

Abstract
Background
Hypertension is a chronic condition associated with an increased risk of mortality and morbidity. Renin is the enzyme responsible for converting angiotensinogen to angiotensin I, which is then converted to angiotensin II. Renin inhibitors are a new class of drugs that decrease blood pressure (BP) by preventing the formation of both angiotensin I and angiotensin II.
Objectives
To quantify the dose‐related BP lowering efficacy of renin inhibitors compared to placebo in the treatment of primary hypertension.
To determine the change in BP variability, pulse pressure, and heart rate and to evaluate adverse events (mortality, non‐fatal serious adverse events, total adverse events, withdrawal due to adverse effects and specific adverse events such as dry cough, diarrhoea and angioedema).
Search methods
The Cochrane Hypertension Information Specialist searched the following databases for randomized controlled trials (RCTs) up to February 2017: the Cochrane Hypertension Specialized Register, the Cochrane Central Register of Controlled Trials (CENTRAL) (2017, Issue 2), MEDLINE (from 1946), Embase (from 1974), the World Health Organization International Clinical Trials Registry Platform, and ClinicalTrials.gov. There was no restriction by language or publication status. We also searched the European Medicines Agency (EMA) for clinical study reports, the Novartis Clinical Study Results Database, bibliographic citations from retrieved references, and contacted authors of relevant papers regarding further published and unpublished work.
Selection criteria
We included randomized, double‐blinded, placebo‐controlled studies evaluating BP lowering efficacy of fixed‐dose monotherapy with renin inhibitor compared with placebo for a minimum duration of three to 12 weeks in adult patients with primary hypertension.
Data collection and analysis
This systematic review is a comprehensive update which includes four additional studies and extensive detail from nine clinical study reports (CSRs) of previously included studies obtained from EMA. The remaining three CSRs are not available.
Two review authors independently assessed study eligibility and extracted data. In all cases where there was a difference between the CSR and the published report, data from the CSR was used. Dichotomous outcomes were reported as risk ratio (RR) with 95% confidence intervals (CIs) and continuous outcomes as mean difference (MD) with 95% CIs.
Main results
12 studies (mean duration of eight weeks) in 7439 mostly Caucasian patients (mean age 54 years) with mild‐to‐moderate uncomplicated hypertension were eligible for inclusion in the review. Aliskiren was the only renin inhibitor evaluated. All included studies were assessed to have high likelihood of attrition, reporting and funding bias.
Aliskiren has a dose‐related systolic/diastolic blood pressure (SBP/DBP) lowering effect as compared with placebo MD with 95% CI: aliskiren 75 mg (MD ‐2.97, 95% CI ‐4.76 to ‐1.18)/(MD ‐2.05, 95% CI ‐3.13 to ‐0.96) mm Hg (moderate‐quality evidence), aliskiren 150 mg (MD ‐5.95, 95% CI ‐6.85 to ‐5.06)/ (MD ‐3.16, 95% CI ‐3.74 to ‐2.58) mm Hg (moderate‐quality evidence), aliskiren 300 mg (MD ‐7.88, 95% CI ‐8.94 to ‐6.82)/ (MD ‐4.49, 95% CI ‐5.17 to ‐3.82) mm Hg (moderate‐quality evidence), aliskiren 600 mg (MD ‐11.35, 95% CI ‐14.43 to ‐8.27)/ (MD ‐5.86, 95% CI ‐7.73 to ‐3.99) mm Hg (low‐quality evidence). There was a dose‐dependent decrease in blood pressure for aliskiren 75 mg, 150 mg and 300 mg. The blood pressure lowering effect of aliskiren 600 mg was not different from 300 mg (MD ‐0.61, 95% CI ‐2.78 to 1.56)/(MD ‐0.68, 95% CI ‐2.03 to 0.67). Aliskiren had no effect on blood pressure variability. Due to very limited information available regarding change in heart rate and pulse pressure, it was not possible to meta‐analyze these outcomes.
Mortality and non‐fatal serious adverse events were not increased. This review found that in studies of eight week duration aliskiren may not increase withdrawal due to adverse events (low‐quality evidence). Diarrhoea was increased in a dose‐dependent manner (RR 7.00, 95% CI 2.48 to 19.72) with aliskiren 600 mg (low‐quality evidence). The most frequent adverse events reported were headache, nasopharyngitis, diarrhoea, dizziness and fatigue.
Authors' conclusions
Compared to placebo, aliskiren lowered BP and this effect is dose‐dependent. This magnitude of BP lowering effect is similar to that for angiotensin‐converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs). There is no difference in mortality, nonfatal serious adverse events or withdrawal due to adverse effects with short term aliskiren monotherapy. Diarrhoea was considerably increased with aliskiren 600 mg.
Plain language summary
Blood pressure lowering efficacy of aliskiren
Review Question(s):
We wanted to determine if aliskiren was better than placebo for reducing blood pressure in adult patients with raised blood pressure, and whether this drug had increased adverse effects. We also wanted to determine if there was a change in blood pressure variability, pulse pressure, heart rate and withdrawal due to side effects.
We searched the available medical literature to find all the trials that had assessed these questions. The data included in this review is up to date as of February 2017.
Background
High blood pressure or hypertension can lead to heart attacks and stroke. A new class of drugs called renin inhibitors is indicated for the treatment of hypertension. Aliskiren is the only drug of the renin inhibitor class that has been studied and approved for treatment of hypertension at this time.
Study characteristics
We found 12 studies, eight weeks in duration, that randomly assigned 7439 adult people with uncomplicated mild‐to‐moderate hypertension to aliskiren at doses ranging from 75 mg to 600 mg or placebo. All studies were funded by the manufacturer Novartis. Detailed information regarding adverse events, obtained from nine clinical study reports submitted to regulators, are included in this update.
Key Results
We concluded that aliskiren is better than placebo at lowering blood pressure and that the magnitude of this effect could be similar to other classes of drugs when 300 mg, which is the maximum recommended dose, is used. Aliskiren 300 mg reduced blood pressure by eight points in the upper number (called systolic blood pressure) and five points in the lower number (called diastolic blood pressure). Aliskiren had no effect on blood pressure changeability. Due to very limited information regarding change in heart rate and pulse pressure (difference between upper and lower number) it was not possible to analyze these outcomes.
The studies were too short in duration to assess side effects. Aliskiren did not increase death, non‐fatal serious adverse events or withdrawal due to side effects. The most common adverse events observed were headache, diarrhoea, dizziness and fatigue. Diarrhoea was considerably increased with aliskiren 600 mg as compared to placebo.
Quality of evidence: The decrease in blood pressure at the recommended dose was rated as moderate‐quality evidence and adverse event data was graded as low‐quality evidence as included studies were assessed to have high likelihood of reporting and funding bias.
Summary of findings
for the main comparison.
| Aliskiren compared to placebo for primary hypertension | ||||||
|
Patient or population: primary hypertension Setting: Outpatient Intervention: Aliskiren Comparison: Placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI)mmHg | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Effect with placebo1 | Effect with Aliskiren2 | |||||
| Systolic BP ‐ Aliskiren 75 mg vs. placebo | 2.9 lower to 10.0 lower | 2.97 lower (4.76 lower to 1.18 lower) | ‐ | 1100 (5 RCTs) | ⊕⊕⊕⊝ MODERATE 3 | |
| Systolic BP ‐ Aliskiren 150 mg vs. placebo | 2.0 lower to 10.0 lower | 5.95 lower (6.85 lower to 5.06 lower) | ‐ | 3786 (12 RCTs) | ⊕⊕⊕⊝ MODERATE 4 | |
| Systolic BP ‐ Aliskiren 300 mg vs. placebo | 2.9 lower to 10.0 lower | 7.88 lower (8.94 lower to 6.82 lower) | ‐ | 3009 (10 RCTs) | ⊕⊕⊕⊝ MODERATE 5 | |
| Systolic BP ‐ Aliskiren 600 mg vs. placebo | 3.8 lower to 5.3 lower | 11.35 lower (14.43 lower to 8.27 lower) | ‐ | 393 (2 RCTs) | ⊕⊕⊝⊝ LOW 6,7 | |
| Diastolic BP ‐ Aliskiren 75 mg vs placebo | 3.2 lower to 8.6 lower | 2.05 lower (3.13 lower to 0.96 lower) | ‐ | 1100 (5 RCTs) | ⊕⊕⊕⊝ MODERATE 3 | |
| Diastolic BP ‐ Aliskiren 150 mg vs placebo | 3.0 lower to 8.6 lower | 3.16 lower (3.74 lower to 2.58 lower) | ‐ | 3783 (12 RCTs) | ⊕⊕⊕⊝ MODERATE 4 | |
| Diastolic BP ‐ Aliskiren 300 mg vs placebo | 3.2 lower to 8.6 lower | 4.49 lower (5.17 lower to 3.82 lower) | ‐ | 3001 (10 RCTs) | ⊕⊕⊕⊝ MODERATE 5 | |
| Diastolic BP ‐ Aliskiren 600 mg vs placebo | 6.2 lower to 6.3 lower | 5.86 lower (7.73 lower to 3.99 lower) | ‐ | 393 (2 RCTs) | ⊕⊕⊝⊝ LOW 6,7 | |
|
Diarrhoea ‐ Aliskiren 600 mg vs placebo |
14 per 1,000 | 95 per 1,000 (34 to 266) | RR 7.00 (2.48 to 19.72) | 592 (2 RCTs) | ⊕⊕⊝⊝ LOW 6,7 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk Ratio; MD: Mean Difference | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1. Range of mean decrease in SBP and DBP mmHg as compared to baseline in the individual trials in the placebo group.
2. Effects in Aliskiren group represent the blood pressure lowering effect in excess of that with placebo.
3. Downgraded by 1 level for serious risk of bias (due to high likelihood of attrition, selective reporting and funding bias in all 5 included studies). Also study CSPP100A2204 had high likelihood of selection bias.
4. Downgraded by one level for serious risk of bias (due to high likelihood of attrition, selective reporting and funding bias in all 12 included studies).
5. Downgraded by one level for serious risk of bias (due to high likelihood of attrition, selective reporting and funding bias in all 10 included studies).
6. Downgraded by 1 level for serious risk of bias (due to high likelihood of attrition, selective reporting and funding bias in both included studies).
7. Downgraded by 1 more level due to wide confidence interval.
Background
Description of the condition
Hypertension is defined as a systolic blood pressure (SBP) of 140 mmHg or greater and/or diastolic blood pressure (DBP) of 90 mmHg or greater in people who are not taking antihypertensive medication. (Poulter 2015). It is a chronic condition associated with an increased risk of mortality and morbidity from stroke, coronary heart disease, congestive heart failure, and renal disease. For patients with established hypertension, blood pressure should first be managed with life‐style and behaviour modification. However, if these measures prove inadequate then pharmacotherapy is indicated.
Description of the intervention
The renin‐angiotensin‐aldosterone system (RAAS) is a hormone system that regulates blood pressure and fluid balance. When renal blood flow is reduced, the kidney converts prorenin to renin and secretes it directly into the circulation. Plasma renin then converts angiotensinogen released by the liver to angiotensin I. Angiotensin I is subsequently converted to angiotensin II by the angiotensin‐converting enzyme (ACE) found in the lungs. This causes blood vessels to constrict, resulting in increased blood pressure. Angiotensin II also stimulates the secretion of the hormone aldosterone from the adrenal cortex, which causes the tubules of the kidney to increase the reabsorption of sodium and water into the blood. This increases the volume of the extracellular fluid in the body, which also increases blood pressure.
RAAS is an important target site for five distinctive antihypertensive drug classes: beta blockers, renin inhibitors, angiotensin‐converting enzyme inhibitors (ACEIs), angiotensin receptor blockers (ARBs) and aldosterone inhibitors.
How the intervention might work
The RAAS is a major regulator of cardiovascular homeostasis, in which the vasoactive peptide, angiotensin II, plays a central role. ACEIs and ARBs block this system by inhibiting the generation and action of angiotensin II, respectively. Although ACEIs and ARBs have proven effective, these agents do not block the RAAS completely. Whereas ACEIs and ARBs block the RAAS further downstream, renin inhibitors prevent the formation of renin, the enzyme that catalyses the conversion of angiotensinogen to angiotensin I, the rate‐limiting step in this cascade. Renin is responsible for all "downstream" events leading to production of angiotensin II and subsequent stimulation of its receptors.
Attempts to block renin began in the 1950s with the use of renin antibodies (Helmer 1958). Issues of potency, bioavailability, duration of action and cost of synthesis have marred the drug development of renin inhibitors. For example, potent renin inhibitors such as remikiren and enalkiren had low oral bioavailability (Frishman 1994). Newer drugs such as zankiren and terlakiren looked more promising, but further development was halted in the mid 1990s with the development of ARBs.
More recent programmes to develop renin inhibitors have been based on X‐ray crystallography of renin's active site with computational modelling rather than based on the structure of angiotensinogen (Krum 2007). This has led to aliskiren, a new non‐peptide, low molecular weight, orally active renin inhibitor, which has been approved in the USA, Canada and other countries for the treatment of hypertension.
Why it is important to do this review
ACEIs and ARBs block the RAAS downstream whereas renin inhibitors prevent the formation of renin, which is responsible for all "downstream" events leading to production of angiotensin II and subsequent stimulation of its receptors. It has been speculated that renin inhibitors might provide a more effective means of blockade of the RAAS than is possible with ACEIs or ARBs (Duprez 2006).
Two Cochrane reviews (Heran 2008a; Heran 2008b) have quantified the dose ranging blood pressure lowering efficacy of both ACEIs and ARBs as compared to placebo using similar methodology. This review was done to determine the dose ranging blood pressure lowering efficacy and adverse effects of renin inhibitors as compared to placebo and to compare the magnitude of reduction in blood pressure with ACEIs and ARBs.
It is important to note that a new methodological approach was undertaken to update this review by actively seeking out information available from all possible sources instead of relying on the very limited information available in the journal‐published trials included in the previous update of this review. For this update, a formal request for all relevant clinical study reports (CSRs) was made to the European Medicines Agency (EMA) under the Access to Documents Policy (0043). Information regarding an individual study was obtained from additional web sites: ClinicalTrials.gov, EU Clinical Trial Register, and Novartis Clinical Trial Results Database.
Cochrane reviews typically rely on journal‐published trials for critical appraisal of study conduct, methods and, if appropriate, for meta‐analysis of study data. However, reliance on journal‐published studies may pose a threat to the validity of a meta‐analysis due to levels of bias introduced by the published versions of these trials (Dwan 2013). Reporting bias is particularly relevant with respect to our decision to use data from regulatory sources, rather than solely rely on journal‐published studies, for this systematic review. At the study level reporting bias may occur when studies are not submitted or are rejected for publication (Chalmers 1990); at the outcome level reporting bias can occur as a result of selective non‐reporting of outcomes. The latter in particular has been described as an under‐recognized level of bias that serves to undermine the validity of systematic reviews (Hodkinson 2013; Kirkham 2010; McGauran 2010). Despite being considered one of the highest forms of evidence, systematic reviews may merely further the misrepresentation of journal‐published study data.
In an effort to correct for reporting bias, there has been an increased effort to access clinical study reports (Gotzsche 2011), which have been described as "the most complete synthesis of the planning, execution, and results of a clinical study" (Doshi 2012). These documents are required by regulatory authorities for market approval purposes and may also be produced for ongoing safety evaluations. Compared to journal‐published trials, CSRs do not require compression for journal publication, but are composed of thousands of pages that include multiple data sets on all pre‐specified outcome measures, numerous tables, figures and patient narratives (Jefferson 2015). A growing number of systematic reviews that rely on CSRs suggest that the data reported in journal‐published trials, as a result of discrepant reporting or non‐reporting, do indeed often lead to a misinformed representation of the evidence (Eyding 2010; Jefferson 2014; Sharma 2016). Additionally, studies that directly compare the reporting of harms between CSRs and journal‐published trials reveal the extent of reporting bias, and the distorted representation of safety within journal publications (Hodkinson 2016; Maund 2014; Wieseler 2012). Lacking access to the full complement of evidence, systematic reviews may serve to influence health policy and prescribing practices that result in harm or lack of benefit, a burden directly borne by the patient.
In light of these considerations, we took a relatively new methodological approach to rely more heavily on regulatory sources of data. We chose to include data from the US Food and Drug Administration medical review (FDA Medical Review 2007) within the 2017 update of this systematic review (Hart 2012; Rising 2008), and CSRs obtained through the European Medicine Agency's Access to Documents policy (0043). Though the process of obtaining CSRs and navigating the large volume of these documents is time‐consuming (Doshi 2016), the motivation to do so stems from a desire to create a review that is based upon transparent and complete data, and is essentially of greater value for our readers.
Objectives
Primary objective
To determine the dose ranging blood pressure lowering efficacy of fixed dose renin inhibitor monotherapy as compared with placebo over a period of three to 12 weeks in adult patients with primary hypertension.
Secondary objectives
To determine whether renin inhibitors affect blood pressure variability, pulse pressure, and heart rate as compared with placebo.
To document adverse events of renin inhibitors as compared with placebo (including mortality, non‐fatal serious adverse events, total adverse events, withdrawal due to adverse effects and specific adverse events such as dry cough, diarrhoea and angioedema).
Methods
Criteria for considering studies for this review
Types of studies
We included published and unpublished study reports of double‐blind, randomized, placebo‐controlled trials that randomly allocated patients to a fixed dose of renin inhibitor monotherapy or parallel placebo with duration of follow‐up of at least three weeks in adult patients with primary hypertension. There were no limits on language or publication status. Studies reported blood pressure measurements at baseline (following washout) and at one or more time points between three and 12 weeks post‐treatment on a fixed dose of drug.
Types of participants
Using a standard method of measurement, such as a calibrated standard mercury sphygmomanometer, adult participants had to have a baseline blood pressure of at least 140 mm Hg systolic and/or a diastolic blood pressure of at least 90 mm Hg. Participants could not have a creatinine levels greater than 1.5 times the normal level. Participants were not restricted by age, gender, baseline risk or any other co‐morbid conditions.
Types of interventions
Intervention included monotherapy with different fixed doses of renin inhibitor. Control group received a placebo. Data from study or arms of study in which titration to a higher dose was based on blood pressure response were not eligible. However, if a lower fixed dose was used for minimum of three weeks and then in all patients aliskiren was force titrated to a fixed higher dose for at least three weeks duration, then data at both doses were used from the same study to evaluate dose response.
Types of outcome measures
Primary outcomes
1. Change from baseline of trough and/or peak systolic and diastolic blood pressure over a minimum time frame of three weeks.
Secondary outcomes
Change in standard deviation of blood pressure.
Change in pulse pressure.
Change in heart rate.
Adverse events (including mortality, non‐fatal serious adverse events, total adverse events, withdrawal due to adverse effects and specific adverse events such as dry cough, diarrhoea and angioedema).
All secondary outcomes were assessed over a minimum time frame of three weeks and measured at the end of follow‐up period.
Search methods for identification of studies
Electronic searches
The Cochrane Hypertension Information Specialist conducted systematic searches in the following databases for randomized controlled trials without language, publication year or publication status restrictions:
the Cochrane Hypertension Specialised Register via the Cochrane Register of Studies (CRS‐Web) (searched 12 February 2017);
the Cochrane Central Register of Controlled Trials (CENTRAL; 2017, Issue 2) via the Cochrane Register of Studies (CRS‐Web) (searched 12 February 2017);
MEDLINE Ovid (from 1946 onwards), MEDLINE Ovid Epub Ahead of Print, and MEDLINE Ovid In‐Process & Other Non‐Indexed Citations (searched 12 February 2017);
Embase Ovid (searched 12 February 2017);
ClinicalTrials.gov (www.clinicaltrials.gov) searched 12 February 2017);
World Health Organization International Clinical Trials Registry Platform (www.who.int/trialsearch) searched 12 February 2017).
The Information Specialist modelled subject strategies for databases on the search strategy designed for MEDLINE. Where appropriate, they were combined with subject strategy adaptations of the highly sensitive search strategy designed by Cochrane for identifying randomized controlled (as described in the Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0, Box 6.4.b. (Higgins 2011)). Search strategies for major databases are provided in Appendix 1.
Searching other resources
The Cochrane Hypertension Information Specialist searched the Hypertension Specialised Register segment (which includes searches of MEDLINE and Epistemonikos for systematic reviews) to retrieve existing systematic reviews relevant to this systematic review, so that we could scan their reference lists for additional trials.
We checked the bibliographies of included studies and any relevant systematic reviews identified for further references to relevant trials.
Where necessary, we contacted authors of key papers and abstracts to request additional information about their trials.
We did not perform a separate search for adverse effects of interventions used for the treatment of hypertension. We considered adverse effects described in included studies only.
In preparation for the 2017 update, a formal request for all relevant clinical study reports (CSRs) was made to the European Medicines Agency (EMA) under the Access to Documents Policy (0043). Additionally, an enquiry for all relevant CSRs was submitted to Clinical Study Data Request (CSDR) (https://clinicalstudydatarequest.com), the data sharing platform to which the manufacturer, Novartis, belongs. We also searched the Novartis Clinical Trial Results Database (https://www.novctrd.com).
Review author KL submitted an enquiry to Novartis on the data sharing platform to which this company belongs, www.clinicalstudydatarequest.com (CSDR) on 17 October 2015 with respect to 12 studies that met the inclusion criteria for this review. Placing an enquiry is a step recommended if studies are not listed on the platform for data‐sharing. The Novartis data holder is expected to respond to the enquiry regarding potential for data‐sharing privileges to be granted. Two separate attempts were made to request a response from the data holder using the CSDR platform on 11 November 2015 and 20 January 2016. Unfortunately, we have not received a response from Novartis to date. CSDR Support has also made repeated attempts to elicit a response from the Novartis data holder to our enquiry without success.
Data collection and analysis
Selection of studies
Two review authors (VM and PF) independently screened the titles and the abstracts resulting from the search strategies from 2008 until October 2014. Two review authors (VM and KL) independently screened the titles and the abstracts resulting from the search strategies from October 2014 until February 2017. Articles were rejected on initial screening if it was judged from the title or the abstract that the article was not a report of a randomized placebo‐controlled study. We retrieved the full text of the remaining articles. The bibliographies of pertinent articles, reviews and texts were searched for additional citations.
Two review authors independently (VM and KL or PF) assessed the eligibility of the studies using a study selection form. We resolved discrepancies by discussion, and when necessary by a third author (JMW or KB).
Data extraction and management
Two review authors (VM and PF or KL) extracted data independently and cross‐checked them. All numeric calculations and graphic interpolations were confirmed by a second person.
This update relied on data from nine CSRs (not including appendices) received to date instead of their respective journal‐published study: 1) CSPP100A2201, 2) CSPP100A1201, 3) CSPP100A2308, 4) CSPP100A2203, 5) CSPP100A2405 6) CSPP100A2204, 7) CSPP100A2327, 8) CSPP100A2323 and 9) CSPP100A1301. The 9th CSR is of the study CSPP100A1301, which to date has no corresponding journal‐published trial.
The EMA does not possess CSRs of the three remaining studies meeting the inclusion criteria.
1. CSPA100A1301: Provided this trial does not have a corresponding journal‐published trial, we have relied on the results available on the ClinicalTrials.gov web site for this study.
2. CSPA100A2305: We have relied on the journal‐published report; additionally, results have been posted on the Novartis Clinical Trial Results Database, as well as results posted on ClinicalTrials.gov web site.
3. CSPP100A2328: We have relied on the journal‐published report; additionally, results have been posted on Novartis Clinical Trial Results database. This study was not registered on ClinicalTrials.gov web site.
In all cases where there was a difference between the CSR and published report, data from the CSR was used.
Assessment of risk of bias in included studies
Two review authors independently (VM and PF or KL) assessed the risk of bias of all included studies and prepared a 'Risk of bias' table.
Since our access to information for each included study differed based on the availability of CSRs, and/or results posted on Novartis Clinical Trial Results Database and/or results posted on ClinicalTrials.gov web site, our approach to evaluating the risk of bias of each study is based on differing availability of information.
1. Within CSRs, we found that the details on methods are generally reported more extensively than in journal‐published article. Since appendices were not included with the CSRs received by the EMA (their arrival is pending), we chose to assess a level of bias as 'unclear risk' when details on methods were insufficiently reported or not reported at all within a CSR. In the future update of this review, we will include information from the CSR appendices which are being requested from the EMA. Provided that Jefferson 2014 had access to CSRs with appendices for review, we could not follow their approach to assess a level of bias as 'high risk' of bias instead of 'unclear risk' when details on methods are insufficiently reported or not reported at all within a CSR.
2. For situations where we did not have access to the corresponding CSR of a journal‐published trial, the 'Risk of bias' assessment was conducted according to the journal‐published trial in accordance with the criteria established in Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
3. As a result of not having access to either the CSR or journal‐published study, we relied on clinical study synopses available on ClinicalTrials.gov for one study that met our inclusion criteria (CSPA100A1301). As these synopses are even more compressed in terms of reporting than journal‐published studies, we did not assume 'high risk' of bias for random sequence generation, allocation concealment or blinding if the methods were not described; rather, we rated them as 'unclear', or if described sufficiently, as 'low risk' of bias.
Measures of treatment effect
Mean difference was used in the meta‐analyses in this version and the previous version to report continuous outcomes such as change in SBP and DBP and the effect size is reported as MD with 95% confidence intervals (CIs).
Relative risk was used in the meta‐analyses in this version and the previous version to report dichotomous outcomes such as mortality, non‐fatal serious adverse events, withdrawal due to adverse effects and specific adverse events and the effect size is reported as RR with 95% CIs.
Unit of analysis issues
In the 12 included studies, although sitting trough SBP and DBP measurements were reported at different time points within each study, we have included data at the longest period of follow‐up on the monotherapy dose. Ten of the 12 included studies were of eight weeks duration and data at this time point was included in the analyses. From one study CSPP100A2323, 26 weeks in duration, data reported at three weeks on aliskiren monotherapy were used. Study CSPP100A1201 was 13 weeks in duration, but data reported at the end of the double‐blind period at eight weeks were included in the analyses.
Dealing with missing data
In the original review (Musini 2008b) and the first update in 2011, in the case of missing values for standard deviation of the change in blood pressure, the standard deviation was imputed based on the information in the same study or from other studies using the same dose. The following hierarchy (listed from high to low preference) was used to impute standard deviation values.
Standard deviation of change in blood pressure data from a different position (standing or supine).
Standard deviation of blood pressure at the end of treatment.
Standard deviation of blood pressure at baseline (except if this measure is used for entry criteria) (Musini 2009).
Mean standard deviation of change in blood pressure from other studies using the same class of drug.
In this 2017 update, we were able to include full reporting of mean sitting SBP (MSSBP) and mean sitting DBP (MSDBP) data with standard deviation (SD), least square means (LSMs) of change in MSSBP and MSDBP with standard error of the mean (SEM) from eight CSRs of published studies (CSPP100A1201; CSPP100A2201; CSPP100A2203; CSPP100A2204; CSPP100A2308; CSPP100A2327; CSPP100A2405; CSPP100A2323), and from one CSR of an unpublished study CSPP100A1301. Therefore, the reduction in LSM of change in SBP and DBP data including SD or SEM reported in this update may vary from previous versions of the review.
In study CSPP100A2323 patients were on aliskiren 150 mg dose until week 3 after which patients were force titrated to 300 mg until week 6. After week 6, patients initially randomized to placebo were re‐randomized to receive aliskiren 300 mg or hydrochlorothiazide (HCTZ) 25 mg in a 1:1 ratio. As there is no parallel placebo group after week 6, only data for change in MSDBP and MSSBP at week 3 for aliskiren 150 mg dose and at week 6 for aliskiren 300 mg dose is useful. The data for mean change in MSDBP with SEM was obtained from (CSR Page 84). The CSR did not report this information at week 3. Change in MSDBP and MSSBP at week 3 and for MSSBP at week 6 were estimated from the graphs (figure 3 in the published journal article). The SD's were imputed from the baseline SD for MSSBP (SD at baseline =11.2) and since the SD's were unusually low as reported in the baseline characteristics for MSDBP SD (3.3) the SD's were imputed from the average SD for all studies using aliskiren 150 mg dose or aliskiren 300 mg dose respectively .
Since CSRs are not available for two published studies CSPP100A2328 and CSPA100A2305 the missing values for standard error were imputed as follows:
In study CSPP100A2328, the change in MSDBP and MSSBP from baseline with standard deviation of change for placebo and aliskiren 75 mg, 150 mg and 300 mg at week 8 are reported from Novartis Clinical Trial Results Database.
In study CSPA100A2305, the change in MSDBP and MSSBP from baseline with standard deviation of change for placebo, aliskiren 150 mg and 300 mg at week 8 are reported from the clinical study synopses as reported on ClinicalTrials.gov (NCT00739973).
For the unpublished study CSPA100A1301 for which CSR is not available with EMA, the change in MSDBP and MSSBP from baseline with standard deviation of change for placebo and aliskiren 150 mg at week 8 are reported from clinical synopses posted on ClinicalTrials.gov (NCT01237223).
Assessment of heterogeneity
Test for heterogeneity of treatment effect between the studies was done using a standard Chi2 statistic for heterogeneity. The fixed‐effect model was applied to obtain summary statistics of pooled studies, unless significant between‐study heterogeneity was present, in which case the random‐effects model was used to test statistical significance.
Assessment of reporting biases
We planned to assess publication bias using a funnel plot if at least 10 studies met the inclusion criteria. In this version of the review we did not use a funnel plot to assess publication bias.
Data synthesis
Data synthesis and analyses were carried out using the Cochrane Review Manager software, RevMan 5.3.
The least square means (LSMs) reporting of blood pressure data is non‐robust to data outliers, particularly if outliers have a skewed distribution. This can cause an increase in type 2 errors, however it is not known if there were significant outliers in the data.
Subgroup analysis and investigation of heterogeneity
The following subgroup analyses were planned a priori.
Race: black, white, other (Asian)
Age: adults (18 to 64 years), elderly (65 years or years old)
Gender
Baseline severity of hypertension: mild, moderate, severe
Since none of the included studies provided data for specific age groups, gender, race or baseline severity of hypertension, we could not perform these analyses. However, the FDA Medical Review 2007 provided additional information regarding mean placebo‐corrected change from baseline in BP by dose and gender (seeTable 2); dose and age (seeTable 3); and dose and race (seeTable 4) from five placebo‐controlled studies (CSPP100A1201; CSPP100A2201; CSPP100A2203; CSPP100A2204; CSPP100A2308). This information is described in Effects of interventions.
1. Reported as table 21 in the FDA medical review (page 51): Reviewers mean placebo‐corrected change from baseline in BP by dose and gender based on 5 placebo controlled studies.
| Dose | Female | Male | ||
| SBP mmHg | DBP mmHg | SBP mmHg | DBP mmHg | |
| 75 mg | ‐1.3 | ‐1.1 | ‐3.6 | ‐2.9 |
| 150 mg | ‐5.5 | ‐2.9 | ‐5.9 | ‐3.5 |
| 300 mg | ‐9.4 | ‐4.8 | ‐9.0 | ‐5.4 |
| 600 mg | ‐12.6 | ‐6.4 | ‐10.7 | ‐6.5 |
| 75 mg | ‐1.3 | ‐1.1 | ‐3.6 | ‐2.9 |
2. Reported as table 22 in the FDA medical review (page 51): Reviewers mean placebo‐corrected change from baseline in BP by dose and age based on 5 placebo controlled studies.
| Dose | Age < 65 years | Age > 65 years | ||
| SBP mmHg | DBP mmHg | SBP mmHg | DBP mmHg | |
| 75 mg | ‐2.7 | ‐1.9 | ‐3.6 | ‐3.4 |
| 150 mg | ‐5.5 | ‐2.9 | ‐6.9 | ‐4.8 |
| 300 mg | ‐9.7 | ‐5.2 | ‐7.1 | ‐4.4 |
| 600 mg | ‐11.5 | ‐6.6 | ‐11.6 | ‐6.4 |
| 75 mg | ‐2.7 | ‐1.9 | ‐3.6 | ‐3.4 |
3. Reported as table 23 in the FDA medical review (page 52): Reviewers mean placebo‐corrected change from baseline in BP by dose and race based on 5 placebo controlled studies.
| Dose | White | Black | Asian | |||
| SBP mmHg | DBP mmHg | SBP mmHg | DBP mmHg | SBP mmHg | DBP mmHg | |
| 75 mg | ‐2.1 | ‐1.7 | ‐2.8 | 0.2 | ‐8.8 | ‐3.2 |
| 150 mg | ‐6.5 | ‐3.5 | ‐5.5 | ‐1.4 | ‐3.7 | ‐2.9 |
| 300 mg | ‐9.6 | ‐4.8 | ‐6.1 | ‐2.6 | ‐12.7 | ‐6.3 |
| 600 mg | ‐12.3 | ‐6.7 | ‐8.7 | ‐3.5 | ‐11.2 | ‐9.4 |
When heterogeneity was significant, we planned to investigate the factors contributing to heterogeneity such as differences in population characteristics or response to placebo that would possibly explain the reason for heterogeneity. However, no significant heterogeneity (I2 > 50%) was found for any outcome measure.
Sensitivity analysis
To test the robustness of the results several sensitivity analyses were planned a priori to be performed.
Studies of high risk of bias versus low risk of bias
Studies that were industry‐sponsored versus non‐industry sponsored
Studies that assess the drug as primary drug of investigation versus those that assess the drug as comparator
Studies with blood pressure data measured in the sitting position versus other measurement positions
Studies with published standard deviations of blood pressure change versus imputed standard deviation
Sensitivity analyses could not be done as all studies had similar risks of bias; all studies were industry sponsored; aliskiren was the primary drug of investigation; all studies reported mean sitting trough SBP and DBP; and standard deviation was imputed only in one study (CSPP100A2323).
We conducted a sensitivity analyses to look for differences in the magnitude of SBP and DBP reduction due to inclusion of information from CSRs in this update compared to previous update in 2011 which included information only from published journal articles. Additional information obtained from CSRs in this update did not result in altering the magnitude of reduction in MSSBP and MSDBP at any dose as compared to placebo from the previous 2011 update as the 95% CIs overlapped. See Table 5
4. Comparing MSSBP and MSDBP effect size of previous review versus present update after obtaining information from Clinical study Reports.
| Dose of Aliskiren mg/day | Previous version without access to information from CSR | Present version with access to information from CSR | ||
|
MSSBP mmHg MD with 95% CI |
MSDBP mmHg MD with 95% CI |
MSSBP mmHg MD with 95% CI |
MSDBP mmHg MD with 95% CI |
|
| Aliskiren 75 mg vs. Placebo | ‐2.64 (‐4.06 to ‐1.23) | ‐2.07 (‐2.94 to ‐1.20) | ‐2.97 (‐4.76 to ‐1.18) | ‐2.05 (‐3.13 to ‐0.96) |
| Aliskiren 150 mg vs. Placebo | ‐5.55 (‐6.39 to ‐4.71) | ‐2.91 (‐3.46 to ‐2.37) | ‐5.95 (‐6.85 to ‐5.06) | ‐3.16 (‐3.74 to ‐2.58) |
| Aliskiren 300 mg vs. Placebo | ‐7.93 (‐8.77 to ‐7.08) | ‐4.76 (‐5.33 to ‐4.19) | ‐7.88 (‐8.94 to ‐6.82) | ‐4.49 (‐5.17 to ‐3.82) |
| Aliskiren 600 mg vs. Placebo | ‐11.36 (‐13.53 to ‐9.19) | ‐6.57 (‐7.92 to ‐5.23) | ‐11.35 (‐14.43 to ‐8.27) | ‐5.86 (‐7.73 to ‐3.99) |
MSSBP: mean sitting systolic blood pressure; MSDBP: mean sitting diastolic blood pressure;
Results
Description of studies
Results of the search
From our search strategy we identified 862 study reports: 250 when the original search was run in 2008, another 172 when the review was updated in June 2011 and another 440 for the update in February 2017. Of these, 634 were duplicates or not relevant to this review.
We retrieved 46 articles for detailed evaluation: 8 for the initial version of this review in 2008, another 10 for the June 2011 update and another 28 for the current update in February 2017.
A total of 12 studies met the inclusion criteria: six published studies for the initial version of this review in 2008 (CSPP100A2201, CSPP100A1201, CSPP100A2308, CSPP100A2327, CSPP100A2203, CSPP100A2204), another two published studies for the June 2011 update (CSPP100A2328, and CSPP100A2323), and another four for the most recent update in February 2017, two of which are published studies (CSPP100A2405, CSPA100A2305), and two are unpublished studies (CSPP100A1301 and CSPA100A1301). See PRISMA Study flow diagram (Figure 1).
1.

PRISMA Study flow diagram.
See Table 6 for detailed information regarding identifier for individual studies meeting the inclusion criteria. All included study have been identified in this update using Novartis Clinical Trial Results Database study identifier number as opposed to the journal‐published author name and year, which were used in the previous two versions of this review.
5. Study identifier(s) in various websites of individual studies meeting the inclusion criteria.
|
Novartis Clinical Trial Results Database identifier used for study identification in this review |
Journal‐Published Author /year |
Registered on Novartis Clinical Trial Results Database |
Registered on ClinicalTrials.gov |
CSR* received from EMA |
Request to EMA was made on November 15th 2015. Weeks waited to obtain CSR |
| CSPA100A1301 | No journal publication | Results NOT posted |
NCT01237223 Results posted |
EMA does NOT possess | ‐ |
| CSPA100A2305 | Littlejohn 2013 | Results posted |
NCT00739973 Results posted |
EMA does NOT possess | ‐ |
| CSPP100A1201 | Kushiro 2006 | Results NOT posted | NOT registered | Mar 11, 2016 | 16 |
| CSPP100A1301 | No journal publication | Results posted |
NCT00344110 Results NOT posted |
May 23, 2016 | 27 |
| CSPP100A2201 | Gradman 2005 | Results NOT posted | NOT registered | Jan 25, 2016 | 10 |
| CSPP100A2203 | Pool 2007 | NOT FOUND | NOT registered | Apr 12, 2016 | 21 |
| CSPP100A2204 | Villamil 2007 | Results NOT posted |
NCT00219024 Results posted |
Apr 6, 2016 | 20 |
| CSPP100A2308 | Oh 2007 | Results NOT posted |
NCT00219128 Results NOT posted |
Feb 20, 2016 | 14 |
| CSPP100A2323 | Schmieder 2009 | Results posted |
NCT00219154 Results NOT posted |
May 27, 2016 | 27 |
| CSPP100A2327 | Oparil 2007 | Results NOT posted | NOT registered | June 16, 2016 | 30 |
| CSPP100A2328 | Puig 2009 | Results posted | NOT registered | EMA does NOT possess | ‐ |
| CSPP100A2405 | Villa 2012 | Results posted |
NCT00706134 Results posted |
December 22, 2015 | 5 |
Two studies were not published in journals but were identified on the Novartis Clinical Trial Results Database.
One published study CSPP100A2203 was not found on the Novartis Clinical Trial Results Database (CSPP100A2203).
Six studies, which are registered on the Novartis Clinical Trial Results Database, did not provide results (CSPP100A1201, CSPP100A2201, CSPP100A1301, CSPP100A2308, CSPP100A2327, and CSPP100A2204).
Five of the included studies are not registered on ClinicalTrials.gov web site ( CSPP100A1201, CSPP100A2201, CSPP100A2203, CSPP100A2327, CSPP100A2328).
Three studies are registered on ClinicalTrials.gov web site (CSPA100A1301, CSPP100A2308, and CSPP100A2323), but the results are not posted.
We have received only partial clinical study reports (CSRs) as appendices and individual patient data were not included. We need to make separate specific requests to the European Medicines Agency (EMA) to obtain this information.
Included studies
All 12 included studies were sponsored by the manufacturer (Novartis).
The 12 included studies had a total of 7439 patients (placebo = 2319; aliskiren = 5120). The majority of included studies were randomized , double‐blind, parallel group multicentre studies of eight weeks duration. The exceptions include the three trials: CSPP100A1201 (13 weeks duration), CSPP100A2327 (four weeks duration per dose), and CSPP100A2323 (three weeks duration per dose).
Six studies (CSPA100A2305CSPA100A1301; CSPP100A2203; CSPP100A2204; CSPP100A2327; CSPP100A2323) included active treatment arms in addition to placebo, comparing arms of aliskiren, combination therapy with valsartan, hydrochlorothiazide (HCTZ), and the respective monotherapies. Details of each included study are provided in Characteristics of included studies.
Following a one‐ to two‐week washout period from any prior antihypertensive medications, all studies had a two ‐to four‐week single‐blind placebo‐controlled run‐in period, followed by three to 13 weeks of randomized treatment. In most published studies, baseline mean sitting diastolic blood pressure (MSDBP) and mean sitting systolic blood pressure (MSSBP) were provided, but no endpoint MSDBP and MSSBP were provided. Consequently, comparisons were based on least square means (LSMs) and standard error (SE), which were converted to standard deviations (SDs). Only two published studies provided baseline and endpoint BP data (CSPP100A2201 and CSPP100A2203). The differences in the change in MSDBP and MSSBP in these two studies were similar to the LSMs provided.
In five of the studies (CSPP100A1201; CSPP100A2203; CSPP100A2204; CSPP100A2328; CSPP100A2405), patients received aliskiren once daily 75 mg, 150 mg and 300 mg.
In two studies (CSPP100A2201; CSPP100A2308), patients received once daily aliskiren 150 mg, 300 mg or 600 mg.
In CSPP100A2327, patients received once daily aliskiren 150 mg and then increased it to 300 mg in all patients after four weeks.
In CSPP100A2323, study patients received aliskiren 150 mg, HCTZ 12.5 mg or placebo for three weeks and all patients were then force titrated to double the dose for an additional three weeks. After six weeks from the start of the study, the placebo group was given active treatment so only data from aliskiren 150 mg data at week three and aliskiren 300 mg data at week six have been included.
In CSPA100A2305, patients were randomized to aliskiren 150 mg, 300 mg and placebo monotherapy as well as several combination therapy groups for a duration of eight weeks.
In CSPA100A1301, study patients were randomized to aliskiren 150 mg and placebo for a duration of eight weeks.
In CSPP100A1301, study patients were randomized to aliskiren 150 mg and placebo for a duration of eight weeks.
Population Characteristics
All studies had similar inclusion and exclusion criteria (see Characteristics of included studies).
Inclusion criteria: Patients were at least 18 years of age with mild‐to‐moderate essential hypertension defined as MSDBP > 95 mm Hg and < 110 mm Hg at baseline. Two studies (CSPP100A1201 and CSPP100A1301) were conducted in Japanese centres with only Asian adult patients (20 to 80 years of age for CSPP100A1201; 20 to 75 years of age for CSPP100A1301). All 12 studies had similar SBP and DBP at baseline ranging from 152 mmHg to 160 mmHg for SBP and 90 mmHg to 100 mmHg DBP. Most studies had Caucasian patients, ranging from 61% to 99% of randomized patients. The mean age in all studies ranged from 52 to 56 years of age except in CSPP100A2405. This study was conducted in elderly hypertensive patients ≥ 65 years of age; mean age of participants was 72 years.
Exclusion criteria: Patients were excluded if pregnant or breast feeding, had MSDBP >110 mm Hg and/or MSSBP > 180 mm Hg, had a history or evidence of secondary hypertension, type 1 or type 2 diabetes with poor glycaemic control, or any surgical or medical condition that might alter the absorption, distribution, metabolism or excretion of study drugs. Patients were also excluded if they had a history of severe cardiovascular, cerebrovascular, hepatic, or renal disease.
Blood Pressure Measurements
The position of the patient during blood pressure measurement may affect the blood pressure lowering effect. When blood pressure measurement data was available in more than one position, data was extracted in accordance with the following order of preference: 1) sitting; 2) standing; and 3) supine. In all studies, MSSBP and MSDBP were available for baseline measurements, and LSMs and SE were available for end of treatment reductions in MSSBP and MSDBP.
If blood pressure measurements were available at more than one time during the 24‐hour period, the trough measurement was used. Peak level is defined as within 12 hours of the dose and trough level is defined as between 12 and 24 hours. All studies reported trough MSSBP and MSDBP levels.
In all studies, sitting blood pressure (BP) measurements were recorded at baseline and regular intervals throughout studies (for example, at weeks one, two, four, six and eight). As well, sitting BP was measured using a calibrated standard mercury sphygmomanometer or an alternative calibrated method, in accordance with the 1988 AHA Committee Report on Blood Pressure Determination. Sitting blood pressure was measured after the patient had rested in sitting position for at least five minutes. The measurement was repeated a total of three times at an interval of one to two minutes. The three values were averaged to obtain a mean sitting blood pressure.
BP was measured at trough (24 + 3 hour post dose). At the first study visit, the patient's arm with the highest sitting DBP became the arm used for all subsequent readings in the study. At each study visit BP was measured three times at one‐ to two‐minute intervals, with the mean used as the BP for that visit. As well, one BP measurement was taken in standing position after the patient stood for two minutes.
Power
In five studies (CSPP100A2201; CSPP100A2203; CSPP100A2308; CSPP100A2328; CSPA100A2305), sample size was calculated to ensure sufficient power for both a primary intention‐to‐treat (ITT) analysis and a per‐protocol analysis. The study assumed a dropout rate of 10% and a standard deviation of 8 mm Hg for MSDBP. The sample size provided a 90% power to detect a treatment difference of at least 3.5 mm Hg for pair‐wise comparisons at a two‐sided significance level of 0.05.
In CSPP100A1201, the sample size provided an 80% power to detect a 3.5 mm Hg in MSDBP.
The study CSPP100A2327 provided 90% power, and CSPP100A2323 provided 95% power, to detect a treatment difference of at least 2 mm Hg in MSDBP.
In CSPP100A2405, the sample size was calculated to provide at least 97% power to detect a difference of 5 mm Hg in MSSBP between at least one aliskiren dose and placebo.
In CSPP100A2204, the sample size provided an 90% power to detect a 3.3 mm Hg in MSDBP and a SD of 8 mmHg.
The synopses of CSPA100A1301 and CSPP100A1301 studies do not provide details of how sample size was calculated.
Outcome Characteristics
The primary outcome of all studies was reduction in MSDBP.
Secondary efficacy variables of the studies included the following.
Mean reduction in MSSBP
Diastolic responder rates (defined as DBP less than 90 mm Hg or equal to 10 mm Hg reduction in DBP)
BP control rates (defined as a BP less than 140/90 mm Hg)
Safety and tolerability of aliskiren
Effects on plasma renin activity and renin concentration.
Excluded studies
Of the 46 articles retrieved for detail reading of study methodology, 31 studies did not meet our inclusion criteria and were excluded: two in 2008, eight in 2011 and 21 in 2017. The reasons for exclusion are reported in Characteristics of excluded studies. The main reasons form exclusion were lack of a parallel placebo control group or no aliskiren monotherapy treatment arm.
Risk of bias in included studies
See Figure 2
2.

Methodological quality summary: review authors' judgments about each methodological quality item for each included study.
Allocation
Random sequence generation (selection bias)
We considered 10 studies to have low risk of bias CSPA100A2305; CSPP100A1201; CSPP100A1301; CSPP100A2201; CSPP100A2203; CSPP100A2308; CSPP100A2323; CSPP100A2327; CSPP100A2328; CSPP100A2405), because they reported adequate sequence generation using computer‐generated random numbers and patients were assigned to treatment groups via central allocation or using validated interactive voice response system that automates random assignment.
We considered two studies to have 'unclear' risk of bias:
Study CSPA100A1301: random sequence generation was not reported within the clinical study synopses. Also, lack of access to their respective CSRs renders us unable to rate this level of bias. We have rated it as 'unclear' at this time.
Study CSPP100A2204: was rated to have unclear risk of bias as randomization was performed by Covance Inc. using validated system, however the detail of the method used was not reported in the CSR and we did not have access to the appendices for further details.
Allocation concealment (selection bias)
We considered nine studies to have low risk of bias for allocation concealment as randomization data were kept strictly confidential until completion of the study and patients, investigators, collaborators and the sponsor were unaware of the treatment assignments throughout the study until the database was locked (CSPP100A1201; CSPP100A1301; CSPP100A2201; CSPP100A2203; CSPP100A2308; CSPP100A2405; CSPP100A2327; CSPP100A2328; CSPP100A2323).
We considered three studies to have unclear risk of bias:
CSPA100A1301: as method of allocation concealment was not described in the clinical synopsis.
CSPP100A2204: randomization number was provided along with a unique medication number for the packages of study drug to be dispensed. We did not have access to the appendices for more detail.
CSPA100A2305: method of allocation concealment was not described in the clinical synopsis. The CSR is not available with EMA.
Blinding
We considered 10 studies to have low risk of bias for blinding as the identity of the treatment was concealed from patients, investigators and staff performing the assessment by the use of drugs that were identical in packaging, labelling, appearance, odour and schedule of administration (CSPA100A1301; CSPP100A1201; CSPP100A1301; CSPP100A2201; CSPP100A2204; CSPP100A2308; CSPP100A2323; CSPP100A2327; CSPP100A2328; CSPP100A2405 ).
We considered two studies to have unclear risk of bias as blinding of outcome assessors was not described (CSPA100A2305 and CSPP100A2203).
Study CSPA100A2305: is described as double‐blind. In order to adequately blind the study, patients were required to take a total of three tablets and two capsules of study medication throughout the study. No further details are provided.
Study CSPP100A2203: aliskiren was supplied as capsule for two doses (75 mg and 150 mg) and as tablet for 300 mg dose and there was insufficient information to determine if blinding was successful.
Incomplete outcome data
We considered four studies to have high risk of bias for incomplete outcome reporting because total withdrawal of randomized patients significantly differed in placebo and aliskiren treatment groups. Also last observation carried forward (LOCF) analysis was done for missing data at study endpoint. CSPA100A1301; CSPA100A2305; CSPP100A2201; CSPP100A2327.
We considered eight studies to have unclear risk of bias as reasons for withdrawal differed between placebo and treatment groups particularly due to unsatisfactory therapeutic effect. Also LOCF analysis was done for missing data at study endpoint (CSPP100A1201; CSPP100A1301; CSPP100A2203; CSPP100A2204; CSPP100A2308; CSPP100A2328; CSPP100A2405; CSPP100A2323.
Selective reporting
We meticulously looked for all possible studies conducted in various databases as well as the manufacturer's web site Novartis Clinical Trial Results Database, FDA web site U.S. Food and Drug Administration 2016 and ClinicalTrials.gov and found reporting bias for two studies (CSPA100A1301; CSPP100A1301), which had no journal publication but were listed on the manufacturer's web site.
We considered all 12 studies to have unclear risk of selective reporting bias. Protocols and CSRs (without appendices) were available for nine included studies. Most primary and secondary outcomes in these studies were reported. However for outcomes such as pulse pressure and heart rate, data were available in appendices which were not provided within CSRs from EMA. For the remaining three studies, since protocols were not available, we were unable to determine selective reporting risk of bias.
Other potential sources of bias
We considered all 12 studies to have high risk of other bias as they were sponsored by the manufacturer. Authors of six studies are employees of Novartis Pharmaceuticals Corporation and are therefore eligible for Novartis stock and stock options (CSPP100A2201; CSPA100A2305; CSPP100A2308; CSPP100A2327; CSPP100A2323; CSPP100A2405).
There was no description available with respect to the trialists' conflict of interest for three studies (CSPA100A1301; CSPP100A2328; CSPP100A2204). From the FDA Medical Review 2007 the regulator comments that financial disclosure forms were obtained for CSPP100A2201, CSPP100A2203, CSPP100A2204, and CSPP100A2308, but the financial disclosure form for CSPP100A1201 was not obtained. Moreover, the returned forms identified "Grants, Honoraria, Travel Expenses" exceeding $25,000 for Study (Study identifier redacted in FDA medical review) and "Consultant, speaker" exceeding $25,000 for Study (Study identifier redacted in FDA medical review). The regulator comments that "the two potential conflicts of interest could not prejudice the results greatly even if there were overt manipulation" due to the fact that the study takes place over multiple centres with small percentages of total study participants, and the study is double‐blinded.
Effects of interventions
See: Table 1
Mean change from baseline of trough systolic blood pressure
SeeTable 1
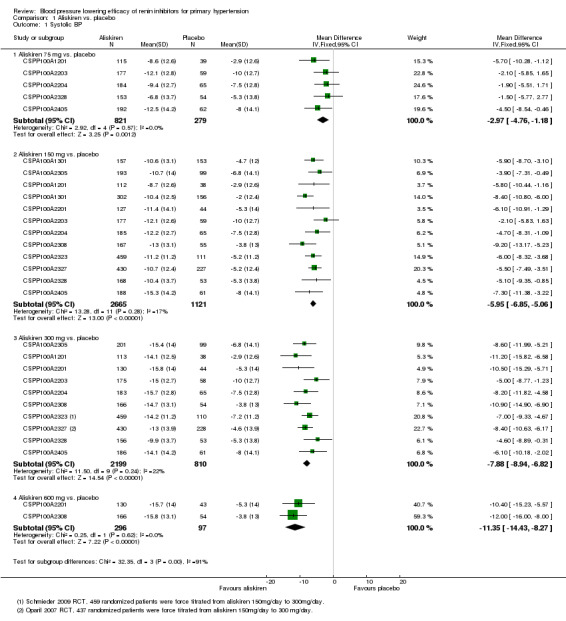
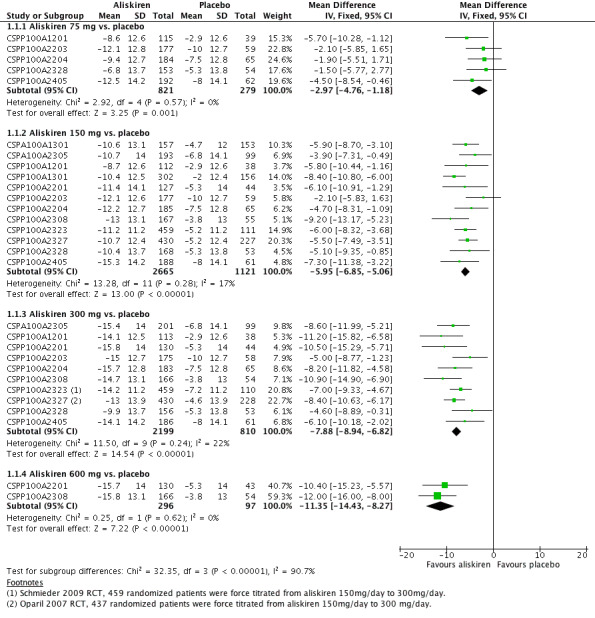
Aliskiren monotherapy was superior to placebo in lowering mean sitting systolic blood pressure (MSSBP). The additional magnitude of blood pressure lowering minus the placebo effect can be seen in Analysis 1.1.
1.1. Analysis.
Comparison 1 Aliskiren vs. placebo, Outcome 1 Systolic BP.
Aliskiren 75 mg versus placebo (mean difference (MD) ‐2.97, 95% confidence interval (CI) ‐4.76 to ‐1.18; participants = 1100; studies = 5; I2 = 0%); aliskiren 150 mg versus placebo (MD ‐5.95, 95% CI ‐6.85 to ‐5.06; participants = 3786; studies = 12; I2 = 17%); aliskiren 300 mg versus placebo (MD ‐7.88, 95% CI ‐8.94 to ‐6.82; participants = 3009; studies = 10; I2 = 22%); aliskiren 600 mg versus placebo (MD ‐11.35, 95% CI ‐14.43 to ‐8.27; participants = 393; studies = 2; I2 = 0%) see Figure 3. Quality of evidence was graded as moderate for 75 mg, 150 mg and 300 mg dose and as low for 600 mg dose.
3.
Forest plot of comparison: 1 Aliskiren vs. placebo, outcome: 1.1 Systolic BP.
Highly significant subgroup differences were observed therefore mean overall effect size across all doses is not shown. The number of placebo group patients are divided equally in dose ranging studies when more than one dose of aliskiren was used.
CSPP100A2308 study the SBP reduction in the treatment and placebo group are reported from the CSR page 61.
CSPP100A2405 study the SD for all treatment groups are calculated from SEM reported on page 7 in the CSR .
Note: Since there was highly significant subgroup differences between aliskiren 75 mg, 150 mg, 300 mg and 600 mg dose for MSSBP reduction (I2 = 90%), effect size is only presented as subtotals for each dose and overall MSSBP reduction is not shown. The number of patients in the placebo group is divided between various aliskiren dose comparisons if the study used more than one dose of aliskiren as compared to placebo.
There was no statistically significant heterogeneity observed for reduction in MSSBP for aliskiren 75 mg, 150 mg, 300 mg or the 600 mg dose. The I2 was zero for the 75 mg and 600 mg doses. For aliskiren 150 mg, one unpublished study which had a low MSSBP reduction in the placebo group ( ‐2.0 mmHg in CSPP100A1301) when removed from the analysis, reduced the I2 from 17% to 0%.
For aliskiren 300 mg, when two published studies, which had a low MSSBP reduction in the placebo group ( ‐2.9 mmHg in CSPP100A1201 and ‐3.8 mmHg in CSPP100A2308) were removed from the analysis, the I2 was reduced from 22% to 0%. Baseline MSSBPs were similar in all studies.
Mean change from baseline of trough diastolic blood pressure
SeeTable 1
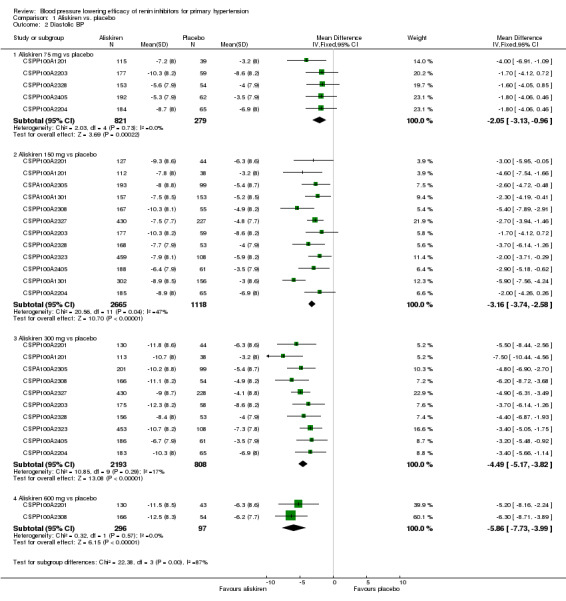
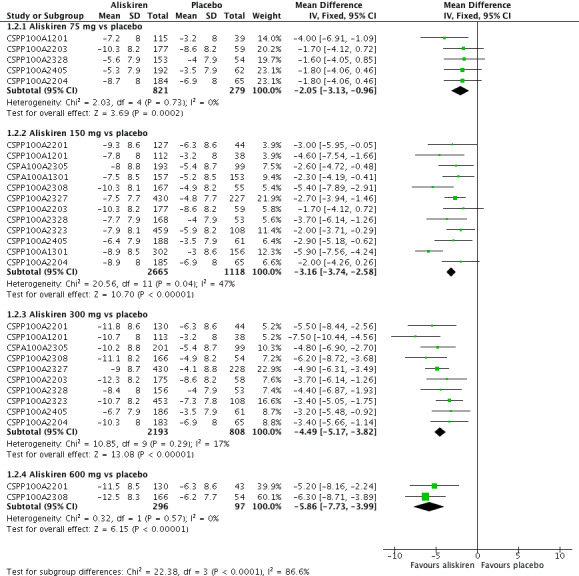
Aliskiren monotherapy was superior to placebo in lowering mean sitting diastolic blood pressure (MSDBP). The additional magnitude of blood pressure lowering minus the placebo effect can be seen in Analysis 1.2.
1.2. Analysis.
Comparison 1 Aliskiren vs. placebo, Outcome 2 Diastolic BP.
Aliskiren 75 mg versus placebo (MD ‐2.05, 95% CI ‐3.13 to ‐0.96; participants = 1100; studies = 5; I2 = 0%); aliskiren 150 mg versus placebo (MD ‐3.16, 95% CI ‐3.74 to ‐2.58; participants = 3783; studies = 12; I2 = 47%); aliskiren 300 mg versus placebo (MD ‐4.49, 95% CI ‐5.17 to ‐3.82; participants = 3001; studies = 10; I2 = 17%); aliskiren 600 mg versus placebo (MD ‐5.86, 95% CI ‐7.73 to ‐3.99; participants = 393; studies = 2; I2 = 0%), see Figure 4. Quality of evidence was graded as moderate for 75 mg, 150 mg and 300 mg dose, and as low for 600 mg dose.
4.
Forest plot of comparison: 1 Aliskiren vs. placebo, outcome: 1.2 Diastolic BP.
Highly significant subgroup differences were observed, therefore mean overall effect size across all doses is not shown. The number of placebo group patients are divided equally in dose ranging studys when more than one dose of aliskiren was used.
CSPP100A2308 study the DBP reduction in the treatment and placebo group are reported from the CSR which differ from those reported in the published article. The previous version of this review used data from the published article.
CSPP100A2405 study the SD for all treatment groups are calculated from SEM reported from the CSR on page 8.
Note: Since there was highly significant subgroup differences between aliskiren 75 mg, 150 mg, 300 mg and 600 mg dose for MSSBP reduction (I2 = 86.6%), effect size is only presented as subtotals for each dose and overall MSDBP reduction is not shown. The number of patients in the placebo group is divided between various aliskiren dose comparisons if the study used more than one dose of aliskiren.
There was no statistically significant heterogeneity observed for MSDBP for aliskiren 75 mg, and 600 mg dose. For aliskiren 150 mg, when one unpublished study which had a low MSDBP reduction in the placebo group (‐3.0 mmHg in CSPP100A1301) was removed from the analysis, the I2 was reduced from 47% to 0%. For aliskiren 300 mg , removing either CSPP100A1201 or CSPP100A2405, which had a low MSDBP reduction in the placebo group ( ‐3.0 mmHg in CSPP100A1201 or ‐3.5 mmHg in CSPP100A2405) from the analysis, reduced the I2 from 4% to 0%. Baseline MSDBPs were similar in all studies.
Comparing MSSBP and MSDBP reduction between aliskiren doses from the same study
Refer Table 7
6. Comparing MSSBP and MSDBP reduction between aliskiren doses from the same study.
| Dose comparison | SBP WMD with 95% CI | DBP WMD with 95% CI | Comment |
| Aliskiren 150 mg vs 75 mg | ‐1.89 (‐3.16 to ‐0.62) | ‐0.80 (‐1.58 to ‐0.03) | Aliskiren 150 mg reduces SBP and DBP more than 75 mg by 2/1 mmHg |
| Aliskiren 300 mg vs 75 mg | ‐5.10 (‐6.79 to ‐3.40) | ‐2.49 (‐3.53 to ‐1.45) | Aliskiren 300 mg reduces SBP and DBP more than 75 mg 5/3 mmHg |
| Aliskiren 300 mg vs 150 mg | ‐2.62 (‐3.38 to ‐1.87) | ‐1.80 (‐2.28 to ‐1.32) | Aliskiren 300 mg reduces SBP and DBP more than 150 mg 3/2 mmHg |
| Aliskiren 600 mg vs 150 mg | ‐3.40 (‐5.58 to ‐1.23) | ‐2.20 (‐3.55 to ‐0.85) | Aliskiren 600 mg reduces SBP and DBP more than 150 mg by 3/2mmHg |
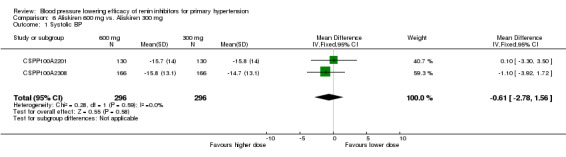
| Aliskiren 600 mg vs 300 mg | ‐0.61 (‐2.78 to 1.56) | ‐0.68 (‐2.03 to 0.67) | No significant difference was observed |
Aliskiren 150 mg lowered MSSBP and MSSDP more than aliskiren 75 mg SBP: (MD ‐1.89, 95% CI ‐3.16 to ‐0.62; participants = 1651; studies = 5; I2 = 23%) Analysis 2.1 DBP: (MD ‐0.80, 95% CI ‐1.58 to ‐0.03; participants = 1651; studies = 5; I2 = 0%) Analysis 2.2
2.1. Analysis.
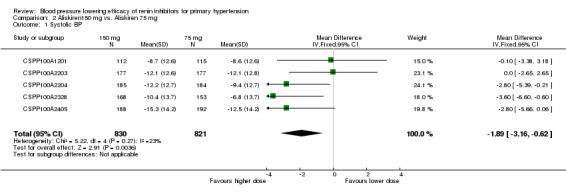
Comparison 2 Aliskiren150 mg vs. Aliskiren 75 mg, Outcome 1 Systolic BP.
2.2. Analysis.
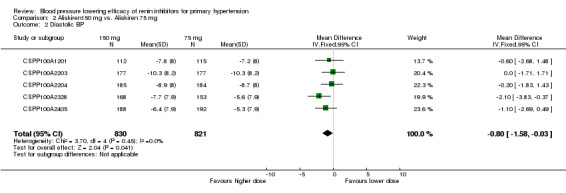
Comparison 2 Aliskiren150 mg vs. Aliskiren 75 mg, Outcome 2 Diastolic BP.
Aliskiren 300 mg lowered MSSBP and MSSDP more than aliskiren 75 mg SBP: (MD ‐5.10, 95% CI ‐6.79 to ‐3.40; participants = 904; studies = 3; I2 = 21%) Analysis 3.1 DBP: (MD ‐2.49, 95% CI ‐3.53 to ‐1.45; participants = 904; studies = 3; I2 = 7%) Analysis 3.2
3.1. Analysis.
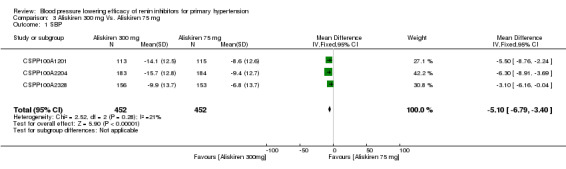
Comparison 3 Aliskiren 300 mg Vs. Aliskiren 75 mg, Outcome 1 SBP.
3.2. Analysis.
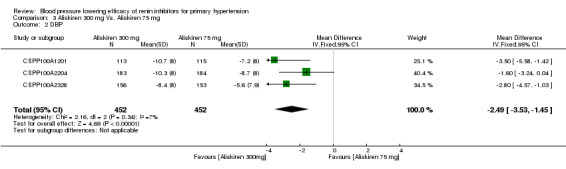
Comparison 3 Aliskiren 300 mg Vs. Aliskiren 75 mg, Outcome 2 DBP.
Aliskiren 300 mg lowered MSSBP and MSSDP more than aliskiren 150 mg SBP: (MD ‐2.62, 95% CI ‐3.38 to ‐1.87; participants = 4405; studies = 10; I2 = 51%) Analysis 4.1 DBP: ((MD ‐1.80, 95% CI ‐2.28 to ‐1.32; participants = 4405; studies = 10; I2 = 33%) Analysis 4.2
4.1. Analysis.
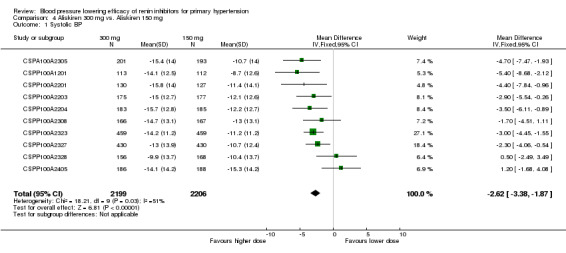
Comparison 4 Aliskiren 300 mg vs. Aliskiren 150 mg, Outcome 1 Systolic BP.
4.2. Analysis.
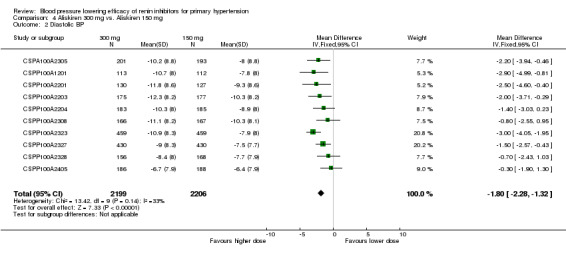
Comparison 4 Aliskiren 300 mg vs. Aliskiren 150 mg, Outcome 2 Diastolic BP.
Aliskiren 600 mg lowered MSSBP and MSSDP more than aliskiren 150 mg SBP: (MD ‐3.40, 95% CI ‐5.58 to ‐1.23; participants = 590; studies = 2; I2 = 0%) Analysis 5.1 DBP: (MD ‐2.20, 95% CI ‐3.55 to ‐0.85; participants = 590; studies = 2; I2 = 0%) Analysis 5.2
5.1. Analysis.
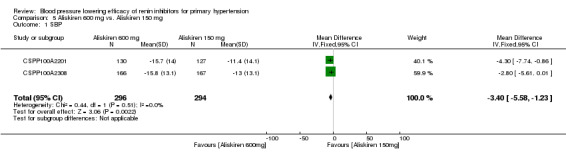
Comparison 5 Aliskiren 600 mg vs. Aliskiren 150 mg, Outcome 1 SBP.
5.2. Analysis.
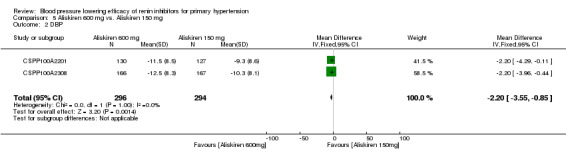
Comparison 5 Aliskiren 600 mg vs. Aliskiren 150 mg, Outcome 2 DBP.
However, there were no differences in MSSBP (MD ‐0.61, 95% CI ‐2.78 to 1.56; participants = 592; studies = 2; I2 = 0%) Analysis 6.1 or MSDBP (MD ‐0.68, 95% CI ‐2.03 to 0.67; participants = 592; studies = 2; I2 = 33%) Analysis 6.2 for aliskiren 600 mg as compared to aliskiren 300 mg.
6.1. Analysis.
Comparison 6 Aliskiren 600 mg vs. Aliskiren 300 mg, Outcome 1 Systolic BP.
6.2. Analysis.

Comparison 6 Aliskiren 600 mg vs. Aliskiren 300 mg, Outcome 2 Diastolic BP.
Change in standard deviation
End of treatment standard deviation of SBP and DBP was similar in the placebo and aliskiren arms in the 12 included studies. Standard deviation of the change in MSSBP ranged from 11.2 to 14.2 in the aliskiren treatment arm as compared to 11.2 to 14.1 in the placebo treatment arm. Standard deviation of the change in MSDBP ranged from 7.7 to 8.8 in aliskiren treatment arm and from 7.7 to 8.7 in the placebo treatment arm. This is consistent with aliskiren having no effect on BP variability.
Due to limited information available on standard error of the mean change in SD, this outcome has not been meta‐analyzed.
Change in pulse pressure
Limited information on pulse pressure is available in the bodies of the clinical study reports (CSRs); rather, this outcome is reported within the CSR appendices to which we do not yet have access.
However, CSPP100A2201 reports a reduction in pulse pressure at endpoint of ‐1.34, ‐3.5 and ‐3.91 for aliskiren 150 mg, 300 mg, and 600 mg, respectively versus +1.4 for placebo. At all study visits, aliskiren 600 mg produced the greatest reduction in sitting pulse pressure.
CSPP100A2203 reports pulse pressure increased (aliskiren 150 mg n = 1) as an adverse event, but the magnitude of this value is reported only in the appendices which are not yet accessible at this time.
No journal‐published study reported on pulse pressure at both baseline or endpoint. Therefore due to very limited information available on change in pulse pressure, this data has not been meta‐analyzed.
Change in heart rate
Five CSRs report on baseline sitting pulse rate but no data has been reported at the end of treatment. This data may be available in appendices which are not yet accessible at this time.
CSPP100A1201 study reports baseline sitting pulse rate (SD) as 72.0 (9.80), 75.0 (9.73), 72.0 (9.68), and 73.8 (8.91) for placebo, aliskiren 75 mg, 150 mg, and 300 mg.
CSPP100A2201 reports baseline sitting pulse as 72.8 (9.24), 72.9 (9.43), 72.2 (8.21) and 73.2 (8.51) for placebo, aliskiren 150 mg, 300 mg, and 600 mg, respectively. Also reported is baseline standing pulse rate as 76.1 (9.00), 76.3 (9.32), 76.0 (8.38) and 76.1 (8.95) for placebo, aliskiren 150 mg, 300 mg, and 600 mg, respectively. Aliskiren 600 mg produced a greater increase in trough sitting pulse than placebo at week eight (1.23 bpm versus ‐0.96 bpm; P = 0.0423). No other statistically significant differences between treatments were observed in either within‐treatment or between‐treatment analyses of trough sitting pulse at any time point.
CSPP100A2203 reports baseline sitting pulse as 71.6 (9.83), 72.0 (9.39), 72.3 (9.48) and 72.7 (9.22) for placebo, aliskiren 75 mg, aliskiren 150 mg, and aliskiren 300 mg, respectively. Also reported is baseline standing pulse rate as 76.3 (10.56), 77.0 (10.11), 76.4 (10.20, and 77.4 (10.02) for placebo, aliskiren 75 mg, aliskiren 150 mg, and aliskiren 300 mg respectively. Changes from baseline were minim al and generally similar across treatment groups.
CSPP100A2204 reports average baseline sitting and standing pulse rate as 72.2 bpm and 75.6bpm, respectively (page 60 of CSR). Summary statistics for baseline average sitting and standing pulse are provided in appendices, but not yet ascertained for the randomized and per protocol population.
CSPP100A2405 reports baseline sitting pulse rate as 73.8 (9.32), 72.8 (9.00), 73.8 (9.07) and 73.3 (9.03) for placebo, aliskiren 75 mg, 150 mg, and 300 mg, respectively (page 67 of CSR). There were no clinically meaningful differences in mean change from baseline in sitting pulse.
The journal‐published studies and clinical study synopses did not report on pulse rate. Therefore, due to very limited information available, these data have not been meta‐analyzed.
Mortality
No deaths were reported in six studies during single‐blind, double‐blind or withdrawal period (CSPA100A1301; CSPA100A2305; CSPP100A1301; CSPP100A2201; CSPP100A2308; CSPP100A2328.
Refer to Table 8 for detail information on cause of death in the following studies:
7. Mortality during single‐blind, double‐blind and withdrawal period in the 12 included studies.
| Study identifier | Single‐blind period | Double‐blind period | Withdrawal period |
| CSPA100A1301 | No deaths | No deaths | No deaths |
| CSPA100A2305 | No deaths | No deaths | No deaths |
| CSPP100A1201 | One death in placebo group during observation period due to pancreatic carcinoma and metastases to liver. | One death occurred in aliskiren 150 mg monotherapy on day 41 of the study, due to drug intoxication. The overdose was attributed to a psychiatric drug prescribed prior to start of the study, and a diagnosis of manic depressive psychosis was recorded. | No deaths |
| CSPP100A1301 | No deaths | No deaths | No deaths |
| CSPP100A2201 | No deaths | No deaths | No deaths |
| CSPP100A2203; | No deaths | One death occurred in the placebo arm on day 16 from "natural causes". One death occurred in the valsartan 160 mg arm (day 26) as a result of motor vehicle accident. |
No deaths |
| CSPP100A2204 | No deaths | One death was reported in the Aliskiren/HCTZ 150/25 mg group due to thoracic trauma from a motor vehicle accident. | No deaths |
| CSPP100A2308 | No deaths | No deaths | No deaths |
| CSPP100A2323 | 1 death occurred in placebo run‐in phase. Patient hospitalized due to supraventricular tachycardia and abdominal pain and died due to stroke after discontinuing from the study. | No deaths | One death occurred in the aliskiren/HCTZ 150/300 mg group from acute bronchopneumonia and associated sepsis during the 30‐day follow‐up period after discontinuing the study. |
| CSPP100A2327 | No deaths | 2 deaths reported. 1 death in aliskiren group on day 41 due to myocardial infarction. 1 death in valsartan group on day 13 and the cause was reported as hypertensive arteriosclerotic cardiovascular disease with SAE of sudden death. There were no deaths in placebo arm or combination therapy group. |
No deaths |
| CSPP100A2328 | No deaths | No deaths | No deaths |
| CSPP100A2405: | One death occurred during the follow‐up period of the study from pneumonia leading to respiratory failure (day of death not provided). Further information regarding this death is described in the patient narratives of the CSR, which we do not have access to yet. Within the CSR body, the arm of the study that this participant belongs to has been redacted. In the 2008 FDA Medical Review report, a death due to colon cancer is reported as occurring within CSPP100A2204 (N = 1, aliskiren 300 mg). The medical review states "no other details were provided". For our comments on this please see Discussion. |
CSR: clinical study report; HTCZ: hydrochlorothiazide; SAE: serious adverse event
During the single‐blind period, there was one death in the placebo group in two studies, CSPP100A1201 and CSPP100A2323.
During the double‐blind period, there was one death in aliskiren 150 mg group in study CSPP100A1201; one death in placebo group in study CSPP100A2203; one death in aliskiren/HCTZ 150 mg/25 mg group in study CSPP100A2204; and one death in aliskiren group in study CSPP100A2327.
During the withdrawal period there was one death in the aliskiren/HCTZ 150 mg/300 mg group in study CSPP100A2323 and one death in aliskiren 300 mg in study CSPP100A2405.
Due to the short duration of these studies and a very low incidence of death, the outcome of death was not meta analyzed.
Non‐fatal serious adverse events (SAE)
During the double‐blind treatment period two SAEs were reported in the aliskiren monotherapy group in study CSPP100A2203; two SAEs in the aliskiren monotherapy group in study CSPP100A2204; four SAEs in aliskiren 150 mg to 600 mg aliskiren monotherapy arms in study CSPP100A2308; eight SAEs in aliskiren monotherapy arms in study CSPP100A2327; four SAEs in aliskiren monotherapy arm in study CSPP100A2328; and five SAEs in aliskiren monotherapy arm in study CSPP100A2405. Refer to Table 9 for detailed information.
8. Non‐fatal serious adverse events during wash out, single‐blind, double‐blind and withdrawal period.
| Study identifier | Wash out period | Single blind period | Double blind period | Withdrawal period |
| CSPA100A1301 | Not reported | Not reported | No SAEs were reported in the aliskiren monotherapy group. 2 in amlodipine 2.5 mg; 1 in amlodipine 5 mg; and 1 in aliskiren 150/amlodipine 5 mg combination arm (detail regarding SAE was not reported) |
Not reported |
| CSPA100A2305 | Not reported | Not reported | 9 SAEs reported. 2 in placebo and 7 in other treatment groups (retinal detachment, abdominal mass, bronchitis, calculus ureteric, gastroenteritis, hand fracture, hydronephrosis, pneumonia, and cerebrovascular accident). None occurred in aliskiren 150 mg or 300 mg groups. |
Not reported |
| CSPP100A1201 | Not reported | Reported 1 SAE in the single‐blind period ‐ infectious enterocolitis/dehydration (N = 1, 55M, placebo). | 2 SAEs in the double‐blind period: cerebral infarction (N = 1, 69F, placebo); acute myocardial infarction (N = 1, 58F, aliskiren 75 mg). |
Not reported |
| CSPP100A1301 | Not reported | Not reported | 2 SAEs reported. 1 SAE in placebo due to myocardial infarction. The other in the losartan 50 mg group due to right medullary infarction. None in aliskiren 150 mg group. |
Not reported |
| CSPP100A2201 | One in washout period ‐ transient Ischaemic attack | Two in the single‐blind run‐in phase ‐ anxiety; and diverticulitis and | 2 SAEs in irbesartan group ‐ Intravertebral disc protrusion; and bipolar depression. | Two in the withdrawal period in placebo group ‐ Left ventricular failure; and gout and infective arthritis). |
| CSPP100A2203; | Not reported | Reported 6 SAE. 1 bile duct cancer, 1 diverticulitis, 1 pregnancy, 1 mild melanorrhagia and gastric pain, 1 myocardial infarction, and 1 squamous cell carcinoma. |
8 SAEs reported. 2 in Placebo: 1 due to natural cause and 1 due to myocardial infarction requiring hospitalization on day 46 resulting in study drug discontinuation. 1 in aliskiren 75 mg on day 4 coronary artery disease requiring hospitalization resulting in study discontinuation. Patient required quadruple bypass surgery. 1 in aliskiren 300 mg on day 42 pregnancy resulting in study discontinuation. 1 in valsartan 160 mg on day 28 ‐ angioneurotic oedema resulting in study discontinuation, dyspnoea and chest pressure. Aliskiren 150 mg/valsartan 160 mg (N = 1, Day 29) motor vehicle accident requiring hospitalization; not discontinued from study. |
1 in aliskiren 75 mg/valsartan 80 mg on day 3 post‐study. |
| CSPP100A2204* | Not reported | 4 SAE due to (lung cancer, fractured leg, angina and urinary tract infection) | 28 patients experienced at least 1 SAE in the monotherapy and combination therapy treatment groups. 1 in A 75 mg ‐ due to renal colic; 1 in aliskiren 150 mg due to haemorrhagic diarrhoea; none in aliskiren 300 mg; 1 in HCTZ 6.25 mg due to neoplasm of skin; 3 in HCTZ 12.5 mg ‐ due to breast cancer, joint injury and pregnancy; 2 in HCTZ 25 mg due to deep vein thrombosis and lymphadenopathy; 5 in aliskiren 75 mg/HCTZ 12.5 mg due to (diplopia, IIIrd nerve paresis, mood disorder, phlebothrombosis, psychotic disorder, small intestinal obstruction and syncope); 4 in aliskiren 75 mg/HCTZ 25 mg due to (cerebral infarction, dysarthria, physical disability, pregnancy and renal colic); 2 in aliskiren 150 mg/ HCTZ 6.25 mg ulcerative colitis, pregnancy); 3 in aliskiren 150 mg/ HCTZ 12.5 mg (non‐cardiac chest pain, pregnancy, syncope); 2 in aliskiren 150 mg/ HCTZ 25 mg (lung neoplasm, road traffic accident); 2 in aliskiren 300 mg/ HCTZ 12.5 mg (diabetes mellitus, lung disorder); 1 in aliskiren 300 mg/HCTZ 25 mg (coronary artery disease). There were no SAEs in the placebo group. |
Not reported |
| CSPP100A2308 | 2 SAEs reported during washout period ‐ 1 due to subarachnoid haemorrhage due to rupture of cerebral aneurysm on day 8; 1 due to partial small bowel obstruction on day 13 |
One SAE ‐ bladder carcinoma prior to randomization and before receiving double‐blind study drug. Patient was randomized to placebo but later discontinued due to unsatisfactory therapeutic effect; | 4 SAEs reported. 1 SAE in aliskiren 150 mg on day 27‐ unstable angina and increased blood pressure; 2 SAEs in aliskiren 300 mg ‐ hospitalization for acute appendicitis on day 34; hospitalization for depression on day 51; 1 SAE in aliskiren 600 mg of hospitalization for pain/bodily injury on day 35. No SAE reported in the placebo group. |
One SAE was reported during withdrawal period in aliskiren 150 mg of serious venous occlusion and thrombosis of the right eye on day 8 withdrawal period). |
|
CSPP100A2323 data at week 3 and week 6 not available |
Not reported | Not reported | During 26 weeks double‐blind treatment period 19 SAEs reported. 10 in aliskiren group and 9 in HCTZ group. |
Not reported |
| CSPP100A2327* | not reported | Not reported | 20 SAEs reported. 5 in placebo group ‐ atrial flutter, cerebrovascular accident, headache, hypertension, hypertensive crisis and ventricular tachycardia); 8 in aliskiren group ‐ 1 gastritis, 1 grand mal convulsions, 1 intestinal poly, 2 myocardial infarction, 1 non‐cardiac chest pain, 1 peripheral vascular disease, 1 acute renal failure); 6 in valsartan group ‐ 1 angina pectoris, 1 arteriosclerosis, 1 breast cancer, 1 bronchitis, 1 COPD, 1 facial paresis, 1 ovarian cancer, 1 pneumonia, 1 pulmonary oedema); 3 in aliskiren/valsartan group ‐ 1 aortic aneurysm, 1 intravertebral disc protrusion, 1 prostate cancer and 1 thyroidectomy). The FDA medical review (2007) also reported two cases of renal carcinoma occurred (N = 1, placebo, day 20 post‐study; N = 1, aliskiren, day 44 post‐study). For our comments on this, please see Discussion. |
Not reported |
| CSPP100A2328 | 11 patients had serious AEs during the washout and placebo run‐in periods (detail of which are not provided). | Not reported | 1 SAE was reported in aliskiren 75 mg group ‐ cardiac chest pain; 2 in aliskiren 150 mg group ‐ rectal bleeding due to anal ulcer and episode of secondary anaemia; basal cell carcinoma; 1 in the 300 mg group ‐TIA with concomitant nausea and dyspnoea; and 2 in the placebo group ‐ prostate cancer; accelerated hypertension with left facial numbness. |
Not reported |
| CSPP100A2405: | Reported 2 SAEs in the washout period; Detail not provided in CSR. | Three in the single‐blind placebo run‐in period. Detail not provided in CSR. | 6 SAEs were reported. 2 in aliskiren 75 mg group ‐ erysipelas (skin infection) and osteoarthritis; 3 in aliskiren 150 mg group ‐ severe glaucoma; moderate haemorrhoids; and mild hemorrhagic stroke; none in aliskiren 300 mg group; and 1 in the placebo group ‐ vertigo, wrist fracture, concussion, and head contusion subsequent to a fall. |
Not reported |
COPD: chronic obstructive pulmonary disease; CSR: clinical study report; HTCZ: hydrochlorothiazide; SAE: serious adverse event; TIA: transient ischaemic attack
In addition, the FDA Medical Review 2007 reports further details for the following studies.
CSPP100A1201 : The FDA medical review reports events of cancer in aliskiren monotherapy arms, and lists two SAEs of rectal cancer (n = 1, in 59 Asian women, aliskiren 75 mg, day 75) and (n = 1, in 54 Asian men, aliskiren 75 mg, day 330). However, the study identifiers within the FDA medical review have been redacted.
The clinical study synopsis for CSR 1202, an open‐label, extension study of CSPP100A1201, lists two events of rectal cancer as reasons for study discontinuation (n = 1, aliskiren monotherapy; n = 1, aliskiren + diuretic), and the corresponding journal‐published study Kushiro 2006 also reports two events of rectal cancer, but does not report the arm of the study in which they occurred, and states "these were considered by the investigators to be present at the commencement of the study." Provided that the regulator reviewed only one study of Japanese participants (CSPP100A1201), and given the rectal cancer outcomes reported in Kushiro 2006, we believe the SAEs pertain to the extension study of CSPP100A1201. Unfortunately, due to the redactions in the FDA medical review and the lack of access to the unabridged CRSs (including patient narratives), we cannot confirm this. These SAEs are not described in the body of CSR 1201.
CSPP100A2327: The FDA Medical Review (2007) reported a case of an isolated seizure (n = 1, aliskiren, day 3). The participant was hospitalized with a diagnosis of grand mal seizure, despite no previous history of seizures. The regulator notes that imaging studies and EEG were not provided in the initial report provided. The FDA medical review (2007) also reported two cases of renal carcinoma occurred (n = 1, placebo, day 20 post‐study; n = 1, aliskiren, day 44 post‐study). For our comments on this, please see Discussion.
CSPP100A2204: The three aliskiren monotherapy groups each had one patient with SAEs. Though these are reported within the CSR body, they are described in greater detail in the 2007 FDA medical review: renal colic necessitating hospitalization (n = 1 in 44 men, aliskiren 75 mg, day 19); hematuria and urinary retention necessitating catheterization to unblock a clot (n = 1 in 80 men, aliskiren 150 mg, day 19); and rectal bleeding from a stenosing tumour, which was confirmed as adenocarcinoma (n = 1 in 69 men, aliskiren 300 mg, day 46) aliskiren 300 mg group). Study medication was not discontinued. We note that CSPP100A2204 reports this latter SAE only as "hemorrhagic diarrhoea". This explains the reason that the FDA medical review (2007) reports an event of colon cancer (n = 1 in 69 men, aliskiren 300 mg, day 59), though this event is not reported in CSPP100A2204. It is unfortunate that we cannot confirm at this time whether this patient is the same patient or different from the reported event of death due to colon cancer within the 2008 FDA medical review.
During the single‐blind period, the FDA medical review (2007) reports chest pain and headache (n = 1 in 42 women, aliskiren 300 mg, day 25), which developed into cardiac ischaemia with diagnosis based on ST depression in V3‐5 not present on the baseline ECG after one dose of blinded study medication. The study drug was discontinued. The patient was subsequently hospitalized for unstable angina. HCTZ monotherapy had oneSAE in the 6.25 mg group; three SAEs in the 12.5 mg group and two SAEs in the 25 mg group. An event of colon cancer is reported in the FDA medical review (2007) (n = 1, HCTZ 25 mg, day 57), though this is not described in CSPP100A2204. Combination treatment groups had a total of 19 SAEs. The FDA medical review (2007) reports a stroke (n = 1, in 67 men, aliskiren 150 mg/HCTZ 6.25 mg) 19 days after the participant discontinued from the study. This SAE is not described within the body of CSPP100A2204.
Any adverse event
Reporting of total adverse events varies between studies since journal‐published studies did not report details regarding adverse events. In the 12 included studies, total adverse events in aliskiren groups ranged from 7.8% to 55% and from 21% to 50% in the placebo group. The most frequent events being ‐ headache ‐ ranged from 1.7% to 7.8% in aliskiren treatment groups and from 3.4 % to 13.5% in the placebo group; diarrhoea ‐ ranged from 0.8% to 11.4% in aliskiren treatment groups and from 0.5 % to 1.7% in the placebo group; dizziness ‐ ranged from 1.2% to 5.3% in aliskiren treatment groups and from 2% to 4.2% in the placebo group; and fatigue ‐ ranged from 0.8% to 3.8% in aliskiren treatment groups and from 1% to 3.1% in the placebo group. Refer Table 10 for additional details.
9. Total adverse events during double blind treatment period.
| Study Identifier |
Total AEs and Common AEs as reported in the corresponding available CSR A = Aliskiren; P = Placebo |
| CSPA100A1301 |
Total AEs: A150 =15(7.8%); A 300 = 18(8.9%) and P = 22(7.7%) Headache: A150 =13( 6.7%); A 300 =15(7.4%) and P = 20(10.1%) Periphaeral edema: A150 = 2(1.0%); A 300 = 3(1.5%) and P = 2(1%) |
| CSPA100A2305 |
Total AEs: A150 = 40.1%; A 300 = 46.7%; A 600 = 52.4%; P = 43% Headache: A150 = 12(7%); A300 = 13(7.7%); A600 = 9(5.4%); P = 16(9.7%) Nasopharyngitis: A 150 = 5(2.9%); A 300 = 6(3.6%); A 600 = 3(1.8%); P = 10(6.1%) Dizziness: A 150 = 2(1.2%); A 300 = 9(5.3%); A 600 = 5(3%); P = 7(4.2%) Diarrhoea:A 150 = 2(1.2%); A 300 = 3(1.8%); A 600 = 19 (11.4%); P = 2(1.2%) Nausea: A 150 = 2(1.2%); A 300 = 3(1.8%); A 600 = 0%; P = 4(2.4%) |
| CSPP100A1201 |
Total AEs: A75 = 61(53%); A150 = 58(51.8%); A 300 = 62(54.9%); P = 58(50.4)% Headache: A75 = 3(2.6%); A 150 = 3(2.7%); A 300 = 6 (5.3%) and P = 4(3.5%) Nasopharyngitis:A75 = 24(20.9%); A150 = 20(17.9%); A300 = 20(17.7%); P = 16(13.9%) Back pain:A75 = 2(1.7%); A150 = 0%; A300 = 0% and P = 0% Diarrhoea: A75 = 1(0.9%); A150 = 1(0.9%); A300 = 4(3.5%) and P = 1(0.9%) Vertigo: A75 =0%; A150 = 0%); A300 = 1(0.9%) and P = 2(1.7%) |
| CSPP100A1301 |
Total AEs: A150 = 152(50.3%), P = 66(42.3%) Nasopharyngitis: A150 = 48(15.9%), P = 13(8.3%) Headache: A150 = 9(3.0%), P = 6(3.8%) Occult blood positive: A150 = 9 (3.0%), P =5 (3.2%) |
| CSPP100A2201 |
Total AEs: A150 = 127 (26.8%); A300 = 130 (36.2%); A600 = 43 (33.1%); P = 32 (32.1%) Headache: A150 = 3 (2.4%); A300 = 8 (6.2%); A600 = 6 (4.6%); P = 7 (5.3%) Diarrhoea: A150 = 2 (1.6%); A300 = 1 (0.8%); A600 = 9 (6.9%); P = 2 (1.5%) Dizziness: A150 = 2 (1.6%); A300 = 4 (3.1%); A600 = 3 (2.3%); P = 5 (3.8%) Fatigue: A150 = 1 (0.8%); A300 = 5 (3.8%); A600 = 2 (1.5%); P = 4 (3.1%) |
| CSPP100A2203; |
Total AEs: A75 = 63(35.2%); A150 = 59(33.1%); A300 = 50(28.6%) and P = 57(32.2%) Headache: A75 = 15(8.4%); A150 = 9(5.1%;) A 300 = 7(4.0%) and P = 15(8.5%) Fatigue: A75 = 7(3.9%); A150 = 4(2.2%;) A 300 = 4(2.3%) and P = 4(2.3%) Back pain: A75 = 2(1.1%); A150 = 4(202%;) A 300 = 3(1.7%) and P = 2(1.1%) Diarrhoea: A75 = 2(1.1%); A150 = 1(0.6%;) A 300 = 5(2.9%) and P = 3(1.7%) Dizziness: A75 = 4(2.2%); A150 = 4(2.2%;) A 300 = 3(1.7%) and P = 2(1.1%) |
| CSPP100A2204 |
Total AEs: A75 = 69(37.5%); A150 = 69(37.3%); A300 = 71 (39.2%); P = 85(44%) Headache: A75 = 13(7.1%); A 150 = 13(7%); A 300 = 10 (5.5%); P = 26 (13.5%) Nasopharyngitis: A75 = 9( 4.9%); A150 = 5(2.7%;) A300 = 3 (1.7%); P = 10 (5.2%) Diarrhoea: A75 = 3(1.6%); A150 = 3(1.6%); A300 = 4(2.2%); P = 1(0.5%) Cough: A75 = 1(0.5%); A150 = 2(1.1%); A300 = 1(0.6%); P = 1(0.5%) Dizziness: A75 = 1(0.5%); A150 = 1(0.5%); A300 = 3(1.7%); P = 2(1.0%) Peripheral edema: A75 = 4(2.2%); A150 = 3(1.6%); A300 = 2(1.1%); P = 1(0.5%) |
| CSPP100A2308 |
Total AEs: A150 = 40.1%; A 300 = 46.7%; A 600 = 52.4%; P = 43% Headache: A150 = 12(7%); A300 = 13(7.7%); A600 = 9(5.4%); P = 16(9.7%) Nasopharyngitis: A 150 = 5(2.9%); A 300 = 6(3.6%); A 600 = 3(1.8%); P = 10(6.1%) Dizziness: A 150 = 2(1.2%); A 300 = 9(5.3%); A 600 = 5(3%); P = 7(4.2%) Diarrhoea: A 150 = 2(1.2%); A 300 = 3(1.8%); A 600 = 19 (11.4%); P = 2(1.2%) Nausea: A 150 =2 (1.2%); A 300 = 3(1.8%); A 600 =0%; P = 4(2.4%) |
| CSPP100A2323 |
Total AEs at 6 weeks : A 26.4%; P = 28.5% (numbers not reported) Other AE were not reported at week 6. |
| CSPP100A2327 |
Total AEs: A150 to 300 = 149(34%) and P = 168(37%) Headache: A150 to 300 = 14(3.2%); P = 41(9%) Diarrhoea*: A150 to 300 = 2.3%; P = not reported Nasopharyngitis: A150 to 300 = 16(3.7%) ; P = 9(2%) Dizziness: A 150 to 300 = 8(1.8%) ; P = 9(2%) Fatigue:A 150 to 300 = 4(0.9%) ; P = 5(1.1%) Nausea: A 150 to 300 = 6(1.4%) ; P = 11(2.4%) |
| CSPP100A2328 |
Total AEs: A75 = 57(37.3%); A150 = 59(34.7%); A300 = 56(35.4%); P = 60(37.5%) Headache: A75 = 12(7.8%); A150 = 7(4.1%); A300 = 10(6.3%) and P = 9(5.6%) Nasopharyngitis: A75 = 2(1.3%); A150 = 2(1.2%); A300 = 5(3.2%) and P = 3(1.9%) Diarrhoea: A75 = 7(4.6%); A150 = 2(1.2%); A300 = 1(0.6%) and P = 1(0.6%) Cough: A75 = 1(0.7%); A150 = 4(2.4%); A300 = 1(0.6%) and P = 3(1.9%) Dizziness: A75 = 1(0.7%); A150 = 5(2.9%); A300 = 1(0.6%) and P = 3(1.9%) |
| CSPP100A2405: |
Total AEs: A75 = 36(18.8%); A150 = 41(21.7%); A300 = 36(19.1%); P = 39(21.0%) Headache: A75 = 4(2.1%);A150 = 2(1.1%); A300 = 1(1.1%); P = 4(2.1%) Influenza: A75 = 1(0.5%); A150 = 4(2.1%); A300 = 1(1.1%); P = 1(0.5%) |
Withdrawals due to adverse events
Withdrawals due to adverse effects between placebo and aliskiren at 75 mg, 300 mg and 600 mg dose did not differ (see Analysis 1.3). Aliskiren 75 mg versus placebo ‐ (RR 0.59, 95% CI 0.33 to 1.07; participants = 1653; studies = 5; I2 = 0%); aliskiren 300 mg versus placebo ‐ (RR 0.70, 95% CI 0.47 to 1.03; participants = 4216; studies = 10; I2 = 0%); and aliskiren 600 mg versus placebo ‐ (RR 0.56, 95% CI 0.19 to 1.64; participants = 592; studies = 2; I2 = 0%).
1.3. Analysis.
Comparison 1 Aliskiren vs. placebo, Outcome 3 Withdrawals due to adverse event.
Compared to aliskiren 150 mg group, withdrawals due to adverse effects were greater in the placebo group mostly due to lack of therapeutic effect. Aliskiren 150 mg versus placebo ‐ (RR 0.46, 95% CI 0.30 to 0.71; participants = 3421; studies = 10; I2 = 16%). CSPP100A2323 was not included in this comparison as the authors only report a combined total withdrawal rate n = 22 (3.9%) for both aliskiren and placebo treatment groups.
For withdrawals due to adverse effects, there was no statistically significant heterogeneity at 75 mg, 150 mg, 300 mg or 600 mg dose. For 75 mg, 300 mg and 600 mg dose, the I2 was 0%. For the 150 mg dose, when CSPP100A2201 was removed from the analyses, the I2 was reduced from 16% to 0%. The reason for heterogeneity could not be determined.
Refer to Table 11 for detail regarding reasons for withdrawal from included studies.
10. Listing reasons for withdrawal due to adverse events.
| Study Identifier | Reasons for increased withdrawal due to adverse events |
| CSPA100A1301 | Not reported. |
| CSPA100A2305 | Not reported. |
| CSPP100A1201 | Significant adverse events occurred for N = 7 patients, leading to discontinuation from study, as described below.
|
| CSPP100A1301 |
|
| CSPP100A2201 | A total of N = 18 patients discontinued from the study due to AEs. It was suspected that 2/3 of these discontinuations were for reasons drug‐related. Headaches resulted in the discontinuation for N = 3 patients. |
| CSPP100A2203; | Most frequent reasons for 12 discontinuations are: fatigue (N = 5, 0.4%), headache (N = 3, 0.3%), diarrhoea (N = 2, 0.2%), and peripheral oedema (N = 2, 0.2%). Reasons for 14 discontinuations from AE (safety population).
The FDA medical review reports facial oedema (N = 1, 0.6%) that appears to be angioedema, resulting in discontinuation from study; however, this is not reported in CSR 2203.
|
| CSPP100A2204 | The range of AEs resulting in study discontinuation is 0% is the aliskiren 150 mg arm of the study to 4.4% in the aliskiren 300 mg arm (N = 7). The most frequent AE resulting in study discontinuation is headache (N = 12, 0.4%). Reasons for discontinuations from adverse events ‐ N = 62 (2.2%).
Additionally four patients discontinued from the study due to abnormal laboratory values.
|
| CSPP100A2308 | Most frequent reasons for 9 discontinuation from study: headache (N = 4), dizziness (N = 3), and diarrhoea (N = 2). Reasons for discontinuations from AE (safety population).
We question that a participant withdrew for the reason specified as "unsatisfactory effect" as opposed to being categorized as a discontinuation from an SAE as the event necessitated hospitalization. On Day 27 in CSPP100A2308, the participant (N = 1, 64F, aliskiren 150 mg) was hospitalized for unstable angina and increased blood pressure. The unstable angina was treated with concomitant medication and her condition resolved within 3 days. |
| CSPP100A2323 | We do not have data at week 3 and week 6 for this study. |
| CSPP100A2327 | The range of AEs resulting in study discontinuation is 8 (1.8%) is the aliskiren arm; 8 (2.6%) in valsartan arm; 6 (1.3%) in aliskiren/valsartan combination arm; and 11 (2.4%) in the placebo arm. Reasons for discontinuations from adverse events ‐ N = 37 (2%).
|
| CSPP100A2328 | Aliskiren 75 mg = 3 (2.0%); aliskiren 150 mg = 5 (2.9%); aliskiren 300 mg = 4(2.5%) . Placebo = 4 (2.5%). Reasons for withdrawal were not reported. |
| CSPP100A2405: | Reasons for 18 discontinuations from AE (safety population)
|
Specific adverse events
Dry cough as an adverse event was reported in the following five studies: refer Analysis 1.4.
1.4. Analysis.
Comparison 1 Aliskiren vs. placebo, Outcome 4 Cough.
CSPP100A1201: aliskiren 150 mg n = 1 (0.9%)
CSPP100A2201: placebo n = 1 (0.8%), aliskiren 150 mg n = 2 (1.6%), aliskiren 300 mg n = 4 (3.1%)
CSPP100A2203: placebo n = 2 (1.1%), aliskiren 75 mg n = 2 (1.1%), aliskiren 150 mg n = 5 (2.8%), aliskiren 300 mg n = 1 (0.6%)
CSPP100A2204: aliskiren 300 mg n = 1 (0.6%)
CSPP100A2328: placebo: n = 3 (1.9%), aliskiren 75 mg n = 1 (0.7%), aliskiren 150 mg: n = 4 (2.4%), aliskiren 300 mg: n = 1 (0.6%).
Cough was not reported in the other seven studies (CSPA100A2305; CSPP100A2308; CSPP100A2405; CSPA100A1301;CSPP100A1301; CSPP100A2327; and CSPP100A2323).
Based on five studies in 2886 patients, the incidence of cough did not differ between aliskiren (75 mg to 600 mg dose) as compared to placebo (RR 1.14, 95% CI 0.49 to 2.64; I2 = 0%).
Diarrhoea
Refer to Analysis 1.5 and to Table 1 graded as low‐quality evidence.
1.5. Analysis.
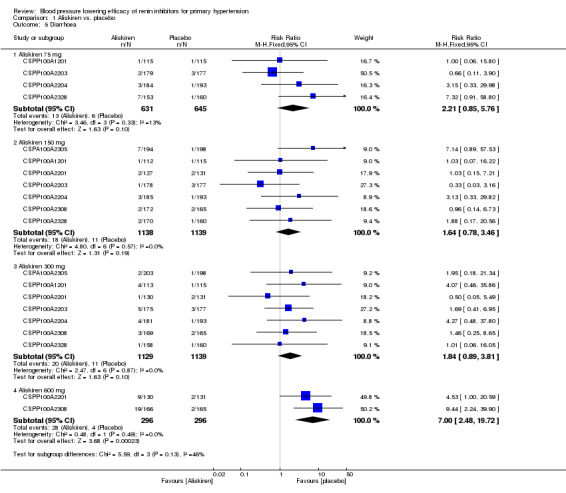
Comparison 1 Aliskiren vs. placebo, Outcome 5 Diarrhoea.
Aliskiren at 75 mg to 300 mg dose as compared to placebo showed a trend towards increase in incidence of diarrhoea. However at a dose of 600 mg, a substantial increase was observed (from 14 per 1000 in placebo group to 95 per 1000 in aliskiren 600 mg group):
aliskiren 75 mg: (RR 2.21, 95% CI 0.85 to 5.76; participants = 1276; studies = 4; I2 = 13%);
aliskiren 150 mg: (RR 1.64, 95% CI 0.78 to 3.46; participants = 2277; studies = 7; I2 = 0%);
aliskiren 300 mg: (RR 1.84, 95% CI 0.89 to 3.81; participants = 2268; studies = 7; I2 = 0%); and
aliskiren 600 mg: (RR 7.00, 95% CI 2.48 to 19.72; participants = 592; studies = 2; I2 = 0%).
Angioedema:
No journal‐published study reported angioedema. The FDA Medical Review 2007 reporting angioedema are the following.
CSPP100A2203 reports angioneurotic oedema occurred in valsartan 160 mg (n = 1, 1.7%). In the FDA medical review, this is described as "moderate facial edema diagnosed as angioedema. Drug was discontinued and the edema resolved. This patient had a similar episode with an ACE inhibitor." The FDA medical review also reports an event of facial oedema (n = 1) that appears to be angioedema, beginning with treatment and resolving eight days after discontinuing treatment. A third event of ankle and periorbital oedema (n = 1) is reported in the aliskiren 300 mg/valsartan 320 mg group beginning five days into treatment and resolving three days after discontinuing treatment.
CSPP100A2204 reports 'peripheral oedema' (n = 1, aliskiren 300 mg) as a reason for study discontinuation. However, the FDA medical review (2007) reports a separate event in greater detail: "A 61 year‐old diabetic white male in the aliskiren 75mg group developed dyspnoea and edema of the lower limb to the knee, hands and face (eyelid but no lip or tongue) on day 22. He also had purplish‐blue lesions with angiomatous aspect on the upper part of his trunk. He completed the study with continuation of the edema and also developed anorexia, weight loss, tachycardia and elevated CRP." The regulator comments that "the participant was diagnosed with scleredema, and that the investigator confirmed that there was no evidence of heart failure at baseline and suspected angioedema."
CSPP100A2308 ‐The FDA medical review describes that a patient in the aliskiren 300 mg group reported "Oedema hands and feet" on day 53. The regulator suggests this case may represent angioedema.
Subgroup analysis from FDA medical review
The FDA Medical Review 2007 provided additional information regarding mean placebo‐corrected change from baseline in BP by dose and gender (Table 2); dose and age (Table 3); and dose and race (Table 4) from five placebo‐controlled studies (CSPP100A1201; CSPP100A2201; CSPP100A2203; CSPP100A2204; CSPP100A2308).
The FDA medical reviewer comments: "In the multivariate regressions aliskiren shows a clear dose‐response from 75 through 600 mg for both SBP and DBP. Blacks show a reduced response and Asians an increased response with statistically significant interaction term with aliskiren use for Asians for DBP and almost statistically significant for Blacks for DBP. The interaction term for age > 65 years and drug is statistically significant for SBP. There appear to be differences in response in various sub groups with aliskiren use. Blacks respond poorly to aliskiren monotherapy while Asians and the elderly may respond to lower doses."
Discussion
To our knowledge this is the first systematic review to use clinical study reports (CSRs) and other regulatory documents to report on a more comprehensive set of efficacy and safety data regarding dose‐related blood pressure lowering efficacy of renin inhibitors versus placebo. As a result, this updated review provides more detailed information regarding mortality, non‐fatal serious adverse events, total adverse events and specific adverse events. Though we found outcome reporting to be far more extensive within CSRs as compared to journal‐published studies, even access to these regulatory documents remain insufficient as a means to correct for reporting bias. Our attention was drawn to the discrepant reporting or non‐reporting of important safety outcomes, discovered by closely reading the FDA medical review of Dr. Thomas Marciniak. These findings have increased our awareness for the need to access the patient narratives on adverse events located within the appendices of CSRs. Further to this, individual patient data and access to all case reports and narratives are likely needed to report more completely on the harms and efficacy of aliskiren. Given the EMA can provide de‐identified patient narratives on serious and non‐serious adverse events, we question the reasons the sponsor, Novartis, has for not providing any case narratives to researchers who are granted data‐sharing privileges through the CSDR platform. Though obtaining the data from CSR bodies has increased our ability to report more extensively on efficacy and safety outcomes, we remain unable to accurately depict the clinical safety, usefulness and value of aliskiren. As to whether aliskiren results in greater benefit than harm to patients remains unclear. This is a direct result of not having access to the entire complement of evidence.
We have not identified any randomized controlled trials (RCTs) evaluating the effectiveness of renin inhibitors as compared to placebo in reducing mortality and morbidity, as well as documenting the long‐term reduction in systolic blood pressure (SBP) and diastolic blood pressure (DBP), which is the goal of antihypertensive therapy.
Zhang 2015 conducted a systematic review of RCTS comparing the effect of aliskiren versus non‐aliskiren therapy on major cardiovascular outcomes in clinically diverse patients with hypertension. Based on six studies (ALTITUDE 2012; AVOID 2011; AVANT GARDE ‐ TIMI 43 2010; ASPIRE 2011 ; ASTRONAUT 2013; and AQUARIUS 2013), reporting data in 12,465 patients, the authors concluded that aliskiren had no effect (RR with 95% CI) on major cardiovascular events 0.93 (0.77 to 1.13), total mortality 1.00 (0.77 to 1.29), cardiac death 1.01 (0.79 to 1.29), myocardial infarction 0.71 (0.36 to 1.38), or stroke 0.87 (0.48 to 1.58).
Summary of main results
Aliskiren at all doses lowered both SBP and DBP as compared to placebo ranging from ‐3.0 to ‐11.4 mmHg for SBP and ‐2.1 to ‐5.9 mmHg for DBP. There was a significant dose response for the 75 mg, 150 mg and 300 mg doses. The effect of 600 mg of aliskiren was not different from 300 mg. Information in this update from nine available CSRs obtained from EMEA did not result in altering the magnitude of reduction in mean sitting systolic blood pressure (MSSBP) and mean sitting diastolic blood pressure (MSDBP) at any dose as compared to placebo from the previous 2011 update as the 95% confidence intervals (CIs) overlapped.
Comparison between various doses within the same study showed that aliskiren 150 mg lowered SBP/DBP more than aliskiren 75 mg (‐1.9/‐0.8 mmHg); aliskiren 300 mg lowered SBP/DBP more than aliskiren 150 mg (‐2.6/‐1.8 mmHg); and aliskiren 600 mg lowered SBP/DBP more than aliskiren 150 mg ‐3.4/‐2.2 mmHg, However, SBP/DBP lowering did not differ between aliskiren 600 mg versus aliskiren 300 mg.
The magnitude of the BP lowering with aliskiren at the maximum recommended dose of 300 mg daily ‐7.9/‐4.5 mm Hg is similar to what has been determined using similar methods in systematic reviews of angiotensin‐converting‐enzyme inhibitors (ACEIs), ‐8/‐5 mm Hg and angiotensin receptor blockers (ARBs) ‐8/‐5 mm Hg (Heran 2008a; Heran 2008b; Law 2003). Direct comparison of renin inhibitor with ARBs from the three placebo‐controlled studies included in this systematic review that compared aliskiren 300 mg with ARBs also support this finding.
CSPP100A2327 study: aliskiren 300 mg lowered MSSBP as well as MSDBP as compared to baseline to a similar extent as valsartan 320 mg at week eight (SBP : ‐13.0 mm Hg versus ‐12.8 mm Hg, respectively and DBP: ‐9.0 mm Hg versus ‐9.7 mm Hg, respectively).
CSPP100A2203 study: aliskiren 300 mg lowered MSSBP as well as MSDBP as compared to baseline to a similar extent as valsartan 320 mg at week eight (SBP: ‐5.08 mm Hg versus ‐6.54 mm Hg and DBP: ‐ 3.67mm Hg versus ‐2.69 mm Hg).
CSPP100A2201 study: aliskiren 150 mg lowered MSSBP as well as MSDBP as compared to baseline to a similar extent as irbesartan 150 mg at week eight (SBP: ‐6.01 mm Hg versus ‐7.21 mm Hg and DBP: ‐2.94 mm Hg versus ‐2.54 mm Hg).
This suggests that inhibiting the renin angiotensin system at different sites does not lead to any clinically different BP lowering effects. Adequately powered RCTs assessing different classes of drugs in head‐to‐head studies are the best way to look for clinically significant differences in BP lowering effect.
The AHRQ review 2011 update by Powers 2011 concluded that studies evaluating the renin inhibitors aliskiren versus ACEIs and ARBs on mortality and morbidity outcomes were relatively short, and few deaths or cardiovascular events occurred, resulting in insufficient evidence to discern differences. They also reported that a random‐effects meta‐analysis of 23 RCTs comparing ACEIs and ARBs found no significant difference in the proportion of patients who achieved successful BP control on a single antihypertensive agent.
This update provides additional details on adverse events from CSRs. During the double‐blind period, there were two (0.04%) deaths and 27 (0.5%) non‐fatal serious adverse events in 5120 patients in the aliskiren monotherapy arm as compared to one (0.04%) death and 14 (0.6%) non‐fatal serious adverse events in 2319 patients in the placebo group. There were no differences in all‐cause mortality and non‐fatal serious adverse events. The durations (eight to 13 weeks) of the included studies were too short and were not adequately powered to detect differences in adverse events between aliskiren and placebo. Journal‐published studies and clinical study summaries listed only a few events, generally those with an incidence greater than 1% to 2.5% in any group, while others reported by broad systems affected by the drugs: cardiac, immune system, respiratory, etc. The most common adverse events reported were headache, nasopharyngitis, dizziness, and back pain, which were similar in placebo groups and aliskiren groups at all doses. Consistent with the FDA medical review, we have determined a statistically significant, dose‐dependent relationship between aliskiren 600 mg and diarrhoea. Due to the limited duration of the studies, it is impossible to determine whether diarrhoea is merely a marker for more serious adverse outcomes that may require longer periods of time to develop. This review found low‐quality evidence that with short‐term use, aliskiren does not increase withdrawal due to adverse effects as compared to placebo.
Overall completeness and applicability of evidence
There are numerous ways we have experienced a lack of access to the entire complement of evidence and encountered limitations accordingly. Although this update is based primarily on CSRs, which are far more comprehensive than journal‐published trials, we were only able to obtain nine CSRs of the 12 studies that met the inclusion criteria of this review, provided the EMA does not possess the other three studies. In addition, of the nine CSRs we have obtained, we do not yet possess the appendices, which contain valuable and revealing information (e.g. patient narratives), as has been previously discussed. Overt publication bias can be noted as two studies do not have a corresponding journal publications (CSPA100A1301 and CSPP100A1301), but can also be demonstrated by a lack of public trial registration. Five of 12 studies (42%) are not registered on ClinicalTrials.gov (CSPP100A1201; CSPP100A2201; CSPP100A2203; CSPP100A2327; CSPP100A2328). Of the seven studies that are registered, three trials (43%) do not have any study results posted on the ClinicalTrials.gov web site. These findings cause us to question the degree to which the 2007 US Food and Drug Administration Amendments Act is followed, as it mandates that all drugs and devices approved for use must be publicly registered at ClinicalTrials.gov within 24 months of trial completion, regardless of whether the trial is published. Of the studies included within this review, we have observed trials that are not journal‐published, not registered publicly, and are not possessed by the European regulatory authority. A combination of all three situations would effectively hide the existence of a trial, which could distort the evidence, mislead health policy and prescribing practices. Although we have tried to diligently include all available information from multiple sources, we have encountered various obstacles that limit our ability to evaluate the safety and efficacy of aliskiren as compared to placebo.
Safety Warning
Although aliskiren has shown a good safety and tolerability profile in short‐term studies in adult patients with hypertension, the Food and Drug Administration (FDA) has recently issued a warning about combining aliskiren with ACEIs and ARBs in patients diagnosed with diabetes or renal impairment.
A recent RCT (ALTITUDE 2012) was halted prematurely due to safety concerns when aliskiren 300 mg daily was used as an adjunctive therapy with an ACEI or an ARB. This study enrolled adults 35 years of age or older with type 2 diabetes and evidence of microalbuminuria, macroalbuminuria, or cardiovascular disease.
-
A second interim efficacy analyses of the data from 8561 participants found that adjunct therapy of aliskiren with ARBs or ACEIs (n = 4274) contributed to an increased rate of adverse events and did not reduce cardiovascular or renal outcomes as compared to placebo (n = 4287) after a median follow up of 32.9 months.
The primary endpoint was cardiovascular death or a first occurrence of cardiac arrest with resuscitation, myocardial infarction, stroke, unplanned hospitalization for heart failure, end stage renal disease, death attributable to kidney failure, or loss of kidney function, and doubling of baseline serum creatinine. It occurred in 783 (18.3%) of patients assigned to aliskiren as compared with 732 (17.1%) in the placebo arm (hazard ratio (HR), 1.08, 95% CI, 0.98 to 1.20; P = 0.12).
Within the primary endpoint, 19 participants experienced cardiac arrest requiring resuscitation versus eight patients assigned to placebo (HR, 2.40, 95% CI 1.05 to 5.48; P = 0.04).
The proportion of patients with hyperkalaemia (serum potassium level, ≥6 mmol/L) was significantly higher in the aliskiren group than in the placebo group (11.2% versus 7.2%), as was the proportion with reported hypotension (12.1% versus 8.3%) and diarrhoea (9.8% versus 7.3%) (P < 0.001 for all comparisons).
During the study, 1445 patients assigned to aliskiren (33.8%) and 1218 assigned to placebo (28.4%) discontinued the study drug permanently for a reason other than death (P = 0.001).
In the aliskiren group, 563 patients (13.2%) discontinued the study drug because of an adverse event, as compared with 437 patients (10.2%) in the placebo group (P < 0.001).
Favourable results for the aliskiren group were seen with respect to surrogate markers such as lower SBP and DBP measurements (1.3 and 0.6 mm Hg, respectively), and reduced urinary albumin excretion as compared to placebo.
However, the authors concluded that "A number of studies of various drugs have shown favourable changes in surrogate markers of disease progression, with subsequent studies of morbidity and mortality documenting a lack of clinical benefit or even harm. The present study documented more adverse events in the aliskiren group than in the placebo group without clinical benefits to offset them, which underscores the need to go beyond surrogate bio‐markers and obtain risk‐benefit data from clinical endpoint studies to better inform clinical decisions" (ALTITUDE 2012).
Harel 2012 conducted a systematic review comparing combined treatment using aliskiren and ACEIs or ARBs with monotherapy in 4814 participants from 10 RCTS. Harel also concluded that use of aliskiren in combination with ACEIs or ARBs is associated with an:
increased risk for hyperkalaemia as compared to ACEI or ARB monotherapy RR with 95% CI 1.58 (1.24 to 2.02) as well as aliskiren monotherapy RR with 95% CI 1.67 (1.01 to 2.79).
The risk of acute kidney injury did not differ significantly between the combined therapy and monotherapy groups (RR 1.14, 0.68 to 1.89).
Makani 2013 conducted a similar systematic review of combined treatment using aliskiren and ACEIs or ARBs with monotherapy with these agents in 68,405 participants from 33 RCTs. Makani concluded that although dual blockade of the renin‐angiotensin system may have seemingly beneficial effects on certain surrogate endpoints, it failed to reduce mortality and was associated with an excessive risk of adverse events such as hyperkalaemia, hypotension, and renal failure compared with monotherapy. The authors concluded that the risk to benefit ratio argues against the use of dual therapy.
Regulatory Marketing Authorization
Rasilez (aliskiren) remains approved with the EMA, but Tekturna (aliskiren) has been withdrawn. Enviage (aliskiren) and Sprimeo (aliskiren) have also been withdrawn with the EMA. But Tekturna (aliskiren) remains approved for marketing with the FDA. Additionally, for both the FDA and EMA, a number of combination treatments have had their marketing authorization withdrawn, through a combination of voluntary withdrawal and forced withdrawal from the regulatory body. U.S. Food and Drug Administration 2016.
Quality of the evidence
We used the GRADE approach to build a 'Summary of Findings' table as suggested in the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2011). We used the GRADE approach to rate the quality of the evidence as 'high', 'moderate' 'low', or 'very low' using the five GRADE considerations.
Risk of bias: not serious; serious; or very serious.
Inconsistency: not serious; serious; or very serious.
Indirectness: not serious; serious; or very serious.
Imprecision: not serious; serious; or very serious.
Publication bias: undetected; or strongly suspected.
We reported 'Summary of Findings' tables for SBP, DBP and diarrhoea. Although the inclusion criteria of the 12 studies was high quality to begin with ‐ randomized double‐blind studies, the overall quality of evidence is graded as moderate‐quality evidence for BP outcome and low‐quality evidence for adverse event outcomes because of a number of limitations in this systematic review. Refer to the 'Risk of bias' assessment of individual studies Figure 2 as well as reasons for downgrading evidence in the Table 1.
Potential biases in the review process
Publication bias is evident as it has come to our attention that several studies were not registered on ClinicalTrials.gov (CSPP100A2201; CSPP100A1201; CSPP100A2327; CSPP100A2203; CSPP100A2328). The EMA does not possess CSRs of CSPP100A2328, CSPA100A2305, or CSPA100A1301.
CSPP100A1301 and CSPA100A1301 do not have corresponding journal‐published studies.
With respect to the dose‐ranging BP lowering effect size as reported above and shown in forest plots, this could be an overestimate of the magnitude of BP lowering due to publication bias and also because wide confidence intervals in effect size for some doses were observed. All included studies were sponsored by the manufacturer, Novartis. Requests for the unpublished studies have not been granted. It is possible that negative studies exist, which have not been published and therefore are not included in this meta‐analysis. In addition, the BP lowering efficacy estimate is over a short duration (eight to 13 weeks).
No study is available as yet evaluating effectiveness of long‐term renin inhibitor monotherapy on clinically relevant outcomes such as (mortality, cardiovascular and cerebrovascular morbidity).
Agreements and disagreements with other studies or reviews
Comparison with other systematic reviews
Several systematic reviews published by Weir 2007; White 2010; Gradman 2010; and Chen 2013 were limited to a smaller subset of included studies and provide very limited information regarding adverse events.
Weir 2007 published pooled analysis of antihypertensive efficacy, safety, and tolerability of the oral direct renin inhibitor aliskiren alone, and in combination with other antihypertensive agents, from seven randomized, multicentre studies in patients with hypertension. Data were available for 7045 patients (mean age 52.5 to 59.8 years, 50.2% to 72.5% men) with mild‐to‐moderate hypertension (mean sitting diastolic blood pressure (MSDBP) 95 mm Hg to 109 mm Hg) over treatment durations of six to eight weeks.
In placebo‐controlled studies, aliskiren reduced MSDBP from baseline by 8.6 to 12.1/7.2 to 10.3 mm Hg (75 mg), 8.7 to 13.0/7.8 to 10.3 mm Hg (150 mg), 14.1 to 15.8/10.3 to 12.3 mm Hg (300 mg), and 15.7 to 15.8/11.5 to 12.5 mm Hg (600 mg), compared with 2.9 to 10.0/3.3 to 8.6 mm Hg for placebo.
Aliskiren demonstrated comparable efficacy in patients aged > 65 years old or < 65 years old, in men and women, and it lowered BP effectively in all racial subgroups.
Rates of discontinuation due to adverse events were low (1.7% to 2.6%).
The most frequently reported adverse events with aliskiren were headache (5.7%), nasopharyngitis (4.4%), diarrhoea (2.6%), dizziness (1.8%) and fatigue (1.6%).
The overall incidence of adverse events with aliskiren monotherapy was similar to placebo (39.8% versus 40.2%, respectively).
The incidence of diarrhoea with aliskiren was higher than placebo due to a significantly higher rate with aliskiren 600 mg (P = .0001 versus placebo).
White 2010 published pooled analysis of safety and tolerability of aliskiren in more than 12,000 patients with hypertension. Twelve randomized double‐blind clinical studies in patients with hypertension were evaluated: eight short‐term (8‐week) placebo‐controlled studies and four long‐term (26‐ to 52‐week) active‐controlled studies.
In short‐term studies:
adverse events occurred in similar proportions of aliskiren 150 mg and 300 mg (33.6% and 31.6%, respectively) and placebo treatment groups (36.8%);
in placebo‐controlled studies, the incidence of angioedema ⁄ urticaria was lower in patients receiving aliskiren 150 mg (0.2%) and aliskiren 300 mg (0.3%) than in the placebo group (0.5%; RR, 0.31; 95% CI, 0.07 to 1.47 for 150 mg; RR, 0.57; 95% CI, 0.17 to 1.89 for 300 mg);
cough was not significantly different for aliskiren (0.7% to 1.4%) versus placebo (0.7%);
hyperkalaemia was 0.1% in aliskiren 300 mg group and 0.1% in the placebo group.
Analysis by individual aliskiren monotherapy dose demonstrated no significant increase in diarrhoea compared with placebo for patients receiving aliskiren 150 mg (RR, 1.18; 95% CI, 0.62 to 2.24) or 300 mg (RR, 1.62; 95% CI, 0.91 to 2.90). No patient receiving short‐term treatment with aliskiren 150 mg or 300 mg discontinued due to diarrhoea. There was no evidence for an increased risk of gastrointestinal bleeding or ulceration with aliskiren treatment over placebo in the short‐term studies (RR, 0.62; 95% CI, 0.06 to 6.87 for aliskiren 150 mg and RR, 0.57; 95% CI, 0.05 to 6.28 for aliskiren 300 mg).
Gradman 2010 published a pooled analysis of efficacy, safety and tolerability of monotherapy with the direct renin inhibitor aliskiren (150 mg and 300 mg) over eight to 12 weeks in women with mild‐to‐moderate hypertension with MSDBP > 95 and < 110 mmHg across eight randomized and double‐blind studies. Safety and tolerability were assessed in the five placebo‐controlled studies in the analysis.
Women participating in the studies tended to be slightly older than men (range of means 55.1–57.3 years versus 52.9 to 55.3 years), with a longer duration of hypertension (7.8 to 8.2 years versus 6.5 to 7.8 years).
There was a higher proportion of Black women (5.0% to 17.7%) than Black men (2.2% to 8.5%); conversely the proportion of Asian women (7.7% to 10.4%) was lower than the proportion of Asian men (12.8% to 25.5%).
Slightly more women than men were obese (40.8% to 42.8% versus 31.1% to 37.7%).
Baseline BP was similar in women and men.
In the 1527 women enrolled in these studies:
aliskiren 150 mg and 300 mg produced significantly greater BP reductions (14.1/11.0 and 16.1/12.3 mmHg, respectively) compared with placebo (7.2/7.6 mmHg; P < 0.0001);
BP reductions with aliskiren monotherapy in women were similar to those observed in men, and consistent across subgroups of age, metabolic syndrome and obesity;
the overall incidence of adverse events in women was similar with aliskiren treatment (150 mg, 42.3%; 300 mg, 46.0%); and placebo (39.0%);
adverse events with aliskiren were more frequent in women than in men, consistent with previous studies of gender differences in drug tolerability;
few patients in this analysis discontinued aliskiren treatment and the incidence was similar in women (150 mg, 7.3%; 300 mg, 5.8%); and men (150 mg, 6.8%; 300 mg, 6.6%);
discontinuation rates with placebo were higher than with aliskiren therapy for both women (12.5%) and men (12.9%), primarily because of the higher numbers of discontinuations due to unsatisfactory therapeutic effect with placebo (5.0% to 5.6%) than with active therapy (1.5% to 2.3%). The reasons for discontinuation from aliskiren treatment were similar across doses and genders, with adverse events (0.8% to 2.7%) and withdrawal of consent (0.9% to 2.3%) being the most commonly reported reasons.
Chen 2013 investigated the antihypertensive effects and tolerability of aliskiren in comparison with other antihypertensive drugs and placebo in patients with hypertension. Nine studies included study arms with placebo, involving 3541 patients. They did not provide dose‐ranging decrease in SBP and DBP. Results were only shown for aliskiren at 300 mg dose and it was significantly superior to placebo in lowering both DBP and SBP WMD ‐4.75, 95% CI ‐5.59 to ‐3.92, P < 0.00001; MD ‐8.12, 95% CI ‐9.61 to ‐6.63, P < 0.00001, respectively). The Chen 2013 meta‐analysis indicates that aliskiren is associated with a similar incidence of adverse events and discontinuation due to adverse events to placebo.
Authors' conclusions
Implications for practice.
The renin inhibitor aliskiren 150 mg and 300 mg lowered systolic and diastolic blood pressure (BP) as compared to placebo in a dose‐dependent manner (moderate‐quality evidence). The findings of this systematic review also suggest that aliskiren 300 mg lowered BP by a magnitude that is similar to what has been determined with angiotensin‐converting‐enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs). This review found that in studies of eight‐week duration, aliskiren may not increase withdrawal due to adverse events as compared to placebo (low‐quality evidence). Diarrhoea was increased in a dose‐dependent manner and a substantial increase was observed with aliskiren 600 mg as compared to placebo (low‐quality evidence). All studies meeting the inclusion criteria of this review excluded high‐risk patients therefore generalizing these results for decision making in clinical practice is limited to only adult patients with uncomplicated primary hypertension.
Implications for research.
The goal of antihypertensive therapy is to reduce mortality and morbidity. No randomized controlled trial (RCT) has been identified that studies the long‐term effects of renin inhibitors monotherapy on mortality and morbidity versus placebo. More RCTs of longer duration are needed to assess the BP lowering efficacy of renin inhibitors as compared to other classes of antihypertensive drugs. All published RCTs must provide data on all outcomes and all negative studies need to be published, in order for this review to provide an accurate summary of the best available evidence.
The Food and Drug Administration (FDA) has recently issued a warning about combining aliskiren with ACEIs and ARBs in patients diagnosed with diabetes or renal impairment. Therefore long‐term mortality and morbidity data in patients with primary hypertension on monotherapy with renin inhibitors should be a priority. There is an urgent need to go beyond surrogate biomarker and obtain harm‐benefit data from clinical endpoint studies to better inform clinical decisions. There is growing evidence that the surrogate marker ‐ the lowering of BP ‐ is inadequate in predicting health outcomes with antihypertensive therapy (Wright 2009).
Feedback
Comment on Blood pressure lowering efficacy of renin inhibitors for primary hypertension, 17 April 2017
Summary
I read with great interest the updated review regarding the efficacy of renin inhibitors in primary uncomplicated hypertension. The review authors finally concluded based on the available studies that aliskiren is better than placebo at lowering blood pressure. The authors also commented that studies were too short in duration to assess side effects. From the available information from studies it was also found that aliskiren did not increase death, non‐fatal serious adverse events or withdrawals due to side effects and that the most common adverse events were headache, diarrhoea, dizziness and fatigue, with diarrhoea being considerably increased with aliskiren 600 mg as compared to placebo. This suggests that there is strong evidence for using this medication in primary hypertension with good proven efficacy.
When going through the whole studies and related information I noticed that there is a section titled 'Additional references' but I could not find the relevant information in the main contents of the review. What is the main purpose of it? I am curious to know its detail in the review process.
One of the additional references is a study authored by Parving et al, 2012. The trial was stopped prematurely after the second interim efficacy analysis. After a median follow‐up of 32.9 months, the primary end point had occurred in 783 patients (18.3%) assigned to aliskiren as compared with 732 (17.1%) assigned to placebo, with hazard ratio of 1.08. Effects on secondary renal end points were similar. Systolic and diastolic blood pressures were lower with aliskiren. The proportion of patients with hyperkalemia was significantly higher in the aliskiren group than in the placebo group, as was the proportion with reported hypotension. I am curious if this was known to the authors and included in the review process before formulating their conclusions in regards to renin inhibitors.
Reply
Thank‐you for your interest in reading this systematic review.
The 'Additional references' section lists articles cited in the text of the review, including those cited in the 'Background' and 'Methods' sections, other than those to studies included in or excluded from the analysis. Each reference listed here is linked to relevant text in the review. For more information, see chapter 4.7.2 in the Cochrane Handbook for Systematic Reviews of Interventionshttp://handbook.cochrane.org
The protocol of this review was published in early 2008 and the completed review was first published at the end of 2008. We published the first update in 2011 and updated it for a second time in 2017. This most recent update includes extensive details from clinical study reports of included studies, obtained from the European Medicine Agency. We have reported and meta‐analysed all the outcomes stated in the protocol and reached conclusions based on all available efficacy and safety information.
The Parving et al 2012 (referenced as ALTITUDE 2012 in our review) was a study found when we updated the search and was excluded from this review as it does not meet our minimum inclusion criteria. It compares aliskiren 300 mg daily to placebo as an adjunct to an ACE inhibitor or an ARB in patients with type 2 diabetes and chronic kidney disease, cardiovascular disease or both. The inclusion criteria for our review was "randomised placebo controlled trials comparing monotherapy with aliskiren at any dose to placebo in adult patients with primary hypertension with a minimum duration of 3 weeks". However, safety information reported in Parving et al 2012 (ALTITUDE 2012) was included in the 'Discussion' section of our review under the heading 'Overall completeness and applicability of evidence / Safety warning' as it was relevant to warn readers of the potential harms that may be caused by this drug. The conclusions of ALTITUDE 2012 are presented in the review as a quote from the authors of this study.
Contributors
Name: Pugazhenthan Thangaraju, MBBS, MD
Email Address:drpugal23@gmail.com
Affiliation: Central Leprosy Teaching and Research Institute, Ministry of Health and Family Welfare, Government of India, Chengalpattu, Tamilnadu, India.
Cochrane Role: none.
Conflict of interest disclosure:I do not have any affiliation with or involvement in any organisation with a financial interest in the subject matter of my comment.
What's new
| Date | Event | Description |
|---|---|---|
| 19 April 2017 | Feedback has been incorporated | Feedback and authors' reply incorporated. |
History
Protocol first published: Issue 2, 2008 Review first published: Issue 4, 2008
| Date | Event | Description |
|---|---|---|
| 12 February 2017 | New citation required and conclusions have changed | This update includes detailed information from nine clinical study reports (CSRs) obtained from the European Medicines Agency (EMA). In addition, data from an unpublished study has been included from a clinical study summary obtained from the Novartis Clinical Trial Results Database, as well as data from four new studies that met the inclusion criteria. The previous version of this review had access to very limited data on harm. The addition of information from CSRs resulted in a significantly improved 'Risk of bias' assessment, as well as provided extensive detailed data on harms including mortality, serious adverse events and other specific adverse events. |
| 12 February 2017 | New search has been performed | This review includes updated search conducted from June 1st 2011 until February 12th 2017. |
| 1 June 2011 | New search has been performed | First updated search included 2 additional studies that met the inclusion criteria. |
| 12 November 2008 | Amended | Contact details updated. |
Acknowledgements
The authors would like to acknowledge the contribution of Doug Salzwedel, the information specialist of the Hypertension Review Group in updating the search strategy and providing search findings from various databases; Stephen Adams in retrieving innumerable articles; Aaushi Rawat in helping with screening the search results; for identifying studies that meet the inclusion criteria; and doing data abstraction for the two additional studies identified in 2011. We would also like to thank Ciprian Jauca who assisted in editing this review.
Appendices
Appendix 1. Search strategies
Database: Ovid MEDLINE(R) 1946 to Present with Daily Update Search Date: 12 February 2017 ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 renin/ai (1869) 2 (aliskiren or ciprokiren or ditekiren or enalkiren or remikiren or rasilez or tekturna or terlakiren or zankiren).mp. (1156) 3 ((RAS or renin) adj2 inhibit$).tw. (5263) 4 or/1‐3 (6117) 5 hypertension/ (214478) 6 (antihypertens$ or hypertens$).tw. (351338) 7 exp blood pressure/ (269022) 8 (blood pressure or bloodpressure).tw. (234885) 9 or/5‐8 (648256) 10 randomized controlled trial.pt. (448646) 11 controlled clinical trial.pt. (91968) 12 randomized.tw. (365905) 13 placebo.tw. (172966) 14 drug therapy.fs. (1936909) 15 randomly.ab. (235372) 16 trial.ab. (357116) 17 groups.ab. (1465387) 18 or/10‐17 (3704199) 19 animals/ not (humans/ and animals/) (4291203) 20 18 not 19 (3162819) 21 4 and 9 and 20 (1293) 22 remove duplicates from 21 (1252) *************************** Database: Cochrane Hypertension Specialised Register Search Date: 12 February 2017 ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ #1 (aliskiren OR ciprokiren OR ditekiren OR enalkiren OR remikiren OR rasilez OR tekturna OR terlakiren OR zankiren) AND INSEGMENT #2 (RAS OR renin) NEAR2 inhibit* AND INSEGMENT #3 #1 OR #2 AND INSEGMENT #4 MESH DESCRIPTOR Hypertension AND INSEGMENT #5 (antihypertens* OR hypertens*) AND INSEGMENT #6 MESH DESCRIPTOR Blood Pressure EXPLODE ALL AND INSEGMENT #7 (blood pressure OR bloodpressure) AND INSEGMENT #8 #4 OR #5 OR #6 OR #7 AND INSEGMENT #9 #3 AND #8 AND INSEGMENT #10 RCT:DE AND INSEGMENT #11 (Review OR Meta‐Analysis):MISC2 AND INSEGMENT #12 #9 AND (#10 OR #11) AND INSEGMENT *************************** Database: Cochrane Central Register of Controlled Trials <2017, Issue 2> via Cochrane Register of Studies (CRS‐Web)
Search Date: 12 February 2017 ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ #1 (aliskiren OR ciprokiren OR ditekiren OR enalkiren OR remikiren OR rasilez OR tekturna OR terlakiren OR zankiren) AND CENTRAL:TARGET #2 (RAS OR renin) NEAR2 inhibit* AND CENTRAL:TARGET #3 #1 OR #2 AND CENTRAL:TARGET #4 MESH DESCRIPTOR Hypertension AND CENTRAL:TARGET #5 (antihypertens* OR hypertens*) AND CENTRAL:TARGET #6 MESH DESCRIPTOR Blood Pressure EXPLODE ALL AND CENTRAL:TARGET #7 (blood pressure OR bloodpressure) AND CENTRAL:TARGET #8 #4 OR #5 OR #6 OR #7 AND CENTRAL:TARGET #9 #3 AND #8 AND CENTRAL:TARGET *************************** Database: Embase <1974 to 2017 February 10> Search Date: 12 February 2017 ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 exp renin inhibitor/ (5481) 2 (aliskiren or ciprokiren or ditekiren or enalkiren or remikiren or rasilez or tekturna or terlakiren or zankiren).mp. (3415) 3 ((RAS or renin) adj2 inhibit$).tw. (7360) 4 or/1‐3 (10648) 5 exp hypertension/ (632467) 6 (antihypertens$ or hypertens$).tw. (546894) 7 (blood pressure or bloodpressure).tw. (346971) 8 or/5‐7 (966045) 9 randomized controlled trial/ (478668) 10 crossover procedure/ (55130) 11 double‐blind procedure/ (140883) 12 (randomi?ed or randomly).tw. (950148) 13 (crossover$ or cross‐over$).tw. (87214) 14 placebo.ab. (244256) 15 (doubl$ adj blind$).tw. (177774) 16 assign$.ab. (302044) 17 allocat$.ab. (110685) 18 or/9‐17 (1413635) 19 (exp animal/ or animal.hw. or nonhuman/) not (exp human/ or human cell/ or (human or humans).ti.) (5888790) 20 18 not 19 (1241949) 21 4 and 8 and 20 (874) 22 remove duplicates from 21 (837) *************************** Database: ClinicalTrials.gov Search Date: 12 February 2017 ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ Search terms: randomized Study type: Interventional Studies Condition: hypertension Interventions: (aliskiren OR ciprokiren OR ditekiren OR enalkiren OR rasilez OR remikiren OR renin inhibitors OR tekturna OR terlakiren OR zankiren) Outcome Measures: blood pressure (86) *************************** Database: WHO International Clinical Trials Registry Platform Search Date: 12 February 2017 ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ Condition: hypertension Intervention: (aliskiren OR ciprokiren OR ditekiren OR enalkiren OR rasilez OR remikiren OR renin inhibitors OR tekturna OR terlakiren OR zankiren) Recruitment status: ALL (182)
Appendix 2. Regulatory information searches
Drugs@FDA: http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm
Data and analyses
Comparison 1. Aliskiren vs. placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Systolic BP | 12 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.1 Aliskiren 75 mg vs. placebo | 5 | 1100 | Mean Difference (IV, Fixed, 95% CI) | ‐2.97 [‐4.76, ‐1.18] |
| 1.2 Aliskiren 150 mg vs. placebo | 12 | 3786 | Mean Difference (IV, Fixed, 95% CI) | ‐5.95 [‐6.85, ‐5.06] |
| 1.3 Aliskiren 300 mg vs. placebo | 10 | 3009 | Mean Difference (IV, Fixed, 95% CI) | ‐7.88 [‐8.94, ‐6.82] |
| 1.4 Aliskiren 600 mg vs. placebo | 2 | 393 | Mean Difference (IV, Fixed, 95% CI) | ‐11.35 [‐14.43, ‐8.27] |
| 2 Diastolic BP | 12 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.1 Aliskiren 75 mg vs placebo | 5 | 1100 | Mean Difference (IV, Fixed, 95% CI) | ‐2.05 [‐3.13, ‐0.96] |
| 2.2 Aliskiren 150 mg vs placebo | 12 | 3783 | Mean Difference (IV, Fixed, 95% CI) | ‐3.16 [‐3.74, ‐2.58] |
| 2.3 Aliskiren 300 mg vs placebo | 10 | 3001 | Mean Difference (IV, Fixed, 95% CI) | ‐4.49 [‐5.17, ‐3.82] |
| 2.4 Aliskiren 600 mg vs placebo | 2 | 393 | Mean Difference (IV, Fixed, 95% CI) | ‐5.86 [‐7.73, ‐3.99] |
| 3 Withdrawals due to adverse event | 12 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 75 mg vs. placebo | 5 | 1653 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.59 [0.33, 1.07] |
| 3.2 150 mg vs. placebo | 10 | 3421 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.30, 0.71] |
| 3.3 300 mg vs. placebo | 10 | 4216 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.70 [0.47, 1.03] |
| 3.4 600 mg vs placebo | 2 | 592 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.19, 1.64] |
| 4 Cough | 5 | 2886 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.49, 2.64] |
| 5 Diarrhoea | 7 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 Aliskiren 75 mg | 4 | 1276 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.21 [0.85, 5.76] |
| 5.2 Aliskiren 150 mg | 7 | 2277 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.64 [0.78, 3.46] |
| 5.3 Aliskiren 300 mg | 7 | 2268 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.84 [0.89, 3.81] |
| 5.4 Aliskiren 600 mg | 2 | 592 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.00 [2.48, 19.72] |
Comparison 2. Aliskiren150 mg vs. Aliskiren 75 mg.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Systolic BP | 5 | 1651 | Mean Difference (IV, Fixed, 95% CI) | ‐1.89 [‐3.16, ‐0.62] |
| 2 Diastolic BP | 5 | 1651 | Mean Difference (IV, Fixed, 95% CI) | ‐0.80 [‐1.58, ‐0.03] |
Comparison 3. Aliskiren 300 mg Vs. Aliskiren 75 mg.
Comparison 4. Aliskiren 300 mg vs. Aliskiren 150 mg.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Systolic BP | 10 | 4405 | Mean Difference (IV, Fixed, 95% CI) | ‐2.62 [‐3.38, ‐1.87] |
| 2 Diastolic BP | 10 | 4405 | Mean Difference (IV, Fixed, 95% CI) | ‐1.80 [‐2.28, ‐1.32] |
Comparison 5. Aliskiren 600 mg vs. Aliskiren 150 mg.
Comparison 6. Aliskiren 600 mg vs. Aliskiren 300 mg.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Systolic BP | 2 | 592 | Mean Difference (IV, Fixed, 95% CI) | ‐0.61 [‐2.78, 1.56] |
| 2 Diastolic BP | 2 | 592 | Mean Difference (IV, Fixed, 95% CI) | ‐0.68 [‐2.03, 0.67] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
CSPA100A1301.
| Methods | Randomized, double‐blind, placebo‐controlled, parallel group study. | |
| Participants | 1342 patients were randomized to 6 different combination treatment groups. Inclusion criteria: Not reported Exclusion criteria: Not reported. Baseline characteristics: Mean age was 55.1 + 10 years; 19% patients were > 65 years old; 73% participants were male; race not reported, mean sitting SBP and DBP, pulse pressure and heart rate was not reported |
|
| Interventions | Placebo (N = 153) and aliskiren 150 mg (N = 157) monotherapy (Other combination therapy groups and amlodipine 2.5 mg and 5 mg monotherapy groups are not described) Treatment duration = 8 weeks |
|
| Outcomes | Primary: Change From Baseline in MSDBP to End of Study (Week 8); Secondary: Change From Baseline in MSDBP to End of Study (Week 8); Percentage of patients achieving goal BP; serious adverse events; total adverse events |
|
| Notes | Study CSPA100A1301 is not published in a journal. It is registered on clinicalTrials.gov. Identifier: NCT01237223 (results are posted) It is registered on Novartis Clinical Trial Results database (results are not posted) EMA does not possess CSR for this study. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Stated as randomized ; no description is available. |
| Allocation concealment (selection bias) | Unclear risk | No description is available. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Clinical study synopsis reported study as double‐blind (Subject, Outcomes Assessor). In order to adequately blind the study, patients were required to take a total of 3 tablets and 2 capsules of study medication throughout the study. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 33 (21.6%) patients in placebo group and 15 (9.6%) patients in aliskiren 150 mg group did not complete the study. Reasons for withdrawal differed (adverse events 12 in placebo and 4 in A 150 mg; unsatisfactory therapeutic effect, 20 in placebo and 6 in A 150 mg). It is not known how missing data were included in the analysis. |
| Selective reporting (reporting bias) | Unclear risk | Protocol is not available to assess selective reporting bias. Clinical study synopsis does not provide details. |
| Other bias | High risk | Study sponsored by Novartis. Study is not published in a journal. NCT01237223 ‐ short clinical synopsis is posted on clinical Trial.gov Novartis Clinical Trial Results database (results are not posted) |
CSPA100A2305.
| Methods | A double‐blind, multicentre, randomized, placebo‐controlled, multifactorial study was conducted at 208 centres across 18 countries (Argentina, Australia, Canada, Colombia, Denmark, Finland, Greece, Italy, Mexico, Panama, Peru, Romania, Russia, South Africa, Spain, Sweden, Taiwan and the USA). | |
| Participants | 1688 patients were randomized to 9 different monotherapy as well as combination treatment groups. 596 were randomized to aliskiren 150 mg, 300 mg and placebo groups. Inclusion criteria: Men and women aged >18 years with primary hypertension; MSDBP > 95 mmHg and <110 mmHg at randomization; an absolute difference of ≤ 10 mmHg in their MSDBP during the last 2 visits of the single‐blind run‐in period. Exclusion criteria: grade III hypertension (MSDBP >110 mmHg or MSSBP > 180 mmHg); secondary hypertension; a history of severe cardiovascular or cerebrovascular disease; type 1 or type 2 diabetes mellitus that was not well‐controlled (glycosylated haemoglobin [HbA1c] 48.0%; severe renal impairment; a history of dialysis; or a history of nephrotic syndrome; or hepatic disease; a history of hepatic encephalopathy; oesophageal varices; or portocaval shunt; pregnant or lactating women. Women of childbearing potential had to be using effective contraceptive methods for inclusion in the study; Baseline characteristics: Mean age 54.1 + 10.7 years; 17.2% of participants aged 65 years or older; Caucasian (62.1%) Black (19.9%), Asian patients (6.6%), others 11.4%; Mean body mass index 30.3 + 5.4 (Kg/m2); 11.0% of participants had diabetes; mean sitting SBP/DBP at baseline was 157 + 11.7/99.5 + 3.8 mmHg; mean sitting pulse pressure or heart rate was not reported. |
|
| Interventions | Aliskiren 150 mg (N = 195), aliskiren 300 mg (N = 203), or placebo (N = 198). (Two amlodipine monotherapy and four aliskiren/amlodipine combination groups) Treatment was administered once daily at 800 hours, except on clinic visit days, when patients were instructed to delay the treatment until all assessments had been completed. Treatment duration = 8 weeks. |
|
| Outcomes | Primary: antihypertensive efficacy of the combination of aliskiren/amlodipine was superior to each of the component monotherapies, as assessed by change in MSDBP from baseline to week‐8 endpoint across doses. Secondary: change in MSSBP from baseline to week‐8 endpoint; proportion of patients achieving BP control (< 140/90mmHg) at week 8; Change in mean 24 h ambulatory BP from baseline to week‐8 endpoint in a subgroup of patients; Changes in PRA from baseline to week‐8 endpoint in a subset of patients; All adverse events (AEs) and serious AEs. |
|
| Notes | Study CSPA100A2305 is published in a journal as Littlejohn 2013 It is registered on clinicalTrials.gov. Identifier: NCT00739973 (results are posted) It is registered on Novartis Clinical Trial Results database (results are posted) EMA does not possess CSR for this study. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomly assigned in an equal ratio, using a validated interactive voice response system, to one of the nine treatment groups. |
| Allocation concealment (selection bias) | Unclear risk | Not described. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Described as double‐blind. In order to adequately blind the study, patients were required to take a total of 3 tablets and 2 capsule of study medication throughout the study. There is no mention of how outcome assessors were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 30 (15.2%) patients discontinued in placebo group; 19 (9.7%) each in aliskiren 150 mg and 300 mg groups. The incidence of withdrawal due to unsatisfactory therapeutic effect was lower in the combination groups (0.5% to 1.1%) than in the placebo (8.6%) and monotherapy (1.1% to 4.1%) groups. Other common reasons for discontinuation included AEs and withdrawal of consent, with the incidence generally similar across treatment groups. Three patients were mis‐randomized, as they were discontinued from the single‐blind period and were not treated in the double‐blind period. For endpoint analyses, the last observation was carried forward for patients who did not have a measurement at week 8. |
| Selective reporting (reporting bias) | Unclear risk | All primary and secondary outcomes were reported. Pulse pressure and heart rate were not reported at baseline or end of treatment. |
| Other bias | High risk | J Zhang, H Hsu and DL Keefe are employees of Novartis Pharmaceuticals Corporation and are therefore eligible for Novartis stock and stock options. The remaining authors declare no conflict of interest. EMA does not possess CSR for this study. |
CSPP100A1201.
| Methods | Multicentre, randomized, placebo‐controlled, double‐blind, parallel group study. | |
| Participants | 615 were recruited from 29 clinical centres in Japan and 455 were randomized. 160 withdrew during the single‐blind period (138 not meeting protocol requirements,15 due to adverse events and 7 withdrawal of consent). Inclusion criteria: Japanese men and women with essential hypertension between the ages of 20 and 80 years; MSDBP > 90 mm Hg and < 110 mm Hg and MSSBP < 180 mmHg Exclusion criteria: Severe hypertension (MSDBP >110 mmHg and/or mean sitting systolic BP (MSSBP) >180 mmHg); secondary hypertension, suspected malignant hypertension; a history of severe cardiovascular or cerebrovascular disease; type 1 or type 2 diabetes mellitus receiving insulin or with poor glucose control (glycosylated haemoglobin [HbA1c] > 8%); serious hepatic or renal disease; history of pancreatitis; malignant tumours in the last 5 years; autoimmune disease; anaemia; gout or hyperthyroidism; apparent dehydration; pregnancy; Patients receiving treatment for gastric or duodenal ulcer or had taken any investigational medications within the last 12 weeks; patients with autoimmune disease, symptomatic anaemia, hypothyroidism, gout, or unable to comply with the protocol. Baseline characteristics: Mean age 53 + 11years; Males: 73%; MSSBP 156 + 11.8 mmHg and MSDBP 99.6 + 4.4 mmHg; Mean sitting pulse: 75 + 9.8 beats per minute; Pulse pressure was not reported. |
|
| Interventions | 455 patients were randomized to Placebo: N = 115; aliskiren 75 mg: N = 115; aliskiren 150 mg: N = 112; aliskiren 300 mg: N = 113 Duration of treatment = 13 weeks |
|
| Outcomes | Primary: Change in trough MSDBP from baseline at endpoint. Secondary: Change in trough MSSBP from baseline; proportion of patients responding to treatment (MSDBP < 90 mmHg and/or with a greater or equal 10 mmHg decrease in MSDBP from baseline to completion; dose‐response relationship in terms of primary and secondary outcomes | |
| Notes | Study CSPP100A1201 is published in a journal as Kushiro 2006 It is not registered on clinicalTrials.gov It is registered on Novartis Clinical Trial Results database (results are not posted) CSR without appendices was received from EMA on March 11th 2016 BP data were reported at the end of the double‐blind period at 8 weeks. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Drug allocation tables were prepared by computer‐generated random numbers and patients were assigned to treatment groups via central allocation." |
| Allocation concealment (selection bias) | Low risk | "The allocation schedule was then concealed until the key code was broken, so all patients, investigators, collaborators and the sponsor were unaware of the treatment assignments throughout the study." |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "The study drugs and placebo were indistinguishable in terms of appearance, shape, packaging form labelling etc". "To maintain blinding, all patients took three tablets a day (two aliskiren tablets plus one placebo, two placebo tablets plus one aliskiren, or three placebo tablets)." "The key code was broken after all CRF were completed and after data for analysis was locked". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Reasons for withdrawal during washout and single‐blind period: 138 (22.4%) ‐not meeting requirement specified in the protocol; 15 (2.4%) due to adverse events and 7(1.1%) due to withdrawal of consent. Of 455 patients randomized, 21 (4.4%) were withdrawn due to major protocol deviations. Most withdrawals 11(9.6%) occurred in the placebo group; 5 (4.3%) withdrew in 75 mg group; 3 (2.7%) in 150 mg group and 2 (1.8%) in the 300 mg group. (page 67 of CSR). Last observation was carried forward in patients with missing data during DB period at week 8. |
| Selective reporting (reporting bias) | Unclear risk | All primary and secondary outcomes were reported as stated in the protocol. Pulse pressure was not reported at baseline or endpoint. Heart rate was reported at baseline but no data are reported on change in heart rate at endpoint. These are available in appendices that were not provided by EMA. |
| Other bias | High risk | Manufacturer sponsored. Conflict of interest of authors has not been provided. Patients could have been selected based on their response to other drugs acting on the renin‐angiotensin system. Patients with adverse events were excluded during washout and single‐blind period. Study was not registered with clinicalTrials.gov. |
CSPP100A1301.
| Methods | A randomized , double‐blind, placebo‐ and active‐controlled, parallel‐group, multicentre, comparative study. | |
| Participants | 1206 patients were enrolled from 53 Japanese centres and 761 were randomized. Inclusion criteria: Adult (20 to 75 years old) Asian outpatients with essential hypertension defined as MSDBP > 90 mmHg and < 110 mmHg at visit 2, and > 95 mmHg and < 110 mmHg at visit 3; difference in MSDBP measured at visit 2 and visit 3 needed to be < 10mmHg. Exclusion criteria: MSSBP measured > 180 mmHg and/or if the MSDBP > 110mmHg at visit 1, 2, or 3; Suspected or confirmed secondary hypertension; malignant hypertension; severe signs of: cardiac disease, renal disease, hepatic disease, cerebrovascular disorder; pancreatitis, or history of pancreatitis; treatment of duodenal or gastric ulcer; over dehydration or electrolyte abnormalities of clinical concern; type I or type II diabetes mellitus (HbA1c greater than 8% at start of run‐in period (visit 1); history of malignant tumours including leukaemia and lymphoma within the past 5 years; history of autoimmune diseases; anaemia; fecal occult blood at start of run‐in period; hypothyroidism; exposure to aliskiren or placebo within 12 weeks of start of run‐in period; history of hypersensitivity to ARBs or drugs with similar chemical structures to aliskiren; alcoholic patients, or those with history of drug abuse within 52 weeks of study start date; patients considered unlikely to comply with the requirements specified in the protocol by the investigator. Baseline characteristics: Mean age 52 + 10.2 years, with 12.6% of participants aged 65 years or older; Oriental (100%); Male 73.2%; Mean BMI 25.6 + 3.8; Diabetes 8.7%; MSSBP/MSDBP 152.0 + 11.5/99.0 + 4.0 mmHg; Mean sitting pulse was 73.0 + 10.4 bpm; mean sitting pulse pressure was not reported. |
|
| Interventions | 761 patients were randomized Placebo (N=156), aliskiren 150 mg (N = 302), losartan 50 mg (N = 303) Duration of treatment = 8 weeks |
|
| Outcomes | Primary: change from baseline in MSDBP Secondary: change from baseline in MSSBP; responder rate (percentage of patients a MSDBP < 90 mmHg and/or at least 10 mmHg reduction from baseline), blood pressure control rate (percentage of patients with a MSDBP < 90 mmHg and MSSBP < 140 mmHg, changes from baseline of the standing and supine systolic and diastolic blood pressures; adverse events, vital signs, laboratory tests, ECG and fecal occult blood. |
|
| Notes | Study CSPP100A1301 is not published in a journal. It is registered on clinicalTrials.gov. Identifier: NCT003344110 (results are not posted) It is registered on Novartis Clinical Trial Results database (results are posted) CSR without appendices was received from EMA on May 23rd 2016. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were assigned to treatment groups by central randomization. Participants were provided with drug numbers by SPP 100 subject registration Centre and drug numbers were recorded in the eCRF by the investigators for use as randomization numbers and study drug was allocated according to the numbers. |
| Allocation concealment (selection bias) | Low risk | The allocation table was prepared using computer‐generated random numbers. The table was sealed by the person responsible for drug allocation and kept in strict confidence through unblinding. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Stated as double‐blind. Study drugs were identical in packaging, labelling, administration schedule, appearance, and odour. Patients were required to take a placebo during the placebo run‐in period (4 weeks) and were given 2 tablets according to the patient's assigned group. Participants, investigators, sub investigators, site staff, evaluators and data analysts remained blind to the identity of the treatment from randomization to database lock. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Discontinuations from the study ‐ placebo: N = 15 (9.6%) and aliskiren 150 mg: N = 14 (4.6%). Last observation was carried forward in patients with missing data during DB period at week 8. Missing data at week 8 had very little effect on analysis of results for MSSBP and MSDBP. |
| Selective reporting (reporting bias) | Unclear risk | Most primary and secondary outcomes were reported. Baseline and endpoint BP data was reported. Pulse rate was reported at baseline but not reported at endpoint. Pulse pressure was not reported. These may be available in appendices that were not provided by EMA. |
| Other bias | High risk | Manufacturer sponsored. Conflicts of interests unknown. Study is not published in a journal. Of the 1206 patients enrolled, 445 were not randomized (286 did not meet protocol requirements; 89 had abnormal laboratory value; 39 had adverse event; 22 withdrew consent and 9 had protocol violation). |
CSPP100A2201.
| Methods | Multicentre randomized, double‐blind, placebo‐controlled parallel‐group study. | |
| Participants | 793 patients were enrolled from 56 centres in USA, Germany and Belgium of which 652 were randomized. 141 patients were excluded (69 due to failure to meet BP criteria; 19 due to withdrawal of consent and 15 due to abnormal test procedure results). Inclusion criteria: Men and women aged 18 years or older, with mild‐to‐moderate essential hypertension (MSDBP > 95 mmHg and < 110 mmHg). Exclusion criteria: Severe hypertension (MSDBP > 110mmHg and/or MSSBP > 180mmHg); secondary hypertension; a history of severe cardiovascular or cerebrovascular disease; type 1 or type 2 diabetes mellitus receiving insulin or with poor glucose control (glycosylated haemoglobin [HbA1c] > 8%); serious hepatic or renal disease; history of malignancy; history of severe or life‐threatening diseases; history of drug or alcohol abuse; apparent dehydration; and pregnancy or nursing mothers. Baseline characteristics: Mean age 56 + 11.5 years; 22.7% patients were > 65 years old; Male 50.2% ; Caucasians 76.8%; Black 17.2%; Other 6%; mean BMI 30.8 + 6.3; MSSBP 152.2 + 11.2 mmHg and MSDBP 99.0 + 3.6 mmHg; Mean sitting pulse: 72.8 + 8.7 bpm; and mean pulse pressure was not reported. |
|
| Interventions | 652 patients randomized Placebo: N = 131; aliskiren 150 mg: N = 127; aliskiren 300 mg: N = 130; aliskiren 600 mg: N = 130; Irbesartan 150 mg: N = 134 Duration of treatment = 8 weeks |
|
| Outcomes | Primary: change from baseline in trough MSDBP Secondary: change in MSSBP; percentage of patients achieving BP control (SBP < 140 mmHg and DBP < 90 mmHg; dose‐response analysis; trough‐to‐peak ratio; withdrawal effect; safety and tolerability assessments. | |
| Notes | Study CSPP100A2201 is published in a journal as Gradman 2005. It is not registered on clinicalTrials.gov It is registered on Novartis Clinical Trial Results database (results are not posted) CSR without appendices was received from EMA on January 25th 2016 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomization with a block size of 5 and stratified by region was performed by the interactive voice response system provider using a validated system that automates the random assignment of treatment groups to randomization numbers." |
| Allocation concealment (selection bias) | Low risk | "Randomization data were kept strictly confidential until completion of the study and blinded data cleaning process. Access during the study was available only to authorized persons who maintained the randomization database and were not involved in the conduct of the study. The database lock procedure was followed to merge clinical data and treatment codes for analyses after the completion of the study and data cleaning." |
| Blinding (performance bias and detection bias) All outcomes | Low risk | All medications were identical in appearance. "All study personnel and participants remained blinded to the treatment assignment for the duration of the study." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Withdrawal rate was 22 (16.8%) in the placebo group; 12 (9.4%) in aliskiren 150 mg; 11 (8.5%) in aliskiren 300 mg and 10 (7.7%) in aliskiren 600 mg. Reason for withdrawal differed ‐ 7.6% in placebo group withdrew due to lack of therapeutic effect and 0.8% to 1.6% withdrawal in aliskiren treatment groups. How missing data were analyzed has not been reported. |
| Selective reporting (reporting bias) | Unclear risk | Most primary and secondary outcomes were reported. Baseline and endpoint BP data was reported. Pulse rate was reported at baseline but not reported at endpoint. Change in pulse pressure was reported without standard deviation on page 53 of the CSR. |
| Other bias | High risk | Manufacturer sponsored. Authors are employees of Novartis Pharmaceuticals Corporation and are therefore eligible for Novartis stock and stock options. Patients could have been selected based on their response to other drugs acting on the renin‐angiotensin system. Study is not registered on www.ClinicalTrials.gov |
CSPP100A2203.
| Methods | Multicentre, randomized, double‐blind placebo‐controlled, multifactorial, parallel‐group study | |
| Participants | 1441 patients were enrolled from 94 centres in the USA, Germany, France, Denmark, and Poland, of which 1123 were randomized to 11 treatment groups. 318 patients discontinued single‐blind period (13.4% due to abnormal test procedure; 3.3% due to abnormal laboratory values; 2.8% due to withdrawal of consent; 1.5% due to adverse events; 0.5% due to protocol violation; 0.1% due to unsatisfactory therapeutic effect; 0.3% due to condition no longer required study drug; 0.1% due to administrative problems and 0.1% due to lost to follow‐up). Inclusion criteria: Adult men and women 18 years or older with mild‐to‐moderate essential hypertension (MSDBP 95 mmHg to < 110 mmHg). Exclusion criteria: Severe hypertension (MSSBP 180 mmHg or more or MSDBP of 110 mmHg or more); secondary hypertension; type I or uncontrolled diabetes mellitus; type 2 diabetes mellitus; history of severe cardiovascular or cerebrovascular disease or other life‐threatening medical conditions. Baseline characteristics: Mean age 56 + 12.2 years; 24.9% were > 65 years of age; Male 55.9%; Caucasians 92.1%; Black 6.28; other 1.2%; mean BMI 29.5 + 5.0; MSSBP 153.3 + 12.0 mmHg and MSDBP 99.0 + 3.5 mmHg; mean sitting pulse 72 + 9.3 bpm; mean pulse pressure is not reported. |
|
| Interventions | 1123 patients were randomized Placebo: N = 177; aliskiren 75 mg: N = 179; aliskiren 150 mg: N = 178; aliskiren 300 mg: N = 175; several valsartan monotherapy groups and combination therapy groups. Duration of treatment = 8 weeks |
|
| Outcomes | Primary: change from baseline to endpoint in MSDBP of monotherapy with aliskiren at all doses versus placebo. Secondary: change from baseline in MSSBP of aliskiren monotherapy as well as in combination therapy groups; safety and tolerability of aliskiren; of aliskiren combination therapy; impact of treatment on selected biomarker. |
|
| Notes | Study CSPP100A2203 is published in a journal as Pool 2007. It is not registered on clinicalTrials.gov It is not registered on Novartis Clinical Trial Results database. CSR without appendices was received from EMA on April 12th 2016. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomization by region was performed by the interactive voice response system provider using a validated system that automates the random assignment of treatment groups to randomization numbers." |
| Allocation concealment (selection bias) | Low risk | Each centre received a supply of medication packs each labelled with a unique number. "Randomization codes were kept strictly confidential until the database was locked." |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Aliskiren 75 mg, aliskiren 150 mg were supplied as capsule and aliskiren 300 mg as tablet. Placebo was supplied both as a capsule and in tablet form. To adequately blind the study, patients had to take 3 tablets/capsules of study medication. Insufficient information to determine if blinding was successful. Blinding of outcome assessor is not reported. Appendices were not provided by EMA. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 15 (8.5%) participants withdrew from placebo group; 21 (11.7%) from aliskiren 75 mg group; 13 (7.3%) from aliskiren 150 mg group and 9 (5.1%) from aliskiren 300 mg group. Reason for withdrawal differed between groups ‐ lack of therapeutic effect was noted in 2.8% in placebo group; 5.6% in aliskiren 75 mg; 2.2% in aliskiren 150 mg and 2.3% in aliskiren 300 mg group. How missing data will be analyzed is not reported. |
| Selective reporting (reporting bias) | Unclear risk | Protocol was available and primary and secondary outcomes were reported. Pulse rate was reported at baseline but no detail provided at endpoint. Pulse pressure is not reported at baseline or endpoint. This information may be available in the appendices that were not provided with the CSR by the EMA. |
| Other bias | High risk | Manufacturer sponsored. Conflict of interest of authors has not been provided. Patients could have been selected based on their response to other drugs acting on the renin‐angiotensin system. Study is not registered with clinicalTrials.gov or Novartis Clinical Trial Results database. |
CSPP100A2204.
| Methods | Multicentre, randomized, double‐blind, double‐dummy, placebo‐ and active‐controlled. multifactorial, parallel‐group study of aliskiren monotherapy compared to placebo and combination therapy of aliskiren with HCTZ compared to component monotherapies. | |
| Participants | 3190 patients were enrolled from 19 countries in Argentina, Brazil, Canada, Columbia, Finland, France, Germany, Guatemala, Italy, the Netherlands, Norway, Peru, Poland, Russia, Slovakia, Spain, Sweden, Taiwan and USA in the single‐blind placebo run‐in period. 2776 were randomized, of which 2558 (92.1%) completed the study and 204 (7.3%) discontinued. Discontinuation rate was highest in the placebo group most often due to unsatisfactory therapeutic effect (2%) and those due to adverse events (2.3%). Inclusion criteria: Women and men aged 18 years or older with mild‐to‐moderate essential hypertension (MSDBP from > 95 mmHg to < 110 mmHg) at visit 3. Female patients were either postmenopausal, surgically sterile or using effective contraceptive methods. Exclusion criteria: Pregnant or nursing women; patients with severe hypertension (DBP > 110 mmHg and /or SBP > 180 mmHg) ; those with secondary hypertension; cerebrovascular accident, MI; coronary bypass surgery; TIA within last 12 months; Keith ‐ Wagener grade III to IV hypertensive retinopathy; malignancy including leukaemia and lymphoma within the past five years but excluding basal cell carcinoma; drug or alcohol abuse; gouty arthritis; current diagnosis of heart failure; angina pectoris requiring pharmacotherapy other than sublingual nitroglycerin; second or third degree heart block without a pace maker; any potential life‐threatening arrhythmia or clinically significant valvular disease; surgical or medical conditions which might significantly alter the absorption, distribution, metabolism, or excretion of study medication; known or suspected contraindications to aliskiren, including history of allergy to study drug and previous participation in an investigation clinical study within 1 month of visit 1 of the study; Participants with type I or 2 diabetes mellitus with poor glycaemic control; laboratory serum sodium or potassium values ≥ 5.5 mEq/L; hepatic disease; or renal impairment; history of major gastrointestinal tract surgery or current or previously active inflammatory bowel disease were excluded. Baseline characteristics: Mean age 54.6 + 11.6 years; 21.1% patients were > 65 years of age; Male 54.8%; Caucasians 85.4%; Black 4.6%; Asian 2.5%; Native American 1.8% and others 5.7%; MSSBP 153.6 + 12.2 mmHg and MSDBP 99.2 + 3.6 mmHg; mean sitting pulse pressure was 72.2 beats/minute |
|
| Interventions | Placebo: N = 195; aliskiren 75 mg: N = 184; aliskiren 150 mg: N = 185; aliskiren 300 mg: N = 183; HCTZ monotherapy groups and aliskiren combination therapy groups. Duration of treatment = 8 weeks |
|
| Outcomes | Primary: Change from baseline in MSDBP. Secondary: change from baseline in MSSBP, assessment of dose‐response efficacy, proportion of patients showing a successful response MSDBP < 90 mmHg or 10 mmHg or greater reduction from baseline, proportion of patients achieving BP control (<140 mmHg/90 mmHg), safety and tolerability, effects of treatment on plasma renin activity and renin concentration. |
|
| Notes | Study CSPP100A2204 is published in a journal as Villamil 2007. It is registered on clinicalTrials.gov. Identifier: NCT00219024 (results are posted). It is registered on Novartis Clinical Trial Results database (results are not posted). CSR without appendices was received from EMA on April 6th 2016 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomization was performed by Covance Inc. using validated system that automates random assignment. The detail of the method used is not reported in the CSR. The randomization method and list is provided in appendix 5.2, which was not available. 14 patients received randomization numbers in error, were not treated with DB treatment and did not provide post baseline data. One patient completed the single‐blind phase and was assigned DB treatment without randomization and medication assignment by IVRS. Two patients received placebo instead of the assigned medication for 9 or 10 days and were subsequently discontinued due to protocol violation. |
| Allocation concealment (selection bias) | Unclear risk | Randomization number was provided along with a unique medication number for the packages of study drug to be dispensed. No further detail is provided. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | The identity of the treatment was concealed by the use of drugs that were identical in packaging, labelling, appearance, odour and schedule of administration. A double‐dummy design was used because the identity of the doses of aliskiren and HCTZ could not be disguised due to their different forms. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Total withdrawals were 22 (11.3%) in placebo group 15 (8.2%) in aliskiren 75 mg group; 16 (8.6%) in aliskiren 150 mg group and 17 (9.3%) in aliskiren 300 mg group. (Page 49 of CSR). The reasons for withdrawal in the placebo group were higher due to unsatisfactory therapeutic effect as compared to aliskiren groups. |
| Selective reporting (reporting bias) | Unclear risk | Protocol was available and primary and secondary outcomes were reported. Pulse rate was reported at baseline but no detail provided at endpoint. Pulse pressure is not reported at baseline or endpoint. This information may be available in the appendices that were not provided with the CSR by the EMA. |
| Other bias | High risk | Manufacturer sponsored. Conflict of interest of authors has not been reported. Patients could have been selected based on their response to other drugs acting on the renin‐angiotensin system. |
CSPP100A2308.
| Methods | Randomized double‐blind placebo‐controlled, parallel‐group, multicentre study. | |
| Participants | Patients were recruited at 68 centres internationally (Canada, Guatemala, Korea, the Netherlands and USA). Of the 833 patients who entered single‐blind period, 672 were randomized to 4 double‐blind treatment groups. 63 (9.4%) discontinued treatment (15.1% due to abnormal test procedure; 0.4% due to abnormal laboratory values; 2.5% due to withdrawal of consent; 1.5% due to adverse events; 0.4% due to protocol violation; 0.1% due to unsatisfactory therapeutic effect; 0.1% due to condition no longer required study drug; 0.1% due to administrative problems and 0.5% due to lost to follow‐up). Inclusion criteria: Men and women aged 18 years or over with mild‐to‐moderate essential hypertension (MSDBP > 95 mmHg and <110 mmHg. Exclusion criteria: Patients who previously entered an aliskiren study; severe hypertension (MSDBP > 110 mmHg and/or MSSBP > 180 mmHg); secondary hypertension; known Keith‐Waegner grade III or IV hypertensive retinopathy; history of hypertensive encephalopathy or cerebrovascular accident; TIA during 12 months prior to visit 1; current diagnosis of heart failure (NYHA class II ‐ Iv); history of MI, coronary bypass surgery or any percutaneous intervention during 6 months prior to visit 1; current angina pectoris requiring pharmacotherapy; second or third degree heart block without a pacemaker; potentially life‐threatening arrhythmia; clinically significant valvular disease; type I or II diabetes mellitus with poor glycaemic control HbA1C > 9%; serum sodium less than lower limit of normal and serum potassium < 3.5 mEq/L or > 5.5 mEq/L or dehydration at visit 1; any surgical or medical condition that might significantly alter absorption, distribution, metabolism or excretion of the drug; history of malignancy including leukaemia or lymphoma; history of drug or alcohol abuse within last 12 months; pregnant or nursing women; and history of non compliance to medical regimens or unwillingness to comply with study protocol. Baseline characteristics: Mean age 53 + 10.5 years; 13.1% were > 65 years; Male 61.6%; Caucasians 61.3%; Black 12.4%; Asian 18%; Others 8.3%; Mean BMI 29 + 5.9 kg/m2; MSDBP 99.6 + 3.7 mmHg and MSSBP 152.1 + 12.4 mmHg. |
|
| Interventions | Placebo: N = 165; aliskiren 150 mg: N = 172; aliskiren 300 mg: N = 169; aliskiren 600 mg: N = 166 Duration of treatment = 8 weeks |
|
| Outcomes | Primary: change in MSDBP from baseline Secondary: change in MSSBP, dose‐response relationship, 24‐h ambulatory BP monitoring profiles and trough‐to‐peak ratios, proportion of patients achieving a successful treatment response (MSDBP < 90 mmHg and /or greater or equal to a 10mm Hg reduction from baseline, BP control (BP < 140/90 mmHg, effects on plasma renin activity, and renin concentration, heart rate, safety and tolerability, and effect of treatment withdrawal on BP, plasma renin activity, and renin concentration. | |
| Notes | Study CSPP100A2308 is published in a journal as Oh 2007 It is registered on clinicalTrials.gov. Identifier: NCT00219128 (results are not posted) It is registered on Novartis Clinical Trial Results database (results are not posted) CSR without appendices was received from EMA on February 20th 2016 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomization list is produced using a validated system that automates the random assignment of treatment groups to randomized numbers in the specified ratios to insure treatment assignment is unbiased and concealed from patients and investigator staff. |
| Allocation concealment (selection bias) | Low risk | Randomization list is produced using a validated system that automates the random assignment of treatment groups to randomized numbers in the specified ratios to insure treatment assignment is unbiased and concealed from patients and investigator staff. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Patients, investigator and staff performing the assessment and data analysis will remain blind to the identity of the treatment from the time of randomization until database lock and kept strictly confidential. The identity of the treatments will be concealed by the use of study drugs that are identical in packaging, labelling, schedule of administration, appearance, taste and odour. Patients were instructed to take 3 tablets of study medication per dose throughout the study at approximately 8.00am. Unblinding will occur in the case of patient emergencies and at the end of the conclusion of the study. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | of the 833 patients enrolled in SB period, 671 (80.6%) completed SB period and were then randomized to DB treatment and 162 (19.4%) discontinued. 30 (18.2%) patients withdrew in placebo group; 10 (5.8%) in aliskiren 150 mg group; 9 (5.3%) in aliskiren 300 mg group and 14 (8.4%) in aliskiren 600 mg group. Reasons for withdrawal differed between treatment groups ‐ unsatisfactory therapeutic effect in 17 (10.3%) in placebo; 3 (1.7%) in aliskirenA 150 mg; 1 (0.6%) in aliskiren 300 mg and 4 (2.4%) in aliskiren 600 mg. For each patient, the last post baseline measurement during double blind‐treatment period was carried forward as endpoint measurement for the variable analyzed. |
| Selective reporting (reporting bias) | Unclear risk | Protocol was available and primary and secondary outcomes were reported. Pulse rate and pulse pressure were not reported at baseline or endpoint. This information may be available in the appendices that were not provided with the CSR by the EMA. |
| Other bias | High risk | Manufacturer sponsored. Author Keefe D is employee of Novartis. Conflict of interest of other authors has not been provided in the published article. Patients could have been selected based on their response to other drugs acting on the renin‐angiotensin system. |
CSPP100A2323.
| Methods | Randomized, double‐blind, parallel‐group, placebo‐ and active‐controlled multicentre dose titration study. After 3 weeks, randomized patients receiving aliskiren 150 mg or HCTZ 12.5 mg underwent forced titration to doubled doses of their respective treatments. After an additional 3 weeks, patients in the placebo group were reassigned to aliskiren 300 mg or hydrochlorothiazide 25 mg for 20 weeks. |
|
| Participants | 1440 patients were screened of which 1275 were enrolled in the single‐blind run‐in period. During this period 151 (11.8%) patients discontinued (most common reasons were abnormal laboratory test 7.1%, which included patients who did not meet the blood pressure criteria for randomization; withdrawal of consent in 2.6% and adverse events 0.9%). 1124 patients were randomized to receive once‐daily treatment with aliskiren 150 mg, hydrochlorothiazide 12.5 mg, or placebo. 978 (87%) completed the double‐blind period. Inclusion criteria: Patients aged ≥18 years with MSDBP > 90 mmHg and < 110 mmHg at the single‐blind placebo run‐in visit. At randomization, patients had to have a MSDBP > 95 mmHg and < 110 mmHg and show a absolute difference of < 10 mmHg in MSDBP from their previous study visit. Exclusion criteria: Patients with were severe hypertension (MSDBP ≥110 mmHg and/or MSSBP ≥180 mmHg); suspected secondary or malignant hypertension; Known Keith‐Waegner grade III or IV hypertensive retinopathy; malignancy excluding basal cell carcinoma within the last 5 years; drug or alcohol abuse in the last 12 months; current diagnosis of heart failure (NYHA class II ‐ IV); angina pectoris requiring pharmacological treatment; second or third degree heart block without a pace maker; clinically significant valvular disease; potentially life‐threatening or symptomatic arrhythmia; TIA; coronary bypass surgery or PCI during 12 months prior to visit 1; type 1 diabetes mellitus (DM); type 2 DM, poorly controlled (glycosylated haemoglobin > 9.0% or microalbuminuria at visit 1); serious hepatic, pancreatic, or renal disease; any surgical or medical condition that might significantly alter absorption, distribution, metabolism or excretion of the drug; history of major gastrointestinal tract surgery such as gastrectomy, gastroenterostomy, or bowel resection; history of active inflammatory bowel disease; currently active gastritis, duodenal or gastric ulcer; gastrointestinal bleeding in the past 3months; any history of pancreatitis, pancreatic injury, or evidence of impaired pancreatic function or abnormal lipase or amylase during past 12 months of visit 1; evidence of hepatic disease, history of hepatic encephalopathy; history of oesophageal varices; history of portocaval shunt; evidence of renal impairment or dialysis or nephrotic syndrome; current treatment with cholesterol absorption inhibitors; proteinuria or serum sodium less than lower limit of normal and/or serum potassium < 3.5 mEq/L or dehydration at visit 1; clinically significant allergy; pregnant and breast feeding women were excluded. Baseline characteristics: Mean age 56 + 11 years; 22.8% were > 65 years of age; male 56%; Caucasians 99%; Asian (0.7%) Black 0.2% and other 0.2%; Mean BMI 29.1 + 4.8 kg/m2;and diabetes in 10.9% patients; MSSBP 154.2 + 11.2 mmHg and MSDBP 99 + 3.4 mmHg |
|
| Interventions | Placebo: N = 221; aliskiren 150 mg N = 459; HCTZ 12.5 mg N = 444; After 3 weeks on therapy, patients underwent forced titration to aliskiren 300 mg and HCTZ 25 mg. After 6 weeks, patients receiving placebo were reassigned to aliskiren 300 mg (N = 108) or HCTZ 25 mg (N = 113). Duration of treatment = 26 weeks (monotherapy data useful at week 3 for 150 mg dose and at week 6 for 300 mg dose) |
|
| Outcomes | Primary: Change from baseline to endpoint in MSDBP. Secondary: changes in MSSBP at week 26 endpoint and MSDBP and MSSBP at week 52 endpoint; comparison of the BP‐lowering efficacy of aliskiren 300 mg monotherapy and hydrochlorothiazide 25 mg monotherapy at week 12 endpoint; evaluation of the proportion of patients with response to treatment and BP control at the week 26 and week 52 endpoints; and comparison of the long‐term safety and tolerability of an aliskiren regimen with a HCTZ regimen. |
|
| Notes | Study CSPP100A323 is published in a journal as Schmieder 2009 It is registered on clinicalTrials.gov. Identifier: NCT00219154 (results are posted) It is registered on Novartis Clinical Trial Results database (results are not posted) CSR without appendices was received on May 27th 2016. BP data at end of the aliskiren monotherapy period is analyzed at the end of 3 weeks for 150 mg and 300 mg dose. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomization by centre was performed by the interactive voice response system provider with the use of a validated system that automates the random assignment of patients to randomization numbers." The IVRS assigned a 2=part patient number. The first part was the centre number and the second part was one of the series of numbers allocated to the centre. |
| Allocation concealment (selection bias) | Low risk | "Randomization was performed using the procedure to ensure that treatment assignment was unbiased and concealed from the patient and investigator staff. Data were kept strictly confidential until the time of unblinding. Novartis Pharmaceuticals Corporation was responsible for managing the database and conducting audits" |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Aliskiren 150 mg and 300 mg were provided as film ‐coated tablets each of a different size, shape and colour. The placebos to aliskiren 150 mg and 300 mg were matched in respective size, shape and colour to the active tablets. HCTZ 12.5 and 25 mg and placebo to HCTZ were provided as identically appearing capsules. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | During the 6‐week double‐blind, placebo‐controlled treatment period, 67 patients (6.0%) discontinued study treatment. Discontinuations were significantly higher (P = 0.05) in the placebo group 21 (9.5%) than in the aliskiren group 20 (4.4%) but were not significantly greater than in the HTCZ group 26 (5.9%). The most common reason for discontinuation in placebo group was adverse events, withdrawal of consent and unsatisfactory therapeutic response each occurring in 2.7% patients. |
| Selective reporting (reporting bias) | Unclear risk | Protocol was available and primary and secondary outcomes were reported at 26 weeks. Pulse rate and pulse pressure are not reported at baseline or at week 3, week 6 and study endpoint. This information may be available in the appendices that were not provided with the CSR by the EMA. Most data in the CSR are reported at 26 weeks. Data that can be used at 3 and 6 weeks for this review have been partially reported. MSDBP data with SEM are reported at week 6 in the CSR. Other MSSBP and MSDBP data have been calculated from the graph. SAE data detail at week 3 and 6 have not been reported. |
| Other bias | High risk | Study supported by Novartis. Authors received research grants, consulting and speaker fees from Novartis. |
CSPP100A2327.
| Methods | Randomized, double‐blind, parallel‐group, placebo‐controlled, dose‐escalation study | |
| Participants | 5133 patients were screened at 194 centres in the USA and Europe. 3980 enrolled in 3‐4 weeks single‐blind placebo run ‐in period. 1797 patients were randomized to once‐daily oral administration of treatment for 4 weeks and all patients underwent a forced titration to double the dose of their treatments, and were treated for another 4 weeks. Inclusion criteria: Men and women aged 18 years or over with stage 1‐2 hypertension (MSDBP > 95 mmHg to < 110 mmHg) and an absolute difference in MSDBP of < 10 mmHg from the prior visit, as well as a mean 8‐hour daytime ABPM DBP > 90 mmHg. Exclusion criteria: Patients who previously entered an aliskiren study; severe hypertension (MSDBP > 110 mmHg and/or MSSBP > 180 mmHg); secondary hypertension; known Keith‐Waegner grade III or IV hypertensive retinopathy; history of hypertensive encephalopathy or cerebrovascular accident, TIA; MI, coronary bypass surgery, or PCI; serum sodium less than lower limit of normal; serum potassium > 5.3 mEq/L at visit 1; history of type I or II diabetes mellitus with poor glycaemic control HbA1C > 8% at visit 1; current angina pectoris requiring pharmacological treatment; second or third degree heart block without a pacemaker; potentially life‐threatening arrhythmia; clinically significant valvular disease; any surgical or medical condition that might significantly alter absorption, distribution, metabolism or excretion of the drug; history of malignancy including leukaemia or lymphoma; history of major gastrointestinal tract surgery such as gastrectomy, gastroenterostomy, or bowel resection; history of active inflammatory bowel disease; currently active gastritis, duodenal or gastric ulcer; gastrointestinal bleeding in the past 3months; any history of pancreatitis, pancreatic injury, or evidence of impaired pancreatic function or abnormal lipase or amylase during past 12 months of visit 1; Evidence of hepatic disease, history of hepatic encephalopathy; history of oesophageal varices; history of portocaval shunt; evidence of renal impairment or dialysis or nephrotic syndrome; Current treatment with cholestyramine or colestipol resins; history of hyper sensitivity to any of the study drugs or those belonging to the same therapeutic class; history of angioedema due to usage of ARB or ACE inhibitors; history of malignancy of any organ system treated or untreated in the past 5 years; history of drug or alcohol abuse within last 12 months; pregnant or nursing women; and history of non‐compliance to medical regimens or unwillingness to comply with study protocol were excluded. Baseline characteristics: Mean age was 52 + 10.4 years with 13.9% > 65 years in placebo group and 11.7% in aliskiren group, respectively; Males 58.4 to 61.2%; Caucasian 74.6 to 76%; 15 to 16% Black; 1.5% Asian and 7 to 9% others; Mean BMI 30.2 + 5.7 kg/m2;Baseline MSSBP was 154.1 + 12.8 mmHg and MSDBP was 100.4 + 4.2 mmHg. |
|
| Interventions | Placebo: N = 459; aliskiren 150 mg to 300 mg: N = 437; valsartan 160 mg to 320 mg N = 455; aliskiren 150 mg/300 mg/valsartan 160 mg/320 mg: N = 446 Duration of treatment = 8 weeks |
|
| Outcomes | Primary: change in MSDBP for baseline to week‐8 endpoint Secondary: change in MSSBP from baseline to week‐8 endpoint, proportion of patients achieving a successful response to treatment (MSDBP < 90 mmHg or a 10 mmHg or greater reduction from baseline, or both, or achieving BP control (<140 mmHg/90 mmHg). Changes from baseline to week 8 in 24‐h ambulatory BP measurements, changes in biomarker (plasma renin concentration, plasma renin activity and plasma aldosterone concentration) | |
| Notes | Study CSPP100A2327 is published in a journal as Oparil 2007. EudraCT no. 2005‐000039‐73 It is not registered on clinicalTrials.gov It is registered on Novartis Clinical Trial Results database (results are not posted). CSR without appendices was received from EMA on June 16th 2016. Data of this study was also reported in the FDA Medical Review. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The randomisation list was generated by Novartis Drug Supply Management (Basel, Switzerland) with a validated system that automated the random assignment of treatment groups to randomisation numbers." |
| Allocation concealment (selection bias) | Low risk | The randomization scheme was reviewed by a biostatistics quality assurance group at Novartis and locked by them after approval. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "Placebo and drug tablets and capsules were matched in size, shape, and colour to maintain blinding." Patients were required to take two tablets and two capsules. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 63 (13.7%) withdrew from placebo group and 53 (12%) in aliskiren group. Reason for withdrawal differed between groups ‐ Unsatisfactory therapeutic effect ‐ 36 (8%) in placebo and 13 (3%) in aliskiren 150 mg group. |
| Selective reporting (reporting bias) | Unclear risk | Protocol was available and primary and secondary outcomes were reported. Pulse rate and pulse pressure are not reported at baseline or endpoint. This information may be available in the appendices that were not provided with the CSR by the EMA. |
| Other bias | High risk | Manufacturer sponsored. Oparil is the recipient of grants‐in‐aid from Abbot Laboratories, Astra Zeneca, Aventis, Bioavail, Boehringher Ingelhiem, Bristol Meyers Squibb, Forest laboratories, GlaxoSmithkline, Novartis, Merck & Co, Pfizer, Sanofi Aventis, and the Salt Institute. Other authors are employees of Novartis and therefore eligible for stock and stock option Patients could have been selected based on their response to other drugs acting on the renin‐angiotensin system. Manufacturer sponsored. The overall incidence of major protocol violation was higher in placebo group (10.9%) than in other aliskiren monotherapy groups (5.4%). |
CSPP100A2328.
| Methods | Multicentre, randomized, double‐blind, parallel‐group, placebo‐controlled study. | |
| Participants | 1508 patients with essential hypertension were screened from 53 centres in the USA and 48 centres in the European Union. 642 eligible patients were randomized, of which 576 (89.7%) completed the double‐blind treatment period. Inclusion criteria: Patients who previously entered an aliskiren study; patients aged ≥18 years with stage 1 or 2 essential uncomplicated hypertension. MSDBP ≥95 and <110 mmHg, with a difference of ≤10 mmHg, on their last 2 visits and mean 8‐hour daytime ambulatory DBP ≥90 mmHg. Patients fulfilling these eligibility criteria then underwent ABPM at visit 5. Only patients with mean 8‐hour daytime ambulatory DBP (ADBP) ≥90 mmHg were entered in the study. Exclusion criteria: Patients with were severe hypertension (MSDBP ≥110 mmHg and/or MSSBP ≥180 mmHg); suspected secondary or malignant hypertension; type 1 diabetes mellitus (DM); type 2 DM, poorly controlled (glycosylated haemoglobin > 8.0%) or requiring insulin; serious cardiac, hepatic, pancreatic, renal, or cerebrovascular disease; serum sodium less than lower limit of normal and/or serum potassium > 5.3 mEq/L or dehydration at visit 1; atrial fibrillation or atrial flutter; clinically significant allergy; and malignant tumours; pregnant and breast‐feeding women were excluded. Baseline characteristics: Mean age 52 + 10.7 years; 13.1% > 65 years in placebo group and 9.8%, 15.8% and 14.6% in aliskiren 75 mg, 150 mg and 300 mg groups respectively; Males 60%; Caucasian 80.8%; Black 14.2%; Asian 2.3% and others 12.6%; Mean BMI 30.4 + 5.6 kg/m2; baseline MSSBP 153.5 + 12.8 mmHg and MSDBP 100.5 + 4.1 mmHg at baseline |
|
| Interventions | N = 642 patients were randomized Placebo: N = 160; aliskiren 75 mg: N =1 53; aliskiren 150 mg: N = 171; aliskiren 300 mg: N = 158 Duration of treatment = 8 weeks |
|
| Outcomes | Primary: Change from baseline in MSDBP at the week‐8 endpoint Secondary: Change from baseline in MSSBP at week‐4 and week‐8 endpoint; change in MSDBP at weeks 4 and 8; proportion of responders (MSDBP < 90 mmHg and/or at least a 10 mmHg reduction from baseline); proportion of patients achieving BP control (MSSBP/MSDBP <140/90 mmHG); dose‐response relationship; safety and tolerability of aliskiren compared to placebo; impact of treatment on plasma renin activity, plasma renin concentration and plasma aldosterone |
|
| Notes | Study CSPP100A2328 is published in a journal as Puig 2009. It is not registered on clinicalTrials.gov It is registered on Novartis Clinical Trial Results database (results are posted). EMA does not possess CSR for this study. CSPP100A2328 synopsis has been used to report mean change from baseline in MSSBP and MSDBP with SD and adverse events. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The randomization list was generated using a validated system that automated the random assignment of treatment groups to randomization numbers. At randomization, eligible patients were assigned the lowest available number on the randomization list and were supplied with a medication pack labelled with the relevant randomization number. The randomization scheme was reviewed by a biostatistics quality assurance group at Novartis Pharma AG and locked by them after approval." |
| Allocation concealment (selection bias) | Low risk | "At randomization eligible patients were assigned the lowest available number on the randomization list and were supplied with a medication pack labelled with he relevant randomization number. Randomization codes were kept confidential until after database lock." |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "Blinding was maintained by providing the study drugs along with matching placebo tablets. All patients took 3 tablets daily at 8:00am, except on the day of visit, when the study drug was taken after the visit procedures were completed." |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | "Of the 642 randomized patients, 576 (89.7%) completed the double‐blind treatment period. The proportion of discontinuations was higher in the aliskiren 150 mg group 21 (12.3%) and the placebo group 19 (11.9%), and lower in the aliskiren 75 mg 12 (7.8%) and aliskiren 300 mg 14 (8.9%) groups." |
| Selective reporting (reporting bias) | Unclear risk | Could not be assessed as protocol is not available. SAE data detail not available. |
| Other bias | High risk | Study funded by manufacturer (Novartis). Medical writers from Novartis drafted manuscript. It is not registered on clinicalTrials.gov. EMA does not possess CSR for this study |
CSPP100A2405.
| Methods | Randomized, double‐blind, parallel‐group, placebo‐controlled study. Randomization was stratified by region and age group (≥ 65 to < 75 years and ≥ 75 years). | |
| Participants | 836 patients were enrolled at 95 centres in 8 countries (Argentina, Czech Republic, Germany, Iceland, Italy, the Netherlands, Poland and Slovakia). 754 (90.2%) completed the single‐blind, run‐in period. However, two patients were mistakenly randomized, creating a randomized set with two additional patients (N = 756). Though these two patients did not meet study criteria, they were included in the placebo group for the randomized set, but excluded from the full analysis set and safety set (N = 754). Inclusion criteria: Men and women in the outpatient setting aged ≥ 65 years with essential hypertension (defined as MSSBP ≥150 mmHg and < 180 mmHg and MSDBP <110 mmHg) and in addition, patients’ MSSBP had to differ by ≤15 mmHg between the last two visits of the placebo run‐in period for inclusion in the study. Exclusion criteria: Patients with severe hypertension; those with secondary hypertension; cardiac dysfunction; diabetes; malignancy including leukaemia and lymphoma within the past five years; surgical or medical conditions which might significantly alter the absorption, distribution, metabolism, or excretion of study medication; known or suspected contraindications to aliskiren including history of allergy to ACE‐inhibitors or ARBS; previous exposure to aliskiren within 3 months of visit 1 of the study; previous participation in an investigation clinical study within 1 month of visit 1 of the study; participants with laboratory serum potassium values ≥ 5.5 mEq/L; with an estimated GFR < 45 mL/min/1.73m2; Women who were pregnant, lactating or women of childbearing potential were also excluded. Baseline characteristics: Mean age was 72 + 6 with 30.6 to 32.3% patients > 75 years old; 43.2 to 47.1% patients were male; 98.4 to 100% were Caucasians; 1.1% Black; 0.5 to 1.6% were others; Mean BMI 29 + 5.0 kg/m2; 16 to 21% patients had diabetes; MSSBP at baseline was 160 + 8 mmHg and MSDBP was 90 + 8 mmHg. |
|
| Interventions | Aliskiren 75 mg ( N = 192), 150 mg (N = 189), or 300 mg (N = 186) and placebo (N = 189) once daily Duration of treatment = 8 weeks. |
|
| Outcomes | Primary: Change in MSSBP from baseline to week‐8 endpoint. Secondary: Change in MSDBP from baseline to week‐8 endpoint; changes in MSSBP and MSDBP at week 4 and 8; mean 24‐hour ambulatory systolic and diastolic blood pressures; the proportion of patients achieving BP control (MSSBP/DBP < 140 mmHg/90 mmHg) at the week‐8 endpoint; serious adverse events and total adverse events |
|
| Notes | Study CSPP100A2405 is published in a journal as Villa 2012. It is registered on clinicalTrials.gov. Identifier: NCT00706134. It is registered on Novartis Clinical Trial Results database (results are posted). CSR without appendices was received from EMA on December 22nd 2015. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | An Interactive Voice Response System (IVRS) was used to randomly assign patients to study medication. The medication randomization list was produced by Novartis Drug Supply Management that automates the random assignment of medication numbers to medication packs containing each of the study drugs. Randomization was stratified by region and by age group > 65 years and < 75 years and > 75 years of age. The treatment groups were generally well‐matched for baseline and demographic characteristics. |
| Allocation concealment (selection bias) | Low risk | The IVRS automated the random assignment of patient numbers to randomization numbers, which are linked to the different treatment arms. The treatment assignment is unbiased and concealed from patients and investigator staff. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Placebo tablets were matched to the active study drug, and a double‐dummy design was used to ensure study blinding. Patients, investigator staff, persons performing assessments and data analysts will remain blind to the identity of the treatment from time of randomization until database lock. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 754 completed the placebo run‐in period and were randomized to study treatment. Overall, 700 patients completed double‐blind treatment, with higher completion rates in the aliskiren 150‐ and 300 mg groups than in the aliskiren 75 mg or placebo groups. 18 (9.5%) discontinued in placebo group; 19 (9.9%) in aliskiren 75 mg; 6 (3.2%) in 150 mg group and 11 (5.9%) in 300 mg group. Reason for withdrawal differed ‐ withdrawal due to lack of therapeutic effect was 4.8% in the placebo group compared to 1.6% in aliskiren 75 mg and 150 mg group. Last‐observation‐carried‐ forward approach was used for the week‐8 endpoint analyses. |
| Selective reporting (reporting bias) | Unclear risk | Protocol was available and primary and secondary outcomes were reported. Pulse rate and pulse pressure were not reported at baseline or endpoint. This information may be available in the appendices that were not provided with the CSR by the EMA.. |
| Other bias | High risk | Study supported by Novartis. Most authors were employees of Novartis and therefore eligible for Novartis stock and stock options. |
ABPM: ambulatory blood pressure monitoring; ACE: angiotensin‐converting enzyme; ARB: angiotensin receptor blockers; BMI: body mass index; CRF: case report form; CSR: clinical study report; DBP: diastolic blood pressure; EMA: European Medicines Agency; GFR: glomerular filtration rate; HTCZ: hydrochlorothiazide; IVRS: Interactive Voice; Response System; MI: myocardial infarction; MSDBP: mean sitting diastolic blood pressure; MSSBP: mean sitting systolic blood pressure; PCI: percutaneous coronary intervention; PRA: plasma renin activity; SAE: serious adverse event; SBP: systolic blood pressure; TIA: transient ischaemic attack
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Andersen 2008 | No placebo monotherapy arm. |
| Dorresteijn 2013 | A randomized, four‐way, double‐blind, single‐centre, cross‐over study was performed in 31 adult white patients. Screening was followed by a 40‐week study period in which patients received each of four once‐daily monotherapies sequentially in a random order: aliskiren 300 mg, moxonidine 0.4 mg, HCTZ 25 mg and matching placebo. Each treatment period began with a 2‐week titration phase during which halved doses were used. Thereafter, patients were force titrated to full medication doses. The efficacy and tolerability of each treatment was assessed after 8 weeks, followed by a 1‐week tapering period and a 1‐week washout period. Therefore, each treatment cycle lasted 10 weeks in total. Patients subsequently crossed over to the next treatment period. The parallel group phase of aliskiren 300 mg monotherapy for 8 weeks fixed dose compared to placebo meets the inclusion criteria but data have not been provided for office systolic and diastolic blood pressure measurements during the first phase of treatment when patients were randomized to aliskiren monotherapy and placebo. |
| Frandesen 2008 | A randomized double blind cross‐over study meets the inclusion criteria but is available as abstract only with no details of methodology to assess risk of bias. Also the study does not provide data on SBP or DBP. |
| Gheorghiade 2013 | No aliskiren monotherapy arm. |
| Jordan 2007 | No placebo monotherapy arm. |
| Jumar 2015 | No aliskiren monotherapy arm. |
| Maser 2013 | Although this study is a double‐blind randomized placebo‐controlled study it included patients untreated for hypertension as well as those patients on antihypertensive medication other than aliskiren and whose systolic BP (SBP) was 115 mmHg to 159 mmHg and diastolic BP (DBP) was 60 mmHg to 99 mmHg at screening. Therefore all patients were not on aliskiren monotherapy. |
| Mihai 2013 | No aliskiren monotherapy arm. |
| NCT00219141 | Aliskiren in combination with losartan compared to losartan on the regression of left ventricular hypertrophy in overweight patients with essential hypertension (ALLAY). There is no parallel placebo group. |
| NCT00417170 | Comparison of aliskiren and amlodipine on insulin resistance and endothelial dysfunction in patients with hypertension and metabolic syndrome. There is no parallel placebo group. |
| NCT00654875 | This study compared the efficacy and safety of once daily dosing of aliskiren (300 mg (qd) once a day) to twice‐daily dosing of aliskiren (150 mg (twice a day) in treating moderate hypertension. There is no parallel placebo group. |
| NCT00777946 | This study evaluated the efficacy and safety of combination aliskiren/amlodipine in patients not adequately responding to aliskiren alone. There is no parallel placebo group. |
| NCT00819767 | Efficacy and safety of aliskiren in patients with mild‐to‐moderate hypertension during exercise. No details of the study are provided. |
| NCT00865020 | This study compared efficacy and safety of aliskiren 300 mg compared to telmisartan 80 mg after 1 week of treatment withdrawal (ASSERTIVE). There is no parallel placebo group. |
| NCT00927394 | This study compared aliskiren and valsartan vs valsartan alone in patients with stage II systolic hypertension and type II diabetes mellitus. There is no parallel placebo group. |
| NCT01042392 | Efficacy of aliskiren compared to ramipril in the treatment of moderate systolic hypertensive patients (ALIAS). No other detail provided. |
| NCT01138423 | Treatment of Adiposity Related hypErTension (TARGET). No details of the study are provided. |
| NCT01318395 | This is a randomized, double‐blind, active‐controlled, parallel study to analyze effects of the combination of aliskiren and valsartan on the vascular structure and function of retinal vessels. It compares aliskiren 150 mg for 1 week then 300 mg/day for 7 weeks to placebo once per day. Results have not been reported. |
| Nicholls 2013 | No aliskiren monotherapy arm. |
| Nussberger 2007 | Dose selection/different formulation of drug than one marketed used in this study. |
| O'Brien 2007 | Open‐label combination therapy. |
| Persson 2009 | It is a double‐blind randomized cross‐over study. Patients were randomized to placebo, aliskiren 300 mg or irbasartan 300 mg or combination therapy. However all patients also received furosemide in a stable dose throughout the study so it was excluded as there is no aliskiren monotherapy arm. |
| Pitt 2007 | Combination therapy study. |
| Scirica 2010 | No aliskiren monotherapy arm. |
| Shah 2012 | No aliskiren monotherapy arm. |
| Solomon 2011 | Also identified as NCT00414609. No aliskiren or placebo monotherapy arm as these drugs were added to standard therapy in patients with acute myocardial infarction. |
| Stanton 2003 | Dose selection study with no placebo control. |
| Strasser 2007 | No placebo control. |
| Teo 2014 | No aliskiren monotherapy arm. Patients were given additional antihypertensives HCTZ 25 mg or amlodipine 5 mg or respective placebos |
| Uresin 2007 | No placebo control. |
| Verdecchia 2007 | No placebo control. |
DBP: diastolic blood pressure;HTCZ: hydrochlorothiazide; SBP: systolic blood pressure
Differences between protocol and review
The previous version of this review included information from only published journal articles of included studies. However, in the 2017 update we were able to obtain nine clinical study reports (CSRs) of included studies and therefore it was possible to report change in mean sitting systolic blood pressure (MSSBP) and mean sitting diastolic blood pressure (MSDBP) with standard deviation of change from 11 studies, and required to impute missing standard deviation data from only one included study. In all cases where there was a difference between the CSR and published report, data from the CSR was used. Therefore, magnitude of blood pressure reduction and standard deviation of change data may vary from previous versions of the review.
This update includes additional information regarding mortality, non‐fatal serious adverse events, total adverse events and any other common specific adverse events (headache, nasopharyngitis, and diarrhoea). We had already included cough and angioedema as secondary outcomes in the published protocol.
We were also able to reassess the risk of bias of each individual study as detailed information was available in the CSR obtained from EMA and therefore judgement may vary from the previous version of this review. The quality of evidence has been downgraded from high quality in the 2011 update to moderate quality for BP data and to low quality for adverse event data in this 2017 update.
We also graded overall evidence using GRADEpro software and included a 'Summary of findings' table.
Contributions of authors
James Wright and Vijaya Musini formulated the idea for the review and developed the basis for the protocol.
Vijaya Musini and Patricia Fortin took the lead roles in searching, identifying, and assessing studies, in data abstraction and analyses, and in writing the review for the original version of this review in 2009 and for the 2011 update.
Vijaya Musini attended to 2017 update by performing a screen to updated search findings, data extraction from clinical study reports, analysis, carrying out a 'Risk of bias' assessment and preparing a 'Summary of findings' table, and editing/writing the review.
Kendra Lawrence attended to the 2017 update of this review by performing a screen to updated search findings, requesting and obtaining CSRs from EMA, data extraction from clinical study reports, analysis, carrying out a 'Risk of bias' assessment, and editing/writing the review.
Patricia Fortin contributed to screening, data extraction from published journal articles, and editing of the review for the 2017 update.
James Wright and Ken Bassett aided in confirming accuracy of data, settling discrepancies in inclusion criteria or data abstraction, and in editing the final version of the review.
Sources of support
Internal sources
Department of Anesthesiology, Pharmacology & Therapeutics, University of British Columbia, Canada.
External sources
Canadian Institues of Health Research, Canada.
Declarations of interest
None known.
Edited (no change to conclusions), comment added to review
References
References to studies included in this review
CSPA100A1301 {unpublished data only}
- Novartis. Efficacy and safety of SPA100 (fixed‐dose combination of aliskiren/amlodipine) in patients with essential hypertension. ClinicalTrials.gov identifier NCT01237223 May 11 2012:1‐27.
CSPA100A2305 {published data only}
- Littlejohn TW 3rd, Jones SW, Zhang J, Hsu H, Keefe DL. Efficacy and safety of aliskiren and amlodipine combination therapy in patients with hypertension: a randomized, double‐blind, multifactorial study. Journal of Human Hypertension 2013;27(5):321‐7. [DOI] [PubMed] [Google Scholar]
- Novartis. An 8‐week double‐blind, multicenter, randomized, multifactorial, placebo‐controlled, parallel group study to evaluate the efficacy and safety of aliskiren administered alone and in combination with amlodipine in patients with essential hypertension. Novartis Clinical Trial Results Database. 26th May 2010:1‐16. [Little John]
CSPP100A1201 {published data only}
- Kushiro T, Hiroshige I, Yoshihisa A, Hiromi G, Shinji T, Keefe D. Aliskiren, a novel oral renin inhibitor, provides dose‐dependent efficacy and placebo‐like tolerability in Japanese patients with hypertension. Hypertension Research ‐ Clinical & Experimental 2006;29(12):997‐1005. [DOI] [PubMed] [Google Scholar]
- Novartis. Dose‐finding study of SPP100 in essential hypertension. Clinical Study Report. European Medicines Agency October 13th 2005:10531‐665.
CSPP100A1301 {unpublished data only}
- Novartis. A randomized, double‐blind, placebo and active‐controlled, multicenter, parallel‐group study comparing SPP100 (Aliskiren) 150 mg to placebo and to Losartan 50 mg to evaluate efficacy, safety and pharmacokinetics in patients with mild to moderate essential hypertension. Clinical Study Report. European Medicines Agency January 10, 2008:1‐137.
CSPP100A2201 {published data only}
- Gradman A, Schmieder R, Lins R, Nussberger J, Chiang Y, Bedigian M. Aliskiren, a novel orally effective renin inhibitor, provides dose‐dependent antihypertensive efficacy and placebo‐like tolerability in hypertensive patients. Circulation 2005;111:1012‐8. [DOI] [PubMed] [Google Scholar]
- Novartis. A multicentre, randomized, double blind, placebo‐controlled, parallel‐group study comparing aliskiren 150 mg, 300 mg, and 600 mg and Irbesartan 150 mg in patients with mild‐to‐moderate essential hypertension. Clinical Study Report. European Medicines Agency April 1st 2004:1‐61.
- Nussberger J, Gradman AH, Schmieder RE, Lins RL, Chiang Y, Prescott MF. Plasma renin and the antihypertensive effect of the orally active renin inhibitor aliskiren in clinical hypertension. International Journal of Clinical Practice 2007;61(9):1461‐1468. [DOI] [PubMed] [Google Scholar]
CSPP100A2203 {published data only}
- Novartis. A randomized, double blind, multicentre, multifactorial, placebo‐controlled, parallel‐group study to confirm the efficacy and safety of aliskiren monotherapy, and evaluate efficacy and safety of combinations of aliskiren and valsartan in hypertensive patients. Clinical Study Report. European Medicines Agency June 8th 2005:1547‐1634.
- Pool J, Roland E, Schmieder M, Aldigier JC, Januszewicz A, Zidek W, Chiang Y, Satlin A. Aliskiren, an orally effective renin inhibitor, provides antihypertensive efficacy alone and in combination with valsartan. American Journal of Hypertension 2007;20(1):11‐20. [DOI] [PubMed] [Google Scholar]
CSPP100A2204 {published data only}
- Novartis. An 8 week, double‐blind, multicenter, randomized, multifactorial, placebo‐controlled, parallel group,study to evaluate the efficacy and safety of aliskiren administered alone and in combination with hydrochlorothiazide in patients with essential hypertension. Clinical Study Report. European Medicines Agency October 25th 2005:3354‐3454.
- Villamil A, Chrysant S, Calhoun D, Schober B, Hsu H, Matrisciano‐Dimichino L, Zhang J. Renin inhibition with aliskiren provides additive antihypertensive efficacy when used in combination with hydrochlorothiazide. Journal of hypertension 2007;25(1):217‐226. [DOI] [PubMed] [Google Scholar]
CSPP100A2308 {published data only}
- Novartis. An 8 week, randomized, double‐blind, placebo‐controlled, parallel group, multicenter, study comparing aliskiren 150 mg, 300 mg, and 600 mg to placebo in patients with essential hypertension. Clinical Study Report. European Medicines Agency November 1st 2005:8458‐543.
- Oh B, Mitchell J, Herron J, Chung J, Khan Ml, Keefe D. Aliskiren, an oral renin inhibitor, provides dose‐dependent efficacy and sustained 24‐hour blood pressure control in patients with hypertension. Journal of the Amarican College of Cardiology 2007;49(11):1157‐63. [DOI] [PubMed] [Google Scholar]
CSPP100A2323 {published data only}
- Novartis. A twenty six‐week, randomized, double‐blind, parallel group, multicenter, active‐controlled, dose titration study to evaluate the efficacy and safety of aliskiren compared to HCTZ with the optional addition of amlodipine, followed by a second twenty six weeks of blinded treatment, in patients with essential hypertension. Clinical Study Report. European Medicines Agency May 16th 2006:28012‐9161.
- Schmieder RE, Philipp T, Guerediaga J, Gorostidi M, Smith B, Weissbach N, et. al. Long‐term antihypertensive efficacy and safety of the Oral direct renin Inhibitor aliskiren. Circulation 2009;119:417‐25. [DOI] [PubMed] [Google Scholar]
CSPP100A2327 {published data only}
- Novartis. An eight‐week randomized, double‐blind, placebo‐controlled, parallel‐group, multicenter study comparing aliskiren 150 mg, 300 mg, and 600 mg to placebo in patients with essential hypertension. Clinical Study Report. European Medicines Agency September 2006:17604‐20246.
- Oparil S, Yarrows S, Patel SF, Ang H, Zhang J, Satlin A. Efficacy and safety of combined use of aliskiren and valsartan in patients with hypertension: a randomised, double‐blind trial. Lancet 2007;370(9583):221‐9. [DOI] [PubMed] [Google Scholar]
CSPP100A2328 {published data only}
- Novartis. A randomized, double‐blind, placebo‐controlled, parallel‐group, multicenter study comparing an eight‐week treatment of aliskiren 75 mg, 150 mg and 300 mg to placebo in patients with essential hypertension. Novartis Clinical Trials Results Database November 4th 2009:1‐14.
- Puig JG, Schunkert H, Taylor AA, Boye S, Jin J, Keefe DL. Evaluation of the dose‐response relationship of aliskiren, a direct renin inhibitor in an 8‐week, multicenter, randomized, double‐blind, parallel‐group, placebo‐controlled study in adult patients with stage 1 or 2 essential hypertension. Clinical Therapeutics 2009;31(12):2839‐50. [DOI] [PubMed] [Google Scholar]
CSPP100A2405 {published data only}
- Novartis. An eight‐week double‐blind, multi‐center, randomized, placebo‐controlled, parallel‐group study to evaluate the efficacy and safety of aliskiren 75 mg, 150 mg and 300 mg in elderly patients with essential hypertension when given with a light meal. Clinical Study Report. European Medicines Agency. Date not reported:1‐100.
- Villa G, Breton S, Ibram G, Keefe D. Efficacy, safety and tolerability of Aliskiren monotherapy administered with a light meal in elderly hypertensive patients: A randomized, double blind, placebo‐controlled, dose‐response evaluation study. Journal of Clinical Pharmacology 2012;52:1901‐11. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Andersen 2008 {published data only}
- Andersen K, Weinberger MH, Egan B, Constance CM, Ali MA, Jin J, et al. Comparative efficacy and safety of aliskiren, an oral direct renin inhibitor, and ramipril in hypertension: A 6‐month, randomized, double‐blind trial. Journal of Hypertension 2008;3:589‐92. [DOI] [PubMed] [Google Scholar]
Dorresteijn 2013 {published data only}
- Dorresteijn JA, Schrover IM, Visseren FL, Scheffer PG, Oey PL, Danser AH, et al. Differential effects of renin‐angiotensin‐aldosterone system inhibition, sympathoinhibition and diuretic therapy on endothelial function and blood pressure in obesity‐related hypertension: a double‐blind, placebo‐controlled cross‐over trial. Journal of Hypertension 2013;31(2):393‐403. [DOI] [PubMed] [Google Scholar]
Frandesen 2008 {published data only}
- Frandsen E, Persson F, Boomsma F, Prescott M, Dole WP, Rossing P, et al. The effect of direct renin inhibition alone or in combination with angiotensin II receptor blockade on markers of the renin‐angiotension aldosterone system. Diabetologica 2008;51(Suppl 1):S 491. [Google Scholar]
Gheorghiade 2013 {published data only}
- Gheorghiade M, Bohm M, Greene SJ, Fonarow GC, Lewis EF, Zannad F, et al. ASTRONAUT Investigators and Coordinators. Effect of aliskiren on postdischarge mortality and heart failure readmissions among patients hospitalized for heart failure: the ASTRONAUT randomized trial.. JAMA 2013 March 20;309(11):1125‐35. [DOI] [PubMed] [Google Scholar]
Jordan 2007 {published data only}
- Jordan J, Engeli S, Boye SW, Breton S, Keefe DL. Direct Renin inhibition with aliskiren in obese patients with arterial hypertension. Hypertension 2007;49(5):1047‐55. [DOI] [PubMed] [Google Scholar]
Jumar 2015 {published data only}
- Jumar A, Ott C, Kistner I, Friedrich S, Schmidt S, Harazny, JM, et al. Effect of aliskiren on vascular remodelling in small retinal circulation. Journal of Hypertension 2015;33(12):2491‐9. [DOI] [PubMed] [Google Scholar]
Maser 2013 {published data only}
- Maser RE, Lenhard MJ, Kolm P, Edwards DG. Direct renin inhibition improves parasympathetic function in diabetes. Diabetes, Obesity and Metabolism 2013;15:28‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mihai 2013 {published data only}
- Mihai G, Varghese J, Kampfrath T, Gushchina L, Hafer L, Deiuliis, et al. Aliskiren effect on plaque progression in established atherosclerosis using high resolution 3D MRI (ALPINE): a double‐blind placebo‐controlled trial. Journal of the American Heart Association 2013 June;2(3):e004879. [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT00219141 {published data only}
- Novartis. Aliskiren in combination with losartan compared to losartan on the regression of left ventricular hypertrophy in overweight patients with essential hypertension (ALLAY). ClinicalTrials.gov identifier NCT00219141 May 2011:1‐31.
NCT00417170 {published data only}
- Novartis. Comparison of aliskiren and amlodipine on insulin resistance and endothelial dysfunction in patients with hypertension and metabolic syndrome. ClinicalTrials.gov Identifier NCT00417170 September 2011:1‐14.
NCT00654875 {published data only}
- Novartis NCT00654875. Efficacy and safety of once daily dosing of aliskiren (300 mg (qd) once a day) to twice daily dosing of aliskiren (150 mg (bid) twice a day) in treating moderate hypertension. ClinicalTrials.gov Identifier NCT00654875 June 2011:1‐18.
NCT00777946 {published data only}
- Novartis. Study to evaluate the efficacy and safety of combination aliskiren/amlodipine in patients not adequately responding to aliskiren alone. ClinicalTrials.gov Identifier NCT00777946 June 2011:1‐18.
NCT00819767 {published data only}
- Novartis. Efficacy and safety of aliskiren in patients with mild to moderate hypertension during exercise. ClinicalTrials.gov identifier NCT00819767 January 2009:1‐13.
NCT00865020 {published data only}
- Novartis. Efficacy and safety of aliskiren 300 mg compared to telmisartan 80 mg after 1 week of treatment withdrawal (ASSERTIVE). ClinicalTrials.gov Identifier NCT00865020 June 2011:1‐17.
NCT00927394 {published data only}
- Novartis. Aliskiren and valsartan vs valsartan alone in patients with stage ii systolic hypertension and type ii diabetes mellitus. ClinicalTrials.gov Identifier NCT00927394 December 2012:1‐29.
NCT01042392 {published data only}
- Novartis. Efficacy of aliskiren compared to ramipril in the treatment of moderate systolic hypertensive patients (ALIAS). ClinicalTrials.gov Identifier NCT01042392 March 2012:1‐26.
NCT01138423 {published data only}
- Novartis. Treatment of Adiposity Related hypErTension (TARGET). ClinicalTrials.gov Identifier NCT01138423 February 2012.
NCT01318395 {published data only}
- Novartis. Randomized, double blind, active‐controlled, parallel study to analyse effects of the combination of aliskiren and valsartan on the vascular structure and function of retinal vessels. ClinicalTrials.gov Identifier NCT01318395 June 2014.
Nicholls 2013 {published data only}
- Nicholls SJ, Bakris GL, Kastelein JJ, Menon V, Williams B, Armbrecht J, et al. Effect of aliskiren on progression of coronary disease in patients with prehypertension: the AQUARIUS randomized clinical trial. JAMA 2013 September 18.;310(11):1135‐44. [DOI] [PubMed] [Google Scholar]
Nussberger 2007 {published data only}
- Nussberger J, Gradman AH, Schmieder RE, LinsRL, Chiang Y, Prescott MF. Plasma renin and the antihypertensive effect of the orally active renin inhibitor aliskiren in clinical hypertension. International Journal ofClinical Practice 2007;61(9):1461‐8. [DOI] [PubMed] [Google Scholar]
O'Brien 2007 {published data only}
- O'Brien E, Barton J, Nussberger J, Mulcahy D, Jensen C, Dicker P, et al. Aliskiren reduces blood pressure and suppresses plasma renin activity in combination with a thiazide diuretic, an angiotensin‐converting enzyme inhibitor, or an angiotensin receptor blocker. Hypertension 2007;49(2):276‐84. [DOI] [PubMed] [Google Scholar]
Persson 2009 {published data only}
- Novartis. Study to assess the optimal renoprotective dose of aliskiren in hypertensive patients with type 2 diabetes and incipient or overt nephropathy. ClinicalTrials.gov identifier NCT00464776 April 2007.
- Persson F, Rossing P, Reinhard H, Juhl T, Stehouwer CDA, Schalkwijk C, et al. Renal effects of aliskiren compared with and in combination with irbesartan in patients with type 2 diabetes, hypertension, and albuminuria. Diabetes Care 2009;10(32):1873‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pitt 2007 {published data only}
- Pitt B, McMurray JJ, Latini R, Maggioni A, Solomon SD, Smith BA, et al. Neurohumoral effects of a new oral direct renin inhibitor in stable heart failure: The Aliskiren Observation of Heart Failure Treatment study (ALOFT. Circulation 2007;116(16 Suppl):S. [Google Scholar]
Scirica 2010 {published data only}
- Scirica BM, Morrow DA, Bode C, Ruzyllo W, Ruda M, Oude Ophuis AJ, et al. Patients with acute coronary syndromes and elevated levels of natriuretic peptides: the results of the AVANT GARDE‐TIMI 43 Trial. European Heart Journal 2010 August;31(16):1993‐2005. [DOI] [PubMed] [Google Scholar]
Shah 2012 {published data only}
- Shah AM, Shin SH, Takeuchi M, Skali H, Desai AS, Kober L, et al. Left ventricular systolic and diastolic function, remodelling, and clinical outcomes among patients with diabetes following myocardial infarction and the influence of direct renin inhibition with aliskiren. European Journal of Heart Failure 2012, February;142(2):185‐92. [DOI] [PubMed] [Google Scholar]
Solomon 2011 {published data only}
- Solomon SD, Shin SH, Shah A, Skali H, Desai A, Kober L, et al. Effect of the direct renin inhibitor aliskiren on left ventricular remodelling following myocardial infarction with systolic dysfunction. European Heart Journal 2011;32(10):1227‐34. [DOI] [PubMed] [Google Scholar]
Stanton 2003 {published data only}
- Stanton A, Jensen C, Nussberger J, O'Brien E. Blood pressure lowering in essential hypertension with an oral renin inhibitor, aliskiren. Hypertension 2003;42(6):1137‐43. [DOI] [PubMed] [Google Scholar]
Strasser 2007 {published data only}
- Strasser RH, Puig JG, Farsang C, Croket M, Li J, Ingen H. A comparison of the tolerability of the direct renin inhibitor aliskiren and lisinopril in patients with severe hypertension. Journal of Human Hypertension 2007;21(10):780‐7. [DOI] [PubMed] [Google Scholar]
Teo 2014 {published data only}
- Teo KK, Pfeffer M, Mancia G, O'Donnell M, Dagenais G, Diaz R, et al. Aliskiren Prevention of Later Life Outcomes trial Investigators. Aliskiren alone or with other antihypertensives in the elderly with borderline and stage 1 hypertension: the APOLLO trial. European Heart Journal 2014;35(26):1743‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Uresin 2007 {published data only}
- Uresin Y, Taylor AA, Kilo C, Tschope D, Santonastaso M, Ibram G, et al. Efficacy and safety of the direct renin inhibitor aliskiren and ramipril alone or in combination in patients with diabetes and hypertension. Journal of the Renin‐Angiotensin‐Aldosterone System 2007;8(4)(4):190‐8. [DOI] [PubMed] [Google Scholar]
Verdecchia 2007 {published data only}
- Verdecchia P, Calvo C, Mockel V, Keeling L, Satlin A. Safety and efficacy of the oral direct renin inhibitor aliskiren in elderly patients with hypertension. Blood Pressure 2007;16(6):381‐91. [DOI] [PubMed] [Google Scholar]
Additional references
ALTITUDE 2012
- Parving HH, Brenner BM, McMurray JH, Zeeuw D, Haffner SM, Solomon, et al. ALTITUDE Investigators. Cardiorenal end points in a trial of aliskiren for type 2 diabetes. New England Journal of Medicine 2012;367:2204‐13. [DOI] [PubMed] [Google Scholar]
AQUARIUS 2013
- Nicholls SJ, Bakris GL, Kastelein JP, et al. Effect of Aliskiren on Progression of Coronary Disease in Patients With PrehypertensionThe AQUARIUS Randomized Clinical Trial. JAMA 2013;310(11):1135‐44. [DOI] [PubMed] [Google Scholar]
ASPIRE 2011
- Solomon SD, Shin SH, Shah A, Skali H, Desai A, Kober L, Maggioni AP, Rouleau JL, Kelly RY, Hester A, McMurray JJ, Pfeffer MA. Aliskiren Study in Post‐MI Patients to Reduce Remodeling (ASPIRE) Investigators. European Heart Journal May 2011;32:1227‐34. [DOI] [PubMed] [Google Scholar]
ASTRONAUT 2013
- Gheorghiade M, Böhm M, Greene SJ, Fonarow GC, Lewis EF, Zannad F, Solomon SD, Baschiera F, Botha J, Hua TA, Gimpelewicz CR, Jaumont X, Lesogor A, Maggioni AP, ASTRONAUT Investigators and Coordinators. Effect of aliskiren on postdischarge mortality and heart failure readmissions among patients hospitalized for heart failure: the ASTRONAUT randomized trial. JAMA March 20 2013;309(11):1125‐35. [DOI] [PubMed] [Google Scholar]
AVANT GARDE ‐ TIMI 43 2010
- Scirica BM, Morrow DA, Bode C, Ruzyllo W, Ruda M, Oude Ophuis AJ, Lopez‐Sendon J, Swedberg K, Ogorek M, Rifai N, Lukashevich V, Maboudian M, Cannon CP, McCabe CH, Braunwald E. Patients with acute coronary syndromes and elevated levels of natriuretic peptides: the results of the AVANT GARDE‐TIMI 43 Trial. European Heart Journal 2010;31(16):1993‐2005. [DOI] [PubMed] [Google Scholar]
AVOID 2011
- Persson F, Lewis JB, Lewis EJ, Rossing P, Hollenberg NK, Parving HH. Aliskiren in combination with losartan reduces albuminuria independent of baseline blood pressure in patients with type 2 diabetes and nephropathy. Clin J Am Soc Nephrol May 2011;6(5):1025‐31. [DOI: 10.2215/CJN.07590810] [DOI] [PMC free article] [PubMed] [Google Scholar]
Chalmers 1990
- Chalmers I. Underreporting research is scientific misconduct. JAMA 1990;263(10):1405‐8. [PubMed] [Google Scholar]
Chen 2013
- Chen Y, Meng L, Shao H, Feng Yu. Aliskiren vs. other antihypertensive drugs in the treatment of hypertension: a meta‐analysis. Hypertension Research 2013;36:252‐61. [DOI] [PubMed] [Google Scholar]
Doshi 2012
- Doshi P, Jefferson T, Mar C. The imperative to share clinical study reports: recommendations from the Tamiflu experience. PLoS Medicine 2012;9(4):1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Doshi 2016
- Doshi P, Jefferson T. Open data 5 years on: a case series of 12 freedom of information requests for regulatory data to the European Medicines Agency. Trials 2016;17(78):1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Duprez 2006
- Duprez D. Role of the renin–angiotensin–aldosterone system in vascular remodeling and inflammation: a clinical review. Journal of Hypertension 2006;24:983‐91. [DOI] [PubMed] [Google Scholar]
Dwan 2013
- Dwan K, Gamble C, Williamson PR, Kirkham JJ, for the Reporting Bias Group. Systematic review of the empirical evidence of study publication bias and outcome reporting bias ‐ an updated review. PloS One 2013;8(7):1‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
Eyding 2010
- Eyding D, Lelgemann M, Grouven U, Harter M, Kromp M, Kaiser T, et al. Reboxetine for acute treatment of major depression: systematic review and meta‐analysis of published and unpublished placebo and selective serotonin reuptake inhibitor controlled trials. BMJ 2010;341:816. [DOI] [PMC free article] [PubMed] [Google Scholar]
FDA Medical Review 2007
- Center for drug evaluation and research. Tekturna (Aliskiren) Medical Review. FDA database March 5th 2007, issue Application number 21‐985:1‐283.
Frishman 1994
- Frishman WH, Fozailoff A, Lin C, Dike C. Renin inhibition: a new approach to cardiovascular therapy. Journal of Clinical Pharmacology 1994;34:873‐80. [DOI] [PubMed] [Google Scholar]
Gotzsche 2011
- Gotzsche P, Jorgenson AW. Opening up data at the European Medicines Agency. BMJ 2011;343:1184‐6. [DOI] [PubMed] [Google Scholar]
Gradman 2005
- Gradman A, Schmieder R, Lins R, Nussberger J, Chiang Y, Bedigian M. Aliskiren, a novel orally effective renin inhibitor, provides dose‐dependent antihypertensive efficacy and placebo‐like tolerability in hypertensive patients. Circulation 2005;111:1012‐8. [DOI] [PubMed] [Google Scholar]
Gradman 2010
- Gradman AH, Weir MR, Wright M, Bush CA, Keefe DL. Efficacy, safety and tolerability of aliskiren, a direct renin inhibitor, in women with hypertension: a pooled analysis of eight studies. Journal of Human Hypertension 2010;24:721‐9. [DOI] [PubMed] [Google Scholar]
Harel 2012
- Harel Z, Gilbert C, Wald R, Bell C, Perl J, Juurlink D, et al. The effect of combination treatment with aliskiren and blockers of the renin‐angiotensin system on hyperkalaemia and acute kidney injury: systematic review and meta‐analysis. BMJ 2012;344(e42):1‐13. [DOI: 10.1136/bmj.e42] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hart 2012
- Hart 2012. Effect of reporting bias on meta‐analyses of drug trials reanalysis of meta‐analyses. BMJ 2012;344:13. [DOI] [PubMed] [Google Scholar]
Helmer 1958
- Helmer OM. Studies on renin antibodies. Circulation 1958;17(4 part 2):648‐52. [DOI] [PubMed] [Google Scholar]
Heran 2008a
- Heran BS, Wong MMY, Heran IK, Wright JM. Dose‐related blood pressure lowering efficacy of angiotensin converting enzyme (ACE) inhibitors in the treatment of primary hypertension. Cochrane Database of Systematic Reviews 2008, Issue 4. [DOI: 10.1002/14651858.CD003823] [DOI] [PMC free article] [PubMed] [Google Scholar]
Heran 2008b
- Heran BS, Wong MMY, Heran IK, Wright JM. Blood pressure lowering efficacy of angiotensin receptor blockers for primary hypertension. Cochrane Database of Systematic Reviews 2008, Issue 4. [DOI: 10.1002/14651858.CD003822.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.10 [updated March 2011]. Available from www.cochrane‐handbook.org: The Cochrane Collaboration, 2011. [Google Scholar]
Hodkinson 2013
- Hodkinson A, Kirkham J, Tudur‐Smith C, Gamble C. Reporting of harms data in RCTs: a systematic review of empirical assessments against the CONSORT harms extension. BMJ 2013;3:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hodkinson 2016
- Hodkinson A, Gamble C, Smith CT. Reporting of harms outcomes: a comparison of journal publications with unpublished clinical study reports of orlistat trials. Trials 2016;17(1):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jefferson 2014
- Jefferson T, Jones MA, Doshi P, Mar CB, Hama R, Thompson MJ, et al. Neuraminidase inhibitors for preventing and treating influenza in adults and children. Cochrane Database of Systematic Reviews 2014, Issue 4. [DOI: 10.1002/14651858.CD008965.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jefferson 2015
- Jefferson T. Facing the unreliability of clinical trials literature. Drug and Therapeutics Bulletin of Navarre, Spain 2015;22(2):1‐11. [Google Scholar]
Kirkham 2010
- Kirkham JJ, Dwan KM, Altman DG, Gamble C, Dodd S, Smyth R, et al. The impact of outcome reporting bias in randomised controlled trials on a cohort of systematic reviews. BMJ 2010;340:637‐40. [DOI] [PubMed] [Google Scholar]
Krum 2007
- Krum H, Gilbert RE. Novel therapies blocking the renin‐angiotensin‐aldosterone system in the management of hypertension and related disorders. Journal of Hypertension 2007;25:25‐35. [DOI] [PubMed] [Google Scholar]
Kushiro 2006
- Kushiro T, Hiroshige I, Yoshihisa A, Hiromi G, Shinji T, Keefe D. Aliskiren, a novel oral renin inhibitor, provides dose‐dependent efficacy and placebo‐like tolerability in Japanese patients with hypertension. Hypertension Research ‐ Clinical & Experimental 2006;29(12):997‐1005. [DOI] [PubMed] [Google Scholar]
Law 2003
- Law MR, Wald NJ, Morris JK, Jordan RE. Value of low dose combination treatment with blood pressure lowering drugs: analysis of 354 randomised trials. BMJ 2003;326(7404):1427. [PUBMED: 12829555] [DOI] [PMC free article] [PubMed] [Google Scholar]
Littlejohn 2013
- Littlejohn TW 3rd, Jones SW, Zhang J, Hsu H, Keefe DL. Efficacy and safety of aliskiren and amlodipine combination therapy in patients with hypertension: a randomized, double‐blind, multifactorial study. Journal of Human Hypertension 2013;27(5):321‐7. [DOI] [PubMed] [Google Scholar]
Makani 2013
- Makani H, Bangalore S, Desouza K, Shah A, Messerli F. Efficacy and safety of dual blockade of the renin‐angiotensin system: meta‐analysis of randomised trials. BMJ 2013;346(f360):1‐15. [DOI: 10.1136/bmj.f360] [DOI] [PMC free article] [PubMed] [Google Scholar]
Maund 2014
- Maund E, Tendal B, Hrobjartsson A, Jorgenson K, Lundh A, Schroll J, et al. Benefits and harms in clinical trials of duloxetine for treatment of major depressive disorder: comparison of clinical study reports, trial registries, and publications. BMJ 2014;348:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
McGauran 2010
- McGauran N, Wieseler B, Kreis J, Schuler YB, Kolsch H, Kaiser T. Reporting bias in medical research ‐ a narrative review. Trials 2010;11:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Musini 2009
- Musini VM, Wright JM. Factors affecting blood pressure variability: lessons learned from two systematic reviews of randomized controlled trials. PLoS One 2009;4(5):e5673. [DOI: 10.1371/journal.pone.0005673] [DOI] [PMC free article] [PubMed] [Google Scholar]
Novartis Clinical Trial Results Database
- Novartis. Novartis Clinical Trials Database https://www.novartisclinicaltrials.com/TrialConnectWeb/home.nov.
Oh 2007
- Oh B, Mitchell J, Herron J, Chung J, Khan Ml, Keefe D. Aliskiren, an oral renin inhibitor, provides dose‐dependent efficacy and sustained 24‐hour blood pressure control in patients with hypertension. Journal of the Amarican College of Cardiology 2007;49(11):1157‐63. [DOI] [PubMed] [Google Scholar]
Oparil 2007
- Oparil S, Yarrows S, Patel SF, Ang H, Zhang J, Satlin A. Efficacy and safety of combined use of aliskiren and valsartan in patients with hypertension: a randomised, double‐blind trial. Lancet 2007;370(9583):221‐9. [DOI] [PubMed] [Google Scholar]
Pool 2007
- Pool J, Roland E, Schmieder M, Aldigier JC, Januszewicz A, Zidek W, Chiang Y, Satlin A. Aliskiren, an orally effective renin inhibitor, provides antihypertensive efficacy alone and in combination with valsartan. American Journal of Hypertension 2007;20(1):11‐20. [DOI] [PubMed] [Google Scholar]
Poulter 2015
- Poulter NR, Prabhakaran D, Caulfield M. Hypertension. Lancet 22nd August 2015;386(9995):801‐12. [DOI] [PubMed] [Google Scholar]
Powers 2011
- Powers B, Greene L, Balfe LM. Updates on the treatment of essential hypertension: a summary of AHRQ's comparativeeffectiveness review of angiotensin‐converting enzyme inhibitors, angiotensin II receptorblockers, and direct renin inhibitors. Journal of Managed Care Pharmacy October 2011;17(8):Supplement S1‐S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Puig 2009
- Puig JG, Schunkert H, Taylor AA, Boye S, Jin J, Keefe DL. Evaluation of the dose‐response relationship of aliskiren, a direct renin inhibitor in an 8‐week, multicenter, randomized, double‐blind, parallel‐group, placebo‐controlled study in adult patients with stage 1 or 2 essential hypertension. Clinical Therapeutics 2009;31(12):2839‐50. [DOI] [PubMed] [Google Scholar]
Rising 2008
- Rising K, Bacchett P, Bero L. Reporting bias in drug trials submitted to the Food and Drug Administration: review of publication and presentation. PLoS Medicine 2008;5(11):1561‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schmieder 2009
- Schmieder RE, Philipp T, Guerediaga J, Gorostidi M, Smith B, Weissbach N, et. al. Long‐term antihypertensive efficacy and safety of the Oral direct renin Inhibitor aliskiren. Circulation 2009;119:417‐25. [DOI] [PubMed] [Google Scholar]
Schünemann 2011
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks Jj, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0 (updated March 2011). Available from www.cochrane‐handbook.org: The Cochrane Collaboration, 2011. [Google Scholar]
Sharma 2016
- Sharma T, Guski LS, Freund N, Gøtzsche P. Suicidality and aggression during antidepressant treatment: systematic review and meta‐analyses based on clinical study reports. BMJ 2016;352(65):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
U.S. Food and Drug Administration 2016
- FDA Drug Safety Communication: New Warning and Contraindication for blood pressure medicines containing aliskiren (Tekturna). http://www.fda.gov/Drugs/DrugSafety/ucm300889.htm#data Last updated on19th January 2016.
Villa 2012
- Villa G, Breton S, Ibram G, Keefe D. Efficacy, safety and tolerability of Aliskiren monotherapy administered with a light meal in elderly hypertensive patients: A randomized, double blind, placebo‐controlled, dose‐response evaluation study. Journal of Clinical Pharmacology 2012;52:1901‐11. [DOI] [PubMed] [Google Scholar]
Villamil 2007
- Villamil A, Chrysant S, Calhoun D, Schober B, Hsu H, Matrisciano‐Dimichino L, Zhang J. Renin inhibition with aliskiren provides additive antihypertensive efficacy when used in combination with hydrochlorothiazide. Journal of hypertension 2007;25(1):217‐226. [DOI] [PubMed] [Google Scholar]
Weir 2007
- Weir MR, Bush C, Anderson DR, Zhang J, Keefe D, Satlin A. Antihypertensive efficacy, safety, and tolerability of the oral direct renin inhibitor aliskiren in patients with hypertension: a pooled analysis. Journal of the American Society of Hypertension 2007;1(4):264‐77. [DOI] [PubMed] [Google Scholar]
White 2010
- White WB, Bresalier R, Kaplan AP, Palmer BF, Riddell RH, Lesogor A, et al. Safety and tolerability of the direct renin Inhibitor Aliskiren: A pooled analysis of clinical experience in more than 12,000 patients with hypertension. Journal of Clinical Hypertension 2010;12(10):765‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wieseler 2012
- Wieseler B, Chrysant SG, Calhoun D, Schober B, Hsu H, Matrisciano‐Dimichino L, et al. Impact of document type on reporting ality of clinical drug trials: a comparison of registry reports, clinical study reports, and journal publications. BMJ 2012;344:1‐11. [DOI] [PubMed] [Google Scholar]
Wright 2009
- Wright JM, Musini VM. First‐line drugs for hypertension. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD001841.pub2] [DOI] [PubMed] [Google Scholar]
Zhang 2015
- Zhang Jing‐Tao, Chen Ke‐Ping, Guan Ting, Zhang Shu. Effect of aliskiren on cardiovascular outcomes in patients with pre ‐hypertension: a meta‐analysis of randomized controlled trials. Drug Design, Development and Therapy 2015;9:1963‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Musini 2009
- Musini VM, Fortin PM, Bassett K, Wright JM. Blood pressure lowering efficacy of renin inhibitors for primary hypertension:a Cochrane systematic review. Journal of Human Hypertension 2009;23(8):495‐502. [DOI] [PubMed] [Google Scholar]
Musini 2008a
- Musini VM, Fortin PM, Bassett K, Wright JM. Blood pressure lowering efficacy of renin inhibitors for primary hypertension. Cochrane Database of Systematic Reviews 2008, Issue 2. [DOI: 10.1002/14651858.CD007066] [DOI] [PubMed] [Google Scholar]
Musini 2008b
- Musini VM, Fortin PM, Bassett K, Wright JM. Blood pressure lowering efficacy of renin inhibitors for primary hypertension. Cochrane Database of Systematic Reviews 2008, Issue 4. [DOI: 10.1002/14651858.CD007066.pub2] [DOI] [PubMed] [Google Scholar]
Musini 2011
- Musini VM, Fortin PM, Bassett K, Wright JM. Blood pressure lowering efficacy of renin inhibitors for primary hypertension. Cochrane Database of Systematic Reviews 2011, Issue 9. [DOI: 10.1002/14651858.CD007066.pub2] [DOI] [PubMed] [Google Scholar]