI decreased) |
| 2.13 Pain intensity 'early' (30 min/1 h) | SMD (IV, Random), 95% CI | 3 | ‐0.51 [‐1.01, ‐0.00] | 2 | ‐0.73 [‐1.05, ‐0.40] | Yes (lower CI: clinically relevant moderate effect) |
| 2.15 Additional analgesia required (escape analgesia) | RR (MH, Random), 95% CI | 3 | 0.76 [0.45, 1.28] | 2 | 0.65 [0.39, 1.09] | No |
| 2.16 Rate of caesarean delivery | RR (MH, Random), 95% CI | 2, all at low risk of bias | ||||
| 3. Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE) | ||||||
| 3.1 Satisfaction with pain relief | SMD (IV, Random), 95% CI | 7 | ‐0.22 [‐0.40, ‐0.04] | 1 | 0.27 [‐0.31, 0.86] | Yes (CI includes 0) |
| 3.3 Respiratory depression (< 9, < 8 breaths/min) | RR (IV, Random), 95% CI, 0/0 cell counts | 3 | 0.91 [0.51, 1.62] | 0 | Not estimable | All studies at high risk |
| 3.4 Oxygen desaturation (SpO2 < 92%) | RR (MH, Random), 95% CI | 3 | 3.24 [1.66, 6.32] | 0 | Not estimable | All studies at high risk |
| 3.5 Oxygen desaturation (SpO2 < 95%) | RR (MH, Random), 95% CI | 3 | 3.27 [2.32, 4.61] | 1 | 11.38 [1.62, 79.78] | Yes (effect and CI increased) |
| 3.6 Hypotension | RR (IV, Random), 95% CI, 0/0 cell counts | 4 | 0.59 [0.37, 0.94] | 0 | Not estimable | All studies at high risk |
| 3.7 Bradycardia | RR (IV, Random), 95% CI, 0/0 cell counts | 2 | 1.0 [0.00, 1.0E12] | 0 | Not estimable | All studies at high risk |
| 3.8 Nausea | RR (MH, Random), 95% CI | 8 | 1.49 [1.19, 1.86] | 1 | 3.94 [0.96, 16.22] | Yes (CI includes 1) |
| 3.9 Vomiting | RR (MH, Random), 95% CI | 6 | 1.63 [1.25, 2.13] | 0 | Not estimable | All studies at high risk |
| 3.10 Pruritus | RR (MH, Random), 95% CI | 7 | 0.75 [0.48, 1.18] | 0 | Not estimable | All studies at high risk |
| 3.11 Sedation (1 h) | MD (IV, Random), 95% CI | 3 | 0.71 [0.03, 1.39] | 0 | Not estimable | All studies at high risk |
| 3.12 Apgarscore ≤ 7 (< 7) at 5 min | RR (IV, Random), 95% CI, 0/0 cell counts | 5 | 1.26 [0.62, 2.57] | 0 | Not estimable | All studies at high risk |
| 3.13 Apgarscore at 5 min | MD (IV,), 95% CI | 3 | 0.06 [‐0.27, 0.39] | 0 | Not estimable | All studies at high risk |
| 3.14 Need for naloxone | RR (IV, Random), 95% CI, 0/0 cell counts | 2 | 0.02 [0.00, 1.6E8] | 0 | Not estimable | All studies at high risk |
| 3.15 FHR/CTG abnormalities, non‐reassuring fetal status | RR (MH, Random), 95% CI | 5 | 1.55 [0.49, 4.92] | 1 | 11.38 [1.62, 79.78] | Yes (CI > 1: favours epidural) |
| 3.16 Pain intensity 'early' (1 h) | SMD (IV, Random), 95% CI | 6 | 0.57 [0.31, 0.84] | 0 | Not estimable | All studies at high risk |
| 3.18 Additional analgesia required | RR (IV, Random), 95% CI, 0/0 cell counts | 6 | 9.27 [3.73, 23.07] | 0 | Not estimable | All studies at high risk |
| 3.19 Rate of caesarean delivery | RR (MH, Random), 95% CI | 9 | 0.99 [0.81, 1.21] | 1 | 0.88 [0.06, 13.14] | Yes (CI increased) |
| 4. Remifentanil (PCA) versus remifentanil (continuous IV) | ||||||
| 4.1 Respiratory depression (< 8 breaths/min) | RR (IV, Random), 95% CI, 0/0 cell counts | 2 | 0.98 [0.00, 1.0E12] | 1 | 0.98 [0.00, ∞] | No |
| 4.3 Hypotension | RR (IV, Random), 95% CI, 0/0 cell counts | 2 | 0.98 [0.00, 1.0E12] | 1 | 0.98 [0.00, ∞] | No |
| 4.4 Bradycardia | RR (IV, Random), 95% CI, 0/0 cell counts | 2 | 0.98 [0.00, 1.0E12] | 1 | 0.98 [0.00, ∞] | No |
| 4.5 Nausea (and vomiting) | RR (MH, Random), 95% CI | 2 | 0.85 [0.28, 2.54] | 1 | 0.53 [0.21, 1.39] | No |
| 4.8 Need for naloxone | RR (IV, Random), 95% CI, 0/0 cell counts | 2 | 0.98 [0.00, 1.0E12] | 1 | 0.98 [0.00, ∞] | No |
All RR for outcomes including 0/0 cell counts (zero/zero event trials) were calculated using TSA (constant continuity correction, 0.01). Review Manager 5 produces computational errors when both the intervention and control group have zero events. By using TSA there is no possibility to choose the MH method (only IV) which may cause small deviations within results.
Abbreviations:
[95% CI]: 95% confidence interval; IV: Inverse Variance; MD: mean difference; MH: Mantel‐Haenszel; n: number of participants; RPCA: Remifentanil PCA; RR: risk ratio; SMD: standardised mean difference
4. Sensitivity analysis: Attrition bias.
|
Sensitivity analysis: Attrition bias |
Statistical method | All studies | 'high risk of bias'‐studies excluded | Impact on robustness (95% CI) | ||
| n | Effect estimate | n | Effect estimate | |||
| 1. Remifentanil (PCA) versus another opioid (IV/IM) | ||||||
| 1.1 Satisfaction with pain relief | SMD (IV, Random), 95% CI | 4, all at low risk of bias | ||||
| 1.3 Oxygen desaturation (SpO2 < 95%) | RR (MH, Random), 95% CI | 2 | 0.48 [0.00, 47.37] | 1 | 3.50 [0.84, 14.61] | Yes (CI + effect moved to favour of opioid) |
| 1.4 Nausea (and vomiting) | RR (MH, Random), 95% CI | 4, all at low risk of bias | ||||
| 1.6 Pruritus | RR (IV, Random), 95% CI, 0/0 cell counts | 2, all at low risk of bias | ||||
| 1.10 FHR/CTG abnormalities, non‐reassuring fetal status | RR (MH, Random), 95% CI | 2, all at low risk of bias | ||||
| 1.11 Pain intensity 'early' (30 min/1 h) | SMD (IV, Random), 95% CI | 3, all at low risk of bias | ||||
| 1.13 Additional analgesia required (escape analgesia) | RR (MH, Random), 95% CI | 3 | 0.57 [0.40, 0.81] | 2 | 0.48 [0.25, 0.91] | No |
| 1.14 Rate of caesarean delivery | RR (MH, Random), 95% CI | 4 | 0.70 [0.34, 1.41] | 3 | 0.60 [0.29, 1.24] | No |
| 2. Remifentanil (PCA) versus another opioid (PCA) | ||||||
| 2.2 Oxygen desaturation (SpO2 < 95%) | RR (MH, Random), 95% CI | 2 | 1.28 [0.49, 3.30] | 0 | Not estimable | All studies at high risk |
| 2.10 Need for naloxone | RR (IV, Random), 95% CI, 0/0 cell counts | 2 | 0.03 [0.00, 1.8E8] | 1 | 0.00 [0.00, 0.06] | Yes (CI moved to favour RPCA) |
| 2.12 NACS at 15/30 min | MD (IV, Random), 95% CI | 2 | 1.11 [‐0.65, 2.87] | 0 | Not estimable | All studies at high risk |
| 2.13 Pain intensity 'early' (30 min/1 h) | SMD (IV, Random), 95% CI | 3, all at low risk of bias | ||||
| 2.15 Additional analgesia required (escape analgesia) | RR (MH, Random), 95% CI | 3, all at low risk of bias | ||||
| 2.16 Rate of caesarean delivery | RR (MH, Random), 95% CI | 2 | 2.78 [0.99, 7.82] | 1 | 1.78 [0.20, 16.10] | Yes (CI increased) |
| 3. Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE) | ||||||
| 3.1 Satisfaction with pain relief | SMD (IV, Random), 95% CI | 7 | ‐0.22 [‐0.40, ‐0.04] | 3 | ‐0.27 [‐0.64, 0.10] | Yes (CI includes 0) |
| 3.3 Respiratory depression (< 9, < 8 breaths/min) | RR (IV, Random), 95% CI, 0/0 cell counts | 3 | 0.91 [0.51, 1.62] | 1 | 0.91 [0.39, 2.10] | No |
| 3.4 Oxygen desaturation (SpO2 < 92%) | RR (MH, Random), 95% CI | 3 | 3.24 [1.66, 6.32] | 0 | Not estimable | All studies at high risk |
| 3.5 Oxygen desaturation (SpO2 < 95%) | RR (MH, Random), 95% CI | 3 | 3.27 [2.32, 4.61] | 1 | 4.33 [1.47, 12.79] | Yes (effect and CI increased) |
| 3.6 Hypotension | RR (IV, Random), 95% CI, 0/0 cell counts | 4 | 0.59 [0.37, 0.94] | 2 | 0.01 [0.00, 7.8E7] | Yes (CI includes 1) |
| 3.7 Bradycardia | RR (IV, Random), 95% CI, 0/0 cell counts | 2 | 1.0 [0.00, 1.0E12] | 1 | 1.0 [0.00, ∞] | No |
| 3.8 Nausea | RR (MH, Random), 95% CI | 8 | 1.49 [1.19, 1.86] | 4 | 1.27 [0.82, 1.98] | Yes (CI includes 1) |
| 3.9 Vomiting | RR (MH, Random), 95% CI | 6 | 1.63 [1.25, 2.13] | 3 | 1.54 [0.75, 3.14] | Yes (CI includes 1) |
| 3.10 Pruritus | RR (MH, Random), 95% CI | 7 | 0.75 [0.48, 1.18] | 5 | 0.86 [0.48, 1.56] | No |
| 3.11 Sedation (1 h) | MD (IV, Random), 95% CI | 3, all at low risk of bias | ||||
| 3.12 Apgarscore ≤ 7 (< 7) at 5 min | RR (IV, Random), 95% CI, 0/0 cell counts | 5 | 1.26 [0.62, 2.57] | 3 | 1.26 [0.62, 2.57] | No |
| 3.13 Apgarscore at 5 min | MD (IV,), 95% CI | 3 | 0.06 [‐0.27, 0.39] | 0 | Not estimable | All studies at high risk |
| 3.14 Need for naloxone | RR (IV, Random), 95% CI, 0/0 cell counts | 2, all at low risk of bias | ||||
| 3.15 FHR/CTG abnormalities, non‐reassuring fetal status | RR (MH, Random), 95% CI | 5 | 1.55 [0.49, 4.92] | 2 | 0.87 [0.41, 1.87] | Yes (CI decreased, effect changed) |
| 3.16 Pain intensity 'early' (1 h) | SMD (IV, Random), 95% CI | 6 | 0.57 [0.31, 0.84] | 3 | 0.57 [0.25, 0.89] | No |
| 3.18 Additional analgesia required | RR (IV, Random), 95% CI, 0/0 cell counts | 6 | 9.27 [3.73, 23.03] | 5 | 5.29 [1.2, 23.3] | No |
| 3.19 Rate of caesarean delivery | RR (MH, Random), 95% CI | 9 | 0.99 [0.81, 1.21] | 6 | 1.02 [0.83, 1.25] | No |
| 4. Remifentanil (PCA) versus remifentanil (continuous IV) | ||||||
| 4.1 Respiratory depression (< 8 breaths/min) | RR (IV, Random), 95% CI, 0/0 cell counts | 2 | 0.98 [0.00, 1.0E12] | 1 | 0.98 [0.00, ∞] | No |
| 4.3 Hypotension | RR (IV, Random), 95% CI, 0/0 cell counts | 2 | 0.98 [0.00, 1.0E12] | 1 | 0.98 [0.00, ∞] | No |
| 4.4 Bradycardia | RR (IV, Random), 95% CI, 0/0 cell counts | 2 | 0.98 [0.00, 1.0E12] | 1 | 0.98 [0.00, ∞] | No |
| 4.5 Nausea (and vomiting) | RR (MH, Random), 95% CI | 2 | 0.85 [0.28, 2.54] | 1 | 1.67 [0.43, 6.52] | No |
| 4.8 Need for naloxone | RR (IV, Random), 95% CI, 0/0 cell counts | 2 | 0.98 [0.00, 1.0E12] | 1 | 0.98 [0.00, ∞] | No |
All RR for outcomes including 0/0 cell counts (zero/zero event trials) were calculated using TSA (constant continuity correction, 0.01). Review Manager 5 produces computational errors when both the intervention and control group have zero events. By using TSA there is no possibility to choose the MH method (only IV) which may cause small deviations within results.
Abbreviations:
[95% CI]: 95% confidence interval; IV: Inverse Variance; MD: mean difference; MH: Mantel‐Haenszel; n: number of participants; RPCA: Remifentanil PCA; RR: risk ratio; SMD: standardised mean difference
Assessing the quality of the body of evidence using the GRADE approach
We assessed the quality of evidence using the GRADE approach as outlined in the GRADE handbook in order to assess the quality of the body of evidence for all comparisons relating to the following outcomes.
Satisfaction with pain relief
Pain intensity at 'early' (30 minutes/one hour) time points
Additional analgesia required (escape analgesia)
Conversion to caesarean delivery
Adverse events for women (apnoea, respiratory depression)
Adverse events for infants (Apgar scores less than seven at five minutes)
We used the GRADEpro Guideline Development Tool to import data from Review Manager 5 (RevMan 2014) in order to create ’Summary of findings’ tables for all main comparisons (if at least two relevant studies were available). All GRADE‐relevant outcomes were listed in the ’Summary of findings’ tables irrespective of whether data were available or not. With the GRADE approach we appraised the quality of evidence on the basis of the extent to which one can be confident that the estimate of effect reflects the item assessed. The quality of the body of evidence reflects within‐study risk of bias (methodological quality), indirectness, heterogeneity of the data (inconsistency), imprecision of effect estimates, and risk of publication bias.
For risk of bias, we judged the quality of evidence as adequate when most information was derived from studies at low risk of bias; we downgraded the quality by one level (serious) when most information was provided by studies at high or unclear risk of bias and we downgraded the quality by two levels (very serious) when the proportion of data from studies at high risk of bias was sufficient to affect interpretation of results (impact on robustness of estimated effect and confidence interval (CI); see Table 7; Table 8; Table 9: sensitivity analyses for selection bias, blinding, attrition bias) (Guyatt 2011a).
For inconsistency, we downgraded the quality of evidence by one level when the I2 statistic was 50% or higher without satisfactory explanation by subgroup analysis (Guyatt 2011b).
We judged the quality of evidence for indirectness as adequate when the outcome data were based on direct comparisons of interest, on the population of interest, and on the outcome of interest (not surrogate markers) (Guyatt 2011c). Otherwise, we downgraded for inconsistency by one level.
If the 95% CI excluded a risk ratio (RR) of 1.0 or a standardised mean difference (SMD) of 0.0, and the total number of participants exceeded the required information size (RIS, in case of RR) or optimal information size (OIS, in case of SMD) criterion (for detailed explanation on RIS and OIS see Data synthesis), precision was judged as adequate (Guyatt 2011d); we also did not downgrade, if the 95% CI was narrow (for RR: lower CI > 0.75, upper CI < 1.25), and included a RR of 1.0 or a SMD of 0.0 (no appreciable difference between treatments), and the total number of participants exceeded the RIS or OIS criterion. We downgraded the quality of evidence for imprecision by one level when the CI around the effect size was large or overlapped an absence of effect and failed to exclude an important benefit or harm and when the number of participants was lower than the required information size (RIS or OIS) or the monitoring boundaries were not crossed (see trial sequential analysis and optimal information size calculation: Data synthesis; Table 10; Table 11; Table 12; Table 13). We downgraded by two levels for very serious imprecision due to a small number of studies (n = 1) with a small sample size (< 150 participants).
5. Trial sequential analysis (low risk of bias‐based) for dichotomous GRADE‐relevant outcomes.
|
EE [95% CI], P value, I2 (%), n |
TSA_Low risk of bias‐based (all low) | |||||
| RRR (%) |
CER (%) |
H (%) |
RIS | evidence | ||
| 1.13 Additional analgesia | 0.58 [0.42, 0.79], 0.0005, 15%, 190 |
51.21 | 58 | 25 | 156 | evidence of effect (intervention) |
| low risk of bias studies: Evron 2005 + Ng 2011 (best) | ||||||
| 1.14 Rate of caesarean delivery | 0.63 [0.30, 1.32], 0.22, 0%,215 |
37.47 | 19 | 25 | 1444 | absence of evidence |
| low risk of bias studies: Evron 2005 + Ng 2011 (best) | ||||||
| 2.15 Additional analgesia | 0.87 [0.74, 1.03], 0.11, 0%, 215 |
35.21 | 28 | 25 | 1024 | absence of evidence |
| low risk of bias studies: Douma 2010 (best) + Volikas 2001 | ||||||
| 2.16 Rate of caesarean delivery | 2.78 [0.99, 7.82], 0.05, 0%, 143 |
‐77.76 | 12.5 | 25 | 852 | absence of evidence |
| only low risk of bias study: Volikas 2001 | ||||||
| 3.3 Respiratory depression | 0.91 [0.51, 1.62], 0.75, 0%,687 |
9.09 | 58 | 25 | 4986 | absence of evidence |
| best study (high risk): Stocki‐2014 | ||||||
|
3.12 Apgarscore < 7 at 5 min |
1.26 [0.62, 2.57], 0.52, 0%, 1322 |
‐26.33 | 3 | 25 | 2.9E4 | absence of evidence |
| not best study (0/0 events), but largest (high risk): Ismail 2012 | ||||||
| 3.18 Additional analgesia | 9.27 [3.73, 23.03], < 0.0001, 0%, 1037 |
‐218.8 | 5 | 25 | 449 | evidence of effect (control) |
| Not best study (0/0 events), but second best (high risk): Stocki 2014 | ||||||
| 3.19 Rate of caesarean delivery | 1.0 [0.82, 1.22], 0.9857, 0%, 1578 |
‐12.5 | 8 | 25 | 4.4E4 | absence of evidence |
| best study (high risk): Evron 2008 clinically relevant (RRR) assumptions: RRR = ‐ 50%, CER (empirical) = 22%, H (empirical) = 0% → IS = 924 (lack of effect) |
||||||
| 4.1 Respiratory depression | 0.98 [0.06, 15.37], 0.9896, 0%, 135 |
4 | 1 | 25 | 3.4E6 | absence of evidence |
| best study (high risk): Shen 2013 | ||||||
TSA (trial sequential analysis): random‐effects modelling; IV (inverse variance); (α = 0.05, power = 90% (ß = 0.10); zero event handling = constant continuity correction, 0.01; H = 25% (mild heterogeneity); calculated with TSA software (http://www.ctu.dk/tsa/)
Abbreviations:
CER: control event rate; EE [95% CI]: estimated effect with 95% confidence interval; EER: experimental event rate; H: heterogeneity adjustment factor; n: number of participants; NA: not applicable; RIS: required information size; RRR: relative risk reduction = (EER‐CER)/CER; TSMB: trial sequential monitoring boundary
6. Trial sequential analysis (empirical) for dichotomous GRADE‐relevant outcomes.
|
EE [95% CI], P value, I2 (%), n |
TSA_Empirical (with all studies) | |||||
|
RRR (%) |
CER (%) |
H (%) |
RIS | evidence | ||
| 1.13 Additional analgesia | 0.58 [0.42, 0.79], 0.0005, 15%, 190 |
42.39 | 62 | 21.39 | 194 | evidence of effect, TSMB, (intervention) |
| 1.14 Rate of caesarean delivery | 0.63 [0.30, 1.32], 0.22, 0%,215 |
30.4 | 15 | 0 | 2245 | absence of evidence |
| 2.15 Additional analgesia | 0.87 [0.74, 1.03], 0.11, 0%, 215 |
12.58 | 38 | 0 | 4218 | absence of evidence |
| 2.16 Rate of caesarean delivery | 2.78 [0.99, 7.82], 0.05, 0%, 143 |
‐177.7 | 6 | 0 | 372 | absence of evidence |
| 3.3 Respiratory depression | 0.91 [0.51, 1.62], 0.75, 0%,687 |
2 | 4 | 0 | 2.5E6 | absence of evidence |
|
3.12 Apgarscore < 7 at 5 min |
1.26 [0.62, 2.57], 0.52, 0%, 1322 |
‐26 | 2 | 0 | 3.4E4 | absence of evidence |
| 3.18 Additional analgesia | 9.27 [3.73, 23.03], < 0.0001, 0%, 1037 |
‐665 | 1 | 0 | 394 | evidence of effect (control) |
| 3.19 Rate of caesarean delivery | 1.0 [0.82, 1.22], 0.9857, 0%, 1578 |
1.18 | 22 | 0 | 1.1E6 | absence of evidence |
| 4.1 Respiratory depression | 0.98 [0.06, 15.37], 0.9896, 0%, 135 |
2 | 1 | 0 | 1.0E7 | absence of evidence |
TSA (trial sequential analysis): random‐effects modelling; IV (inverse variance); (α = 0.05, power = 90% (ß = 0.10); zero event handling = constant continuity correction, 0.01; H = 25% (mild heterogeneity); calculated with TSA software (http://www.ctu.dk/tsa/)
Abbreviations:
CER: control event rate; EE [95% CI]: estimated effect with 95% confidence interval; EER: experimental event rate; H: heterogeneity adjustment factor; n: number of participants; NA: not applicable; RIS: required information size; RRR: relative risk reduction = (EER‐CER)/CER; TSMB: trial sequential monitoring boundary
7. Optimal information size calculation (minimal clinically relevant difference) for GRADE‐relevant continuous outcomes.
|
EE [95% CI], P value, I2, n |
OIS_minimal clinically relevant difference1 | |||||
| mean1 | mean2 | SDlargest | OIS | evidence | ||
| 1.1 Satisfaction with pain relief | 2.11 [0.72, 3.49], 0.003, 93%, 216 |
7 | 6 | 2.22 | 208 | evidence of effect (intervention) |
| best low risk of bias study: Ng 2011 | ||||||
| 1.11 Pain intensity 'early' | ‐1.58 [‐2.69, ‐0.48], 0.005, 89%, 180 |
25.6 | 35.6 | 26.6 | 298 | absence of evidence |
| best low risk of bias study: Ng 2011 | ||||||
| 2.13 Pain intensity 'early' | ‐0.51 [‐1.01, ‐0.00], 0.05, 52%, 215 | 5.282 | 6.282 | 2.414 | 246 | absence of evidence |
| best low risk of bias study: Douma 2010 | ||||||
| 3.1 Satisfaction with pain relief | ‐0.22 [‐0.40, ‐0.04], 0.02, 52%, 2135 |
8.1 | 9.1 | 1.5 | 96 | evidence of effect (control) |
| best study (high risk): Stocki 2014 | ||||||
| 3.16 Pain intensity 'early' | 0.57 [0.31, 0.84], < 0.0001, 0%, 235 |
3.3 | 2.3 | 3.3 | 458 | absence of evidence |
| best study (high risk): Stocki 2014 | ||||||
The summary statistics for the GRADE‐relevant continuous outcomes was SMD (standardised mean difference). The TSA software (version 0.9 Beta) did not support trial sequential analysis of SMD. Therefore, we conducted OIS (optimal information size) calculations (http://stat.ubc.ca/˜rollin/stats/ssize/n2.html) which corresponds to a sample size calculation for an individual trial with the following general assumptions on α = 0.05 and ß = 0.10 (power = 90%).
1The assumed minimal clinically relevant difference was 1.0 cm (10 mm) on a VAS 0 to 10 cm (0 to 100 mm) scale. The mean2 was derived from the control group (low risk of bias (best) trial).
Abbreviations:
EE [95% CI]: estimated effect with 95% confidence interval; mean1: intervention group; mean2: control group; n: number of participants; SDlargest: largest standard deviation of the pooled studies was assumed
8. Optimal information size calculation (low risk of bias‐based) for GRADE‐relevant continuous outcomes.
|
EE [95% CI], P value, I2, n |
OIS_low risk of bias‐based (best) | |||||
| mean1 | mean2 | SDlargest | OIS | evidence | ||
| 1.1 Satisfaction with pain relief | 2.11 [0.72, 3.49], 0.003, 93%, 216 |
8 | 6 | 2.22 | 52 | evidence of effect (intervention) |
| best low risk of bias study: Ng 2011 | ||||||
| 1.11 Pain intensity 'early' | ‐1.58 [‐2.69, ‐0.48], 0.005, 89%, 180 |
22.1 | 35.6 | 26.6 | 164 | evidence of effect (intervention) |
| best low risk of bias study: Ng 2011 | ||||||
| 2.13 Pain intensity 'early' | ‐0.51 [‐1.01, ‐0.00], 0.05, 52%, 215 | 4.56 | 6.282 | 2.414 | 82 | lack of effect |
| best low risk of bias study: Douma 2010 | ||||||
| 3.1 Satisfaction with pain relief | ‐0.22 [‐0.40, ‐0.04], 0.02, 52%, 2135 |
8.6 | 9.1 | 1.5 | 380 | evidence of effect (control) |
| best study (high risk): Stocki 2014 | ||||||
| 3.16 Pain intensity 'early' | 0.57 [0.31, 0.84], < 0.0001, 0%, 235 |
4 | 2.3 | 3.3 | 160 | evidence of effect (control) |
| best study (high risk): Stocki 2014 | ||||||
The summary statistics for the GRADE‐relevant continuous outcomes was SMD (standardised mean difference). The TSA software (version 0.9 Beta) did not support trial sequential analysis of SMD. Therefore, we conducted OIS (optimal information size) calculations (http://stat.ubc.ca/˜rollin/stats/ssize/n2.html) which corresponds to a sample size calculation for an individual trial with the following general assumptions on α = 0.05 and ß = 0.10 (power = 90%).
The mean2 was derived from the control group (low risk of bias (best) trial).
Abbreviations:
EE [95% CI]: estimated effect with 95% confidence interval; mean1: intervention group; mean2: control group; n: number of participants; SDlargest: largest standard deviation of the pooled studies was assumed
For publication bias (Guyatt 2011e), we downgraded the quality of evidence by one level if the statistical test for funnel plot asymmetry suggested publication bias and the adjustment for small‐study effects as assessed by Duval and Tweedie’s trim and fill analysis changed the conclusion (see Assessment of reporting biases). We downgraded the level of evidence for publication bias by two levels, if most of the trials were small and industry‐ sponsored (Guyatt 2011e).
The GRADE assessment resulted in one of four levels of 'quality', and these expressed our confidence in the estimate of effect (Balshem 2011).
High quality: we are very confident that the true effect lies close to that of the estimate of the effect
Moderate quality: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different
Low quality: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect
Very low quality: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect
Measures of treatment effect
Dichotomous data
For dichotomous data, we presented results as summary RR with 95% CIs which were obtained from the intervention and control event rates.
Continuous data
For continuous data the mean difference (MD) was obtained from the difference between the intervention and the control group mean values with associated standard deviations (SD) if outcomes were measured the same way in the trials. We used the SMD to combine trials that measured the same outcome, but used different methods (outcomes: satisfaction with pain relief, pain intensity). Back‐transformation of SMD values into absolute values on a scale between 0 to 10 cm (visual analogue scale (VAS)) was performed for the outcomes satisfaction and pain to facilitate clinical interpretation. Therefore, the smallest as well as the largest SD of the pooled studies were used for back‐transformation (SMD * SD) to reflect the range of possible effects.
We included data reported as median and interquartile range (IQR) with a symmetric distribution (data with asymmetric distribution were not pooled) in addition to mean values and SD in the analysis. In the case of a symmetric distribution, we obtained the mean and SD from median and IQR values in accordance with Higgins 2011. If SD was missing, we calculated the SD from the CIs for group means by using the appropriate formula (Higgins 2011).
Unit of analysis issues
Cluster‐randomised trials
We planned to include cluster‐randomised trials in the analyses along with individually‐randomised trials. However, for the present review we did not identify any relevant cluster‐randomised trials. For further updates, we plan to adjust their standard errors (SE) using the methods described in the Handbook using an estimate of the intra‐cluster correlation co‐efficient (ICC) derived from the trial (if possible), from a similar trial or from a study of a similar population. If we have to use ICCs from other sources, we plan to report this and conduct sensitivity analyses to investigate the effect of variation in the ICC. If we will identify both cluster‐randomised trials and individually‐randomised trials for future updates, we plan to synthesise the relevant information. We will consider it reasonable to combine the results from both if there is little heterogeneity between the study designs and the interaction between the effect of intervention and the choice of randomisation unit is considered to be unlikely.
We also plan to acknowledge heterogeneity in the randomisation unit and will perform a sensitivity analysis to investigate the effects of the randomisation unit.
Multi‐armed studies
We overcame a unit‐of‐analysis error for studies that contributed multiple comparisons by combining groups (by using the appropriate formula for adding SDs when required) to create a single pair‐wise comparison, if the presented data in the trials allow us to do so (Higgins 2011).
Dealing with missing data
For included studies, we noted levels of attrition. We used only published data and did not contact the trials' authors for missing outcome data (e.g. reasons for missing data). We explored the impact of including studies with high levels of missing data in the overall assessment of treatment effect by using sensitivity analysis (Table 9).
For all outcomes, we carried out analyses, as far as possible, on an intention‐to‐treat (ITT) basis, i.e. we attempted to include all participants randomised to each group in the analyses, and all participants were analysed in the group to which they were allocated, regardless of whether or not they received the allocated intervention. The denominator for each outcome in each trial was the number randomised minus any participants whose outcomes are known to be missing. Full application of the ITT principle was possible only if complete outcome data were available for all randomly assigned participants.
In the case of missing data, we used an 'available‐case analysis' by excluding all participants for whom the outcome was missing from the analysis.
Assessment of heterogeneity
We assessed clinical and methodological heterogeneity of included studies to decide if the studies were sufficiently homogeneous (eligibility criteria) to be combined. We used clinical judgement, not heterogeneity statistics, to decide whether the studies could be combined.
We assessed statistical heterogeneity in each meta‐analysis using the Tau2, I2 and Chi2 statistics. We regarded heterogeneity as substantial if an I2 was greater than 50% and either a Tau2 was greater than zero, or there was a low P value (less than 0.10) in the Chi2 test for heterogeneity.
Assessment of reporting biases
We planned to investigate reporting biases (such as publication bias) using funnel plots, if there were 10 or more studies in the meta‐analysis. However, in the present review none of the outcomes included 10 or more studies. For further updates, if the number of studies increases, we plan to assess funnel plot asymmetry visually. If asymmetry is suggested by a visual assessment, we will perform exploratory analyses (e.g. Eggers regression test for continuous data, Arcsine test for dichotomous data) to further investigate funnel plot asymmetry and to adjust for small‐study effects by use of the Duval and Tweedie’s trim and fill analysis.
Data synthesis
We carried out meta‐analysis using the Review Manager software (RevMan 2014). We used the random‐effects meta‐analysis to produce an overall summary estimate since there was sufficient clinical heterogeneity to expect that the underlying treatment effects differed between trials. The random‐effects summary was treated as the average of the range of possible treatment effects and we discussed the clinical implications of treatment effects differing between trials. We performed a fixed‐effect meta‐analysis (which assumes that the pooled studies are sufficiently similar and estimating the same underlying treatment effect) as a sensitivity analysis (Table 14).
9. Sensitivity analysis: Random‐effects versus fixed‐effect model.
|
Sensitivity analysis: Random‐effects versus fixed‐effect model |
Statistical method | Random‐effects model | Fixed‐effect model |
Impact on robustness (95% CI) (fixed‐effect model) |
|||
| n | Effect estimate | n | Effect estimate | ||||
| 1. Remifentanil (PCA) versus another opioid (IV/IM) | |||||||
| 1.1 Satisfaction with pain relief | SMD (IV), 95% CI | 4 | 2.11 [0.72, 3.49] | 4 | 1.85 [1.51, 2.19] | Yes (CI decreased, large effect) | |
| 1.3 Oxygen desaturation (SpO2 < 95%) | RR (MH), 95% CI | 2 | 0.48 [0.00, 47.37] | 2 | 0.66 [0.28, 1.57] | Yes (CI decreased) | |
| 1.4 Nausea (and vomiting) | RR (MH), 95% CI | 4 | 0.54 [0.29, 0.99] | 4 | 0.51 [0.28, 0.95] | No | |
| 1.6 Pruritus | RR (IV), 95% CI, 0/0 cell counts |
2 | 1.02 [0.00, 1.1E12] | 2 | 1.02 [0.00, 1.1E12] | No | |
| 1.10 FHR/CTG abnormalities, non‐reassuring fetal status | RR (MH), 95% CI | 2 | 0.30 [0.10, 0.90] | 2 | 0.30 [0.10, 0.85] | No | |
| 1.11 Pain intensity 'early' (30 min/1 h) | SMD (IV), 95% CI | 3 | ‐1.58 [‐2.69, ‐0.48] | 3 | ‐1.35 [‐1.68, ‐1.01] | Yes (CI decreased, large effect) | |
| 1.13 Additional analgesia required (escape analgesia) | RR (MH), 95% CI | 3 | 0.57 [0.40, 0.81] | 3 | 0.53 [0.39, 0.71] | No | |
| 1.14 Rate of caesarean delivery | RR (MH), 95% CI | 4 | 0.70 [0.34, 1.41] | 4 | 0.77 [0.39, 1.49] | No | |
| 2. Remifentanil (PCA) versus another opioid (PCA) | |||||||
| 2.2 Oxygen desaturation (SpO2 < 95%) | RR (MH), 95% CI | 2 | 1.28 [0.49, 3.30] | 2 | 1.39 [1.16, 1.67] | Yes (CI > 1: favours opioid) | |
| 2.10 Need for naloxone | RR (IV,), 95% CI, 0/0 cell counts |
2 | 0.03 [0.00, 1.8E8] | 2 | 0.01 [0.00, 2.4E6] | No | |
| 2.12 NACS at 15/30 min | MD (IV), 95% CI | 2 | 1.11 [‐0.65, 2.87] | 2 | 1.15 [0.38, 1.93] | Yes (CI > 0: favours RPCA) | |
| 2.13 Pain intensity 'early' (30 min/1 h) | SMD (IV), 95% CI | 3 | ‐0.51 [‐1.01, ‐0.00] | 3 | ‐0.57 [‐0.86, ‐0.29] | Yes (CI < 0: favours RPCA) | |
| 2.15 Additional analgesia required (escape analgesia) | RR (MH), 95% CI | 3 | 0.76 [0.45, 1.28] | 3 | 0.74 [0.55, 1.00] | No | |
| 2.16 Rate of caesarean delivery | RR (MH), 95% CI | 2 | 2.78 [0.99, 7.82] | 2 | 2.78 [0.99, 7.77] | No | |
| 3. Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE) | |||||||
| 3.1 Satisfaction with pain relief | SMD (IV), 95% CI | 7 | ‐0.22 [‐0.40, ‐0.04] | 7 | ‐0.29 [‐0.38, ‐0.20] | No | |
| 3.3 Respiratory depression (< 9, < 8 breaths/min) | RR (IV), 95% CI, 0/0 cell counts |
3 | 0.91 [0.51, 1.62] | 3 | 1.2 [0.67, 2.17] | No | |
| 3.4 Oxygen desaturation (SpO2 < 92%) | RR (MH), 95% CI | 3 | 3.24 [1.66, 6.32] | 3 | 3.46 [2.32, 5.16] | No | |
| 3.5 Oxygen desaturation (SpO2 < 95%) | RR (MH), 95% CI | 3 | 3.27 [2.32, 4.61] | 3 | 3.30 [2.43, 4.49] | No | |
| 3.6 Hypotension | RR (IV,), 95% CI, 0/0 cell counts |
4 | 0.59 [0.37, 0.94] | 4 | 0.57 [0.36, 0.89] | No | |
| 3.7 Bradycardia | RR (IV,), 95% CI, 0/0 cell counts |
2 | 1.0 [0.00, 1.0E12] | 2 | 1.0 [0.00, 1.0E12] | No | |
| 3.8 Nausea | RR (MH), 95% CI | 8 | 1.49 [1.19, 1.86] | 8 | 1.53 [1.22, 1.91] | No | |
| 3.9 Vomiting | RR (MH), 95% CI | 6 | 1.63 [1.25, 2.13] | 6 | 1.62 [1.24, 2.10] | No | |
| 3.10 Pruritus | RR (MH), 95% CI | 7 | 0.75 [0.48, 1.18] | 7 | 0.76 [0.54, 1.07] | No | |
| 3.11 Sedation (1 h) | MD (IV), 95% CI | 3 | 0.71 [0.03, 1.39] | 3 | 0.91 [0.57, 1.25] | No | |
| 3.12 Apgarscore ≤ 7 (< 7) at 5 min | RR (IV,), 95% CI, 0/0 cell counts |
5 | 1.26 [0.62, 2.57] | 5 | 1.22 [0.67, 2.62] | No | |
| 3.13 Apgarscore at 5 min | MD (IV,), 95% CI | 3 | 0.06 [‐0.27, 0.39] | 3 | 0.06 [‐0.27, 0.39] | No | |
| 3.14 Need for naloxone | RR (IV,), 95% CI, 0/0 cell counts |
2 | 0.02 [0.00, 1.6E8] | 2 | 0.01 [0.00, 4.6E5] | No | |
| 3.15 FHR/CTG abnormalities, non‐reassuring fetal status | RR (MH), 95% CI | 5 | 1.55 [0.49, 4.92] | 5 | 1.38 [0.84, 2.25] | No | |
| 3.16 Pain intensity 'early' (1 h) | SMD (IV), 95% CI | 6 | 0.57 [0.31, 0.84] | 6 | 0.57 [0.31, 0.84] | No | |
| 3.18 Additional analgesia required | RR (IV,), 95% CI, 0/0 cell counts |
6 | 9.27 [3.73, 23.03] | 6 | 10.86 [4.37, 26.95] | No | |
| 3.19 Rate of caesarean delivery | RR (MH), 95% CI | 9 | 0.99 [0.81, 1.21] | 9 | 0.96 [0.79, 1.18] | No | |
| 4. Remifentanil (PCA) versus remifentanil (continuous IV) | |||||||
| 4.1 Respiratory depression (< 8 breaths/min) | RR (IV,), 95% CI, 0/0 cell counts |
2 | 0.98 [0.00, 1.0E12] | 2 | 0.98 [0.00, 1.0E12]] | No | |
| 4.3 Hypotension | RR (IV,), 95% CI, 0/0 cell counts |
2 | 0.98 [0.00, 1.0E12] | 2 | 0.98 [0.00, 1.0E12] | No | |
| 4.4 Bradycardia | RR (IV,), 95% CI, 0/0 cell counts |
2 | 0.98 [0.00, 1.0E12] | 2 | 0.98 [0.00, 1.0E12] | No | |
| 4.5 Nausea (and vomiting) | RR (MH), 95% CI | 2 | 0.85 [0.28, 2.54] | 2 | 0.81 [0.38, 1.73] | No | |
| 4.8 Need for naloxone | RR (IV,), 95% CI, 0/0 cell counts |
2 | 0.98 [0.00, 1.0E12] | 2 | 0.98 [0.00, 1.0E12] | No | |
All RR for outcomes including 0/0 cell counts (zero/zero event trials) were calculated using TSA (constant continuity correction, 0.01). Review Manager 5 produces computational errors when both the intervention and control group have zero events. By using TSA there is no possibility to choose the MH method (only IV) which may cause small deviations within results.
Abbreviations:
[95% CI]: 95% confidence interval; IV: Inverse Variance; MD: mean difference; MH: Mantel‐Haenszel; n: number of participants; RPCA: Remifentanil PCA; RR: risk ratio; SMD: standardised mean difference
For random‐effects analyses, the results were presented as the average treatment effect with 95% CIs, and the estimates of Tau2 and I2.
Meta‐analysis of adverse events frequently requires a synthesis of data with sparse event rates. Combining such data can be challenging especially when zero events exist in one or both arms of the study, which may lead to computational problems. Review Manager 5 (RevMan 2014) automatically checks for studies with problematic zero counts in one arm, and adds a constant value (0.5) to all cells of study results tables where the problems occur (constant continuity correction (ccc) 1.0) (Higgins 2011). However, Review Manager 5 (RevMan 2014) does not include options for analyses when included studies have zero counts in both arms. Removing these studies from the meta‐analysis creates the risk of inflating the magnitude of the pooled effect. Thus, we performed a sensitivity analysis (Table 15) applying three different approaches to implement continuity correction factors of 1.0 and 0.01 (constant, reciprocal, and empirical continuity correction) in a meta‐analysis model including studies with zero events in both arms as proposed by Sweeting and colleagues (Sweeting 2004). Briefly, the reciprocal approach adds a continuity correction factor proportional to the reciprocal of the size of the opposite treatment arm, which was found preferable when arm sizes were not balanced; with the empirical approach a continuity correction factor is calculated which 'pulls' the estimate in the direction of the pooled effect size estimate obtained in the analysis (Sweeting 2004). We used the TSA software which allows inclusion of zero/zero event trials with all three approaches for meta‐analysis with two or more trials. If there were no differences between the results of the different approaches, we reported in the Effects of interventions section only the pooled effect estimates calculated by the constant continuity correction (0.01) approach for zero event handling in both arms as single sensitivity analysis.
10. Zero event handling: Continuity corrections.
| Data |
0‐ and 0/0‐event trials included (TSA) |
0‐event trials included and 0/0‐event trials excluded (RevMan)1 | |||||||
|
Outcome (n, studies) |
0‐events, 0/0‐events, imbalance (Yes/No) |
Summary statistic | Reciprocal (1.0) | Reciprocal (0.01) | Empirical (1.0) | Empirical (0.01) | Constant (1.0) | Constant (0.01) |
Constant (1.0) |
|
1.3 Oxygen desaturation (2) |
1, 0 (Yes) |
RR [95% CI], P value, I2 |
0.51 [0.01, 30.22], 0.7471, 86% |
3.41 [0.82, 14.22] 0.0918, 0% |
0.57 [0.01, 24.87] 0.7699, 87% |
3.39 [0.81, 14.10], 0.0938, 0% |
0.5 [0.01, 31.95], 0.7421, 86% |
3.42 [0.82, 14.25], 0.0914, 0% |
0.5 [0.01, 31.95], 0.7421, 86% |
|
1.4 Nausea (and vomiting) (4) |
1, 0 (No) |
RR [95% CI], P value, I2 |
0.54 [0.29, 0.99], 0.0460, 0% |
0.56 [0.30, 1.04], 0.0665, 0% |
0.54 [0.29, 0.99], 0.0463, 0% |
0.56 [0.30, 1.04], 0.0667, 0% |
0.54 [0.29, 0.99], 0.0461, 0% |
0.56 [0.30, 1.04], 0.0664, 0% |
0.54 [0.29, 0.99], 0.0461, 0% |
|
1.6 Pruritus (2) |
0, 2 (No) |
RR [95% CI], P value, I2 |
1.0 [0.06, 15.71], 1.0, 0% |
1.0 [0.00, 1.0E12], 1.0, 0% |
NA | NA | 1.02 [0.07, 16.06], 0.9874, 0% |
1.02 [0.00, 1.1E12], 0.9987, 0% |
NA |
|
1.14 Rate of caesarean delivery (4) |
2, 0 (No) |
RR [95% CI], P value, I2 |
0.69 [0.34, 1,40], 0.3084, 0% |
0.63 [0.30, 1.32], 0.2164, 0% |
0.7 [0.34, 1.43], 0.3268, 1% |
0.63 [0.30, 1.32], 0.2182, 0% |
0.70 [0.35, 1.40], 0.3103, 0% |
0.63 [0.30, 1.32], 0.2165, 0% |
0.70 [0.35, 1.40], 0.3103, 0% |
| 2.10 Need for naloxone (2) | 1, 1 (No) |
RR [95% CI], P value, I2 |
0.49 [0.05, 5.29], 0.5580, 0%, |
0.03 [0.00, 1.1E8], 0.7484, 0% |
NA | NA | 0.48 [0.04, 5.30], 0.5473, 0% |
0.03 [0.00, 1.8E8], 0.7549, 0% |
0.3 [0.03, 2.72], 0.2847, 0% |
|
3.3 Respiratory depression (3) |
1, 1 (Yes) |
RR [95% CI], P value, I2 |
0.97 [0.56, 1.70] 0.9206, 0% |
0.91 [0.51, 1.62] 0.7506, 0% |
0.98 [0.57, 1.71] 0.9550, 0% |
0.91 [0.51, 1.62] 0.7532, 0% |
0.98 [0.56, 1.71] 0.9424, 0% |
0.91 [0.51, 1.62] 0.7518, 0% |
1.35 [0.30, 6.18], 0.6967, 37% |
|
3.4 Oxygen desaturation (3) |
1, 0 (Yes) |
RR [95% CI], P value, I2 |
3.2 [1.72, 5.94], 0.0002, 46% |
2.88 [1.94, 4.27], < 0.0001, 0% |
3.04 [1.70, 5.43], 0.0002, 38% |
2.88 [1.94, 4.27], < 0.0001, 0% |
3.19 [1.72, 5.91], 0.0002, 46% |
2.88 [1.94, 4.27], < 0.0001, 0% |
3.19 [1.72, 5.91], 0.0002, 46% |
|
3.6 Hypotension (4) |
2, 1 (No) |
RR [95% CI], P value, I2 |
0.59 [0.38, 0.93], 0.0225, 0% |
0.59 [0.37, 0.94], 0.0271, 0% |
0.59 [0.38, 0.93], 0.0219, 0% |
0.59 [0.38, 0.94], 0.0273, 0% |
0.59 [0.38, 0.93], 0.0225, 0% |
0.59 [0.37, 0.94], 0.0271, 0% |
0.58 [0.23, 1.48], 0.2517, 16% |
|
3.7 Bradycardia (2) |
0, 2 (No) |
RR [95% CI], P value, I2 |
1.0 [0.07, 15.07], 1.0, 0% |
1.0 [0.00, 1.0E12], 1.0, 0% |
NA | NA | 1.0 [0.07, 15.07], 1.0, 0% |
1.0 [0.00, 1.0E12], 1.0, 0% |
NA |
|
3.10 Pruritus (7) |
1, 0 (Yes) |
RR [95% CI], P value, I2 |
0.75 [0.48, 1.18], 0.2182, 29% |
0.78 [0.51, 1.18], 0.2366, 21% |
0.75 [0.48, 1.18], 0.2170, 29% |
0.78 [0.51, 1.18], 0.2368, 21% |
0.75 [0.48, 1.18], 0.2154, 29% |
0.78 [0.51, 1.18], 0.2370, 21% |
0.75 [0.48, 1.18], 0.2154, 29% |
| 3.12 Apgarscore < 7 at 5 min (5) | 2, 2 (No) |
RR [95% CI], P value, I2 |
1.26 [0.65, 2.43], 0.4944, 0% |
1.26 [0.62, 2.57], 0.5193, 0% |
1.28 [0.66, 2.47], 0.4596, 0% |
1.26 [0.62, 2.57], 0.5209, 0% |
1.26 [0.65, 2.43], 0.4904, 0% |
1.26 [0.62, 2.57], 0.5197, 0% |
1.28 [0.65, 2.51], 0.4801, 0% |
|
3.14 Need for naloxone (2) |
1, 1 (Yes) |
RR [95% CI], P value, I2 |
0.34 [0.03, 3.82], 0.3846, 0% |
0.02 [0.00, 1.6E8], 0.7247, 0% |
NA | NA | 0.46 [0.04, 4.88], 0.5200, 0% |
0.02 [0.00, 1.6E8], 0.7447, 0% |
0.2 [0.03, 1.15], 0.0720, 0% |
|
3.15 FHR/CTG abnormalities (5) |
1, 0 (No) |
RR [95% CI], P value, I2 |
1.54 [0.50, 4.75], 0.4499, 46% |
1.88 [0.63, 5.61], 0.2578, 35% |
1.53 [0.52, 4.54], 0.4410, 44% |
1.88 [0.63, 5.64], 0.2600, 35% |
1.54 [0.50, 4.75], 0.4499, 46% |
1.88 [0.63, 5.61], 0.2578, 35% |
1.54 [0.50, 4.75], 0.4499, 46% |
|
3.18 Additional analgesia required (6) |
2, 1 (No) |
RR [95% CI], P value, I2 |
7.47 [3.28, 16.99] < 0.0001, 0% |
9.26 [3.73, 23.03] < 0.0001, 0% |
9.66 [3.97, 23.52] < 0.0001, 0% |
9.23 [3.71, 22.95] < 0.0001, 0% |
7.65 [3.37, 17.38] < 0.0001, 0% |
9.27 [3.73, 23.03] < 0.0001, 0% |
8.1 [3.5, 18.75], < 0.0001, 0% |
|
3.19 Rate of caesarean delivery (9) |
1, 0 (No) |
RR [95% CI], P value, I2 |
0.99 [0.81, 1.21], 0.9076, 0% |
1.0 [0.82, 1.22], 0.9858, 0% |
0.99 [0.81, 1.21], 0.9058, 0% |
1.0 [0.82, 1.22], 0.9857, 0% |
0.99 [0.81, 1.21], 0.9067, 0% |
1.0 [0.82, 1.22], 0.9857, 0% |
0.99 [0.81, 1.21], 0.9067, 0% |
|
3.20 Rate of assisted birth (8) |
1, 0 (No) |
RR [95% CI], P value, I2 |
0.92 [0.66, 1.26], 0.5914, 0% |
0.94 [0.68, 1.30], 0.6917, 0% |
0.92 [0.66, 1.26], 0.5926, 0% |
0.94 [0.68, 1.30], 0.6918, 0% |
0.92 [0.66, 1.26], 0.5914, 0% |
0.94 [0.68, 1.30], 0.6917, 0% |
0.92 [0.66, 1.26], 0.5914, 0% |
|
3.26 Neonatal resuscitation (2) |
2, 0 (No) |
RR [95% CI], P value, I2 |
1.01 [0.04, 24.25], 0.9933, 57% |
1.09 [0.00, 3.1E8], 0.9929, 0% |
NA | NA | 1.02 [0.04, 25.09], 0.9901, 57% |
1.03 [0.00, 3.4E8], 0.9980, 0% |
1.02 [0.04, 25.09], 0.9901, 57% |
|
4.1 Respiratory depression (2) |
0, 2 (No) |
RR [95% CI], P value, I2 |
1.0 [0.06, 15.66], 1.0, 0% |
1.0 [0.00, 1.0E12], 1.0, 0% |
NA | NA | 0.98 [0.06, 15.37], 0.9896, 0% |
0.98 [0.00, 1.0E12], 0.9989, 0% |
NA |
|
4.3 Hypotension (2) |
0, 2 (No) |
RR [95% CI], P value, I2 |
1.0 [0.06, 15.66], 1.0, 0% |
1.0 [0.00, 1.0E12], 1.0, 0% |
NA | NA | 0.98 [0.06, 15.37], 0.9896, 0% |
0.98 [0.00, 1.0E12], 0.9989, 0% |
NA |
|
4.4 Bradycardia (2) |
0, 2 (No) |
RR [95% CI], P value, I2 |
1.0 [0.06, 15.66], 1.0, 0% |
1.0 [0.00, 1.0E12], 1.0, 0% |
NA | NA | 0.98 [0.06, 15.37], 0.9896, 0% |
0.98 [0.00, 1.0E12], 0.9989, 0% |
NA |
| 4.8 Need for naloxone (2) | 0, 2 (No) |
RR [95% CI], P value, I2 |
1.0 [0.06, 15.66], 1.0, 0% |
1.0 [0.00, 1.0E12], 1.0, 0% |
NA | NA | 0.98 [0.06, 15.37], 0.9896, 0% |
0.98 [0.00, 1.0E12], 0.9989, 0% |
NA |
1Review Manager 5 ignores zero/zero events trials and uses a constant continuity correction of 0.5 for studies with zero events in 1 arm. For the reciprocal, the empirical, and the constant approach including zero/zero‐event trials we used the TSA software. By using TSA there is no possibility to choose the Mantel‐Haenszel method (only inverse variance possible) which may cause small deviations within results.
We performed sensitivity analyses by using different approaches for handling of zero event trials (reciprocal, empirical, and constant approach) in meta‐analysis with two or more studies.
A) reciprocal approach ; value (k): 1.0, 0.01
Adds a factor of the reciprocal of the size of the opposite treatment arm to the cells which accounts for imbalance in group sizes.
B) empirical approach ; value (k): 1.0, 0.01
All studies without zero events are used to calculate a pooled effect estimate. Using this effect estimate a continuity correction factor can be calculated which produces an estimated effect close to the pooled estimated effect in the studies with zero events in both arms.
C) constant approach ; value (k): 1.0, 0.01
A value of 0.5 or 0.005, respectively, is added to each group in a 2 x 2 table; thus 1 participant is added to each intervention arm.
Abbreviations:
NA: not applicable; RR: risk ratio
Meta‐analyses are at risk of producing type I errors ('false positive') and type II errors ('false negative') as a result of sparse data and repetitive significance testing following updates with new trials (Brok 2008; Thorlund 2009; Wetterslev 2008; Wetterslev 2009). Trial sequential analysis (TSA) is a statistical approach that adjusts for random‐error risk (Wetterslev 2008). TSA reveals us the required number of participants (required information size (RIS)) needed in a meta‐analysis to detect or reject a certain intervention effect, and displays the trial sequential monitoring boundaries (TSMB), which allows testing for statistical significance before the RIS has been reached. The TSMB adjust the P value that is required for obtaining statistical significance according to the number of participants and events in a meta‐analysis (the fewer participants and events, the more restrictive the TSMB are and a lower P value is required to obtain statistical significance) (Brok 2008). In a post‐hoc sensitivity analysis, we applied TSA and calculated the RIS and the TSMB for each GRADE‐relevant dichotomous outcome on the basis of a risk for a type I error of 5%, a type II error of 10% (90% power), and a relative risk reduction (RRR) and control event rate based either on the representative estimate of all 'low risk of bias' trials (TSA 'low risk of bias'‐based; Table 10), or on the empirical estimate of the meta‐analysis (TSA 'empirical'; Table 11); we further adjusted for heterogeneity by using the heterogeneity‐adjustment factor (H; Thorlund 2011) assuming mild heterogeneity (H = 25%) for TSA 'low risk of bias'‐based and a model variance‐based correction for TSA 'empirical'. TSA cannot adjust for risk of bias, therefore, generally speaking such analyses should be restricted only to low risk of bias trials. However, due to limitations in the number and quality of studies, we performed TSA on all trials (low and high risk of bias trials). TSA was performed by using TSA software.
For GRADE‐relevant continuous outcomes we calculated the optimal information size (OIS) by a traditional sample size calculation used for individual trials (http://stat.ubc.ca/˜rollin/stats/ssize/n2.html), because the TSA software does not support meta‐analyses using SMDs as summary statistics. In a post‐hoc sensitivity analysis, we calculated the OIS based on a risk for a type I error of 5%, a type II error of 10% (90% power), and a mean difference based either on the minimal clinically relevant difference (1 cm on VAS 0 to 10 cm scale for satisfaction and pain) (Table 12) or on data of the 'low risk of bias' (or best) trial (Table 13). We performed OIS calculations on all trials (low and high risk of bias trials). OIS calculations do not adjust for heterogeneity. In general, OIS considerations are less conservative than the TSA approach. We used both calculations the 'OIS_minimally clinically relevant difference' and the 'OIS_low risk of bias (or best) trial' as sensitivity analyses and used the more conservative result for judgment on imprecision (GRADE).
Both RIS/TSMB and OIS provide us relevant information to estimate the level of evidence reached for the experimental intervention.
Subgroup analysis and investigation of heterogeneity
In the event of substantial heterogeneity (I2 > 50%), we planned to investigate heterogeneity using subgroup analyses and sensitivity analyses based on the comparators described above (Types of interventions). For the present review, none of the planned subgroup analyses were carried out because of sparse data. If sufficient data in future updates are present, we plan to perform subgroup analyses and compare subgroups by a mixed‐effects meta‐regression. We plan to use the R packages Metafor 2015 for meta‐regression and mixed‐effects model analysis.
We will carry out the following subgroup analyses.
Different methods and doses of remifentanil patient‐controlled analgesia (bolus versus only continuous infusion, regimen with a fixed dose versus dose‐escalating regimen, etc.).
Different parenteral opioids (e.g. pethidine (meperidine) versus fentanyl).
Planned subgroup analysis will be restricted in future updates to the review's primary and GRADE‐relevant outcomes.
We plan to assess subgroup differences by interaction tests available within Review Manager 5 (RevMan 2014), and will report the results of subgroup analyses quoting the χ2 statistic and P value, and the interaction test I2 value.
Sensitivity analysis
We performed sensitivity analyses to assess the robustness of the pooled estimates focusing on the following issues.
Risk of bias: we explored the impact of studies with high risk of selection bias (Table 7), performance and detection bias (Table 8), attrition bias (Table 9) on the robustness of the estimated effect
TSA/OIS: information size considerations based on 'low risk of bias'‐based and empirical assumptions for all GRADE‐relevant outcomes (Table 10; Table 11; Table 12; Table 13)
Random‐effects model versus fixed‐effect model (Table 14)
Zero event handling: different approaches to implement continuity correction factors of 1.0 and 0.01 (constant, reciprocal, and empirical continuity correction) (Table 15)
Statistical heterogeneity (I2 > 50%): we explored the effect of exclusion of individual studies from the analysis on the I2 value
For future updates if cluster‐randomised trials are included, we plan to conduct a sensitivity analysis (see Unit of analysis issues) to investigate the robustness of the results.
All sensitivity analyses were restricted to the primary and/or the GRADE‐relevant outcomes with two or more studies. Results of sensitivity analyses were reported in the Effects of interventions section when relevant differences affecting robustness of the estimated effects were recognised.
Results
Description of studies
Results of the search
The results of our search are displayed in a PRISMA flow chart (Figure 1). The search was performed in November and December 2015 (see Methods).
1.
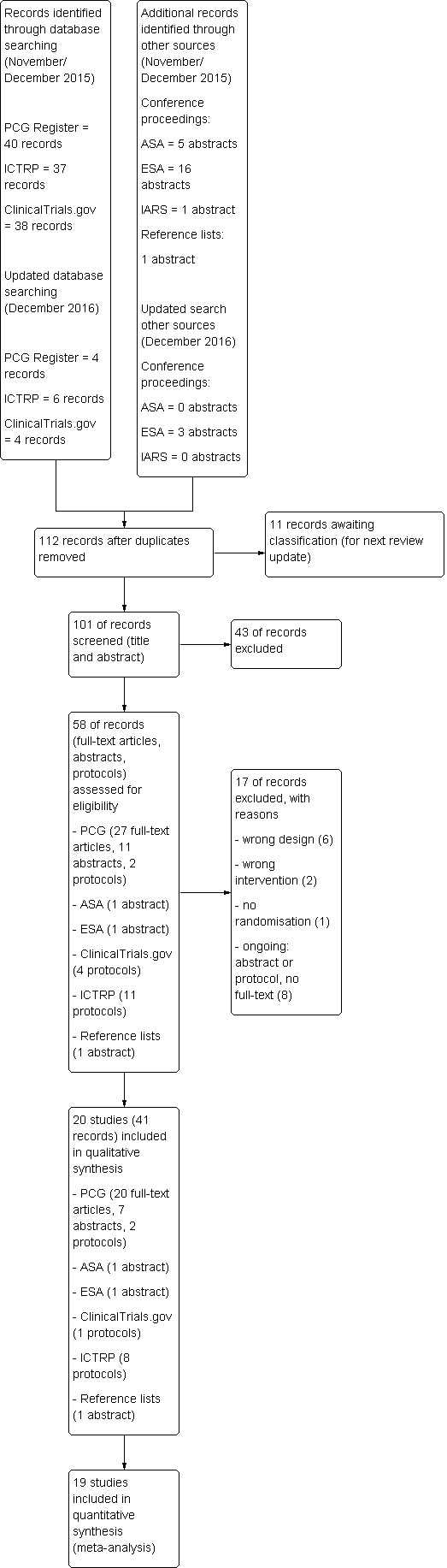
Study flow diagram.
We identified 115 records through database searching and another 23 by handsearching ASA, ESA and IARS congress abstracts as well as the reference lists of included articles. One‐hundred and one records remained after duplicates had been removed. These were screened independently by two review authors (SW, YJ) regarding title and abstract. Fifty‐eight remaining records were assessed for eligibility by reviewing the full texts and protocols. Seventeen records did not fulfil the eligibility criteria and had to be excluded. Finally, 41 records (full‐text articles, abstracts, and protocols) which could be allocated to 20 studies were included in the qualitative synthesis, and 19 of these studies were used to perform the meta‐analyses.
One trial was published in Spanish (Calderon 2006), all other studies were written in English. We did not include any abstracts or protocols without full texts in our final analysis since we could not retrieve enough information from these studies for eligibility assessment despite contacting the respective authors.
An updated search in December 2016 retrieved a further four trial reports from Cochrane Pregnancy and Childbirth's Trials Register and 13 reports (including duplicates) in ASA, ESA, IARS congress abstracts, ClinicalTrials.gov and the WHO International Clinical Trials Registry Platform (ICTRP). Eleven unique reports will be analysed for eligibility in the next review update (Abdalla 2015; Godinho 2016; Gunes 2014; Karadjova 2016; Kondoh 2016; Leong 2015; Logtenberg 2016; Moreira 2016; Pinar 2016; Pintaric 2016; Weiniger 2016).
Included studies
Trial characteristics
All included studies were published between 2001 (Volikas 2001) and 2015 (Douma 2015) and were randomised, controlled trials that reported on women in labour scheduled for vaginal delivery and requesting analgesia. In the present version of the review neither cluster‐randomised trials nor trials published in abstract form only were included. A detailed description of all included studies is presented in the Characteristics of included studies table.
Eleven trials were conducted in Europe (Blair 2005; Calderon 2006; Douma 2010; Douma 2011; Douma 2015; Freeman 2015; Stourac 2014; Thurlow 2002; Tveit 2012; Volikas 2001; Volmanen 2008), six in the Middle East (El‐Kerdawy 2010; Evron 2005; Evron 2008; Ismail 2012; Khooshideh 2015; Stocki 2014), two in Asia (Ng 2011; Shen 2013) and one in North America (Balki 2007).
A total of 3713 participants was randomised in the included studies with 3569 being analysed. Of these participants, 1523 received remifentanil patient‐controlled analgesia (PCA) and 2046 were assigned to a control intervention. The exact time point of randomisation remained unclear in some cases (Balki 2007; Blair 2005; Calderon 2006; Douma 2010; Douma 2011; El‐Kerdawy 2010; Thurlow 2002). In one trial, it is reported that randomisation took place before the start of actual labour and thus before analgesia request (Freeman 2015). In all other studies participants were assigned to the remifentanil PCA group or the control intervention as soon as labour had started and the request for analgesia was made (Douma 2015; Evron 2005; Evron 2008; Ismail 2012; Khooshideh 2015; Ng 2011; Shen 2013; Stocki 2014; Stourac 2014; Tveit 2012; Volikas 2001; Volmanen 2008).
The largest randomised sample size was 1414 (Freeman 2015) with 38% of the total number of women. Regarding this study, it has to be pointed out that this huge sample size also included women who did not receive any labour analgesia but were analysed for several important outcomes. We just considered participants with request for analgesic agents.
The smallest sample size was 17 (Volikas 2001). With the exception of three trials (Evron 2008; Freeman 2015; Ismail 2012), all studies had small sample sizes with fewer than 200 participants.
All trials except one (women with pre‐eclampsia, El‐Kerdawy 2010) excluded women and pregnancies with high risk (e.g. obesity, pre‐eclampsia, substance abuse, insulin‐dependent diabetes). Freeman 2015 and El‐Kerdawy 2010 included women from 32 weeks of gestation; in all other trials women had a term pregnancy.
All studies reported at least one outcome of interest. We could not identify any studies reporting on 'postpartum haemorrhage', 'depressed baby', 'satisfaction with childbirth experience', 'sense of control in labour', 'effect on mother/baby interaction', 'long‐term childhood development', and 'costs'.
None of the trials was funded by industry.
Comparisons and interventions
We had planned to analyse seven different comparators against remifentanil (PCA). For two of them, namely nitrous oxide (or other forms of inhalational analgesia, comparison 6) and placebo (or no treatment, comparison 7), no eligible studies could be retrieved.
Four studies investigated remifentanil (PCA) versus another opioid (IV/IM) (comparison 1, Calderon 2006; Evron 2005; Ng 2011; Thurlow 2002); three studies dealt with remifentanil (PCA) versus another opioid (PCA) (comparison 2, Blair 2005; Douma 2010; Volikas 2001); 10 studies compared remifentanil (PCA) with epidural analgesia/combined spinal‐epidural analgesia (CSE) (comparison 3, Douma 2011; Douma 2015; Evron 2008; El‐Kerdawy 2010; Freeman 2015; Ismail 2012; Stocki 2014; Stourac 2014; Tveit 2012; Volmanen 2008); two studies made a comparison between remifentanil (PCA) and remifentanil (continuous IV) (comparison 4, Khooshideh 2015; Shen 2013); and one study analysed remifentanil versus remifentanil using the same mode (PCA), but different regimen (variable bolus doses with fixed infusion dose versus variable infusion dose with fixed bolus doses) (comparison 5,Balki 2007).
With regard to comparison 1, three studies used pethidine/meperidine IM as a control intervention (Calderon 2006; Thurlow 2002; Ng 2011) and one study compared remifentanil (PCA) with meperidine infusion IV (Evron 2005). No study investigated a subcutaneous administration of other opioids as a comparator.
Looking at comparison 2, the control intervention was pethidine (PCA) for two trials (Blair 2005; Volikas 2001) whereas one trial compared remifentanil (PCA) both with meperidine (PCA) and fentanyl (PCA) (Douma 2010).
All trials in comparison 3 chose epidural analgesia in any way as the control intervention. In seven studies epidural analgesia with different combinations of opioids was offered to the participants (Douma 2011 (ropivacaine/sufentanil); Douma 2015 (ropivacaine/sufentanil); El‐Kerdawy 2010 (bupivacaine/fentanyl); Freeman 2015 (ropivacaine/sufentanil, bupivacaine/sufentanil, levobupivacaine/sufentanil, bupivacaine/fentanyl); Stourac 2014 (bupivacaine/sufentanil); Tveit 2012 (ropivacaine/fentanyl); Volmanen 2008 (levobupivacaine/fentanyl)). One study added combined spinal‐epidural as a second control intervention (Ismail 2012 (both levobupivacaine/fentanyl)). The two remaining studies compared remifentanil (PCA) with patient‐controlled epidural analgesia (PCEA) (Stocki 2014 (bupivacaine/fentanyl)) or different combinations of remifentanil (PCA) and PCEA (Evron 2008:(1) PCEA ropivacaine, (2) PCEA ropivacaine plus remifentanil (PCA), (3) PCEA ropivacaine plus acetaminophen IV)).
The investigated interventions ranged from 35 minutes (Blair 2005) to 594 minutes (Volikas 2001). The lockout times of remifentanil (PCA) used in the included trials ranged from one minute (Ismail 2012; Stocki 2014; Volmanen 2008) to 30 minutes (Calderon 2006) with bolus doses ranging from 0.1 µg/kg (Shen 2013) to 0.5 µg/kg (Volikas 2001) or from 5 µg (Thurlow 2002) to 50 µg (Calderon 2006). Supplementary remifentanil background infusion was used in four studies (Balki 2007; Calderon 2006; El‐Kerdawy 2010; Evron 2008). A detailed description is provided in Table 16.
11. Interventions.
| Study | Comparator | Analgesia duration (mean ± SD, median (range)) [min] | Background infusion [µg/(kg*min)] | Bolus dose | Bolus application speed (calculated) | Bolus dose escalation on request | Lockout time [min] | Maximum dose | Total dose administered (mean ± SD, median (range [IQR]) |
| Balki 2007 | Remifentanil variable infusion, fixed bolus | 463 | 0.025 | 0.25 µg/kg | NA | 0.5 ‐ 1 µg/kg, every 15 min | 2 | 3000 µg in 4 h | 474 (188 ‐ 925) µg/h |
| Blair 2005 | Pethidine PCA | 147.5 ± 79 | no | 40 µg | 133.33 µg/min | no | 2 | NA | NA |
| Calderon 2006 | Meperidine IM | 280 ± 55 | 0.025 | 50 µg | 2 µg/min | no | 30 | NA | NA |
| Douma 2010 | (1) Meperidine PCA (2) Fentanyl PCA |
234 ± 136 | no | 40 µg | NA | no | 2 | 1200 µg/h | 1840 ± 1090 µg |
| Douma 2011 | epidural | 286 ± 145 | no | 40µg | 66.67 µg/min | no | 2 | 1200 µg/h | 2817 ± 1564 µg |
| Douma 2015 | epidural | 192 ± 116 | no | 40µg | 66.67 µg/min | no | 2 | 1200 µg/h | 1417 µg |
| El‐Kerdawy 2010 | epidural | NA | 0.0 | 0.25 µg/kg | 1.5 µg/(kg*min) | no | 5 | 3000 µg in 4 h | NA |
| Evron 2005 | Meperidine IV | NA | no | 20 µg | NA | 5 µg increments, every 15 ‐ 20 min | 3 | 1500 µg/h | 1034.5 (133 ‐ 4021) µg |
| Evron 2008 | epidural | NA | 0.025 | 20 µg | NA | 25% increase every 15 ‐ 20 min | 3 | NA | 8.5 ± 2.2 µg/(kg*h) |
| Freeman 2015 | epidural | 236 (128 ‐ 376) | no | 30 µg | NA | increase to 40 µg or decrease to 20 µg | 3 | 40 µg per bolus | NA |
| Ismail 2012 | epidural/CSE | NA | no | 25 µg | 25 µg/min | escalation scheme (0.1 – 0.2 – 0.3 – 0.5 – 0.7 – 0.9 µg/kg) until the maximum dose of 0.9 µg/kg | 1 | 25 µg/mL + 0.9 µg/kg per bolus | NA |
| Khooshideh 2015 | Remifentanil IV | NA | no | 0.25 µg/kg | NA | increased to 0.4 µg/kg (if VNRS ≥ 7) | 4 | 0.4 µg/kg per bolus | 942.6 ± 86.4 µg |
| Ng 2011 | Pethidine IM | NA | no | 25 µg (< 60 kg) or 30 µg (≥ 60 kg) | 6.67 µg/min | no | 3.75‐4.50 | 500 µg/h (calculated) | NA |
| Shen 2013 | Remifentanil IV | 1511 | no | 0.1 µg/kg | 0.2 µg/(kg*min) | increments of 0.1 µg/kg to 0.4 µg/kg | 2 | 0.4 µg/kg per bolus | 1340 (1220 ‐ 1480 [890 ‐ 1680]) µg |
| Stocki 2014 | epidural | NA | no | 20 µg | NA | up to 60 µg | 2 min, 1 min on request | 60 µg per bolus | 1725 ± 1392 µg |
| Stourac 2014 | epidural | 162.75 ± 77.15 | no | 20 µg | NA | 10 µg increments (if VAS decrease < 2) | 3 | NA | NA |
| Thurlow 2002 | Meperidine IM | NA | no | 20 µg | 60 µg/min | NA | 3 | NA | NA |
| Tveit 2012 | epidural | 225 ± 117.2 | no | 0.15 µg/kg | 100 µg/min | 0.15 µg/kg increments every 15 min | 2 | No limit | NA |
| Volikas 2001 | Pethidine PCA | 334 ± 260 | no | 0.5 µg/kg | NA | no | 2 | No limit | 3670 (120 ‐ 4880) µg (mean (range)) |
| Volmanen 2008 | epidural | Max. 60 | no | 25 µg | 25 µg/min | escalation scheme (0.1 – 0.2 – 0.33 – 0.5 – 0.7 – 0.9 µg/kg) until the maximum dose of 0.9 µg/kg | 1 | 25 µg/mL + 0.9 µg/kg per bolus | 0.14 (0.08 ‐ 0.18 [0.03 ‐ 0.32]) µg/(kg*min) |
1Time from the start of remifentanil analgesia until the final dose increment, 50% survival (Kaplan‐Meier cumulative event curve)
Abbreviations:
NA: not applicable
Co‐interventions/Co‐analgesics
In four studies additional Entonox was offered to all women in labour (Blair 2005; Ng 2011; Thurlow 2002; Volikas 2001); in one study pethidine IM was provided on top of that (Ng 2011). Epidural analgesia was used as rescue analgesia in six trials (Balki 2007; Douma 2010; Evron 2005; Thurlow 2002; Shen 2013; Volikas 2001). One trial offered unknown additional analgesia after one hour (Stocki 2014).
Excluded studies
Nine studies were excluded from qualitative analysis (Figure 1). Six of them were cross‐over trials (Jost 2013; Varposhti 2013; Volmanen 2004; Volmanen 2005; Volmanen 2009; Volmanen 2011), one study did not randomise the participants (Solek‐Pastuszka 2009), one study dealt with an intervention that was not of interest for this review (Balcioglu 2007), and one did not provide PCA (Shahriari 2007) (see Characteristics of excluded studies).
Ongoing studies
Eight trials were classified as ongoing and were therefore not included in our current review (Ongoing studies). We plan to use these data for further updates. More information is provided in Characteristics of ongoing studies.
Studies awaiting classification
There are 11 studies awaiting classification identified in our updated search from December 2016 (Abdalla 2015; Godinho 2016; Gunes 2014; Karadjova 2016; Kondoh 2016; Leong 2015; Logtenberg 2016; Moreira 2016; Pinar 2016; Pintaric 2016; Weiniger 2016). See:Characteristics of studies awaiting classification). These studies are not included in the current review.
Risk of bias in included studies
The risk of bias in each included study was rated as presented in Figure 2 and described in the Characteristics of included studies.
2.
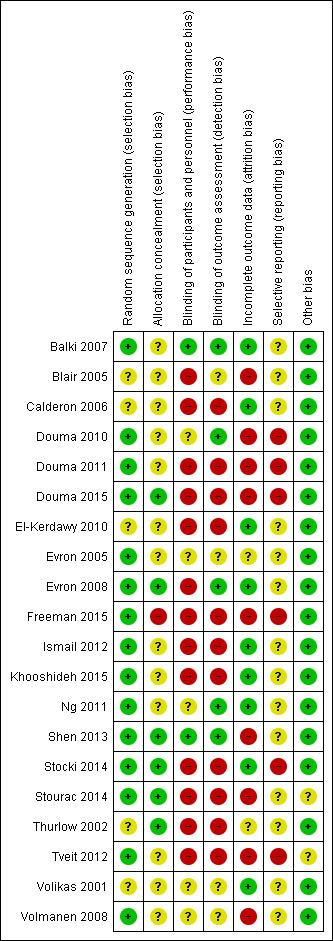
'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
With regard to Figure 3, the categories 'random sequence generation' and 'other bias' showed low risk of bias across all included studies in 75% and 90% of cases, respectively. Selective reporting and allocation concealment remained at unclear risk of bias in most cases. In terms of blinding the majority of studies (65%) revealed high risk of bias. In the domain 'attrition bias' 45% of all studies were classified as low or high risk of bias, respectively.
3.
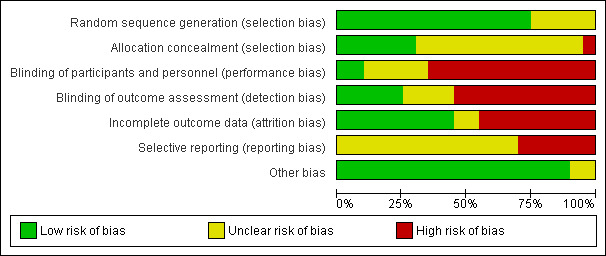
'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
Random sequence generation
In 15 studies randomisation was achieved by computer‐generated codes (Balki 2007; Douma 2010; Douma 2011; Douma 2015; Evron 2005; Evron 2008; Freeman 2015; Ismail 2012; Khooshideh 2015; Ng 2011; Shen 2013; Tveit 2012; Volmanen 2008), shuffling cards (Stocki 2014), or throwing dice (Stourac 2014). They were therefore estimated to have low risk of bias.
In five trials it was not sufficiently described which randomisation method was used (Blair 2005; Calderon 2006; El‐Kerdawy 2010; Thurlow 2002), or if the method worked appropriately (Volikas 2001), thus we considered them as having an unclear risk of bias.
No study had high risk of bias regarding random sequence generation.
Allocation concealment
Six studies were judged to have a low risk of bias since allocation concealment was achieved either by using sequentially numbered opaque sealed envelopes (SNOSE) (Douma 2015; Evron 2008; Shen 2013; Stocki 2014; Thurlow 2002), or allocation could not be foreseen due to the method used for randomisation (throwing dice) (Stourac 2014).
In 10 trials it was not clear from the description if SNOSE was correctly applied to cover allocation (Balki 2007; Douma 2010; Douma 2011; Evron 2005; Ismail 2012; Khooshideh 2015; Ng 2011; Tveit 2012; Volikas 2001; Volmanen 2008). Three trials did not describe any method for allocation concealment (Blair 2005; Calderon 2006; El‐Kerdawy 2010). Thus, we estimated these trials to have an unclear risk of bias.
One study was assigned to the category high risk of bias, because allocation concealment was uncovered for women and personnel before the start of treatment (Freeman 2015).
Blinding
Blinding of participants and personnel (performance bias)
Two trials were considered to have a low risk of bias (Balki 2007; Shen 2013) because it was clearly stated that all physicians and participants were blinded adequately.
We judged five studies to have an unclear risk of bias (Douma 2010; Evron 2005; Ng 2011; Volikas 2001; Volmanen 2008). In these trials, blinding attempts were made but we assumed that there was the possibility to uncover blinding due to the different pharmacokinetics of the compared interventions (pharmacological half‐life and clinical effects following bolus request).
Thirteen trials had high risk of bias regarding performance bias. In four of these trials it was pointed out that participants and personnel were not blinded (Douma 2015; Freeman 2015; Stocki 2014; Tveit 2012). Six of the high‐risk studies did not address this issue (Calderon 2006; Douma 2011; El‐Kerdawy 2010; Ismail 2012; Stourac 2014; Thurlow 2002), but on the basis of the methods described we assumed that blinding did not occur due to technical reasons. The remaining three trials were single‐blinded trials (Blair 2005; Evron 2008; Khooshideh 2015). Hence, either participants or personnel or even both of them were not blinded, and in addition to that it is uncertain that single‐blinding worked adequately because of the nature of the intervention.
Blinding of outcome assessor (detection bias)
Five studies were estimated to have a low risk of bias (Balki 2007; Douma 2010; Evron 2008; Ng 2011; Shen 2013) because blinding of outcome assessors was appropriate.
In four studies the risk of bias remained unclear (Blair 2005; Evron 2005; Volikas 2001; Volmanen 2008) since information was insufficient to judge whether all outcome assessments were adequately blinded or not. Blinding attempts were made for several outcomes. Nevertheless, subjective outcomes or outcome measurements could likely be influenced by lack of blinding.
The remaining 11 studies were considered having a high risk of bias. In three of them it was reported that blinding was not performed (Douma 2015; Freeman 2015; Stocki 2014). In eight studies this issue was not addressed for most relevant outcomes (Calderon 2006; Douma 2011; El‐Kerdawy 2010; Ismail 2012; Khooshideh 2015; Stourac 2014; Thurlow 2002; Tveit 2012). Due to the description of the interventions we inferred that at least subjective outcomes or outcome measurements were likely to be influenced by lack of blinding.
Incomplete outcome data
Nine studies had a low risk of bias (Balki 2007; Calderon 2006; El‐Kerdawy 2010; Evron 2008; Ismail 2012; Khooshideh 2015; Ng 2011; Stocki 2014; Volikas 2001). In seven of these trials no missing outcome data were detected (Balki 2007; Calderon 2006; El‐Kerdawy 2010; Ismail 2012; Khooshideh 2015; Ng 2011; Volikas 2001), whereas two trials described reasons for their missing data (less than 15%, respectively) that were unlikely to be related to true outcome (Evron 2008; Stocki 2014). Full‐intention‐to‐treat (ITT)/partial‐ITT analysis was used in all studies except two; in one study, data were analysed per‐protocol (Evron 2008), and the other study did not define the method of data analysis (El‐Kerdawy 2010).
In two studies attrition bias remained unclear (Evron 2005; Thurlow 2002). One trial did not report on reasons for missing data (up to 22%) with regard to the outcome adverse events for women (Evron 2005). Additionally, these missing data were imbalanced between the groups and the rate of escape analgesia amounted to 38%. As a result, it was uncertain if this outcome was biased. Similar reasons applied to the second trial (Thurlow 2002) with incomplete outcome data without reasons declared and high escape analgesia rates (up to 81%). Both trials used partial‐ITT‐analysis.
High risk of bias was assigned to nine studies (Blair 2005; Douma 2010; Douma 2011; Douma 2015; Freeman 2015; Shen 2013; Stourac 2014; Tveit 2012; Volmanen 2008). A large amount of data (more than 15%) were missing for many important outcomes, partly with reasons stated (Stourac 2014), partly without reasons declared (Blair 2005; Douma 2010; Douma 2011; Douma 2015; Freeman 2015; Shen 2013; Tveit 2012; Volmanen 2008). However, reasons for missing outcome data were likely to be related to true outcome. One study used partial‐ITT for data analysis (Freeman 2015). All other studies with high risk of bias concerning attrition bias used per‐protocol analysis.
Selective reporting
No study was considered to have a low risk of bias.
Fourteen trials were assessed having an unclear risk of bias. There were no references to trial registries and no published study protocols in 13 cases (Balki 2007; Blair 2005; Calderon 2006; El‐Kerdawy 2010; Evron 2005; Evron 2008; Ismail 2012; Ng 2011; Shen 2013; Stourac 2014; Thurlow 2002; Volikas 2001; Volmanen 2008). One retrospectively registered study protocol was available and all reported outcomes were specified. Nonetheless, several outcomes were reported that were not defined in the study protocol (Khooshideh 2015).
The remaining six studies were judged to have high risk of bias (Douma 2010; Douma 2011; Douma 2015; Freeman 2015; Stocki 2014; Tveit 2012). The corresponding protocols were available that revealed several deviations in the definitions of primary and secondary outcomes. Additionally, some pre‐defined outcomes were not reported at all. Three protocols were registered prospectively (Douma 2011; Douma 2015; Stocki 2014), two retrospectively (Douma 2010; Tveit 2012), and one study had two different protocols that were published both prospectively and retrospectively (Freeman 2015).
Other potential sources of bias
Eighteen studies appeared to be free of other sources of bias und were therefore estimated having low risk of bias (Balki 2007; Blair 2005; Calderon 2006; Douma 2010; Douma 2011; Douma 2015; El‐Kerdawy 2010; Evron 2005; Evron 2008; Freeman 2015; Ismail 2012; Khooshideh 2015; Ng 2011; Shen 2013; Stocki 2014; Thurlow 2002; Volikas 2001; Volmanen 2008).
In one study enrolment stopped early due to high participating refusal (Stourac 2014). In another trial technical problems with infusion pumps occurred and as a consequence the study had to be closed (Tveit 2012). Both studies were underpowered and were considered to have unclear risk of bias.
No study had high risk of bias.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5
Summary of findings for the main comparison. Remifentanil (PCA) compared to another opioid (IV/IM) for pain management in labour.
| Remifentanil (PCA) compared to another opioid (IV/IM) for pain management in labour | ||||||
| Patient or population: women in labour with planned vaginal delivery Setting: labour wards in Europe (two studies), Middle East (one study), and Asia (one study) Intervention: remifentanil (PCA) Comparison: another opioid (IV/IM) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with another opioid (IV/IM) | Risk with remifentanil (PCA) | |||||
| Satisfaction (overall) with pain relief (VAS 0 to 10 cm, NRS 1 to 4, NRS 0 to 10, VRS 0 to 5) |
see comment | The standardised mean satisfaction score in the intervention group was 2.11 higher (0.72 higher to 3.49 higher)** | ‐ | 216 (4 RCTs) | ⊕⊝⊝⊝ VERY LOW 1 2 | A SMD of 2.11 higher is equivalent to a range of 2.74 cm higher (SD = 1.3) to 4.68 cm higher (SD = 2.22) on a VAS 0 to 10 cm scale in the intervention group. The mean satisfaction scores in the control group range from 4.23 to 6.0 cm.# ** |
| Pain intensity 'early' (30 min/1 h) (VAS 0 to 10 cm, VAS 0 to 100 cm) |
see comment | The standardised mean pain score 'early' in the intervention group was 1.58 fewer (2.69 fewer to 0.48 fewer)*** | ‐ | 180 (3 RCTs) | ⊕⊝⊝⊝ VERY LOW 1 2 3 | A SMD of 1.58 fewer is equivalent to a range of 1.26 cm fewer (SD = 0.8) to 2.8 cm fewer (SD = 1.77) on a VAS 0 to 10 cm scale in the intervention group. The mean pain scores in the control group range from 3.56 to 6.3 cm (VAS 0 to 10 cm).# *** |
| Additional analgesia required (escape analgesia) | Study population | RR 0.57 (0.40 to 0.81) | 190 (3 RCTs) | ⊕⊕⊕⊝ MODERATE 4 | ||
| 621 per 1.000 | 354 per 1.000 (248 to 503) | |||||
| Rate of caesarean delivery | Study population | RR 0.63 (0.30 to 1.32) | 215 (4 RCTs) | ⊕⊕⊝⊝ LOW 4 5 | Two studies includes zero events in one arm (constant continuity correction of 0.01).7 | |
| 148 per 1.000 | 93 per 1.000 (44 to 195) | |||||
| Maternal apnoea | see comment | see comment | ‐ | (0 studies) | ‐ | No trial assessed this outcome. |
| Maternal respiratory depression (< 8 breaths/min) | None out of 18 women in the control group and three out of 18 in the remifentanil group had a respiratory depression. | not estimable | 36 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 4 6 | Only one trial assessed this outcome. | |
| Apgar score < 7 at 5 min | None of the newborns in both groups had an Apgar score < 7 at 5 min. | not estimable | 88 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 4 6 | Only one trial assessed this outcome. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; SMD: standardised mean difference; SD: standard deviation; RoB: Risk of bias; RIS: required information size; OIS: optimal information size | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 RoB ‐ downgrading (very serious): Substantial information is derived from studies at high risk of bias. After exclusion of high risk trials the CI crosses the line of no effect.
2 Inconsistency ‐ downgrading (serious): I2 > 50%.
3 Imprecision ‐ downgrading (serious): The number of women is insufficient to demonstrate the anticipated effect (OIS not reached).
4 RoB ‐ downgrading (serious): Substantial information is derived by high risk of bias studies (If more than one study: Exclusion of high risk of bias trials has no substantial effect on robustness of the results).
5 Imprecision ‐ downgrading (serious): The number of women is insufficient to demonstrate the anticipated effect (RIS not reached). The result is imprecise including appreciable benefit and harm.
6 Imprecision ‐ downgrading (very serious): Only one study with small sample size (< 150 participants) reported this outcome.
7 Estimated effect with zero/zero event handling (constant continuity correction of 1.0), Analysis 1.14: RR = 0.70 [0.34, 1.41], I2 = 1%.
# The SMD was back‐transformed into the VAS 0 to 10 cm scale to facilitate the interpretation. The smallest as well as the largest SD of the studies were used for back‐transformation to reflect the range of effect.
** Higher values indicate higher levels of satisfaction.
*** Lower values indicate less pain.
Summary of findings 2. Remifentanil (PCA) compared to another opioid (PCA) for pain management in labour.
| Remifentanil (PCA) compared to another opioid (PCA) for pain management in labour | ||||||
| Patient or population: women in labour with planned vaginal delivery Setting: labour wards in Europe (three studies) Intervention: remifentanil (PCA) Comparison: another opioid (PCA) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with another opioid (PCA) | Risk with remifentanil (PCA) | |||||
| Satisfaction (overall) with pain relief (VRS 1 to 10) |
The mean satisfaction in the combined (meperidine + fentanyl) control group was 7.1 on a VRS 1 to 10 scale | Mean satisfaction in the remifentanil group was 0.92 VRS higher (0.46 to 1.39 higher).** | ‐ | 110 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 6 | Only one trial assessed this outcome. |
| Pain intensity 'early' (30 min/1 h) (VAS 0 to 10 cm, VAS 0 to 100 cm) |
see comment | The standardised mean pain score 'early' in the intervention group was 0.51 fewer (1.01 fewer to 0)*** | ‐ | 215 (3 RCTs) | ⊕⊝⊝⊝ VERY LOW 2 3 4 | A SMD of 0.51 fewer is equivalent to a range of 1.13 cm fewer (SD = 2.22) to 1.46 cm fewer (SD = 2.875) on a VAS 0 to 10 cm scale in the intervention group. Mean pain scores in the control groups range from 5.13 cm to 7.0 cm (VAS 0 to 10 cm).# *** |
| Additional analgesia required (escape analgesia) | Study population | RR 0.76 (0.45 to 1.28) | 215 (3 RCTs) | ⊕⊕⊝⊝ LOW 3 4 | ||
| 381 per 1.000 | 289 per 1.000 (171 to 487) | |||||
| Rate of caesarean delivery | Study population | RR 2.78 (0.99 to 7.82) | 143 (2 RCTs) | ⊕⊝⊝⊝ VERY LOW 4 5 | ||
| 56 per 1.000 | 156 per 1.000 (56 to 439) | |||||
| Maternal apnoea | see comment | see comment | ‐ | (0 studies) | ‐ | No trial assessed this outcome. |
| Maternal respiratory depression | see comment | see comment | ‐ | (0 studies) | ‐ | No trial assessed this outcome. |
| Apgar score ≤ 7 (< 7) at 5 min | Three out of eight newborns in the control group and none out of nine in the remifentanil group had an Apgar score < 7 at 5 min. | not estimable | 17 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 6 7 | Only one trial assessed this outcome. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OIS: optimal information size; RIS: required information size; RoB: Risk of Bias; RR: risk ratio; SD: standard deviation; SMD: standardised mean difference | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 RoB ‐ downgrading (serious): Information is derived from a high risk of bias trial.
2 RoB ‐ downgrading (serious): After exclusion of 1 high risk of bias trial (blinding) the estimated effect with CI reached clinically relevance ‐0.73 [‐1.05, ‐0.40]
3 Inconsistency ‐ downgrading (serious): I2 > 50%
4 Imprecision ‐ downgrading (serious): The number of women is insufficient to demonstrate the anticipated effect (RIS/OIS not reached). The result is imprecise including appreciable and no appreciable effect.
5 RoB ‐ downgrading (very serious): Substantial information is derived from studies at high risk of bias. Exclusion of high risk of bias trials widened the CI including appreciable benefit and harm.
6 Imprecision ‐ downgrading (very serious): Only one study with small sample size (< 150 participants) reported this outcome.
7 RoB ‐ downgrading (serious): Information is derived from a trial with unclear risk of bias.
# The SMD was back‐transformed into the VAS 0 to 10 cm scale to facilitate the interpretation. The smallest as well as the largest SD of the studies were used for back‐transformation to reflect the range of effect.
** Higher values indicate higher levels of satisfaction.
*** Lower values indicate less pain.
Summary of findings 3. Remifentanil (PCA) compared to epidural/CSE for pain management in labour.
| Remifentanil (PCA) compared to epidural/CSE for pain management in labour | ||||||
| Patient or population: women in labour with planned vaginal delivery Setting: labour wards in Europe (six studies) and Middle East (four studies) Intervention: remifentanil (PCA) Comparison: epidural analgesia/central neuraxial blocks (CSE) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with epidural analgesia/central neuraxial blocks (CSE) | Risk with remifentanil (PCA) | |||||
| Satisfaction (overall) with pain relief (NRS 0 to 4, 1 to 4, 0 to 10, 1 to 10, VRS 1 to 4) |
see comment | The standardised mean satisfaction score in the intervention group was 0.22 fewer (0.40 fewer to 0.04 fewer)** | ‐ | 2135 (7 RCTs) | ⊕⊝⊝⊝ VERY LOW 1 2 | A SMD of 0.22 fewer is equivalent to a range of 0.15 cm fewer (SD = 0.7) to 0.61 cm fewer (SD = 2.78) on a VAS 0 to 10 cm scale in the intervention group. Mean satisfaction scores in the control group range from 6.7 to 9.1 cm (VAS 0 to 10 cm).# ** |
| Pain intensity 'early' (1 h) (VAS 0 to 10 cm, VAS 0 to 100 cm, NRS 0 to 10) |
see comment | The standardised mean pain score 'early' in the intervention group was 0.57 higher (0.31 higher to 0.84 higher)*** | ‐ | 235 (6 RCTs) | ⊕⊕⊝⊝ LOW 3 4 | A SMD of 0.57 higher is equivalent to a range of 0.57 cm higher (SD = 1.0) to 1.43 cm higher (SD = 2.5) on a VAS 0 to 10 cm scale in the intervention group. The mean pain scores in the control group range from 1.6 to 4.14 cm (VAS 0 to 10 cm).# *** |
| Additional analgesia required | Study population | RR 9.27 (3.73 to 23.03) | 1037 (6 RCTs) | ⊕⊕⊕⊝ MODERATE 3 | One study includes zero events in both arms; two studies include zero events in one arm (constant continuity correction of 0.01). 8 | |
| 10 per 1.000 | 93 per 1.000 (34 to 230) | |||||
| Rate of caesarean delivery | Study population | RR 1.0 (0.82 to 1.22) | 1578 (9 RCTs) | ⊕⊕⊕⊝ MODERATE 3 | One study includes zero events in one arm (constant continuity correction of 0.01). 9 | |
| 215 per 1.000 | 215 per 1.000 (176 to 262) | |||||
| Maternal apnoea | None out of 19 women in the control group and nine out of 19 in the remifentanil group had an apnoea. | not estimable | 38 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 5 7 | Only one trial assessed this outcome. | |
| Maternal respiratory depression (< 9, < 8 breaths/min) | Study population | RR 0.91 (0.51 to 1.62) | 687 (3 RCTs) | ⊕⊕⊝⊝ LOW 3 6 | One study includes zero events in both arms; one study includes zero events in one arm (constant continuity correction of 0.01). 10 | |
| 38 per 1.000 | 35 per 1.000 (19 to 62) | |||||
| Apgar score ≤ 7 (< 7) at 5 min | Study population | RR 1.26 (0.62 to 2.57) | 1322 (5 RCTs) | ⊕⊕⊝⊝ LOW 3 6 | Two studies include zero events in both arms; two studies include zero events in one arm (constant continuity correction of 0.01). 11 | |
| 23 per 1.000 | 30 per 1.000 (14 to 59) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OIS: optimal information size; RIS: required information size; RoB: Risk of Bias; RR: risk ratio; SD: standard deviation; SMD: standardised mean difference | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 RoB ‐ downgrading (very serious): Substantial information is derived from studies at high risk of bias. After exclusion of high risk trials the CI crosses the line of no effect.
2 Inconsistency ‐ downgrading (serious): I2 > 50%
3 RoB ‐ downgrading (serious): Substantial information is derived from studies at high risk of bias. Exclusion of high risk of bias trials has no substantial impact on robustness of the results.
4 Imprecision ‐ downgrading (serious): The number of women is insufficient to demonstrate the anticipated effect (OIS not reached).
5 RoB ‐ downgrading (serious): Information is derived from a high risk of bias trial.
6 Imprecision ‐ downgrading (serious): The number of women is insufficient do demonstrate the anticipated effect (RIS/OIS not reached). The result is imprecise including appreciable benefit and harm.
7 Imprecision ‐ downgrading (very serious): Only one study with small sample size (< 150 participants) reported this outcome.
8 Estimated effect with zero/zero event handling (constant continuity correction of 1.0), Analysis 3.18: RR = 8.1 [3.5, 18.75], I2 = 0%.
9 Estimated effect with zero/zero event handling (constant continuity correction of 1.0), Analysis 3.19: RR = 0.99 [0.81, 1.21], I2 = 0%.
10 Estimated effect with zero/zero event handling (constant continuity correction of 1.0), Analysis 3.3: RR = 1.52 [0.23, 9.90], I2 = 50%.
11 Estimated effect with zero/zero event handling (constant continuity correction of 1.0), Analysis 3.12: RR = 1.28 [0.65, 2.51], I2 = 0%.
# The SMD was back‐transformed into the VAS 0 to 10 cm scale to facilitate the interpretation. The smallest as well as the largest SD of the studies were used for back‐transformation to reflect the range of effect.
** Higher values indicate higher levels of satisfaction.
*** Lower values indicate less pain.
Summary of findings 4. Remifentanil (PCA) compared to remifentanil (continuous IV) for pain management in labour.
| Remifentanil (PCA) compared to remifentanil (continuous IV) for pain management in labour | ||||||
| Patient or population: women in labour with planned vaginal delivery Setting: labour wards in Asia (one study) and Middle East (one study) Intervention: remifentanil (PCA) Comparison: remifentanil (continuous IV) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with remifentanil (continuous IV) | Risk with remifentanil (PCA) | |||||
| Satisfaction (overall) with pain relief | see comment | see comment | ‐ | (0 studies) | ‐ | No trial assessed this outcome. |
| Pain intensity 'early' (30 min/1 h) (VAS 0 to 10 cm) |
The mean pain score in the remifentanil (continuous IV) group was 4.0 cm on a VAS 0 to 10 cm scale. | Mean pain score in the remifentanil (PCA) group was 1.0 cm fewer (1.8 fewer to 0.2 fewer).*** | not estimable | 53 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | Only one trial assessed this outcome. |
| Additional analgesia required (escape analgesia) | Two out of 29 women in the remifentanil (PCA) group and four out of 30 participants in the remifentanil (continuous IV) group required additional epidural analgesia. | not estimable | 59 (1 RCT) |
⊕⊝⊝⊝ VERY LOW 1 2 | Only one trial assessed this outcome. | |
| Rate of caesarean delivery | see comment | see comment | ‐ | (0 studies) | ‐ | No trial assessed this outcome. |
| Maternal apnoea | see comment | see comment | ‐ | (0 studies) | ‐ | No trial assessed this outcome. |
| Maternal respiratory depression (< 8 breaths/min) | see comment | see comment | RR 0.98 (0.00 to 1.0E12) | 135 (2 RCTs) | ⊕⊕⊝⊝ LOW 3 4 | All study arms include zero events (constant continuity correction of 0.01). 5 |
| Apgar score < 7 at 5 min | see comment | see comment | ‐ | (0 studies) | ‐ | No trial assessed this outcome. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OIS: optimal information size; RIS: required information size; RoB: Risk of bias | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 RoB ‐ downgrading (serious): Information is derived from a high risk of bias trial.
2 Imprecision ‐ downgrading (very serious): Only one study with small sample size (< 150 participants) reported this outcome.
3 RoB ‐ downgrading (serious): Substantial information is derived from studies at high risk of bias. Exclusion of high risk of bias trials has no substantial impact on robustness of the results.
4 Imprecision ‐ downgrading (serious): The number of women is insufficient to demonstrate the anticipated effect (RIS/OIS not reached). The result is imprecise including appreciable benefit and harm.
5 Estimated effect with zero/zero event handling (constant continuity correction of 1.0), Analysis 4.1: RR = not estimable
*** Lower values indicate less pain.
Summary of findings 5. Remifentanil (PCA, increasing bolus dose) compared to remifentanil (PCA, increasing infusion dose) for pain management in labour.
| Remifentanil (PCA, increasing bolus dose) compared to remifentanil (PCA, increasing infusion dose) for pain management in labour | ||||||
| Patient or population: women in labour with planned vaginal delivery Setting: labour ward in North America (one study) Intervention: remifentanil (PCA, IB (increasing bolus dose)) Comparison: remifentanil (PCA, IF (increasing infusion dose)) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with remifentanil (continuous IV) | Risk with remifentanil (PCA) | |||||
| Satisfaction (overall) with pain relief (VNRS 0 to 10) |
The mean satisfaction scores in the remifentanil (PCA, IF) group was 8.4 on a VNRS 0 to 10 scale. | Mean satisfaction scores in the remifentanil (PCA, IB) group was 0.2 higher (0.81 fewer to 1.21 higher).** | not estimable | 20 (1 RCT) |
⊕⊕⊝⊝ LOW 1 | Only one trial assessed this outcome. |
| Pain intensity 'early' (30 min/1 h) | see comment | see comment | ‐ | (0 studies) | ‐ | No trial assessed this outcome. |
| Additional analgesia required (escape analgesia) | Only one out of 10 woman in the remifentanil (PCA, IF) group crossed over to the epidural group. | not estimable | 20 (1 RCT) |
⊕⊕⊝⊝ LOW 1 | Only one trial assessed this outcome. | |
| Rate of caesarean delivery | Four out of 10 women in each group delivered by caesarean section. | not estimable | 20 (1 RCT) |
⊕⊕⊝⊝ LOW 1 | Only one trial assessed this outcome. | |
| Maternal apnoea | see comment | see comment | ‐ | (0 studies) | ‐ | No trial assessed this outcome. |
| Maternal respiratory depression (< 8 breaths/min) | see comment | see comment | ‐ | (0 studies) | ‐ | No trial assessed this outcome. |
| Apgar score < 7 at 5 min | None of the newborns in both groups had an Apgar score < 7 at 5 min. | not estimable | 20 (1 RCT) |
⊕⊕⊝⊝ LOW 1 | Only 1 trial assessed this outcome. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; IB: increasing bolus dose; IF: increasing infusion dose; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Imprecision ‐ downgrading (very serious): Only one study with small sample size (< 150 participants) reported this outcome.
** Higher values indicate higher levels of satisfaction.
To get an overview about the meta‐analyses of all comparisons in this review, we summarised the direction of effect estimates (favours remifentanil (patient‐controlled analgesia (PCA)), favours control, no favour of remifentanil (PCA) or control) for all outcomes and the GRADE's level of evidence for the predefined GRADE‐relevant outcomes (Figure 4).
4.
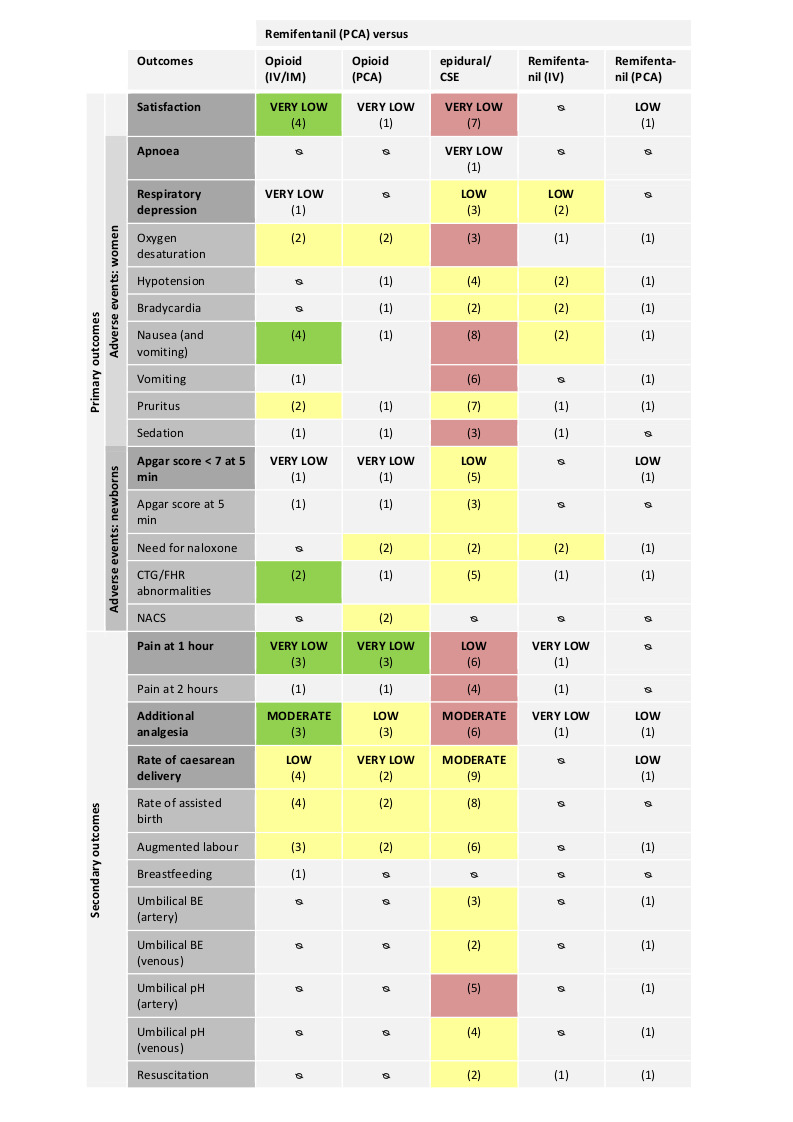
Abbreviations:
IV: intravenous; IM: intramuscular; PCA: patient‐controlled analgesia; CTG: cardiotocography; FHR: fetal heart rate; NACS: neonatal neurologic and adaptive capacity score; BE: base excess.
Direction of estimated effects (results of meta‐analyses) for all primary and secondary outcomes with two or more studies and level of evidence (GRADE) for all GRADE‐relevant, pre‐defined outcomes:
The direction of the estimated effects were labelled as green (favours remifentanil (PCA)), red (favours control), yellow (neither favour of remifentanil (PCA) nor control), (1) (only one RCT, no meta‐analysis performed), ∅ (no RCTs available).
The GRADE levels of the evidence were expressed as VERY LOW, LOW, MODERATE, and HIGH for all GRADE‐relevant outcomes (dark grey, bold). For details on GRADE levels of evidence see the summary of findings tables (Table 1; Table 2; Table 3; Table 4; Table 5).
Remifentanil (PCA) compared to another opioid (IV/IM)
Four trials compared remifentanil (PCA) to another opioid (IV/IM) (Calderon 2006; Evron 2005; Ng 2011; Thurlow 2002).
Primary outcomes
Satisfaction with pain relief
All four trials with 216 participants reported data on overall satisfaction with pain relief (Calderon 2006; Evron 2005; Ng 2011; Thurlow 2002). Random‐effects meta‐analysis revealed a strongly increased standardised mean satisfaction score in women receiving remifentanil (PCA) when compared to another opioid (IV/IM) (standardised mean difference (SMD) 2.11, 95% confidence interval (CI) 0.72 to 3.49; I2 = 93%, Analysis 1.1; fixed‐effect model SMD 1.85, 95% CI 1.51 to 2.19, Table 14). We detected substantial statistical heterogeneity (I2 = 93%). Due to the small number of studies no subgroup analyses were performed. Excluding the trial Evron 2005 that provided another opioid intravenously and not intramuscularly like the remaining three studies decreased heterogeneity from 93% to 55%. 'Risk of bias' assessment for satisfaction with pain relief resulted in two trials at high risk of bias for blinding (Calderon 2006; Thurlow 2002). In trials with an overall low or unclear risk of bias (Evron 2005; Ng 2011), no evidence of effect for remifentanil (PCA) to increase satisfaction was found (SMD 2.46, 95% CI ‐0.34 to 5.26, Table 8). Optimal information size (OIS) considerations showed that with an anticipated minimal clinically relevant difference of 1 cm (visual analogue scale (VAS) 0 to 10 cm), and a control mean satisfaction score of 6 cm the OIS was estimated at 208 participants (Table 12). Including all four trials (n = 216), independent of the 'Risk of bias' assessment, sufficient information was retained to confirm evidence of effect for remifentanil (PCA) to increase overall satisfaction with pain relief.
1.1. Analysis.
Comparison 1 Remifentanil (PCA) versus another opioid (IV/IM), Outcome 1 Satisfaction with pain relief.
We graded the quality of evidence for the outcome 'satisfaction with pain relief' as 'very low' (double‐downgrade for quality and downgrade for inconsistency; Table 1).
Adverse events for women
We could not identify any studies reporting on 'apnoea', 'hypotension', and 'bradycardia'.
Respiratory depression
One trial reported on the incidence of women with respiratory depression (< 8 breaths/minute) (Thurlow 2002). Three out of 18 women in the remifentanil (PCA) group and none out of 18 women in the meperidine IM group had a respiratory depression during labour (Analysis 1.2). Because only one small trial (very serious imprecision) with high risk of bias assessed this outcome and evidence is strongly limited, we graded the quality of the evidence as 'very low' (Table 1).
1.2. Analysis.
Comparison 1 Remifentanil (PCA) versus another opioid (IV/IM), Outcome 2 Respiratory depression (< 8 breaths/min).
Oxygen desaturation
Two studies with 113 women were pooled which reported oxygen desaturation defined as SpO2 < 95% (Evron 2005; Thurlow 2002). Overall, in both trials there was no evidence of effect for a decreased risk of oxygen desaturation in the remifentanil (PCA) group when compared to the other opioid (IV/IM) group in a random‐effects model (risk ratio (RR) 0.48, 95% CI 0.00 to 47.37; I2 = 88%, Analysis 1.3; fixed‐effect model RR 0.66, 95% CI 0.28 to 1.57, Table 14). Since we detected substantial statistical heterogeneity (I2 = 88%) and the individual trials have markedly different results, this meta‐analysis was not reliable. One trial reported zero events in the remifentanil (PCA) group (Evron 2005). The estimated effect and the I2 statistic was not robust when using a constant continuity correction of 0.01 to handle zero event trials (RR 3.42, 95% CI 0.82 to 14.25; I2 = 0%, Table 15). The estimated effect was not robust in terms of risk of bias, because one trial was assessed as high risk of bias for blinding (Thurlow 2002, sensitivity analysis: RR 0.05, 95% CI 0.00 to 0.82, Table 8), and the other trial for attrition bias (Evron 2005, sensitivity analysis: RR 3.50, 95% CI 0.84 to 14.61, Table 9).
1.3. Analysis.
Comparison 1 Remifentanil (PCA) versus another opioid (IV/IM), Outcome 3 Oxygen desaturation (SpO2 < 95%).
Nausea (and vomiting)
All four trials including 216 women reported either on combined nausea and vomiting (Calderon 2006; Evron 2005; Thurlow 2002) or on separate nausea or vomiting (Ng 2011). Random‐effects meta‐analysis revealed a decreased risk for women to suffer from nausea (and vomiting) in the remifentanil (PCA) group when compared to the other opioid (IV/IM) group (RR 0.54, 95% CI 0.29 to 0.99; I2 = 0%, Analysis 1.4). One trial reported zero events in the remifentanil (PCA) group (Evron 2005). The estimated effect was not robust when using a constant continuity correction of 0.01 to handle zero event trials (RR 0.56, 95% CI 0.30 to 1.04, Table 15). Two trials were assessed as high risk of bias for blinding (Calderon 2006; Thurlow 2002). Exclusion of those two trials no longer revealed evidence of effect for remifentanil (PCA) to decrease the risk for nausea (and vomiting) in women when compared with the administration of another opioid (IV/IM) (RR 0.36, 95% CI 0.06 to 2.29, Table 8).
1.4. Analysis.
Comparison 1 Remifentanil (PCA) versus another opioid (IV/IM), Outcome 4 Nausea (and vomiting).
Vomiting
One trial with 68 women reported on vomiting (Ng 2011). One out of 34 women vomited in the remifentanil (PCA) group and two out of 34 vomited in the pethidine (IM) group (P = 0.55) (Analysis 1.5).
1.5. Analysis.
Comparison 1 Remifentanil (PCA) versus another opioid (IV/IM), Outcome 5 Vomiting.
Pruritus
Two trials including 156 participants analysed the occurrence of pruritus in both groups (Evron 2005; Ng 2011). None of the participants in either group of both trials reported to suffer from pruritus (Analysis 1.6). The pooled effect could be estimated by using the trial sequential analysis (TSA) software which allows a constant continuity correction of 0.01 for zero event handling in both arms, which yielded an unreliably wide CI (RR 1.02, 95% CI 0.00 to 1.1E12, Table 15). Both trials were assessed as low or unclear risk of bias for the domains selection bias, blinding, and attrition bias (Table 7; Table 8; Table 9).
1.6. Analysis.
Comparison 1 Remifentanil (PCA) versus another opioid (IV/IM), Outcome 6 Pruritus.
Sedation
One trial with 77 women reported on sedation scores one hour after onset of analgesia in which women in the remifentanil (PCA) group were less sedated than women in the meperidine (IV) group (1.1 +/‐ 0.2 versus 2.6 +/‐ 0.2, mean +/‐ standard deviation (SD), Ramsay sedation score, P < 0.001) (Analysis 1.7) (Evron 2005).
1.7. Analysis.
Comparison 1 Remifentanil (PCA) versus another opioid (IV/IM), Outcome 7 Sedation (1 h).
Adverse events for the newborn
We could not identify any studies reporting on 'need for naloxone' and 'NACS' (neurologic and adaptive capacity score).
Apgar score less than seven at five minutes
One trial with 88 newborns assessed this outcome and none of the newborns in either group had an Apgar score less than seven at five minutes (Analysis 1.8) (Evron 2005). Because only one small trial (very serious imprecision) with unclear risk of bias reported on this outcome which strongly limited evidence, we graded the quality of the evidence as 'very low' (Table 1).
1.8. Analysis.
Comparison 1 Remifentanil (PCA) versus another opioid (IV/IM), Outcome 8 Apgar score < 7 at 5 min.
Apgar score at five minutes
One trial with 68 newborns reported on average Apgar scores at five minutes with no difference in the remifentanil (PCA) and the meperidine (IV) group (median Apgar score of 9, IQR 9 to 9 in both groups) (Analysis 1.9) (Ng 2011).
1.9. Analysis.
Comparison 1 Remifentanil (PCA) versus another opioid (IV/IM), Outcome 9 Apgar score at 5 min.
FHR/CTG abnormalities, non‐reassuring fetal status
Two trials including 156 newborns reported on either opioid‐induced loss of fetal heart rate (FHR) (Evron 2005) or on fetal distress with impaired cardiotocography (CTG) (Ng 2011). The pooled meta‐analysis revealed evidence of effect for a decreased risk of FHR/CTG abnormalities in the remifentanil (PCA) group when compared to the other opioid (IV/IM) group (RR 0.30, 95% CI 0.10 to 0.90; I2 = 0%, Analysis 1.10). This estimated effect was robust with respect to the fixed‐effect model sensitivity analysis (Table 14). All trials were assessed as low or unclear risk for selection bias, attrition bias, and low risk of blinding (Table 7; Table 8; Table 9).
1.10. Analysis.
Comparison 1 Remifentanil (PCA) versus another opioid (IV/IM), Outcome 10 FHR/CTG abnormalities, non‐reassuring fetal status.
Secondary outcomes
We could not identify any studies reporting on 'umbilical cord base excess/pH' and 'need for neonatal resuscitation'.
Pain intensity (pain score 'early' at one hour)
Three trials including 180 women assessed pain intensity at one hour after onset of analgesia (Calderon 2006; Evron 2005; Ng 2011). Random‐effects meta‐analysis showed that remifentanil (PCA) had a moderate to strong effect on the reduction of standardised mean pain scores at one hour when compared to other opioids (IV/IM) (SMD ‐1.58, 95% CI ‐2.69 to ‐0.48; I2 = 89%, Analysis 1.11; fixed‐effect model SMD ‐1.35, 95% CI ‐1.68 to ‐1.01). There was substantial statistical heterogeneity (I2 = 89%). Excluding the trial Ng 2011, heterogeneity decreased to 0% without clinical explanation. One trial was assessed as high risk of bias for blinding (Calderon 2006). In trials with overall low or unclear risk of bias (Evron 2005; Ng 2011), evidence of effect was no longer present for remifentanil (PCA) to decrease pain scores when compared to other opioids (IV/IM) (SMD ‐1.28, 95% CI ‐2.62 to 0.07, Table 8). The OIS was estimated at 298 participants using optimal information size considerations anticipating a minimal clinically relevant reduction of 10 mm (VAS 0 to 100 mm), and a control mean pain score of 35.6 mm (Table 12). Including all three trials (n = 180), independent of the 'Risk of bias' assessment, sufficient information was not available to confirm evidence of effect for remifentanil (PCA) to decrease pain intensity when compared to other opioids (IV/IM).
1.11. Analysis.
Comparison 1 Remifentanil (PCA) versus another opioid (IV/IM), Outcome 11 Pain intensity 'early' (1 h).
We graded the quality of evidence for the outcome 'pain score 'early'' as 'very low' (double‐downgrade for quality, downgrade for inconsistency, and downgrade for imprecision; Table 1).
Pain intensity (pain score 'late' at two hours)
One trial with 68 women provided data on pain scores at two hours after onset of analgesia (Ng 2011). Women receiving remifentanil (PCA) reported less pain (20.0 +/‐ 17.7, mean +/‐ SD, VAS 0 to 100 mm) compared to women receiving pethidine (IM) (36.66 +/‐ 26.66 mm, P < 0.001) (Analysis 1.12).
1.12. Analysis.
Comparison 1 Remifentanil (PCA) versus another opioid (IV/IM), Outcome 12 Pain intensity 'late' (2 h).
Additional analgesia required (escape analgesia)
Three studies including 190 women offered and reported on additional analgesia on request to women in labour. One trial offered epidural analgesia (Evron 2005), one pethidine IM and Entonox (Ng 2011), and one first Entonox and later an epidural (Thurlow 2002). Overall, in all trials the administration of remifentanil (PCA) was associated with a lower requirement for additional escape analgesia when compared to the administration of other opioids (IV/IM) in a random‐effects meta‐analysis (RR 0.57, 95% CI 0.40 to 0.81; I2 = 28%, Analysis 1.13). There was no substantial statistical heterogeneity in the analysis (I2 = 28%). One trial was assessed as high risk of bias for both blinding and incomplete outcome data (Thurlow 2002). Exclusion of this trial had no impact on the robustness of the estimated effect (RR 0.48, 95% CI 0.25 to 0.91, Table 8, Table 9). Trial sequential analysis on all three trials, independent of the 'Risk of bias' assessment, showed that with 'low risk of bias'‐based and with 'empirical' assumptions the RIS was 156 (Table 10) and 194 (Table 11) participants, respectively. In case of TSA 'empirical' the trial sequential monitoring boundaries (TSMB) was crossed (revealing statistical significance before the RIS has been reached and) indicating that sufficient information was retained to confirm evidence of effect for remifentanil (PCA) to decrease the requirements for additional analgesia compared to other opioids (IV/IM).
1.13. Analysis.
Comparison 1 Remifentanil (PCA) versus another opioid (IV/IM), Outcome 13 Additional analgesia required (escape analgesia).
We graded the quality of evidence for the outcome 'additional analgesia required' as 'moderate' (downgrade for quality; Table 1).
Rate of caesarean delivery
All four trials including 215 women reported on the rate of caesarean delivery (Calderon 2006; Evron 2005; Ng 2011; Thurlow 2002). Overall, in all trials there was no evidence of effect for remifentanil (PCA) to decrease the risk for caesarean delivery compared to the other opioid (IV/IM) group when analysed in a random‐effects meta‐analysis (RR 0.70, 95% CI 0.34 to 1.41; I2 = 1%, Analysis 1.14). There was almost no statistical heterogeneity in the analysis detectable. Two trials reported zero events in either the remifentanil (PCA) group (Calderon 2006) or the opioid (IV/IM) group (Thurlow 2002). The estimated effect was robust when using a constant continuity correction of 0.01 to handle zero event trials (RR 0.63, 95% CI 0.30 to 1.32, Table 15). Two trials were assessed as high risk of bias for blinding (Calderon 2006; Thurlow 2002) and one trial for incomplete outcome data (Thurlow 2002). Sensitivity analyses revealed no impact on the robustness of the estimated effects for both blinding (RR 0.63, 95% CI 0.30 to 1.31, Table 8) and attrition bias (RR 0.60, 95% CI 0.29 to 1.24, Table 9). Trial sequential analysis on all three trials, independent of the 'Risk of bias' assessment, showed that with 'low risk of bias'‐based and with 'empirical' assumptions the RIS was 1444 (Table 10) and 2245 participants (Table 11), respectively. Therefore, information was insufficient to demonstrate evidence of no effect.
1.14. Analysis.
Comparison 1 Remifentanil (PCA) versus another opioid (IV/IM), Outcome 14 Rate of caesarean delivery.
We graded the quality of evidence for the outcome 'rate of caesarean delivery' as 'low' (downgrade for quality, downgrade for imprecision; Table 1).
Rate of assisted birth
All four trials with 215 women reported on rate of assisted birth; two trials reported on ventouse delivery (Ng 2011; Thurlow 2002), one on non‐defined instrumental delivery (Calderon 2006), and one on vacuum extraction and forceps delivery (Evron 2005). Random‐effects meta‐analysis showed no evidence of effect for the remifentanil (PCA) group to reduce the risk for assisted birth compared to the other opioid (IV/IM) group (RR 0.82, 95% CI 0.32 to 2.09; I2 = 0%, Analysis 1.15).
1.15. Analysis.
Comparison 1 Remifentanil (PCA) versus another opioid (IV/IM), Outcome 15 Rate of assisted birth.
Augmented labour
Three trials including 190 women analysed augmentation of labour by use of oxytocin (Evron 2005; Ng 2011; Thurlow 2002). The pooled meta‐analysis revealed no difference in the rate of augmented labour between the remifentanil (PCA) and the other opioid (IV/IM) group (RR 0.97, 95% CI 0.72 to 1.29; I2 = 17%, Analysis 1.16).
1.16. Analysis.
Comparison 1 Remifentanil (PCA) versus another opioid (IV/IM), Outcome 16 Augmented labour.
Breastfeeding initiation
One trial assessed the outcome 'breastfeeding initiation' as 'feeding difficulties' (Evron 2005). The study reported that three out of 43 women in the remifentanil (PCA) group and six out of 45 in the meperidine (IV) group had difficulties with breastfeeding (P > 0.05) (Analysis 1.17).
1.17. Analysis.
Comparison 1 Remifentanil (PCA) versus another opioid (IV/IM), Outcome 17 Breastfeeding initiation (feeding difficulties).
Remifentanil (PCA) compared to another opioid (PCA)
Three trials compared remifentanil (PCA) to another opioid (PCA) (Blair 2005; Douma 2010; Volikas 2001).
Primary outcomes
Satisfaction with pain relief
One trial including 110 women, 38 women in the remifentanil (PCA) group and 72 women in the combined control group (meperidine (PCA): 30 women, fentanyl (PCA): 42 women), provided data on overall satisfaction with pain relief (Douma 2010). Women in the remifentanil (PCA) group (8.1 +/‐ 1.1, mean +/‐ SD, verbal rating scale (VRS) 1 to 10) were more satisfied than women in the combined control group (7.175 +/‐ 1.331, Analysis 2.1) (single groups: meperidine (PCA) (7.0 +/‐ 1.5, P < 0.05) and fentanyl (PCA) group (7.3 +/‐ 1.2, P > 0.05)). Because only one small trial assessed this outcome (very serious imprecision), with high risk of attrition bias which strongly limits the evidence, we graded the quality of the evidence as 'very low' (Table 2).
2.1. Analysis.
Comparison 2 Remifentanil (PCA) versus another opioid (PCA), Outcome 1 Satisfaction with pain relief.
Adverse events for women
We could not identify any studies reporting on 'apnoea', and 'respiratory depression'.
Oxygen desaturation
Two studies with 190 women were pooled which reported oxygen desaturation defined as either SpO2 < 95% (Douma 2010) or SpO2 < 94% (Blair 2005). In a random‐effects meta‐analysis there was no evidence of effect that administration of remifentanil (PCA) was associated with a higher risk for oxygen desaturation when compared to other opioids (PCA) (RR 1.28, 95% CI 0.49 to 3.30; I2 = 98%, Analysis 2.2). Under the fixed‐effect model the remifentanil (PCA) group was associated with a higher risk for oxygen desaturation (RR 1.39, 95% CI 1.16 to 1.67, Table 14). However, due to substantial statistical heterogeneity (I2 = 98%), and the different results of the individual trials this meta‐analysis was not reliable. The estimated effect was not robust in terms of risk of bias, because one trial was assessed as high risk of bias for blinding (Blair 2005, sensitivity analysis: RR 1.64, 95% CI 1.25 to 2.15, Table 8), and both trials were assessed as high risk of attrition bias (Table 9).
2.2. Analysis.
Comparison 2 Remifentanil (PCA) versus another opioid (PCA), Outcome 2 Oxygen desaturation (SpO2 < 95%).
Hypotension
One trial with 17 women assessed the outcome 'hypotension' and reported that there were no episodes of hypotension in neither the remifentanil (PCA) group nor the pethidine (PCA) group (Analysis 2.3) (Volikas 2001).
2.3. Analysis.
Comparison 2 Remifentanil (PCA) versus another opioid (PCA), Outcome 3 Hypotension.
Bradycardia
One trial including 17 women assessed the outcome 'bradycardia' and reported that there were no episodes of bradycardia in either group (Analysis 2.4) (Volikas 2001).
2.4. Analysis.
Comparison 2 Remifentanil (PCA) versus another opioid (PCA), Outcome 4 Bradycardia.
Nausea (and vomiting)
One trial with 153 participants, 51 women in either group (remifentanil, meperidine, and fentanyl), reported on nausea and vomiting (Douma 2010). There was no difference between the groups with respect to the risk of nausea and vomiting as 20 out of 51 women in both the remifentanil and the fentanyl group, and 23 out of 51 women in the meperidine group suffered from nausea and vomiting (Analysis 2.5).
2.5. Analysis.
Comparison 2 Remifentanil (PCA) versus another opioid (PCA), Outcome 5 Nausea (and vomiting).
Pruritus
One trial including 152 women assessed the risk of pruritus (Douma 2010). Pruritus occurred more frequently in the remifentanil group (eight out of 51 women) than in the meperidine group (three out of 51) or the fentanyl group (one out of 50) (P < 0.05) (Analysis 2.6).
2.6. Analysis.
Comparison 2 Remifentanil (PCA) versus another opioid (PCA), Outcome 6 Pruritus.
Sedation
One trial including 159 women reported on sedation scores one hour after onset of analgesia in which women in the remifentanil (PCA) group (1.85 +/‐ 0.8, mean +/‐ SD, Observer sedation score 1 to 5) were more sedated than women in the combined control group (1.42 +/‐ 1.414, Analysis 2.7), (single groups: meperidine (PCA) (1.45 +/‐ 0.5, P < 0.05) and fentanyl (PCA) group (1.39 +/‐ 0.5, P < 0.01)) (Douma 2010).
2.7. Analysis.
Comparison 2 Remifentanil (PCA) versus another opioid (PCA), Outcome 7 Sedation (1 h).
Adverse events for the newborn
Apgar score less than seven at five minutes
One trial comparing remifentanil (PCA) versus pethidine (PCA) provided data on Apgar score less than seven at five minutes (Volikas 2001). This study had been terminated after 17 participants completed the trial, on agreement with the local ethics committee, due to concerns with the poor Apgar scores in the pethidine group. None of the nine newborns in the remifentanil (PCA) group and three out of eight newborns in the pethidine (PCA) group had an Apgar score less than seven at five minutes (Analysis 2.8). Because only one small trial assessed this outcome (very serious imprecision) with unclear risk of selection bias and blinding, which strongly limits the evidence, we graded the quality of the evidence as 'very low' (Table 2).
2.8. Analysis.
Comparison 2 Remifentanil (PCA) versus another opioid (PCA), Outcome 8 Apgar score < 7 at 5 min.
Apgar score at five minutes
One trial with 115 newborns reported on average Apgar score at five minutes with no difference between the remifentanil (PCA) (9.9 +/‐ 0.3, mean +/‐ SD) and the combined control group (9.642 +/‐ 0.619, Analysis 2.9), (single groups: meperidine (PCA) (9.7 +/‐ 0.6) and fentanyl (PCA) group (9.6 +/‐ 0.6)) (Douma 2010). This trial of Douma 2010 was assessed as high risk of attrition bias because about 30% of the data on newborns were not reported, without giving appropriate reasons for that.
2.9. Analysis.
Comparison 2 Remifentanil (PCA) versus another opioid (PCA), Outcome 9 Apgar score at 5 min.
Need for naloxone
Two trials with 55 newborns provided data on the need for naloxone (Blair 2005; Volikas 2001). Only one event for the need of naloxone was reported in the control pethidine (PCA) group of one trial (Volikas 2001); the other trial included zero events in both arms, which was not estimable with Review Manager 5 (RR 0.30, 95% CI 0.01 to 6.47; I2 = 0%, Analysis 2.10). A pooled effect could be estimated by using the TSA software, which allows a constant continuity correction of 0.01 for zero event handling in both arms, and which yielded an unreliably wide CI (RR 0.03, 95% CI 0.00 to 1.8E8, Table 15). The study from Blair 2005 was assessed as high risk of performance and attrition bias. Exclusion of this trial had no impact on robustness of the estimated effect with respect to all sensitivity analyses performed (Table 7; Table 8; Table 9).
2.10. Analysis.
Comparison 2 Remifentanil (PCA) versus another opioid (PCA), Outcome 10 Need for naloxone.
FHR/CTG abnormalities, non‐reassuring fetal status
None of the included studies comparing remifentanil (PCA) to another opioid (PCA) assessed 'FHR/CTG abnormalities or non‐reassuring fetal status'. However, Douma 2010 reported on the incidence of newborns with reactive CTG and derived no difference between the remifentanil (44 out of 52), the meperidine (44 out of 53), and the fentanyl group (48 out of 54); vice versa 15%, 17%, and 11% of the newborns, respectively, must have shown a non‐reactive CTG (Analysis 2.11).
2.11. Analysis.
Comparison 2 Remifentanil (PCA) versus another opioid (PCA), Outcome 11 FHR/CTG abnormalities, non‐reassuring fetal status.
NACS at 15/30 minutes
Two trials including 94 newborns provided data on NACS at either 15 minutes (Douma 2010) or 30 minutes postpartum (Blair 2005). In a random‐effects meta‐analysis no evidence of effect was found that remifentanil (PCA) was associated with higher NACS compared to another opioid (PCA) (mean difference (MD) 1.11, 95% CI ‐0.65 to 2.87; I2 = 81%, Analysis 2.12). Under the fixed‐effect model the remifentanil (PCA) group was associated with a higher NACS when compared to another opioid (PCA) (MD 1.15, 95% CI 0.38 to 1.93, Table 14). However, due to substantial statistical heterogeneity (I2 = 81%) the fixed‐effect model was not reliable. 'Risk of bias' assessment for NACS resulted in one trial assessed as high risk of performance bias (Blair 2005); sensitivity analysis changed the direction of the estimated effect (RR 0.20, 95% CI ‐0.93 to 1.33, Table 8). Both trials were assessed as high risk of attrition bias.
2.12. Analysis.
Comparison 2 Remifentanil (PCA) versus another opioid (PCA), Outcome 12 NACS at 15/30 min.
Secondary outcomes
We could not identify any studies reporting on 'breastfeeding initiation', 'umbilical cord base excess/pH', and 'need for neonatal resuscitation'.
Pain intensity (pain score 'early' at 30 minutes/one hour)
Three trials including 215 women provided data on pain intensity at one hour after onset of analgesia (Blair 2005; Douma 2010; Volikas 2001). In the case of Blair 2005 which reported pain intensity as median with IQR, we used the '30 minutes' time point instead of the 'one hour' time point because of asymmetric data. In a random‐effects meta‐analysis remifentanil (PCA) reduced the standardised mean pain intensity when compared to other opioid (PCA), however, the upper CI limit reached the line of no effect (SMD ‐0.51, 95% CI ‐1.01 to ‐0.00; I2 = 52%, Analysis 2.13). Under the fixed‐effect model, evidence of effect was found for remifentanil (PCA) to decrease pain scores when compared to other opioids (PCA) (SMD ‐0.57, 95% CI ‐0.86 to ‐ 0.29, Table 14). However, substantial statistical heterogeneity (I2 = 52%) reduced the reliability of the fixed‐effect model. One trial was assessed as high risk of bias for blinding (Blair 2005). Exclusion of this trial revealed a moderate to strong (clinically relevant) reduction in pain intensity of women after administration of remifentanil (PCA) when compared to another opioid (PCA) (SMD ‐0.73, 95% CI ‐1.05 to ‐0.40, Table 8), and decreased the heterogeneity to I2 = 0% without any other clinical explanation. The OIS was calculated at 246 participants using optimal information size considerations anticipating a minimal clinically relevant reduction of 10 cm (VAS 0 to 10 cm) and a control mean pain score of 6.282 cm (Table 12). Including all three trials (n = 215), independent of the 'Risk of bias' assessment, sufficient information was not available to confirm evidence of effect for remifentanil (PCA) to decrease pain intensity when compared to other opioids (PCA).
2.13. Analysis.
Comparison 2 Remifentanil (PCA) versus another opioid (PCA), Outcome 13 Pain intensity 'early' (30 min/1 h).
We graded the quality of evidence for the outcome 'pain score 'early'' as 'very low' (downgrade for quality, downgrade for inconsistency, and downgrade for imprecision; Table 2.
Pain intensity (pain score 'late' at two hours)
One trial with 108 women reported on pain intensity at two hours with mean pain scores in the remifentanil (PCA) group of 5.7 +/‐ 2.7 cm (mean +/‐ SD, VAS 0 to 10 cm) and the combined control group of 6.598 +/‐ 2.233 (Analysis 2.14), (single groups: meperidine (PCA) group wih6.76 +/‐ 2.3 cm and the fentanyl (PCA) group with 6.47 +/‐ 2.2 cm) (Douma 2010).
2.14. Analysis.
Comparison 2 Remifentanil (PCA) versus another opioid (PCA), Outcome 14 Pain intensity 'late' (2 h).
Additional analgesia required (escape analgesia)
Three studies including 215 women offered and reported on additional analgesia on request to women in labour. One trial offered Entonox (Blair 2005), one trial offered an epidural (Douma 2010), and one trial provided both Entonox and epidural analgesia (Volikas 2001). Random‐effects meta‐analysis revealed no evidence of effect for remifentanil (PCA) to reduce requirements for additional analgesia when compared to other opioids (PCA) (RR 0.76, 95% CI 0.45 to 1.28; I2 = 64%, Analysis 2.15). We detected substantial statistical heterogeneity (I2 = 64%). Excluding Blair 2005 or Douma 2010 decreased the heterogeneity to 0%, respectively, without clinical explanation. One trial was assessed as high risk of bias for blinding (Blair 2005). Exclusion of this trial had no impact on robustness of the estimated effect (Table 8). Trial sequential analysis on all three trials, independent of the 'Risk of bias' assessment, showed that with 'low risk of bias'‐based and with 'empirical' assumptions the RIS was 1024 (Table 10) and 4218 participants (Table 11), respectively. The RIS was not reached and the TSMB were not crossed indicating that insufficient information was retained to confirm evidence of no effect for remifentanil (PCA) on the requirements for additional analgesia compared to other opioids (PCA).
2.15. Analysis.
Comparison 2 Remifentanil (PCA) versus another opioid (PCA), Outcome 15 Additional analgesia required (escape analgesia).
We graded the quality of evidence for the outcome 'additional analgesia required' as 'low' (downgrade for inconsistency and imprecision; Table 2).
Rate of caesarean delivery
Two trials with 143 women provided data on rate of caesarean delivery (Douma 2010; Volikas 2001). Pooled meta‐analysis revealed an increased risk for caesarean section under remifentanil (PCA) analgesia when compared to other opioids (PCA) (RR 2.78, 95% CI 0.99 to 7.82; I2 = 0%, Analysis 2.16). However, the lower CI limit crossed the line of no effect whereby a wide range of treatment effects ‐ clinically relevant and non‐relevant – is compatible with this result. One trial was assessed as high risk of attrition bias (Douma 2010). Exclusion of the high risk of bias trial widened the CI including appreciable benefit and harm (RR 1.78, 95% CI 0.20 to 16.10, Table 9). Trial sequential analysis on both trials, independent of the 'Risk of bias' assessment, showed that with 'low risk of bias'‐based and with 'empirical' assumptions, the RIS was 852 (Table 10) and 372 participants (Table 11), respectively. The RIS was not reached and the TSMB were not crossed indicating that insufficient information was retained to confirm evidence of effect for remifentanil (PCA) to increase the rate of caesarean deliveries compared to other opioids (PCA).
2.16. Analysis.
Comparison 2 Remifentanil (PCA) versus another opioid (PCA), Outcome 16 Rate of caesarean delivery.
We graded the quality of evidence for the outcome 'rate of caesarean delivery' as 'very low' (double‐downgrade for quality, and downgrade for imprecision; Table 2).
Rate of assisted birth
Two trials with 143 women reported on rate of assisted birth; one trial by ventouse and forceps delivery (Volikas 2001) and the other one by non‐defined instrumental delivery (Douma 2010). Random‐effects meta‐analysis showed no evidence of effect for remifentanil (PCA) to increase the risk for assisted birth compared to the other opioid (PCA) group (RR 1.22, 95% CI 0.62 to 2.37; I2 = 0%, Analysis 2.17).
2.17. Analysis.
Comparison 2 Remifentanil (PCA) versus another opioid (PCA), Outcome 17 Rate of assisted birth.
Augmented labour
Two trials including 152 women analysed augmentation of labour by use of oxytocin (Douma 2010; Volikas 2001). The pooled meta‐analysis revealed no evidence of effect for remifentanil (PCA) to increase the risk for augmentation of labour compared to the other opioid (PCA) group (RR 1.37, 95% CI 0.59 to 3.15; I2 = 70%, Analysis 2.18). Since we detected substantial statistical heterogeneity (I2 = 70%), and the individual trials have markedly different results, this meta‐analysis was not reliable.
2.18. Analysis.
Comparison 2 Remifentanil (PCA) versus another opioid (PCA), Outcome 18 Augmented labour.
Remifentanil (PCA) compared to epidural analgesia/combined spinal‐epidural analgesia (CSE)
Ten trials compared remifentanil (PCA) to either epidural analgesia (Douma 2011; Douma 2015; El‐Kerdawy 2010; Evron 2008; Freeman 2015; Stocki 2014; Stourac 2014; Tveit 2012; Volmanen 2008) or both epidural and CSE (Ismail 2012). For the latter trial, we combined both control groups (epidural and CSE) into one control group.
Primary outcomes
Satisfaction with pain relief
Seven trials including 2135 participants, with 931 in the remifentanil (PCA) and 1204 in the control epidural/CSE group provided data on overall satisfaction with pain relief (Douma 2011; Douma 2015; El‐Kerdawy 2010; Freeman 2015; Ismail 2012; Stocki 2014; Volmanen 2008). Overall, when all trials were pooled in a random‐effects meta‐analysis, women in the epidural/CSE group were slightly more satisfied with pain relief than women in the remifentanil (PCA) group (SMD ‐0.22, 95% CI ‐0.40 to ‐0.04; I2 = 52%, Analysis 3.1, fixed‐effect model SMD ‐0.29, 95% CI ‐0.38 to ‐0.20, Table 14). We detected substantial statistical heterogeneity (I2 = 52%). Excluding Ismail 2012 that not only investigated epidural analgesia but also CSE, decreased the heterogeneity to 0%. 'Risk of bias' assessment for satisfaction with pain relief resulted in one trial assessed as high risk of selection bias (Freeman 2015), six trials as high risk of bias for blinding (Douma 2011; Douma 2015; El‐Kerdawy 2010; Freeman 2015; Ismail 2012; Stocki 2014), and four trials as high risk of attrition bias (Douma 2011; Douma 2015; Freeman 2015; Volmanen 2008). In trials with low or unclear risk of bias evidence of effect for remifentanil (PCA) to decrease satisfaction was no longer found (selection bias: SMD ‐0.20, 95% CI ‐0.46 to 0.07 (Table 7), blinding: SMD 0.27, 95% CI ‐0.31 to 0.86 (Table 8), attrition bias: SMD ‐0.27, 95% CI ‐0.64 to 0.10 (Table 9)). Optimal information size considerations revealed that with an anticipated difference of 0.5 cm (VAS 0 to 10 cm) and a control mean satisfaction score of 9.1 cm (both assumptions were based on the 'best' trial), the OIS was estimated at 380 participants (Table 13). Including all trials (n = 2135), independent of the 'Risk of bias' assessment, sufficient information was retained to confirm evidence of effect for epidural analgesia to increase overall satisfaction with pain relief compared to remifentanil (PCA).
3.1. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 1 Satisfaction with pain relief.
We graded the quality of evidence for the outcome 'satisfaction with pain relief' as 'very low' (double‐downgrade for quality and downgrade for inconsistency; Table 3).
Adverse events for women
Apnoea
One trial including 38 women provided data on apnoea defined as a respiratory rate of zero for at least 20 s (Stocki 2014). The study reported that five women during the first hour of analgesia and nine out of 19 women during the whole study period in the remifentanil (PCA) group had one or more apnoea events, whereas none of the 19 women in the epidural group had an apnoea (one hour: P = 0.045) (Analysis 3.2). Because only one small trial assessed this outcome (very serious imprecision), which was assessed as high risk of bias for blinding, we graded the quality of the evidence as 'very low' (Table 3).
3.2. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 2 Apnoea.
Respiratory depression
Three trials with 687 women (400 remifentanil, 287 epidural) investigated the occurrence of respiratory depression defined as either less than eight breaths/minute (Freeman 2015; Stocki 2014) or less than nine breaths/minute (Tveit 2012). The trial from Tveit 2012 did not detect any event in either group and Freeman 2015 did not detect any event in the epidural group. Zero events in both arms were not estimable with Review Manager 5 and were ignored in the meta‐analysis, which revealed no evidence of effect for remifentanil to increase the risk for respiratory depression when compared to epidural analgesia (RR 1.52, 95% CI 0.23 to 9.90; I2 = 50%, Analysis 3.3). A pooled effect of all three trials that demonstrated no difference between both interventions in terms of risk of respiratory depression could be estimated by using the TSA software and the application of a constant continuity correction of 0.01 for zero event handling in both arms (RR 0.91, 95% CI 0.51 to 1.62; I2 = 0%, Table 15). 'Risk of bias' assessment resulted in one trial judged as high risk for selection bias (Freeman 2015), all trials as high risk for blinding, and two trials as high risk for attrition bias (Freeman 2015; Tveit 2012). In trials with low or unclear risk of bias, no difference between both interventions in terms of risk of respiratory depression was obtained (selection bias: RR 0.91, 95% CI 0.52 to 1.61 (Table 7) and attrition bias: RR 0.91, 95% CI 0.39 to 2.10 (Table 9)). Trial sequential analysis on all three trials, independent of the 'Risk of bias' assessment, showed that with 'low risk of bias'‐based and with 'empirical' assumptions the RIS was 4986 (Table 10) and 2.5 E6 participants (Table 11), respectively. The RIS was not reached and the TSMB were not crossed indicating that insufficient information was retained to confirm evidence of no effect.
3.3. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 3 Respiratory depression (< 9, < 8 breaths/min).
We graded the quality of evidence for the outcome 'respiratory depression' as 'low' (downgrade for quality and downgrade for imprecision; Table 3).
Oxygen desaturation (SpO2 < 92%)
Three trials with 774 women, 446 in the remifentanil (PCA) and 328 in the epidural group, reported on oxygen desaturation defined as SpO2 < 92% (Douma 2015; Freeman 2015; Tveit 2012). Random‐effects meta‐analysis revealed a strongly increased risk for oxygen desaturation in women with remifentanil (PCA) analgesia when compared to women with an epidural (RR 3.24, 95% CI 1.66 to 6.32; I2 = 52%, Analysis 3.4, fixed‐effect model RR 3.46, 95% CI 2.32 to 5.16, Table 14). We detected substantial statistical heterogeneity. The I2 was decreased to 24% when excluding Tveit 2012, which had no limit regarding remifentanil administration. One trial reported zero events in the epidural group (Tveit 2012). The estimated effect was robust when using a constant continuity correction of 0.01 to handle zero event trials; however, the I2 was reduced to 0% (RR 2.88, 95% CI 1.94 to 4.27; I2 = 0%, Table 15). One trial was assessed as high risk for allocation concealment (Freeman 2015). Exclusion of this trial impacted on the robustness of the results in which evidence of effect for the high risk of oxygen desaturation in the remifentanil (group) was no longer present (RR 5.83, 95% CI 0.40 to 84.06, Table 7). Moreover, all three trials were assessed as high risk of bias for blinding and incomplete outcome data (Table 8; Table 9).
3.4. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 4 Oxygen desaturation (SpO2 < 92%).
Oxygen desaturation (SpO2 < 95%, < 94%)
Three trials including 800 women, 458 in the remifentanil (PCA) and 342 in the epidural group, reported on oxygen desaturation defined as either SpO2 < 94% (Stocki 2014) or SpO2 < 95% (Freeman 2015; Volmanen 2008). In Stocki 2014, all women in both groups received continuous supplementary oxygen (2 L/min) throughout the respiratory monitoring period. In a random‐effects meta‐analysis a strongly increased risk of oxygen desaturation in women with remifentanil (PCA) analgesia was found when compared to women with epidural analgesia (RR 3.27, 95% CI 2.32 to 4.61; I2 = 3%, Analysis 3.5 fixed‐effect model RR 3.30, 95% CI 2.43 to 4.49, Table 14). 'Risk of bias' assessment for oxygen desaturation resulted in one trial assessed as high risk of selection bias (Freeman 2015), two trials as high risk of bias for blinding (Freeman 2015; Stocki 2014), and two trials as high risk of attrition bias (Freeman 2015; Volmanen 2008). In trials with low or unclear risk of bias evidence of effect for remifentanil (PCA) to increase the risk of oxygen desaturation when compared to epidural/CSE was more increased (selection bias: RR 5.44, 95% CI 2.11 to 14.02 (Table 7), blinding: RR 11.38, 95% CI 1.62 to 79.78 (Table 8), attrition bias: RR 4.33, 95% CI 1.47 to 12.79 (Table 9)).
3.5. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 5 Oxygen desaturation (SpO2 < 95%).
Hypotension
Four trials including 823 women (458 remifentanil, 365 epidural) reported on hypotension defined either as systolic blood pressure of < 90 mmHg (Freeman 2015) or as > 25% decrease from baseline systolic blood pressure (Stourac 2014); two trials did not define hypotension (Douma 2011; El‐Kerdawy 2010). Two trials detected events in either the remifentanil (PCA) (Stourac 2014) or the epidural group (El‐Kerdawy 2010), and one trial did not detect any event of hypotension in both arms (Douma 2011). Using Review Manager 5 which ignores studies with zero events in both arms revealed no evidence of effect that remifentanil (PCA) was associated with a decreased risk for hypotension compared to epidural analgesia (RR 0.58, 95% CI 0.22 to 1.49; I2 = 17%, Analysis 3.6). However, when meta‐analysis was performed by using the TSA software, which allows a constant continuity correction of 0.01 for zero event handling in both arms, evidence of effect was found for remifentanil (PCA) to decrease the risk for hypotension in comparison to epidural analgesia (RR 0.59, 95% CI 0.37 to 0.94, Table 15). One trial was assessed as high risk for selection and attrition bias (Freeman 2015), one for attrition bias (Stourac 2014); all trials were judged as high risk for blinding. Exclusion of high risk of bias trials revealed no evidence of effect for remifentanil (PCA) to decrease the risk for hypotension compared to epidural analgesia, however, with an unreliably wide CI (selection bias: RR 0.57, 95% CI 0.00 to 2.4E7 (Table 7), attrition bias: RR 0.01, 95% CI 0.00 to 7.8E7 (Table 9)).
3.6. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 6 Hypotension.
Bradycardia
Two trials with 44 women reported on bradycardia defined either as a heart rate of less than 50 beats/minute (Stourac 2014) or without definition (Douma 2011). In none of the women in either group of both trials was bradycardia detected (Analysis 3.7); the pooled effect could be estimated by using the TSA software, which allows a constant continuity correction of 0.01 for zero event handling in both arms, which yielded an unreliably wide CI (RR 1.00, 95% CI 0.00 to 1.0E12, Table 15). One trial was assessed as high risk for attrition bias (Stourac 2014) and both trials were judged as high risk for blinding; the estimated effect was robust with respect to all sensitivity analyses performed (Table 14; Table 8; Table 9).
3.7. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 7 Bradycardia.
Nausea
Eight trials including 1909 women (807 remifentanil, 1102 epidural/CSE) provided data on nausea (Douma 2011; Douma 2015; El‐Kerdawy 2010; Freeman 2015; Ismail 2012; Stocki 2014; Tveit 2012; Volmanen 2008). Random‐effects meta‐analysis showed that remifentanil (PCA) was associated with a increased risk of suffering from nausea compared to epidural/CSE (RR 1.49, 95% CI 1.19 to 1.86; I2 = 0%, Analysis 3.8, fixed‐effect model RR 1.53, 95% CI 1.22 to 1.91, Table 14). 'Risk of bias' assessment resulted in one trial judged as high risk for selection bias (Freeman 2015), seven trials as high risk for blinding, (Douma 2011; Douma 2015; El‐Kerdawy 2010; Freeman 2015; Ismail 2012; Stocki 2014; Tveit 2012), and four trials as high risk for attrition bias (Douma 2015; Freeman 2015; Tveit 2012; Volmanen 2008). The effect estimate was robust with respect to the 'selection bias' sensitivity analysis (RR 1.41, 95% CI 1.09 to 1.83 (Table 7). However, evidence of effect for remifentanil (PCA) to increase the risk for nausea compared to epidural analgesia was no longer present when only trials with low or unclear risk for blinding (RR 3.94, 95% CI 0.96 to 16.22 (Table 8)) or attrition bias were pooled (RR 1.27, 95% CI 0.82 to 1.98, Table 9).
3.8. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 8 Nausea.
Vomiting
Six trials with 1840 women (773 remifentanil, 1067 epidural/CSE) reported data on vomiting (Douma 2011; Douma 2015; El‐Kerdawy 2010; Freeman 2015; Ismail 2012; Tveit 2012). The random‐effects meta‐analysis revealed that remifentanil (PCA) was associated with a higher risk of vomiting compared to epidural/CSE (RR 1.63, 95% CI 1.25 to 2.13; I2 = 0%, Analysis 3.9, fixed‐effect model RR 1.62, 95% CI 1.24 to 2.10, Table 14). 'Risk of bias' assessment resulted in one trial judged as high risk for selection bias (Freeman 2015), and four trials as high risk for attrition bias (Douma 2015; Freeman 2015; Tveit 2012; Volmanen 2008); all trials were assessed as high risk of bias for blinding. The effect estimate was robust with respect to the 'selection bias' sensitivity analysis (RR 1.82, 95% CI 1.29 to 2.57 (Table 7). However, evidence of effect for remifentanil (PCA) to increase the risk of vomiting compared to epidural analgesia was no longer present when only trials with low or unclear risk for attrition bias were meta‐analysed (RR 1.54, 95% CI 0.75 to 3.14, Table 9).
3.9. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 9 Vomiting.
Pruritus
Seven trials including 1852 women (777 remifentanil, 1075 epidural/CSE) provided data on pruritus (Douma 2011; Douma 2015; El‐Kerdawy 2010; Freeman 2015; Ismail 2012; Stocki 2014; Tveit 2012). Meta‐analysis showed no evidence of effect for remifentanil (PCA) to reduce the risk to suffer from pruritus (random‐effects model RR 0.75, 95% CI 0.48 to 1.18; I2 = 29%, Analysis 3.10, fixed‐effect model RR 0.76, 95% CI 0.54 to 1.07, Table 14). One trial reported zero events in the remifentanil (PCA) group (Tveit 2012). The estimated effect was robust when using a constant continuity correction of 0.01 to handle zero event trials (RR 0.78, 95% CI 0.51 to 1.18, Table 15). 'Risk of bias' assessment for pruritus resulted in one trial assessed as high risk of selection bias (Freeman 2015) and two trials as high risk of attrition bias (Freeman 2015; Tveit 2012); sensitivity analysis has no impact on robustness of the estimated effect (Table 7; Table 9). All trials were assessed as high risk of bias for blinding (Table 8).
3.10. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 10 Pruritus.
Sedation
Three trials including 148 women reported on sedation scores one hour after onset of analgesia (Douma 2011; Douma 2015; El‐Kerdawy 2010). Random‐effects meta‐analysis revealed evidence of effect for remifentanil (PCA) to increase mean sedation scores when compared with epidural analgesia (SMD 0.71, 95% CI 0.03 to 1.39; I2 = 68%, Analysis 3.11). Substantial statistical heterogeneity was detected (I2 = 68%) and decreased to 0% when excluding Douma 2015 without clinical explanation. All studies were assessed as low or unclear risk of bias for selection and attrition bias (Table 7; Table 9); and all trials were judged as high risk of bias for blinding (Table 8).
3.11. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 11 Sedation (1 h).
Adverse events for the newborn
We could not identify any studies reporting on the outcome 'NACS'.
Apgar score ≤ seven at five minutes
Five trials with 1322 newborns (470 remifentanil, 852 epidural/CSE) provided data on Apgar scores at five minutes; four of the five trials reported the number of newborns with an Apgar score ≤ seven (Douma 2011; Douma 2015; El‐Kerdawy 2010) or less than seven (Ismail 2012) at five minutes; one trial was dichotomised for the present meta‐analysis because Apgar scores at five minutes were reported as median with IQR along with the information in the text that all newborns had Apgar scores less than eight at five minutes (Stocki 2014). Two trials detected events in either the remifentanil (PCA) (Douma 2015) or the epidural group (Douma 2011) and two trials did not detect any newborn in both groups with an Apgar score ≤ seven (El‐Kerdawy 2010; Stocki 2014). Using Review Manager 5 which ignores studies with zero events in both arms, there was no evidence of effect that remifentanil (PCA) was associated with an increased risk for newborns to have Apgar scores ≤ seven compared to epidural analgesia (RR 1.28, 95% CI 0.65 to 2.51; I2 = 0%, Analysis 3.12). The estimated effect was robust with respect to inclusion of studies with zero events in both arms and a constant continuity correction of 0.01 (RR 1.26, 95% CI 0.62 to 2.57, Table 15). Two trials were assessed as high risk of attrition bias (Douma 2011; Douma 2015); sensitivity analysis did not reveal an impact on the robustness of the estimated effect (Table 9). All trials were judged as low risk for selection bias (Table 7) and as high risk of bias for blinding (Table 8).
3.12. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 12 Apgar score ≤ 7 (< 7) at 5 min.
Trial sequential analysis on all five trials, independent of the 'Risk of bias' assessment, showed that with 'low risk of bias'‐based and with 'empirical' assumptions, the RIS was 29,000 (Table 10) and 34,000 newborns (Table 11), respectively. With 1322 newborns the RIS was not reached and the TSMB were not crossed indicating that insufficient information was retained to confirm evidence of no effect.
We graded the quality of evidence for the outcome 'Apgar score ≤ seven at five minutes' as 'low' (downgrade for quality and downgrade for imprecision; Table 3).
Apgar score at five minutes
Three trials with 137 newborns reported on mean Apgar scores at five minutes (Douma 2011; Douma 2015; Stourac 2014). When all trials were pooled in a random‐effects meta‐analysis there was no difference between remifentanil (PCA) and epidural analgesia with respect to mean Apgar scores at five minutes postpartum (MD 0.06, 95% CI ‐0.27 to 0.39; I2 = 0%, Analysis 3.13). 'Risk of bias' assessment for Apgar score at five minutes resulted in all three trials assessed as high risk of bias for blinding (Table 8) and incomplete outcome data (Table 9).
3.13. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 13 Apgar score at 5 min.
Need for naloxone
Two trials including 1170 newborns (395 remifentanil, 775 epidural/CSE) analysed the rate of need for naloxone (El‐Kerdawy 2010; Ismail 2012). One trial did not detect any event in the remifentanil group (El‐Kerdawy 2010), and the other trial did not detect any event of naloxone usage in both arms (Douma 2011), which was not estimable with Review Manager 5 (RR 0.20, 95% CI 0.01 to 3.85; I2 = 0%, Analysis 3.14). A pooled effect of both trials could be estimated by using the TSA software, which allows a constant continuity correction of 0.01 for zero event handling in both arms; there was no evidence of effect that remifentanil (PCA) was associated with a decreased risk of need for naloxone compared to epidural analgesia, however, with an unreliably wide CI (RR 0.02, 95% CI 0.00 to 1.6E8, Table 15). Both trials were assessed for 'need for naloxone' as low or unclear risk of selection and attrition bias (Table 7; Table 9); all trials were judged as high risk of bias for blinding (Table 8).
3.14. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 14 Need for naloxone.
FHR/CTG abnormalities, non‐reassuring fetal status
Five studies including 1280 newborns (449 remifentanil, 831 epidural/CSE) provided data on FHR/CTG abnormalities (El‐Kerdawy 2010; Stourac 2014; Tveit 2012; Volmanen 2008) or non‐reassuring fetal status (Ismail 2012). Random‐effects meta‐analysis revealed no evidence of effect for remifentanil (PCA) to increase the risk for FHR/CTG abnormalities or non‐reassuring fetal status (RR 1.55, 95% CI 0.49 to 4.92; I2 = 48%, Analysis 3.15, fixed‐effect model RR 1.38, 95% CI 0.84 to 2.25, Table 14). One trial reported zero events in the remifentanil (PCA) group (El‐Kerdawy 2010). The estimated effect was robust when using a constant continuity correction of 0.01 to handle zero event trials (RR 1.88, 95% CI 0.63 to 5.61, Table 15). 'Risk of bias' assessment resulted in four trials assessed as high risk of bias for blinding (El‐Kerdawy 2010; Ismail 2012; Stourac 2014; Tveit 2012) and three trials as high risk for attrition bias (Stourac 2014; Tveit 2012; Volmanen 2008). The estimated effect was not robust when trials with unclear risk of bias for blinding (RR 11.38, 95% CI 1.62 to 79.78, Table 8) or low risk for attrition bias were pooled (RR 0.87, 95% CI 0.41 to 1.87, Table 9).
3.15. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 15 FHR/CTG abnormalities, non‐reassuring fetal status.
Secondary outcomes
We could not identify any studies reporting on 'breastfeeding initiation'.
Pain intensity (pain score 'early' at one hour)
Six trials including 235 women (115 remifentanil, 120 epidural) provided data on pain intensity at one hour after onset of analgesia (Douma 2011; Douma 2015; El‐Kerdawy 2010; Stocki 2014; Stourac 2014; Tveit 2012). Random‐effects meta‐analysis showed that epidural analgesia was more favourable in lowering the standardised mean pain scores at one hour when compared to remifentanil (PCA) analgesia (SMD 0.57, 95% CI 0.31 to 0.84; I2 = 0%, Analysis 3.16, fixed‐effect model SMD 0.57, 95% CI 0.31 to 0.84, Table 14). 'Risk of bias' assessment resulted in three trials assessed as high risk for attrition bias (Douma 2011; Stourac 2014; Tveit 2012). The effect estimate was robust with respect to sensitivity analysis (Table 9). All trials were judged as high risk of bias for blinding (Table 8). The OIS was estimated at 458 women using optimal information size considerations anticipating a minimal clinically relevant difference of 1 cm (VAS 0 to 10 cm) and a control mean pain score of 2.3 cm (Table 12). Including all six trials with 235 women, independent of the 'Risk of bias' assessment, sufficient information was not available to confirm evidence of effect for epidural analgesia to decrease pain intensity when compared to remifentanil (PCA).
3.16. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 16 Pain intensity 'early' (1 h).
We graded the quality of evidence for the outcome 'pain score 'early'' as 'low' (downgrade for quality and downgrade for imprecision; Table 3).
Pain intensity (pain score 'late' at two hours)
Four trials with 143 women reported on pain intensity at two hours after onset of analgesia (Douma 2011; Douma 2015; Stocki 2014; Tveit 2012). In a random‐effects meta‐analysis epidural analgesia was associated with a strong effect on pain reduction when compared to remifentanil (PCA) (SMD 1.46, 95% CI 0.66 to 2.26; I2 = 71%, Analysis 3.17). There was substantial statistical heterogeneity in the analysis (I2 = 71%). Excluding Douma 2011 decreased the heterogeneity to 0% without clinical explanation.
3.17. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 17 Pain intensity 'late' (2 h).
Additional analgesia required (escape analgesia)
Six studies including 1037 women (543 remifentanil, 494 epidural) offered participants the possibility on request to cross‐over to the other treatment arm and provided data suitable for meta‐analysis (Douma 2011; Douma 2015; Evron 2008; Freeman 2015; Stocki 2014; Tveit 2012). Overall, in all trials the risk for women in the remifentanil (PCA) group was remarkably higher to cross‐over to the epidural than the other way around in a random‐effects meta‐analysis (RR 8.10, 95% CI 3.50 to 18.75; I2 = 0%, Analysis 3.18, fixed‐effect model RR 10.86, 95% CI 4.37 to 26.95, Table 14). One trial (Evron 2008) reported zero events in both arm and two trials reported zero events in the epidural group (Douma 2011; Tveit 2012); the estimated effect was robust when random‐effects meta‐analysis was performed by using the TSA software, which allows a constant continuity correction of 0.01 for zero event handling in both arms (RR 9.27, 95% CI 3.73 to 23.03, Table 15). 'Risk of bias' assessment for this outcome resulted in one trial assessed as high risk of selection and attrition bias (Freeman 2015); the estimated effect was robust with respect to corresponding sensitivity analyses (RR 5.29, 95% CI 1.2 to 23.3, Table 7; Table 9). All trials were assessed as high risk of bias for blinding (Table 8). Trial sequential analysis on all six trials, independent of the 'Risk of bias' assessment, showed that with 'low risk of bias'‐based and with 'empirical' assumptions the RIS was 449 (Table 10) and 394 participants (Table 11), respectively. In both cases the RIS was crossed with 1037 women indicating that sufficient information was retained to confirm evidence of effect for remifentanil (PCA) to be associated with a higher risk to cross‐over to the epidural group compared to cross‐over in the opposite direction.
3.18. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 18 Additional analgesia required.
We graded the quality of evidence for the outcome 'Additional analgesia required (escape analgesia)' as 'moderate' (downgrade for quality; Table 3).
Rate of caesarean delivery
Nine trials with 1578 women (570 remifentanil, 1008 epidural/CSE) provided data on the rates of caesarean delivery (Douma 2011; Douma 2015; El‐Kerdawy 2010; Evron 2008; Ismail 2012; Stocki 2014; Stourac 2014; Tveit 2012; Volmanen 2008). Random‐effects meta‐analysis revealed no difference in the risk for caesarean delivery associated with both interventions (RR 0.99, 95% CI 0.81 to 1.21; I2 = 0%, Analysis 3.19, fixed‐effect model RR 0.96, 95% CI 0.79 to 1.18, Table 14). One trial reported zero events in the remifentanil (PCA) group (Stocki 2014). The estimated effect was robust when using a constant continuity correction of 0.01 to handle zero event trials (RR 1.0, 95% CI 0.82 to 1.22, Table 15). 'Risk of bias' assessment resulted in eight trials judged as high risk for blinding (Douma 2011; Douma 2015; El‐Kerdawy 2010; Evron 2008; Ismail 2012; Stocki 2014; Stourac 2014; Tveit 2012) and five trials as high risk for attrition bias (Douma 2011; Douma 2015; Stourac 2014; Tveit 2012; Volmanen 2008). The estimated effect was robust with respect to all sensitivity analyses performed (blinding: RR 0.88 95% CI 0.06 to 13.14 (Table 8); attrition bias: RR 1.02 95% CI 0.83 to 1.25 (Table 9). Trial sequential analysis on all trials, independent of the 'Risk of bias' assessment, showed that with 'clinically relevant' assumptions, the RIS was calculated to be at 924 participants (Table 10). The RIS was crossed with 1578 women indicating that sufficient information was retained to confirm lack of effect for remifentanil (PCA) to increase or decrease the rate of caesarean deliveries compared to an epidural.
3.19. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 19 Rate of caesarean delivery.
We graded the quality of evidence for the outcome 'rate of caesarean delivery' as 'moderate' (downgrade for quality; Table 3).
Rate of assisted birth
Eight trials with 1550 women (557 remifentanil, 993 epidural/CSE) reported on the rate of assisted birth; one trial reported on forceps delivery (Evron 2008), one on ventouse and forceps delivery (Tveit 2012), two on vacuum extraction (Stocki 2014; Volmanen 2008), and four on non‐defined instrumental delivery (Douma 2011; Douma 2015; El‐Kerdawy 2010; Ismail 2012). Random‐effects meta‐analysis showed no evidence of effect for remifentanil (PCA) to decrease the risk for assisted birth when compared to the epidural/CSE group (RR 0.92, 95% CI 0.66 to 1.26; I2 = 0%, Analysis 3.20). One trial reported zero events in the remifentanil (PCA) group (El‐Kerdawy 2010). The estimated effect was robust when using a constant continuity correction of 0.01 to handle zero event trials (RR 0.94, 95% CI 0.68 to 1.30, Table 15).
3.20. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 20 Rate of assisted birth.
Augmented labour
Six trials including 1379 women analysed augmentation of labour by use of oxytocin (Douma 2011; Douma 2015; Ismail 2012; Stocki 2014; Tveit 2012; Volmanen 2008). The pooled meta‐analysis revealed no significant effect for remifentanil (PCA) to lower the risk for augmentation of labour when compared with epidural/CSE (RR 0.91, 95% CI 0.82 to 1.02; I2 = 0%, Analysis 3.21). However, the CI and the distribution of the studies in the forest plot revealed at least a good chance for a reduced risk of labour augmentation in women with remifentanil (PCA).
3.21. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 21 Augmented labour.
Umbilical cord base excess (artery)
Three trials with 75 participants reported on umbilical cord base excess (artery) (Douma 2011; Stocki 2014; Tveit 2012). The normal range of arterial cord blood base excess is defined as ‐8.6 to ‐2.6 mmol/L (base deficit: 2.6 to 8.6 mmol/L) (Victory 2004). Only one trial reported a mean base deficit outside the range (Douma 2011; remifentanil (PCA): 11.1 mmol/L, epidural: 8.8 mmol/L); all other reported data were within normal limits. Random‐effects meta‐analysis revealed a larger mean base deficit under remifentanil (PCA) when compared to epidural analgesia (MD ‐0.97, 95% CI ‐2.65 to 0.72; I2 = 29%, Analysis 3.22).
3.22. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 22 Umbilical cord base excess (artery).
Umbilical cord base excess (venous)
Two trials with 129 women provided data on umbilical cord base excess (venous) (Douma 2015; Tveit 2012). The normal range of venous cord blood base excess is defined as ‐6.9 to ‐2.1 mmol/L (base deficit: 2.1 to 6.9 mmol/L) (Victory 2004). All reported mean base deficits were in the normal range. Random‐effects meta‐analysis revealed no difference in the mean base deficit under remifentanil (PCA) when compared to epidural analgesia with substantial statistical heterogeneity (MD ‐0.05, 95% CI ‐2.39 to 2.30; I2 = 74%, Analysis 3.23).
3.23. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 23 Umbilical cord base excess (venous).
Umbilical cord pH (artery)
Five trials including 1245 women reported on umbilical cord pH (artery) (Douma 2011; El‐Kerdawy 2010; Ismail 2012; Stocki 2014; Tveit 2012). The normal range of arterial cord blood pH is defined as 7.17 to 7.31 (Victory 2004). Only one trial reported a mean cord blood pH outside the range (Douma 2011; remifentanil (PCA): 7.14); all other reported data lay in between. Random‐effects meta‐analysis revealed lower mean pH values under remifentanil (PCA) when compared to epidural (MD ‐0.01, 95% CI ‐0.02 to ‐0.00; I2 = 0%, Analysis 3.24). However, the upper CI reached the line of no effect and a mean difference of ‐0.01 was not considered as clinically relevant.
3.24. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 24 Umbilical cord pH (artery).
Umbilical cord pH (venous)
Four trials including 1299 women provided data on umbilical cord pH (venous) (Douma 2015; El‐Kerdawy 2010; Ismail 2012; Tveit 2012). The normal range of venous cord blood pH is defined as 7.27 to 7.39 (Victory 2004). Only one trial reported mean cord pH values outside the range (Douma 2011; remifentanil (PCA): 7.23, epidural: 7.21); all other reported data lay in between. Random‐effects meta‐analysis revealed no evidence of effect that mean pH values were higher under remifentanil (PCA) when compared to epidural analgesia (MD 0.01, 95% CI ‐0.01 to 0.02; I2 = 57%, Analysis 3.25). There is substantial statistical heterogeneity (I2 = 57%).
3.25. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 25 Umbilical cord pH (venous).
Need for neonatal resuscitation
Two trials with 69 newborns reported on neonatal resuscitation with either mechanical ventilation (El‐Kerdawy 2010) or manual ventilation (Stocki 2014). A random‐effects meta‐analysis showed no difference in the risk for neonatal resuscitation between remifentanil (PCA) and epidural analgesia (RR 1.02, 95% CI 0.04 to 25.09; I2 = 57%, Analysis 3.26). There is substantial statistical heterogeneity in the analysis (I2 = 57%). Two trials reported zero events in either the remifentanil (PCA) group (El‐Kerdawy 2010) or the epidural group (Stocki 2014). The estimated effect was robust when using a constant continuity correction of 0.01 to handle zero event trials; however, the I2 was reduced to 0% (RR 1.03, 95% CI 0.00 to 3.4E8; I2 = 0%, Table 15).
3.26. Analysis.
Comparison 3 Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE), Outcome 26 Neonatal resuscitation.
Remifentanil (PCA) compared to remifentanil (continuous IV)
Two trials compared remifentanil (PCA) to remifentanil (continuous IV) (Khooshideh 2015; Shen 2013).
Primary outcomes
Satisfaction with pain relief
None of the trials reported data on overall satisfaction with pain relief which were suitable for quantitative meta‐analysis of the present review.
Adverse events for women
We could not identify any studies reporting on 'apnoea'.
Respiratory depression
Two trials with 135 participants provided data on respiratory depression defined as a respiratory rate of less than eight breaths/minute (Khooshideh 2015; Shen 2013). Both trials reported that none of the participants in either group had a respiratory depression during the study period (Analysis 4.1); the pooled effect could be estimated by using the TSA software, which allows a constant continuity correction of 0.01 for zero event handling in both arms (RR 0.98, 95% CI 0.00 to 1.0E12, I2 = 0%, Table 15). Both trials were assessed as low or unclear risk of selection bias; one trial was at high risk of bias for blinding (Khooshideh 2015); and the other trial was assessed as high risk of attrition bias (Shen 2013); the estimated effect was robust with respect to all sensitivity analyses performed (Table 8; Table 9). Trial sequential analysis on both trials, independent of the 'Risk of bias' assessment, showed that with 'low risk of bias'‐based and with 'empirical' assumptions the RIS was 3.4 E6 (Table 10) and 1.0 E7 participants (Table 11), respectively. The RIS was not reached and the TSMB were not crossed indicating that insufficient information was retained to confirm evidence of no effect.
4.1. Analysis.
Comparison 4 Remifentanil (PCA) versus remifentanil (continuous IV), Outcome 1 Respiratory depression (< 8 breaths/min).
We graded the quality of evidence for the outcome 'respiratory depression' as 'low' (downgrade for quality, downgrade for imprecision; Table 4).
Oxygen desaturation (SpO2 < 95%)
One trial with 53 women assessed oxygen desaturation defined as SpO2 < 95% (Shen 2013). Three out of 27 women in the remifentanil (PCA) group and five out of 26 women in the remifentanil (continuous IV) group had an oxygen saturation below 95% (P = 0.659) (Analysis 4.2).
4.2. Analysis.
Comparison 4 Remifentanil (PCA) versus remifentanil (continuous IV), Outcome 2 Oxygen desaturation (SpO2 < 95%).
Hypotension
Two trials with 135 women reported on hypotension defined either as a systolic blood pressure of less than 90 mm Hg (Khooshideh 2015) or without definition (Shen 2013). Both trials reported that none of the women in either group had hypotension during the study period (Analysis 4.3). The pooled effect could be estimated by using the TSA software, which allows a constant continuity correction of 0.01 for zero event handling in both arms which yielded an unreliably wide CI (RR 0.98, 95% CI 0.00 to 1.0E12, I2 = 0%, Table 15). Both trials were assessed as low or unclear risk of selection bias; one trial was at high risk of bias for blinding (Khooshideh 2015); and the other trial was assessed as high risk of attrition bias (Shen 2013); the estimated effect was robust with respect to all sensitivity analyses performed (Table 8; Table 9).
4.3. Analysis.
Comparison 4 Remifentanil (PCA) versus remifentanil (continuous IV), Outcome 3 Hypotension.
Bradycardia
Two trials including 135 women reported on bradycardia defined as a heart rate of less than 50 beats/minute (Khooshideh 2015) or without definition (Shen 2013). Both trials reported that none of the participants in either group suffered from bradycardia during the study period (Analysis 4.4). The pooled effect could be estimated by using the TSA software, which allows a constant continuity correction of 0.01 for zero event handling in both arms which yielded an unreliably wide CI (RR 0.98, 95% CI 0.00 to 1.0E12, I2 = 0%, Table 15). Both trials were assessed as low or unclear risk of selection bias; one trial was at high risk of bias for blinding (Khooshideh 2015); and the other trial was assessed as high risk of attrition bias (Shen 2013); the estimated effect was robust with respect to all sensitivity analyses performed (Table 8; Table 9).
4.4. Analysis.
Comparison 4 Remifentanil (PCA) versus remifentanil (continuous IV), Outcome 4 Bradycardia.
Nausea (and vomiting)
Two studies with 135 women provided data on either nausea (Shen 2013) or (combined) nausea and vomiting (Khooshideh 2015). Random‐effects meta‐analysis revealed no evidence of effect for remifentanil (PCA) to reduce the risk for nausea (and vomiting) in women compared to remifentanil (continuous IV) (RR 0.85, 95% CI 0.28 to 2.54; I2 = 45%, Analysis 4.5). Both trials were assessed as low or unclear risk of selection bias; one trial was at high risk of bias for blinding (Khooshideh 2015); and the other trial was assessed as high risk of attrition bias (Shen 2013); the estimated effect was not robust with respect to the sensitivity analyses performed (blinding: RR 0.53, 95% CI 0.21 to 1.39, Table 8; attrition bias: RR 1.67, 95% CI 0.43 to 6.52, Table 9).
4.5. Analysis.
Comparison 4 Remifentanil (PCA) versus remifentanil (continuous IV), Outcome 5 Nausea (and vomiting).
Pruritus
One trial including 53 women assessed the risk of pruritus (Shen 2013). There was no difference in the occurrence of pruritus between remifentanil (PCA) (one out of 27 women) compared to remifentanil (continuous IV) (two out of 26 women) (P = 0.973) (Analysis 4.6).
4.6. Analysis.
Comparison 4 Remifentanil (PCA) versus remifentanil (continuous IV), Outcome 6 Pruritus.
Sedation
One trial including 53 women reported on sedation scores one hour after onset of analgesia (Shen 2013). The median Ramsay sedation score with IQR in both groups was reported as 3 (3 ‐ 3) (P = 0.573) (Analysis 4.7).
4.7. Analysis.
Comparison 4 Remifentanil (PCA) versus remifentanil (continuous IV), Outcome 7 Sedation (1 h).
Adverse events for the newborn
We could not identify any studies reporting on the outcomes 'Apgar score ≤ seven at five minutes', 'Apgar score at five minutes', and 'NACS'.
Need for naloxone
Two trials with 135 women reported on the rate of newborns with need for naloxone (Khooshideh 2015; Shen 2013). Both trials reported that none of the newborns in either group required naloxone (Analysis 4.8). Tthe pooled effect could be estimated by using the TSA software, which allows a constant continuity correction of 0.01 for zero event handling in both arms which yielded an unreliably wide CI (RR 0.98, 95% CI 0.00 to 1.0E12, I2 = 0%, Table 15). Both trials were assessed as low or unclear risk of selection bias; one trial was at high risk of bias for blinding (Khooshideh 2015); and the other trial was assessed as high risk of attrition bias (Shen 2013); the estimated effect was robust with respect to all sensitivity analyses performed (Table 8; Table 9).
4.8. Analysis.
Comparison 4 Remifentanil (PCA) versus remifentanil (continuous IV), Outcome 8 Need for naloxone.
FHR/CTG abnormalities, non‐reassuring fetal status
One trial with 53 newborns provided data on non‐reassuring fetal status (Shen 2013). Four cases in the remifentanil (PCA) group and five in the remifentanil (continuous IV) group had non‐reassuring FHR tracings (transient fetal bradycardia) (P = 0.950) (Analysis 4.9).
4.9. Analysis.
Comparison 4 Remifentanil (PCA) versus remifentanil (continuous IV), Outcome 9 FHR/CTG abnormalities, non‐reassuring fetal status.
Secondary outcomes
We could not identify any studies reporting on 'rate of caesarean delivery', 'rate of assisted birth', 'breastfeeding initiation', 'umbilical cord base excess/pH', and 'augmented labour'.
Pain intensity (pain score 'early' at one hour)
One trial reported on median pain scores (with IQR) at one hour after onset of analgesia (Shen 2013). Women in the remifentanil (continuous IV) group had higher pain scores (4 (3 ‐ 5), VAS 0 to 10 cm) compared to women in the remifentanil (PCA) group (3 (2 ‐ 4)) (P < 0.01) (Analysis 4.10). Because only one small trial assessed this outcome (very serious imprecision) with high risk of attrition bias which strongly limits the evidence, we graded the quality of the evidence as 'very low' (Table 4).
4.10. Analysis.
Comparison 4 Remifentanil (PCA) versus remifentanil (continuous IV), Outcome 10 Pain intensity 'early' (1 h).
Pain intensity (pain score 'late' at two hours)
One trial with 53 women provided data on pain scores at two hours after onset of analgesia (Shen 2013). There was no significant difference in pain scores between the remifentanil (PCA) group (4 (3 ‐ 5), median (IQR), VAS 0 to 10 cm) and the remifentanil (continuous IV) group (5 (4 ‐ 6), P > 0.01) (Analysis 4.11).
4.11. Analysis.
Comparison 4 Remifentanil (PCA) versus remifentanil (continuous IV), Outcome 11 Pain intensity 'late' (2 h).
Additional analgesia required (escape analgesia)
One trial with 59 women reported on the rate of women requiring additional analgesia (Shen 2013). Two women in the remifentanil (PCA) group and four women in the remifentanil (continuous IV) group required an additional epidural because of inadequate analgesia (Analysis 4.12). Because only one small trial (very serious imprecision) with high risk of attrition bias assessed this outcome which strongly limits the evidence, we graded the quality of the evidence as 'very low' (Table 4).
4.12. Analysis.
Comparison 4 Remifentanil (PCA) versus remifentanil (continuous IV), Outcome 12 Additional analgesia required (escape analgesia).
Neonatal resuscitation
One study with 53 newborns assessed the outcome 'neonatal resuscitation' and reported that none of the newborns in either group required resuscitation (Analysis 4.13) (Shen 2013).
4.13. Analysis.
Comparison 4 Remifentanil (PCA) versus remifentanil (continuous IV), Outcome 13 Neonatal resuscitation.
Remifentanil (PCA, increasing bolus, fixed infusion dose) compared to remifentanil (PCA, increasing infusion, fixed bolus dose)
One trial compared remifentanil (PCA, IB (increasing bolus dose, fixed infusion dose)) to remifentanil (PCA, IF (increasing infusion dose, fixed bolus dose)) (Balki 2007).
Primary outcomes
Satisfaction with pain relief
One trial with 20 women reported on overall satisfaction with pain relief (Balki 2007); woman's satisfaction scores were similar in the remifentanil (PCA, IB) group (8.6 +/‐ 1.2, verbal numerical rating scale (VNRS) 0 to 10, mean +/‐ SD) and the remifentanil (PCA, IF) group (8.4 +/‐ 1.1) (P = 0.77) (Analysis 5.1). Because only one small trial assessed this outcome (very serious imprecision), which strongly limits the evidence, we graded the quality of the evidence as 'low' (Table 5).
5.1. Analysis.
Comparison 5 Remifentanil (PCA, increasing bolus, fixed infusion dose) versus remifentanil (PCA, increasing infusion, fixed bolus dose), Outcome 1 Satisfaction with pain relief.
Adverse events for women
We could not identify any study reporting on the outcome 'apnoea', 'respiratory depression', and 'sedation'.
Oxygen desaturation (SpO2 < 95%)
One trial with 20 women reported on oxygen desaturation defined as SpO2 < 95% (Balki 2007); six out of 10 women in the remifentanil (PCA, IB) group and four out of 10 women in the remifentanil (PCA, IF) group had oxygen saturation levels below 95% (P = 0.42) (Analysis 5.2).
5.2. Analysis.
Comparison 5 Remifentanil (PCA, increasing bolus, fixed infusion dose) versus remifentanil (PCA, increasing infusion, fixed bolus dose), Outcome 2 Oxygen desaturation (SpO2 < 95%).
Hypotension
One trial with 20 women reported on hypotension (Balki 2007); none of the women suffered from hypotension in both groups (Analysis 5.3).
5.3. Analysis.
Comparison 5 Remifentanil (PCA, increasing bolus, fixed infusion dose) versus remifentanil (PCA, increasing infusion, fixed bolus dose), Outcome 3 Hypotension.
Bradycardia
One trial with 20 women reported on bradycardia (Balki 2007); none of the women had bradycardia in both groups (Analysis 5.4).
5.4. Analysis.
Comparison 5 Remifentanil (PCA, increasing bolus, fixed infusion dose) versus remifentanil (PCA, increasing infusion, fixed bolus dose), Outcome 4 Bradycardia.
Nausea
One trial with 20 women reported on nausea (Balki 2007); six out of 10 women in the remifentanil (PCA, IB) group and two out of 10 women in the remifentanil (PCA, IF) group suffered from nausea (P = 0.095) (Analysis 5.5).
5.5. Analysis.
Comparison 5 Remifentanil (PCA, increasing bolus, fixed infusion dose) versus remifentanil (PCA, increasing infusion, fixed bolus dose), Outcome 5 Nausea.
Vomiting
One trial with 20 women reported on vomiting (Balki 2007); four out of 10 women in the remifentanil (PCA, IB) group and one out of 10 women in the remifentanil (PCA, IF) group vomited (P = 0.17) (Analysis 5.6).
5.6. Analysis.
Comparison 5 Remifentanil (PCA, increasing bolus, fixed infusion dose) versus remifentanil (PCA, increasing infusion, fixed bolus dose), Outcome 6 Vomiting.
Pruritus
One trial with 20 women reported on the occurrence of pruritus (Balki 2007); only one woman in the remifentanil (PCA, IB) group suffered from pruritus (P = 0.5) (Analysis 5.7).
5.7. Analysis.
Comparison 5 Remifentanil (PCA, increasing bolus, fixed infusion dose) versus remifentanil (PCA, increasing infusion, fixed bolus dose), Outcome 7 Pruritus.
Adverse events for the newborn
We could not identify any studies reporting on the outcomes 'Apgar score at five minutes' and 'NACS'.
Apgar score ≤ seven at five minutes
Balki 2007 reported that all 20 newborns had an Apgar score ≥ seven at five minutes (Analysis 5.8). Because only one small trial assessed this outcome (very serious imprecision), which strongly limits the evidence, we graded the quality of the evidence as 'low' (Table 5).
5.8. Analysis.
Comparison 5 Remifentanil (PCA, increasing bolus, fixed infusion dose) versus remifentanil (PCA, increasing infusion, fixed bolus dose), Outcome 8 Apgar score < 7 at 5 min.
Need for naloxone
One trial with 20 newborns reported on requirement for naloxone (Balki 2007); none of the newborns in either group required naloxone (Analysis 5.9).
5.9. Analysis.
Comparison 5 Remifentanil (PCA, increasing bolus, fixed infusion dose) versus remifentanil (PCA, increasing infusion, fixed bolus dose), Outcome 9 Need for naloxone.
FHR/CTG abnormalities, non‐reassuring fetal status
One trial with 20 women reported on non‐reassuring FHR (Balki 2007); two out of 10 newborns in the remifentanil (PCA, IB) group and one out of 10 newborns in the remifentanil (PCA, IF) group showed non‐reassuring FHR traces (P = 0.61) (Analysis 5.10).
5.10. Analysis.
Comparison 5 Remifentanil (PCA, increasing bolus, fixed infusion dose) versus remifentanil (PCA, increasing infusion, fixed bolus dose), Outcome 10 FHR/CTG abnormalities, non‐reassuring fetal status.
Secondary outcomes
We could not identify any studies reporting on 'pain intensity (pain score 'early' at one hour)', 'pain intensity (pain score 'late' at two hours)', 'rate of assisted birth', and 'breastfeeding initiation'.
Additional analgesia required (escape analgesia)
One trial with 20 women reported on the need for an additional epidural (Balki 2007); only one woman in the remifentanil (PCA, IF) group crossed over to the epidural (Analysis 5.11). Because only one small trial assessed this outcome (very serious imprecision), which strongly limits the evidence, we graded the quality of the evidence as 'low' (Table 5).
5.11. Analysis.
Comparison 5 Remifentanil (PCA, increasing bolus, fixed infusion dose) versus remifentanil (PCA, increasing infusion, fixed bolus dose), Outcome 11 Additional analgesia required (escape analgesia).
Rate of caesarean delivery
One trial with 20 women provided data on the rate of caesarean delivery (Balki 2007); four women in each group delivered by caesarean section (Analysis 5.12). Because only one small trial assessed this outcome (very serious imprecision), which strongly limits the evidence, we graded the quality of the evidence as 'low' (Table 5).
5.12. Analysis.
Comparison 5 Remifentanil (PCA, increasing bolus, fixed infusion dose) versus remifentanil (PCA, increasing infusion, fixed bolus dose), Outcome 12 Rate of caesarean delivery.
Augmented labour
One trial with 20 women reported on augmentation of labour (Balki 2007); three out of 10 women in the remifentanil (PCA, IB) group and seven out of 10 women in the remifentanil (PCA, IF) group needed augmentation of labour (P = 0.14) (Analysis 5.13).
5.13. Analysis.
Comparison 5 Remifentanil (PCA, increasing bolus, fixed infusion dose) versus remifentanil (PCA, increasing infusion, fixed bolus dose), Outcome 13 Augmented labour.
Umbilical cord base excess (artery)
One trial with 20 women reported on umbilical cord base excess (artery) (Balki 2007); there were no differences between the remifentanil (PCA, IB) group (‐4.3 +/‐ 3.2 mmol/L) and the remifentanil (PCA, IF) group (‐4.6 +/‐ 2.0 mmol/L) and the mean values lay in normal ranges (Victory 2004) (P = 0.60) (Analysis 5.14).
5.14. Analysis.
Comparison 5 Remifentanil (PCA, increasing bolus, fixed infusion dose) versus remifentanil (PCA, increasing infusion, fixed bolus dose), Outcome 14 Umbilical cord base excess (artery).
Umbilical cord base excess (venous)
One trial with 20 women reported on umbilical cord base excess (venous) (Balki 2007); there were no differences between the remifentanil (PCA, IB) group (‐4.7 +/‐ 3.5 mmol/L) and the remifentanil (PCA, IF) group (‐4.1 +/‐ 2.3 mmol/L) and the mean values lay in normal ranges (Victory 2004) (P = 0.91) (Analysis 5.15).
5.15. Analysis.
Comparison 5 Remifentanil (PCA, increasing bolus, fixed infusion dose) versus remifentanil (PCA, increasing infusion, fixed bolus dose), Outcome 15 Umbilical cord base excess (venous).
Umbilical cord pH (artery)
One trial with 20 women reported on umbilical cord pH (artery) (Balki 2007); there were no differences between the remifentanil (PCA, IB) group (7.24 +/‐ 0.08) and the remifentanil (PCA, IF) group (7.25 +/‐ 0.05) and the mean values lay in normal ranges (Victory 2004) (P = 0.70) (Analysis 5.16).
5.16. Analysis.
Comparison 5 Remifentanil (PCA, increasing bolus, fixed infusion dose) versus remifentanil (PCA, increasing infusion, fixed bolus dose), Outcome 16 Umbilical cord pH (artery).
Umbilical cord pH (venous)
One trial with 20 women reported on umbilical cord pH (venous) (Balki 2007); there were no differences between the remifentanil (PCA, IB) group (7.27 +/‐ 0.08) and the remifentanil (PCA, IF) group (7.29 +/‐ 0.05) and the mean values lay in normal ranges (Victory 2004) (P = 0.92) (Analysis 5.17).
5.17. Analysis.
Comparison 5 Remifentanil (PCA, increasing bolus, fixed infusion dose) versus remifentanil (PCA, increasing infusion, fixed bolus dose), Outcome 17 Umbilical cord pH (venous).
Need for neonatal resuscitation
One trial with 20 newborns reported on the need for neonatal resuscitation (Balki 2007); one newborn in the remifentanil (PCA, IF) group had to be resuscitated (P = 0.50) (Analysis 5.18).
5.18. Analysis.
Comparison 5 Remifentanil (PCA, increasing bolus, fixed infusion dose) versus remifentanil (PCA, increasing infusion, fixed bolus dose), Outcome 18 Neonatal resuscitation.
Remifentanil (PCA) compared to nitrous oxide (or other forms of inhalational analgesia)
No trials were identified which compared remifentanil (PCA) to nitrous oxide (or other forms of inhalational analgesia).
Remifentanil (PCA) compared to placebo or no treatment
No trials were identified which compared remifentanil (PCA) to placebo or no treatment.
Discussion
Summary of main results
The present systematic review reveals several important findings about the administration of remifentanil (PCA) for labour analgesia when compared to different control interventions with respect to the general superiority or inferiority across all analysed outcomes. The results suggest that remifentanil (PCA) is superior to the administration of other opioids (IV/IM) or other opioids (PCA) and inferior to epidural or combined spinal‐epidural analgesia (CSE) with regard to the overall direction of estimated effects for most outcomes which are of interest for this review (Figure 4). However, there are outcome‐specific variations in the quality level of evidence (GRADE) ranging from 'very low' to 'moderate' by which the confidence in the estimated effects varies from 'very little confidence and the true effect is likely to be substantially different from the estimate of effect' to 'moderately confident that the true effect lies close the estimated effect'. In the case of the other comparators, namely remifentanil (continuous IV) and remifentanil (different administration mode) there is currently only a limited number of studies available for which reason we are not able to reliably estimate the direction of effects, which limits the quality of evidence (Figure 4). We could not identify any randomised controlled trial (RCT) eligible for inclusion that compares remifentanil (PCA) to either inhalational anaesthesia or placebo treatment. Therefore, this review does not provide reliable evidence for those comparisons.
For the main comparison of the current review, remifentanil (PCA) versus another opioid (IV/IM), we were able to include four studies and the quality levels of evidence for GRADE‐relevant outcomes ranged from 'very low' to 'moderate' (Table 1). Satisfaction with pain relief was higher (SMD 2.11, 95% CI 0.72 to 3.49) and pain intensity 'early' was lower (SMD ‐1.58, 95% CI ‐2.69 to ‐0.48) in the remifentanil group compared to the other opioid (IV/IM) group. Superiority was clinically relevant in both cases with a SMD of 2.11 higher for satisfaction and 1.58 lower for pain intensity, which is equivalent to a range of 2.74 to 4.68 cm and 1.26 to 2.8 cm on a visual analogue scale (VAS) 0 to 10 cm scale, respectively. However, the quality of evidence was 'very low' for both satisfaction and pain intensity 'early'. Women in the remifentanil group had a reduced requirement for additional analgesia (RR 0.57, 95% CI 0.40 to 0.81) with moderate‐quality evidence. From the meta‐analysis for the risk of caesarean delivery, there was no evidence of effect for remifentanil (PCA) to reduce the risk for caesarean section (RR 0.63, 95% CI 0.30 to 1.32, constant continuity correction (ccc) = 0.01). Quality of evidence for rate of caesarean section was graded as 'low'. Sparse data (one study) were available describing adverse events for women as well as for newborns. In one trial of remifentanil (PCA) versus another opioid (IM), three out of 18 women in the remifentanil and none out of 18 in the control group had a respiratory depression. Another trial of remifentanil (PCA) compared to another opioid (IV) reported the risk for newborns with Apgar scores less than seven at five minutes with zero events in both study arms and no reliable conclusion could be reached. Quality of evidence was graded as 'very low' for both 'maternal respiratory depression' and 'Apgar score less than seven at five minutes'; no study investigating the comparison of interest reported on the risk for apnoea associated with both interventions.
For the second comparison remifentanil (PCA) versus another opioid (PCA), we included three trials and the quality levels of evidence for the GRADE‐relevant outcomes were graded as 'very low' or 'low' (Table 2). For the outcome satisfaction with pain relief we identified only one relevant study reporting higher satisfaction under remifentanil (PCA) and the quality of evidence was graded as 'very low'. Pain intensity 'early' was lower in the remifentanil (PCA) group when compared to the other opioid (PCA) group (SMD ‐0.51, 95% CI ‐1.01 to ‐0.00). The effect was equivalent to a range of 1.13 to 1.46 cm on a VAS 0 to 10 cm scale and was assessed as clinically relevant. However, the quality of evidence was graded as 'very low'. There is no evidence of effect that remifentanil (PCA) reduced the requirements for additional analgesia when compared to other opioids (PCA) (RR 0.76, 95% CI 0.45 to 1.28). Quality of evidence was graded as 'low'. The meta‐analysis on the risk of caesarean delivery suggested that remifentanil (PCA) strongly increased the risk for caesarean sections compared to other opioids (PCA) (RR 2.78, 95% CI 0.99 to 7.82). However, the 95% CI crossed the line of no effect (RR = 1). Quality of evidence for rate of caesarean delivery was graded as 'very low'. There were very few data available on adverse events for women and newborns. No study could be identified that investigated 'apnoea' or 'maternal respiratory depression' in labouring women for this comparison. Only one trial analysed 'Apgar score less than seven at five minutes' and reported that three out of eight newborns in the pethidine (PCA) group and none in the remifentanil (PCA) group had an Apgar score of less than seven at five minutes. Quality of evidence was graded as 'very low'.
Ten trials were included in the comparison of remifentanil (PCA) versus epidural/CSE, which is the highest number of identified trials for a comparison in the current review. The quality of evidence for GRADE‐relevant outcomes ranged from 'very low' to 'moderate' (Table 3). Satisfaction with pain relief was lower (SMD ‐0.22, 95% CI ‐0.40 to ‐0.04) and pain intensity 'early' was higher (SMD 0.57, 95% CI 0.31 to 0.84) in the remifentanil (PCA) group compared to the epidural/CSE group with a quality of evidence level of 'very low' and 'low', respectively. A SMD of 0.22 lower for satisfaction and 0.57 higher for pain intensity is equivalent to a range of 0.15 to 0.61 cm and 0.57 to 1.43 cm on a VAS 0 to 10 cm scale, respectively, which can be regarded as moderate effects at best. Women using epidural/CSE for pain relief seemed to profit longer from analgesia compared to women in the remifentanil (PCA) group since pain intensity 'late' was lower in the control group with a SMD of 1.46 (95% CI 0.66 to 2.26), which is equivalent to a range of 1.9 to 4.1 cm on a VAS 0 to 10 cm scale. Moreover, women in the remifentanil (PCA) group had a strongly increased risk for requirement of additional analgesia (escape analgesia) (RR 9.27, 95% CI 3.73 to 23.03, ccc = 0.01) moderate‐quality evidence. There was evidence of no effect for remifentanil to increase or decrease the risk for caesarean delivery when compared to epidural/CSE (RR 1.0, 95% CI 0.82 to 1.22, ccc = 0.01), and we graded the quality of evidence as 'moderate'. For the GRADE‐relevant outcomes relevant to adverse events for women, maternal apnoea and respiratory depression, we could only include one and three trials, respectively. The only available study that investigated the risk for apnoea reported that half of the women in the remifentanil and none in the epidural group had an apnoea. The quality of evidence was graded as 'very low' for apnoea. For the risk of maternal respiratory depression, when trials that reported zero events in both arms were included, there was no difference between both interventions (RR 0.91, 95% CI 0.51 to 1.62, ccc = 0.01). We graded the quality of evidence as 'low' for respiratory depression. For the outcomes 'oxygen desaturation', 'nausea', 'vomiting', and 'sedation' epidural/CSE was found to be superior to remifentanil (PCA); whereas for 'hypotension', when all trials with zero events in both arms were included, remifentanil (PCA) was superior to epidural/CSE; for 'bradycardia' and 'pruritus', there was no evidence of effect for one of the two treatment alternatives. For all outcomes relevant to assess the adverse events on newborns associated with the interventions, there was no significant difference between remifentanil (PCA) and epidural/CSE detectable. However, we graded the quality of evidence for the estimated effect on 'Apgar score less than seven at five minutes' (RR 1.26, 95% CI 0.62 to 2.57, ccc = 0.01) as 'low'.
For the comparison remifentanil (PCA) versus remifentanil (continuous IV), we identified two relevant trials and the quality levels of evidence for the GRADE‐relevant outcomes were 'very low' or 'low' (Table 4). No trial reported on satisfaction with pain relief and only one trial provided data on both pain intensity 'early' (less pain in the remifentanil (PCA) group) and 'additional analgesia' (more women required additional analgesia in the remifentanil (continuous IV) group). For the last two outcomes the quality of evidence was graded as 'very low'. Sparse data were available describing adverse events for women as well as for newborns. No study investigating the comparison of interest reported on the risk for apnoea for women or risk for newborns to have an Apgar score less than seven at five minutes associated with both interventions. There is no difference in the risk for maternal respiratory depression between both interventions when all trials including those with zero events in both arms were meta‐analysed (RR 0.98, 95% CI 0.00 to 1.0E12, ccc = 0.01). Quality of evidence was graded as 'low' for 'respiratory depression'. For the outcomes 'hypotension', 'bradycardia', 'nausea and vomiting', and 'need for naloxone' neither of these interventions could be identified as being superior to the other.
The comparison remifentanil (PCA with increasing bolus dose) versus remifentanil (PCA with increasing infusion dose) was reported by only one small trial that suggested remifentanil (PCA with increasing infusion dose) to be associated with fewer side effects for women. Nevertheless, the quality level of evidence was 'low' for the reported outcomes 'satisfaction', 'additional analgesia', 'rate of caesarean delivery', and 'Apgar scores less than seven at five minutes' (Table 5).
Overall completeness and applicability of evidence
The investigated groups of participants were relatively homogeneous. High‐risk parturients and pregnancies were excluded in all studies except one (women with pre‐eclampsia, El‐Kerdawy 2010) that were examined in the current review. Women had to be healthy (without systemic or serious diseases) and had to have an uncomplicated cephalic presentation. Pregnancies were all at full term with the exception of two studies that included women from 32 weeks of gestation (El‐Kerdawy 2010; Freeman 2015). Thus, results can be adapted to low‐risk, but not equally to high‐risk groups which we also planned to analyse.
Most studies were conducted in Europe (11 trials) or the Middle East (six trials). All other geographical regions were underrepresented. Results may differ across the globe due to various clinical standards and settings. More studies in other parts of the world have to be carried out to detect differences or similarities regarding outcomes with remifentanil (PCA).
In half of all studies remifentanil PCA was compared to epidural analgesia so that this comparison group provided reliable results at least for some outcomes. For other interventions results have to be considered with caution because only a few studies investigated alternatives for labour analgesia.
Furthermore, all included trials displayed many differences regarding the conduct of their studies, despite investigating the same intervention. When analysing these and comparing the conclusions it has to be taken into account that ‘remifentanil (PCA)’ may not be ‘remifentanil (PCA)’ due to widely differing dosing regimen. Bolus applications, lockout times and also concomitant medications (e.g. Entonox) varied across all studies. The discrepancies may appear small since several trials have investigated regimens for remifentanil application (e.g. dose‐finding studies) which functioned as guidance for the studies included in the current review. Nevertheless, there was heterogeneity in conducting the studies that cannot be neglected. No general conclusion can be drawn and further studies with comparable designs have to be conducted. It seems essential in order to obtain more reliable effect estimates, to include at least a core set of outcomes of interest for efficacy and harm (GRADE‐relevant outcomes), while concomitantly looking for more sophisticated study‐endpoints.
Quality of the evidence
The body of evidence identified by this review is based on 20 studies with 3569 participating women. Fifty per cent (10 studies: 2983 participants) of the identified trials compared remifentanil (PCA) to epidural/CSE, 20% (four studies: 216 parturients) to another opioid (IV/IM), 15% (three studies: 215 parturients) to another opioid (PCA), 10% (two studies: 135 parturients) to remifentanil (IV), and 5% (one study: 20 parturients) to remifentanil (different administration regimen). No trials were identified that analysed remifentanil (PCA) versus inhalational analgesia other than in a cross‐over approach or placebo/no treatment. Trial sequential analysis (TSA) and optimal information size (OIS) considerations for all analysable GRADE‐relevant outcomes across all comparisons identified five out of 14 outcomes ('1.1 Satisfaction with pain relief', '1.7 Additional analgesia required', '3.1 Satisfaction with pain relief', '3.17 Additional analgesia required', '3.18 Rate of caesarean delivery') for which sufficient information was obtained based on the assumptions made in the current review. For the remaining nine outcomes 13% to 95% of information is still lacking and, therefore, effect as well as lack of effect cannot be excluded.
From a strict methodological point of view the majority of included studies must be considered of rather poor quality. However, looking at the challenges for trials in the labour setting, many attempts need to be acknowledged to achieve as much of the suggested quality criteria as possible to reduce the risk of bias. Especially, blinding and incomplete outcome data reporting are problematic aspects, for which 65% and 45% of the included studies, respectively, were judged to be at 'high risk of bias'. When looking at the rather poor quality assessments based on the lack of efficient blinding, it has to be taken into account that the high rate of unblinded or not efficiently blinded studies is mostly attributable to the different natures of the compared interventions (e.g. IV PCA application versus epidural) and, therefore, not a direct sign for poor study quality (even though there seems to be room for improvement; see Implications for research). Nonetheless, risk of bias is present in those studies and study results may be influenced by lack of efficient blinding. Selection bias plays a role for the largest study (Freeman 2015), because allocation concealment was uncovered for participants and personnel before the start of treatment. To take into account the limitations in study quality, we performed sensitivity analyses for selection bias, performance and detection bias, as well as for attrition bias and downgraded the quality of evidence for GRADE‐relevant outcomes by one level if substantial information was derived from studies at high risk of bias, and by two levels if there also was an impact on the robustness of the estimated effect.
Substantial statistical heterogeneity (I2 > 50%) between studies was detected in 11 out of 36 (30%) primary and GRADE‐relevant outcomes with at least two included trials across all comparisons. As only one of those outcomes included seven studies and all other outcomes included ≤ four studies, we decided not to perform subgroup analyses as an attempt to explain heterogeneity because we wanted to avoid spurious findings. In all cases of substantial statistical heterogeneity of GRADE‐relevant outcomes we downgraded the quality of evidence for inconsistency.
Publication bias could not be investigated in the current version of this review since the largest number of studies included in a single outcome was nine ('rate of caesarean delivery'). The predefined requirement in the protocol to perform further analysis on publication bias and small‐study effects was a number of at least 10 studies per outcome. For future updates, if the number of included studies is increased, we will analyse publication bias with funnel plots and regression tests.
Potential biases in the review process
This review was performed according to procedures described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). In addition to the search of the Cochrane Pregnancy and Childbirth Group’s Trials Register, we searched ClinicalTrials.gov, the WHO International Clinical Trials Registry Platform (ICTRP) and congress proceedings for unpublished, planned and ongoing trial reports and abstracts. We contacted authors with published study protocols and asked for the actual status and if there were data available for inclusion in the current review. Therefore, we can be confident that all trials that fit our criteria were identified.
All processes in the review were checked twice by two independent authors. In case of disagreement, a third and fourth review author were involved. The author review group consists of several experts in the field (PK, LE, AA, NP) who are in contact with those performing clinical research in the field. The two authors who were responsible for independent data extraction and critical appraisal (SW, YJ) come from various areas of research which are not directly related to interventions of interest (research associate, physician), whereby potential prejudice was minimised. All authors were not blinded regarding authorship of the included trials.
If there were missing outcome data in the trial without reasons declared, we did not contact the authors for further information (e.g. reasons for missing outcome data) since we wanted to prevent reporting bias. We just used published outcome data. Contact with authors was made in case of unknown sample sizes (e.g. sample size was not reported for satisfaction or pain scores at one hour or two hours).
Several studies reported their data as median and interquartile ranges rather than as mean and standard deviation. We included these data if they were symmetric and converted them to mean and standard deviation by using the calculation described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
Some data had to be extracted graphically (satisfaction, pain scores) and were checked independently by two review authors for correctness to ensure that deviations from actual findings were minimised.
At the protocol stage we did not plan to perform trial sequential analysis (TSA) and optimal information size (OIS) considerations to calculate the required information size (RIS) or OIS, respectively. However, we believe that those considerations help us to more reliably assess the quality of the evidence, especially in view of rather limited numbers of trials and participants which introduce a risk for spurious findings in the meta‐analyses. Therefore, we have incorporated the TSA and OIS approach into the assessment of 'imprecision' (GRADE). Since the assumptions for TSA and OIS calculations were made in a post‐hoc manner, we adopted the assumptions from the pooled estimates obtained from either 'low risk of bias' trials or all meta‐analysed trials ('empirical'). The assumptions may not perfectly meet the clinical practice and relevant differences in outcomes in every case, and occasionally may take into account a too large difference between the groups which does not match clinical experience. However, we considered it to be the most objective approach to set the basic conditions, especially in view of the fact that we retrospectively decided to include measures to assess the OIS.
Moreover, TSA or OIS considerations cannot consider risks of bias, wherefore trials at 'high risk of bias' should ideally not be included in the analyses. Due to the limited number of studies in this review in general and with a high proportion of studies at 'high risk of bias' (mainly performance, detection, and attrition bias), we decided to include all available studies independent of their risks of bias in the meta‐analysis. Furthermore, TSA in this review was based on assumptions gained from either all 'low risk of bias' studies or from the 'best' study (overall risk of bias) if no 'low risk of bias' study was available, and from all studies ('empirical') available for the respective outcome. Therefore, assumptions may itself be affected by bias and it is possible that smaller intervention effects may be more realistic whereby the required information sizes would be increased. These are undoubtedly limitations of the current review.
In the current review, we included all studies into meta‐analyses even if they had reported zero events in both arms. By inclusion of those studies we wanted to avoid creating a risk of inflating the magnitude of the pooled effect. The inclusion of zero total event trials enabled the estimation of a pooled effect by using the TSA software for six outcomes ('1.4 Pruritus', '3.6 Bradycardia', '4.1 Respiratory depression', '4.2 Hypotension', '4.3 Bradycardia', '4.5 Need for naloxone'), which were not estimable using Review Manager 5 (RevMan 2014). For two outcomes, either the 95% CI and the P value ('3.5 Hypotension') or the direction of the estimated effect ('3.4 Respiratory depression') were noticeably changed by inclusion of zero total event trials.
Agreements and disagreements with other studies or reviews
There are five other systematic reviews with or without meta‐analysis dealing with remifentanil for labour analgesia which have been published up to March 2016 (Leong 2011; Liu 2014; Schnabel 2011; Stourac 2016; Van de Velde 2015).
Leong 2011 searched five databases (MEDLINE, CINAHL, Embase, CENTRAL and Maternity and Infant Care databases) and handsearched from 1998 to 2010 for RCTs with women in labour comparing remifentanil (patient‐controlled or physician‐controlled) with meperidine (IM, IV or PCA). In contrast to the current review, no distinction was made regarding the way of remifentanil or meperidine administration. Reduction in pain scores was selected as the primary outcome (VAS 0 to 100 mm). Further outcomes were maternal side effects (sedation, oxygen desaturation, bradypnoea) and effects on the neonate (Apgar scores, umbilical cord pH, neurologic and adaptive capacity score (NACS)). Seven studies with 349 women met the inclusion criteria and three studies with 233 participants were meta‐analysed. All studies except one (Shahriari 2007), which dealt with anaesthetist‐administered remifentanil were also included in the present review. As a result Leong and colleagues reported that remifentanil decreased the mean VAS score at one hour by 25 mm compared to meperidine. This corresponds with the current result (Table 1), which revealed a pain reduction of 1.26 cm to 2.8 cm on a VAS 0 to 10 cm scale at 30 minutes/one hour. Both Leong's and the present review could not draw definite conclusions with regard to maternal and neonatal side effects due to insufficient data. Leong and colleagues performed qualitative analysis while the current review conducted quantitative analysis. The authors also used the 'Risk of bias' assessment according to the Cochrane Handbook, therefore the current review contains a more critical judgement of 'Risk of bias'.
Liu 2014 performed a search in three databases (PubMed, Embase, and the Cochrane Library), as well as a handsearch until November 2012 for RCTs with women in labour comparing remifentanil (PCA) with epidural analgesia. The primary outcomes were pain scores at one and two hours. Nausea, vomiting, pruritus and umbilical cord artery pH values were defined as secondary outcomes. Five studies with 886 participants were included in qualitative and quantitative analyses, which were all the subject of the current review. The authors concluded that epidural analgesia led to greater pain relief than remifentanil (mean difference at one hour: 1.9 cm on a VAS 0 to 10 cm), but for secondary outcomes no definite results could be presented. The results on pain relief are more optimistic than the results in the present review with a range of pain increase from 0.57 cm to 1.43 cm on a VAS 0 to 10 cm when looking at remifentanil (PCA). The Cochrane 'Risk of bias' assessment as well as GRADE was performed. In these cases judgement of risk of bias as well as the quality of evidence was more critical in the current review.
Schnabel 2011 conducted a systematic search in two databases (the Cochrane Library and MEDLINE) until August 2011 for RCTs comparing remifentanil (PCA) with any other labour analgesia. The primary outcome was conversion to epidural analgesia. Pain scores after one hour were defined as secondary outcomes. Additionally, type of delivery, maternal satisfaction, and maternal and neonatal adverse events were examined. Twelve RCTs with 593 participants were included in the systematic review and 11 studies were subject to meta‐analyses. Two of the 12 trials were not included in the current review because of the cross‐over design (Volmanen 2005) and no patient‐controlled analgesia (Shahriari 2007). The authors drew the conclusion that remifentanil administration correlated with lower rates of conversion to epidural analgesia, lower mean pain scores at one hour (mean difference ‐2.17 cm) and higher satisfaction scores when compared to pethidine administration which was also shown in the current review. In comparison to epidural analgesia, remifentanil (PCA) was associated with higher pain scores after one hour (mean difference 1.89 cm), which is more optimistic than the results in the present review with a pain increase of 0.57 cm to 1.43 cm on a VAS 0 to 10 cm when looking at remifentanil (PCA). For all other outcomes mentioned above, no definite result could be found. Critical appraisal was made with the Oxford scale which has several drawbacks compared to the Cochrane 'Risk of bias' assessment tool.
Stourac 2016 searched four databases (US National Library of Medicine, PubMed, SCOPUS and Web of Science database) until December 2014 for RCTs that reported on remifentanil administration (PCA or continuous IV). There were 44 articles eligible and included in the review; 15 RCTs which reported VAS pain scores at 0 and one hours after the start of analgesia were analysed. Two of the 15 randomised trials were not included in the present review because of wrong intervention (PCA versus PCA, Balcioglu 2007) and cross‐over design (Volmanen 2005). Stourac's meta‐analysis revealed a significant decrease in VAS from 0 to one hour in the remifentanil group (summary fixed model ‐2.8). There was no comparison drawn between remifentanil and other interventions. There were no other outcomes meta‐analysed. It is not possible to make a point regarding agreements or disagreements with the current review.
Van de Velde 2015 found 36 studies which investigated remifentanil (PCA) for labour analgesia when performing a search in January 2015. No meta‐analysis was conducted, but results regarding analgesic efficacy, modalities of PCA delivery and maternal safety were qualitatively described. Three of the already mentioned reviews were included (Leong 2011; Liu 2014; Schnabel 2011) together with two RCTs (Shen 2013; Stocki 2014) which were also analysed in the present review. It was concluded that remifentanil PCA provided better pain relief than other opioids but was inferior to epidural analgesia. This result is consistent with the findings we made with quantitative analysis.
In summary, the previous reviews revealed similar effects of interventions (direction of estimated effects). However, the current review is more critical concerning the quality of the available evidence than any other previous review.
Inclusion of more recent studies on remifentanil for labour analgesia improved the precision and the external validity of the present review. In addition, the current review included zero total event trials into the meta‐analyses, analysed imprecision for each GRADE‐relevant outcome by TSA or OIS considerations, investigated robustness of the estimated effects by sensitivity analyses based on the result of the 'Risk of bias' assessment, and provides sufficient background information to the studies' details.
Authors' conclusions
Implications for practice.
Based on the current systematic review there is mostly very low‐ to low‐quality evidence to inform practice and the following conclusions are only relevant to healthy women with an uncomplicated pregnancy who are at full term.
Remifentanil (patient‐controlled analgesia, PCA) provides stronger pain relief 'early' and women are more satisfied with pain relief compared to other opioids administered either IV/IM or using PCA. This finding is based on all doses and across all regimens combined in the remifentanil (PCA) and IV/IM opioid groups. In contrast, remifentanil (PCA) is inferior to epidural/combined spinal‐epidural analgesia (CSE) with respect to pain reduction ('early') and satisfaction with pain relief. Information to assess other comparators with respect to efficacy is insufficient (remifentanil (different PCA regimen)) or lacking (remifentanil (IV), inhalational analgesia or placebo).
There is insufficient information available to communicate assured information to the practice concerning safety aspects for both the mother and the newborn, especially for the relevant 'safety' outcomes 'maternal apnoea', 'maternal respiratory depression', and 'Apgar score less than seven at five minutes'. Basing on the available data, we conclude that remifentanil (PCA) is inferior to an epidural with respect to maternal apnoea, oxygen desaturation and opioid‐induced side effects such as nausea, vomiting, and sedation. For newborns there is no evidence of effect that remifentanil (PCA) increase the risk for Apgar scores less than seven at five minutes compared to an epidural. Information to assess other comparators with respect to safety is insufficient (remifentanil (IM/IM), remifentanil (PCA), remifentanil (continuous IV), remifentanil (different PCA regimen)) or still lacking (inhalational analgesia and placebo).
There is moderate‐quality evidence that remifentanil (PCA) is associated with lower risk for the requirement of escape analgesia when compared to the administration of other opioids (IV/IM) and that the administration of remifentanil (PCA) is associated with higher risks for the requirement of additional analgesia compared to an epidural. Other opioids administered via PCA were associated with similar risks for requirement of escape analgesia, however, the quality of evidence is low for this information.
There is no difference in the risk for caesarean delivery between remifentanil (PCA) and other opioids administered IV/IM. However, remifentanil (PCA) might be associated with an increased risk for caesarean section compared to another opioid (PCA). Finally, there is moderate‐quality evidence that there is no difference in the risk for caesarean delivery between remifentanil (PCA) and epidural/CSE.
More research is needed, especially, on maternal and neonatal safety aspects. Future research may significantly alter the current situation.
Implications for research.
In order to reliably inform women on analgesic effectiveness and the risk of adverse events for both women and newborns when remifentanil (PCA) is used for management of labour pain, we need additional information based on the findings of the current review. In the following paragraph, we point out the remaining gaps in knowledge and propose ways and possibilities to reach higher‐quality levels of evidence.
In general, more participants including high‐risk groups in prospective randomised controlled trials of all comparisons are needed for most of the relevant outcomes. Especially trials investigating remifentanil (PCA) versus remifentanil (IV) or remifentanil using different administration regimens are needed to uncover the optimal mode and regimen of remifentanil administration with respect to efficacy and safety. In this light, dose‐response studies would also be informative. Studies investigating the same regimen should be more standardised and more comparable. Relevant adverse events such as 'apnoea' and 'respiratory depression' are underreported and more systematic interventional trials are needed to reliably assess safety for mothers. The same is true for all outcomes summarised as adverse events for newborns.
Moreover, some patient‐relevant outcomes, such as 'sense of control in labour', 'satisfaction with childbirth‐experience', 'effect on mother‐baby interaction' were not investigated by the included studies. However, those outcomes may be of interest for women who have to choose between different options for labour analgesia.
With respect to methodological aspects and studies' quality, some relevant points should be considered when planning and conducting future high‐quality trials.
To avoid selection bias randomisation should occur after the request for analgesia.
In scenarios when blinding of participants and attending personnel is difficult or even impossible due to the different nature of the interventions under investigation (e.g. IV PCA device versus epidural, or if pharmacokinetic profiles of the investigated interventions differ to a large extent), attempts should be made to blind the outcome assessment, whenever feasible. For some outcomes such as 'overall satisfaction with pain relief', which can take place after the intervention was terminated, rating could be made by outcome assessors who are not otherwise involved in the study. Other theoretical options include the attempt of blinding observers by using 'dummy' epidural and 'dummy' PCA devices in combination with evaluators not otherwise involved in the study. However, due to the authors’ experience in the field such interventions may in the end not be reliable considering the pharmacokinetic and dynamic profile of the competing interventions. When blinding is deemed feasible and sensible, efficacy of blinding can be judged by asking participants a couple of days later which intervention they actually thought they were allocated to and compare it to the real allocation. According to the agreement, one can judge whether blinding for participants has worked.
The current review identifies attrition bias as a further issue at a half of all included studies. Numerous trials have more than 15% of missing data for some outcomes without reporting reasons for it, so that we could not assess whether the lack of reporting is related to the outcome of interest or not. Therefore, attempts should be made to minimise the amount of missing data and if missing data are not avoidable, the reasons for missing data should be reported. Since occasionally also scientific journals propose to shorten research papers, which may lead to incomplete outcome reporting, the use of an abridged paper version with additional material (complete tables) on the web should be encouraged.
In this context we suggest a rigorous prospective trial registration (Weibel 2016).
Trialists of included studies often conducted data analysis on a per‐protocol basis. However, for interventional trials aiming to establish superiority of one group an intention‐to‐treat (ITT) analysis is often preferable.
It maintains allocation and comparability of the intervention groups and thus reveals potential shortcomings in the analgesia method or other interventions applied. A per‐protocol analysis can be used as an additional analysis, since the true effect can be underestimated by ITT analysis. In case of high cross‐over rates, data‐analysis for adverse events may be additionally conducted as an as‐treated analysis to uncover the frequency and severity of side effects of the interventions of interest.
Our review identifies a substantial heterogeneity in the definition, measurement, and reporting of several outcomes. Standardised definitions for outcomes would be useful as for instance for apnoea (respiratory rate of zero for at least 20 s), respiratory depression (less than eight breaths/minute), and oxygen desaturation (≤ 95% and ≤ 92% SpO2 for one, two, and five minutes, respectively). The assessment of 'satisfaction with pain relief' and 'pain intensity' should be standardised by using the same scale (e.g. VAS 0 to 10 cm) and the same time points (within 24 hours of delivery for 'satisfaction', and hourly for 'pain'). 'Pain' should be optimally assessed at an early (e.g. at one hour after initiation of analgesia) and late time point (e.g. three to four hours after initiation of analgesia). Assessments regarding 'satisfaction' should be repeated several days postpartum to avoid the influence of immediate birth experience.
Concomitantly administered interventions especially those with likely influence on pain levels or the occurrence of side effects, need to be reported and – if applicable – kept to a minimum. This applies to systemic and inhalational analgesia as well as to interventions with influence on the occurrence of adverse effects (i.e. supplemental oxygen).
Acknowledgements
We would like to thank Elisabeth Friedrich‐Würstlein (University library, Wuerzburg) for her assistance during the literature search and organising full texts of the relevant publications.
This project was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to Cochrane Pregnancy and Childbirth. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
As part of the pre‐publication editorial process, this review has been commented on by three peers (an editor and two referees who are external to the editorial team) and the Group's Statistical Adviser.
Peter Kranke: received a Meta‐Analysis grant supporting this review from the European Society of Anaesthesiology.
Appendices
Appendix 1. ClinicalTrials.gov and ICTRP search strategy
remifentanil AND labor
remifentanil AND labour
Appendix 2. Study eligibility form
| Author (Year) | ||
| Public Title | ||
| Scientific Title | ||
| Identified through | Main search/references/additional searches. | |
| Study status | Active/recruiting/ongoing/finished | |
| Relevant study design | Randomised controlled trial | Yes/No/Unclear |
| Relevant population | Women in labour with planned vaginal delivery including women of high‐risk groups. Specifically excluded are women undergoing caesarean section. | Yes/No/Unclear |
| Relevant intervention | Experimental group must have received remifentanil administered via a patient‐controlled analgesia device for pain relief in labour. No other analgesics are allowed for simultaneous administration. Control group must have placebo treatment, no treatment, or any other intervention for pain relief in labour. | Yes/No/Unclear |
| Intervention experimental | ||
| Intervention control | ||
| If you have not answered Yes to all of the questions, please exclude the study. If you answered Yes to all questions, please continue to data extraction form and critical appraisal. | ||
Appendix 3. Data extraction form
| Author (Year) | ||
| Study design/Methods | RCT, blinding, randomisation, purpose, when and were was the study conducted?, trial identifier | |
| Participant flow | ||
| Experimental | Control | |
| Number of participants assessed for eligibility | ||
| Number of participants randomised | ||
| Number of participants receiving treatment | ||
| Number of participants analysed | ||
| Inclusion criteria | ||
| Exclusion criteria | ||
| Population/Baseline details | Experimental | Control |
| Mean age/Median age | ||
| Mean weight/Median weight | ||
| ASA I/II | ||
| Type of delivery (spontaneous delivery/instrumental/caesarean section) | ||
| Week of gestation | ||
| Singleton, twin, multiple pregnancy | ||
| Parity | ||
| Duration of labour (first stage of labour, second stage of labour) | ||
| Interventions | ||
| Outcomes (primary endpoint, dichotomous, continuous) | ||
| Notes | sample size, power analysis, concomitant medications, funding | |
| Intervention | |
| Analgesia duration | |
| Background infusion | |
| Bolus dose | |
| Bolus application speed | |
| Bolus dose escalation on request | |
| Lockout time | |
| Maximum dose | |
| Total administered dose of remifentanil (if stated) |
| Dichotomous outcome data | Experimental | Control | ||
|
(n) Number assessed |
(n) Number with outcome |
(n) Number assessed |
(n) Number with outcome |
|
| Additional analgesia required (e.g. conversion to epidural analgesia) | ||||
| Conversion to caesarean delivery | ||||
| Breastfeeding initiation | ||||
| Assisted vaginal birth | ||||
| Need for neonatal resuscitation | ||||
| Adverse events for women (e.g. apnoea, respiratory depression, oxygen desaturation, hypotension, bradycardia, nausea, vomiting, postpartum haemorrhage, maternal somnolence) | ||||
| Adverse events for the newborn (e.g. Apgar scores less than 7 at 5 minutes, need for naloxone, depressed baby, opioid‐induced loss of fetal heart rate variability) | ||||
| Continuous outcome data (Unit of measurement) | Experimental | Control | ||
| n | Mean (SD) | n | Mean (SD) | |
| Satisfaction with pain relief | ||||
| Sense of control in labour | ||||
| Effect (negative) on mother/baby interaction | ||||
| Pain scores | ||||
| Satisfaction with childbirth experience | ||||
| Long‐term childhood development | ||||
| Cost | ||||
| Umbilical cord base excess (arterial) | ||||
| Umbilical cord base excess (venous) | ||||
| Umbilical cord pH (arterial) | ||||
| Umbilical cord pH (venous) | ||||
| Apgar scores at 1, 5, 10 min | ||||
| Sedation scores | ||||
Appendix 4. Critical appraisal form
| Author (Year) | ||
| Journal | ||
| Title | ||
| Random sequence generation (selection bias) | ||
| State here the method used to generate allocation and reasons for grading | Adequate/Inadequate/Unclear | |
| Allocation concealment (selection bias) | ||
| State here the method used to conceal allocation and reasons for grading | Adequate/Inadequate/Unclear | |
| Blinding of participants and personnel (performance bias) | ||
| Person responsible for participants care | Low risk/High risk/Unclear | |
| Participant | Low risk/High risk/Unclear | |
| Blinding of outcome assessment (detection bias) | ||
| Outcome assessor | Low risk/High risk/Unclear | |
| Incomplete outcome data (attrition bias) | ||
| Drop‐out rate > 15% | Yes/No/Unclear | |
| Missing values reported, balanced across groups, and unrelated to true outcome | Yes/No/Unclear | |
| Escape rate > 15% | Yes/No/Unclear | |
| Cross‐over rate > 15% | Yes/No/Unclear | |
| Data analysis described | Yes/No/Unclear | |
| Imputation methods correct | Yes/No/Unclear | |
| Selective reporting (reporting bias) | ||
| Free of selective outcome reporting? | Yes/No/Unclear | |
| Other bias | ||
| Other sources of bias | Yes/No/Unclear | |
Data and analyses
Comparison 1. Remifentanil (PCA) versus another opioid (IV/IM).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Satisfaction with pain relief | 4 | 216 | Std. Mean Difference (IV, Random, 95% CI) | 2.11 [0.72, 3.49] |
| 2 Respiratory depression (< 8 breaths/min) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Oxygen desaturation (SpO2 < 95%) | 2 | 113 | Risk Ratio (M‐H, Random, 95% CI) | 0.48 [0.00, 47.37] |
| 4 Nausea (and vomiting) | 4 | 216 | Risk Ratio (M‐H, Random, 95% CI) | 0.54 [0.29, 0.99] |
| 5 Vomiting | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6 Pruritus | 2 | 156 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 7 Sedation (1 h) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 8 Apgar score < 7 at 5 min | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 Apgar score at 5 min | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 10 FHR/CTG abnormalities, non‐reassuring fetal status | 2 | 156 | Risk Ratio (M‐H, Random, 95% CI) | 0.30 [0.10, 0.90] |
| 11 Pain intensity 'early' (1 h) | 3 | 180 | Std. Mean Difference (IV, Random, 95% CI) | ‐1.58 [‐2.69, ‐0.48] |
| 12 Pain intensity 'late' (2 h) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 13 Additional analgesia required (escape analgesia) | 3 | 190 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.40, 0.81] |
| 14 Rate of caesarean delivery | 4 | 215 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 [0.34, 1.41] |
| 15 Rate of assisted birth | 4 | 215 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.32, 2.09] |
| 16 Augmented labour | 3 | 190 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.72, 1.29] |
| 17 Breastfeeding initiation (feeding difficulties) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 2. Remifentanil (PCA) versus another opioid (PCA).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Satisfaction with pain relief | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2 Oxygen desaturation (SpO2 < 95%) | 2 | 190 | Risk Ratio (M‐H, Random, 95% CI) | 1.28 [0.49, 3.30] |
| 3 Hypotension | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Bradycardia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Nausea (and vomiting) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6 Pruritus | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7 Sedation (1 h) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 8 Apgar score < 7 at 5 min | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 Apgar score at 5 min | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 10 Need for naloxone | 2 | 55 | Risk Ratio (M‐H, Random, 95% CI) | 0.30 [0.01, 6.47] |
| 11 FHR/CTG abnormalities, non‐reassuring fetal status | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12 NACS at 15/30 min | 2 | 94 | Mean Difference (IV, Random, 95% CI) | 1.11 [‐0.65, 2.87] |
| 13 Pain intensity 'early' (30 min/1 h) | 3 | 215 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.51 [‐1.01, ‐0.00] |
| 14 Pain intensity 'late' (2 h) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 15 Additional analgesia required (escape analgesia) | 3 | 215 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.45, 1.28] |
| 16 Rate of caesarean delivery | 2 | 143 | Risk Ratio (M‐H, Random, 95% CI) | 2.78 [0.99, 7.82] |
| 17 Rate of assisted birth | 2 | 143 | Risk Ratio (M‐H, Random, 95% CI) | 1.22 [0.62, 2.37] |
| 18 Augmented labour | 2 | 152 | Risk Ratio (M‐H, Random, 95% CI) | 1.37 [0.59, 3.15] |
Comparison 3. Remifentanil (PCA) versus epidural/combined spinal‐epidural analgesia (CSE).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Satisfaction with pain relief | 7 | 2135 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.22 [‐0.40, ‐0.04] |
| 2 Apnoea | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Respiratory depression (< 9, < 8 breaths/min) | 3 | 687 | Risk Ratio (M‐H, Random, 95% CI) | 1.52 [0.23, 9.90] |
| 4 Oxygen desaturation (SpO2 < 92%) | 3 | 774 | Risk Ratio (M‐H, Random, 95% CI) | 3.24 [1.66, 6.32] |
| 5 Oxygen desaturation (SpO2 < 95%) | 3 | 800 | Risk Ratio (M‐H, Random, 95% CI) | 3.27 [2.32, 4.61] |
| 6 Hypotension | 4 | 823 | Risk Ratio (M‐H, Random, 95% CI) | 0.58 [0.22, 1.49] |
| 7 Bradycardia | 2 | 44 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 8 Nausea | 8 | 1909 | Risk Ratio (M‐H, Random, 95% CI) | 1.49 [1.19, 1.86] |
| 9 Vomiting | 6 | 1840 | Risk Ratio (M‐H, Random, 95% CI) | 1.63 [1.25, 2.13] |
| 10 Pruritus | 7 | 1852 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.48, 1.18] |
| 11 Sedation (1 h) | 3 | 148 | Std. Mean Difference (IV, Random, 95% CI) | 0.71 [0.03, 1.39] |
| 12 Apgar score ≤ 7 (< 7) at 5 min | 5 | 1322 | Risk Ratio (M‐H, Random, 95% CI) | 1.28 [0.65, 2.51] |
| 13 Apgar score at 5 min | 3 | 137 | Mean Difference (IV, Random, 95% CI) | 0.06 [‐0.27, 0.39] |
| 14 Need for naloxone | 2 | 1170 | Risk Ratio (M‐H, Random, 95% CI) | 0.20 [0.01, 3.85] |
| 15 FHR/CTG abnormalities, non‐reassuring fetal status | 5 | 1280 | Risk Ratio (M‐H, Random, 95% CI) | 1.55 [0.49, 4.92] |
| 16 Pain intensity 'early' (1 h) | 6 | 235 | Std. Mean Difference (IV, Random, 95% CI) | 0.57 [0.31, 0.84] |
| 17 Pain intensity 'late' (2 h) | 4 | 143 | Std. Mean Difference (IV, Random, 95% CI) | 1.46 [0.66, 2.26] |
| 18 Additional analgesia required | 6 | 1037 | Risk Ratio (M‐H, Random, 95% CI) | 8.10 [3.50, 18.75] |
| 19 Rate of caesarean delivery | 9 | 1578 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.81, 1.21] |
| 20 Rate of assisted birth | 8 | 1550 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.66, 1.26] |
| 21 Augmented labour | 6 | 1379 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.82, 1.02] |
| 22 Umbilical cord base excess (artery) | 3 | 75 | Mean Difference (IV, Random, 95% CI) | ‐0.97 [‐2.65, 0.72] |
| 23 Umbilical cord base excess (venous) | 2 | 129 | Mean Difference (IV, Random, 95% CI) | ‐0.05 [‐2.39, 2.30] |
| 24 Umbilical cord pH (artery) | 5 | 1245 | Mean Difference (IV, Random, 95% CI) | ‐0.01 [‐0.02, ‐0.00] |
| 25 Umbilical cord pH (venous) | 4 | 1299 | Mean Difference (IV, Random, 95% CI) | 0.01 [‐0.01, 0.02] |
| 26 Neonatal resuscitation | 2 | 69 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.04, 25.09] |
Comparison 4. Remifentanil (PCA) versus remifentanil (continuous IV).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Respiratory depression (< 8 breaths/min) | 2 | 135 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 2 Oxygen desaturation (SpO2 < 95%) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Hypotension | 2 | 135 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 4 Bradycardia | 2 | 135 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5 Nausea (and vomiting) | 2 | 135 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.28, 2.54] |
| 6 Pruritus | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7 Sedation (1 h) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 8 Need for naloxone | 2 | 135 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 9 FHR/CTG abnormalities, non‐reassuring fetal status | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10 Pain intensity 'early' (1 h) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 11 Pain intensity 'late' (2 h) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 12 Additional analgesia required (escape analgesia) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13 Neonatal resuscitation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 5. Remifentanil (PCA, increasing bolus, fixed infusion dose) versus remifentanil (PCA, increasing infusion, fixed bolus dose).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Satisfaction with pain relief | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2 Oxygen desaturation (SpO2 < 95%) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Hypotension | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Bradycardia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Nausea | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6 Vomiting | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7 Pruritus | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8 Apgar score < 7 at 5 min | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 Need for naloxone | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10 FHR/CTG abnormalities, non‐reassuring fetal status | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11 Additional analgesia required (escape analgesia) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12 Rate of caesarean delivery | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13 Augmented labour | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14 Umbilical cord base excess (artery) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 15 Umbilical cord base excess (venous) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 16 Umbilical cord pH (artery) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 17 Umbilical cord pH (venous) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 18 Neonatal resuscitation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Balki 2007.
| Methods | Randomised, controlled trial. Double‐blinded. No statement on time of randomisation. The purpose of this pilot study was to compare two regimens of IV remifentanil PCA, along with continuous background infusion, for labour analgesia. The study was conducted in Mount Sinai Hospital, Toronto, Canada, from September 2005 to December 2006. Trial Identifier: NA |
|
| Participants |
Participant flow: Number assessed for eligibility: 22 Number randomised: 20 (10/10) Number receiving treatment: 20 (10/10) Number analysed: 20 (10/10) Inclusion criteria: Term pregnancy, ASA I and II women in active labour, who requested systemic analgesia with or without contraindications to epidural analgesia Exclusion criteria: Allergy or hypersensitivity to remifentanil, opioid dependence or addiction, consumption of narcotics within 24 h of the study period, FHR abnormalities, fetal compromise and/or language barrier Baseline details: Fixed bolus group (n = 10): Age (years, mean (SD)): 32.7 (5.9) Weight (kg, mean (SD)): 85 (30) ASA I/II (n/n): NA Type of delivery (n): vaginal (6), CS (4) Week of gestation: 39.2 ± 1.5 Singleton, twin, multiple pregnancy: NA Parity (n): Primipara (5) Duration of labour: ‐ First stage of labour (min, mean (SD)): NA ‐ Second stage of labour (min, mean (SD)): NA Fixed infusion group (n = 10): Age (years, mean (SD)): 30.4 (5.8) Weight (kg, mean (SD)): 77.1 (14.1) ASA I/II (n/n): NA Type of delivery (n): vaginal (6), CS (4) Week of gestation: 39.0 ± 1.4 Singleton, twin, multiple pregnancy: NA Parity (n): Primipara (7) Duration of labour: ‐ First stage of labour (min, mean (SD)): NA ‐ Second stage of labour (min, mean (SD)): NA |
|
| Interventions | Initially, all women received a standard regimen of remifentanil with an infusion of 0.025 µg/(kg*min) and a PCA bolus of 0.25 µg/kg. The PCA lockout interval was set at 2 min, and the 4 h limit was 3 mg. As labour progressed and women required additional analgesia, they received higher doses of either the infusion (group: constant bolus) or the PCA boluses (group: constant infusion). (At the woman’s request, if there was either no change or worsening of pain scores; each step was maintained for at least 15 min before progressing to the subsequent one.) Fixed bolus group (n = 10): The infusion rate was increased stepwise from 0.025 µg/(kg*min) to 0.05 µg/(kg*min), 0.075 µg/(kg*min) and 0.1 µg/(kg*min), while the bolus of 0.25 µg/kg was maintained. Fixed infusion group (n = 10): The bolus dose was increased stepwise from 0.25 µg/kg to 0.5 µg/kg, 0.75 µg/kg and 1 µg/kg, while the infusion rate of 0.025 µg/(kg*min) was kept constant. |
|
| Outcomes | The primary outcome variables were maternal pain and desaturation. Continuous: ‐ overall satisfaction (VNRS 0 to 10, within 2 h after delivery) ‐ pain intensity (VNRS 0 to 10, at 0, every 30 min until delivery), overall pain score (VNRS 0 to 10, within 2 h of delivery) ‐ umbilical cord BE (artery, vein), umbilical cord pH (artery, vein) ‐ sedation score (observer, 5 to 0, at baseline and lowest sedation) Dichotomous: ‐ additional analgesia (epidural) ‐ rate of CS ‐ need for neonatal resuscitation ‐ augmented labour (no substance indicated) ‐ women: oxygen desaturation (< 95%, < 90%), hypotension, bradycardia, nausea, vomiting, pruritus, drowsiness, dizziness, confusion ‐ newborns: Apgar score ≥ 7 at 1 and 5 min, non‐reassuring FHR, need for naloxone |
|
| Notes | ‐ Small trial sample size (< 200 participants) ‐ Power analysis performed (VNRS pain, n = 10 per group) Concomitant medication: Nausea or vomiting was treated with dimenhydrinate, and diphenhydramine was administered for pruritus. The woman could choose to cross over to epidural analgesia at any time during labour, unless there was a contraindication to a regional technique. Funding: NA |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “The patients were randomized, via a computer‐generated randomisation scheme, into one of the two study groups.” |
| Allocation concealment (selection bias) | Unclear risk | Quote: “The group allocation was blinded via sealed envelopes until the time of PCA administration.” Not specifically mentioned sequentially numbered, opaque envelopes (SNOSE). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: “The patient and the obstetrician, as well as the registered nurse collecting the data, were all blinded to the study group. The group allocation was known only to the anaesthesiologist who was making changes to the pump settings when needed.” Blinding for participants and personnel adequate. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: “The patient and the obstetrician, as well as the registered nurse collecting the data, were all blinded to the study group. The group allocation was known only to the anaesthesiologist who was making changes to the pump settings when needed.”, “Fetal heart rate tracings were analysed by an obstetrician (P.B.) who was blinded to the study group.” Blinding for outcome assessment adequate. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ‐ No missing outcome data after randomisation. ‐ Rate of escape (epidural): 10%/0% ‐ Rate of cross‐over: NA ‐ Data‐analysis: Full‐ITT |
| Selective reporting (reporting bias) | Unclear risk | There is no reference to a trial registry and no published study protocol. |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Blair 2005.
| Methods | Randomised, controlled trial. Single‐blinded. No statement on time of randomisation. The purpose of this trial was to determine the analgesic efficacy and safety of remifentanil versus pethidine via PCA for women in established uncomplicated labour. There are no details where or when the study was conducted. The authors’ origin is United Kingdom. Trial Identifier: NA |
|
| Participants |
Participant flow: Number assessed for eligibility: NA Number randomised: 40 (20/20) Number receiving treatment: 40 (20/20) Number analysed: 39 (20/19) Inclusion criteria: Women with ASA I or II, either before the onset of labour in the antenatal ward or in early labour before any analgesia had been requested Exclusion criteria: Women were excluded from the study if they planned to use epidural analgesia or had pre‐eclampsia, multiple pregnancy, premature labour or allergy to any agent under investigation. Baseline details: Remifentanil group (n = 20): Age (years, mean (SD)): 29 (5.2) Weight (kg, mean (SD)): 76 (9) ASA I/II (n/n): NA Type of delivery (n): spontaneous (NA) Week of gestation: NA Singleton, twin, multiple pregnancy: NA Parity (median (IQR [range])): 1 (1 ‐ 2 [0 ‐ 5]) Duration of labour: ‐ before PCA use (min, mean (SD)): 125 (98) ‐ first stage of labour (min, mean (SD)): 260 (97) ‐ second stage of labour (min, mean (SD)): 22 (22) Pethidine group (n = 19): Age (years, mean (SD)): 29 (5.4) Weight (kg, mean (SD)): 76 (12) ASA I/II (n/n): NA Type of delivery (n): spontaneous labour (NA) Week of gestation: NA Singleton, twin, multiple pregnancy: NA Parity (median (IQR [range])): 1 (0 ‐ 2 [0 ‐ 3]) Duration of labour: ‐ before PCA use (min, mean (SD)): 191 (205) ‐ first stage of labour (min, mean (SD)): 296 (158) ‐ second stage of labour (min, mean (SD)): 20 (12) |
|
| Interventions |
Remifentanil group (n = 20): Women received a remifentanil PCA using 40 µg remifentanil with a lockout of 2 min. The bolus dose and lockout period for the remifentanil PCA were based on an average maternal weight (80 kg) and a bolus of 0.5 µg/kg. Pethidine group (n = 19): Control participants received PCA using pethidine 15 mg with a lockout of 10 min. |
|
| Outcomes | The primary endpoint of the study was overall pain. Continuous: ‐ satisfaction with analgesia (VAS 0 to 10, at 0, 30 min until 120 min, median + IQR + range (symmetric)) ‐ pain intensity (VAS 0 to 10, at 0, every 30 min until 120 min, median + IQR (asymmetric)), overall pain score (VAS 0 to 10, at 2 h after delivery) ‐ umbilical cord pH (not specified) ‐ sedation score (observer, 5 to 1, and parturient score, VAS 0 to 10, at 0, 30, 60, 90, 120 min, respectively, median + IQR + range (asymmetric)) ‐ women: mean respiratory rate, mean SBP, mean HR, median nausea score (VAS 0 to 10) (at 0, 30, 60, 90, 120 min, respectively) ‐ newborns: mean FHR (at 0, 30, 60, 90, 120 min), Apgar score at 1 and 5 min (median + IQR + range (asymmetric)), NACS at 30 and 120 min (median + IQR + range (symmetric)) Dichotomous: ‐ additional analgesia (Entonox) ‐ women: oxygen desaturation (% total PCA time spent with < 94% and < 90%, diagrammed) ‐ newborns: need for naloxone |
|
| Notes | ‐ Small trial sample size (< 200 participants) ‐ Power analysis performed (VAS overall pain, n = 20 per group) Concomitant medication: All women were free to change to regional analgesia at any time if so desired. Entonox was available to all women throughout the study. Funding: NA |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “The women were randomly allocated to receive PCA using either remifentanil […] or pethidine […].” No method described. |
| Allocation concealment (selection bias) | Unclear risk | No statement on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: “[…] women, who were unaware of which treatment they were receiving.” No statement on whether key study personnel were blinded. We assume that participants and attending personnel might be able to uncover group allocation due to the different pharmacokinetics of the two interventions. The used method of blinding may only work for the outcome assessors (which was not mentioned in the published report for most of the relevant outcomes). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: “Baseline non‐invasive blood pressure, heart rate, SpO2, respiratory rate, observer sedation score […] and fetal heart rate were recorded by a blinded investigator. Baseline visual analogue scale (VAS) measurements were also recorded for the pain of contractions, satisfaction with current analgesia, nausea, anxiety and sedation.”, “At delivery, Apgar scores at 1 and 5 min were recorded, cord blood was taken for blood gas analysis and the fetal requirement for naloxone was noted.”, "The cardiotocograph (CTG) was recorded for a minimum of 1 h after starting the PCA and was subsequently analysed by a blinded obstetrician […].” It is unclear from the description whether assessment for most of the outcomes after treatment (during study) was blinded. The study did address this issue only for the assessment of FHR patterns. We do not know who was responsible for outcome assessment and participants and attending personnel may be able to uncover group allocation. The subjective1 outcomes or outcome measurements are likely to be influenced by lack of blinding. Therefore, insufficient information exists to judge "yes" or "no". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | ‐ Dropout rate: 0%/5% Quote: “There was one protocol violation in the pethidine group and no data were included from this patient.” The protocol violation was not described. ‐ No statement on why 1 woman in the Remifentanil group was not analysed for all outcomes (satisfaction with pain relief, AE for newborn) ‐ Rate of escape (Entonox): 90%/100% (may influence data on AE, satisfaction and pain) ‐ Rate of cross‐over: NA ‐ Data‐analysis: Per‐protocol (protocol violation) |
| Selective reporting (reporting bias) | Unclear risk | There is no reference to a trial registry and no published study protocol. |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Calderon 2006.
| Methods | Randomised, controlled trial. No statement on blinding. No statement on time of randomisation. The purpose of this trial was to evaluate the effectiveness and security of remifentanil administered by means of elastomeric infusor with PCA IV compared with IM meperidine in obstetric women with contraindication for epidural analgesia. There are no details where or when the study was conducted. The authors’ origin is Spain. Trial Identifier: NA |
|
| Participants |
Participant flow: Number assessed for eligibility: NA Number randomised: 24 (12/12) Number receiving treatment: 24 (12/12) Number analysed: 24 (12/12) Inclusion criteria: ASA I to III, aged 20 to 40 years, requesting analgesia Exclusion criteria: NA Baseline details: Remifentanil group (n = 12): Age (years, mean (SD)): 28 (5) Weight (kg, mean (SD)): 72 (8) ASA I/II (n/n): 8/2 Type of delivery (n): spontaneous (NA), instrumental (1), CS (0) Week of gestation: NA Singleton, twin, multiple pregnancy: NA Parity: NA Duration of labour: ‐ First stage of labour (min, mean (SD)): NA ‐ Second stage of labour (min, mean (SD)): NA Meperidine group (n = 12): Age (years, mean (SD)): 30 (3) Weight (kg, mean (SD)): 75 (6) ASA I/II (n/n): 7/3 Type of delivery (n): spontaneous (NA), instrumental (2), CS (1) Week of gestation: NA Singleton, twin, multiple pregnancy: NA Parity: NA Duration of labour: ‐ First stage of labour (min, mean (SD)): NA ‐ Second stage of labour (min, mean (SD)): NA |
|
| Interventions |
Remifentanil group (n = 12): An elastomeric infusor with a capacity of 250 mL was filled with 2.5 mg of remifentanil and a 12 mL/h was started (average infusion of 0.025 µg/(kg*min) of remifentanil and boluses of 5 mL with a time of closing of 30 min). Meperidine group (n = 12): Women were given 1 mg/kg of meperidine and 2.5 mg of haloperidol every 4 h by IM route. |
|
| Outcomes | The primary endpoint of the study was not defined. Continuous: ‐ overall satisfaction (VAS 0 to 10, time point unclear) ‐ pain intensity (VAS 0 to 100, at 0, every 30 min until 280 min, "expulsivo") ‐ newborns: Apgar score at 1 and 5 min Dichotomous: ‐ rate of CS, rate of assisted birth (instrumental) ‐ women: nausea + vomiting |
|
| Notes | ‐ Small trial sample size (< 200 participants) ‐ Power analysis not performed Concomitant medication: NA Funding: NA Intervention: Lockout time of 30 min seems too long for adequate analgesia. Contact to the authors: We contacted Dr. Torres via e‐mail (23 June 2016) to inquire the number of women who reported 'pain intensity at 2 hours'. We did not receive any answer. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “24 patients were randomized […].” No method described. |
| Allocation concealment (selection bias) | Unclear risk | No statement on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The study did not address this issue. However, we assume that blinding of parturients and personnel did not occur due to technical reasons and at least the subjective1 outcomes or outcome measurements are likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | The study did not address this issue. However, we assume that blinding of outcome assessment did not occur due to technical reasons and at least the subjective1 outcomes or outcome measurements are likely to be influenced by lack of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ‐ No missing outcome data after randomisation ‐ Rate of escape: NA ‐ Rate of cross‐over: NA ‐ Data‐analysis: Full‐ITT |
| Selective reporting (reporting bias) | Unclear risk | There is no reference to a trial registry and no published study protocol. |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Douma 2010.
| Methods | Randomised, controlled trial. Double‐blinded. No statement on time of randomisation. The purpose of this trial was to compare the analgesic efficacy of remifentanil with meperidine and fentanyl in a patient‐controlled setting (PCA). There are no details where or when the study was conducted. The authors’ origin is the Netherlands. Trial Identifier: NTR543 |
|
| Participants |
Participant flow: Number assessed for eligibility: 180 Number randomised: 180 (60/60/60) Number receiving treatment: 159 (52/53/54) Number analysed: 159 (21 excluded, delivery within 1 h after randomisation) Inclusion criteria: ASA physical status I or II, singleton cephalic presentation in active labour Exclusion criteria: Obesity (BMI (body mass index) ≥ 40 kg/m²), opioid allergy, substance abuse history, and women at high‐risk (pre‐eclampsia, severe asthma, insulin‐dependent diabetes mellitus, hepatic insufficiency, or renal failure) Baseline details: Remifentanil group (n = 52): Age (years, mean (SD)): 33.1 (5.0) Weight (kg, mean (SD)): 81 (13) ASA I/II (n/n): NA Type of delivery: spontaneous (62%), instrumental (22%), CS (16%) Week of gestation: 40 Singleton, twin, multiple pregnancy: singleton pregnancy Parity: Primiparity 58% Duration of labour: ‐ First stage of labour (min, mean (SD)): 363 (191) ‐ Second stage of labour (min, mean (SD)): 36 (30) Meperidine group (n = 53): Age (years, mean (SD)): 33.6 (5.5) Weight (kg, mean (SD)): 84 (14) ASA I/II (n/n): NA Type of delivery: spontaneous (69%), instrumental (23%), CS (9%) Week of gestation: 40 Singleton, twin, multiple pregnancy: singleton pregnancy Parity: Primiparity 66% Duration of labour: ‐ First stage of labour (min, mean (SD)): 293 (155) ‐ Second stage of labour (min, mean (SD)): 42 (35) Fentanyl group (n = 54): Age (years, mean (SD)): 33.5 (4.1) Weight (kg, mean (SD)): 79 (12) ASA I/II (n/n): NA Type of delivery: spontaneous (85%), instrumental (13%), CS (2%) Week of gestation: 40 Singleton, twin, multiple pregnancy: just singleton pregnancy Parity: Primiparity 68% Duration of labour: ‐ First stage of labour (min, mean (SD)): 348 (175) ‐ Second stage of labour (min, mean (SD)): 38 (26) |
|
| Interventions |
Remifentanil group (n = 52): Women received a 40 µg loading dose and remifentanil 40 µg per bolus with a lockout of 2 min and a maximum dose limit of 1200 µg/h. Meperidine group (n = 53): Women received a 49.5 mg loading dose and 5 mg boluses with a lockout of 10 min and a maximum overall dose limit of 200 mg. Fentanyl group (n = 54): Women received a 50 µg loading dose and boluses of 20 µg with a lockout of 5 min and a maximum dose limit of 240 µg/h. |
|
| Outcomes | The primary endpoint was not clearly stated but power analysis was performed for average pain score. Continuous: ‐ overall satisfaction (VRS 1 to 10, at 2 h after delivery) ‐ pain intensity (VAS 0 to 10, at 0 h, every 1 h until 6 h) ‐ umbilical cord BE (not specified), umbilical cord pH (not specified) ‐ sedation score (observer, 1 to 5, at 0, 1, 2, 3 h) ‐ newborns: Apgar score at 1 and 5 min, NACS at 15 and 120 min Dichotomous: ‐ additional analgesia (epidural) ‐ rate of CS, rate of assisted birth (instrumental) ‐ oxytocin use ‐ women: oxygen desaturation (< 95%), nausea + vomiting, pruritus ‐ newborns: CTG reactive |
|
| Notes | ‐ Small trial sample size (< 200 participants) ‐ Power analysis performed (average pain score, n = 60 per group) Concomitant medication: All women were free to change to epidural analgesia at any time. The PCA device was discontinued at full cervical dilatation. Hypotension (systolic arterial pressure < 90 mmHg or > 25% below baseline) was treated with IV fluids and ephedrine 5 mg IV. When oxygen saturation decreased below 95%, oxygen 6 L/min was administered by facemask. If labour failed to progress (first or second stage), oxytocin was given, according to the hospital protocol. Funding: This work was supported by the Bronovo Research Fund. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Randomization was established by using a computer‐generated random sequence […].” |
| Allocation concealment (selection bias) | Unclear risk | Quote: “[…] random sequence in numbered envelopes.” Not specifically mentioned opaque and sealed envelopes (SNOSE). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: “Study medication was prepared and blinded by the hospital pharmacy.”, “Observants and medical personnel attending to the parturient were unaware of the drug assignment.” We assume that participants and attending personnel might be able to uncover group allocation due to the different pharmacokinetics of the 2 interventions. The used method of blinding may only work for the outcome assessors. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: “Study medication was prepared and blinded by the hospital pharmacy.”, “With the exception of baseline data, all observations and measurements were made by blinded observers. Observants entered the delivery room only after the PCA device had been connected […]. This way the observants were unable to notice time differences in the administration […], which might have jeopardized blinding. Observants had no knowledge of the differences in programming of the PCA devices.”, “Observants and medical personnel attending to the parturient were unaware of the drug assignment.”, “Fetal heart rate patterns were scored as reactive or non‐reactive at regular intervals by an obstetrician who was blinded to the treatment groups.” An attempt was made and reported in the method section to blind the outcome assessor. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | ‐ Dropout rate: 15%/12%/10% Quote: “[…] 21 were excluded due to delivery within 1h after randomisation.” No statement on time of randomisation. ‐ Large amount (up to 50%) of outcome data (satisfaction with pain relief, AE newborn/AE mother, rate of CS, rate of assisted birth, umbilical blood pH/base excess) not reported. No reasons declared. ‐ Rate of escape (epidural): 13%/34%/15% (may influence data on AE, satisfaction and pain) ‐ Rate of cross‐over: NA ‐ Data‐analysis: Per‐protocol (women who delivered within 1 hour were excluded) |
| Selective reporting (reporting bias) | High risk | The protocol is available (NTR543, ISRCTN12122492) and there are several deviations. In the protocol the primary outcomes were amongst others requirement for naloxone as fetal outcome and presence of opioid substances in umbilical and maternal blood samples. In the published report both outcomes were mentioned within the methods but results were not reported. The observer sedation score was not pre‐specified in the protocol. The study protocol was retrospectively registered: Study registration (NTR543): 12/2005 First enrolment: 08/2005 |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Douma 2011.
| Methods | Randomised, controlled trial. No statement on blinding. No statement on time of randomisation. The purpose of this trial was to compare the efficacy of IV remifentanil PCA with epidural ropivacaine/sufentanil during labour. There are no details where or when the study was conducted. The authors’ origin is the Netherlands. Trial Identifiers: NTR1127 or EUCTR2007‐000808‐32‐NL |
|
| Participants |
Participant flow: Number assessed for eligibility: 147 Number randomised: 26 (14/12) Number receiving treatment: 25 (14/11) Number analysed: 20 (10/10 at 1 h, 9/8 at 2h, 6/6 at 3 h) Inclusion criteria: singleton pregnancy, ASA I or II, without prior use of opioid analgesics Exclusion criteria: Cervical dilation > 5 cm, pre‐eclampsia, insulin‐dependent diabetes, substance abuse, opioid allergy and morbid obesity (BMI ≥ 40 kg/m²) Baseline details: Remifentanil group (n = 10): Age (years, mean (SD)): 32.7 (5.9) Weight (kg, mean (SD)): 83.3 (16.7) ASA I/II (n/n): NA Type of delivery (n): spontaneous (7), instrumental (1), CS (2) Week of gestation: NA Singleton, twin, multiple pregnancy: singleton Parity (n): Primiparity (5) Duration of labour: ‐ First stage of labour (min, mean (SD)): 488 (277) ‐ Second stage of labour (min, mean (SD)): 71 (40) Epidural group (n = 10): Age (years, mean (SD)): 31.0 (5.2) Weight (kg, mean (SD)): 78.9 (11.9) ASA I/II (n/n): NA Type of delivery (n): spontaneous (4), instrumental (4), CS (2) Week of gestation: NA Singleton, twin, multiple pregnancy: singleton Parity (n): Primiparity (7) Duration of labour: ‐ First stage of labour (min, mean (SD)): 410 (173) ‐ Second stage of labour (min, mean (SD)): 32 (14) |
|
| Interventions |
Remifentanil group (n = 10): Parturients randomised to the IV remifentanil group received a 40 µg loading dose and boluses of 40 µg with a 2 min lockout time and bolus duration of 36 s using a Graseby 3300 syringe pump (Smiths Medical International, Ashford, Kent, UK). Maximum dose limit was 1200 µg/h. The PCA device was discontinued when parturients reached full cervical dilation. No further analgesia was provided during the second stage. Epidural group (n = 10): Women randomised to receive epidural analgesia were pre‐hydrated with 500 mL IV crystalloid solution before an epidural catheter was placed using a midline paramedian approach with a 17‐gauge Tuohy needle and loss‐of‐resistance to saline at L2–3 or L3–4. A loading dose of 0.2% ropivacaine 12.5 mL was given through the epidural catheter, followed by a continuous infusion of 0.1% ropivacaine with sufentanil 0.5 µg/mL at 10 mL/h. If analgesia was inadequate, additional boluses of the epidural solution were given. At full cervical dilation the epidural infusion was discontinued according to local hospital policy. |
|
| Outcomes | The primary outcome parameter of this study was the VAS pain score. Continuous: ‐ satisfaction with analgesia (VAS 0 to 10, at 0, 1, 2, 3 h after starting analgesia), overall satisfaction (NRS 1 to 10, at 2 h after delivery) ‐ pain intensity (VAS 0 to 10, at 0, 1, 2, 3 h after starting analgesia) ‐ umbilical cord BE (artery), umbilical cord pH (artery) ‐ sedation score (observer, 1 to 5, at 0, 1, 2, 3 h after starting analgesia) ‐ newborns: Apgar score at 1 and 5 min Dichotomous: ‐ additional analgesia (conversion remifentanil (PCA) to epidural) ‐ rate of CS, rate of assisted birth (instrumental) ‐ oxytocin use ‐ women: oxygen desaturation (< 95%), hypotension, bradycardia, nausea, vomiting, pruritus ‐ newborns: Apgar score ≤ 7 at 5 min, CTG reactive |
|
| Notes | ‐ Small trial sample size (< 200 participants) ‐ Power analysis performed (VAS pain score, n = 10 per group) Concomitant medication: If pain relief was inadequate at any time, the woman could request epidural analgesia. Hypotension (SBP < 90 mmHg or > 25% below baseline) was treated with IV fluids and IV ephedrine 5 mg or phenylephrine 100 µg. Supplemental oxygen was administered if maternal oxygen saturation (SpO2) levels remained below 95% for more than 60 s. Funding: NA |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “[…] numbered envelopes that had been randomised using a computer‐generated random sequence […].” |
| Allocation concealment (selection bias) | Unclear risk | Quote: “[…] numbered envelopes.” Not specifically mentioned opaque and sealed envelopes (SNOSE). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The study did not address this issue. However, we assume that blinding of parturients and personnel did not occur due to technical reasons and at least the subjective1 outcomes or outcome measurements are likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: “Fetal heart rate patterns were scored as reactive or non‐reactive by an obstetrician who was blinded to study allocation.” The study did not address this issue for most of the relevant outcomes with exception of the assessment of FHR patterns. However, we assume that blinding of outcome assessment did not occurred due to technical reasons and at least all other subjective1 outcomes or outcome measurements are likely to be influenced by lack of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | ‐ Dropout rate: 29%/17% Quote: “Twenty‐six parturients were enrolled of whom 20 completed the study; 10 subjects received remifentanil, 10 received epidural analgesia. Six parturients were excluded because of either delivery within one hour of randomisation (n = 5) or unsuccessful placement of the epidural catheter (n = 1).” No statement on time of randomisation. ‐ 30%/20% of outcome data were not reported for neonatal outcomes. No reasons declared. One woman in the remifentanil group required an epidural 2 h after initiation of the intervention. Outcome data of this woman were excluded from the analysis for satisfaction, pain, neonatal outcomes. Reason for exclusion may be related to true outcome. ‐ Rate of escape: NA ‐ Rate of cross‐over: 7%/NA, cross‐over participants were analysed as randomised ‐ Data‐analysis: Per‐protocol (women who delivered within 1 h were excluded) |
| Selective reporting (reporting bias) | High risk | The protocol is available (NTR1127, EUCTR2007‐000808‐32‐NL) and there are several deviations. In the protocol pain scores, woman's satisfaction, and fetal outcome (Apgar scores, umbilical cord pH, NACS, and requirement for naloxone) were defined as primary outcomes. No secondary outcomes were defined. In the published report the only primary outcome was pain score. Woman's satisfaction, sedation score, and SpO2 were defined as secondary outcomes. NACS and requirement for naloxone were not reported. Sedation, nausea and vomiting, SpO2 were not pre‐specified in the protocol. The study protocol was prospectively registered: Study registration (NTR1127): 17/11/2007 First enrolment: 26/11/2007 |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Douma 2015.
| Methods | 2‐arm randomised, controlled trial with a third‐arm observational cohort. Not blinded. Randomisation after onset of labour. The purpose of this trial was to compare the incidence of maternal fever (temperature ≥ 38°C) in parturients receiving IV remifentanil by PCA, with those receiving either epidural analgesia or no analgesia. The study was conducted in Leiden University Medical Center (period unknown). Trial Identifier: NTR1498 |
|
| Participants |
Participant flow: Number assessed for eligibility: 250 Number randomised: 116 (57/59) Number receiving treatment: 114 (57/57) Number analysed: 98 (49/49) + 42 control group Inclusion criteria: ASA I or II parturients with a singleton pregnancy, between 37 and 42 weeks of gestation Exclusion criteria: BMI ≥ 40 kg/m², insulin‐dependent diabetes, severe pre‐eclampsia (proteinuria ≥ 5 g/24 h), use of antibiotics during delivery, initial maternal SpO2 < 98%, initial maternal temperature ≥ 38°C, cervical dilation of > 7 cm and ruptured membranes for > 24 h at the time of inclusion. If delivery occurred within 1 h of starting the study, women were excluded from analysis. Baseline details: Remifentanil group (n = 49): Age (years, mean (SD)): 32 (4.8) Weight (kg, mean (SD)): 81 (17.2) ASA I/II (n/n): NA Type of delivery (n): spontaneous (32), instrumental (9), CS (7), missing (1) Week of gestation: 39 Singleton, twin, multiple pregnancy: singleton Parity (n): Nulliparous (25) Duration of labour: ‐ First stage of labour (min, mean (SD)): 355 (179) ‐ Second stage of labour (min, mean (SD)): 35 (29.9) Epidural group (n = 49): Age (years, mean (SD)): 31 (5.6) Weight (kg, mean (SD)): 81 (12.6) ASA I/II (n/n): NA Type of delivery(n): spontaneous (29), instrumental (9), CS (10), missing (1) Week of gestation: 40 Singleton, twin, multiple pregnancy: singleton Parity (n): Nulliparous (27) Duration of labour: ‐ First stage of labour (min, mean (SD)): 434 (158) ‐ Second stage of labour (min, mean (SD)): 40 (28.9) Control group (n = 42): Age (years, mean (SD)): 33 (4.5) Weight (kg, mean (SD)): 83 (13.3) ASA I/II (n/n): NA Type of delivery (n): spontaneous (34), instrumental (3), CS (5) Week of gestation: 40 Singleton, twin, multiple pregnancy: singleton Parity (n): Nulliparous (11) Duration of labour: ‐ First stage of labour (min, mean (SD)): 224 (131) ‐ Second stage of labour (min, mean (SD)): 24 (24.1) |
|
| Interventions |
Remifentanil group (n = 49): Women in the remifentanil (PCA) group received a 40 µg bolus (lockout 2 min, bolus duration 36 s) using a Graseby 3300 syringe pump (Smiths Medical Int., Luton, UK). The maximum dose permitted was 1200 µg/h. No background infusion was added. Because of concerns about the potential for neonatal respiratory depression, the pump was stopped when the woman reached full cervical dilatation. Epidural group (n = 49): A catheter was inserted at the L2–3 or L3–4 interspace using a 17‐gauge Tuohy needle. Parturients received a loading dose of ropivacaine 25 mg (0.2% ropivacaine 12.5 mL), followed by a continuous infusion of 0.1% ropivacaine and sufentanil 0.5 µg/mL at 10 mL/h. In case of inadequate analgesia, additional 10 mL boluses could be given. In case of epidural catheter dislodgement, the catheter was replaced. Control group (n = 42): No intervention |
|
| Outcomes | The primary outcome variable was the proportion of women who developed a temperature ≥ 38°C before delivery. Continuous: ‐ overall satisfaction (NRS 1 to 10, after delivery) ‐ pain intensity (VAS 0 to 10, at 0, 1, 2, 3, 4 h after starting analgesia, diagrammed) ‐ umbilical cord BE (venous), umbilical cord pH (venous) ‐ sedation score (observer, 1 to 5, at 0, 1, 2, 3, 4 h after starting analgesia) ‐ newborns: Apgar score at 1 and 5 min Dichotomous: ‐ additional analgesia (escape analgesia) ‐ rate of CS, rate of assisted birth (instrumental) ‐ oxytocin use ‐ women: oxygen desaturation (< 92%, < 90%, for 1, 2 or 5 min), nausea, vomiting, pruritus ‐ newborns: Apgar score ≤ 7 at 5 min, CTG reactive |
|
| Notes | ‐ Small trial sample size (< 200 participants) ‐ Power analysis performed (incidence of fever, n = 175 in total) Concomitant medication: When parturients were dissatisfied with analgesia, an epidural was offered as alternative. When SpO2 dropped < 92% for more than 60 s, oxygen was administered by facemask. Intrapartum fever was defined as maternal tympanic temperature ≥ 38°C. In cases of fever, antibiotics could be administered to the parturient. Hypotension (SBP < 90 mmHg or > 25% below pre‐analgesia values) was treated with IV fluids and/or IV ephedrine or phenylephrine. Funding: NA Contact to the authors: We contacted Dr. Douma via e‐mail (15 March 2016) to inquire the number of women who reported 'satisfaction with pain relief' and 'pain intensity at 1, 2, 3, and 4 hours'. We received the missing data. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Randomisation was performed using a computer‐ generated randomisation list and treatments (RPCA or EA) […]” |
| Allocation concealment (selection bias) | Low risk | Quote: “…were presented in a numbered opaque sealed envelope that was opened upon the request for analgesia.” |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: “The study was not blinded.” No blinding and at least the subjective1 outcomes or outcome measurements are likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: “The study was not blinded.” No blinding and at least the subjective1 outcomes or outcome measurements are likely to be influenced by lack of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | ‐ Dropout rate: 14%/17% Quote: “We assessed the eligibility of 250 women, of whom 164 were enrolled in the study […]. After excluding women who delivered within one hour, [2 failed epidural, 2 withdrawals, 13 delivered within 1 hour, 5 exclusion criterion] 140 women were analysed, 49 received RPCA, 49 received EA and 42 were in the observational control group." No reasons declared for exclusion of the 5 women (exclusion criterion). ‐ Quote: “Due to technical difficulties, continuous saturation data were not always available and this information is reported for only 114 women.”; Data on type of delivery from one woman each group were missing with no reasons declared. ‐ Rate of escape: NA ‐ Rate of cross‐over: 16%/2%, cross‐over participants were analysed as randomised ‐ Data‐analysis: Per‐protocol (women who delivered within 1 h were excluded) |
| Selective reporting (reporting bias) | High risk | 2 protocols are available (NTR1498 and EUCTR2008‐002792‐28‐NL) and there are several deviations. In the protocol maternal temperature and maternal saturation were defined as primary outcomes. In the published report, however, maternal saturation was reported as a secondary outcome. The secondary outcomes pain and overall satisfaction were not pre‐specified in the protocol. The pre‐specified outcomes NACS and requirement for naloxone were not reported in the published report. The study protocol was prospectively registered: Study registration (NTR1498): 10/2008. First enrolment: 11/2008 |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
El‐Kerdawy 2010.
| Methods | Randomised, controlled trial. No statement on blinding. No statement on time of randomisation. The study was planned to compare the use of remifentanil patient‐controlled IV analgesia (PCIA) to epidural bupivacaine plus fentanyl for labour analgesia in pre‐eclamptic women. There are no details where or when the study was conducted. The authors’ origin is Egypt/Saudi Arabia. Trial Identifier: NA |
|
| Participants |
Participant flow: Number assessed for eligibility: NA Number randomised: NA Number receiving treatment: NA Number analysed: 30 (15/15) Inclusion criteria: ≥ 32 weeks of gestation, normal cephalic presentation, < 5 cm cervical dilatation, clinical diagnosis of pre‐eclampsia Exclusion criteria: Remifentanil allergy, progression to eclampsia, evidence of increased intracranial pressure or focal neurologic deficit, platelet count of less than 80*10⁹/L, evidence of pulmonary oedema, non‐reassuring FHR tracing requiring imminent delivery Baseline details: Remifentanil group (n = 15): Age (years, mean (SD)): 26 (8) Weight (kg, mean (SD)): 79 (22) ASA I/II (n/n): NA Type of delivery (n): spontaneous (10), instrumental (0), CS (3) Week of gestation: 36 ± 4 Singleton, twin, multiple pregnancy: NA Parity: NA Duration of labour: ‐ First stage of labour (min, mean (SD)): NA ‐ Second stage of labour (min, mean (SD)): NA Epidural group (n = 15): Age (years, mean (SD)): 28 (9) Weight (kg, mean (SD)): 84 (37) ASA I/II (n/n): NA Type of delivery (n): spontaneous (8), instrumental (3), CS (4) Week of gestation: 35 ± 3 Singleton, twin, multiple pregnancy: NA Parity: NA Duration of labour: ‐ First stage of labour (min, mean (SD)): NA ‐ Second stage of labour (min, mean (SD)): NA |
|
| Interventions |
Remifentanil group (n = 15): The remifentanil hydrochloride concentration used was 50 µg/mL (3 mg diluted to 60 mL of normal saline). The PCA was set to deliver 0.5 µg/kg as a loading bolus infused over 10 s, lockout time of 5 min, PCA bolus of 0.25 µg/kg, continuous background infusion of 0.05 µg/(kg*min), and maximum dose was 3 mg in 4 h. Women were advised to start the PCA bolus when they felt the signs of a coming uterine contraction. Epidural group (n = 15): An epidural catheter was placed under complete aseptic technique at the L3‐L4 or L4‐L5 interspaces. A test dose of 0.25% bupivacaine was administered, and epidural analgesia was established with initial bolus of 10 mL to 15 mL of 0.25% bupivacaine plus 1 µg/kg fentanyl. Analgesia was maintained by continuous infusion of 0.125% bupivacaine plus 2 µg/mL fentanyl at a rate of 10 mL to 12 mL per hour aiming to obtain a T‐10 sensory level. |
|
| Outcomes | The primary endpoint of the study was not defined. Continuous: ‐ overall satisfaction (NRS 1 to 4, 24 h after delivery) ‐ pain intensity (VAS 0 to 10, at 0, 1 h and after delivery) ‐ umbilical cord pH (artery, vein) ‐ sedation score (observer, 1 to 4, 0, 1 h and after delivery) ‐ women: mean respiratory rate, mean SpO2, mean HR (at 0, 1 h and after delivery, respectively) Dichotomous: ‐ rate of CS, rate of assisted birth (instrumental) ‐ need for neonatal mechanical ventilation ‐ women: hypotension (at 0, 1 h and after delivery), nausea, vomiting, pruritus ‐ newborns: Apgar score ≤ 7 at 1 and 5 min, need for naloxone, FHR abnormalities 1 h after analgesia |
|
| Notes | ‐ Small trial sample size (< 200 participants) ‐ Power analysis not performed ‐ Satisfactory analgesia was considered if VAS ≤ 3 Concomitant medication: If the assigned analgesia was inadequate for the woman at any time, an alternative was offered and further study recording was discontinued. Hypotension (defined as reduction of > 25% of baseline level) was treated by either additional IV crystalloid or IV bolus doses (e.g. 2.5 to 5.0 mg) of ephedrine. Funding: NA Intervention: Lockout time of 5 min seems too long for adequate analgesia. Ethics: The study did not report that the study protocol was approved by the local Ethics committee. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “30 preeclamptic patients were randomly assigned….” No method described. |
| Allocation concealment (selection bias) | Unclear risk | No statement on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The study did not address this issue. However, we assume that blinding of parturients and personnel did not occur due to technical reasons and at least the subjective1 outcomes or outcome measurements are likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | The study did not address this issue. However, we assume that blinding of outcome assessment did not occur due to technical reasons and at least the subjective1 outcomes or outcome measurements are likely to be influenced by lack of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ‐ No missing outcome data after randomisation ‐ Rate of escape: NA ‐ Rate of cross‐over: NA ‐ Data‐analysis: NA |
| Selective reporting (reporting bias) | Unclear risk | There is no reference to a trial registry and no published study protocol. |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Evron 2005.
| Methods | Randomised, controlled trial. Double‐blinded. Randomisation after onset of labour. The purpose of this trial was to compare the analgesic effect of remifentanil in PCIA during labour and delivery with the effect of an IV infusion of meperidine. There are no details where or when the study was conducted. The authors’ origin is Israel. Trial Identifier: NA |
|
| Participants |
Participant flow: Number assessed for eligibility: NA Number randomised: 88 (43/45) Number receiving treatment: 88 (43/45) Number analysed: 88 (43/45) Inclusion criteria: Term parturients with singleton cephalic presentation requesting systemic analgesia, ASA I or II, active labour (cervical dilation of 3 cm to 6 cm) Exclusion criteria: ASA III or more, obesity (more than 100 kg or BMI ≥ 40kg/m²), history of drug (including analgesic chronic use or large doses) or alcohol abuse, smoking more than 10 cigarettes per day, and abnormal liver, renal, or haematological function Baseline details: Remifentanil group (n = 43): Age (years, mean (SD)): 29.5 (5.3) Weight (kg, mean (SD)): 75.1 (16) ASA I/II (n/n): NA Type of delivery (n): spontaneous (40), vacuum extraction (1), forceps delivery (0), CS (2) Week of gestation: NA Singleton, twin, multiple pregnancy: NA Parity (n): Primiparity (22) Duration of labour: ‐ First stage of labour (min, mean (SD)): active phase 245.2 (150.8) ‐ Second stage of labour (min, mean (SD)): 38.0 (32.2) Meperidine group (n = 45): Age (years, mean (SD)): 29.2 (5.2) Weight (kg, mean (SD)): 74.8 (11.27) ASA I/II (n/n): NA Type of delivery (n): spontaneous (38), vacuum extraction (2), forceps delivery (0), CS (5) Week of gestation: NA Singleton, twin, multiple pregnancy: NA Parity (n): Primiparity (19) Duration of labour: ‐ First stage of labour (min, mean (SD)): active phase 251.4 (118.8) ‐ Second stage of labour (min, mean (SD)): 42.2 (45.6) |
|
| Interventions |
Remifentanil group (n = 43): Participants randomised to receive remifentanil were connected to PCIA by a 1‐way infusion line (PCAM Syringe Pump Model P500; IVAC Medical Systems, NH) with patient‐controlled boluses of 20 µg each as a starting dose, regardless of parturient weight, and a 3 min lockout interval without basal infusion. The dose was increased by the attending anaesthesiologist every 15 to 20 min by 5 µg increments, on woman's request, to a maximum dose limit of 1500 µg/h. If any parturient had reached the maximum dose, a single bolus would have had 70 µg (0.93 µg/kg). Meperidine group (n = 45): Parturients in the control group received 75 mg of meperidine in 100 mL of normal saline over 30 min (approximately 1 mg/kg in a single bolus). In case of insufficient analgesia, another dose of 75 mg, followed by 50 mg when necessary, was administered, to a maximum dose of 200 mg of meperidine. |
|
| Outcomes | The primary endpoint of the study was not explicitly stated but power analysis was performed for pain score. Continuous: ‐ overall satisfaction (NRS 1 to 4, within 24 h after delivery) ‐ pain intensity (VAS 0 to 100, at 0, 1 h and end of first stage of labour) ‐ umbilical cord pH (not specified) ‐ sedation score (observer, Ramsey sedation score, at 1 h and end oft 1st stage of labour) ‐ women: mean respiratory rate, mean SBP, mean HR (at 0, 1 h and end oft 1st stage of labour, respectively) Dichtomous: ‐ additional analgesia (epidural) ‐ rate of CS, rate of assisted birth (vacuum, forceps) ‐ feeding difficulties ‐ oxytocin use ‐ women: oxygen desaturation (< 95%), nausea + vomiting, pruritus ‐ newborns: Apgar score < 7 at 1 and 5 min, opioid‐induced loss of FHR, FHR reactive |
|
| Notes | ‐ Small trial sample size (< 200 participants) ‐ Power analysis performed (VAS pain scores, n = 88 in total) Concomitant medication: For inadequate analgesia, adverse effects due to opioids, or failure of the technique (VAS > 40), epidural analgesia was offered. The decision to cross‐over from systemic opioids to epidural analgesia was made by the parturient in corroboration with the anaesthesiologist and after an additional trial of increasing the dose of the analgesic and a repeat VAS score of > 40. A VAS of 40 was considered an indication for cross‐over to epidural analgesia. To avoid possible hypoxaemia, supplemental oxygen was administered to the parturients whenever SpO2 decreased to less than 95%. Funding: NA Intervention: Lockout time 3 min seems too long for adequate analgesia (borderline). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Randomization was based on computer‐generated codes […]” |
| Allocation concealment (selection bias) | Unclear risk | Quote: “[…] kept in sequentially numbered opaque envelopes until just before use.” Not specifically mentioned sealed envelopes (SNOSE). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: “For blinding, PCIA remifentanil parturients were connected to a dummy IV saline bag, and IV meperidine parturients were connected to a dummy saline PCIA.”, “A senior anaesthesiologist, not involved in data recording, attended each parturient throughout labor.” Blinding was attempted to achieve by insertion of both an IV catheter and a PCA pump. However, we assume that participants and attending personnel might be able to uncover group allocation due to the different pharmacokinetics of the two interventions. The used method of blinding may only work for the outcome assessors (which was not explicitly mentioned in the published report for all outcomes). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: “For blinding, PCIA remifentanil parturients were connected to a dummy IV saline bag, and IV meperidine parturients were connected to a dummy saline PCIA.”, “A senior anaesthesiologist, not involved in data recording, attended each parturient throughout labor.”, "Patient satisfaction [...] was assessed 24 h after delivery by an anaesthesiologist blinded to the mode of labor analgesia." The study did not address this issue for most of the relevant outcomes with exception of the assessment of woman's satisfaction. We do not know who was responsible for other outcome assessment and attending personnel and parturients may be able to uncover group allocation. The subjective1 outcomes or outcome measurements are likely to be influenced by lack of blinding. Therefore, insufficient information exists to judge "yes" or "no". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | ‐ Dropout rate: 0% ‐ Large amount (2%/22%) of outcome data (AE for women: SpO2) not reported and imbalanced between groups. No reasons declared. ‐ Rate of escape (epidural): 12%/38% (may influence data on AE, satisfaction and pain) ‐ Rate of cross‐over: NA ‐ Data‐analysis: Partial‐ITT |
| Selective reporting (reporting bias) | Unclear risk | There is no reference to a trial registry and no published study protocol. |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Evron 2008.
| Methods | Randomised, controlled trial. Single‐blinded. Randomisation after onset of labour (VAS pain score ≥ 30 mm). The purpose of this trial was to test the hypothesis whether labour can induce hyperthermia during epidural analgesia, and to assess the effects of analgesic doses of the IV opioid remifentanil or antipyretic doses of acetaminophen in the prevention of hyperthermia during labour. The study was conducted in Wolfson Medical Center affiliated to Tel‐Aviv University (period unknown). Trial Identifier: NA |
|
| Participants |
Participant flow: Number assessed for eligibility: NA Number randomised: 213 Number receiving treatment: 201 (12 women which did not receive any analgesia were excluded) Number analysed: 192 (44/50/49/49), only women with at least 2 h of labour Inclusion criteria: Healthy women with singleton cephalic presentation at term, spontaneous active labour Exclusion criteria: Fever (oral temperature ≥ 38°C), signs of infection, ruptured membranes for more than 24 h, CS Baseline details: IV Remifentanil group (n = 44): Age (years, mean (SD)): 29 (7) Weight (kg, mean (SD)): 75 (11) ASA I/II (n/n): NA Type of delivery (n): spontaneous (NA), forceps (1), CS (4) Week of gestation: NA Singleton, twin, multiple pregnancy: singleton Number of birth (n)(1/2/3/≥ 4): 20/7/11/11 Duration of labour: ‐ First stage of labour (min, mean (SD)): NA ‐ Second stage of labour (min, mean (SD)): NA Epidural ropivacaine group (n = 50): Age (years, mean (SD)): 28 (5) Weight (kg, mean (SD)): 79 (14) ASA I/II (n/n): NA Type of delivery (n): spontaneous (NA), forceps (3), CS (5) Week of gestation: NA Singleton, twin, multiple pregnancy: singleton Number of birth (n) (1/2/3/≥ 4): 28/12/5/5 Duration of labour: ‐ First stage of labour (min, mean (SD)): NA ‐ Second stage of labour (min, mean (SD)): NA Epidural ropivacaine and IV Remifentanil group (n = 49): Age (years, mean (SD)): 27 (5) Weight (kg, mean (SD)): 78 (10) ASA I/II (n/n): NA Type of delivery (n): spontaneous (NA), forceps (3), CS (11) Week of gestation: NA Singleton, twin, multiple pregnancy: singleton Number of birth (n) (1/2/3/≥ 4): 25/9/4/7 Duration of labour: ‐ First stage of labour (min, mean (SD)): NA ‐ Second stage of labour (min, mean (SD)): NA Epidural ropivacaine and IV acetaminophen group (n = 49): Age (years, mean (SD)): 27 (4) Weight (kg, mean (SD)): 74 (14) ASA I/II (n/n): NA Type of delivery (n): spontaneous (NA), forceps (3), CS (3) Week of gestation: NA Singleton, twin, multiple pregnancy: singleton Number of birth (1/2/3/≥ 4): 29/13/4/3 Duration of labour: ‐ First stage of labour (min, mean (SD)): NA ‐ Second stage of labour (min, mean (SD)): NA |
|
| Interventions |
Remifentanil group (n = 44): Parturients randomised to PCIA with remifentanil initially received a basal infusion of 0.025 µg/(kg*min) combined with 20 µg bolus doses with a lockout of 3 min. The dose was increased by 25% every 15 to 20 min as required. Acetaminophen was administered by continuous infusion 30 min after the initiation of epidural analgesia with the PCIA machine device at a rate of 0.47 mg/(kg*min) to a maximal dose of 2 g. Epidural ropivacaine group (n = 50): Epidural analgesia was administered after pre‐hydration with 500 mL Ringer’s lactate solution. A test dose of 3 mL lidocaine (2% without epinephrine) was followed by increments of 5 mL to 10 mL of 0.2% ropivacaine; maintenance was provided with the same solution via patient‐controlled epidural analgesia (PCEA) with a background infusion of 10 mg/h and a 10 mg patient‐activated bolus with 20 min lockout. The maximal dose of ropivacaine was 20 mL/h. The same ropivacaine dose was administered to women in all epidural groups. Epidural ropivacaine and IV Remifentanil group (n = 49): Epidural analgesia was administered after pre‐hydration with 500 mL Ringer’s lactate solution. A test dose of 3 mL lidocaine (2% without epinephrine) was followed by increments of 5 mL to 10 mL of 0.2% ropivacaine; maintenance was provided with the same solution via PCEA with a background infusion of 10 mg/h and a 10 mg patient‐activated bolus with 20 min lockout. The maximal dose of ropivacaine was 20 mL/h. Parturients initially received a basal infusion of 0.025 µg/(kg*min) combined with 20 µg bolus doses with a lockout of 3 min. The dose was increased by 25% every 15 to 20 min as required. Epidural ropivacaine and IV acetaminophen group (n = 49): Epidural analgesia was administered after pre‐hydration with 500 mL Ringer’s lactate solution. A test dose of 3 mL lidocaine (2% without epinephrine) was followed by increments of 5 mL to 10 mL of 0.2% ropivacaine; maintenance was provided with the same solution via PCEA with a background infusion of 10 mg/h and a 10 mg patient‐activated bolus with 20 min lockout. The maximal dose of ropivacaine was 20 mL/h. Acetaminophen was administered by continuous infusion 30 min after the initiation of epidural analgesia with the PCIA machine device at a rate of 0.47 mg/(kg*min) to a maximal dose of 2 g. |
|
| Outcomes | The primary outcome was the incidence of hyperthermia. Continuous: ‐ pain intensity (VAS 0 to 100 mm, averaged over study period) Dichotomous: ‐ additional analgesia (conversion remifentanil (PCA) to epidural) ‐ rate of CS, rate of assisted birth (forceps) |
|
| Notes | ‐ Power analysis not described Concomitant medication: Women with breakthrough pain (VAPS > 30 mm) were given rescue analgesia: women in the ropivacaine or ropivacaine and acetaminophen groups were given up to 4 additional boluses of 8 mL ropivacaine (0.2%), even if they had reached the maximum dose specified above; and women in either remifentanil group had their baseline infusion and bolus doses increased, as necessary, in 25% increments. If 4 increases proved insufficient, women assigned to IV remifentanil were switched to epidural analgesia. Oxytocin augmentation was applied when the rate of cervical dilatation was less than 1 cm/h. Funding: Supported by NIH Grant GM 061655 (Bethesda, MD), the Gheens Foundation (Louisville, KY), the Joseph Drown Foundation (Los Angeles, CA), and the Commonwealth of Kentucky Research Challenge Trust Fund (Louisville, KY). Mallinckrodt Anesthesiology Products, Inc (St. Louis, MO) donated the thermocouples. Exergen, Inc (Boston, MA) donated the infrared skin‐temperature thermometer. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was based on computer‐generated codes […].” |
| Allocation concealment (selection bias) | Low risk | Quote: “Randomization was based on computer‐generated codes that were maintained in sequentially numbered opaque envelopes until just prior to use. The randomisation envelopes were opened and the designated treatment started when the visual analogue pain score (VAPS) reached 30 mm.” |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: “The treatment regimen was blinded for the evaluator anaesthesiologist by using two patient‐controlled analgesia machine devices (PCIA and PCEA) for every patient. A “dummy” IV saline infusion (PCIA) was attached to parturients with patient‐controlled epidural analgesia (PCEA) and the other was a “dummy” epidural catheter attached superficially to the skin and connected to a PCEA syringe in the group with patient‐controlled IV analgesia (PCIA) with remifentanil.” The study seemed to be not blinded for parturients and personnel and we assume that at least the subjective1 outcomes or outcome measurements are likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: “The treatment regimen was blinded for the evaluator anaesthesiologist by using two patient‐controlled analgesia machine devices (PCIA and PCEA) for every patient. A “dummy” IV saline infusion (PCIA) was attached to parturients with patient‐controlled epidural analgesia (PCEA) and the other was a “dummy” epidural catheter attached superficially to the skin and connected to a PCEA syringe in the group with patient‐controlled IV analgesia (PCIA) with remifentanil.” An attempt was made and reported in the method section to blind the outcome assessor. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ‐ Dropout rate: 10% (overall) Reasons for missing outcome data were described (12 women delivered quickly without requirement for analgesia and nine women with labour < 2 h were excluded). Reasons may be unlikely to be related to true outcome. ‐ Rate of escape: NA ‐ Rate of cross‐over: 0% (remifentanil (PCA) to epidural) ‐ Data‐analysis: Per‐protocol (women without requirement for analgesia or with labour < 2 h were excluded) |
| Selective reporting (reporting bias) | Unclear risk | There is no reference to a trial registry and no published study protocol. |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Freeman 2015.
| Methods | Multicentre randomised, controlled equivalence trial. No blinding. Randomisation before start of actual labour. The purpose of this trial was to determine women’s satisfaction with pain relief using PCA with remifentanil compared with epidural analgesia during labour. The study was conducted in three academic hospitals, 11 teaching hospitals, and one general hospital in the Netherlands from 30 May 2011 to 24 October 2012. Trial Identifier: NTR2551 |
|
| Participants |
Participant flow: Number assessed for eligibility: NA Number randomised: 1414 (709/705) Number receiving treatment (of interest): 698 (402/296) Number analysed: 1358 (687/671) Inclusion criteria: Healthy women or those who have a mild systemic disease (ASA I or II), aged 18 or older, scheduled to deliver vaginally after 32 weeks Exclusion criteria: Contraindications for epidural analgesia or hypersensitivity to one of the drugs used Baseline details: Remifentanil group (n = 687): Age (years, mean (SD)): 31.5 (5.1) BMI (kg/m², median (IQR)): 23.7 (21.5‐26.9) ASA I/II (n/n): 491/196 Type of delivery (n): spontaneous (518), instrumental (63), CS (106) Week of gestation (median (IQR)): 37.8 (35.5 ‐ 39.2) Singleton, twin, multiple pregnancy (n): multiple pregnancy (24) Parity (n): 0 (323), ≥ 1 (364) Duration of labour: ‐ First stage of labour (min, mean (IQR)): 236 (128 ‐ 376) ‐ Second stage of labour (min, mean (IQR)): 20 (10 ‐ 46) Epidural group (n = 671): Age (years, mean (SD)): 31.7 (4.8) BMI (kg/m², median (IQR)): 23.8 (21.4 ‐ 27.6) ASA I/II (n/n): 461/210 Type of delivery (n): spontaneous (501), instrumental (70), CS (100) Week of gestation (median (IQR)): 37.1 (35.3 ‐ 39.0) Singleton, twin, multiple pregnancy (n): multiple pregnancy (30) Parity (n): 0 (329), ≥ 1 (342) Duration of labour: ‐ First stage of labour (min, mean (IQR)): 309 (181 ‐ 454) ‐ Second stage of labour (min, mean (IQR)): 24 (10 ‐ 53) |
|
| Interventions |
Remifentanil group (n = 687): The patient‐controlled device was programmed to deliver 30 µg remifentanil (solution 20 µg/mL) on request with a lockout time of 3 min. The dose could be increased to 40 µg in case of insufficient pain relief or decreased to 20 µg in case of excessive side effects. No background infusion was allowed. Women who were treated with patient‐controlled remifentanil were instructed on how to use the device and to maximise analgesia by pressing the device’s button in anticipation of the next contraction. Epidural group (n = 671): Women randomised to epidural analgesia received this when they requested pain relief, according to local protocol. |
|
| Outcomes | The primary outcome measure was satisfaction with pain relief measured on a VAS ranging from 0 to 100 mm. Continuous: ‐ satisfaction with pain relief (VAS scale unclear, during active labour, after pain relief, at request, averaged), overall satisfaction (NRS 0 to 10, after birth) ‐ pain intensity (VAS scale unclear, during active labour, after pain relief, at request, averaged) Dichotomous: ‐ additional analgesia (escape analgesia) ‐ rate of CS, rate of assisted birth (instrumental) ‐ need for neonatal admission ‐ oxytocin use ‐ umbilical cord pH (artery), pH < 7.1 (twin 1) ‐ women: respiratory depression (< 8 breaths/min), oxygen desaturation (< 95%, < 92%), hypotension (< 90 mmHg), nausea, vomiting, pruritus, postpartum haemorrhage (≥ 1000 mL) ‐ newborns: Apgar score ≤ 7 at 5 min (twin 1) |
|
| Notes | ‐ Power analysis performed (satisfaction with pain relief, n = 102 per group) Concomitant medication: If pain relief was inadequate, women could request epidural analgesia. They were advised to discontinue using the device during the second stage of labour to minimise the risk of neonatal side effects. If pain relief after epidural analgesia was judged inadequate by the woman, she could receive patient‐controlled remifentanil instead of epidural analgesia. No advice was given regarding continuing epidural analgesia during the second stage of labour. Funding: This study was funded by a grant from ZonMW (Dutch Organization for Health Care Research and Development) project No 80‐82310‐97‐11039. Intervention: Lockout time of 3 min seems too long for adequate analgesia (borderline). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Randomisation was performed through a web based randomisation program. We randomised in fixed blocks of three, stratified for centre and parity.” |
| Allocation concealment (selection bias) | High risk | Quote (full‐text publication): “The allocation code appeared after a patient’s initials were entered into the randomisation program.” Quote (protocol: BMC Pregnancy and Childbirth 2012, 12: 63): "The consent form must be signed before performance of any study‐related activity. After obtaining informed consent women will be randomized and will be informed on the assigned method of pain relief before labour starts (as in usual care). They are only given pain relief during labour at their request or if a medical reason should arise." Allocation concealment was uncovered for participants and personnel before the start of treatment. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: “Blinding was not possible because of the nature of the two interventions.” No blinding and at least the subjective1 outcomes or outcome measurements are likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: “Blinding was not possible because of the nature of the two interventions.” No blinding and at least the subjective1 outcomes or outcome measurements are likely to be influenced by lack of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | ‐ Dropout rate: 3%/5% ‐ Large amount (5.5% to 41.5%) of data for maternal side effects and neonatal data were missing. No reasons reported. ‐ Randomisation occurred before onset of labour and only 65% and 52% received an analgesic intervention in the remifentanil and epidural group, respectively. Only 57% of women in the remifentanil group and only 42% in the epidural group received the allocated intervention. ‐ Rate of women with other pain relief (not escape, immediate use): 10%/15% ‐ Rate of cross‐over (insufficient pain relief): 13%/1% ‐ Rate of cross‐over (protocol violation): 9%/10% ‐ Data analysis: A partial ITT analysis with all women (including those without pain relief) was performed for rate of CS, assisted vaginal birth, postpartum haemorrhage, Apgar score < 7 at 5 min, and neonatal admission. A partial‐ITT with only women received pain relief (cross‐over participants as randomised) was performed for satisfaction, pain, cross‐over rate, and maternal side effects (SpO2, BP, respiratory depression, PONV, pruritus). ‐ Study design: equivalence study (per‐protocol analysis recommended) ‐ Multiple imputation was used to correct missing primary outcome data |
| Selective reporting (reporting bias) | High risk | A published protocol (BMC Pregnancy and Childbirth 2012, 12: 63) and a registered protocol (NTR2551) are available and there are several deviations. In the registered protocol cost‐effectiveness was defined as the primary outcome and pain relief, woman's satisfaction, pain scores, and maternal and neonatal side effects were defined as secondary outcomes. In the published report, however, the authors stated: “Our published protocol stated that both effectiveness and cost effectiveness were primary outcome measures. Satisfaction with pain relief was the primary outcome measure for effectiveness from the start of the study [which was not reported in the protocol]. We planned to perform a cost effectiveness analysis as well, taking into account the primary outcome for effectiveness. Because this was not made clear enough in the original protocol and registry it was changed in the last amended protocol. This last amended protocol was submitted before the last randomised woman delivered and as a result we did not have access to the data.” The first protocol (NTR2551) was prospectively registered: Study registration: 10/2010 First enrolment: 05/2011 The second protocol (BMC Pregnancy and Childbirth 2012, 12: 63) was retrospectively registered: Protocol received: 04/2012 First enrolment: 05/2011 |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Ismail 2012.
| Methods | Randomised, controlled trial. No statement on blinding. Randomisation after onset of labour. The purpose of this trial was to assess if there is a difference in duration of labour, the mode of delivery, average VAS pain scores, maternal overall satisfaction with analgesia, side effects and neonatal outcomes in nulliparous women who received early labour analgesia with either epidural, PCIA with remifentanil or combined spinal–epidural (CSE) techniques. The study was conducted in TAIBA Hospital in Kuwait during the period from September 2009 to August 2011. Trial Identifier: NA |
|
| Participants |
Participant flow: Number assessed for eligibility: 1460 Number randomised: 1140 (380/380/380), 320 women were excluded due to cervical dilation of 4 cm or more Number receiving treatment: 1140 (380/380/380) Number analysed: 1140 (380/380/380) Inclusion criteria: Spontaneous labour (with at least two painful uterine contractions in 10 min and the cervix is at least 80 % effaced and up to 3 cm dilated) and requesting labour analgesia Exclusion criteria: Allergy to opioids, a history of the use of centrally‐acting drugs of any sort, chronic pain, psychiatric diseases records, participants younger than 18 years or older than 40 years, not willing to, or could not finish the whole study, alcohol‐ or opioid‐dependent women, non‐vertex presentation or scheduled induction of labour, diabetes mellitus and pregnancy‐induced hypertension, twin gestation and breech presentation, any contraindication to neuraxial or systemic opioid analgesia, cervical dilation of 4 cm or more, estimated fetal weight above 4000 g and abnormal FHR tracing on admission Baseline details: Remifentanil group (n = 380): Age (years, mean (SD)): 28.35 (5.54) Weight (kg, mean (SD)): 81 (13) ASA I/II (n/n): NA Type of delivery (n): spontaneous (250), instrumental (35), CS (95) Week of gestation (mean (SD)): 39.2 (1.1) Singleton, twin, multiple pregnancy: singleton Parity: NA Duration of labour: ‐ First stage of labour (h, mean (SD)): latent phase: 7.7 (0.8), active phase: 1.80 (0.6) ‐ Second stage of labour (h, mean (SD)): 0.95 (0.4) Epidural group (n = 380): Age (years, mean (SD)): 28.6 (5.49) Weight (kg, mean (SD)): 83 (15) ASA I/II (n/n): NA Type of delivery (n): spontaneous (249), instrumental (36), CS (95) Week of gestation (mean (SD)): 39.0 (1.3) Singleton, twin, multiple pregnancy: singleton Parity: NA Duration of labour: ‐ First stage of labour (h, mean (SD)): latent phase: 7.8 (0.9), active phase: 1.88 (0.7) ‐ Second stage of labour (h, mean (SD)): 1.0 (0.5) Spinal‐epidural group (n = 380): Age (years, mean (SD)): 28.8 (5.50) Weight (kg, mean (SD)): 82 (14) ASA I/II (n/n): NA Type of delivery (n): spontaneous (255), instrumental (38), CS (87) Week of gestation (mean (SD)): 39.1 (1.2) Singleton, twin, multiple pregnancy: singleton Parity: NA Duration of labour: ‐ First stage of labour (min, mean (SD)): latent phase: 6.6 (0.7), active phase: 1.55 (0.4) ‐ Second stage of labour (h, mean (SD)): 0.80 (0.3) |
|
| Interventions |
Remifentanil group (n = 380): The PCIA device was set to deliver 0.1 µg/kg of Ultiva (remifentanil hydrochloride, Glaxo Operations UK Ltd, Barnard Castle, Durham, UK), diluted with saline and given as a solution of 25 µg/mL as a bolus infused during a period of 1 min, with a lockout time of 1 min, into an IV catheter attached to a 1‐way line providing continuous infusion of saline at approximately 100 mL/h. During the study, the IV PCIA bolus was increased following a dose escalation scheme (0.1 – 0.2 – 0.3 – 0.5 – 0.7 – 0.9 µg/kg) after every second contraction until the parturient answered ‘no’ to the question whether she would like to get more efficient pain relief or until a maximum dose of 0.9 µg/kg was achieved. Epidural group (n = 380): Blocks were performed in the sitting position. The epidural space was located at the L3–L4 interspace using loss of resistance to air (an 18‐gauge Tuohy needle was used). A 3 mL epidural test dose of 2% lidocaine was given through the epidural catheter. After the test dose, an 8 mL dose of 0.125% levobupivacaine with 2 µg/mL fentanyl was administered through the epidural catheter. Then the catheter was connected to an electronic pump set to deliver a continuous infusion of 8 mL/h of 0.125% levobupivacaine and 2 µg/mL fentanyl. Further boluses of 5 mL to 10 mL of 0.125% levobupivacaine were given by the attending anaesthesiologist upon request. Spinal‐epidural group (n = 380): Blocks were performed in the sitting position. The epidural space was located at the L3–L4 interspace using loss of resistance to air (an 18‐gauge Tuohy needle was used). A 3 mL epidural test dose of 2% lidocaine was given through the epidural catheter. A needle‐through‐needle technique was performed with 2 mg levobupivacaine and 15 µg fentanyl (total volume of 2 mL) was injected intrathecally and the spinal needle was removed. Then the epidural catheter was inserted and connected to an electronic pump set to deliver the same previously mentioned mixture. |
|
| Outcomes | The primary outcome was the rate of caesarean delivery. Continuous: ‐ overall satisfaction (VRS 1 to 4, 24 h after delivery) ‐ pain intensity (VAS 0 to 100, averaged) ‐ umbilical cord pH (artery, vein) ‐ women: mean respiratory rate, mean systolic and diastolic blood pressure, mean HR (averaged, respectively) Dichotomous: ‐ rate of CS, rate of assisted birth (instrumental) ‐ umbilical cord pH (artery), pH < 7.2 ‐ oxytocin use after analgesia ‐ women: nausea, vomiting, pruritus ‐ newborns: Apgar score < 7 at 1 and 5 min, non‐reassuring fetal status (indication for CS), need for naloxone |
|
| Notes | ‐ Power analysis not performed Concomitant medication: NA Funding: NA Intervention: Maximum dose might be too high (might cause adverse effects). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “[…] the participants were randomized (in 3 blocks of 380 participants per block) through a computer‐generated, random‐number list […]” |
| Allocation concealment (selection bias) | Unclear risk | Quote: “The group assignment numbers were sealed in an envelope and kept by the study supervisor.” Not specifically mentioned sequentially numbered, opaque envelopes (SNOSE). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The study did not address this issue. However, we assume that blinding of parturients and personnel did not occur due to technical reasons and at least the subjective1 outcomes or outcome measurements are likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | The study did not address this issue. However, we assume that blinding of outcome assessment did not occur due to technical reasons and at least the subjective1 outcomes or outcome measurements are likely to be influenced by lack of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ‐ No missing outcome data after randomisation ‐ Rate of escape: NA ‐ Rate of cross‐over: NA ‐ Data‐analysis: Full‐ITT |
| Selective reporting (reporting bias) | Unclear risk | There is no reference to a trial registry and no published study protocol. The study reported only significant positive results in the abstract. The non‐significant primary outcome was not there reported. |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Khooshideh 2015.
| Methods | Randomised, controlled trial. Single‐blinded. Randomisation after onset of labour. The purpose of this trial was to compare the efficacy and adverse maternal and neonatal effects of remifentanil given by bolus PCA versus continuous IV infusion for labour analgesia. The study was conducted at the Department of Anesthesiology of Ali Ebn‐e Abitaleb Hospital, Zahedan, Iran from January 2010 to March 2013. Trial Identifier: IRCT2012100811020N2 |
|
| Participants |
Participant flow: Number assessed for eligibility: NA Number randomised: 82 (41/41) Number receiving treatment: 82 (41/41) Number analysed: 82 (41/41) Inclusion criteria: Aged 18 to 35 years, gestational ages of 37 to 40 weeks Exclusion criteria: BMI > 30 or < 20 kg/m², pre‐eclampsia, using psychiatric drugs, opioid or alcohol consumption, occurrence of antenatal haemorrhage, fetal distress and requesting epidural analgesia Baseline details: Remifentanil infusion group (n = 41): Age (years, mean (SD)): 24.83 (4.67) BMI (kg/m², mean (SD)): 24.07 (2.21) ASA I/II (n/n): NA Type of delivery: NA Week of gestation (mean (SD)): 38.61 (1.16) Singleton, twin, multiple pregnancy: NA Parity: NA Duration of labour: ‐ First stage of labour (min, mean (SD)): 165.3 (38.7) ‐ Second stage of labour (min, mean (SD)): 42.1 (12) Remifentanil bolus group (n = 41): Age (years, mean (SD)): 24.83 (4.67) BMI (kg/m², mean (SD)): 24.07 (2.21) ASA I/II (n/n): NA Type of delivery: NA Week of gestation (mean (SD)): 38.49 (1.23) Singleton, twin, multiple pregnancy: NA Parity: NA Duration of labour: ‐ First stage of labour (min, mean (SD)): 153.2 (34.2) ‐ Second stage of labour (min, mean (SD)): 40 (10.3) |
|
| Interventions |
Remifentanil infusion group (n = 41): Remifentanil was incrementally infused with the starting dosage of 0.025 µg/(kg*min) and as required the infusion rate was increased to reach the doses of 0.05, 0.075 and 0.1 µg/(kg*min). Remifentanil bolus group (n = 41): Remifentanil was given by bolus PCA using an IVAC PCAM model P5000 pump, with the starting dosage of 0.25 µg/kg and as required increased to reach the dose of 0.4 µg/kg with a lockout time of 4 min. |
|
| Outcomes | The primary endpoint of the study was reduction of labour pain. Continuous: ‐ pain intensity (VNRS 0 to 10, at baseline, averaged at stage 1 (every 15 min) and at stage 2 (every 15 min)) ‐ averaged oxytocin use ‐ sedation score (observer, MOAA/S 1 to 5, after remifentanil administration) Dichotomous: ‐ overall satisfaction (good to excellent, time point unclear) ‐ women: respiratory depression (< 8 breaths/min), oxygen desaturation (< 90 %), hypotension (< 90 mmHg), bradycardia (< 50 beats/min), nausea + vomiting ‐ newborns: Apgar score < 7 at 1 min, need for naloxone |
|
| Notes | ‐ Small trial sample size (< 200 participants) ‐ Power analysis not performed Concomitant medication: Remifentanil dosage was increased when VNRS was ≥ 7. Remifentanil was discontinued if any of the following criteria were detected: HR < 50 beats/min, SBP < 90 mmHg, SPO2 < 90% and respiratory rate (RR) < 8 breaths/min. Based on the standard protocols, oxytocin was infused in cases with inappropriate labour progress. Funding: Zahedan University of Medical Sciences Intervention: Lockout time 4 min (bolus group) seems too long for adequate analgesia. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “A computerized random number generator was used for sequence generation. Simple randomisation with a 1:1 allocation ratio was used in this study.” |
| Allocation concealment (selection bias) | Unclear risk | Quote: “We used the consecutive opaque envelopes for the allocation concealment. The envelopes were opaque when held to the light and opened sequentially and only participant’s name and other details were written on the appropriate envelope.” |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: “This randomized, single‐blind clinical trial…” The authors did not describe how they have blinded the parturients and personnel. We assume from the description of the intervention that blinding was not possible since a PCA pump was used only in the remifentanil PCA group. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | No statement on blinding of outcome assessors. However, we assume from the description of the intervention that blinding was not possible since a PCA pump was used only in the remifentanil PCA group. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ‐ No missing outcome data after randomisation ‐ Rate of escape: NA ‐ Rate of cross‐over: NA ‐ Data‐analysis: Full‐ITT |
| Selective reporting (reporting bias) | Unclear risk | The study protocol (IRCT2012100811020N2) is available and all pre‐specified primary and secondary outcomes have been reported in the final report. However, the outcomes remifentanil dose, maternal satisfaction, and maternal and neonatal side effects have not been pre‐specified in the protocol. The protocol was retrospectively registered: Protocol registration: 02/2013 First enrolment: 01/2012 |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Ng 2011.
| Methods | Randomised, controlled trial. Double‐blinded. Randomisation after onset of labour. The purpose of this trial was to compare the efficacy of PCA remifentanil with IM pethidine for labour analgesia. There are no details where or when the study was conducted. The authors’ origin is China. Trial Identifier: NA |
|
| Participants |
Participant flow: Number assessed for eligibility: 69 Number randomised: 68 (34/34) Number receiving treatment: 68 (34/34) Number analysed: 68 (34/34) Inclusion criteria: Full term (36 to 40 weeks' gestation) parturients, ASA I and II, in the first stage of spontaneous labour, who requested parenteral opioid for labour analgesia Exclusion criteria: Complicated obstetric history (such as gestational diabetes, pregnancy induced hypertension or antepartum haemorrhage), multiple pregnancies, non‐cephalic presentation Baseline details: Remifentanil group (n = 34): Age (years, mean (SD)): 28 (5) Weight (kg, mean (SD)): 68.2 (11.1) ASA I/II (n/n): 24/10 Type of delivery: NA Week of gestation (median (IQR [range])): 39 (38 ‐ 40 [37 ‐ 41]) Singleton, twin, multiple pregnancy: singleton Parity (n): 0 (30), ≥ 1 (4) Duration of labour: ‐ First stage of labour (min, mean (SD)): NA ‐ Second stage of labour (min, mean (SD)): NA Pethidine group (n = 34): Age (years, mean (SD)): 29 (5) Weight (kg, mean (SD)): 68.0 (8.9) ASA I/II (n/n): 27/7 Type of delivery: NA Week of gestation (median (IQR [range])): 39 (39 ‐ 40 [37 ‐ 41]) Singleton, twin, multiple pregnancy: singleton Parity (n): 0 (28), ≥1 (6) Duration of labour: ‐ First stage of labour (min, mean (SD)): NA ‐ Second stage of labour (min, mean (SD)): NA |
|
| Interventions |
Remifentanil group (n = 34): All parturients were provided with a PCA device (Omnifuse PCA, Smiths Medical, Kent, UK). The machine was loaded with a 50 mL syringe of study drug containing remifentanil 20 µg/mL. For each successful PCA demand, an IV bolus of 1.25 mL study drug (remifentanil 25 µg) was delivered to parturients weighing < 60 kg and 1.5 mL (remifentanil 30 µg) for those weighing ≥ 60 kg. The bolus was delivered at a rate of 20 mL/h. The effective lockout interval was 3.75 – 4.50 min with an hourly limit of 25 mL. A background infusion was not used. Parturients in the remifentanil group received an IM injection of 1.5 mL saline 0.9%. Parturients were then instructed to press the PCA demand button as soon as they felt the start of uterine contraction. Pethidine group (n = 34): All parturients were provided with a PCA device (Omnifuse PCA, Smiths Medical, Kent, UK). The machine was loaded with a 50 mL syringe of study drug containing 0.9% saline. For each successful PCA demand, an IV bolus of 1.25 mL study drug (saline) was delivered to parturients weighing < 60 kg and 1.5 mL (saline) for those weighing ≥ 60 kg. The bolus was delivered at a rate of 20 mL/h. The effective lockout interval was 3.75 to 4.50 min with an hourly limit of 25 mL. A background infusion was not used. Parturients were then instructed to press the PCA demand button as soon as they felt the start of uterine contraction. A single IM injection of pethidine 50 mg diluted to 1.5 mL with saline was given to parturients weighing < 60 kg and pethidine 75 mg in 1.5 mL to those weighing ≥ 60 kg. |
|
| Outcomes | The primary endpoint of the study was VAS pain score during the entire duration of the study. Continuous: ‐ overall satisfaction (NRS 0 to 10, after delivery, median + IQR + range (symmetric)) ‐ pain intensity (VAS 0 to 100, at 0 h, every hour until 4 h, > 5 h, diagrammed) ‐ women: mean highest respiratory rate, oxygen saturation, SBP and pulse rate ‐ newborns: Apgar score at 1 and 5 min (median + IQR + range (symmetric)), mean FHR Dichotomous: ‐ additional analgesia (Entonox, pethidine IM) ‐ rate of CS, rate of assisted birth (ventouse) ‐ syntocinon use ‐ women: nausea, vomiting, pruritus (women reporting VAS ≥ 30 mm or requiring treatment, respectively), dizziness, sedation score = 1 = alert ‐ newborns: fetal distress with impaired CTG |
|
| Notes | ‐ Small trial sample size (< 200 participants) ‐ Power analysis performed (VAS pain, n = 34 per group) Concomitant medication: Rescue analgesia with IM pethidine and Entonox was offered to parturients with VAS > 50 mm or upon request. The time from the start of the study to the first request for rescue analgesia was recorded. Funding: NA Intervention: Bolus application time seems too slow (0.11 µg/s). Contact to the authors: We contacted Dr. Ng via e‐mail (23 June 2016) to inquire the number of women who reported 'pain intensity at 2 hours'. We received the missing data. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “parturients were randomly assigned to receive either PCA remifentanil or intramuscular pethidine for labour analgesia according to a computer‐generated code […].” |
| Allocation concealment (selection bias) | Unclear risk | Quote: “[…] code concealed in an opaque envelope.” Not specifically mentioned sequentially numbered, sealed envelopes (SNOSE). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: “All parturients were provided with a PCA device (Omnifuse PCA, Smiths Medical, Kent, UK). The machine was loaded with a 50‐ml syringe of study drug containing either remifentanil 20 µg/ml or 0.9% saline.”, “Parturients in the remifentanil group received an intramuscular injection of 1.5 ml saline 0.9%.”, “Study drugs were prepared by an anaesthetist not otherwise involved in the study.”, “The parturient, the attending obstetricians, midwives and research staff responsible for data collection and outcome assessment were blinded to the group identity.” Blinding was attempted to achieve by application of both a PCA pump and an IM injection. However, we assume that participants and attending personnel might be able to uncover group allocation due to the different pharmacokinetics of the two interventions. The used method of blinding may only work for the outcome assessors. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: “All parturients were provided with a PCA device (Omnifuse PCA, Smiths Medical, Kent, UK). The machine was loaded with a 50‐mL syringe of study drug containing either remifentanil 20 µg/mL or 0.9% saline.”, “The parturient, the attending obstetricians, midwives and research staff responsible for data collection and outcome assessment were blinded to the group identity.” An attempt was made and reported in the method section to blind the outcome assessor. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ‐ No missing outcome data after randomisation ‐ Rate of escape (pethidine IM or Entonox): 50%/85% (may influence data on AE, satisfaction and pain) ‐ Rate of cross‐over: NA ‐ Data‐analysis: Full‐ITT |
| Selective reporting (reporting bias) | Unclear risk | There is no reference to a trial registry and no published study protocol. |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Shen 2013.
| Methods | Randomised, controlled trial. Double‐blinded. Randomisation after onset of labour. The purpose of this trial was to compare the maternal and neonatal effects of remifentanil given by PCA or continuous infusion for labour analgesia. The study was conducted in China from July 2008 and September 2009. Trial Identifier: NA |
|
| Participants |
Participant flow: Number assessed for eligibility: 60 Number randomised: 60 (30/30) Number receiving treatment: 60 (30/30) Number analysed: 53 (27/26) Inclusion criteria: ASA I to II, singleton term pregnancy, a cervical dilation of 1 to 3 cm, healthy fetus with a cephalic presentation, normal FHR pattern Exclusion criteria: Inability to understand PCA and VAS score, age less than 18 years or more than 45 years, morbid obesity, prior administration of regional or systemic analgesia, alcohol abuse, diabetes mellitus, pregnancy‐induced hypertension, pre‐eclampsia, severe disease of brain, heart, lung, liver or kidney Baseline details: Remifentanil bolus group (n = 27): Age (years, mean (SD)): NA Weight (kg, mean (SD)): NA ASA I/II (n/n): NA Type of delivery: NA Week of gestation: NA Singleton, twin, multiple pregnancy: NA Parity: NA Duration of labour: ‐ First stage of labour (min, mean (SD)): NA ‐ Second stage of labour (min, mean (SD)): NA Remifentanil infusion group (n = 26): Age (years, mean (SD)): NA Weight (kg, mean (SD)): NA ASA I/II (n/n): NA Type of delivery: NA Week of gestation: NA Singleton, twin, multiple pregnancy: NA Parity: NA Duration of labour: ‐ First stage of labour (min, mean (SD)): NA ‐ Second stage of labour (min, mean (SD)): NA |
|
| Interventions |
Remifentanil bolus group (n = 30): Two syringe pumps (Graseby 3300; Graseby Medical Ltd., Watford, UK) were connected, one set up for PCA and the other for continuous infusion. A nurse made up two labelled syringes, one with remifentanil and the other with 0.9% saline. Remifentanil (Yichang Humanwell Pharmaceutical Co., Ltd., Yichang, Hubei, China) was diluted to 10 µg/mL with saline 0.9%. The initial PCA set‐up was with a bolus of 0.1 µg/kg given over 30 s with a 2‐min lockout interval. The parturients were advised to press the PCA button at the start of a uterine contraction. No additional training was given to parturients in respect of how to recognise the beginning of a contraction, and the decision of whether to press the PCA button depended on the parturient alone. The initial continuous infusion was with a dose of 0.05 µg/(kg*min). The PCA bolus dose was increased in increments of 0.1 µg/kg from 0.1 to 0.4 µg/kg. The continuous infusion pump was increased stepwise from 0.05 to 0.2 µg/(kg*min) with an increment of 0.05 µg/(kg*min). The two pumps were disconnected at the time of delivery, and the final dose of remifentanil was recorded. Remifentanil infusion group (n = 30): Two syringe pumps (Graseby 3300; Graseby Medical Ltd., Watford, UK) were connected, one set up for PCA and the other for continuous infusion. A nurse made up two labelled syringes, one with remifentanil and the other with 0.9% saline. The continuous infusion pump was increased stepwise from 0.05 to 0.2 µg/(kg*min) with an increment of 0.05 µg/(kg*min). The two pumps were disconnected at the time of delivery, and the final dose of remifentanil was recorded. |
|
| Outcomes | The primary endpoint of the study was not explicitly stated but power analysis was performed for pain score. Continuous: ‐ overall satisfaction (NRS 0 to 10, 1 h after delivery, median + IQR + range (asymmetric)) ‐ pain intensity (VAS 0 to 10, 0, every 30 min until 120 min, delivery, diagrammed), median pain relief score (NRS 0 to 5, 0, every 30 min until 120 min, delivery, diagrammed) (median + IQR + range, respectively (symmetric)) ‐ sedation score (observer, Ramsey sedation score, at 0, 30, 60, 120 min and after delivery, median + IQR + range (symmetric)) Dichotomous: ‐ additional analgesia (epidural) ‐ need for neonatal resuscitation ‐ women: respiratory depression (< 8 breaths/min), oxygen desaturation (< 95%), hypotension, bradycardia, nausea, vomiting, pruritus ‐ newborns: non‐reassuring FHR/transient fetal bradycardia, need for naloxone, respiratory depression |
|
| Notes | ‐ Small trial sample size (< 200 participants) ‐ Power analysis performed (pain score, n = 30 per group) Concomitant medication: During labour, if the parturient requested a higher dose of analgesia then both pumps were adjusted simultaneously. To avoid potential hypoxaemia, parturients received oxygen by nasal catheter with 2 L/min oxygen flow when there was an episode of desaturation, which was defined as SpO2 < 95%, and the dose of remifentanil was not increased. The bolus or the infusion was discontinued in the following circumstances: HR < 50 beats/min, respiratory rate < 8 breaths/min, SpO2 < 90%, mean arterial pressure > 25% decrease from baseline, FHR < 110 beats/min, or sedation score > 4. When these variables returned to a normal level, the remifentanil administration was restarted at a dose one step lower than that preceding the event. If any adverse reactions persisted, the trial was stopped, the parturient was excluded and the appropriate treatment was followed. For adverse reactions, unsatisfactory analgesia, or unwillingness to continue the trial, epidural analgesia was provided unless there was a contraindication. The decision to cross‐over to epidural analgesia was made by the parturient in collaboration with the anaesthesiologist. Adverse reactions were recorded throughout the labour. Funding: This study was supported by grant YKK08119 and YKK11058 from Medical Science and Technology Development Foundation, Nanjing Department of Health, Nanjing, Jiangsu, China, grant CSZ00838 from Wuxi Municipal Science and Technology Projects, Wuxi, Jiangsu, China and grant 08NMUM063 from the Science & Technology Development Foundation of Nanjing Medical University, Nanjing, Jiangsu, China. Intervention: Application time of remifentanil seems too long (0.003 µg/(kg*s). Contact to the authors: We contacted Dr. Shen via e‐mail (23 June 2016) to inquire the number of women who reported 'pain intensity at 2 hours'. We received the missing data. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “The randomisation code was generated by computer […].” |
| Allocation concealment (selection bias) | Low risk | Quote: “[…] and then sealed in sequentially numbered, opaque envelopes before the beginning of the trial.”, “Only one investigator who selected the envelope knew the grouping.” |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: “The anaesthesiologists who set up the analgesia and recorded the data, the nurses who prepared the medication, midwives, obstetricians and mothers were all blinded to the group allocation. Only one investigator who selected the envelope knew the grouping.”, “Two syringe pumps […] were connected to the cannula, one set up for PCA and the other for continuous infusion. A nurse made up two labelled syringes, one with remifentanil and the other with 0.9% saline. Remifentanil […] was diluted to 10 µg/ml with saline 0.9%. The investigator opened the randomisation envelope and then put the syringes into the appropriate pumps according to the group allocation. The syringe label was then covered with black paper.” Blinding for participants and personnel adequate. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: “The anaesthesiologists who set up the analgesia and recorded the data, the nurses who prepared the medication, midwives, obstetricians and mothers were all blinded to the group allocation. Only one investigator who selected the envelope knew the grouping.”, “Two syringe pumps […] were connected to the cannula, one set up for PCA and the other for continuous infusion. A nurse made up two labelled syringes, one with remifentanil and the other with 0.9% saline. Remifentanil … was diluted to 10 µg/ml with saline 0.9%. The investigator opened the randomisation envelope and then put the syringes into the appropriate pumps according to the group allocation. The syringe label was then covered with black paper.” Blinding for outcome assessment adequate. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | ‐ Dropout rate: 10%/13% Quote: “Two women in the PCA group and four subjects in the infusion group chose to cross over to epidural analgesia because of inadequate analgesia despite using the maximum dose, and there was one protocol violation in the PCA group. These mothers delivered their babies spontaneously, with Apgar scores > 9 at 1 min and 10 at 5 min, and no data were included from these subjects.” The protocol violation was not described. Reasons for missing outcome data likely to be related to true outcome. ‐ Rate of escape (epidural): 7%/13% ‐ Rate of cross‐over: NA ‐ Data‐analysis: Per‐protocol |
| Selective reporting (reporting bias) | Unclear risk | There is no reference to a trial registry and no published study protocol. |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Stocki 2014.
| Methods | Randomised, controlled trial. Not blinded. Randomisation after onset of labour. The purpose of this trial was to compare the analgesia efficacy and maternal satisfaction of remifentanil labour analgesia with standard treatment (epidural analgesia). The study was conducted in a Jerusalem tertiary hospital labour and delivery suite from February 2010 to August 2010. Trial Identifier: NCT00801047 |
|
| Participants |
Participant flow: Number assessed for eligibility: 144 Number randomised: 40 (20/20) Number receiving treatment: 39 (19/20) Number analysed: 39 (19/20) Inclusion criteria: Healthy, ASA I or II, age 18 to 40 years, body weight < 110 kg, gestational age > 36 completed weeks, with singleton pregnancy and vertex presentation Exclusion criteria: Contraindication to epidural analgesia, opioid administration in the previous 2 h, previous uterine surgery, pre‐eclampsia, inability to understand the consent form, nasal obstruction for any reason, medical indication for epidural analgesia (e.g. cardiac disease, suspected difficult airway), or non‐reassuring FHR tracing Baseline details: Remifentanil group (n = 19): Age (years, mean (SD)): 31 (5) Weight (kg, mean (SD)): 72 (10) ASA I/II (n/n): NA Type of delivery (n): spontaneous (NA), vacuum delivery (2), CS (0) Week of gestation: NA Singleton, twin, multiple pregnancy: NA Parity (median (IQR) or n): 1 (1‐1), Nulliparous (4) Duration of labour: ‐ First stage of labour (min, mean (SD)): 329 (215) ‐ Second stage of labour (min, mean (SD)): 35 (41) Epidural group (n = 20): Age (years, mean (SD)): 30 (6) Weight (kg, mean (SD)): 73 (13) ASA I/II (n/n): NA Type of delivery (n): spontaneous (NA), vacuum delivery (1), CS (4) Week of gestation: NA Singleton, twin, multiple pregnancy: NA Parity (median (IQR) or n): 1 (0‐1), Nulliparous (6) Duration of labour: ‐ First stage of labour (min, mean (SD)): 404 (259) ‐ Second stage of labour (min, mean (SD)): 69 (81) |
|
| Interventions |
Remifentanil group (n = 19): PCA: The bolus dose was titrated to effect from 20 µg up to a maximum of 60 µg as required; the lockout interval was initially set at 2 min, without a background infusion. The PCIA bolus/lockout interval was titrated to an endpoint of either woman's comfort or a maximal bolus dose of 60 µg/minimal lockout interval of 1 min by the recruiting anaesthesiologist at any time during labour. The PCIA pump tubing was “piggybacked” into the distal most port of the mainline IV fluid tubing. The mainline tubing contained an antireflux valve designed to prevent remifentanil inadvertently backing up in the IV line during administration. Epidural group (n = 20): A 17‐gauge Tuohy needle was inserted by the midline approach using loss of resistance to air at intervertebral space L3‐4 or L2‐3. An incremental initial loading dose of 15 mL of 0.1% bupivacaine with 50 µg fentanyl was administered followed by patient‐controlled epidural analgesia infusion of 0.1% bupivacaine with 2 µg/mL fentanyl: basal infusion of 5 mL/h, patient‐controlled bolus 10 mL, and lockout interval 20 min. Additional epidural bolus doses (either 0.1% bupivacaine 10 mL during the first stage of labour or 1% lidocaine 8 mL during the second stage of labour) were administered by the anaesthesiologist to treat breakthrough pain. If epidural analgesia failed, the epidural catheter was reinserted. |
|
| Outcomes | The primary endpoint of the study was to demonstrate non‐inferiority of remifentanil labour analgesia compared with epidural analgesia in labouring women, measured by hourly assessment of NRS for pain throughout the duration of labour and maternal satisfaction. Continuous: ‐ satisfaction with pain relief (NRS 0 to 10, at 10 min and postpartum) ‐ pain intensity (NRS 0 to 10, at 0, 30 min, 1, 2, 3, 4, 5, 6 h) ‐ newborns: Apgar score at 1 and 5 min (median + IQR + range (symmetric), data for Apgar score at 5 min dichotomised: all newborn had an Apgar score of 10 at 5 min) Dichotomous: ‐ additional analgesia after 1 h (rescue), additional analgesia (cross‐over to the other treatment arm) ‐ rate of CS, rate for assisted birth (vacuum) ‐ need for neonatal resuscitation ‐ umbilical cord BE (artery), umbilical cord pH (artery) ‐ oxytocin use ‐ sedation score (observer, awake or easily arousable at 1 h) ‐ women: apnoea (> 20 s of zero respiratory rate), respiratory depression (< 8 breaths/min), oxygen desaturation (< 94%), nausea (at 1 h and postpartum), pruritus (at 1 h and postpartum) |
|
| Notes | ‐ Small trial sample size (< 200 participants) ‐ Power analysis performed (NRS pain, n = 17 per group) Concomitant medication: Women were informed before study enrolment that conversion to the other treatment would be possible at any time during labour beginning 30 min after analgesia initiation with the study technique. All women received continuous supplementary oxygen (2 L/min) through an oral‐nasal cannula. Funding: Hadassah Medical Organisation Contact to the authors: We contacted Dr. Weiniger via e‐mail (16 March 2016) to inquire the number of women who reported 'pain intensity at 30 min, 1, and 2 hours'. We received the missing data. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Randomization and group allocation were determined: cards were divided into groups of 8 cards. Each group contained 4 allocation cards for remifentanil and 4 allocation cards for epidural analgesia (ratio 1:1), and 8 opaque envelopes numbered in groups from 1 – 8, 9 – 16, etc. were assigned to each group of cards. The cards were placed face down, manually shuffled, randomly selected, and then inserted into the numbered, opaque envelopes by a person not involved in the study. These envelopes were then sealed. Treatment assignment was revealed by breaking the seal of an envelope in consecutive order from number 1.” |
| Allocation concealment (selection bias) | Low risk | Quote: “…inserted into the numbered, opaque envelopes by a person not involved in the study. These envelopes were then sealed. Treatment assignment was revealed by breaking the seal of an envelope in consecutive order from number 1.” |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: “This randomized nonblinded controlled noninferiority study […].” The study was not blinded due to technical reasons and at least the subjective1 outcomes or outcome measurements are likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: “The investigator inquired whether opioid side effects (i.e., pruritus and/or nausea and vomiting) were present or absent.”, “After delivery, face‐to‐face follow‐up was performed on the first postpartum day for both mother and child by one of the investigators”, “Hence, the mathematician was not blinded to group allocation. However, she was blinded to our study hypothesis that remifentanil was noninferior to epidural analgesia and that remifentanil may have respiratory effects different from those seen with epidural analgesia.” The study did not adequately report on blinding of outcome assessors. Therefore, we assume that the study was not blinded due to technical reasons and at least the subjective1 outcomes or outcome measurements are likely to be influenced by lack of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ‐ Dropout rate: 5%/0% One woman was excluded after enrolment but before analgesia due to obstetrician request. Reasons for missing outcome data unlikely to be related to true outcome. ‐ Rate of escape (other pain relief after 1 h): 0%/17% (may influence data on AE, satisfaction and pain) ‐ Rate of cross‐over: 16%/5% ‐ Data‐analysis: Partial‐ITT ‐ Study design: non‐inferiority study (per‐protocol analysis recommended) |
| Selective reporting (reporting bias) | High risk | A registered protocol (NCT00801047) is available and there are several deviations. The VAS pain score was defined as primary outcome and no other primary or secondary outcomes were defined in the protocol. In the published report, however, the authors defined maternal satisfaction as another primary endpoint, and incidence of apnoea was defined as secondary outcome. The primary outcome pain score was reported on a 11‐point NRS scale in the final report. The authors further reported on SpO2, sedation, pruritus, nausea, and adverse effects on the newborn. The protocol was prospectively registered: Protocol registration: 12/2008 First enrolment: 02/2010 |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Stourac 2014.
| Methods | Randomised, controlled trial. No statement on blinding. Randomisation after onset of labour. The purpose of this trial was to compare the analgesic efficacy of parturient‐controlled IV remifentanil administration with epidural analgesia during first stage of labour with regard to maternal and early neonatal side effects. The study was conducted in Czech Republic from March 2010 to May 2010. Trial Identifier: NA |
|
| Participants |
Participant flow: Number assessed for eligibility: 81 Number randomised: 28 (13/15) Number receiving treatment: 28 (13/15) Number analysed: 24 (12/12) Inclusion criteria: No comorbidity (ASA I), singleton head‐down full term pregnancy, spontaneous or induced labour, request for labour analgesia Exclusion criteria: NA Baseline details: Remifentanil group (n = 12): Age (years, mean (SD)): 27.9 (2.95) Weight (kg, mean (SD)): 84 (16.75) ASA I/II (n/n): NA Type of delivery (n): spontaneous (2), induced (10) Week of gestation (weeks + days, mean (SD)): 39 + 3 (1 + 3) Singleton, twin, multiple pregnancy: singleton Parity (n): 1 (10), 2 (2), 3 (0) Duration of labour: ‐ First stage of labour (min, mean (SD)): 260.8 (65.5) ‐ Second stage of labour (min, mean (SD)): 11.25 (10.01) Epidural group (n = 12): Age (years, mean (SD)): 29.4 (2.05) Weight (kg, mean (SD)): 85.83 (16.75) ASA I/II (n/n): NA Type of delivery (n): spontaneous (4), induced (8) Week of gestation (weeks + days, mean (SD)): 40 + 0 (1 + 0) Singleton, twin, multiple pregnancy: singleton Parity (n): 1 (8), 2 (3), 3 (1) Duration of labour: ‐ First stage of labour (min, mean (SD)): 246.7 (76.3) ‐ Second stage of labour (min, mean (SD)): 13.75 (13.51) |
|
| Interventions |
Remifentanil group (n = 13): After the parturient's consent, her peripheral vein was cannulated and infusion of normal saline up to 2 mL/(kg*h) was started. The PCA device was connected to the same cannula. Remifentanil (Ultiva, Glaxo‐Smith‐Kline, Great Britain) was then administered via the PCA device (Technic 1, AMV Technics, Czech Republic) from a 50 mL syringe in a concentration of 20 µg/mL. On demand, the parturient received an IV bolus of 20 µg (1 mL) of remifentanil. Lockout interval was set to 3 min. The significant analgesic effect was set to a minimal VAS score decrease of 2, based on existing evidence. In case of inadequate analgesic VAS decrease (< 2), it was possible for the anaesthesiologist to increase the dose in 10 µg steps. Epidural group (n = 15): The parturients undergoing EA had an epidural catheter inserted into the epidural space by an anaesthesiologist. The dosage regimen of bupivacaine and sufentanil in the EA group was rigidly set to induction dose of 12.5 mg of bupivacaine and 5 µg of sufentanil in 10 mL of normal saline; top‐up boluses of half‐size dose were repeated in 60‐min to 90‐min intervals. The epidural catheter was extracted after the delivery, at least 2 hours before the parturient's transport to post‐natal care ward. |
|
| Outcomes | The primary endpoint of the study was not defined. Continuous: ‐ overall satisfaction (mean % score + range, at 2 ‐ 24 h after delivery) ‐ pain intensity (VAS 0 to 10, 0, 30 min, until delivery (4 h), median + IQR (symmetric)) ‐ umbilical cord pH (not specified) ‐ newborns: Apgar score at 1, 5 and 10 min Dichotomous: ‐ rate of CS ‐ women: hypotension (> 25% decrease from baseline), bradycardia (< 50 beats/min), nausea + vomiting, drowsiness + dizziness ‐ newborns: pathological CTG (conversion to CS) |
|
| Notes | ‐ Small trial sample size (< 200 participants) ‐ Power analysis not performed Concomitant medication: If any signs of sedation (drowsiness), dizziness, muscle rigidity or bradypnoea (< 10 breaths/min) were observed, the lockout interval could be extended by 1 min. Funding: NA Contact to the authors: We contacted Dr. Stourac via e‐mail (29 June 2016) to inquire details on the method used for randomisation. We received the missing information. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Women who had asked for obstetric analgesia and met the inclusion criteria were offered the PCA using remifentanil (randomisation by the parturient)." On request the trial author offered the following information on the randomisation method: "Then she rolled the dice and based on the result she was assigned into remi (even) or epi (odd) groups". |
| Allocation concealment (selection bias) | Low risk | Participants and investigators enrolling participants could not foresee assignment because of the method used for randomisation (throwing dice). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The study did not address this issue. However, we assume that blinding of parturients and personnel did not occur due to technical reasons and at least the subjective1 outcomes or outcome measurements are likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | The study did not address this issue. However, we assume that blinding of outcome assessment did not occur due to technical reasons and at least the subjective1 outcomes or outcome measurements are likely to be influenced by lack of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | ‐ Dropout rate: 8%/20% Quote: “The data of the parturients whose pregnancies were terminated by Caesarean Section were excluded from statistical evaluation. Four pregnancies were terminated by Caesarean Section (2 of them due to labour progress stagnancy, 2 because of a pathological cardiotocography (CTG) record (1 in each branch)).” Reasons for missing outcome data likely to be related to true outcome. ‐ Rate of escape: NA ‐ Rate of cross‐over: NA ‐ Data‐analysis: Per‐protocol |
| Selective reporting (reporting bias) | Unclear risk | There is no reference to a trial registry and no published study protocol. |
| Other bias | Unclear risk | Early stopping. Quote: “28 parturients met the requirements to take part in this prospective randomised trial. This low count was caused by high parturient refusal to participate in the study (N=53) despite agreement with labour analgesia. We are aware of the decreased power with the lower sample size; nevertheless the 95% confidence interval for endpoint estimate in the groups showed clinically non‐significant results within its whole range which supports our findings from the statistical testing. Therefore we decide to terminate the enrolment.” |
Thurlow 2002.
| Methods | Randomised, controlled trial. No statement on blinding. No statement on time of randomisation. The purpose of this trial was to compare the analgesic effect of remifentanil given as PCA with IM meperidine during labour. There are no details where or when the study was conducted. The authors’ origin is the UK. Trial Identifier: NA |
|
| Participants |
Participant flow: Number assessed for eligibility: 36 Number randomised: 36 (18/18) Number receiving treatment: 36 (18/18) Number analysed: 36 (18/18) Inclusion criteria: Aged between 18 and 40 years, between 38 to 42 weeks of gestation in early labour Exclusion criteria: < 50 kg or >100 kg Baseline details: Remifentanil group (n = 18): Age (years, median (IQR)): 28 (22 ‐ 32) Weight (kg, median (IQR)): 66.5 (58 ‐ 78) ASA I/II (n/n): NA Type of delivery (n): spontaneous (11), ventouse (4), CS (3) Week of gestation (median (IQR)): 40.1 (39.25 ‐ 41) Singleton, twin, multiple pregnancy: NA Parity (n): 1 (13), 2 (0), 3 (3), 4 + (2) Duration of labour: ‐ First stage of labour (min, mean (SD)): NA ‐ Second stage of labour (min, mean (SD)): NA Meperidine group (n = 18): Age (years, median (IQR)): 29 (25 ‐ 30) Weight (kg, median (IQR)): 64 (58 ‐ 76) ASA I/II (n/n): NA Type of delivery (n/n): spontaneous (16/17), ventouse (1/17), CS (0/17) Week of gestation (median (IQR)): 39.6 (39 ‐ 40) Singleton, twin, multiple pregnancy: NA Parity (n): 1 (13), 2 (0), 3 (2), 4 + (0) Duration of labour: ‐ First stage of labour (min, mean (SD)): NA ‐ Second stage of labour (min, mean (SD)): NA |
|
| Interventions |
Remifentanil group (n = 18): PCA: starting with a 5 µg bolus, an increasing dose of remifentanil was given at the beginning of each painful contraction until the contraction was pain‐free, and the next painful contraction was noted. Meperidine group (n = 18): IM Meperidine 100 mg |
|
| Outcomes | The primary endpoint of the study was not explicitly defined but power analysis was performed for pain score. Continuous: ‐ midwives' and mother's assessment of overall effective analgesia (VRS 1 to 5, within 2 h after delivery, individual patient data) ‐ pain intensity (VAS 0 to 100, at 0 and 1 h), max. pain score over 2 h (VAS 0 to 100) (median + IQR, respectively (asymmetric)) ‐ sedation score (unclear assessor, VAS 0 to 100, at baseline, median + IQR) Dichotomous: ‐ additional analgesia (Entonox, epidural) ‐ rate of CS, rate of assisted birth (ventouse) ‐ syntocinon use ‐ women: respiratory depression (< 8 breaths/min), oxygen desaturation (< 94%), nausea + vomiting |
|
| Notes | ‐ Small trial sample size (< 200 participants) ‐ Power analysis performed (pain score, n = 30 in total) ‐ Abstract: fewer number of women ‐ The exact intervention in the remifentanil group remains unclear (“20 µg bolus over 20 s, 3 min lockout and no background infusion”) Concomitant medication: If the assigned analgesia was inadequate for the woman at any time, an alternative was offered (epidural analgesia) and further study recordings were discontinued. All women had access to Entonox at all times. Remifentanil group: No antiemetic was given unless indicated clinically. Meperidine group: antiemetic was given (promethazine 25 mg or prochlorperazine 12.5 mg) Funding: NA Intervention: Lockout time of 3 min seems too long for adequate analgesia (borderline). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “[…] women were assigned randomly to one of two groups by sequentially numbered, sealed opaque envelopes prepared by an independent practitioner.” The method used for randomisation was not described. |
| Allocation concealment (selection bias) | Low risk | Quote: “[…] sequentially numbered, sealed opaque envelopes…” |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The study did not address this issue. However, we assume that blinding of parturients and personnel did not occur due to technical reasons and at least the subjective1 outcomes or outcome measurements are likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | The study did not address this issue. However, we assume that blinding of outcome assessment did not occur due to technical reasons and at least the subjective1 outcomes or outcome measurements are likely to be influenced by lack of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | ‐ Dropout rate: 0% ‐ There were no withdrawals. However, there are some outcome data (additional analgesia, mode of delivery) incomplete (missing from notes) without reasons. ‐ Rate of escape (Entonox): 55%/81% (may influence data on AE, satisfaction and pain) ‐ Rate of escape (epidural): 39%/17% ‐ Rate of cross‐over: NA ‐ Data‐analysis: Partial‐ITT |
| Selective reporting (reporting bias) | Unclear risk | There is no reference to a trial registry and no published study protocol. |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Tveit 2012.
| Methods | Randomised, controlled trial. Not blinded. Randomisation after onset of labour. The purpose of this trial was to compare the analgesic efficacy and side effects of remifentanil (PCA) with walking epidural analgesia during labour, with regard to maternal and early neonatal side effects. There are no details where or when the study was conducted. The authors’ origin is Norway. Trial Identifier: NCT00202722 |
|
| Participants |
Participant flow: Number assessed for eligibility: 62 Number randomised: 39 (19/20) Number receiving treatment: 39 (19/20) Number analysed: 37 (17/20) Inclusion criteria: Mixed parity, ASA I or II with normal singleton pregnancies, regular uterine contractions, cervical dilatation more than 2 cm, anticipated vaginal delivery, fetus without suspected abnormality, normal fetal cardiotocographic pattern, no complications during pregnancy, gestation age 37 to 40 weeks Exclusion criteria: Request for an epidural, had received pethidine less than 8 h before the study period, contraindications to remifentanil Baseline details: Remifentanil group (n = 17): Age (years, mean (range)): 26 (20 ‐ 33) Weight (kg, mean (SD)): 79 (13.7) ASA I/II (n/n): NA Type of delivery (n): spontaneous (NA), forceps/ventouse (2), CS (1) Week of gestation: NA Singleton, twin, multiple pregnancy: NA Parity (n): Primiparous (12), Multiparous (5) Duration of labour: ‐ First stage of labour (min, mean (SD)): 360 (185.9) ‐ Second stage of labour (min, mean (SD)): 51 (33.5) Epidural group (n = 20): Age (years, mean (range)): 27 (20 ‐ 37) Weight (kg, mean (SD)): 80 (8.7) ASA I/II (n/n): NA Type of delivery (n): spontaneous (NA), forceps/ventouse (3), CS (3) Week of gestation: NA Singleton, twin, multiple pregnancy: NA Parity (n): Primiparous (11), Multiparous (9) Duration of labour: ‐ First stage of labour (min, mean (SD)): 359 (165.5) ‐ Second stage of labour (min, mean (SD)): 42 (32.2) |
|
| Interventions |
Remifentanil group (n = 17): Those randomised to the RA group received remifentanil hydrochloride (Ultiva, GlaxoSmithKline, Oslo, Norway) diluted in physiological saline to a concentration of 50 µg/mL, given as stepwise bolus doses with no background infusion. The starting bolus dose was 0.15 µg/kg, with increasing dose steps of 0.15 µg/kg and no maximum limit. The dose was allowed to be increased or decreased every 15 min according to the woman’s request for dose adjustment, VAS pain score and side effects. The lockout period was 2 min. Remifentanil was administered using a PCA pump (Baxter 6060 Multi‐Therapy infusion pump, Baxter Healthcare Corporation, Kista, Sweden) with a bolus infusion speed of 2 mL/min (100 µg/min). Calculation of PCA doses was based on estimated bodyweight using the following formula: body height in centimetres ‐ 100 = estimated weight (kg). Epidural group (n = 20): Parturient women randomised to the epidural group had an epidural catheter inserted in the midline at L2–3/L3–4 by the investigator. They received a continuous epidural infusion of ropivacaine 1 mg/mL and fentanyl 2 µg/mL (‘walking epidural’). An initial bolus dose of 10 mL, followed by a 5 mL top‐up after 5 min (total 15 mL) was given before the start of infusion (10 mL/h). Thereafter, the midwife was allowed to adjust the infusion dose (5 mL/h to 15 mL/h) and give rescue doses (5 mL) if needed. If pain relief was inadequate, the position of the epidural catheter was adjusted or a new catheter was placed if necessary. |
|
| Outcomes | The primary endpoint of the study was not defined but power analysis was performed for VAS pain reduction. Continuous: ‐ pain intensity (VAS 0 to 100, at 0, 30 min, until 240 min) ‐ sense of control in labour (VRS 1 to 5, within 24 h after delivery, median + range) ‐ umbilical cord BE (artery, vein), umbilical cord pH (artery, vein) ‐ newborns: Apgar score at 1, 5 and 10 min (median + range) Dichotomous: ‐ satisfaction with pain relief (very satisfied, recommend analgesia, same analgesia next time) ‐ additional analgesia (rescue, epidural group: additional bolus), additional analgesia (conversion to EA) ‐ rate of CS, rate of assisted birth (ventouse, forceps) ‐ neonatal abnormalities ‐ oxytocin use ‐ sedation score (parturient, sedated or very sedated, discomfort by sedation, amnesia from labour) ‐ women: respiratory depression (< eight breaths/min), oxygen desaturation (< 92%, supplemental oxygen), nausea, vomiting, pruritus, drowsiness ‐ newborns: Apgar score < 8 at 1 min, pathological FHR changes |
|
| Notes | ‐ Small trial sample size (< 200 participants) ‐ Power analysis performed (VAS pain reduction, n = 26 per group) ‐ If delivery took place within 30 min of analgesia, the woman was excluded. Concomitant medication: Oxytocin, metoclopramide, ephedrine and IV fluids were available if needed. Supplemental oxygen (4 L/min) was given immediately via nasal cannula if SaO2 was less than 92%. Remifentanil analgesia was temporarily stopped if SaO2 less than 92% persisted or respiratory frequency was less than nine breaths/min, SBP was less than 90 mmHg or HR was less than 50 beats/min. When physiological variables returned to normal, pain therapy was continued on a dose 1 step lower. Funding: The work was funded by Sørlandet Hospital HF, Sørlandets Kompetansefond and Helse Sør‐Øst, Norway. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Randomisation was based on a computer‐generated list [...].” |
| Allocation concealment (selection bias) | Unclear risk | Quote: “[…] codes were kept in sealed envelopes until recruitment..” Not specifically mentioned sequentially numbered, opaque envelopes (SNOSE). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: “Our study has some limitations; it was not blinded, […].” The study was not blinded due to technical reasons and at least the subjective1 outcomes or outcome measurements are likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: “[…] for observer sedation score […] the anaesthesiologist and attending midwife scored independently.”, “After the study, two additional obstetricians, blinded to the analgesia method and neonatal outcome, independently evaluated FHR recordings.” The study was not blinded due to technical reasons and at least all other subjective1 outcomes or outcome measurements are likely to be influenced by lack of blinding. However, the authors attempted to minimise bias by duplicate outcome assessment for the outcomes observer sedation score and FHR. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | ‐ Dropout rate: 10%/0% Two women (10%) were excluded (per‐protocol analysis) from the remifentanil group due to cross‐over to the epidural group (reasons: suspicious FHR changes and inadequate analgesia). However, there are five women in the epidural group who received an extra bolus dose (rescue medication) due to unsatisfactory analgesia and were not excluded. ‐ There were missing outcome data (15% to 70%) regarding neonatal outcomes without reasons. ‐ Rate of escape (additional epidural bolus): NA/25% ‐ Rate of cross‐over: 10%/NA, ‐ Data‐analysis: Per‐protocol |
| Selective reporting (reporting bias) | High risk | A registered protocol (NCT00202722) is available. However, there are large discrepancies between the protocol and the published study report. The protocol deals only with a single group assignment (remifentanil PCA) and 41 enrolled women (39 within the published report), whereupon we contacted the study author. The author confirmed on request by mail that this protocol matches the published study. The protocol was retrospectively registered: Protocol registration: 09/2005 First enrolment: 01/2004 |
| Other bias | Unclear risk | Early stopping. Quote: “We had a technical problem with our infusion pumps after inclusion of 39 patients, and were not able to find new pumps with exactly the same technical specifications. Therefore, we closed the study at this point, leaving us with a number of participants close to the estimation from the power calculation.” |
Volikas 2001.
| Methods | Randomised, controlled trial. Double‐blinded. Randomisation after onset of labour. The purpose of this trial was to compare the efficacy of analgesia and side effects of a remifentanil PCA and pethidine PCA, using a VAS scoring system, throughout the first and second stages of labour. There are no details where or when the study was conducted. The authors’ origin is the UK. Trial Identifier: NA |
|
| Participants |
Participant flow: Number assessed for eligibility: 17 Number randomised: 17 (9/8) Number receiving treatment: 17 (9/8) Number analysed: 17 (9/8) Inclusion criteria: Healthy women at 36 to 40 weeks’ gestation, ASA I or II, with no known obstetric complications, and requesting pethidine analgesia Exclusion criteria: Any contraindication to pethidine or remifentanil and a request for early epidural analgesia in labour Baseline details: Remifentanil group (n = 9): Age (years, mean (SD)): 28.6 (4.7) Weight (kg, mean (SD)): 81.1 (24.1) ASA I/II (n/n): NA Type of delivery (n): spontaneous (5), forceps/ventouse (2), CS (2) Week of gestation: NA Singleton, twin, multiple pregnancy: NA Parity (n): primiparous (5), multiparous (4) Duration of labour (min, mean (SD)): 362 (300) ‐ First stage of labour (min, mean (SD)): NA ‐ Second stage of labour (min, mean (SD)): NA Pethidine group (n = 8): Age (years, mean (SD)): 28.9 (4.6) Weight (kg, mean (SD)): 67.2 (14.3) ASA I/II (n/n): NA Type of delivery (n): spontaneous (5), forceps/ventouse (2), CS (1) Week of gestation: NA Singleton, twin, multiple pregnancy: NA Parity (n): primiparous (4), multiparous (4) Duration of labour (min, mean (SD)): 348 (283) ‐ First stage of labour (min, mean (SD)): NA ‐ Second stage of labour (min, mean (SD)): NA |
|
| Interventions |
Remifentanil group (n = 9): PCA (IVAC PCAM model P5000 pump (ALARIS Medical Systems, UK)) with a remifentanil bolus of 0.5 µg/kg, a lockout period of 2 min and no hourly maximum limit. The remifentanil bolus was calculated using the maternal weight recorded at the antenatal booking clinic. Pethidine group (n = 8): PCA (IVAC PCAM model P5000 pump (ALARIS Medical Systems, UK)) with a pethidine bolus 10 mg, a lockout period of 5 min and a maximum limit of 100 mg per h. |
|
| Outcomes | The primary endpoint of the study was not defined but power analysis was performed for VAS pain. Continuous: ‐ pain intensity (VAS 0 to 100, at 0 h, every hour until 5 h, diagrammed) ‐ women: mean nausea and pruritus (VAS 0 to 100, initial and mean hourly) ‐ newborns: Apgar score at 1 and 5 min (median + range) Dichotomous: ‐ additional analgesia (Entonox and epidural) ‐ rate of CS, rate of assisted birth (forceps/ventouse) ‐ need for neonatal admission ‐ syntocinon use before and after PCA ‐ women: respiratory depression (< 12 breaths/min), hypotension, bradycardia ‐ newborns: Apgar score < 7 at 5 min, need for naloxone |
|
| Notes | ‐ Small trial sample size (< 200 participants) ‐ Power analysis performed (VAS pain, n = 17 in total) Concomitant medication: All the women in both groups were given metoclopramide 10 mg IV every 8 h. They received IV ondansetron 8 mg for persisting nausea and vomiting. The use of Entonox for any period during labour was noted. The PCA was stopped if epidural analgesia was requested but the VAS scores to that point were included in the analysis. Funding: NA Intervention: high bolus starting dose (0.5 mg/kg) Contact to the authors: We contacted Dr. Volikas via e‐mail (23 June 2016) to inquire the number of women who reported 'pain intensity at 2 hours'. We did not receive any answer. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “The women were randomly allocated to one of two groups by selecting the next in a series of sealed envelopes prepared by our pharmacy department.” It is not specifically mentioned that the envelopes were opaque. Therefore, it might be possible that sequence generation is influenced by specifically selecting the preferred group. |
| Allocation concealment (selection bias) | Unclear risk | Quote: “…in a series of sealed envelopes prepared by our pharmacy department.” Not specifically mentioned sequentially numbered, opaque envelopes (SNOSE). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: “The women and observer were blind to the treatment. This was achieved by having two investigators. One investigator selected the envelope and prepared the PCA pump with the appropriate drug. The pump was covered so that the other investigator, the observer, was unable to see which drug the woman was receiving.” We assume that participants and attending personnel might be able to uncover group allocation due to the different pharmacokinetics of the two interventions. The used method of blinding may only work for the outcome assessors. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: “The women and observer were blind to the treatment. This was achieved by having two investigators. One investigator selected the envelope and prepared the PCA pump with the appropriate drug. The pump was covered so that the other investigator, the observer, was unable to see which drug the woman was receiving. The observer, who was an anaesthetist, stayed on the delivery suite at all times to record the visual analogue scale (VAS) scores and provide continuous monitoring of the individual under study. The observer did not have any other commitments on the labour ward.” An attempt was made to blind the outcome assessor. However, we assume, that the outcome assessor who stayed on the delivery suite all the time might be able to uncover group allocation due to the different pharmacokinetics of the two interventions. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ‐ No missing outcome data after randomisation. ‐ Rate of escape (Entonox): 44%/63% (may influence data on AE, satisfaction and pain) ‐ Rate of escape (epidural): 11%/13% ‐ Rate of cross‐over: NA ‐ Data‐analysis: Full‐ITT |
| Selective reporting (reporting bias) | Unclear risk | There is no reference to a trial registry and no published study protocol. |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Volmanen 2008.
| Methods | Randomised, controlled trial. Double‐blinded. Randomisation after onset of labour. The purpose of this trial was to evaluate if IV PCA with remifentanil could provide as satisfactory pain relief for labour as epidural analgesia. There are no details where or when the study was conducted. The authors’ origin is Finland. Trial Identifier: NA |
|
| Participants |
Participant flow: Number assessed for eligibility: 52 Number randomised: 52 (27/25) Number receiving treatment: 51 (27/24) Number analysed: 45 (24/21) Inclusion criteria: Healthy term parturients with uncomplicated singleton pregnancies, normal cephalic presentation, no prior administration of opioid analgesia for at least 4 h or regional analgesia Exclusion criteria: NA Baseline details: Remifentanil group (n = 24): Age (years, median (IQR)): 27 (24 ‐ 32) Weight (kg, median (IQR)): 80 (73 ‐ 86) ASA I/II (n/n): NA Type of delivery (n): spontaneous (NA), vacuum extraction (4), CS (1) Week of gestation: NA Singleton, twin, multiple pregnancy: singleton Parity (n): primiparous (17), multiparous (7) Duration of labour: ‐ First stage of labour (min, mean (SD)): NA ‐ Second stage of labour (min, mean (SD)): NA Epidural group (n = 21): Age (years, median (IQR)): 28 (27 ‐ 31) Weight (kg, median (IQR)): 79 (75 ‐ 87) ASA I/II (n/n): NA Type of delivery (n): spontaneous (NA), vacuum extraction (1), CS (1) Week of gestation: NA Singleton, twin, multiple pregnancy: singleton Parity (n): primiparous (16), multiparous (5) Duration of labour: ‐ First stage of labour (min, mean (SD)): NA ‐ Second stage of labour (min, mean (SD)): NA |
|
| Interventions |
Remifentanil group (n = 24): The PCA (Graseby 3300, Graseby Medical Ltd, Watford, UK) device was set to deliver 0.1 mg/kg of Ultiva (remifentanil hydrochloride, Glaxo Operations UK Ltd, Barnard Castle, Durham, UK), diluted with saline and given as a solution of 25 µg/mL as a bolus infused during a period of 1 min, with a lockout time of 1 min, into an IV catheter attached to a 1‐way line providing continuous infusion of saline at approximately 100 mL/h. In order to mimic a normal clinical situation, the subjective signs of anticipating the next uterine contraction were not specified, and no attempts were made to train the parturient in early recognition of the onset of contractions. The decision as to whether to start the PCA dose was left solely to the woman. Epidural saline was used for blinding during remifentanil administration. The first epidural bolus was given simultaneously with the first PCA bolus. During the study, the IV PCA bolus was increased following a dose escalation scheme (0.1 – 0.2 – 0.33 – 0.5 – 0.7 – 0.9 µg/kg) after every second contraction until the parturient answered ‘no’ to the question whether she would like to get more efficient pain relief or until a maximum dose of 0.9 mg/kg was achieved. If she answered ‘no’ and her pain score was higher than what she had estimated before as acceptable for herself, she was asked why she did not want to get more efficient pain relief. The study period was terminated if the parturient answered ‘yes’ when she was asked whether she wanted more efficient pain relief although the 0.9 µg/kg bolus had been reached, if the parturient wished to stop participation for any reason, or if the midwife noted that the parturient had reached full cervical opening. Epidural group (n = 21): An epidural catheter was sited at the L2/3 lumbar interspace. The parturient received epidurally 20 mL of levobupivacaine (0.625 mg/mL) and fentanyl 2 µg/mL in saline divided into 2 10 mL boluses given by the researcher manually with a 5‐min interval. IV saline was used for blinding in the epidural group. The first epidural bolus was given simultaneously with the first PCA bolus. After the study period was over, an epidural bolus was given when the parturient requested more efficient pain relief. |
|
| Outcomes | The primary endpoint of the study was not defined but power analysis was performed for pain score. Continuous: ‐ pain relief score (NRS 0 to 4, every 10 min until 60 min), median pain relief score (NRS 0 to 4, averaged, median + IQR (symmetric)) ‐ pain intensity (NRS 0 to 10, every 10 min until 60 min, median + IQR (asymmetric)) ‐ umbilical cord pH (artery, median + IQR (asymmetric)) ‐ sedation score (VRS 0 to 3, average over 60 min every 10 min) ‐ women: mean blood pressure, mean HR ‐ newborns: Apgar score at 1 min (median + IQR (asymmetric)) Dichotomous: ‐ rate of CS, rate of assisted birth (vacuum) ‐ oxytocin use (started or increased during the study) ‐ women: oxygen desaturation (< 95%, supplemental oxygen), nausea (before and during the study) ‐ newborns: abnormal FHR (during and 30 min after the study period) |
|
| Notes | ‐ Small trial sample size (< 200 participants) ‐ Power analysis performed (Pain score, n = 20 per group) ‐ The parturients were requested to use sunglasses in order to hide the miosis peculiar to systemic opioid treatment. ‐ The women used the study medicines during a 60‐min study period, which was thought to be long enough for a complete dose escalation for the IV PCA medication. Concomitant medication: SaO2 < 95% was treated with supplemental oxygen at a rate of 2 L/min via nasal cannula. Funding: NA |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “The patients were randomly allocated to two groups using sealed envelopes numbered according to a computer‐generated list that was stratified according to parity.” |
| Allocation concealment (selection bias) | Unclear risk | Quote: “[…] using sealed envelopes numbered according to a computer‐generated list.” Not specifically mentioned opaque envelopes (SNOSE). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: “[…] epidural saline was used for blinding during remifentanil administration. IV saline was used for blinding in the epidural group.”, “Midwives and nurses not involved in the study prepared the medications and placebo syringes. Both the parturient and all the personnel present during the study were blinded as to which medication was used during the study. The parturients were requested to use sunglasses in order to hide the miosis peculiar to systemic opioid treatment.” Blinding was attempted to achieve by insertion of both an epidural catheter and a PCA pump. However, we assume that participants and attending personnel might be able to uncover group allocation due to the different pharmacokinetics of the two interventions. The used method of blinding may only work for the outcome assessors (which was not mentioned in the published report). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: “The FHR tracings were analysed afterwards by an obstetrician who was blinded to the method of analgesia and the outcome of the newborn.” The study did not address this issue for most of the relevant outcomes with exception of the assessment of FHR patterns. We do not know who was responsible for outcome assessment and attending personnel may be able to uncover group allocation. The subjective1 outcomes or outcome measurements are likely to be influenced by lack of blinding. Therefore, insufficient information exists to judge "yes" or "no". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | ‐ Dropout rate: 11%/16% It is unclear from the description why and how many women in each group were excluded: two vs. four (flow diagram: three vs. three) women discontinued the intervention due to insufficient pain relief (flow diagram: due to entering second stage of labour); one woman (epidural) did not receive allocated intervention (dural tap). Exclusion of parturients due to insufficient pain relief is likely to be related to true outcome. ‐ Rate of escape: NA ‐ Rate of cross‐over: NA ‐ Data‐analysis: Per‐protocol |
| Selective reporting (reporting bias) | Unclear risk | There is no reference to a trial registry and no published study protocol. |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
1Subjective outcomes: satisfaction with pain relief, adverse events for women, adverse events for the newborn, pain intensity, additional analgesia required, rate of caesarean delivery, rate of assisted vaginal birth, satisfaction with childbirth experience, sense of control in labour, effect (negative) on mother/baby interaction, breastfeeding initiation, need for neonatal resuscitation, long‐term childhood development, cost.
Objective outcomes: umbilical cord base excess (arterial and venous), umbilical cord pH (arterial and venous), vomiting, postpartum haemorrhage
Abbreviations:
AE: adverse events; ASA: American Society of Anesthesiologists; BE : base excess ; BMI: Body‐Mass‐Index; CS: caesarean section; CSE : combined spinal‐epidural analgesia ; CTG: cardiotocography; EA: epidural analgesia ; FHR: fetal heart rate; HR: heart rate;IM: intramuscular; IQR: interquartile range; ITT: intention‐to‐treat; IV: intravenous; MOAAS: Modified Observer's Assessment of Alertness/Sedation ; NA: not applicable; NACS: neurologic and adaptive capacity score ; NRS: numerical rating scale; PCA: patient‐controlled analgesia; PCIA: patient‐controlled IV analgesia ; PONV: p ostoperative nausea and vomiting ; RPCA: remifentanil patient‐controlled analgesia ; SBP: systolic blood pressure; SD: stand ard deviation; VAS: visual analogue scale; VNRS: verbal numerical rating scale ; VRS: verbal rating scale .
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Balcioglu 2007 | Wrong intervention (PCA versus PCA) |
| Jost 2013 | Cross‐over trial |
| Shahriari 2007 | No patient‐controlled anaesthesia |
| Solek‐Pastuszka 2009 | No randomisation |
| Varposhti 2013 | Cross‐over trial |
| Volmanen 2004 | Cross‐over trial |
| Volmanen 2005 | Cross‐over trial |
| Volmanen 2009 | Cross‐over trial |
| Volmanen 2011 | Cross‐over trial |
PCA: patient‐controlled analgesia
Characteristics of studies awaiting assessment [ordered by study ID]
Abdalla 2015.
| Methods | Randomised, controlled trial, double‐blinded. The purpose of this trial is to assess whether the combination of dexmedetomidine (DMET) with remifentanil produces a synergistic effect that results in lower analgesic requirements. The study was conducted in Ain Shams University Hospital, Cairo, Egypt. There are no details when the study was conducted. Trial identifier: NA |
| Participants |
Inclusion criteria: pregnant women, ASA I‐II, full term (37‐40 weeks), singleton fetus with cephalic presentation, first stage of spontaneous labour Exclusion criteria: known relevant drug allergy, significant respiratory depression from previous exposure to opioids, obstetric complications |
| Interventions | Remifentanil PCA + DMET versus remifentanil PCA + placebo |
| Outcomes | VAS pain scores, maternal and fetal complications, patients' satisfaction |
| Notes | This study is expected to be excluded due to wrong intervention (PCA versus PCA, same regimen). |
Godinho 2016.
| Methods | Case report: "Labour analgesia challenge ‐ when epidural is not possible, what can we do?" The author's origin is Portugal. |
| Participants | Pregnant 31 year‐old female, 86 kg, 1.63 m, with severe scoliosis |
| Interventions | Remifentanil PCA |
| Outcomes | Pain score, occurrence of vomiting, neonatal outcome (Apgar score) |
| Notes | This case report is expected to be excluded (not RCT). |
Gunes 2014.
| Methods | Randomised, controlled trial. The purpose of this study is to compare two different remifentanil PCA protocols (bolus and bolus + infusion) with intramuscular meperidine for labour analgesia. The study was conducted in Cukurova University Medical Faculty, Turkey. There are no details when the study was conducted. Trial identifier: NA |
| Participants |
Inclusion criteria: pregnant women, ASA I‐II, mean gestational age of 270 ± 10 days, planned for vaginal delivery Exclusion criteria: gestational pathology, obesity (BMI > 40 kg/m2), high risk cases (pre‐eclampsia, severe asthma, insulin‐dependent diabetes, hepatorenal disease), history of opioid allergy, long‐term opioid use or chronic pain |
| Interventions | remifentanil PCA bolus versus remifentanil PCA bolus + infusion versus meperidine IM |
| Outcomes | pain‐comfort and sedation scores, remifentanil consumption, side effects, Apgar scores |
| Notes | NA |
Karadjova 2016.
| Methods | Insufficient information about study design. |
| Participants | Inclusion criteria: pregnant women, spontaneous labour, ASA I |
| Interventions | Continuous epidural infusion versus remifentanil PCA |
| Outcomes |
The primary outcome is maternal and neonatal safety. The secondary outcome is efficacy evaluated through hourly pain and satisfaction scores. |
| Notes | There is no full text available (abstract only). |
Kondoh 2016.
| Methods | Parallel randomised trial investigating mosaprid in patients receiving remifentanil for labour analgesia. The study is not yet recruiting. The author's origin is Japan. Trial identifier: JPRN‐UMIN000021322 |
| Participants |
Inclusion criteria: pregnant women with the wish for labour pain relief, age > 20 years. Exclusion criteria: patients who can not consent, < 22 weeks of pregnancy, history of a high degree of hypersensitivity reactions to other drugs, impaired consciousness. |
| Interventions | Mosaprid administration versus mosaprid 5 mg every 4 h during the first labour phase versus no treatment |
| Outcomes |
The primary outcome is pain control and the presence or absence of maternal respiratory depression. Secondary outcomes are drug administration time, drug bolus administration number of times, the presence or absence of labour induction, patient satisfaction, final delivery mode, Apgar score, the presence or absence of umbilical cord arterial blood pH, birthweight, neonatal complications. |
| Notes | This study is expected to be excluded (wrong intervention). |
Leong 2015.
| Methods | Safety study with single‐group assignment investigating vital‐sign patient‐assisted intravenous analgesia with remifentanil. The study is not yet recruiting. The author's origin is Singapore. Trial identifier: NCT02733835 |
| Participants |
Inclusion criteria: patients who choose to use parenteral opioid for pain relief, written informed consent, refuse labour epidural analgesia, contraindication to epidural analgesia, gestational age ≥ 36 weeks. Exclusion criteria: inability to understand instructions or to self‐administer PCA boluses, language differences, hypersensitivity to remifentanil or any component of its formulation or to other fentanyl analogue, severe respiratory disease, history of drug dependence or recreational drug abuse, unmanaged fetal bradycardia. |
| Interventions | patient‐assisted IV remifentanil |
| Outcomes |
The primary outcome is maternal desaturation. The secondary outcomes are apnoea/hypopnoea and maternal bradycardia. |
| Notes | This study is expected to be excluded (no control intervention). |
Logtenberg 2016.
| Methods | Randomised, controlled equivalence trial. Not blinded. The purpose of this study is to compare pain appreciation during labour between RPCA and EA. The study was conducted in the Academic Medical Center, Amsterdam, NL from September 2012 to May 2013. Trial identifier: NTR3687 |
| Participants |
Inclusion criteria: age > 18 years, ASA I or II, low‐risk pregnant women Exclusion criteria: drug allergy: history of hypersensitivity to opioid or local anaesthetic, substances, labour before 32 weeks or after 42 weeks of gestation, initial maternal SpO2 of less than 95%, initial maternal temperature of 38°C or higher, prior administration of regional of opioid analgesia (during this delivery) |
| Interventions | Epidural anaesthesia versus remifentanil PCA |
| Outcomes |
The primary endpoint of this study is pain appreciation, expressed by women's satisfaction with pain on a VAS scale, measured hourly from the onset of active labour. Secondary outcomes are overall satisfaction with pain during delivery judged 2 h and 6 weeks after delivery, pain scores during labour and maternal and neonatal side effects. |
| Notes | This publication belongs to the ongoing study abstract Logtenberg 2014. |
Moreira 2016.
| Methods | Case report: "General anesthesia with target controlled infusion of propofol and remifentanil for planned caesarean section in a parturient with unrupted intracranial aneurysm" The authors' origin is Portugal. |
| Participants | NA. |
| Interventions | Remifentanil target‐controlled intravenous infusion and propofol for general anaesthesia. |
| Outcomes | Mean arterial blood pressure, neonatal outcomes, time of assisted mask ventilation. |
| Notes | This case report is expected to be excluded (not RCT). |
Pinar 2016.
| Methods | Randomised parallel trial investigating the effects of different opioids on emergence from general anaesthesia for short gynaecological surgery. The study recruitment is completed. The author's origin is Turkey. Trial identifier: ISRCTN23443592 |
| Participants |
Inclusion criteria: female patients, aged 18 ‐ 60, ASA I ‐ II, have undergone dilatation curettage and/or endometrial biopsy procedures. Exclusion criteria: psychiatric disorder, opioid drug abuse. |
| Interventions | IV remifentanil versus IV fentanyl for general anaesthesia |
| Outcomes |
The primary outcomes are emergence time from general anaesthesia and discharge time from post‐anaesthesia care unit. Secondary outcomes are pain scores, additional analgesia requirement, patient's satisfaction and intra‐operative dreaming. |
| Notes | This study is expected to be excluded (wrong intervention). |
Pintaric 2016.
| Methods | Observational study investigating remifentanil and neuraxial anaesthesia for labour in multiparous women. The study is not yet recruiting. The author's origin is Slovenia. Trial identifier: NCT02963337 |
| Participants |
Inclusion criteria: patient's request for pain relief, ASA I ‐ III, aged 18 ‐ 55 years, uncomplicated singleton pregnancy with cephalic presentation, gestation age > 37 weeks, regular uterine contractions, cervical dilation 2 cm to 6 cm, anticipated vaginal delivery, fetus without suspected abnormality and normal CTG. Exclusion criteria: contraindications for remifentanil use or for CSE. |
| Interventions | Remifentanil PCA versus CSE. |
| Outcomes |
The primary outcome is pain relief. The secondary outcomes are duration of the first and second stage of labour and patient's satisfaction with pain relief. |
| Notes | This study is expected to be excluded (observational study). |
Weiniger 2016.
| Methods | Safety analysis of a prospective IRB‐approved study of healthy women receiving IV patient‐controlled boluses of remifentanil. The purpose was to detect respiratory depression in labouring women receiving remifentanil. The authors' origin is Israel. |
| Participants | Healthy women in labour |
| Interventions | Remifentanil PCA |
| Outcomes | Number of apneic episodes |
| Notes | This safety analysis is expected to be excluded (not RCT). |
ASA: American Society of Anesthesiologists; CSE: combined spinal‐epidural analgesia; CTG: cardiotocography; EA: epidural analgesia; IRB: institutional review board; IV: intravenous; NA: not applicable; PCA: patient‐controlled analgesia; RCT: randomised controlled trial; RPCA: remifentanil patient‐controlled analgesia; VAS: visual analogue scale
Characteristics of ongoing studies [ordered by study ID]
EUCTR2007‐000736‐10‐NL.
| Trial name or title | A comparison of pethidine/meperidine intramuscularly and remifentanil patient‐controlled analgesia during labour in Westfriesgasthuis ‐ PCA remifentanil during labour |
| Methods | Randomised, controlled trial, not blinded. The purpose of this trial is to compare woman's satisfaction using pethidine/meperidine IM and PCA remifentanil for pain relief during labour and to assess the safety of remifentanil for parturient and fetus. There are no details where the study is conducted. The authors' origin is the Netherlands. Trial identifier: EUCTR2007‐000736‐10‐NL |
| Participants |
Inclusion criteria: pregnant women in labour aged > 18 years in Westfriesgasthuis, ASA I, planned vaginal delivery, informed consent, term pregnancy (37 + 0 to 42 + 0 weeks), head‐down position, no congenital abnormalities Exclusion criteria: requesting or undergoing epidural analgesia, known allergy for remifentanil, other fetal positions than head down, parturient who feels she does not have right amount of time to consider enrolling in this study, fetal congenital abnormalities |
| Interventions | Pethidine/meperidine IM and remifentanil PCA |
| Outcomes |
The primary endpoints of this study are woman's satisfaction measured by the women's view of birth labour satisfaction questionnaire, pain relief measured by VAS scores, parturient and fetal safety. The secondary objective is the question if there is a difference in the pain perception of primipara and multipara. |
| Starting date | Date of registration: 11/09/2008 Date of first enrolment: 05/11/2007 |
| Contact information | NA |
| Notes | We did not contact the authors due to lack of contact information. |
EUCTR2007‐005424‐33‐NL.
| Trial name or title | Epidural analgesia versus remifentanil PCA during labour ‐ OER‐study |
| Methods | Randomised, controlled trial, not blinded. The purpose of this trial is to compare remifentanil PCA with epidural anaesthesia among healthy nulligravida during labour. There are no details where the study is conducted. The authors' origin is the Netherlands. Trial identifier: EUCTR2007‐005424‐33‐NL |
| Participants |
Inclusion criteria: nulligravida, without serious systemic disease, in partu, less than 6 cm dilatation, in labour, aged > 18 years Exclusion criteria: ASA > II, (pre)eclampsia, HELLP‐syndrome, serious diabetic gravidarum, infection, placenta praevia, psychiatric disorder |
| Interventions | Epidural analgesia versus remifentanil PCA |
| Outcomes |
The primary endpoint of this study is woman's satisfaction. The secondary objective is the outcome of the infant (Apgar score, vacuum or forceps deliveries). |
| Starting date | Date of registration: 10/10/2007 Date of first enrolment: 06/02/2008 |
| Contact information | NA |
| Notes | We did not contact the authors due to lack of contact information. |
Logtenberg 2014.
| Trial name or title | Remifentanil patient‐controlled analgesia (RPCA) versus epidural analgesia (EA) during labour. A randomised multicenter trial |
| Methods | Randomised, controlled trial. Not blinded. The purpose of this study is to compare pain appreciation during labour between RPCA and EA. The study was conducted in the Academic Medical Center, Amsterdam, the Netherlands from September 2012 to May 2013 (abstract). The study has been completed according to the authors. Trial identifier: NTR3687 |
| Participants |
Inclusion criteria: age > 18 years, ASA I or II, low‐risk pregnant women Exclusion criteria: drug allergy: history of hypersensitivity to opioid or local anaesthetic, substances, labour before 32 weeks or after 42 weeks of gestation, initial maternal SpO2 of less than 95%, initial maternal temperature of 38°C or higher, prior administration of regional of opioid analgesia (during this delivery) |
| Interventions | Epidural anaesthesia versus remifentanil PCA |
| Outcomes |
The primary endpoint of this study is pain appreciation, expressed by women's satisfaction with pain on a VAS scale, measured hourly from the onset of active labour. Secondary outcomes are overall satisfaction with pain during delivery judged 2 h and 6 weeks after delivery, pain scores during labour and maternal and neonatal side effects. |
| Starting date | Date of protocol registration: 05/11/2012 Date of first enrolment (protocol): 10/10/2012 |
| Contact information | Sabine Logtenberg: slmlogtenberg@gamil.com/raveleerstelijnstudie@gmail.com, B.W. Mol: b.w.mol@amc.nl |
| Notes | A published abstract is available Logtenberg 2014. We contacted the authors but no additional data could be provided yet. |
NCT00710086.
| Trial name or title | Intravenous remifentanil for labour analgesia (IRELAN) |
| Methods | Randomised, controlled trial. Double‐blinded. The purpose of this trial is to assess the safety and efficacy of IV remifentanil with patient‐controlled technique for labour analgesia. The study was conducted in Nanjing Maternal and Child Health Care Hospital in Nanjing, Jiangsu, China from July 2008 to September 2009. The study has been completed. Trial identifier: NCT00710086 |
| Participants |
Inclusion criteria: nulliparous women, > 18 years and < 45 years, spontaneous labour, analgesia request, epidural puncture contraindications, tendency to bleeding Exclusion criteria: allergy to opioids, a history of the use of centrally‐acting drugs of any sort, chronic pain and psychiatric diseases records, those who were not willing to or could not finish the whole study at any time, using or used in the past 14 days of the monoamine oxidase inhibitors, alcohol‐addictive or narcotic‐dependent women were excluded for their influence on the analgesic efficacy of the epidural analgesics, participants with a non‐vertex presentation or scheduled induction of labour, cervical dilation was 5 cm or greater before performing epidural puncture and catheterisation, diagnosed diabetes mellitus and pregnancy‐induced hypertension, twin gestation and breech presentation |
| Interventions | Hydromorphone intravenous (1 mg at the woman's request if they felt uterine contraction pain) versus remifentanil PCA (0.2 µg/kg, lockout time 2 min, continuous infusion rate 0.2 ‐ 0.8 µg/(kg*min) |
| Outcomes |
The primary endpoint of this study was the maternal VAS rating of pain. Secondary outcome measures: rate and indications of caesarean delivery, rate of instrument‐assisted delivery, duration of analgesia, maternal satisfaction with analgesia, maternal oral temperature, use of oxytocin after analgesia, maximal oxytocin dose, breastfeeding success at six weeks after vaginal delivery, neonatal Apgar score at 1 and 5 min, umbilical cord gas analysis, neonatal sepsis evaluation, neonatal antibiotic treatment, incidence of maternal side effects |
| Starting date | Date of registration: 02/07/2008 Date of first enrolment: July 2008 |
| Contact information | XiaoFeng Shen, Nanjing Medical University |
| Notes | We contacted the authors for further information without any response. |
NCT01563939.
| Trial name or title | Patient‐controlled intravenous analgesia with remifentanil infusion for labour |
| Methods | Randomised, controlled trial. Double‐blinded. The purpose of this trial is to assess the effectiveness of two methods remifentanil administration in the form of either an infusion or PCA demand bolus. The study was conducted in Mount Sinai Hospital, Toronto, Ontario, Kanada from February 2012 to December 2014. It has been terminated due to difficult recruitment. Trial identifier: NCT01563939 |
| Participants |
Inclusion criteria: age 18 ‐ 50 years, written informed consent, term pregnancy in labour with singleton fetus in cephalic presentation, women requesting systemic analgesia, women with contraindication for regional anaesthesia without fetal compromise (coagulopathy, thrombocytopenia, refusal, etc.) Exclusion criteria: refusal to sign written informed consent, inability to communicate in English, opioid dependence or addiction, women on methadone, allergy or hypersensitivity to remifentanil, fetal heart rate abnormalities, fetal congenital anomalies |
| Interventions | Continuous remifentanil IV infusion (stepwise increase in infusion rates and placebo demand bolus of normal saline) versus demand bolus of remifentanil (stepwise increase in bolus dose and placebo continuous infusion of normal saline) |
| Outcomes |
The primary endpoint of the study was the pain score (VNRS from 0 to 10). Secondary outcome measures: maternal satisfaction, consumption of remifentanil, cross‐over to epidural, side effects, fetal and neonatal outcomes (non‐reassuring fetal heart rate as determined by obstetrician, neonatal weight, Apgar scores, naloxone administration, need for resuscitation, NICU admission) |
| Starting date | Date of registration: 23/03/2012 Date of first enrolment: February 2012 |
| Contact information | Mrinalini Balki, Mount Sinai Hospital, New York; Samuel Lunenfeld Research Institute, Mount Sinai Hospital |
| Notes | We contacted the authors for further information without any response. |
NCT02179294.
| Trial name or title | A randomised controlled trial of remifentanil intravenous patient‐controlled analgesia (PCA) versus intramuscular pethidine for pain relief in labour (RESPITE) |
| Methods | Randomised, controlled trial. Not blinded. The purpose of this study is to conduct a RCT to determine if remifentanil (PCA) reduces the proportion of women who subsequently require an epidural for pain relief in comparison to intermittent pethidine IM. The study was conducted in Birmingham, United Kingdom. The study has been completed according to the authors. Trial identifier: EUCTR2012‐005257‐22‐GB and NCT02179294 |
| Participants |
Inclusion criteria: requesting systemic opioid analgesia, aged > 16 years, beyond 37 weeks' gestation, in established labour with vaginal birth intended, able to understand all information (written and oral) presented (using an interpreter if necessary), not participating in any other clinical trial of a medical product, live singleton pregnancy with cephalic presentation Exclusion criteria: contraindication to epidural analgesia, contraindication to intramuscular injection, history of drug sensitivity to pethidine or remifentanil, women taking long‐term opioid therapy including methadone, systemic pain relief opioid in the last 4 hours |
| Interventions | Pethidine 100 mg IM (up to 4 hourly in frequency, maximum of 4 doses, maximum dose being 400 mg in 24 h) versus remifentanil PCA (bolus 40 µg, lockout 2 min) |
| Outcomes |
The primary endpoint is the proportion of women who receive epidural analgesia for pain relief in labour, in each group, after randomisation. Secondary outcome measures: effectiveness of pain relief, incidence of maternal side effects (excessive sedation score, oxygen saturation < 94% whilst breathing room air, nausea requiring anti‐emetic administration, requirement for supplemental oxygen, respiratory depression (< 8 breaths/min)), delivery mode, incidence of fetal distress requiring delivery, neonatal status at delivery (Apgar score at 5 min, incidence of fetal acidosis determined by umbilical cord gas analysis, requirement for neonatal resuscitation, incidence of admission to Special Care Baby Unit), rate of initiation of breast feeding within the first hour of birth, maternal satisfaction with childbirth experience determined by postpartum questionnaire prior to discharge from the delivery ward, resources used intra‐ and postoperatively, including PCA consumables, anaesthetist attendance, costs of staff training, service procurement, provision of care |
| Starting date | Date of registration: 21/06/2013 Date of first enrolment: 12/07/2013 |
| Contact information | Leanne Homer, l.e.homer@bham.ac.uk |
| Notes | We contacted the authors but no additional data could be provided yet. |
Rabie 2006.
| Trial name or title | Remifentanil by patient‐controlled analgesia compared with epidural analgesia for pain relief in labour |
| Methods | Randomised, controlled trial. No statement on blinding. The purpose of this trial is to compare the use of PCA remifentanil to epidural analgesia in labour. There is no statement when or where the study is conducted. The authors' origin is Riyadh, Kingdom of Saudi Arabia. Trial identifier: NA |
| Participants |
Inclusion criteria: healthy pregnant women, ASA I or II, no obstetric complications or contraindication to remifentanil or epidural analgesia Exclusion criteria: NA |
| Interventions | Epidural infusion (bupivacaine 1% plus 2 µg/mL fentanyl) versus remifentanil PCA (bolus of 0.4 µg/kg over 20 s, lockout 1 min) |
| Outcomes | There is no primary outcome defined. General outcomes: pain relief, safety of the mother and the fetus, side effects (bradycardia, hypotension, desaturation, nausea, fetal heart rate changes, Apgar scores at 1 min and 5 min, umbilical cord gas analysis and lactate levels), overall parturient's satisfaction, sedation scores |
| Starting date | Date of registration: NA Date of first enrolment: NA |
| Contact information | M.E. Rabie, H. H. Negmi, A. M. Moustafa, H. Al Oufi, Anesthesia Department, King Faisal Specialist Hospital & Research Centerm Riyadh, KSA; hnegmi@hotmail.com |
| Notes | A published abstract is available Rabie 2006. We contacted the authors for further information without any response. |
ASA: American Society of Anesthesiologists; HELLP: Haemolysis, ElevatedLiver enzymes, and Low Platelet count; IM: intramuscular; IV: intravenous; NICU: neonatal intensive care unit; OER: Open Educational Resources; PCA: patient‐controlled analgesia; VAS: visual analogue scale; VNRS: verbal numerical rating scale
Differences between protocol and review
Change to the authors of the review since publication of the protocol (Jokinen 2015)
The order of the authors list was changed in the current review in accordance to their contributions as described in the Contributions of authors section.
Difference in the methods used between the protocol and the review
1. One comparison was introduced: we have introduced as a new comparator 'remifentanil using the same mode (PCA), but different regimen (e.g. increasing bolus versus constant bolus)' since we could identify one relevant trial and we believe that the administration regimen of remifentanil (PCA) might be relevant for several safety aspects of this intervention.
2. Order of comparisons was changed: we re‐ordered the comparators. In the protocol 'placebo or no treatment' was set as the main comparator. However, studies for this comparison were not available and were also considered to be ethically not feasible. 'Remifentanil (PCA)' versus 'another opioid (IV/IM)' was set as main comparison since the usage of other opioids administered either IV or IM was from the global point of view the most used analgesia for labour pain today.
2. Two outcomes were introduced: we introduced 'neonatal neurologic and adaptive score (NACS)' as an outcome within the domain 'adverse events for newborns' and 'augmented labour (e.g. use of oxytocin)'.
3. GRADE approach: a detailed description of applying the GRADE approach was not given at the protocol stage. However, assessment of the quality of evidence in the current review followed the GRADE guidelines and is now described in detail in the Assessment of risk of bias in included studies section.
4. Handling of median and IQR was changed: at the protocol stage we planned to include all data reported as median with IQR and transform those into mean with SD in accordance to Higgins 2011 followed by a sensitivity analysis to test robustness of the estimated effect with respect to exclusion of trials reporting median and IQR data. In the current review, we decided to include only median and IQR values with a symmetric distribution and data with an asymmetric distribution were not included into the meta‐analysis. Since under a symmetric situation the assumption of 'the median is equal to the mean' is given, we renounced performing a sensitivity analysis.
5. Handling of zero total event trials: we did not plan to include trials reporting zero events in both arms at the protocol stage. However, we think that inclusion of trials with total zero events reduces the risk of inflating the magnitude of the pooled effect. We performed a sensitivity analysis to investigate the impact of inclusion of total zero event trials by different approaches on the robustness of the estimated effects. Handling of zero event trials is described in detail in the Data synthesis section.
5. Trial sequential analysis (TSA) and OIS considerations were introduced: at the protocol stage we did not plan to perform TSA or OIS considerations to calculate the required or optimal information size, respectively. However, we think that those considerations help us to more reliably assess the quality of the evidence. Therefore, we have incorporated the TSA and OIS approach into the assessment of 'imprecision' (GRADE). Since the assumptions for TSA and OIS calculations were made in a post‐hoc manner, we adopted the assumptions from the pooled estimates obtained from either 'low risk of bias' trials or all meta‐analysed trials ('empirical'). The assumptions may not in every case perfectly meet the clinical practice, however, it seems to us to be the most objective approach to set the basic conditions.
6. Restriction of subgroup and sensitivity analyses on designated outcomes was extended: in the protocol we specified that all subgroup and sensitivity analyses should be restricted to the primary outcomes. During preparation of the review we extended the restriction to all GRADE‐relevant outcomes from which two are secondary outcomes. Sensitivity analyses are essential for the assessment of the quality of evidence by the GRADE approach.
Contributions of authors
Stephanie Weibel (SW), Yvonne Jelting (YJ), Arash Afshari (AA), Nathan Leon Pace (NLP), Leopold HJ Eberhart (LE), Johanna Jokinen (JJ), Thorsten Artmann (TA), Peter Kranke (PK)
Conceiving the review: PK
Co‐ordinating the review: PK, SW, YJ
Undertaking manual searches: YJ, SW
Screening search results: SW, YJ
Organizing retrieval of papers: SW, YJ
Screening retrieved papers against inclusion criteria: SW, YJ
Appraising quality of papers: SW, YJ, PK, AA
Abstracting data from papers: SW, YJ
Writing to authors of papers for additional information: YJ, SW
Data management for the review: SW
Entering data into Review Manager 5 (RevMan 5.3): SW
RevMan statistical data: SW, NLP
Other statistical analysis not using RevMan: SW, NLP, AA
Double entry of data: (data entered by person one: SW; data checked by person two: YJ)
Interpretation of data: PK, SW, YJ, AA, NLP, LE, JJ, TA
Statistical inferences: SW, PK, NLP, AA
Writing the review: SW, YJ, PK
Securing funding for the review: PK
Performing previous work that was the foundation of the present study: PK, JJ, TA
Guarantor for the review (one author): PK
Person responsible for reading and checking review before submission: PK
All authors reviewed the final and previous drafts.
Sources of support
Internal sources
-
Stephanie Weibel, Germany.
Department of Anaesthesia and Critical Care, University of Wuerzburg
-
Yvonne Jelting, Germany.
Department of Anaesthesia and Critical Care, University of Wuerzburg
-
Arash Afshari, Denmark.
Juliane Marie Centre ‐ Anaesthesia and Surgical Clinic Department 4013, Copenhagen University Hospital
-
Nathan L Pace, USA.
Department of Anesthesiology, University of Utah
-
Leopold HJ Eberhart, Germany.
Department of Anaesthesiology & Intensive Care Medicine, Philipps‐University Marburg
-
Johanna Jokinen, Germany.
Department of Anaesthesia and Critical Care, University of Wuerzburg
-
Thorsten Artmann, Germany.
Department of Anaesthesia and Intensive Care Medicine, Cnopf Children´s Hospital, Hospital Hallerwiese, Nuernberg
-
Peter Kranke, Germany.
Department of Anaesthesia and Critical Care, University of Wuerzburg
External sources
-
Meta‐Analysis Grant of the European Society of Anaesthesiology, Belgium.
Peter Kranke received a grant supporting this review from the European Society of Anaesthesiology
Declarations of interest
Stephanie Weibel: has no conflict of interest regarding the topic of this review. Stephanie Weibel is an academic researcher. She has received personal payments for consultancies and lecture fees from Genelux Corporation, San Diego, USA (ended March 2014). Genelux Corp does not produce any products of the intervention of interest of this review.
Yvonne Jelting: none known.
Arash Afshari: none known.
Nathan L Pace: has no conflict of interest regarding the topic of this review. Nathan L Pace has received payment for development of educational presentations (Barash, Cullen, Stoelting Clinical Anesthesia 8th edition) and provided consultancy (St Marks Hospital, Salt Lake City, UT) on topics not related to the current review. He has received supplements to attend Cochrane meetings. He also has stocks and shares in companies which have no interest in the topic of this review (TIAA‐CREF, Fidelity, Vanguard, USAA, MorganStanley).
Leopold HJ Eberhart: has no conflict of interest regarding the topic of this review. Leopold HJ Eberhart has received lecture fees (from Baxter GmbH and Fresenius GmbH), payment for lectures (from Grünenthal GmbH, Baxter GmbH and Fresenius, GmbH) and has provided consultancy (for Grünenthal GmbH, Baxter GmbH, ratiopharm GmbH) for topics not related to the current review. He holds a board membership (with Grünenthal GmbH Deutschland) who do not have an interest in the topic of this review.
Johanna Jokinen: none known.
Thorsten Artmann: none known.
Peter Kranke: has no conflict of interest regarding the topic of this review. Peter Kranke has received lecture fees (from FreseniusKabi, MSD, Ratiopharm, Covidien) and has provided consultancy (to MSD, FreseniusKabi, Ratiopharm, Covidien) on topics not related to the current review. He has been involved in the conduct of Phase II and phase III clinical trials not related to the current review. He has published a case series on remifentanil for labour analgesia and has published research reports and editorial views on the topic under review. He has received a Meta‐Analysis grant supporting this review from the European Society of Anaesthesiology (ESA).
New
References
References to studies included in this review
Balki 2007 {published data only}
- Balki M, Kasodekar S, Dhumne S, Bernstein P, Carvalho J. Patient controlled analgesia with remifentanil for labor pain. Canadian Journal of Anesthesia 2006;53(1):26346. [Google Scholar]
- Balki M, Kasodekar S, Dhumne S, Bernstein P, Carvalho J. Patient‐controlled analgesia with background remifentanil infusion for labor pain [abstract]. Anesthesiology 2006;104(Suppl 1):13. [Google Scholar]
- Balki M, Kasodekar S, Dhumne S, Bernstein P, Carvalho JC. Remifentanil patient‐controlled analgesia for labour: optimizing drug delivery regimens. Canadian Journal of Anaesthesia 2007;54(8):626‐33. [PubMed] [Google Scholar]
Blair 2005 {published data only}
- Blair JM, Dobson GT, Hill DA, Fee JPH. Patient‐controlled analgesia for labor: a comparison of remifentanil and pethidine [abstract]. Anesthesiology 2001;95:Abstract no: A1063. [Google Scholar]
- Blair JM, Dobson GT, Hill DA, McCracken GR, Fee JPH. Patient controlled analgesia for labour: a comparison of remifentanil with pethidine. Anaesthesia 2005;60:22‐7. [DOI] [PubMed] [Google Scholar]
Calderon 2006 {published data only}
- Calderon E, Martinez E, Roman MD, Pernia A, Garcia‐Hernandez R, Torres LM. Intravenous remifentanil delivered through an elastomeric device versus intramuscular meperidine comparative study for obstetric analgesia [Remifentalino intravenoso mediante infusor elastomerico frente a meperidina intramuscular. Estudio comparativo en analgesia obstetrica]. Revista de la Sociedad Espanola del Dolor 2006;13(7):462‐7. [Google Scholar]
Douma 2010 {published data only}
- Douma MR, Verwey RA, Kam‐Endtz CE, Linden PD, Stienstra R. Obstetric analgesia: a comparison of patient‐controlled meperidine, remifentanil, and fentanyl in labour. British Journal of Anaesthesia 2010;104(2):209‐15. [DOI] [PubMed] [Google Scholar]
- ISRCTN12122492. Obstetric analgesia: a comparison of patient controlled pethidine, remifentanil and fentanyl in labour. isrctn.com/ISRCTN12122492 Date first received: 14 February 2006.
- NTR543. Obstetric analgesia: a comparison of patient controlled pethidine, remifentanil and fentanyl in labour. trialregister.nl/trialreg/admin/rctview.asp?TC=543 Date first received: 4 December 2005.
Douma 2011 {published data only}
- Douma MR, Middeldorp JM, Verwey RA, Dahan A, Stienstra R. A randomised comparison of intravenous remifentanil patient‐controlled analgesia with epidural ropivacaine/sufentanil during labour. International Journal of Obstetric Anesthesia 2011;20(2):118‐23. [DOI] [PubMed] [Google Scholar]
- EUCTR2007‐000808‐32‐NL. A comparison of remifentanil patient‐controlled analgesia with epidural analgesia during labor. clinicaltrialsregister.eu/ctr‐search/search?query=eudract_number:2007‐000808‐32 Date first received: 17 April 2007.
- NTR1127. A comparison of remifentanil patient‐controlled analgesia with epidural analgesia during labor. trialregister.nl/trialreg/admin/rctview.asp?TC=1127 Date first received: 17 November 2007.
Douma 2015 {published data only}
- Douma MR, Stienstra R, Middeldorp JM, Arbous MS, Dahan A. Differences in maternal temperature during labour with remifentanil patient‐controlled analgesia or epidural analgesia: a randomised controlled trial. International Journal of Obstetric Anesthesia 2015;24:313‐22. [DOI] [PubMed] [Google Scholar]
- EUCTR2008‐002792‐28‐NL. Differences in maternal temperature and saturation after administration of remifentanil PCA or epidural analgesia during labor. clinicaltrialsregister.eu/ctr‐search/search?query=eudract_number:2008‐002792‐28 Date first received 4 June 2008.
- NTR1498. Differences in maternal temperature and saturation after administration of remifentanil PCA or epidural analgesia during labor. trialregister.nl/trialreg/admin/rctview.asp?TC=1498 Date first received 20 October 2008.
El‐Kerdawy 2010 {published data only}
- El‐Kerdawy H, Farouk A. Labor analgesia in preeclampsia: remifentanil patient controlled intravenous analgesia versus epidural analgesia. Middle East Journal of Anesthesiology 2010;20(4):539‐45. [PubMed] [Google Scholar]
Evron 2005 {published data only}
- Evron S, Glezerman M, Sadan O, Boaz M. Remifentanil patient controlled analgesia for labor pain. Anesthesiology 2002;96 Suppl:Abstract no: A1032. [Google Scholar]
- Evron S, Glezerman M, Sadan O, Boaz M, Ezri T. Remifentanil: a novel systematic analgesic for labor pain. Anesthesia & Analgesia 2005;100:233‐8. [DOI] [PubMed] [Google Scholar]
- Evron S, Sadan O, Ezri T, Boaz M, Glezerman M. Remifentanil: a new systemic analgesic for labor pain and an alternative to dolestine [abstract]. American Journal of Obstetrics and Gynecology 2001;185(6 Suppl):S210. [Google Scholar]
Evron 2008 {published data only}
- Evron S, Ezri T, Protianov M, Muzikant G, Sadan O, Herman A, et al. The effects of remifentanil or acetaminophen with epidural ropivacaine on body temperature during labor. Journal of Anesthesia 2008;22(2):105‐11. [DOI] [PubMed] [Google Scholar]
Freeman 2015 {published data only}
- Freeman L, Bloemenkamp K, Franssen M, Papatsonis D, Hollmann M, Woiski M, et al. Remifentanil patient controlled analgesia versus epidural analgesia in labor; a randomized controlled trial. American Journal of Obstetrics and Gynecology 2014;210(1 Suppl):S37. [Google Scholar]
- Freeman LM, Bloemenkamp KW, Franssen MT, Papatsonis DN, Hajenius PJ, Hollmann MW, et al. Patient controlled analgesia with remifentanil versus epidural analgesia in labour: randomised multicentre equivalence trial. BMJ (Clinical Research Ed.) 2015;350:h846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman LM, Bloemenkamp KWM, Franssen MTM, Papatsonis DNM, Hajenius PJ, Huizen ME, et al. Remifentanil patient controlled analgesia versus epidural analgesia in labour. A multicentre randomized controlled trial. BMC Pregnancy and Childbirth 2012;12:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NTR2551. Remifentanil patient controlled analgesia versus epidural analgesia during labor. A randomized multicenter trial ‐ RAVEL [Remifentanil patient gecontroleerde pijnstilling versus epidurale pijnstilling tijdens de bevalling]. trialregister.nl/trialreg/admin/rctview.asp?TC=2551 Date first received: 4 October 2010.
Ismail 2012 {published data only}
- Ismail MT, Hassanin MZ. Neuraxial analgesia versus intravenous remifentanil for pain relief in early labor in nulliparous women. Archives of Gynecology and Obstetrics 2012;286(6):1375‐81. [DOI] [PubMed] [Google Scholar]
Khooshideh 2015 {published data only}
- Khooshide M. Effect of different methods of remifentanil administration in labor pain. en.search.irct.ir/view/11026 Date first received: 18 February 2013.
- Khooshideh M, Shahriari A, Sheikh M. Comparison of the effect of incremental bolus and incremental infusion regimens of remifentanil on labour pain. Shiraz E‐Medical Journal 2015;16(5):e25626. [Google Scholar]
Ng 2011 {published data only}
- Ng TKT, Cheng BCP, Chan WS, Lam KK, Chan MTV. A double‐blind randomised comparison of intravenous patient‐controlled remifentanil with intramuscular pethidine for labour analgesia. Anaesthesia 2011;66(9):796‐801. [DOI] [PubMed] [Google Scholar]
Shen 2013 {published data only}
- Shen MK, Wu ZF, Zhu AB, He LL, Shen XF, Yang JJ, et al. Remifentanil for labour analgesia: a double‐blinded, randomised controlled trial of maternal and neonatal effects of patient‐controlled analgesia versus continuous infusion. Anaesthesia 2013;68(3):236‐44. [DOI] [PubMed] [Google Scholar]
Stocki 2014 {published data only}
- NCT00801047. Intravenous remifentanil patient‐controlled analgesia (PCA) and epidural patient controlled epidural analgesia (PCEA) for labor analgesia. clinicaltrials.gov/show/NCT00801047 Date first received: 2 December 2008.
- Stocki D, Matot I, Einav S, Eventov‐Friedman S, Ginosar Y, Weiniger CF. A randomized controlled trial of the efficacy and respiratory effects of patient‐controlled intravenous remifentanil analgesia and patient‐controlled epidural analgesia in laboring women. Anesthesia & Analgesia 2014;118(3):589‐97. [DOI] [PubMed] [Google Scholar]
- Stocki D, Matot I, Weiniger CF. A prospective randomized controlled trial to compare the efficacy and safety of remifentanil IV PCA to epidural PECA in labor analgesia. European Journal of Anaesthesiology 2011;28 Suppl:164‐5. [Google Scholar]
Stourac 2014 {published data only}
- Stourac P, Suchomelova H, Stodulkova M, Huser M, Krikava I, Janku P, et al. Comparison of parturient‐controlled remifentanil with epidural bupivacain and sufentanil for labour analgesia: randomised controlled trial. Biomedical Papers: Journal of the Palacky University 2014;158(2):227‐32. [DOI] [PubMed] [Google Scholar]
- Stourac P, Suchomelova H, Stodulkova M, Huser M, Krikava I, Stoudek R, et al. Comparison of parturient controlled remifentanil with epidural bupivacain and sufentanil for labour analgesia: randomised controlled trial. Anesthesiology Annual Meeting; 2012 Oct 15; Boston. 2012:A1058. [DOI] [PubMed]
Thurlow 2002 {published data only}
- Thurlow JA, Laxton CH, Dick A, Waterhouse P. A comparison of patient controlled analgesia (PCA) using remifentanil with intramuscular pethidine for pain relief in labour [abstract]. International Journal of Obstetric Anesthesia 2000;9:200. [Google Scholar]
- Thurlow JA, Laxton CH, Dick A, Waterhouse P, Sherman L, Goodman NW. Remifentanil by patient‐controlled analgesia compared with intramuscular meperidine for pain relief in labour. British Journal of Anaesthesia 2002;88(3):374‐8. [DOI] [PubMed] [Google Scholar]
Tveit 2012 {published data only}
- EUCTR2005‐004577‐23‐NO. Remifentanil as pain relief during labour. clinicaltrialsregister.eu/ctr‐search/search?query=eudract_number:2005‐004577‐23 Date first received: 31 October 2005.
- NCT00202722. Remifentanil as intravenous patient‐controlled analgesia (IVPCA) during labour. clinicaltrials.gov/show/NCT00202722 Date first received: 12 September 2005.
- Tveit TO, Seiler S, Halvorsen A, Rosland JH. Labour analgesia: A randomised, controlled trial comparing intravenous remifentanil and epidural analgesia with ropivacaine and fentanyl. European Journal of Anaesthesiology 2012;29(3):129‐36. [DOI] [PubMed] [Google Scholar]
Volikas 2001 {published data only}
- Volikas I, Male D. A comparison of pethidine and remifentanil patient‐controlled analgesia in labour. International Journal of Obstetric Anesthesia 2001;10:86‐90. [DOI] [PubMed] [Google Scholar]
Volmanen 2008 {published data only}
- Volmanen P, Akural EI, Raudaskoski T, Alahuhta S, Sarvela J, Korttila K. Intravenous remifentanil versus epidural levobupivacaine with fentanyl for pain relief in early labour: a randomised, controlled, double blind study. European Journal of Anaesthesiology 2005;Volume 22, Supplement 34:A‐582. [Google Scholar]
- Volmanen P, Sarvela J, Akural EI, Raudaskoski T, Korttila K, Alahuhta S. Intravenous remifentanil vs. epidural levobupivacaine with fentanyl for pain relief in early labour: a randomised, controlled, double‐blinded study. Acta Anaesthesiologica Scandinavica 2008;52(2):249‐55. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Balcioglu 2007 {published data only}
- Balcioglu O, Akin S, Demir S, Aribogan A. Patient‐controlled intravenous analgesia with remifentanil in nulliparous subjects in labor. Expert Opinion on Pharmacotherapy 2007;8(18):3089‐96. [DOI] [PubMed] [Google Scholar]
Jost 2013 {published data only}
- Jost A, Ban B, Kamenik M. Modified patient‐controlled remifentanil bolus delivery regimen for labour pain*. Anaesthesia 2013;68(3):245‐52. [DOI] [PubMed] [Google Scholar]
Shahriari 2007 {published data only}
- Shahriari A, Khooshideh M. A randomized controlled trial of intravenous remifentanil compared with intramuscular meperidine for pain relief in labor. Journal of Medical Sciences 2007;7(4):635‐9. [Google Scholar]
Solek‐Pastuszka 2009 {published data only}
- Solek‐Pastuszka J, Kepinski S, Makowski A, Celewicz Z, Zukowski M, Safranow K, et al. Patient‐controlled continuous epidural analgesia vs intravenous remifentanil infusion for labour anaesthesia [Porownanie jakosci znieczulenia u rodzacych otrzymujacych remifentanil metoda analgezji dozylnej sterowanej przez pacjenta lub ciaglego znieczulenia zewnatrzoponowego]. Anestezjologia Intensywna Terapia 2009;41(2):84‐8. [PubMed] [Google Scholar]
Varposhti 2013 {published data only}
- Varposhti MR, Ahmadi N, Masoodifar M, Shahshahan Z, Tabatabaie MH. Comparison of remifentanil: entonox with entonox alone in labor analgesia. Advanced Biomedical Research 2013;2:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
Volmanen 2004 {published data only}
- Volmanen P, Akural E, Raudaskoski, Alahuhta S. Comparison of maternal haemodynamic effects and respiratory indices during remifentanil and nitrous oxide labour analgesia. International Journal of Obstetric Anesthesia 2004;13(3):S19. [Google Scholar]
Volmanen 2005 {published data only}
- Volmanen P, Akural E, Raudakoski T, Ohtonen P, Alahuhta S. Comparison of remifentanil and nitrous oxide in labour analgesia. Acta Anaesthesiologica Scandinavica 2005;49:453‐8. [DOI] [PubMed] [Google Scholar]
Volmanen 2009 {published data only}
- Volmanen P, Akural E, Alahuhta S. In early labour IVPCA remifentanil bolus during the contraction pause does not improve the analgesic effect but reduces sedation compared with bolus given during the uterine contraction. European Journal of Anaesthesiology 2009;26(Suppl 45):148. [Google Scholar]
Volmanen 2011 {published data only}
References to studies awaiting assessment
Abdalla 2015 {published data only}
- Abdalla W, Ammar MA, Tharwat AI. Combination of dexmedetomidine and remifentanil for labor analgesia: A double‐blinded, randomized, controlled study. Saudi Journal of Anaesthesia 2015;9(4):433‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Godinho 2016 {published data only}
- Godinho P, Lavado J, Goncalves L, Macedo I, Carlos T, Valente E. Labour analgesia challenge ‐ when epidural is not possible, what can we do?. European Journal of Anaesthesiology 2016;33(e‐Suppl 54):159. [Google Scholar]
Gunes 2014 {published data only}
- Gunes S, Turktan M, Gulec UK, Hatipoglu Z, Unlugenc H, Isik G. The comparison of patient‐controlled remifentanil administered by two different protocols (bolus and bolus+infusion) and intramuscular meperidine for labor analgesia. Turkish Journal of Anaesthesiology and Reanimation 2014;42(5):264‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Karadjova 2016 {published data only}
- Karadjova D, Spasovski S, Ivanov E, Sivevski A, Zlatkova M, Churlinov K, et al. Intravenous patient controlled analgesia with remifentanil versus continuous epidural for labor analgesia. Anesthesia and Analgesia 2016;123(3):257. [Google Scholar]
Kondoh 2016 {published data only}
- JPRN‐UMIN000021322. Intravenous remifentanil analgesia during labor and decreasing maternal respiratory depression with mosaprid. https://upload.umin.ac.jp/cgi‐open‐bin/ctr_e/ctr_view.cgi?recptno=R000024594 Date first received: 1 April 2016.
Leong 2015 {published data only}
- NCT02733835. VPIA remifentanil for labour pain to reduce maternal desaturation and improve analgesic titration. clinicaltrials.gov/show/NCT02733835 Date first received: 30 December 2015.
Logtenberg 2016 {published data only}
- Logtenberg S, Oude Rengerink K, Verhoeven CJ, Freeman LM, Akker E, Godfried MB, et al. Labour pain with remifentanil patient‐controlled analgesia versus epidural analgesia: a randomised equivalence trial. BJOG: an International Journal of Obstetrics and Gynaecology 2016 [Epub ahead of print]. [DOI] [PubMed]
Moreira 2016 {published data only}
- Moreira J, Pinheiro F, Amorim P. General anesthesia with target controlled infusion of propofol and remifentanil for planned caesarean section in a parturient with unrupted intracranial aneurysm. European Journal of Anaesthesiology 2016;33(e‐Suppl 54):161. [Google Scholar]
Pinar 2016 {published data only}
- ISRCTN23443592. The effects of remifentanil or fentanyl administration on emergence from general anesthesia with mask after dilatation and curettage or endometrial biopsy procedures in ASA I‐II patients. isrctn.com/ISRCTN23443592 Date first received: 12 January 2016.
Pintaric 2016 {published data only}
- NCT02963337. Remifentanil vs. combined spinal‐epidural analgesia for labor analgesia and progress of labor in multiparous. clinicaltrials.gov/show/NCT02963337 Date first received: 7 November 2016.
Weiniger 2016 {published data only}
- Weiniger C, Carvalho B, Stocki D, Einav S. Respiratory rate and capnography are superior to pulse oximetry in detecting respiratory depression among laboring women receiving remifentanil. European Journal of Anaesthesiology 2016;33(e‐Suppl 54):157. [Google Scholar]
References to ongoing studies
EUCTR2007‐000736‐10‐NL {published data only}
- EUCTR2007‐000736‐10‐NL. A comparison of pethidine/meperidine intramusculary and remifentanil patient‐controlled analgesia during labor in Westfriesgasthuis. clinicaltrialsregister.eu/ctr‐search/search?query=EUCTR2007‐005424‐33‐NL Date first received: 11 September 2008.
EUCTR2007‐005424‐33‐NL {published data only}
- EUCTR2007‐005424‐33‐NL. Epidural analgesia versus remifentanil PCA during labour. clinicaltrialsregister.eu/ctr‐search/search?query=EUCTR2007‐005424‐33‐NL Date first received: 10 October 2007.
Logtenberg 2014 {published data only}
- Logtenberg A, Oude Rengerink K, Verhoeven C, Mol BW. Remifentanil patient controlled analgesia versus epidural analgesia during labour: a randomised trial (NTR3687). Regional Anesthesia and Pain Medicine 2014;39(5 Suppl 1):E241‐E242. [Google Scholar]
- NTR3687. Comparing patient satisfaction and costs between epidural and remifentanil during labour in low risk pregnant women. trialregister.nl/trialreg/admin/rctview.asp?TC=3687 Date first received: 5 November 2012.
NCT00710086 {published data only}
- NCT00710086. Intravenous remifentanil for labor analgesia (IRELAN). clinicaltrials.gov/ct2/show/NCT00710086 Date first received: 2 July 2008.
NCT01563939 {published data only}
- NCT01563939. Patient‐controlled intravenous analgesia with remifentanil infusion for labour. clinicaltrials.gov/show/NCT01563939 Date first received: 23 March 2012.
NCT02179294 {published data only}
- NCT02179294. A randomised controlled trial of remifentanil intravenous patient controlled analgesia (PCA) versus intramuscular pethidine for pain relief in labour (RESPITE). clinicaltrials.gov/show/NCT02179294 Date first received: 11 June 2014.
Rabie 2006 {published data only}
- Rabie ME, Negmi HH, Moustafa AM, Al Oufi H. Remifentanil by patient controlled analgesia compared with epidural analgesia for pain relief in labour [abstract]. Regional Anesthesia and Pain Management 2006;31(5 Suppl 1):52. [Google Scholar]
Additional references
AWMF guidelines 2009
- AWMF. AWMF Online [S3‐Leitlinie "Behandlung akuter perioperativer und posttraumatischer Schmerzen"]. http://www.awmf.org/leitlinien/detail/ll/001‐025.html [accessed 29.11.2011] 2009.
Balshem 2011
- Balshem H, Helfand M, Schünemann HJ, Oxman AD, Kunz R, Brozek J, et al. GRADE guidelines: 3. Rating the quality of evidence. Journal of Clinical Epidemiology 2011;64(4):401‐6. [DOI] [PubMed] [Google Scholar]
Bonner 2012
- Bonner JC, McClymont W. Respiratory arrest in an obstetric patient using remifentanil patient‐controlled analgesia. Anaesthesia 2012;67:538‐40. [DOI] [PubMed] [Google Scholar]
Bosilkovska 2012
- Bosilkovska M, Walder B, Besson M, Daali Y, Desmeules J. Analgesics in patients with hepatic impairment: pharmacology and clinical implications. Drugs 2012;72:1645‐69. [DOI] [PubMed] [Google Scholar]
Brok 2008
- Brok J, Thorlund K, Gluud C, Wetterslev J. Trial sequential analysis reveals insufficient information size and potentially false positive results in many meta‐analyses. Journal of Clinical Epidemiology 2008;61:763‐9. [DOI] [PubMed] [Google Scholar]
Callister 2003
- Callister LC, Khalaf I, Semenic S, Kartchner R, Vehvilainen‐Julkunen K. The pain of childbirth: perceptions of culturally diverse women. Pain Management Nursing 2003;4:145‐54. [DOI] [PubMed] [Google Scholar]
Callister 2010
- Callister LC, Holt ST, Kuhre MW. Giving birth: the voices of Australian women. Journal of Perinatal & Neonatal Nursing 2010;24:128‐36. [DOI] [PubMed] [Google Scholar]
Egan 1993
- Egan TD, Lemmens HJ, Fiset P, Hermann DJ, Muir KT, Stanski DR, et al. The pharmacokinetics of the new short‐acting opioid remifentanil (GI87084B) in healthy adult male volunteers. Anesthesiology 1993;79:881‐92. [DOI] [PubMed] [Google Scholar]
Goetzl 2002
- Goetzl LM. ACOG Practice Bulletin. Clinical Management Guidelines for Obstetrician‐Gynecologists Number 36, July 2002. Obstetric analgesia and anesthesia. Obstetrics and Gynecology 2002;100:177‐91. [DOI] [PubMed] [Google Scholar]
Guyatt 2011a
- Guyatt GH, Oxman AD, Vist G, Kunz R, Brozek J, Alonso‐Coello P, et al. GRADE guidelines: 4. Rating the quality of evidence—study limitations (risk of bias). Journal of Clinical Epidemiology 2011;64(4):407‐15. [DOI] [PubMed] [Google Scholar]
Guyatt 2011b
- Guyatt GH, Oxman AD, Kunz R, Woodcock J, Brozek J, Helfand M, et al. GRADE guidelines: 7. Rating the quality of evidence—inconsistency. Journal of Clinical Epidemiology 2011;64(12):1294‐302. [DOI] [PubMed] [Google Scholar]
Guyatt 2011c
- Guyatt GH, Oxman AD, Kunz R, Woodcock J, Brozek J, Helfand M, et al. GRADE guidelines: 8. Rating the quality of evidence—indirectness. Journal of Clinical Epidemiology 2011;64(12):1303‐10. [DOI] [PubMed] [Google Scholar]
Guyatt 2011d
- Guyatt GH, Oxman AD, Kunz R, Brozek J, Alonso‐Coello P, Rind D, et al. GRADE guidelines 6. Rating the quality of evidence—imprecision. Journal of Clinical Epidemiology 2011;64(12):1283‐93. [DOI] [PubMed] [Google Scholar]
Guyatt 2011e
- Guyatt GH, Oxman AD, Montori V, Vist G, Kunz R, Brozek J, et al. GRADE guidelines: 5. Rating the quality of evidence—publication bias. Journal of Clinical Epidemiology 2011;64(12):1277‐82. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hill 2008
- Hill D. Remifentanil patient‐controlled analgesia should be routinely available for use in labour. International Journal of Obstetric Anesthesia 2008;17:336‐9. [DOI] [PubMed] [Google Scholar]
Hohne 2004
- Hohne C, Donaubauer B, Kaisers U. Opioids during anesthesia in liver and renal failure. Anaesthesist 2004;53:291‐303. [DOI] [PubMed] [Google Scholar]
Irestedt 1994
- Irestedt L. Current status of nitrous oxide for obstetric pain relief. Acta Anaesthesiologica Scandinavica 1994;38:771‐2. [DOI] [PubMed] [Google Scholar]
James 1991
- James MK, Feldman PL, Schuster SV, Bilotta JM, Brackeen MF, Leighton HJ. Opioid receptor activity of GI 87084B, a novel ultra‐short acting analgesic, in isolated tissues. Journal of Pharmacology and Experimental Therapeutics 1991;259:712‐8. [PubMed] [Google Scholar]
Jones 2012
- Jones L, Othman M, Dowswell T, Alfirevic Z, Gates S, Newburn M, et al. Pain management for women in labour: an overview of systematic reviews. Cochrane Database of Systematic Reviews 2012, Issue 3. [DOI: 10.1002/14651858.CD009234.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kartchner 2003
- Kartchner R, Callister L. Giving birth. Voices of Chinese women. Journal of Holistic Nursing 2003;21:100‐16. [DOI] [PubMed] [Google Scholar]
Klomp 2012
- Klomp T, Poppel M, Jones L, Lazet J, Nisio M, Lagro‐Janssen AL. Inhaled analgesia for pain management in labour. Cochrane Database of Systematic Reviews 2012, Issue 9. [DOI: 10.1002/14651858.CD009351.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kranke 2013
- Kranke P, Girard T, Lavand'homme P, Melber A, Jokinen J, Muellenbach RM, et al. Must we press on until a young mother dies? Remifentanil patient controlled analgesia in labour may not be suited as a "poor man's epidural". BMC Pregnancy and Childbirth 2013;13:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lavand'homme 2009
- Lavand'homme P, Roelants F. Patient‐controlled intravenous analgesia as an alternative to epidural analgesia during labor: questioning the use of the short‐acting opioid remifentanil. Survey in the French part of Belgium (Wallonia and Brussels). Acta Anaesthesiologica Belgica 2009;60:75‐82. [PubMed] [Google Scholar]
Leong 2011
- Leong WL, Sng BL, Sia AT. A comparison between remifentanil and meperidine for labor analgesia: a systematic review. Anesthesia and Analgesia 2011;113:818‐25. [DOI] [PubMed] [Google Scholar]
Liu 2014
- Liu ZQ, Chen XB, Li HB, Qiu MT, Duan T. A comparison of remifentanil parturient‐controlled intravenous analgesia with epidural analgesia: a meta‐analysis of randomized controlled trials. Anesthesia and Analgesia 2014;118:598‐603. [DOI] [PubMed] [Google Scholar]
Melzack 1984
- Melzack R. The myth of painless childbirth (the John J. Bonica lecture). Pain 1984;19:321‐37. [DOI] [PubMed] [Google Scholar]
Metafor 2015
- Viechtbauer W. Metafor. The metafor package: a meta‐analysis package for R (http://www.metafor‐project.org/doku.php) (accessed 20 October 2015) 2015.
Moghbeli 2008
- Moghbeli N, Pare E, Webb G. Practical assessment of maternal cardiovascular risk in pregnancy. Congenital Heart Disease 2008;3:308‐16. [DOI] [PubMed] [Google Scholar]
Pruefer 2012
- Pruefer C, Bewlay A. Respiratory arrest with remifentanil patient‐controlled analgesia‐‐another case. Anaesthesia 2012;67:1044‐5. [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager 5 (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Reynolds 2011
- Reynolds F. Labour analgesia and the baby: good news is no news. International Journal of Obstetric Anesthesia 2011;20:38‐50. [DOI] [PubMed] [Google Scholar]
Rosen 2002
- Rosen MA. Nitrous oxide for relief of labor pain: a systematic review. American Journal of Obstetrics and Gynecology 2002;186:S110‐26. [DOI] [PubMed] [Google Scholar]
Saravanakumar 2007
- Saravanakumar K, Garstang JS, Hasan K. Intravenous patient‐controlled analgesia for labour: a survey of UK practice. International Journal of Obstetric Anesthesia 2007;16:221‐5. [DOI] [PubMed] [Google Scholar]
Schnabel 2011
- Schnabel A, Hahn N, Muellenbach R, Frambach T, Hoenig A, Roewer N, et al. Obstetric analgesia in German clinics. Remifentanil as alternative to regional analgesia. Anaesthesist 2011;60:995‐1001. [DOI] [PubMed] [Google Scholar]
Semenic 2004
- Semenic SE, Callister LC, Feldman P. Giving birth: the voices of Orthodox Jewish women living in Canada. Journal of Obstetric, Gynecologic, and Neonatal Nursing 2004;33:80‐7. [DOI] [PubMed] [Google Scholar]
Stourac 2016
- Stourac P, Kosinova M, Harazim H, Huser M, Janku P, Littnerova S, et al. The analgesic efficacy of remifentanil for labour. Systematic review of the recent literature. Biomedical Papers: Journal of the Palacky University 2016;160:30‐8. [DOI] [PubMed] [Google Scholar]
Sweeting 2004
- Sweeting MJ, Sutton AJ, Lambert PC. What to add to nothing? Use and avoidance of continuity corrections in meta‐analysis of sparse data. Statistics in Medicine 2004;23(9):1351‐75. [DOI] [PubMed] [Google Scholar]
Thorlund 2009
- Thorlund K, Devereaux PJ, Wetterslev J, Guyatt G, Ioannidis JP, Thabane L, et al. Can trial sequential monitoring boundaries reduce spurious inferences from meta‐analyses?. International Journal of Epidemiology 2009;38:276‐86. [DOI] [PubMed] [Google Scholar]
Thorlund 2011
- Thorlund K EJ, Wetterslev J, Brok J, Imberger G, Gluud C. User manual for trial sequential analysis (TSA). Copenhagen Trial Unit, Centre for Clinical Intervention Research, Copenhagen, Denmark 2011; Vol. 1:1‐115.
TSA software [Computer program]
- Copenhagen Trial Unit’s Trial Sequential Analysis Software. TSA software 0.9 Beta. Copenhagen: Copenhagen Trial Unit’s Trial Sequential Analysis Software, 2011.
Tuckey 2008
- Tuckey JP, Prout RE, Wee MY. Prescribing intramuscular opioids for labour analgesia in consultant‐led maternity units: a survey of UK practice. International Journal of Obstetric Anesthesia 2008;17:3‐8. [DOI] [PubMed] [Google Scholar]
Van de Velde 2008
- Velde M. Controversy. Remifentanil patient‐controlled analgesia should be routinely available for use in labour. International Journal of Obstetric Anesthesia 2008;17:339‐42. [DOI] [PubMed] [Google Scholar]
Van de Velde 2015
- Velde M. Patient‐controlled intravenous analgesia remifentanil for labor analgesia: time to stop, think and reconsider. Current Opinion in Anaesthesiology 2015;28:237‐9. [DOI] [PubMed] [Google Scholar]
Victory 2004
- Victory R, Penava D, Silva O, Natale R, Richardson B. Umbilical cord pH and base excess values in relation to adverse outcome events for infants delivering at term. American Journal of Obstetrics and Gynecology 2004;191:2021‐8. [DOI] [PubMed] [Google Scholar]
Weber 1996
- Weber SE. Cultural aspects of pain in childbearing women. Journal of Obstetric, Gynecologic, and Neonatal Nursing 1996;25:67‐72. [DOI] [PubMed] [Google Scholar]
Weibel 2016
- Weibel S, Elia N, Kranke P. The transparent clinical trial: Why we need complete and informative prospective trial registration. European Journal of Anaesthesiology 2016;33(2):72‐4. [DOI] [PubMed] [Google Scholar]
Westmoreland 1993
- Westmoreland CL, Hoke JF, Sebel PS, Hug CC Jr, Muir KT. Pharmacokinetics of remifentanil (GI87084B) and its major metabolite (GI90291) in patients undergoing elective inpatient surgery. Anesthesiology 1993;79:893‐903. [DOI] [PubMed] [Google Scholar]
Wetterslev 2008
- Wetterslev J, Thorlund K, Brok J, Gluud C. Trial sequential analysis may establish when firm evidence is reached in cumulative meta‐analysis. Journal of Clinical Epidemiology 2008;61:64‐75. [DOI] [PubMed] [Google Scholar]
Wetterslev 2009
- Wetterslev J, Thorlund K, Brok J, Gluud C. Estimating required information size by quantifying diversity in random‐effects model meta‐analyses. BMC Medical Research Methodology 2009;9:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wilkinson 2010
- Wilkinson SE, Callister LC. Giving birth: the voices of Ghanaian women. Health Care for Women International 2010;31:201‐20. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Jokinen 2015
- Jokinen J, Weibel S, Afshari A, Artmann T, Eberhart LHJ, Pace NL, et al. Patient‐controlled analgesia with remifentanil versus alternative parenteral methods for pain management in labour. Cochrane Database of Systematic Reviews 2015, Issue 12. [DOI: 10.1002/14651858.CD011989] [DOI] [PMC free article] [PubMed] [Google Scholar]