

Abstract
Background
Sickle cell disease (SCD) is one of the most common inherited diseases worldwide. It is associated with lifelong morbidity and a reduced life expectancy. Hydroxyurea (hydroxycarbamide), an oral chemotherapeutic drug, ameliorates some of the clinical problems of SCD, in particular that of pain, by raising fetal haemoglobin. This is an update of a previously published Cochrane Review.
Objectives
To assess the effects of hydroxyurea therapy in people with SCD (all genotypes), of any age, regardless of setting.
Search methods
We searched the Cochrane Cystic Fibrosis and Genetic Disorders Group Haemoglobinopathies Register, comprising of references identified from comprehensive electronic database searches and handsearches of relevant journals and abstract books of conference proceedings. We also searched online trial registries.
Date of the most recent search: 16 January 2017.
Selection criteria
Randomised and quasi‐randomised controlled trials, of one month or longer, comparing hydroxyurea with placebo, standard therapy or other interventions for people with SCD.
Data collection and analysis
Authors independently assessed studies for inclusion, carried out data extraction and assessed the risk of bias.
Main results
Seventeen studies were identified in the searches; eight randomised controlled trials were included, recruiting 899 adults and children with SCD (haemoglobin SS (HbSS), haemoglobin SC (HbSC) or haemoglobin Sβºthalassaemia (HbSβºthal) genotypes). Studies lasted from six to 30 months.
Four studies (577 adults and children with HbSS or HbSβºthal) compared hydroxyurea to placebo; three recruited individuals with only severe disease and one recruited individuals with all disease severities. There were statistically significant improvements in terms of pain alteration (using measures such as pain crisis frequency, duration, intensity, hospital admissions and opoid use), measures of fetal haemoglobin and neutrophil counts and fewer occurrences of acute chest syndrome and blood transfusions in the hydroxyurea groups. There were no consistent statistically significant differences in terms of quality of life and adverse events (including serious or life‐threatening events). Seven deaths occurred during the studies, but the rates by treatment group were not statistically significantly different.
Two studies (254 children with HbSS or HbSβºthal also with risk of primary or secondary stroke) compared hydroxyurea and phlebotomy to transfusion and chelation; there were statistically significant improvements in terms of measures of fetal haemoglobin and neutrophil counts, but more occurrences of acute chest syndrome and infections in the hydroxyurea and phlebotomy group. There were no consistent statistically significant differences in terms of pain alteration and adverse events (including serious or life‐threatening events). Two deaths occurred during the studies (one in a the hydroxyurea treatment arm and one in the control arm), but the rates by treatment group were not statistically significantly different. In the primary prevention study, no strokes occurred in either treatment group but in the secondary prevention study, seven strokes occurred in the hydroxyurea and phlebotomy group (none in the transfusion and chelation group) and the study was terminated early.
The quality of the evidence for the above two comparisons was judged as moderate to low as the studies contributing to these comparisons were mostly large and well designed (and at low risk of bias); however evidence was limited and imprecise for some outcomes such as quality of life, deaths during the studies and adverse events and results are applicable only to individuals with HbSS and HbSβºthal genotypes.
Of the remaining two studies, one (22 children with HbSS or HbSβºthal also at risk of stoke) compared hydroxyurea to observation; there were statistically significant improvements in terms of measures of fetal haemoglobin and neutrophil counts but no statistically significant differences in terms of adverse events (including serious or life‐threatening events).
The final study (44 adults and children with HbSC) compared treatment regimens with and without hydroxyurea – there was statistically significant improvement in terms of measures of fetal haemoglobin, but no statistically significant differences in terms of adverse events (including serious or life‐threatening events). No participants died in either of these studies and other outcomes relevant to the review were not reported.
The quality of the evidence for the above two comparisons was judged to be very low due to the limited number of participants, the lack of statistical power (as both studies were terminated early with approximately only 20% of their target sample size recruited) and the lack of applicability to all age groups and genotypes.
Authors' conclusions
There is evidence to suggest that hydroxyurea is effective in decreasing the frequency of pain episodes and other acute complications in adults and children with sickle cell anaemia of HbSS or HbSβºthal genotypes and in preventing life‐threatening neurological events in those with sickle cell anaemia at risk of primary stroke by maintaining transcranial doppler velocities. However, there is still insufficient evidence on the long‐term benefits of hydroxyurea, particularly in preventing chronic complications of SCD, recommending a standard dose or dose escalation to maximum tolerated dose. There is also insufficient evidence about the long‐term risks of hydroxyurea, including its effects on fertility and reproduction. Evidence is also limited on the effects of hydroxyurea on individuals with HbSC genotype. Future studies should be designed to address such uncertainties.
Plain language summary
Hydroxyurea (also known as hydroxycarbamide) for people with sickle cell disease
Review question
What is the effect of hydroxyurea on clinical outcomes (changes in pain crises, life‐threatening illnesses, survival, haemoglobin levels, quality of life and side effects) in people with sickle cell disease (SCD) of any genotype?
Background
SCD is an inherited genetic disorder that creates problems with haemoglobin (the substance in red blood cells that carries oxygen around the body). The disease can be inherited in different ways; people can inherit two sickle genes (HbSS genotype) or they can inherit the sickle gene from one parent and a different haemoglobin gene (such as haemoglobin C (HbSC genotype) or a beta thalassaemia gene (HbSβ+ or HbSβºthal genotype)) from the second parent.
In people with SCD the abnormal sickle haemoglobin forms long polymers (chains) within the red blood cells when they become de‐oxygenated. This damages the red blood cells and makes them stickier, leading to blockages and reduced blood flow, causing pain and organ damage. Fetal haemoglobin stops these polymers forming in the sickle haemoglobin within the red blood cell. The drug hydroxyurea is used to raise fetal haemoglobin and can reduce the effects of the disease. This is an update of a previously published Cochrane Review.
Search date
The evidence is current to 16 January 2017.
Study characteristics
We included eight studies (899 adults and children with SCD (HbSS, HbSC or HbSβºthal genotypes)). Studies lasted from six to 30 months.
Key results and quality of the evidence
In four studies, 577 adults and children with SCD were randomly selected to receive hydroxyurea or placebo. In two studies, 254 children with SCD, who were also at an increased risk of having a first or second stroke, were randomly selected to receive hydroxyurea and phlebotomy (collection of blood) or blood transfusion and chelation (administration of agents to remove excess iron from the body). These six studies only recruited people with HbSS or HbSβºthal genotypes so results do not apply to people with the HbSC genotype.
There was moderate quality evidence from these six studies that those receiving hydroxyurea experienced significant reductions in the frequency of pain crises, increases in fetal haemoglobin and decreases in neutrophil (white blood cell) counts compared to the comparator treatment. There was no difference between people receiving hydroxyurea or other treatments in terms of quality of life, deaths during the studies and side effects (including serious and life‐threatening side effects); however, there is less information about these outcomes in the studies, so the quality of this evidence is low.
Two further studies were included in the review. In one study, 22 children with SCD, who were also at an increased risk of having a stroke, were randomly selected to receive hydroxyurea or no treatment (observation only) and in one study 44 adults and children were randomly selected to receive treatments with or without adding hydroxyurea. Both studies showed an increase in fetal haemoglobin for people receiving hydroxyurea compared to the comparator treatment and there were no deaths during the studies. There was no difference between people receiving hydroxyurea or other treatments in terms of pain crises and side effects (including serious or life‐threatening side effects) and these studies did not measure quality of life. The quality of the evidence from these studies is very low, given the studies were very small and only recruited around 20% of the intended number of people and results do not apply to all people with SCD (different genotypes).
Conclusions
The evidence shows that hydroxyurea is likely to be effective in the short term at decreasing the frequency of painful episodes and raising fetal haemoglobin levels in the blood in people with SCD. Hydroxyurea is also likely to be effective in preventing first strokes for those at an increased risk of stroke and does not seem to be associated with an increase in any side effects (including serious and life‐threatening side effects).
There is currently not much evidence on whether hydroxyurea is beneficial over a long period of time, what the best dose to take is, or whether treatment causes any long‐term or serious side effects. More studies are needed to answer these questions.
Summary of findings
Background
Description of the condition
Haemoglobinopathies, including sickle cell disease (SCD), are among the most common inherited disorders in the world. SCD affects people originating from sub‐Saharan Africa, Arab countries, the Mediterranean, the Indian subcontinent, the Caribbean and South America, as well as African‐Americans and descendants from immigrants from the above countries in other parts of the world. The genetic mutation gives carriers some advantage against malaria and for this reason has persisted. There is an estimated global birth rate of around 300,000 affected individuals per year, the majority of which are in Africa (Piel 2013). There are an estimated 90,000 to 100,000 individuals with SCD in the USA and approximately 12,000 to 15,000 in the UK (Brousseau 2010; Hassell 2010; NICE 2012).
Haemoglobin is responsible for transporting oxygen around the body packaged in red blood cells. SCD is caused by the recessive inheritance of abnormal haemoglobins. People who inherit only one sickle gene and the normal gene for adult haemoglobin (HbA) are sickle cell carriers (sickle cell trait or AS) and are healthy. When people are homozygous, because they have inherited two sickle genes (SS), they have sickle cell anaemia, which is a variable clinical condition, with the vast majority of individuals suffering some pain attacks and a reduced life expectancy. Clinically significant SCD also arises when people inherit the sickle gene from one parent and another variant haemoglobin gene from the second parent such as haemoglobin C (SC) or a beta thalassaemia gene (Sβ+ or Sβ0). Sickle haemoglobin, when not carrying oxygen, polymerises distorting the red blood cell into the classic 'sickle' shape. The clinical problems arise predominantly as a result of chronic anaemia and the blockage of small blood vessels, which stops oxygen delivery to the tissues causing pain or organ damage or both.
The most frequent clinical problem is pain which causes over 90% of acute hospital admissions (Brozovic 1987) and significant morbidity in the community (Fuggle 1996). Other acute complications include acute chest syndrome, stroke, splenic sequestration, priapism and an increased risk of infection. SCD is a multi‐organ disease with a high chronic disease burden which increases with increasing age. The majority of individuals are anaemic with haemoglobin levels of 60 to 90g/L in HbSS. Common chronic complications include chronic sickle lung damage, pulmonary hypertension, renal dysfunction, chronic bony damage (avascular necrosis), retinopathy and leg ulceration (Howard 2015).
Whilst the majority of those affected used to die in childhood, and still do within parts of the developing world (Chakravorty 2015; Grosse 2011), death rates in children in higher‐income countries have fallen over the past decades and the majority of children (over 95%) now survive to adulthood (Quinn 2010; Telfer 2007; van der Plas 2011). Data from the USA reviewing national mortality data from 1979 to 2005 has confirmed this decrease in mortality in children, but showed an increased mortality rate in adults with a median age of death of 42 years for women and 38 years for men (Lanzkron 2013). Earlier data from the USA showed a median survival for adults with HbSS in the USA of 42 years in men and 46 years in women and in HbSC of 60 years and 68 years, respectively (Platt 1994). This contrasts with Jamaican figures which show median survival for HbSS of 53 years for men and 58.5 years for women (Wierenga 2001) and recent single‐centre data from the UK has shown median survival of 67 years in people with HbSS and higher in those with HbSC (Gardner 2016). Data from the USA has also shown an increase in deaths following transition from paediatric to adult services (Quinn 2010). Death is generally SCD‐related and is caused either by chronic organ failure consequent to the sickling process (e.g. renal failure, pulmonary hypertension, hepatic failure) (Manci 2003) or as a result of an acute catastrophic event, such as stroke (Powars 1983), acute sickle chest syndrome, splenic sequestration (Rogers 1978), sepsis (Manci 2003; van der Plas 2011) or other complications (Gray 1991; Lanzkron 2013; Perronne 2002). The UK National Confidential Enquiry into Patient Outcome and Death (NCEPOD) report reviewed UK mortality data over 48 months and found six paediatric and 40 adult deaths. One paediatric death was due to pneumococcal sepsis and two were due to sub‐arachnoid haemorrhage. In adults the main causes of death were stroke, multi‐organ failure, acute chest syndrome, renal failure and non‐sickle causes (NCEPOD 2008).
Improvements in paediatric outcomes are related to the introduction of neonatal screening, early enrolment in comprehensive paediatric care, penicillin prophylaxis, vaccination to decrease life‐threatening infection and primary stroke prevention with transcranial Doppler screening. In adults, treatment has relied on the avoidance of factors that precipitate crisis (including dehydration, infection and cold), and symptomatic treatment of the acute painful episodes (Davies 1997a; Davies 1997b; Steinberg 1999; STOP 2006). Hydroxyurea is currently the only licensed treatment for SCD. Blood transfusion is often required and may be used to treat acute complications or in the long term to treat or prevent disease complications. Haemopoeitic stem cell transplantation is the only currently available curative treatment option and is offered to children with severe disease phenotype and an human leukocyte antigen (HLA)‐matched sibling donor. Other donor transplant options and adult transplant are currently only available in the context of clinical trials. Gene therapy offers another potential curative treatment and is currently being investigated in clinical trials.
Description of the intervention
Hydroxyurea (also known as hydroxycarbamide) is an anti‐neoplastic oral drug and an inhibitor of ribonucleotide reductase (BABY HUG 2011). The drug has been shown to have many beneficial effects for treating SCD; including increasing fetal haemoglobin (HbF) concentration in red blood cells, improving nitric oxide metabolism, reducing red cell‐endothelial interaction and erythrocyte density (Ware 2010). Such disease‐modifying effects have been shown to decrease episodes of pain, acute chest syndrome, hospital admissions and the need for transfusions among people with SCD (MSH 1995; Strouse 2008); however, frequent monitoring of the person's blood count is required, in addition to monitoring of toxicity. There are recognised, but mostly reversible, side effects of hydroxyurea, such as low neutrophil count, low platelet count, anaemia, rash, headache and occasionally nausea. Furthermore, it may have teratogenic effects and may have an effect on male fertility (Strouse 2008;Zimmerman 2004). Long‐term or serious adverse effects (or both) of hydroxyurea are rare (Steinberg 2010; Strouse 2008) and observational data suggest a survival advantage for those treated with hydroxyurea (Steinberg 2010; Voskaridou 2010).
How the intervention might work
It has long been recognised that raised HbF levels can ameliorate the clinical effects of SCD (Perrine 1978; Platt 1994). HbF levels are high at birth and decrease over the first year of life and and hence clinical manifestations are often delayed until the HbF levels decrease. In addition, individuals who inherit high levels of HbF display a milder disease phenotype. This is because the HbF interferes with the polymer formation of the sickle haemoglobin within the red blood cell. This polymerisation is the underlying pathology in SCD. The more HbF there is, the greater the inhibition. Hydroxyurea was first shown to raise HbF levels in SCD in the 1980s (Platt 1984; Veith 1985). Its intermittent toxicity on the bone marrow leads to a stress response and enhanced erythropoiesis and levels of HbF. In addition to its effects on SCD via HbF enhancement, hydroxyurea also improves blood flow and reduces vaso‐occlusion via other mechanisms including decrease of adhesion molecules and stimulation of nitric oxide production (Green 2014).
Why it is important to do this review
It is important to examine as a whole, the body of work relating to the use of hydroxyurea in SCD, to evaluate the drug's effectiveness and tolerability in adults and children, and in the different types of SCD, the dosage regimens and whether the setting appears to influence the outcome, i.e. high‐income versus low‐ and middle‐income countries. We aim to review any evidence relating to the impact of hydroxyurea on the natural history of SCD and life expectancy.
Objectives
The aims of this review are to determine whether the use of hydroxyurea in people with SCD:
alters the pattern of acute events, including pain;
prevents, delays or reverses organ dysfunction;
alters mortality and quality of life;
is associated with adverse effects.
In addition we hoped to assess whether the response to hydroxyurea in SCD varies with type of SCD, age of the individual, duration and dose of treatment and healthcare setting.
Methods
Criteria for considering studies for this review
Types of studies
All randomised or quasi‐randomised studies, irrespective of language. Studies with quasi‐randomised methods, such as alternation, were included if there was sufficient evidence that the treatment and control groups were similar at baseline.
Types of participants
This review is limited to studies of hydroxyurea in SCD only. In order to fully quantify the potential harm and toxicity of this drug, a further review may need to be undertaken in all patient groups treated with hydroxyurea.
People of any age with SCD (SS, Sβ₀, SC, Sβ+) proven by electrophoresis and sickle solubility test, with family studies or DNA tests as appropriate.
Types of interventions
Hydroxyurea in any formulation at all doses, compared to either placebo or standard treatment (no placebo) for periods of one month or longer.
Types of outcome measures
Primary outcomes
-
Pain alteration
frequency, duration, severity measured on self‐reported patient scales
health service utilisation (e.g. inpatient days, outpatient or accident and emergency department visits)
opioid use
Life‐threatening illness (e.g. acute chest syndrome, stroke and acute splenic sequestration)*
Death during the study
* In the 2017 update of the review, serious adverse events reported in included studies (whether treatment‐related or not) were included under the definition of 'Life‐threatening illness'.
Secondary outcomes
Measures of fetal haemoglobin (HbF or F cells) and neutrophil counts
Other surrogate markers of response (e.g. haemoglobin, mean cell volume, platelet count and growth)
Quality of life, time loss to school or employment, integration into society, scales recording feeling of well‐being and global function (e.g. Karnofsky)
Measures of organ damage (e.g. spleen (pitted red cells), chronic sickle lung disease (transfer factor), liver, chronic renal failure (creatinine), priapism, leg ulcer, neurological damage (e.g. intelligence quotient (IQ)))
Any reported adverse effects or toxicity
Search methods for identification of studies
We searched for all relevant published and unpublished trials without restrictions on language, year or publication status.
Electronic searches
Relevant studies were identified from the Cochrane Cystic Fibrosis and Genetic Disorders Group's Haemoglobinopathies Trials Register using the terms: sickle cell AND hydroxyurea.
The Haemoglobinopathies Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (Clinical Trials) (updated each new issue of the Cochrane Library) and weekly searches of MEDLINE. Unpublished work is identified by searching the abstract books of five major conferences: the European Haematology Association conference; the American Society of Hematology conference; the British Society for Haematology Annual Scientific Meeting; the Caribbean Public Health Agency Annual Scientific Meeting (formerly the Caribbean Health Research Council Meeting); and the National Sickle Cell Disease Program Annual Meeting. For full details of all searching activities for the register, please see the relevant section of the Cochrane Cystic Fibrosis and Genetic Disorders Group's website.
Date of the most recent search of the Group's Haemoglobinopathies Trials Register: 16 January 2017.
We also searched the following trials registries on 20 March 2017.
ClinicalTrials.gov using the following search terms: hydroxyurea AND sickle.
WHO International Clinical Trials Registry Platform (ICTRP) using the following search terms: hydroxyurea AND sickle.
Searching other resources
The bibliographic references of all retrieved studies and reviews were assessed for additional reports of studies.
Data collection and analysis
Selection of studies
For the initial review, two authors (SCD and AO) independently applied inclusion criteria. For the updates to this review, two authors (AJ and SJN) performed this task. There were no discrepancies between the authors' assessments.
Data extraction and management
For the initial review, two authors (SCD and AO) independently extracted the data. For the updates of the review, two authors (AJ and SJN) performed this task. There were no discrepancies between the authors' assessments.
We intended to group outcome data into those measured at three, six, twelve months and annually thereafter. When outcome data were recorded at other time periods, then consideration was given to examining these as well.
Assessment of risk of bias in included studies
For the initial review, two authors (SCD and AO) independently assessed the methodological quality of each trial using the Cochrane Risk of Bias tool (Higgins 2011). For the updates of the review, two authors (AJ and SJN) performed these tasks. The authors assessed methodological quality on the methods of concealment and generation of randomisation sequence, blinding, whether data were available to analyse on intention‐to‐treat basis and whether all randomised participants were included in the analysis. There were no discrepancies between the authors' assessments.
Measures of treatment effect
For binary outcomes, we aimed to calculate a pooled estimate of the treatment effect for each outcome across studies, (the risk of an outcome among treatment allocated participants to the corresponding risk among controls). For each study, we calculated risk ratios (RR) with 95% confidence intervals (CI) for all important, dichotomous outcomes. We present RR in preference to odds ratios (OR), as OR give an inflated impression of the size of effect where event rates are high, as is the case of these studies. For the continuous outcomes, we aimed to calculate a pooled estimate of treatment effect, using the mean difference (MD).
Unit of analysis issues
One study was cross‐over in design (Belgian Study 1996). We planned to analyse data from this study using the approach recommended by Elbourne (Elbourne 2002); extracting and analysing data from paired analyses if possible.
Outcomes were measured at different time points throughout the course of the MSH study. Methods to analyse aggregate longitudinal data if individual patient data are not available are discussed by Jones (Jones 2009); however, data presented did not allow the use of these methods therefore we have carried out analysis at each individual time point reported.
Dealing with missing data
In order to allow an intention‐to‐treat analysis, we collected data by allocated treatment groups, irrespective of compliance, later exclusion (regardless of cause) or loss to follow‐up.
Assessment of heterogeneity
We assessed clinical heterogeneity by reviewing the differences across trials in the characteristics of recruited participants and treatment protocols. We assessed statistical heterogeneity using a Chi² test for heterogeneity. We assessed heterogeneity using the Q test (P < 0.10 for significance) and the I² statistic (greater than 50% indicating considerable heterogeneity (Higgins 2003)) and visually by inspecting forest plots.
Data synthesis
We analysed data using the fixed‐effect model, if we had found considerable heterogeneity (I² statistic > 50%) then we would have examined it using a random‐effects model and subgroup analyses.
Subgroup analysis and investigation of heterogeneity
We intended to perform subgroup analyses according to age (infant, child, adult etc), type of SCD (SS, Sβ₀, SC, Sβ+), dosage regimen (study specific) and setting (community, hospital, outpatient, etc), however, available data did not allow for these analyses.
Sensitivity analysis
We planned a sensitivity analysis based on the methodological quality of the studies, including and excluding quasi‐randomised studies. However, no quasi‐randomised studies were included in the review, therefore, no sensitivity analyses were performed.
Summary of findings and quality of the evidence (GRADE)
In a post hoc change from the protocol, we have presented four summary of findings tables, one for each comparison of the review (Table 1; Table 2; Table 3; Table 4).
Summary of findings for the main comparison. Summary of findings ‐ Hydroxyurea compared with placebo for sickle cell disease.
| Hydroxyurea compared with placebo for sickle cell disease | ||||||
|
Patient or population: adults and children with sickle cell disease Settings: outpatients Intervention: hydroxyurea Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Hydroxyurea | |||||
|
Pain alteration1 Follow‐up: 6 ‐ 24 months |
See comment | See comment | NA | 577 (4 studies)2 |
⊕⊕⊕⊝ moderate5 | All studies showed a significant advantage to hydroxyurea compared to placebo (different measures of pain alteration presented)1. |
|
Life‐threatening illness Follow‐up: 6 ‐ 24 months |
See comment | See comment | NA | 552 (3 studies) |
⊕⊕⊕⊝ moderate5 | Significantly fewer occurrences of ACS (2 studies) and transfusions (3 studies) on hydroxyurea compared to placebo. No significant differences in terms of stroke, hepatic or splenic sequestration (two studies). |
|
Death during the study (all deaths) Follow‐up: 6 ‐ 24 months |
26 per 1000 | 10 per 1000 (0 to 51 per 1000) |
RR 0.39 (0.08 to 1.96) | 577 (4 studies)2 |
⊕⊕⊕⊝ moderate5 | There was also no significant difference between groups in terms of deaths related to SCD. |
|
Measures of HbF (%) Follow‐up: 6 ‐ 24 months |
See comment | See comment | NA | 577 (4 studies)2 |
⊕⊕⊕⊝ moderate5 | There was a significant increase in HbF(%) in the hydroxyurea group compared to the placebo group in all studies (different measures presented)3. |
|
Measures of ANC Follow‐up: 6 ‐ 24 months |
See comment | See comment | NA | 517 (3 studies)2 |
⊕⊕⊕⊝ moderate5 | There was a significant decrease in ANC in the hydroxyurea group compared to the placebo group in all studies (different measures presented)3. |
|
Quality of life: 'Health Status Survey' the 'Profile of Mood States' and the 'Ladder of Life' Follow‐up: 24 months |
See comment | See comment | NA | up to 277 (1 study) |
⊕⊕⊝⊝ low5,6 |
No significant difference in terms of any domain of any scale except for pain recall at 18 months (MD 0.70, 95% CI 0.11 to 1.29, P = 0.02)4. |
|
Adverse events or toxicity: differences in rates of specific adverse events Follow‐up: 6 ‐ 24 months |
See comment | See comment | NA | 577 (4 studies)2 |
⊕⊕⊝⊝ low5,7 |
Significantly fewer events of dactylitis and gastroenteritis on hydroxyurea compared to placebo. No significant differences between groups in terms of all other events. |
| The basis for the assumed risk is the event rate in the control group unless otherwise stated in the comments and footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ACS: acute chest syndrome; ANC: absolute neutrophil counts; CI: confidence interval; HbF: fetal haemoglobin; MD: mean difference;NA: not applicable; RR: risk ratio; SCD: sickle cell disease. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
- Pain alteration measured by mean annual pain crisis rate, time to initiation of treatment to first, second or third crisis, number of vaso‐occlusive crises, proportion of participants experiencing pain, proportion of hospitalisation for painful episodes.
- One study of 25 participants is of a cross‐over design (Belgian Study 1996). Participants are counted only once in this total.
- Different measures presented ‐ change from baseline or post intervention measures ‐ therefore, data from all studies could not be pooled.
- Within the study (MSH 1995, reported in Ballas 2006), to allow for multiple statistical testing of the quality of life domains, a P value < 0.01 was considered significant. Therefore this result not interpreted as significant in the study publication.
- Downgraded once due to applicability: only individuals with HbSS or HbSβº‐thalassemia genotypes were included therefore results are not applicable to individuals with HbSC genotype.
- Downgraded once due to imprecision/uncertainty: caution is encouraged regarding the interpretation of these results as not all participants contributed data to all quality of life domains and the study publication defines statistical significance differently to this review.
- Downgraded once due to imprecision/uncertainty: caution is encouraged regarding the interpretation of these results due to the number of separate outcomes considered in analysis and the increased probability of a statistical type I error.
Summary of findings 2. Summary of findings ‐ Hydroxyurea and phlebotomy compared to transfusion and chelation for people with sickle cell disease and an increased risk of stroke.
| Hydroxyurea and phlebotomy compared to transfusion and chelation for people with sickle cell disease and an increased risk of stroke | ||||||
|
Patient or population: adults and children with sickle cell disease and an increased risk of stroke Settings: outpatients Intervention: hydroxyurea and phlebotomy Comparison: transfusion and chelation | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Transfusion and chelation | Hydroxyurea and phlebotomy | |||||
|
Pain alteration: proportion experiencing serious VOC or sickle‐related pain events Follow‐up: 24 to 30 months |
213 per 1000 | 718 per 1000 (339 to 1000 per 1000) |
RR 3.37 (95% CI 1.59 to 7.11) | 254 (2 studies) |
⊕⊕⊝⊝ low4,5 |
No significant difference between treatment groups in terms of all pain events (serious and non‐serious) in 1 study (RR 1.03, 95% CI 0.81 to 1.30). |
|
Life‐threatening illness Follow‐up: 24 ‐ 30 months |
See comment | See comment | NA | 254 (2 studies) |
⊕⊕⊕⊝ moderate4 | No significant difference between groups in life‐threatening neurological events, hepatobiliary disease and splenic sequestration; significantly more ACS and infections and infestations in the hydroxyurea and phlebotomy compared to the transfusion and chelation group. |
|
Death during the study (all deaths) Follow‐up: 24 ‐ 30 months |
1 death occurred in the transfusion and chelation group of 1 study1. | 1 death occurred in the hydroxyurea and phlebotomy group of 1 study1. | RR 0.99 (95% CI 0.06 to 15.42) | 254 (2 studies) |
⊕⊕⊝⊝ low4,5 |
|
|
Measures of HbF (%) Follow‐up: 24 to 30 months |
See comment | See comment | NA | 254 (2 studies) |
⊕⊕⊕⊝ moderate4 | There was a significant increase in HbF(%) in the hydroxyurea and phlebotomy group compared to the transfusion and chelation group for both studies (different measures presented)2. |
|
Measures of ANC Follow‐up: 24 to 30 months |
See comment | See comment | NA | 254 (2 studies) |
⊕⊕⊕⊝ moderate4 | There was a significant decrease in ANC in the hydroxyurea and phlebotomy group compared to the transfusion and chelation group for both studies (different measures presented)2. |
| Quality of life | Outcome not reported3 | NA | ||||
| Adverse events or toxicity: differences in rates of specific adverse events | See comment | See comment | NA | 254 (2 studies) |
⊕⊕⊝⊝ low4,6 |
There was a statistically significant difference in terms of immune disorders (more in transfusion and chelation group), reticulocytopenia, neutropenia and anaemia (more in hydroxyurea and phlebotomy group) in 1 study and the rate of adverse events was balanced across groups in the other study. |
| The basis for the assumed risk is the event rate in the control group unless otherwise stated in the comments and footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ACS: acute chest syndrome; ANC: absolute neutrophil counts; CI: confidence interval; HbF: fetal haemoglobin; NA: not applicable; RR: risk ratio; VOC: vaso‐occlusive crisis. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
1. Absolute data presented for number of deaths as the confidence interval of the relative effect very large due to the small number of events. 2. Different measures presented ‐ mean or median change from baseline ‐ therefore data from all studies could not be pooled. 3. Quality of life data was collected in TWiTCH 2016; to date, primary results of this study have been published but not quality of life data. When available, quality of life data will be included in an update of this review. 4. Downgraded once due to applicability: only children with HbSS or HbSβº‐thalassemia were included therefore results are not applicable to adults or individuals with HbSC genotype. 5. Downgraded once due to imprecision: small number of events and large CI around the relative effect. 6. Downgraded once due to imprecision/uncertainty: caution is encouraged regarding the interpretation of these results due to the number of separate outcomes considered in analysis and the increased probability of a statistical type I error.
Summary of findings 3. Summary of findings ‐ Hydroxyurea compared with observation for people with sickle cell disease and an increased risk of stroke.
| Hydroxyurea compared with observation for people with sickle cell disease and an increased risk of stroke | ||||||
|
Patient or population: adults and children with sickle cell disease and an increased risk of stroke Settings: outpatients Intervention: hydroxyurea Comparison: observation | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Observation | Hydroxyurea | |||||
|
Pain alteration Follow‐up: NA |
Outcome not reported | NA | ||||
|
Life‐threatening illness Follow‐up: 15 months |
See comment | See comment | NA | 22 (1 study) |
⊕⊝⊝⊝ very low3,4 | No significant differences between groups in terms of ACS, blood transfusions required or acute splenic sequestration. |
|
Death during the study Follow‐up: 15 months |
No deaths occurred | No deaths occurred | NA | 22 (1 study) |
⊕⊝⊝⊝ very low3,4 | |
|
Measures of HbF Follow‐up: 15 months |
See comment | See comment | NA | 22 (1 study) |
⊕⊝⊝⊝ very low3,4 | There was a significant increase in HbF in the hydroxyurea group compared to the observation group1. |
|
Measures of ANC Follow‐up: 15 months |
See comment | See comment | NA | 22 (1 study) |
⊕⊝⊝⊝ very low3,4 | There was a significant decrease in ANC in the hydroxyurea group compared to the observation group1. |
|
Quality of Life Follow‐up: NA |
Outcome not reported2 | NA | ||||
|
Adverse events or toxicity: differences in rates of specific adverse events Follow‐up: 15 months |
See comment | See comment | NA | 22 (1 study) |
⊕⊝⊝⊝ very low3,4 | No significant differences between groups in terms of transient neutropenia, reticulocytopenia, parasite infestation, headache and dizziness. |
| The basis for the assumed risk is the event rate in the control group unless otherwise stated in the comments and footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ACS: acute chest syndrome; ANC: absolute neutrophil counts; CI: confidence interval; HbF: fetal haemoglobin; NA: not applicable; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
1. Median values reported so data cannot be entered into analysis. 2. Outcome was not collected or presented due to early termination of study. 3. Downgraded twice due to serious imprecision: study terminated early with only 22 of target 100 participants recruited. Small number of participants included in final analyses which are likely to be underpowered. 4. Downgraded once due to applicability: only children with HbSS or HbSβº‐thalassemia were included therefore results are not applicable to adults or individuals with HbSC genotype.
Summary of findings 4. Summary of findings ‐ Hydroxyurea compared with no hydroxyurea for people with sickle cell disease.
| Hydroxyurea compared with no hydroxyurea for sickle cell disease | ||||||
|
Patient or population: adults and children with sickle cell disease Settings: outpatients Intervention: hydroxyurea Comparison: no hydroxyurea | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| No hydroxyurea | Hydroxyurea | |||||
|
Pain alteration Follow‐up: NA |
Outcome not reported | NA | ||||
|
Life‐threatening illness Follow‐up: NA |
Outcome not reported | NA | ||||
|
Death during the study Follow‐up: 11 months |
No deaths occurred | No deaths occurred | NA | up to 44 (1 study)1 |
⊕⊝⊝⊝ very low2,3,4 | |
|
Measures of HbF Follow‐up: 24 weeks |
See comment | See comment | NA | up to 44 (1 study)1 |
⊕⊝⊝⊝ very low2,3,4 | There was a significant increase in HbF in the hydroxyurea group compared to the no hydroxyurea group1. |
|
Measures of ANC Follow‐up: 24 weeks |
See comment | See comment | NA | up to 44 (1 study)1 |
⊕⊝⊝⊝ very low2,3,4 | There was no significant difference in ANC between treatment groups1. |
|
Quality of life Follow‐up: NA |
Outcome not reported | NA | ||||
|
Adverse events or toxicity Follow‐up: 11 months |
See comment | See comment | NA | up to 44 (1 study)1 |
⊕⊝⊝⊝ very low2,3,4 | Vaso‐occlusive pain crises, headache / migraine, upper respiratory infection, skin rash diarrhoea and abdominal pain were the most common adverse events during the trial and these events were evenly distributed across treatment groups (not separated by group). |
| The basis for the assumed risk is the event rate in the control group unless otherwise stated in the comments and footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ANC: absolute neutrophil counts; CI: confidence interval; HbF: fetal haemoglobin; NA: not applicable; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
1. Due to the factorial design of the study (22 participants randomised to a treatment arm including hydroxyurea and 22 randomised to a treatment arm without hydroxyurea), results are not entered into analysis. All results of this trial are considered exploratory (CHAMPS 2011). 2. Downgraded once due to indirectness: factorial design of the study makes comparison of hydroxyurea to no hydroxyurea indirect. 3. Downgraded once due to imprecision and risk of bias: study was terminated early with only 44 out of 188 participants recruited and outcome data is presented for only those who completed each follow‐up time. 4. Downgraded once due to applicability: participants with HbSC were included therefore results are not applicable to individuals with HbSS or HbSβº‐thalassemia genotypes.
Hydroxyurea compared to placebo for participants with SCD
Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and an increased risk of stroke
Hydroxyurea compared to observation for participants with SCD and an increased risk of stroke
Hydroxyurea compared to no hydroxyurea for participants with SCD
The following outcomes were reported in all tables (chosen based on relevance to clinicians and consumers): pain alteration; life‐threatening illnesses; deaths during the study; measures of fetal haemoglobin (HbF or F cells) and neutrophil counts, quality of life and adverse events or toxicity.
We determined the quality of the evidence using the GRADE approach; and downgraded evidence in the presence of a high risk of bias in at least one study, indirectness of the evidence, unexplained heterogeneity or inconsistency, imprecision of results, high probability of publication bias. We downgraded evidence by one level if we considered the limitation to be serious and by two levels if very serious.
For clarity in the tables, where outcomes were presented using different measures (e.g. pain alteration) or different domains (e.g. quality of life or adverse events), a general statement is made in the table regarding the summary of findings for these outcomes and the evidence is graded based on all of the measures or sub domains combined.
Results
Description of studies
Results of the search
See Figure 1 for PRISMA study flow diagram.
1.
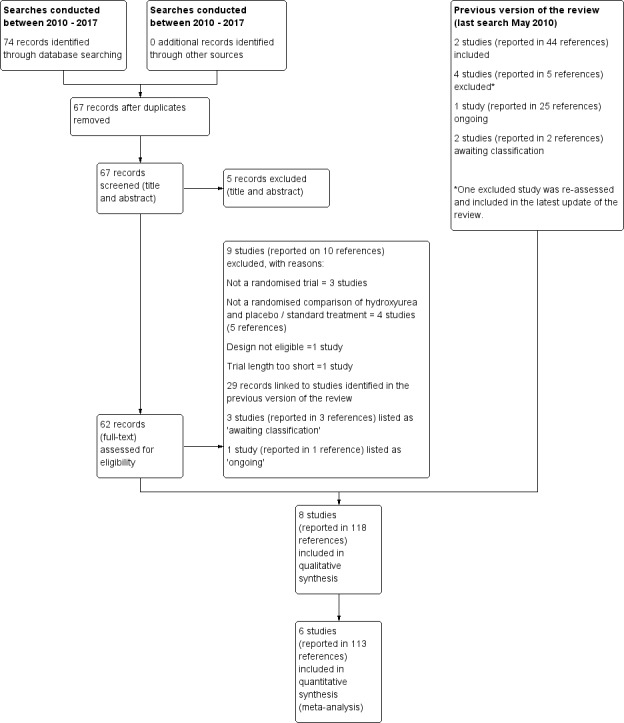
Study flow diagram.
The previous versions of the review (Jones 2001, latest search conducted May 2010) included two studies reported in 44 references (Belgian Study 1996; MSH 1995); additionally, one ongoing study (23 references) was identified (now completed) (BABY HUG 2011). Furthermore, two studies (two references) were listed as awaiting classification (CHAMPS 2011; Jain 2012).
Searches conducted between June 2010 and January 2017 identified 74 records, seven of which were duplicates. Following removal of these, 67 records were screened and five clearly irrelevant records were excluded based on title and abstract. The remaining 62 records were further screened, full texts were accessed where possible. A total of 29 records were linked to studies previously identified (BABY HUG 2011; Belgian Study 1996; CHAMPS 2011; Jain 2012; MSH 1995), nine studies (reported in 10 references) were excluded (see Excluded studies for further details), three studies (reported in three references) were listed as 'awaiting classification' due to uncertainties around the study design and population (see Characteristics of studies awaiting classification for further details) and one study (reported in one reference) was listed as 'ongoing' (see Characteristics of ongoing studies for further details).
The remaining 19 references corresponded to three new studies which were eligible for inclusion (SCATE 2015; SWiTCH 2012; TWiTCH 2016) and one reference previously excluded from the previous version of the review (Ware 2006) was re‐assessed and included under the SWiTCH study (SWiTCH 2012). The three studies previously identified as ongoing or awaiting classification are now also eligible for inclusion in the review (BABY HUG 2011; CHAMPS 2011; Jain 2012)
In total, eight studies (reported in 118 references) are included in the review (BABY HUG 2011; Belgian Study 1996; CHAMPS 2011; Jain 2012; MSH 1995; SCATE 2015; SWiTCH 2012; TWiTCH 2016).
Included studies
Eight studies are included, with a total of 899 children and adults (BABY HUG 2011; Belgian Study 1996; CHAMPS 2011; Jain 2012; MSH 1995; SCATE 2015; SWiTCH 2012; TWiTCH 2016)
The BABY HUG study was a multicentre, randomised, controlled study conducted in 13 centres in the USA (BABY HUG 2011). It was conducted in children aged nine months to 18 months who had haemoglobin SS (HbSS) or haemoglobin Sβºthalassaemia (HbSβºthal). Participants received liquid hydroxyurea (20 mg/kg per day) or matching placebo for two years administered as an oral syrup. A total of 193 children were randomised, 96 were randomised to the hydroxyurea group and 97 were randomised to the placebo group. Participants, caregivers and medical coordinating centre staff were masked to treatment allocation and an unmasked "primary end‐point person" monitored laboratory values and assisted in clinical management.
The Belgian Study was conduced in a single centre in Belgium (Belgian Study 1996). This involved 25 children and young adults with HbSS genotype with the aim of reviewing the impact on pain events, hospitalisation and also on fetal haemoglobin reactivation. This study was also placebo‐controlled, randomised and blinded to the participant but not to the caring physician. In addition, this was a cross‐over study which started at 20 mg/kg per day and, unless cytopenia developed this was raised to a maximum of 25 mg/kg per day. In this study the participants were randomised to either hydroxyurea or placebo for the initial six months and then crossed over to the other arm. There was no statistically significant period or carry‐over effect present for the outcomes of the number of hospitalisations and the number of days in hospital (period and carry‐over effects assessed by the Wilcoxon Rank Sum test).
The CHAMPS study was a randomised multicentre, phase II, double blind placebo‐controlled study with a 2x2 factorial design (CHAMPS 2011). Eligible participants were over the age of five years with HbSC and at least one vaso‐occlusive event in the previous 12 months (but none in the four weeks prior to study entry). A total of 44 participants were randomised equally across four treatment groups for 44 weeks: hydroxyurea (20 mg/kg per day) and magnesium (0.6 mmol/kg per day in two doses), hydroxyurea (20 mg/kg per day) and placebo, placebo and magnesium (0.6 mmol/kg per day in two doses), placebo and placebo. The study was not designed to measure efficacy and measured only laboratory measures.The study was terminated early due to low enrolment after 44 participants had been randomised (target 188).
The Jain study was a double blind (participants, personnel and outcome assessors) randomised controlled study conducted in a tertiary hospital in Nagpur City, India (Jain 2012). The study was conducted in children with sickle cell anaemia (proportion with each genotype not stated) between the ages of five and 18 years with three or more blood transfusions or vaso‐occlusive crises requiring hospitalisation per year despite high HbF. A total of 60 participants were randomised; 30 to hydroxyurea (fixed dose 10 mg/mg per day) and 30 to a matched placebo for 18 months.
The MSH study was a multicentre North American randomised and double‐blind study, which compared hydroxyurea with placebo over two years in adults with sickle cell anemia (SS genotype only) with the objective of reducing the frequency pain crises (MSH 1995). A total of 299 participants were randomised; 152 to hydroxyurea and 147 to matching placebo. Hydroxyurea was started at low dose (15 mg/kg per day) and increased at 12‐weekly intervals by 5 mg/kg per day until mild bone marrow depression, as judged by either neutropenia or thrombocytopenia, at that point the treatment was stopped (as reported for the MSH study) (Handy 1996). Once the blood count had recovered, treatment was restarted at 2.5 mg/kg per day less than the toxic dose. The study was therefore aiming for the maximum tolerated dose (MTD) for each individual within the study. The study was blinded and the study centre recorded and held the mean corpuscular volume (MCV) and HbF levels, which were not looked at by the caring physicians as the MCV and HbF levels rise in most people with SS taking hydroxyurea. As a result of the beneficial effects observed in terms of the primary pain outcome, as reported for the MSH study (Barton 1996), the study was stopped by the National Heart, Lung and Blood Institute of the USA at a mean follow‐up of 21 months, before the planned 24 months of treatment had been completed for all participants (MSH 1995). Long‐term follow‐up continued for the study sample, with all participants offered treatment with hydroxyurea.
The SCATE (Sparing Conversion to Abnormal transcranial doppler (TCD) Elevation) study was a Phase III multicentre, randomised, controlled study conducted in three centres in the USA, Jamaica and Brazil (SCATE 2015). The study was conducted in children with sickle cell anemia (SS, Sβº, HbSOArab, HbSD), haemoglobin SS (HbSS) or haemoglobin Sβº thalassaemia and conditional TCD ultrasound velocities (170 cm to 199 cm per second). A total of 22 participants were randomised; 11 to hydroxyurea at 20 mg/kg with escalation to maximum dose of 35 mg/kg and 11 to standard treatment (observation). The primary aim of the study was to establish whether treatment with hydroxyurea could prevent conversion from conditional to abnormal time averaged mean velocity (TAMV) and subsequent stroke in these children. The planned length of follow‐up was 30 months but the study was terminated after 15 months of follow‐up due to slow participant accrual and the unlikelihood of meeting the trial recruitment target (100) and the primary endpoint.
The SWiTCH study was a non‐inferiority study, comparing hydroxyurea and phlebotomy to standard treatment (transfusion and chelation) using a composite endpoint including secondary stroke prevention and improved management of iron overload. It included children with SCA (HbSS and HbSβºthal, HbSOArab) and previous stroke, who had been receiving chronic transfusions for at least 18 months (SWiTCH 2012). It was conducted in 26 sickle cell centres across the USA and a total of 134 children were randomised (67 to the standard treatment and 67 to the hydroxyurea and phlebotomy group). Participants randomised to hydroxyurea and phlebotomy commenced hydroxyurea at 20 mg/kg with escalation to MTD (defined by dose causing mild myelosuppression). Transfusion continued for four to nine months during hydroxyurea dose escalation. Once MTD was reached and transfusion stopped, phlebotomy of 10 mL/kg monthly was performed, if haemoglobin was sufficient.
The TWiTCH study was a multicentre phase III randomised open label (partially masked) non‐inferiority study conducted at 26 paediatric hospitals and health centres in the USA and Canada (TWiTCH 2016). Eligible participants were children aged four to 16 years with SCA (HbSS, HbSβºthal, HbSOArab) and abnormal TCD ultrasound velocities (≥ 200 cm per second) if they had received 12 months of chronic transfusions. A total of 121 participants were randomised; 60 to hydroxyurea starting at 20 mg/kg per day escalated to the MTD compared to standard treatment (transfusions) for 24 months. Children randomised to the standard treatment group received their usual chelation therapy or deferasirox to manage iron overload. Children randomised to the hydroxyurea group continued to receive transfusions until hydroxyurea was escalated to the MTD and following the discontinuation of transfusions, children received serial phlebotomy to manage iron overload. The primary aim of the study was to establish whether treatment with hydroxyurea could prevent primary stroke in these children. The study was terminated at the first interim analysis when non‐inferiority was demonstrated; the target sample size had been recruited by this point.
Excluded studies
Following the 2010 search for the previous version of the review (Jones 2001), four studies (reported in five references) were excluded from the review (De Montalembert 2006; Silva‐Pinto 2007; Voskaridou 2005; Ware 2006). However, as described above (Results of the search), for the present version of the review, the Ware reference was reassessed and included under the SWiTCH study (SWiTCH 2012).
In total, 12 studies (14 references) were excluded from the current review; five were not randomised (Al‐Nood 2011; Pushi 2000; Silva‐Pinto 2007; Silva‐Pinto 2014; Voskaridou 2005), four did not make a randomised comparison of hydroxyurea and placebo or standard treatment (George 2013; NCT00004492; NCT01960413; Vichinsky 2013), two were of one week of less duration (De Montalembert 2006; NCT01848925) and one was an inappropriate design for measuring the effectiveness of hydroxyurea (NCT02149537).
Risk of bias in included studies
See Figure 2 and Characteristics of included studies for more information.
2.
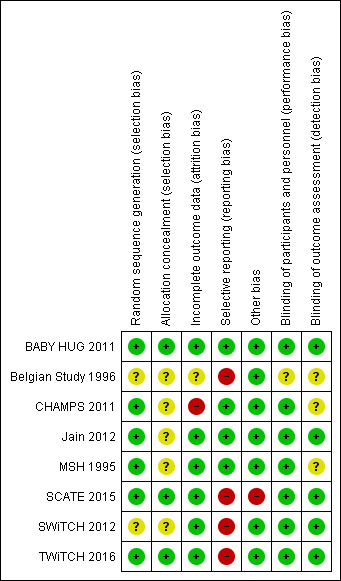
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
One study used an automated telephone response system to randomise participants using lists that had been produced by the medical co‐ordinating centre (BABY HUG 2011). The SCATE and TWiTCH studies used blocked randomisation and central pharmacy allocation (SCATE 2015;TWiTCH 2016 ). All three studies have been assessed as having a low risk of bias.
The MSH study used computerised block randomisation, the Jain trial used random number tables and the CHAMPS study used a sequential allocation algorithm for randomisation (low risk of bias) but none of these studies clearly stated any method of allocation concealment (unclear risk of bias) (CHAMPS 2011; Jain 2012; MSH 1995).
The Belgian study described the randomisation of participants as "drawing sealed envelopes, patients were randomly allocated to one of the following treatment sequences", therefore, the generation and the allocation of the treatment sequence are not clear from this statement (Belgian Study 1996). The hospital pharmacy provided the treatment and placebo for each participant and both were described as "indistinguishable." The SWiTCH trial did not discuss the method for randomisation or allocation concealment and therefore this has been graded as unclear (SWiTCH 2012).
Blinding
The BABY HUG study was described as 'double blind', the study paper stated that "participants, caregivers and medical coordinating centre staff were masked to treatment allocation" (BABY HUG 2011). The hydroxyurea and placebo powders were identical in terms of appearance and packaging and the liquid formulations had the same appearance and taste. In the Jain study, participants, clinicians and outcome assessors including laboratory technicians were blinded to treatment allocation with placebo capsules of identical appearance (Jain 2012). These studies were assessed as low risk of performance and detection bias.
In three studies, the blinding of participants and personnel was also not possible due to the differences between the treatments (hydroxyurea and standard treatment) (SCATE 2015; SWiTCH 2012; TWiTCH 2016). Outcome assessors were partially masked in these studies for assessments around the primary outcomes related to neurological events such as stroke. Given the objective nature of the primary outcomes of the studies judged by the blinded outcome assessors, these studies were assessed as low risk of detection bias.
The MSH study was described as double blind (physician and participant) (MSH 1995) and in the CHAMPS study, treatment was assigned in combinations of identically appearing capsules and blinding was achieved by 'over‐capsulating' tablets (CHAMPS 2011). These two studies were assessed as low risk of performance bias. The Belgian study was single‐blinded (the participant was unaware of the treatment schedule but the physician was aware of the treatment schedule) because of the difficulty of blinding the attending physician to the treatment received (Belgian Study 1996). It was not stated whether outcome assessors were blinded in any of these three studies.
Incomplete outcome data
In four studies, the risk of bias was assessed as low since withdrawals from treatment were documented, an intention‐to‐treat analysis approach in primary analyses was used and all randomised participants were included in the analysis (Jain 2012; MSH 1995; SWiTCH 2012; TWiTCH 2016).
In the BABY HUG study, an intention‐to‐treat analysis was undertaken for primary outcomes (via multiple imputation methods to account for missing data) and for safety outcomes (BABY HUG 2011). Some of the secondary outcomes were reported for only the individuals who completed the study or had data recorded for specific measurements, but given that primary and important safety outcomes were reported using an intention‐to‐treat analysis, this study was judged to be at a low risk of bias.
In the SCATE study, due to the early termination of the study, two randomised participants did not receive their allocated treatment (SCATE 2015). The primary analysis of this study used an intention‐to‐treat approach and a sensitivity analysis considered the actual treatment received so this study was also assessed as having a low risk of bias.
In the Belgian study, three participants were excluded from the analysis due to their failure to attend their monthly evaluation at four to five months (Belgian Study 1996). There was no discussion of whether or not an intention‐to‐treat analysis was used, therefore, the risk of bias was judged to be unclear.
In the CHAMPS study, only participants who completed eight weeks (primary outcome) or 44 weeks (secondary outcomes) of follow‐up were included in the results (CHAMPS 2011). This is not an intention‐to‐treat approach so this trial was judged to be at high risk of bias.
Selective reporting
Three studies defined outcomes in their methods sections, which were reported in the results, and were thus assessed as having a low risk of bias (BABY HUG 2011; Jain 2012; MSH 1995). The CHAMPS study was not designed to measure efficacy and reported only laboratory‐based outcomes; all of these outcomes were well‐defined in the methods and reported in the results, so this study was assessed to be at a low risk of bias (CHAMPS 2011).
In the primary publication of the TWiTCH study, it was stated the the secondary outcomes of neuropsychological status, quality of life and growth would be published at a later date (TWiTCH 2016). For the SWiTCH study, some outcomes (such as growth and development, functional evaluations, neurocognitive evaluations) do not yet seem to have been reported (SWiTCH 2012). As we are not able to include the results at this time, we have judged these studies to be at a high risk of selective reporting bias. If these results can be included at a later date then this judgement will be reconsidered (SWiTCH 2012; TWiTCH 2016). The final two studies planned to measure outcomes which were not reported due to difficulty in collecting the information to inform these outcomes; these studies are also judged to be at a high risk of bias (Belgian Study 1996; SCATE 2015).
Other potential sources of bias
One study was likely to be underpowered due to the early termination of the study with only 22% of the target sample size recruited (SCATE 2015). Another study recruited only 23% of the target sample size (CHAMPS 2011); however; this study was not designed to measure efficacy and analyses were intended to be exploratory, so we did not consider this study to be at high risk of bias. Another study was also terminated early at the first interim analysis, but the target sample size had been recruited at this point so we did not consider this study to be at a high risk of bias (TWiTCH 2016).
No other bias was identified for the remaining studies (BABY HUG 2011; Belgian Study 1996; Jain 2012; MSH 1995; SWiTCH 2012).
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4
For the 2017 update of the review six new studies were included (BABY HUG 2011; Jain 2012; CHAMPS 2011; SCATE 2015; SWiTCH 2012; TWiTCH 2016). Due to differences in the eligible populations and treatments in these new studies, we have made the following comparisons.
1. Hydroxyurea compared to placebo for participants with SCD
This comparison includes four studies (BABY HUG 2011; Belgian Study 1996; Jain 2012; MSH 1995).
2. Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and an increased risk of stroke
This comparison includes two studies (SWiTCH 2012; TWiTCH 2016).
3. Hydroxyurea compared to observation for participants with SCD and an increased risk of stroke
This comparison includes a single study (SCATE 2015).
4. Hydroxyurea compared to no hydroxyurea for participants with SCD
This comparison includes a single study (CHAMPS 2011). We note that this study recruits only individuals with HbSC; however, this comparison (and other comparisons) are worded to allow for future studies to be included in updates of this review which recruit individuals of all genotypes to contribute to this comparison.
We have conducted meta‐analyses for comparisons 1 and 2 (above) where appropriate and presented results narratively or in additional tables (Table 5; Table 6; Table 7; Table 8). Data are not entered into analysis for the Belgian and CHAMPS studies due to the presentation of results from cross‐over and factorial designs respectively; results are reported narratively (Belgian Study 1996; CHAMPS 2011). Long‐term follow‐up of the participants in MSH study continued for up to 17 years and many publications presented results of long‐term follow‐up (see linked reference list of the MSH study) (MSH 1995). After two years of double‐blind, placebo‐controlled therapy, all participants were offered hydroxyurea therapy, so any results reported after the MSH study period are uncontrolled. The long‐term results are not therefore analysed in this review.
1. Clinical events and markers of response in Jain 2012.
| Hydroxyurea | Placebo | P value | |||
| Baseline | 18 months | Baseline | 18 months | ||
| Clinical events (number of events per participant per year) | |||||
| VOC | 12.13 (8.56) | 0.60 (1.37) | 11.46 (3.01) | 10.2 (3.24) | < 0.001 |
| Blood transfusions | 2.43 (0.69) | 0.13 (0.43) | 2.13 (0.98) | 1.98 (0.82) | < 0.001 |
| Hospitalisations | 10.13 (6.56) | 0.70 (1.28) | 9.56 (2.91) | 9.59 (2.94) | < 0.001 |
| Haematological parameters | |||||
| Hb (g/dL) | 8.1 (0.68) | 9.29 (0.55) | 8.21 (0.68) | 7.90 (0.58) | < 0.001 |
| HbF(%) | 19.8 (0.9) | 24 (5.9) | 19.21 (6.37) | 18.92 (5.77) | < 0.001 |
| Reticulocytes (x10⁵ /mm³) | 1.83 (0.96) | 1.15 (0.1) | 1.73 (0.49) | 1.81 (0.67) | < 0.001 |
| Leucocytes (x10³ /mm³) | 7.36 (6.03) | 6.54 (5.54) | 7.26 (4.91) | 7.38 (2.85) | < 0.001 |
| Platelets (x10³ /mm³) | 1.78 (0.26) | 2.01 (0.18) | 1.91 (0.21) | 2.06 (0.26) | 0.28 |
| RBC (x10⁶ /mm³) | 2.89 (0.57) | 1.98 (0.22) | 1.84 (0.47) | 3.11 (0.20) | 0.05 |
| Total bilirubin (mg/dL) | 2.32 (1.42) | 1.10 (0.42) | 2.27 (1.28) | 2.71 (0.93) | < 0.001 |
Values are mean (standard deviation) P values are calculated using independent t‐test.
Hb: haemoglobin HbF: fetal haemoglobin RBC: red blood count VOC: vaso‐occlusive crises WBC: white blood count
2. Laboratory measurements from MSH 1995.
| Baseline | Hyrdoxyurea | Placebo | P value | ||
| 2 years | Baseline | 2 years | 2 years | ||
| WBC (10⁹/L) | 12.6 (3.4) | 9.9 (3.1) | 12.3 (3.2) | 12.2 (2.8) | 0.0001 |
| Neutrophils (10⁹/L) | 6.9 (2.4) | 4.9 (2.0) | 6.7 (2.3) | 6.4 (2.0) | 0.0001 |
| Platelets (10⁹/L) | 468 (147) | 399 (124) | 457 (130) | 423 (122) | 0.12 |
| Hb (g/dL) | 8.5 (1.4) | 9.1 (1.5) | 8.5 (1.2) | 8.5 (1.3) | 0.0009 |
| PCV (%) | 24.9 (4.4) | 27 (5) | 25.2 (4.0) | 25.1 (4.2) | 0.0007 |
| MCV (fl) | 94 (9) | 103 (14) | 93 (9) | 93 (9) | 0.0001 |
| Reticulocytes (10⁹/L) | 327 (98) | 231 (100) | 325 (94) | 300 (99) | 0.0001 |
| HbF (%) | 5 (3.5) | 8.6 (6.8) | 5.2 (3.4) | 4.7 (3.3) | 0.0001 |
| F cells (%) | 33 (17) | 48 (23) | 33 (17) | 35 (18) | 0.0001 |
| F reticulocytes | 15 (8) | 17 (9) | 15 (8) | 15 (7) | 0.0036 |
| Dense cells (%) | 14 (6) | 11 (6) | 14 (7) | 13 (7) | 0.004 |
| Creatinine (mg/dL) | 0.9 (0.3) | 1.0 (0.5) | 0.9 (0.2) | 1.0 (0.5) | 0.64 |
| Total bilirubin (mg/dL) | 3.7 (2.4) | 2.9 (2.5) | 3.7 (2.5) | 4.2 (4.6) | 0.004 |
| Direct bilirubin (mg/dL) | 0.5 (0.3) | 0.4 (0.3) | 0.5 (0.4) | 0.7 (2.2) | 0.08 |
| Aspartate aminotransferase | 44 (23) | 39 (20) | 41 (21) | 43 (27) | 0.16 |
| Alkaline phosphatase | 120 (59) | 117 (48) | 119 (67) | 119 (71) | 0.71 |
Values are mean (standard deviation) P values are calculated using independent t‐test.
Hb: haemoglobin HbF: fetal haemoglobin MCV: mean corpuscular volume PCV: packed cell volume WBC: white blood count
3. Laboratory evaluations from the SWiTCH trial.
| Outcome |
Hydroxyurea and phlebotomy group (n = 67) |
Transfusions and chelation group (n = 66) |
P value |
| HbF (%) | 17.9 (92 to 22.9) | ‐0.2 (‐0.8 to 0.4) | < 0.001 |
| ANC (x10⁹/L) | ‐3.3 (‐5.1 to ‐1.4) | 0.8 (‐1.3 to 2.4) | < 0.001 |
| Hb (g /dL) | 0.0 (‐0.7 to 0.7) | 0.0 (‐0.5 to 0.6) | 0.898 |
| HbA (%) | ‐50.9 (‐66.8 to ‐33.7) | 0.0 (‐12.7 to 6.7) | < 0.001 |
| HbS (%) | 35.0 (21.7 to 46.2) | 0.3 (‐7.5 to 12.3) | < 0.001 |
| MCV (fL) | 19.5 (7.5 to 28.5) | 0.1 (‐2.0 to 2.5) | < 0.001 |
| WBC (x10⁹/L) | ‐5.4 (‐8.1 to ‐2.2) | 0.2 (‐2.0 to 2.3) | < 0.001 |
| ARC (x10⁹/L) | ‐149.1 (‐231.0 to ‐19.0) | ‐11.8 (‐88.2 to 93.2) | < 0.001 |
| Platelets (x10⁹/L) | ‐83.0 (‐171.0 to ‐8.0) | ‐28.0 (‐70.0 to 18.0) | 0.0022 |
| Total bilirubin (mg/dL) | ‐1.1 (‐1.9 to ‐0.6) | 0.4 (‐0.3 to 1.2) | < 0.001 |
| LIC (mg Fe/g) | ‐1.2 (‐2.8 to 7.2) | ‐2.2 (‐5.5 to 4.9) | 0.48888 |
| Serum ferritin (ng/mL) | ‐966.0 (‐1629.0 to 49.0) | 1159.5 (‐662.0 to 2724.0) | < 0.001 |
| LDH (U/L) | ‐67.0 (‐143.0 to 7.0) | ‐8.5 (‐74.0 to 74.0) | 0.0015 |
ANC: absolute neutrophil count ARC: absolute reticulocyte count Hb: haemoglobin HbA: adult haemoglobin HbF: fetal haemoglobin HbS: sickle haemoglobin LDH: lactate dehydrogenase LIC: liver iron concentration MCV: mean corpuscular volume WBC: white blood count
Values are median change from baseline and interquartile range. P values are calculated using Wilcoxon rank sum test.
4. Laboratory evaluations from the SCATE trial.
| Outcome | Hydroxyurea (n = 11) | Observation (n = 11) | P value |
| Hb (g/dL) | 1.6 | ‐0.5 | < 0.0001 |
| MCV (fL) | 8.7 | 1 | 0.0001 |
| ARC ( x10⁹/L) | 22.7 | ‐33.2 | 0.76 |
| WBC ( x10⁹/L) | ‐4.6 | 1.3 | 0.07 |
| ANC ( x10⁹/L) | ‐2.2 | 1.4 | 0.05 |
| Platelets ( x10⁹/L) | ‐76 | ‐35 | 0.56 |
| HbF (%) | 8.9 | 0.3 | 0.002 |
| Weight (kg) | 2.5 | 1.8 | 0.51 |
| Height (cm) | 6.8 | 3.8 | 0.22 |
ANC: absolute neutrophil count ARC: absolute reticulocyte count Hb: haemoglobin HbF: fetal haemoglobin MCV: mean corpuscular volume WBC: white blood count.
Values are median change from baseline and P values are calculated using Wilcoxon rank sum test.
Primary outcomes
1. Pain alteration
Hydroxyurea compared to placebo for participants with SCD
The MSH study defined pain crisis as a visit to a medical facility, lasting four or more hours, requiring opiate analgesia (MSH 1995). There was a statistically significant difference between the hydroxyurea group and placebo group in the mean annual crisis rate (all crises), MD ‐2.80 (95% CI ‐4.74 to ‐0.86) (P = 0.005) and for crises requiring hospitalisation, MD ‐1.50 (95% CI ‐2.58 to ‐0.42) (P = 0.007) (Analysis 1.1).
1.1. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 1 Pain crises.
The MSH study also showed a reduction in median time (Kaplan‐Meier life table estimate) from the initiation of treatment to first painful crisis (2.76 months in the hydroxyurea arm compared with 1.35 months on placebo (Cox regression P value = 0.014), second painful crisis (6.58 months in the hydroxyurea group compared with 4.13 months on placebo (Cox regression P value < 0.0024), and third painful crisis (11.9 months in the hydroxyurea group compared with 7.04 months on placebo (Cox regression P value = 0.0002) (reported in Charache 1995). We note that the analysis of time‐to‐first, second and third painful crisis seems to treat crisis events independently which is unlikely to be a realistic assumption. Furthermore, information relating to the analgesia used by participants in this study has been reported, but has not been presented according to the groups to which the participants were randomised (reported in Ballas 1996).
The Jain study presented the number of clinical events (vaso‐occlusive crises) before and after intervention (Jain 2012). We could not calculate change from baseline in the number of clinical events so we have analysed between group data at 18 months and presented the before and after data in an additional table (Table 5). After 18 months of treatment, there was a statistically significant difference between the hydroxyurea group and placebo group, MD ‐9.60 (95% CI ‐10.86 to ‐8.34) (P < 0.00001) (Analysis 1.1).
The BABY HUG study showed a statistically significantly lower proportion of participants experiencing pain in the hydroxyurea group compared to the placebo group, RR 0.68 (95% CI 0.5 to 0.92) (BABY HUG 2011) (Analysis 1.2).
1.2. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 2 Proportion experiencing pain.
The Belgian study stated that "16 patients out of 22 (73%) did not require any hospitalisation for painful episodes when treated with hydroxyurea as compared with only 3 of 22 (14%) when treated with placebo" (Belgian Study 1996). In addition, it showed as reduction in mean hospital stay, with a stay of 5.3 days in the hydroxyurea group and 15.2 days in the placebo group. A planned outcome of 'number of days in pain' was not reported in the trial publication due to difficulties in collecting this information from participants.
Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and an increased risk of stroke
Both studies reported the number of participants experiencing 'serious' vaso‐occlusive or sickle cell (SCA)‐related pain events (SWiTCH 2012; TWiTCH 2016) and one study also reported the number of participants experiencing any vaso‐occlusive or sickle cell (SCA)‐related pain event (SWiTCH 2012)
There was a statistically significantly higher proportion of participants experiencing serious vaso‐occlusive or sickle cell (SCA)‐related pain in the hydroxyurea and phlebotomy group compared to the transfusion and chelation groups in the SWiTCH and TWiTCH studies, pooled RR 3.37 (95% CI 1.59 to 7.11) (P = 0.001) (SWiTCH 2012; TWiTCH 2016), but there was no significant difference between the groups in terms of all SCA pain events in the SWiTCH study, RR 1.03 (95% CI 0.81 to 1.30) (P = 0.81) (SWiTCH 2012) (Analysis 2.1).
2.1. Analysis.
Comparison 2 Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and risk of stroke, Outcome 1 Proportion experiencing pain.
Hydroxyurea compared to observation for participants with SCD and an increased risk of stroke
No information was reported for this outcome in the SCATE study (SCATE 2015).
Hydroxyurea compared to no hydroxyurea for participants with SCD
No information was reported for this outcome in the CHAMPS study (CHAMPS 2011).
2. Life‐threatening illness
Hydroxyurea compared to placebo for participants with SCD
The BABY HUG and the MSH studies provided data on the occurrence of acute chest syndrome, stroke and participants transfused (BABY HUG 2011; MSH 1995). Statistically significant advantages in the hydroxyurea group was the reduction in the occurrence of the acute sickle chest syndrome, pooled RR 0.43 (95% CI 0.29 to 0.63) (P < 0.0001), and also that fewer participants on hydroxyurea underwent transfusions, pooled RR 0.66 (95% CI 0.52 to 0.82) (P = 0.0003). There was no statistically significant difference in terms of stroke, pooled RR 0.54 (95% CI 0.12 to 2.53) (P = 0.44) (Analysis 1.3).
1.3. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 3 Proportion experiencing life threatening events during study.
The MSH study reported data on hepatic sequestration and the BABY HUG study reported data on splenic sequestration, the differences between the groups were not statistically significant, RR 0.32 (95% CI 0.03 to 3.06) (P = 0.32) and RR 0.90 (95% CI 0.36 to 2.23) (P = 0.82), respectively (MSH 1995; BABY HUG 2011) (Analysis 1.3).
The Jain study presented the number of clinical events (blood transfusions and hospitalisations) before and after intervention (Jain 2012). We could not calculate change from baseline the number of clinical events so we have analysed between group data at 18 months and presented the before and after data in an additional table (Table 5). After 18 months of treatment, there was a statistically significant difference between the hydroxyurea group and placebo group in the number of blood transfusions, MD ‐1.85 (95% CI ‐2.18 to ‐1.52) (P < 0.00001) and in the number of hospitalisations, MD ‐8.89 (95% CI ‐10.04 to ‐7.74) (P < 0.00001). The duration of hospitalisation was also significantly less in the hydroxyurea group than the placebo group, MD ‐4.00 days (95% CI ‐4.87 to ‐3.13) (P < 0.00001) (Analysis 1.4).
1.4. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 4 Number of life‐threatening events during study.
No information was reported for this outcome in the Belgian study (Belgian Study 1996).
Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and an increased risk of stroke
The primary aim of the TWiTCH study was to prevent primary stroke; 29 neurologic events occurred during the study, 17 in the hydroxyurea group and 12 in the standard treatment group; but none were deemed to be stroke or cerebral infarcts (TWiTCH 2016). Three events in each group were deemed to be transient ischaemic attacks and worsened vasculopathy developed in one participant in the standard treatment group.
The aim of the SWiTCH study was to prevent secondary stroke; 91 new‐neurologic events were assessed for stroke and seven participants (all in the hydroxyurea treatment group) has positive stroke adjudication, one of which was fatal (SWiTCH 2012). There were also 20 transient ischaemic attack events in 15 participants (nine on transfusion and chelation and six on hydroxyurea and phlebotomy).
There was no statistically significant difference in life‐threatening neurological events between the treatment groups in either study from the analysis conducted in this review (Analysis 2.2); however, following the seven events in the hydroxyurea and phlebotomy group, the SWiTCH study was terminated following an interim analysis demonstrating the inability of the primary outcome to reduce secondary stroke occurrence while managing iron overload "within the non‐inferiority stroke margin" (SWiTCH 2012).
2.2. Analysis.
Comparison 2 Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and risk of stroke, Outcome 2 Proportion experiencing life‐threatening events during study.
The SWiTCH and TWiTCH studies reported serious adverse events (including life‐threatening events) and other adverse events or toxicities separately (see Secondary outcomes for Adverse events) (SWiTCH 2012; TWiTCH 2016). Thirty‐eight participants experienced 81 serious adverse events in the SWiTCH study and 15 participants experienced 33 serious adverse events in the TWiTCH study (SWiTCH 2012; TWiTCH 2016). There were significantly more participants experiencing serious adverse events and SCD‐related adverse events in the hydroxyurea treatment groups compared to the transfusions groups; pooled RR 1.93 (95% CI 1.17 to 3.20) (P = 0.01) and RR 3.10 (95% CI 1.42 to 6.75) (P = 0.004), respectively (Analysis 2.2). There was no significant difference between groups in terms of hepatobiliary disease and splenic sequestration but serious acute chest syndrome and infections and infestations were significantly more common on hydroxyurea and phlebotomy treatment than on transfusion and chelation, pooled RR 2.84 (95% CI 1.25 to 6.42) (P = 0.01) and pooled RR 3.65 (95% CI 1.05 to 12.76) (P = 0.04), respectively (Analysis 2.2).
As the control arm of the two studies in this comparison involved blood transfusions, we did not consider the number of participants requiring blood transfusion to be a life‐threatening event for this comparison.
Hydroxyurea compared to observation for participants with SCD and an increased risk of stroke
The primary aim of the SCATE study was to prevent stroke and no strokes or transient ischaemic attacks occurred during the study. Vaso‐occlusive events (pain and acute chest syndrome) occurred more commonly in the observation arm than the hydroxyurea arm, but this was not statistically significant (SCATE 2015). There was also no statistically significant difference in cases of acute splenic sequestration or the number of participants requiring blood transfusion (Analysis 3.1).
3.1. Analysis.
Comparison 3 Hydroxyurea compared to observation for participants with SCD and risk of stroke, Outcome 1 Proportion experiencing life‐threatening events during the study.
Hydroxyurea compared to no hydroxyurea for participants with SCD
No information was reported for this outcome in the CHAMPS study (CHAMPS 2011).
3. Death during the study
Hydroxyurea compared to placebo for participants with SCD
The MSH study reported death and causes of death (MSH 1995); no deaths were reported to be related to hydroxyurea treatment. There were two deaths in the treated group and five in the placebo group including one homicide. There was no statistically significant difference between treatment and control groups for this comparison (Analysis 1.5).
1.5. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 5 Deaths during the study.
There were no deaths in the remaining three studies (BABY HUG 2011; Belgian Study 1996; Jain 2012).
Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and an increased risk of stroke
In the SWiTCH study, there was one death in the standard treatment arm (pulmonary embolism) and one death in the hydroxyurea treatment arm (fatal haemorrhagic stroke) (SWiTCH 2012). There were no deaths reported in the TWiTCH study (TWiTCH 2016). There was no statistically significant difference between treatment and control groups for this comparison (Analysis 2.3).
2.3. Analysis.
Comparison 2 Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and risk of stroke, Outcome 3 Deaths during the study.
Hydroxyurea compared to observation for participants with SCD and an increased risk of stroke
There were no deaths reported in the SCATE study (SCATE 2015).
Hydroxyurea compared to no hydroxyurea for participants with SCD
There were no deaths reported in the CHAMPS study (CHAMPS 2011).
Secondary outcomes
1. Measures of fetal haemoglobin and neutrophil counts
Hydroxyurea compared to placebo for participants with SCD
The BABY HUG study reported the change from baseline in HbF (%) and absolute neutrophil count (ANC); there was a statistically significant increase in HbF in the hydroxyurea group compared to the placebo group, MD 6.70 (95% CI 4.75 to 8.65) (P < 0.00001) (BABY HUG 2011) (Analysis 1.6) and a statistically significant decrease in ANC in the hydroxyurea group compared to the placebo group, MD ‐1.70 (95% CI ‐2.90 to ‐0.50) (P < 0.00001) (Analysis 1.8).
1.6. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 6 Change from baseline in fetal haemoglobin (HbF %).
1.8. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 8 Change from baseline in absolute neutrophil count (x10³ per μL).
The Jain and MSH studies presented results before and after intervention (Jain 2012; MSH 1995). We could not calculate change from baseline so we have analysed between group data at the end of the study and presented the before and after data in additional tables (Table 5; Table 6). After the intervention, there was a statistically significant increase in HbF in the hydroxyurea group compared to the placebo group in the two studies, pooled MD 4.07 (95% CI 2.95 to 5.18) (P < 0.0001) (Analysis 1.7).
1.7. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 7 Fetal haemoglobin (HbF %) after treatment.
The MSH study also reported the neutrophil response after treatment; at 10 weeks and at two years there was a statistically significant decrease in neutrophil response in the hydroxyurea group compared to the placebo group, MD ‐1.90 (95% CI ‐2.51 to ‐1.29) (P < 0.0001) and MD ‐1.50 (95% CI ‐2.01 to ‐0.99) (P < 0.0001), respectively (MSH 1995) (Analysis 1.9).
1.9. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 9 Neutrophil response (10⁹/L) after treatment.
One study reported an increase in HbF and a decrease in ANC after six months of hydroxyurea treatment, but did not make a comparison to the placebo group (Belgian Study 1996).
Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and an increased risk of stroke
The SWiTCH study reported the change from baseline in HbF (%) and ANC; there was a statistically significant difference between treatment groups for both of these measures (SWiTCH 2012). The results were presented as median and interquartile ranges and therefore could not be entered into a meta‐analysis; see an additional table for a summary of these results (Table 7).
The TWiTCH study reported the change from baseline in HbF (%) and ANC; there was a statistically significant increase in HbF in the hydroxyurea group compared to the placebo group, MD 15.60 (95% CI 13.41 to 17.79) (P < 0.00001) (Analysis 2.4) and a statistically significant decrease in ANC in the hydroxyurea group compared to the placebo group, MD ‐4.40 (95% CI ‐5.44 to ‐3.36) (P < 0.00001) (TWiTCH 2016) (Analysis 2.5).
2.4. Analysis.
Comparison 2 Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and risk of stroke, Outcome 4 Change from baseline in fetal haemoglobin (HbF %).
2.5. Analysis.
Comparison 2 Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and risk of stroke, Outcome 5 Change from baseline in absolute neutrophil count (x10⁹/L).
Hydroxyurea compared to observation for participants with SCD and an increased risk of stroke
The SCATE study reported a significant increase in HbF and a significant decrease in ANC at last follow‐up in the hydroxyurea group compared to observation (SCATE 2015). Results were reported as median values so could not be included in analysis, see an additional table for a summary of these results (Table 8).
Hydroxyurea compared to no hydroxyurea for participants with SCD
The CHAMPS study, which included participants with HbSC only, reported a statistically significant increase in HbF for hydroxyurea treatment compared to no hydroxyurea treatment at eight weeks and 24 weeks but no statistically significant difference was observed for ANC (CHAMPS 2011). Due to the presentation of results from factorial design of the trial, results cannot be entered into analysis.
2. Other surrogate markers of response
Hydroxyurea compared to placebo for participants with SCD
The BABY HUG study reported change from baseline in haemoglobin (Hb), mean cell volume (MCV), white blood count (WBC), platelet count, absolute reticulocyte count (ARC), reticulocytes and total bilirubin (BABY HUG 2011).
There was a statistically significant increase in Hb (Analysis 1.10), MCV (Analysis 1.11) and a statistically significant decrease in WBC (Analysis 1.12), ARC (Analysis 1.13), reticulocytes (Analysis 1.14) and total bilirubin (Analysis 1.15) in the hydroxyurea group compared to the placebo group. There was no statistically significant difference between groups in platelet count (Analysis 1.16).
1.10. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 10 Change from baseline in haemoglobin (g/L).
1.11. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 11 Change from baseline in m corpuscular volume (fL).
1.12. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 12 Change from baseline in white blood cells (x10³ per μL).
1.13. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 13 Change from baseline in absolute reticulocyte count (x10³ per μL).
1.14. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 14 Change from baseline in reticulocytes (%).
1.15. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 15 Change from baseline in total bilirubin (mg/L).
1.16. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 16 Change from baseline in platelet count (x10³ per μL).
The Jain and MSH studies presented results before and after intervention (MSH 1995; Jain 2012). We could not calculate change from baseline so we have analysed between‐group data at the end of the study and presented the before and after data in additional tables (Table 5; Table 6). After the intervention comparing hydroxyurea and placebo groups, there was a statistically significant increase in Hb and MCV at 10 weeks and at the end of the studies (Analysis 1.17, Analysis 1.18), a statistically significant decrease in total bilirubin at the end of the studies (Analysis 1.19) reticulocytes at 10 weeks, 18 months and two years (Analysis 1.20) and a decrease in platelet count at 10 weeks, 18 months and two years which was not statistically significant (Analysis 1.21).
1.17. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 17 Haemoglobin (g/dL).
1.18. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 18 Mean corpuscular volume (fL).
1.19. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 19 Total bilirubin (mg/L).
1.20. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 20 Reticulocytes.
1.21. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 21 Platelet count.
In terms of other laboratory measures comparing hydroxyurea and placebo groups at the end of the studies, there was a statistically significant increase in packed cell volume (Analysis 1.22), F reticulocytes (Analysis 1.23) and F cells (Analysis 1.24), a statistically significant decrease in red blood count (Analysis 1.25), white blood count (Analysis 1.26) and dense cells (Analysis 1.27) and no difference between groups in leucocytes (Analysis 1.28), creatinine (Analysis 1.29), aspartate aminotransferase (Analysis 1.30) and alkaline phosphatase (Analysis 1.31).
1.22. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 22 Packed cell volume.
1.23. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 23 F reticulocytes.
1.24. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 24 F cells.
1.25. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 25 Red blood count.
1.26. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 26 White blood cells.
1.27. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 27 Dense cells.
1.28. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 28 Leucocytes.
1.29. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 29 Creatinine.
1.30. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 30 Aspartate aminotransferase.
1.31. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 31 Alkaline phosphatase.
One study reported differences in haematologic values after six months of treatment such as Hb, MCV (both significantly increased), platelet count and WBC (both significantly decreased) but only in the hydroxyurea group without comparison to the placebo group (Belgian Study 1996).
The BABY HUG study reported change from baseline in growth, there were no significant differences between the groups in terms of height, weight or head circumference (BABY HUG 2011) (Analysis 1.32). The MSH study reported on weight gain after two years was reported in the MSH study as a mean rise of 3% in the hydroxyurea treated group and 6% in the placebo group, which was not statistically significant (MSH 1995).
1.32. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 32 Change from baseline in growth.
Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and an increased risk of stroke
The SWiTCH study reported the change from baseline several laboratory and haematologic measurements such as Hb, MCV, WBC, platelet count and total bilirubin (SWiTCH 2012). There was a statistically significant difference between treatment groups for most of the measurements (increase in HbF, HbS, MCV and decrease in ANC, HbA, WBC, ARC, platelets, serum ferritin and LDH), except for Hb and liver iron concentration; the results were presented as median and interquartile ranges and therefore could not be entered into a meta‐analysis; see an additional table for summary of these results (Table 7).
The TWiTCH study reported the change from baseline in Hb, MCV, WBC, ARC, platelet count, total bilirubin, sickle haemoglobin, liver iron concentration, serum ferritin and lactate dehydrogenase (TWiTCH 2016). There was a statistically significant increase in MCV (Analysis 2.6) and sickle haemoglobin (Analysis 2.7) and a statistically significant decrease in Hb (Analysis 2.8), ARC (Analysis 2.9), WBC (Analysis 2.10), platelets (Analysis 2.11), total bilirubin (Analysis 2.12), liver iron concentration (Analysis 2.13), serum ferritin (Analysis 2.14) and lactate dehydrogenase (Analysis 2.15) in the hydroxyurea and phlebotomy group compared to the transfusion and chelation group,
2.6. Analysis.
Comparison 2 Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and risk of stroke, Outcome 6 Change from baseline in mean corpuscular volume (fL).
2.7. Analysis.
Comparison 2 Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and risk of stroke, Outcome 7 Change from baseline in sickle haemoglobin (%).
2.8. Analysis.
Comparison 2 Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and risk of stroke, Outcome 8 Change from baseline in haemoglobin (g/L).
2.9. Analysis.
Comparison 2 Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and risk of stroke, Outcome 9 Change from baseline in absolute reticulocyte count (10⁹ / L).
2.10. Analysis.
Comparison 2 Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and risk of stroke, Outcome 10 Change from baseline in white blood count (10⁹ / L).
2.11. Analysis.
Comparison 2 Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and risk of stroke, Outcome 11 Change from baseline in platelets (10⁹ / L).
2.12. Analysis.
Comparison 2 Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and risk of stroke, Outcome 12 Change from baseline in total bilirubin (mg/L).
2.13. Analysis.
Comparison 2 Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and risk of stroke, Outcome 13 Change from baseline in liver iron concentration.
2.14. Analysis.
Comparison 2 Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and risk of stroke, Outcome 14 Change from baseline in serum ferritin (ng/mL).
2.15. Analysis.
Comparison 2 Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and risk of stroke, Outcome 15 Change from baseline in lactate dehydrogenase (U/L).
Hydroxyurea compared to observation for participants with SCD and an increased risk of stroke
The SCATE study reported differences in haematologic values and growth values at last follow‐up (SCATE 2015). There was a statistically significant increase in Hb and MCV in the hydroxyurea group compared to the observation group. Other values were not statistically significantly different (differences the hydroxyurea group and observation group in Hb and MCV were statistically significant and other values were not significant (WBC, ANC, Platelets, ARC, HbF, weight and height)). Results were reported as median values so could not be included in analysis, see an additional table for a summary of these results (Table 8).
Hydroxyurea compared to no hydroxyurea for participants with SCD
The CHAMPS study reported statistically significant increases in MCV and Hb and a statistically significant decrease in WBC and platelets between hydroxyurea treatment and no hydroxyurea treatment at eight weeks and 24 weeks (CHAMPS 2011). No statistically significant difference was observed in red blood cell count (RBC), mean corpuscular haemoglobin concentration (MCHC) and ARC. Due to the presentation of results from factorial design of the study, results cannot be entered into analysis.
3. Quality of life
Hydroxyurea compared to placebo for participants with SCD
The MSH study has published in abstract limited quality of life data collected at six‐monthly intervals (MSH 1995, reported in Terrin 1999 and Ballas 2006). Health‐related quality of life (HQoL) was measured using three measures from the 'Health Status Survey' (nine scales), the 'Profile of Mood States' (four scales) and the 'Ladder of Life'. A lower score for each measure equated to a lower quality of life.
Changes from baseline are presented for only three measures: General Health Perception,' 'Social Function' and 'Changes in Ladder of Life' and four week score was reported for 'Pain Recall'.
There was a statistically significant advantage to hydroxyurea compared to placebo in terms of general health perception at 18 months MD 0.90 (95% CI 0.08, 1.72) (P = 0.03) (Analysis 1.33) and pain recall at 18 months, MD 0.70 (95% CI 0.11 to 1.29) (P = 0.02) (Analysis 1.34) but not at any other measured time point for either measure.
1.33. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 33 Quality of life: general health perception.
1.34. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 34 Quality of life: pain recall.
There were no statistically significant differences between the hydroxyurea and placebo groups at six months, one year, 18 months and two years for 'Social Function' and 'Changes in Ladder of Life' (Analysis 1.35, Analysis 1.36).
1.35. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 35 Quality of life: social function.
1.36. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 36 Changes in 'Ladder of Life'.
For the MSH study, it was reported that there were no significant differences between groups in other measures of HQoL (using a P value cut‐off for statistical significance of P < 0.01 to allow for statistical testing of multiple HQoL domains) (MSH 1995 reported in Ballas 2006). Results are presented as final scores rather than change from baseline so are not entered into analysis in this review. We encourage caution when interpreting results of these quality of life scales as not all participants contributed data towards these analyses.
No information was reported for this outcome in the remaining three studies (BABY HUG 2011; Belgian Study 1996; Jain 2012).
Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and an increased risk of stroke
Quality of life was measured in the TWiTCH study, but results have not yet been published; if results are published at a later date, they will be included in an update of this review (TWiTCH 2016). No information was reported for this outcome in the remaining study (SWiTCH 2012).
Hydroxyurea compared to observation for participants with SCD and an increased risk of stroke
Health‐related quality of life was a planned outcome in the SCATE study but was not reported due to difficulties in collecting information from participants at the early study termination date (SCATE 2015).
Hydroxyurea compared to no hydroxyurea for participants with SCD
This outcome was not reported in the CHAMPS study (CHAMPS 2011).
4. Measure of chronic organ damage
Hydroxyurea compared to placebo for participants with SCD
The MSH study reported similar rates of new leg ulcers, RR 0.85 (95% CI 0.44 to 1.64) (P = 0.64) and avascular necrosis of femur and humerus, RR 0.97 (95% CI 0.39 to 2.37) (P = 0.64), in both groups (MSH 1995). The BABY HUG study reported more participants with decreased spleen function at exit in the placebo group than the hydroxyurea group but this was not statistically significant, RR 0.72 (95% CI 0.44 to 1.16) ( P = 0.18) (BABY HUG 2011) (Analysis 1.37).
1.37. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 37 Proportion of participants with signs of organ damage.
The BABY HUG study reported several measures of:
splenic function: diethylenetriaminepentaacetic acid glomerular filtration rate (Analysis 1.38); Howell‐Jolly body (HJB) (Analysis 1.39); pitted cells (Analysis 1.40); spleen to liver ratio of counts (Analysis 1.41); and spleen volume (Analysis 1.42);
renal function: creatinine (Analysis 1.43); Schwartz glomerular filtration rate (GFR) (Analysis 1.44); cystatin C (Analysis 1.45); urine osmolality (Analysis 1.46); urine pH (Analysis 1.47); urine‐specific gravity (Analysis 1.48); and total kidney volume (Analysis 1.49); and
central nervous system function: TCD velocity (Analysis 1.50); Bayley mental developmental index (MDI) and Bayley motor performance developmental index (PDI) (Analysis 1.51).
1.38. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 38 Signs of organ damage ‐ change from baseline in DTPA GFR.
1.39. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 39 Signs of organ damage ‐ change from baseline in Howell‐Jolley body (per 106 red blood cells).
1.40. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 40 Signs of organ damage ‐ change from baseline in pitted cells (%).
1.41. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 41 Signs of organ damage ‐ change from baseline in spleen: liver ratio of counts.
1.42. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 42 Signs of organ damage ‐ change from baseline in spleen volume (cm3).
1.43. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 43 Signs of organ damage ‐ change from baseline in creatinine (mg/L).
1.44. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 44 Signs of organ damage ‐ change from baseline in Schwartz GFR.
1.45. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 45 Signs of organ damage ‐ change from baseline in cystatin C.
1.46. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 46 Signs of organ damage ‐ change from baseline in urine osmolality.
1.47. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 47 Signs of organ damage ‐ change from baseline in urine pH.
1.48. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 48 Signs of organ damage ‐ change from baseline in urine‐specific gravity.
1.49. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 49 Signs of organ damage ‐ change from baseline in total kidney volume.
1.50. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 50 Signs of organ damage ‐ change from baseline in TCD ultrasound velocity (time‐averaged mean maximum velocity).
1.51. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 51 Signs of organ damage ‐ change from baseline in CNS measures.
Several of the outcomes showed significantly fewer indications of chronic organ damage on hydroxyurea compared to placebo (Analysis 1.39; Analysis 1.40; Analysis 1.41; Analysis 1.43; Analysis 1.46; Analysis 1.48, Analysis 1.49; Analysis 1.50); however, given the number of outcomes measured and the increased probability of type I statistical error and spurious result, the statistical significance of these secondary endpoint comparisons must be carefully interpreted (BABY HUG 2011).
No information was reported for this outcome in the remaining two studies (Belgian Study 1996; Jain 2012).
Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and an increased risk of stroke
The TWiTCH study reported a measure of central nervous system function (TWiTCH 2016). Eligible participants had abnormal TCD ultrasound velocities (> 200 cm per second). The final TCD velocity was significantly lower in the hydroxyurea and phlebotomy group compared to the transfusion and chelation group, MD ‐5.00 (95% CI ‐9.16 to ‐0.84) (P = 0.02) (Analysis 2.16). This outcome was not reported in the SWiTCH study (SWiTCH 2012).
2.16. Analysis.
Comparison 2 Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and risk of stroke, Outcome 16 Signs of organ damage ‐ CNS measures at the end of the study.
Hydroxyurea compared to observation for participants with SCD and an increased risk of stroke
The aim of the SCATE study was to prevent conversion from conditional to abnormal time averaged mean velocity (TAMV) and subsequent stroke; one participant in the hydroxyurea group and five participants in the observation group converted, this difference between groups was not statistically significant (Analysis 3.2) but there was a significantly lower TAMV in the hydroxyurea group compared to the observation group, MD ‐25.70 (95% CI ‐45.38 to ‐6.02) (P = 0.01) (SCATE 2015) (Analysis 3.3). No strokes occurred during the study.
3.2. Analysis.
Comparison 3 Hydroxyurea compared to observation for participants with SCD and risk of stroke, Outcome 2 Signs of organ damage ‐ proportion of participants with a change in CNS measures.
3.3. Analysis.
Comparison 3 Hydroxyurea compared to observation for participants with SCD and risk of stroke, Outcome 3 Signs of organ damage ‐ change from baseline in CNS measures.
Hydroxyurea compared to no hydroxyurea for participants with SCD
This outcome was not reported in the CHAMPS study (CHAMPS 2011).
5. Adverse effects or toxicity
As it is difficult to distinguish between adverse reactions (i.e. adverse events which are definitely drug‐related) and other adverse events which may be sickle‐ or transfusion‐related, we have reported all adverse events (or adverse effects) as they are reported in the study publications. Where possible, we have noted which adverse effects are defined as drug‐related, sickle‐related or transfusion‐related from the study publications.
Hydroxyurea compared to placebo for participants with SCD
The possible adverse effects reported in the MSH study were hair loss, skin rash, fever, gastro‐intestinal disturbance, and 'other' reported events (MSH 1995). There was no statistically significant difference between the groups with these measures (Analysis 1.52). In the MSH study, toxicity relating to the blood count was defined as less than 2500 x 10⁹ neutrophils/L, less than 95,000 x 10⁹ platelets/L, and haemoglobin concentration less than 5.3 g/dL (MSH 1995). Using these definitions 120 of the hydroxyurea‐treated participants (79%) and 54 placebo participants (37%) became 'toxic' at least once, resulting in a dose modification. Importantly, no infections were related to neutropenia and no 'bleeding' episode could be related to thrombocytopenia.
1.52. Analysis.
Comparison 1 Hydroxyurea versus placebo for participants with sickle cell disease, Outcome 52 Proportion of participants experiencing adverse events and toxicity.
The BABY HUG study reported the proportion of participants experiencing a range of adverse events such as dactylitis, priapism, sepsis or bacteraemia, splenomegaly, ANC below 500, ANC 500 to 1250, thrombocytopaenia, severe anaemia, alanine transaminase over 150, bilirubin, creatinine, skin and subcutaneous disorders and splenic sequestration (BABY HUG 2011). The rates of these specific events were generally quite similar with only dactylitis and gastroenteritis showing any differences between the two treatment groups, with statistically significantly fewer events in the hydroxyurea group (Analysis 1.52); however, given the number of outcomes measured and the increased probability of type I statistical error and spurious group differences, the statistical significance must be carefully interpreted.
No major adverse events occurred in either group in the Jain study (Jain 2012); three participants in the hydroxyurea group experienced skin rash and two experienced nausea, but this was not a statistically significant difference compared to the placebo group (Analysis 1.52). No participants experienced alopecia, leucopenia, neutropenia, renal or hepatic toxicity.
It was stated that two children developed transient mild thrombocytopenia in the Belgian study but the treatment allocation of these children was not stated (Belgian Study 1996).
Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and an increased risk of stroke
Life‐threatening adverse events are presented above in primary outcome 2 (Analysis 2.2).
In the TWiTCH study it was stated that adverse events were balanced between the two groups, in the transfusion and chelation group there were 287 sickle‐related adverse events and 279 sickle‐related adverse events in the hydroxyurea and phlebotomy group (TWiTCH 2016). There were 19 adverse events related to chelation treatment in nine children in the control group (aminotransferases, gastrointestinal symptoms, increased serum creatinine, increased serum bilirubin, rash) and 18 adverse events related to phlebotomy in 14 children in the hydroxyurea treatment group (details not stated). No events related to hydroxyurea were reported. There was insufficient information to enter into the analysis, we hope to include any future information in an update.
In the SWiTCH study, there were a total of 1253 non‐neurological adverse events in 128 individuals, there was no statistically significant difference between groups in the number of participants experiencing any adverse event, RR 0.99 (95% CI 0.92 to 1.05) (P = 0.66) (SWiTCH 2012) (Analysis 2.17). In terms of specific events, there was no statistically significant difference between treatment arms for most events. There was a statistically significant difference in terms of immune disorders (more in the transfusion and chelation group), reticulocytopenia, neutropenia and anaemia (more in the hydroxyurea and phlebotomy group) (Analysis 2.17); however, given the number of outcomes measured and the increased probability of type I statistical error and spurious group differences, the statistical significance must be carefully interpreted.
2.17. Analysis.
Comparison 2 Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and risk of stroke, Outcome 17 Proportion of participants experiencing non‐neurological adverse events and toxicity.
Hydroxyurea compared to observation for participants with SCD and an increased risk of stroke
Adverse events reported in the SCATE study included transient neutropenia, reticulocytopenia, parasite infestation, headache and dizziness (SCATE 2015). There was no significant difference between treatment arms for any adverse event (Analysis 3.4)
3.4. Analysis.
Comparison 3 Hydroxyurea compared to observation for participants with SCD and risk of stroke, Outcome 4 Adverse events and toxicity.
Hydroxyurea compared to no hydroxyurea for participants with SCD
In the CHAMPS study, adverse events and toxicity were only reported for all randomised participants and not by treatment group (CHAMPS 2011). A total of 22 serious adverse events were observed in 10 participants and 293 adverse events in 38 participants. It was reported that vaso‐occlusive pain crises, headache or migraine, upper respiratory infection, skin rash diarrhoea and abdominal pain were the most common adverse events during the trial and these events were evenly distributed across treatment groups. Two individuals, one receiving hydroxyurea and one not receiving hydroxyurea experienced mild neutropenia.
Discussion
Summary of main results
Eight randomised controlled studies were eligible for inclusion in this review, with a total of 899 children and adults with sickle cell disease (SCD) (BABY HUG 2011; Belgian Study 1996; CHAMPS 2011; Jain 2012; MSH 1995; SCATE 2015; SWiTCH 2012; TWiTCH 2016). The studies included in the review made the following comparisons: hydroxyurea compared to placebo (BABY HUG 2011; Belgian Study 1996; Jain 2012; MSH 1995); hydroxyurea compared to no hydroxyurea (CHAMPS 2011); hydroxyurea and phlebotomy compared to transfusion and chelation for the prevention of primary or secondary stroke (SWiTCH 2012, TWiTCH 2016); and hydroxyurea compared to observation for participants at an increased risk of stroke (SCATE 2015).
The randomised studies comparing hydroxyurea with placebo (Belgian Study 1996; Jain 2012; MSH 1995) all showed a decrease in pain crises (albeit with slightly different criteria) in adults and children with sickle cell anaemia. The BABY HUG study also showed decreased rates of pain in the hydroxyurea group compared to placebo. This widening of inclusion criteria has had an important role in increasing indications for and use of hydroxyurea. Two studies comparing transfusion and hydroxyurea showed that transfusion was more effective in preventing 'serious' sickle‐related pain (SWiTCH 2012; TWiTCH 2016), but there was no difference in all pain events between the two groups, as reported by one study (SWiTCH 2012).
Rates of acute chest syndrome (ACS) were decreased in those taking hydroxyurea when compared with those taking placebo and this was true in adults (MSH 1995) and children (BABY HUG 2011; Jain 2012). Similar decreases were seen in transfusion rates, a surrogate for life‐threatening illness (BABY HUG 2011; Jain 2012; MSH 1995). Hydroxyurea did not prevent rates of hepatic or splenic sequestration when compared with placebo (BABY HUG 2011; MSH 1995).
The role of hydroxyurea compared with transfusion in the prevention of stroke has been investigated in two studies (SWiTCH 2012;TWiTCH 2016). The SWiTCH study included people with a previous stroke who had received at least 18 months of transfusion and randomised between ongoing transfusion therapy and hydroxyurea and phlebotomy (SWiTCH 2012). Treatment with hydroxyurea and phlebotomy was not expected to be as effective as transfusion in the reduction of secondary stroke but was expected to improve liver iron loads so a composite primary end point was used including stroke recurrence and liver iron concentration. In the 133 participants there were no strokes in the transfusion and chelation group but seven strokes (10%) in the hydroxyurea and phlebotomy group without concomitant improvement liver iron concentration leading to early trial closure. Hydroxyurea and phlebotomy were not as effective as transfusion for secondary stroke prevention. In the TWiTCH study, hydroxyurea was compared with transfusion in children with abnormal transcranial doppler (TCD) velocities who had received transfusion for at least one year and had no evidence of vasculopathy on magnetic resonance angiography (MRA) (TWiTCH 2016). Hydroxyurea was as effective as transfusion in preventing further stroke and final TCD velocity was lower in the hydroxyurea group. These findings are supported by observational data showing that in a non‐trial situation in children with abnormal TCDs who have normalised with transfusion, hydroxyurea can be used to maintain normal TCDs (Bernaudin 2016).
In the randomised studies included in this analysis there was no statistically significant differences in mortality between any treatment and control group. Follow‐up from cohorts from the MSH study have been published at nine and 17.5 years (MSH 1995), although randomisation ceased after trial closure and participants could choose whether to take hydroxyurea or not (Steinberg 2003; Steinberg 2010). These follow‐up studies both confirmed reduced mortality in the group taking hydroxyurea although the analysis was according to cumulative exposure to hydroxyurea and not the original randomisation. Further supporting evidence for reduced mortality with hydroxyurea comes from a prospective non‐randomized trial (LasHS) from Greece with 17 years of follow‐up which showed improved probability of 10‐year survival in participants who were treated with hydroxyurea (Voskaridou 2010). These results should be treated cautiously as the participants were not randomised and the analysis of mortality in an observational study is more complex.
In terms of chronic organ damage; there were no statistically significant differences in renal and splenic function (the primary outcomes of BABY HUG 2011). Hydroxyurea had no significant effect on glomerular filtration rate over the two years of treatment, but it did improve urine concentrating ability and there was less renal enlargement in the hydroxyurea‐treated groups.
Several of the randomised studies showed a statistically significant increase in HbF levels in the groups treated with hydroxyurea, in keeping with the mechanism of action of the drug. This was seen both in participants treated with the maximum‐tolerated dose (MTD) of hydroxyurea (MSH 1995;SWiTCH 2012; TWiTCH 2016) and those treated with a standard dose (BABY HUG 2011;Jain 2012). A statistically significant increase in Hb level was seen in studies which compared hydroxyurea to placebo (BABY HUG 2011; Belgian Study 1996; CHAMPS 2011; MSH 1995;Jain 2012;SCATE 2015) and a decrease in haemolytic markers with hydroxyurea treatment was seen in some of the studies (BABY HUG 2011;Jain 2012;MSH 1995).
Those studies which used dose escalation to the MTD (MSH 1995; SCATE 2015; SWiTCH 2012; TWiTCH 2016), showed greater rates of cytopenias, which are expected as the dose of the drug is raised. The MSH study showed increased haematological toxicity resulting in a dose reduction in the hydroxyurea group as compared with placebo, but there were no infections related to neutropenia or bleeding episodes due to thrombocytopenia (MSH 1995). Similarly, the SWiTCH study showed significantly increased rates of reticulocytopenia, neutropenia and anaemia in the hydroxyurea group, compared with transfusion (SWiTCH 2012). The BABY HUG study which used a standard dose of 20 mg/kg of hydroxyurea showed an increase in neutropenia (in terms of absolute neurophil count) in the hydroxyurea group but no increase in events of thrombocytopenia and no increase in the number of infections (BABY HUG 2011). There are no randomised studies comparing the effect of using a standard dose versus dose escalation to the MTD. Clearly, with careful laboratory monitoring and appropriate patient education, cytopenia should rarely represent a major issue as demonstrated.
In the randomised studies comparing hydroxyurea with placebo there was no increase in adverse events related to hydroxyurea including hair loss, skin rash, fever and gastro‐intestinal disturbance. In addition, in the MSH study there was no increase in the incidence of leg ulcers and avascular necrosis in those treated with hydroxyurea. This is important as they have both been associated with hydroxyurea use in observational reports and leg ulcers are associated with hydroxyurea use in people with myeloproliferative disease. Furthermore, observational data looking at 15 years of hydroxyurea use from infancy has not shown any concerns about safety (Hankins 2014), although as with the mortality data this is not based on randomised data and should be treated with caution.
Overall completeness and applicability of evidence
Evidence from the eight included studies is extensive and for four of the studies it is reported across many publications (BABY HUG 2011;MSH 1995; SWiTCH 2012; TWiTCH 2016). Furthermore as discussed in the Summary of main results, evidence from these randomised studies is generally supported by longer‐term or observation evidence (or both).
There are still questions about the impact of hydroxyurea on the chronic organ damage associated with SCD. Results from the BABY HUG study showed improved urine concentrating ability and less renal enlargement in the hydroxyurea‐treated groups. These results suggest that hydroxyurea may have some beneficial effect on renal function, but these results do not automatically translate into clinical benefit (BABY HUG 2011). The long‐term effect of hydroxyurea therefore, would benefit from further study, including effects on brain development, IQ, growth and development, as well as the prevention and management of chronic organ damage.
As a chemotherapeutic agent, the cytostatic effects of hydroxyurea are different from those of radiation, alkylating agents and other anti‐cancer drugs, many of which are known to increase the risk of development of either leukaemia or cancer. Long‐term follow‐up of people taking hydroxyurea for other diagnoses, including polycythaemia rubra vera (Berk 1995; Fruchtman 1994; Najean 1997), essential thrombocythaemia (Sterkers 1998) and cyanotic heart disease (Triadou 1994), has shown no increase in malignancies in those with normal bone marrows and no significant increase in the other groups. There has been no evidence of malignancy in those with SCD treated with hydroxyurea in large observational studies (Steinberg 2010; Voskaridou 2010). Small numbers of case reports have reported myelodysplasia or acute leukaemia in people with SCD treated with hydroxyurea and two of these have been associated with 17p deletions. This is of interest as 17p deletions have also been associated with the development of myelodysplasia or acute leukaemia in people with myeloproliferative disease treated with hydroxyurea (Aumont 2015; Baz 2012). In vitro work has not shown increased chromosome breakages or decreased repair in people with SCD on hydroxyurea (McGann 2011).
Concerns have been raised about whether hydroxyurea is associated with abnormal spermatogenesis and teratogenic effects but there are few data to confirm or refute that. There is a theoretical risk of hydroxyurea affecting sperm development and abnormal spermatogenesis has been shown in animals treated with hydroxyurea. Sperm abnormalities, including oligospermia and azoospermia, have been shown in males with SCD taking hydroxyurea, but it is not clear if these are due to the hydroxyurea as similar abnormalities have been described in males with SCD not taking hydroxyurea. One small study compared semen analysis before, during and after hydroxyurea treatment and showed semen parameters were affected whilst on hydroxyurea and did not seem to recover quickly (Berthaut 2008). This included only small numbers of individuals and needs further investigation.
Evidence of adverse outcomes and teratogenic effects in humans exposed to hydroxyurea in utero is limited. There is case report evidence of women with malignant disease taking hydroxyurea through pregnancy with no adverse effects on the fetus and 17‐year follow‐up of the MSH study has shown no adverse fetal outcomes in males or females taking hydroxyurea at conception (Ballas 2009) (Table 9) . An expert panel report evaluating the toxicity of hydroxyurea has led to recommendations that hydroxyurea is stopped in men and women who are trying to conceive and in breastfeeding women (National Toxicology Program 2008). The risks of stopping hydroxyurea whilst attempting conception should be carefully discussed with the individual.
5. Pregnancies in the MSH Study.
| Pregnancy | Hydroxyurea | Placebo | |
| Patients | Normal full‐term delivery | 1 | 2 |
| Elective termination | 2 | 1 | |
| Partners of patients | Normal full‐term delivery | 2 | 0 |
| Spontaneous abortion | 1 | 0 | |
| Still pregnant | 0 | 1 | |
| TOTAL | 6 | 4 | |
The CHAMPS study was the only study to include participants with HbSC (CHAMPS 2011). It was not powered to measure efficacy and terminated early due to low enrolment so it is not possible to make conclusions about the efficacy of hydroxyurea in prevention of pain episodes in people with HbSC. Observational data suggest it may be safe and effective in selected individuals, but this needs further study with a phase 3 randomised study (Luchtman‐Jones 2016). A total of 165 people with HbS/B+thalassaemia were included in one observational study but only 44 received hydroxyurea and efficacy on this subgroup was not presented separately (Voskaridou 2010).
Quality of the evidence
We judged the quality of the evidence for the comparison 'Hydroxyurea compared to placebo' to be moderate for most outcomes and low for quality of life and adverse events, where data were more limited and imprecise (Table 1).
Similarly, we judged the quality of the evidence for the comparison 'Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and risk of stroke' to be moderate (life‐threatening illness and measures of fetal haemoglobin and neutrophil counts) or low quality (pain alteration, deaths during the study and adverse events) where data were more limited and imprecise (Table 2).
The studies contributing the majority of the evidence to these comparison were large, generally well designed and of low risk of bias (BABY HUG 2011; MSH 1995; SWiTCH 2012; TWiTCH 2016). These studies recruited only individuals with haemoglobin SS genotype (HbSS) or haemoglobin Sβº (HbSβº) thalassemia genotypes, therefore, results are not applicable to those with haemoglobin SC genotype (HbSC) genotypes. Furthermore, for the two most recently completed studies, results for secondary outcomes such as neuropsychological status, growth and development, functional evaluations and neurocognitive evaluations have not yet been reported. Any results relevant to this review from further publications will be included in this review.
For the other two comparisons in the review 'Hydroxyurea compared to observation for participants with SCD and risk of stroke' and 'Hydroxyurea compared to no hydroxyurea for participants with SCD,' evidence was limited and of very low quality (Table 3; Table 4). Only a single study contributed to each outcome and both of these studies are likely underpowered due to early termination after recruiting only around 20% of their target participants. Furthermore, due to the factorial design of one of the studies (CHAMPS 2011), a direct comparison between hydroxyurea and a control treatment was not possible so all results for this comparison must be interpreted as indirect.
Potential biases in the review process
A rigorous methodological approach was applied to the review and a comprehensive search strategy was employed as outlined in Electronic searches, therefore, we do not believe that our methodological approach has introduced any bias into the review.
Given the designs of some of the studies included in this review (e.g. cross‐over, factorial) and differing study‐defined definitions of the outcomes of interest to this review, in many instances we felt it would be more appropriate to summarise results narratively or perform separate analyses rather than attempting to adjust original results in order to perform meta‐analysis.
We encourage particular caution when interpreting results of the outcome 'adverse events or toxicity', as it is difficult to distinguish between adverse reactions (i.e. adverse events which are definitely drug‐related) and other adverse events which may be sickle‐ or transfusion‐related. For completeness, we have reported all "adverse events" or "adverse effects" as they are reported in the study publications, regardless of any noted relationship to treatment.
Agreements and disagreements with other studies or reviews
As described earlier in this section, there have been several studies supporting the findings of this review. In addition to these, numerous reviews and other studies discussing the role of hydroxyurea in SCD are available but this is the only Cochrane Review of the topic. A report from the National Institutes of Health (NIH) reviewed 414 studies reporting on the role of hydroxyurea in SCD (published up until July 2014) included five of the randomised studies included in this review (NIH 2014). In some cases in the NIH review, where a literature search was not conducted or was insufficient, the panel relied on their cumulative expertise and knowledge to make consensus‐panel recommendations as outlined below.
Strong recommendations with high‐quality evidence
In adults with sickle cell anaemia who have three or more sickle cell‐associated moderate to severe pain crises in a 12‐month period, treat with hydroxyurea.
In infants of nine to 42 months with sickle cell anaemia, offer treatment with hydroxyurea regardless of clinical severity to reduce SCD‐related complications (e.g. pain, dactylitis, ACS, anaemia).
To ensure proper use of hydroxyurea and maximize benefits and safety, use an established prescribing and monitoring protocol.
Strong recommendations with moderate quality evidence
In adults with sickle cell anaemia who have sickle cell‐associated pain that interferes with daily activities and quality of life, treat with hydroxyurea.
In adults with sickle cell anaemia who have a history of severe or recurrent ACS (or both), treat with hydroxyurea.
In adults with sickle cell anaemia who have severe symptomatic chronic anaemia that interferes with daily activities or quality of life, treat with hydroxyurea.
The group also made an additional moderate recommendation with moderate‐quality evidence that children over 42 months and adolescents with sickle cell anaemia should be offered treatment with hydroxyurea regardless of clinical severity to reduce SCD‐related complications (e.g. pain, dactylitis, ACS, anaemia). There were additional moderate and weak recommendations with low‐ and very low‐quality evidence and a consensus panel recommendation.
Authors' conclusions
Implications for practice.
The initial randomised studies showed efficacy of hydroxyurea in decreasing pain episodes and other acute complications in adults and children with HbSS and HbSߺ‐thalassemia (Belgian Study 1996; Jain 2012; MSH 1995). This led to recommendations that hydroxyurea should be offered to this group of individuals. The evidence of similar efficacy in a group of infants not selected for severity (BABY HUG 2011) is suggestive that all infants with sickle cell anaemia may benefit from hydroxyurea. This has led to recommendations that it should be offered to all children with sickle cell anaemia regardless of clinical severity (NIH 2014) and has led to an increased use of hydroxyurea, particularly in children. There is still insufficient evidence on the long‐term benefits of hydroxyurea, particularly in the prevention of the chronic complications of SCD and also insufficient evidence about the long‐term risks of hydroxyurea including its effects on fertility and reproduction. Hydroxyurea is the only licensed drug for the treatment of SCD and its efficacy in reducing the acute complications of the disease and potential risks of use should be discussed with all individuals with SCD and the families of children with SCD, allowing them to take an informed decision about its use.
There is insufficient evidence to recommend its routine use in those individuals with genotypes other than sickle cell anaemia, but its potential benefits and risks of use should be discussed with those with a severe disease phenotype.
Randomised comparisons of hydroxyurea and transfusion showed that hydroxyurea seems to be as effective as transfusion in preventing stroke in children with sickle cell anaemia and abnormal TCD velocities who have received at least one year of transfusion therapy and have no evidence of vasculopathy on MRA. Hydroxyurea does not seem to be as effective as transfusion therapy in preventing stroke in children with sickle cell anaemia who have already experienced a stroke. Hydroxyurea should be offered to children in the former group (sickle cell anaemia, abnormal TCD velocity, normal MRA, received at least one year of transfusion therapy) as an alternative to long‐term transfusion therapy.
There is insufficient evidence on whether to prescribe a standard dose or dose escalation to the MTD. All individuals treated with hydroxyurea should remain in long‐term follow‐up, which should include regular blood monitoring.
Implications for research.
Many questions remain unanswered relating to the role of hydroxyurea in SCD (as discussed below). All of these questions need to be addressed by appropriately structured randomised controlled studies. However, there is a need for international agreement on a baseline common data set which should be collected in such studies so that they can be analysed later using meta‐analysis techniques, as well as the need for standardised and validated measures for sickle painful crisis and quality of life in people with SCD.
The continued long‐term follow‐up of the participants of the MSH and BABY HUG studies, with regular reporting of outcomes, is welcomed (BABY HUG 2011; MSH 1995). Particular issues for future studies include:
the impact of hydroxyurea on the chronic complications of SCD;
the role of hydroxyurea in the management of specific hard to treat complications of SCD of all ages;
study of the long‐term toxicity profile of hydroxyurea in SCD;
the impact of low (15 mg/kg per day) or medium dose (20 mg/kg per day) hydroxyurea dosage in SCD versus dose escalation to maximum tolerated dose;
role of hydroxyurea in developing countries;
the impact of hydroxyurea in genotypes other than HbSS.
What's new
| Date | Event | Description |
|---|---|---|
| 10 April 2017 | New search has been performed | Six new studies have been added to the review (BABY HUG 2011; Jain 2012; CHAMPS 2011; SCATE 2015; SWiTCH 2012; TWiTCH 2016). Summary of findings tables are now included in the review. |
| 10 April 2017 | New citation required and conclusions have changed | Six new trials have been included in the review (BABY HUG 2011; Jain 2012; CHAMPS 2011; SCATE 2015; SWiTCH 2012; TWiTCH 2016). These include participants with different genotypes, different disease severities and participants at risk of primary and secondary stroke. The inclusion of these new trials has impacted upon the conclusions of the review. |
History
Protocol first published: Issue 3, 2000 Review first published: Issue 2, 2001
| Date | Event | Description |
|---|---|---|
| 10 November 2010 | New search has been performed | A search of the Group's Haemoglobinopathies Trials Register identified 25 new references potentially eligible for inclusion in this review. Fifteen of these were additional references to the BABY HUG study which is listed as ongoing (Baby Hug 2007a). Eight references were to the already included MSH trial and provide no further data or information not already included within the review (MSH 1995). The remaining two references (each published in abstract form) have been added to 'Studies awaiting classification', and we await publication of the full papers in order to accurately assess eligibility (Jain 2010; CHAMPS 2011a). |
| 5 November 2008 | New search has been performed | No new trials were identified by the search of the Group's Haemoglobinopathies trials Register. |
| 1 July 2008 | Amended | Converted to new review format. |
| 1 February 2008 | New search has been performed | The search identified four new references. One was an additional reference to the already included MSH 1995 study (Ballas 2006), however, this did not include any new data that can be added to the review. A further reference was to the already excluded De Montalembert 2006 study (De Montalembert 2006). The third reference was an additional reference to the Baby Hug 2007a study now listed in Ongoing studies (Wynn 2007); and the final reference has been added to Excluded studies (Silva‐Pinto 2007). In addition, the Plain Language Summary has been updated in line with latest guidance from The Cochrane Collaboration. |
| 1 February 2007 | New search has been performed | The search identified 12 new references to four studies. Two of the new references were additional references to the already included MSH 1995 study; however, no further data have been included in the review. Seven of the references were to one study, which is listed in 'Studies awaiting assessment' (Baby Hug 2005). The remaining three new references have been excluded (De Montalembert 2005; Voskaridou 2005; Ware 2006). |
| 1 February 2006 | New search has been performed | The search identified one new reference (Charache 1993) which is an additional reference to the already included MSH 1995 trial. This reference provided no additional data. Ashley Jones became lead author in September 2005 and acts as guarantor of the review. |
| 1 November 2004 | New search has been performed | The search identified one new reference to the already included MSH 1995 trial: Orringer EP, Jones S, Strayhorn D, Hoffman E, Parker J, Greenberg CS. The effect of hydroxyurea (HU) administration on circulating d‐dimer levels in patients with sickle cell anemia [abstract]. Blood 1996;88(10 Suppl 1):496a. Following on from the inclusion of this reference, a further outcome has been added to the review: cost effectiveness of hydroxyurea. |
| 1 November 2003 | New search has been performed | Two additional references to the already included MSH 1995 study have been incorporated into the review. |
| 1 February 2002 | New search has been performed | Quality of life data, presented in an abstract by Terrin (Terrin 1999), which reports on the MSH study (MSH 1995), has now been incorporated into the review. |
Acknowledgements
We note the previous contribution of Adebayo Olujohungbe (AO) to previous versions of the review.
This project was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
Data and analyses
Comparison 1. Hydroxyurea versus placebo for participants with sickle cell disease.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Pain crises | 2 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 1.1 Mean annual crisis rate at 2 years (all crises) | 1 | Mean Difference (IV, Fixed, 95% CI) | ‐2.80 [‐4.74, ‐0.86] | |
| 1.2 Mean annual crisis rate at 2 years (all crises requiring hospitalisation) | 1 | Mean Difference (IV, Fixed, 95% CI) | ‐1.50 [‐2.58, ‐0.42] | |
| 1.3 Number of vaso‐occlusive crises after 18 months of treatment | 1 | Mean Difference (IV, Fixed, 95% CI) | ‐9.6 [‐10.86, ‐8.34] | |
| 2 Proportion experiencing pain | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3 Proportion experiencing life threatening events during study | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Acute chest syndrome | 2 | 492 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.43 [0.29, 0.63] |
| 3.2 Hepatic sequestration | 1 | 299 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.03, 3.06] |
| 3.3 Stroke | 2 | 492 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.54 [0.12, 2.53] |
| 3.4 Patients transfused | 2 | 492 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.66 [0.52, 0.82] |
| 3.5 Splenic sequestration | 1 | 193 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.36, 2.23] |
| 4 Number of life‐threatening events during study | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 4.1 Blood transfusions | 1 | Mean Difference (IV, Fixed, 95% CI) | ‐1.85 [‐2.18, ‐1.52] | |
| 4.2 Hospitalisations | 1 | Mean Difference (IV, Fixed, 95% CI) | ‐8.89 [‐10.04, ‐7.74] | |
| 4.3 Duration of hospitalisation (days) | 1 | Mean Difference (IV, Fixed, 95% CI) | ‐2.00 [‐4.87, ‐3.13] | |
| 5 Deaths during the study | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 All deaths | 3 | 552 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.39 [0.08, 1.96] |
| 5.2 Deaths related to SCD | 3 | 552 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.48 [0.09, 2.60] |
| 6 Change from baseline in fetal haemoglobin (HbF %) | 1 | Mean Difference (Fixed, 95% CI) | Totals not selected | |
| 7 Fetal haemoglobin (HbF %) after treatment | 2 | 359 | Mean Difference (IV, Fixed, 95% CI) | 4.07 [2.95, 5.18] |
| 8 Change from baseline in absolute neutrophil count (x10³ per μL) | 1 | Mean Difference (Fixed, 95% CI) | Totals not selected | |
| 9 Neutrophil response (10⁹/L) after treatment | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 9.1 Neutrophils (x10 9/l) at 10 weeks | 1 | Mean Difference (IV, Fixed, 95% CI) | ‐1.90 [‐2.51, ‐1.29] | |
| 9.2 Neutrophils (x10 9/l) at 2 years | 1 | Mean Difference (IV, Fixed, 95% CI) | ‐1.50 [‐2.01, ‐0.99] | |
| 10 Change from baseline in haemoglobin (g/L) | 1 | Mean Difference (Fixed, 95% CI) | Totals not selected | |
| 11 Change from baseline in m corpuscular volume (fL) | 1 | Mean Difference (Fixed, 95% CI) | Totals not selected | |
| 12 Change from baseline in white blood cells (x10³ per μL) | 1 | Mean Difference (Fixed, 95% CI) | Totals not selected | |
| 13 Change from baseline in absolute reticulocyte count (x10³ per μL) | 1 | Mean Difference (Fixed, 95% CI) | Totals not selected | |
| 14 Change from baseline in reticulocytes (%) | 1 | Mean Difference (Fixed, 95% CI) | Totals not selected | |
| 15 Change from baseline in total bilirubin (mg/L) | 1 | Mean Difference (Fixed, 95% CI) | Totals not selected | |
| 16 Change from baseline in platelet count (x10³ per μL) | 1 | Mean Difference (Fixed, 95% CI) | Totals not selected | |
| 17 Haemoglobin (g/dL) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 17.1 At 10 weeks | 1 | 299 | Mean Difference (IV, Fixed, 95% CI) | 0.5 [0.19, 0.81] |
| 17.2 At the end of the study | 2 | 359 | Mean Difference (IV, Fixed, 95% CI) | 1.04 [0.82, 1.25] |
| 18 Mean corpuscular volume (fL) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 18.1 At 10 weeks | 1 | Mean Difference (IV, Fixed, 95% CI) | 12.30 [9.69, 14.91] | |
| 18.2 At 2 years | 1 | Mean Difference (IV, Fixed, 95% CI) | 10.0 [7.34, 12.66] | |
| 19 Total bilirubin (mg/L) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 19.1 At the end of the study | 2 | 359 | Mean Difference (IV, Fixed, 95% CI) | ‐1.56 [‐1.90, ‐1.23] |
| 20 Reticulocytes | 2 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 20.1 Reticulocytes (10⁵/mm³) at 18 months | 1 | Mean Difference (IV, Fixed, 95% CI) | ‐0.66 [‐0.90, ‐0.42] | |
| 20.2 Reticulocytes (10⁹/L) at 10 weeks | 1 | Mean Difference (IV, Fixed, 95% CI) | ‐130.0 [‐152.17, ‐107.83] | |
| 20.3 Reticulocytes (10⁹/L) at 2 years | 1 | Mean Difference (IV, Fixed, 95% CI) | ‐69.0 [‐91.56, ‐46.44] | |
| 21 Platelet count | 2 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 21.1 Platelet count (10³/mm³) at 18 months | 1 | Mean Difference (IV, Fixed, 95% CI) | ‐0.05 [‐0.16, 0.06] | |
| 21.2 Platelet count (x10⁹/L) at 10 weeks | 1 | Mean Difference (IV, Fixed, 95% CI) | ‐35.0 [‐75.19, 5.19] | |
| 21.3 Platelet count (x10⁹/L) at 2 years | 1 | Mean Difference (IV, Fixed, 95% CI) | ‐24.0 [‐51.88, 3.88] | |
| 22 Packed cell volume | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 22.1 Packed cell volume (%) at 2 years | 1 | Mean Difference (IV, Fixed, 95% CI) | 1.90 [0.85, 2.95] | |
| 23 F reticulocytes | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 23.1 F reticulocytes at 2 years | 1 | Mean Difference (IV, Fixed, 95% CI) | 2.0 [0.18, 3.82] | |
| 24 F cells | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 24.1 F cells (%) at 2 years | 1 | Mean Difference (IV, Fixed, 95% CI) | 13.0 [8.33, 17.67] | |
| 25 Red blood count | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 25.1 Red blood count (10⁶/mm³) at 18 months | 1 | Mean Difference (IV, Fixed, 95% CI) | ‐1.13 [‐1.24, ‐1.02] | |
| 26 White blood cells | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 26.1 White blood cells (109/L) at 2 years | 1 | Mean Difference (IV, Fixed, 95% CI) | ‐2.30 [‐2.97, ‐1.63] | |
| 27 Dense cells | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 27.1 Dense cells (%) at 2 years | 1 | Mean Difference (IV, Fixed, 95% CI) | ‐2.0 [‐3.48, ‐0.52] | |
| 28 Leucocytes | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 28.1 Leucocytes (10³/mm³) at 18 months | 1 | Mean Difference (IV, Fixed, 95% CI) | ‐0.84 [‐3.07, 1.39] | |
| 29 Creatinine | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 29.1 Creatinine (mg/dL) at 2 years | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [‐0.11, 0.11] | |
| 30 Aspartate aminotransferase | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 30.1 Aspartate aminotransferase at 2 years | 1 | Mean Difference (IV, Fixed, 95% CI) | ‐4.0 [‐9.40, 1.40] | |
| 31 Alkaline phosphatase | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 31.1 Alkaline phosphatase at 2 years | 1 | Mean Difference (IV, Fixed, 95% CI) | ‐2.0 [‐15.78, 11.78] | |
| 32 Change from baseline in growth | 1 | Mean Difference (Fixed, 95% CI) | Totals not selected | |
| 32.1 Height (cm) | 1 | Mean Difference (Fixed, 95% CI) | ‐0.2 [1.00, 0.60] | |
| 32.2 Weight (kg) | 1 | Mean Difference (Fixed, 95% CI) | 0.10 [‐0.20, 0.40] | |
| 32.3 Head circumference (cm) | 1 | Mean Difference (Fixed, 95% CI) | ‐0.2 [‐0.60, 0.20] | |
| 33 Quality of life: general health perception | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 33.1 General health perception at 6 months | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.30 [‐0.54, 1.14] | |
| 33.2 General health perception at 1 year | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.60 [‐0.18, 1.38] | |
| 33.3 General health perception at 18 months | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.9 [0.08, 1.72] | |
| 33.4 General health perception at 2 years | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.4 [‐0.51, 1.31] | |
| 34 Quality of life: pain recall | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 34.1 Pain recall at 6 months | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.40 [‐0.13, 0.93] | |
| 34.2 Pain recall at 1 year | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.40 [‐0.18, 0.98] | |
| 34.3 Pain recall at 18 months | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.70 [0.11, 1.29] | |
| 34.4 Pain recall at 2 years | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.3 [‐0.30, 0.90] | |
| 35 Quality of life: social function | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 35.1 Social function at 6 months | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.4 [‐0.15, 0.95] | |
| 35.2 Social function at 1 year | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.2 [‐0.36, 0.76] | |
| 35.3 Social function at 18 months | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.40 [‐0.21, 1.01] | |
| 35.4 Social function at 2 years | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.3 [‐0.31, 0.91] | |
| 36 Changes in 'Ladder of Life' | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 36.1 Changes in 'Ladder of Life' at 6 months | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [‐0.52, 0.52] | |
| 36.2 Changes in 'Ladder of Life' at 1 year | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.4 [‐0.15, 0.95] | |
| 36.3 Changes in 'Ladder of Life' at 18 months | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.30 [‐0.22, 0.82] | |
| 36.4 Changes in 'Ladder of Life' at 2 years | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.3 [‐0.23, 0.83] | |
| 37 Proportion of participants with signs of organ damage | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 37.1 New leg ulcers | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.44, 1.64] | |
| 37.2 Aseptic necrosis (humerus or femur) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.39, 2.37] | |
| 37.3 Decreased spleen function at exit (compared to baseline) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.72 [0.44, 1.16] | |
| 38 Signs of organ damage ‐ change from baseline in DTPA GFR | 1 | Mean Difference (Fixed, 95% CI) | Totals not selected | |
| 39 Signs of organ damage ‐ change from baseline in Howell‐Jolley body (per 106 red blood cells) | 1 | Mean Difference (Fixed, 95% CI) | Totals not selected | |
| 40 Signs of organ damage ‐ change from baseline in pitted cells (%) | 1 | Mean Difference (Fixed, 95% CI) | Totals not selected | |
| 41 Signs of organ damage ‐ change from baseline in spleen: liver ratio of counts | 1 | Mean Difference (Fixed, 95% CI) | Totals not selected | |
| 42 Signs of organ damage ‐ change from baseline in spleen volume (cm3) | 1 | Mean Difference (Fixed, 95% CI) | Totals not selected | |
| 43 Signs of organ damage ‐ change from baseline in creatinine (mg/L) | 1 | Mean Difference (Fixed, 95% CI) | Totals not selected | |
| 44 Signs of organ damage ‐ change from baseline in Schwartz GFR | 1 | Mean Difference (Fixed, 95% CI) | Totals not selected | |
| 44.1 Schwartz glomerular filtration rate (mL/min per 1.73m2) | 1 | Mean Difference (Fixed, 95% CI) | ‐8.0 [‐35.00, 21.00] | |
| 45 Signs of organ damage ‐ change from baseline in cystatin C | 1 | Mean Difference (Fixed, 95% CI) | Totals not selected | |
| 46 Signs of organ damage ‐ change from baseline in urine osmolality | 1 | Mean Difference (Fixed, 95% CI) | Totals not selected | |
| 47 Signs of organ damage ‐ change from baseline in urine pH | 1 | Mean Difference (Fixed, 95% CI) | Totals not selected | |
| 48 Signs of organ damage ‐ change from baseline in urine‐specific gravity | 1 | Mean Difference (Fixed, 95% CI) | Totals not selected | |
| 49 Signs of organ damage ‐ change from baseline in total kidney volume | 1 | Mean Difference (Fixed, 95% CI) | Totals not selected | |
| 50 Signs of organ damage ‐ change from baseline in TCD ultrasound velocity (time‐averaged mean maximum velocity) | 1 | Mean Difference (Fixed, 95% CI) | Totals not selected | |
| 51 Signs of organ damage ‐ change from baseline in CNS measures | 1 | Mean Difference (Fixed, 95% CI) | Totals not selected | |
| 51.1 Bayley Mental Development Index | 1 | Mean Difference (Fixed, 95% CI) | 3.0 [0.00, 8.00] | |
| 51.2 Bayley motor performance development index | 1 | Mean Difference (Fixed, 95% CI) | 2.0 [‐1.00, 7.00] | |
| 52 Proportion of participants experiencing adverse events and toxicity | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 52.1 hair loss at 1 or 2 visits | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.69, 2.26] | |
| 52.2 hair loss at 3 or more visits | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.61 [0.31, 1.21] | |
| 52.3 skin rash at 1 or 2 visits | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.62, 1.51] | |
| 52.4 skin rash at 3 or more visits | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.58, 1.60] | |
| 52.5 fever at 1 or 2 visits | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.55, 1.69] | |
| 52.6 fever at 3 or more visits | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.68, 1.17] | |
| 52.7 Gastroinestinal disturbance at 1 or 2 visits | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.50, 1.36] | |
| 52.8 Gastrointestinal disturbance at 3 or more visits | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.74, 1.31] | |
| 52.9 Other abnormalities at 1 or 2 visits | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.60 [0.33, 1.11] | |
| 52.10 Other abnormalities at 3 or more visits | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.83, 1.40] | |
| 52.11 Hospitalisation (for any reason) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.72, 0.96] | |
| 52.12 Dactylitis | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.34 [0.20, 0.58] | |
| 52.13 Priapism | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.52 [0.26, 8.87] | |
| 52.14 Sepsis or bacteraemia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.40 [0.08, 2.03] | |
| 52.15 Splenomegaly | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.61, 1.32] | |
| 52.16 Absolute Neutrophil Count < 500 | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.53 [0.50, 12.71] | |
| 52.17 Absolute Neutrophil 500 ‐ 1250 | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.53 [1.58, 4.03] | |
| 52.18 Thrombocytopaenia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.59 [0.64, 3.92] | |
| 52.19 Alanine transaminase > 150 U/L | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.02 [0.19, 21.92] | |
| 52.20 Severe anaemia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.51 [0.05, 5.48] | |
| 52.21 Bilirubin | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.34 [0.01, 8.17] | |
| 52.22 Creatinine | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 52.23 Skin and subcutaneous disorders | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.75, 1.10] | |
| 52.24 Splenic sequestration | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.36, 2.23] | |
| 52.25 Gastroenteritis | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.44 [0.28, 0.71] | |
| 52.26 Nausea | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.0 [0.25, 99.95] | |
| 52.27 Skin Rash | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.0 [0.38, 129.93] |
Comparison 2. Hydroxyurea and phlebotomy compared to transfusion and chelation for participants with SCD and risk of stroke.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Proportion experiencing pain | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Vaso‐occlusive or sickle cell‐related pain (all) | 1 | 133 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.81, 1.30] |
| 1.2 Vaso‐occlusive or sickle cell‐related pain (serious) | 2 | 254 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.37 [1.59, 7.11] |
| 2 Proportion experiencing life‐threatening events during study | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 Stroke (secondary) | 1 | 133 | Risk Ratio (M‐H, Fixed, 95% CI) | 14.78 [0.86, 253.66] |
| 2.2 Transient ischaemic attack (primary) | 1 | 121 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.21, 4.84] |
| 2.3 Transient ischaemic attack (secondary) | 1 | 133 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.66 [0.25, 1.74] |
| 2.4 Other neurological event (primary) | 1 | 121 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.07, 15.88] |
| 2.5 Acute chest syndrome | 2 | 254 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.84 [1.25, 6.42] |
| 2.6 Infections and Infestations | 2 | 254 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.65 [1.05, 12.76] |
| 2.7 Splenic sequestration or splenectomy | 2 | 254 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.34 [0.01, 8.16] |
| 2.8 Hepatobiliary disease | 1 | 121 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.07, 15.88] |
| 2.9 Total with serious adverse events | 2 | 254 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.93 [1.17, 3.20] |
| 2.10 Total with sickle cell related, non‐neurological adverse events | 1 | 133 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.10 [1.42, 6.75] |
| 3 Deaths during the study | 2 | 254 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.06, 15.42] |
| 4 Change from baseline in fetal haemoglobin (HbF %) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 5 Change from baseline in absolute neutrophil count (x10⁹/L) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 6 Change from baseline in mean corpuscular volume (fL) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 7 Change from baseline in sickle haemoglobin (%) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 8 Change from baseline in haemoglobin (g/L) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 9 Change from baseline in absolute reticulocyte count (10⁹ / L) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 10 Change from baseline in white blood count (10⁹ / L) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 11 Change from baseline in platelets (10⁹ / L) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 12 Change from baseline in total bilirubin (mg/L) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 13 Change from baseline in liver iron concentration | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 14 Change from baseline in serum ferritin (ng/mL) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 15 Change from baseline in lactate dehydrogenase (U/L) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 16 Signs of organ damage ‐ CNS measures at the end of the study | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 16.1 Final TCD ultrasound velocity | 1 | Mean Difference (IV, Fixed, 95% CI) | ‐5.0 [‐9.16, ‐0.84] | |
| 17 Proportion of participants experiencing non‐neurological adverse events and toxicity | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 17.1 Infections and infestations | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.24 [0.89, 1.72] | |
| 17.2 Gastrointestinal disorders | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.74, 1.80] | |
| 17.3 Fever | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.66, 1.75] | |
| 17.4 Musculoskelatal disorders | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.59, 2.20] | |
| 17.5 Immune system disorders | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.12 [0.02, 0.96] | |
| 17.6 Cholelithiasis | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.74 [0.17, 3.17] | |
| 17.7 Cholecystitis | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.97 [0.18, 21.21] | |
| 17.8 Asthma | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.74 [0.17, 3.17] | |
| 17.9 Acute chest syndrome | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.72 [0.53, 5.61] | |
| 17.10 Renal or urinary disorder | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.66 [0.11, 3.80] | |
| 17.11 Priapism | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.97 [0.18, 21.21] | |
| 17.12 Cathether‐related complications | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.48 [0.26, 8.56] | |
| 17.13 Cardiac disorder | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.06, 15.42] | |
| 17.14 Hyderbilirubianemia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.24, 1.02] | |
| 17.15 Alanine transaminase increase | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.55, 2.01] | |
| 17.16 Aspartate transaminase increase | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.38 [0.66, 2.88] | |
| 17.17 Serum creatinine increase | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.97 [0.51, 7.55] | |
| 17.18 Thrombocytopenia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.90 [0.87, 54.51] | |
| 17.19 Reticuloctopenia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.90 [2.16, 22.02] | |
| 17.20 Neutropenia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.85 [1.30, 74.80] | |
| 17.21 Anaemia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.57 [2.05, 21.05] | |
| 17.22 Sickle cell pain | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.29 [0.92, 1.82] | |
| 17.23 Sickle cell‐related events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.81, 1.30] | |
| 17.24 All adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.92, 1.05] |
Comparison 3. Hydroxyurea compared to observation for participants with SCD and risk of stroke.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Proportion experiencing life‐threatening events during the study | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Vaso‐occlusive events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.4 [0.10, 1.64] | |
| 1.2 Acute splenic sequestration | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.07, 14.05] | |
| 1.3 Blood transfusions required | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.04, 2.73] | |
| 2 Signs of organ damage ‐ proportion of participants with a change in CNS measures | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.1 Participants converting from conditional to abnormal TCD ultrasound velocity | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.2 [0.03, 1.45] | |
| 3 Signs of organ damage ‐ change from baseline in CNS measures | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3.1 TCD ultrasound velocity (time‐averaged mean maximum velocity) | 1 | Mean Difference (IV, Fixed, 95% CI) | ‐25.7 [‐45.38, ‐6.02] | |
| 4 Adverse events and toxicity | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 Dizziness | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.14, 66.53] | |
| 4.2 Headaches | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.07, 14.05] | |
| 4.3 Transient neutropenia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.14, 66.53] | |
| 4.4 Reticulocytopenia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.07, 14.05] | |
| 4.5 Parasite infection | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.07, 14.05] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
BABY HUG 2011.
| Methods | Randomised, double blind (participant, investigator), placebo control, parallel assignment, efficacy study conducted at 13 centres in the USA. | |
| Participants | Children aged 9 months to 18 months with SCD (HbSS or HbSߺ‐thalassemia) irrespective of clinical severity. Exclusion criteria were transfusion within 2 months, height, weight or head circumference below the 5th percentile, mental development index less than 70 or abnormal TCD ultrasound velocity. 193 randomised, 96 participants to hydroxyurea and 97 to placebo. Mean (SD) age: hydroxyurea group : 13.6 (2.7) months, placebo group: 13.5 (2.8) months 187 (97%) with HbSS genotype, 109 (56%) females |
|
| Interventions | Hydroxyurea (20 mg/kg/day) versus placebo for 24 months. | |
| Outcomes | Primary outcomes: spleen function (decline in splenic uptake); and glomerular filtration rate with 99mTc‐diethyl‐enetriaminepentaacetic acid plasma clearance. Secondary outcomes: ratio of nuclear decay counts in the spleen and liver; proportion of red blood cells containing pits or Howell‐Jolly bodies; renal function; growth; and development (including neuro‐developmental assessment). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The telephone randomisation schedule was developed by the medical co‐ordinating centre. |
| Allocation concealment (selection bias) | Low risk | Centralised telephone randomisation was used. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Some secondary outcomes reported for only the individuals who completed the study but ITT approach taken for primary outcomes (via multiple imputation) and safety outcomes so risk of bias judged to be low. |
| Selective reporting (reporting bias) | Low risk | All outcomes well defined in the methods and reported well in the results. |
| Other bias | Low risk | None identified. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants, caregivers and medical coordinating centre staff were masked to treatment allocation via packaging and treatment of the same appearance. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | An unmasked "primary endpoint person" monitored laboratory values and assisted clinical management. Other outcome assessors (e.g. those reading the liver‐spleen scans). |
Belgian Study 1996.
| Methods | Randomised placebo‐controlled cross‐over study conducted in a single centre in Belgium. | |
| Participants | 25 children with HbSS genotype, age 2 ‐ 22 years (median 9 years) with > 3 vaso‐occlusive events reported in preceding 12 months and/or history of CVA off transfusion (severe alloimmunisation or compliance), ACS, ASS. | |
| Interventions | Hydroxyurea 20 mg/kg/day rising to 25 (unless toxic cytopenia) versus placebo. Treatment period was for 6 months. | |
| Outcomes | Number of hospitalisations. Number of inpatient days. FBC. HbF%. | |
| Notes | Data are not presented in a way that results can be included in the review so results are presented narratively. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as "drawing sealed envelopes, patients were randomly allocated to one of the following treatment sequences", therefore the generation of the treatment sequence are not clear from this statement. |
| Allocation concealment (selection bias) | Unclear risk | Described as "drawing sealed envelopes, patients were randomly allocated to one of the following treatment sequences", therefore the allocation of the treatment sequence are not clear from this statement. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 3 participants who were excluded from the analysis due to their failure to attend the monthly evaluation at 4 ‐ 5 months. There was no discussion of whether or not an ITT analysis was used so the risk is unclear. |
| Selective reporting (reporting bias) | High risk | A planned outcome (number of days in pain) was dropped from analysis due to difficulty in obtaining information to inform this analysis from participants |
| Other bias | Low risk | Unclear if a washout period was used in this cross‐over study but tests for period effects and carry‐over effect were not significant so the risk of bias in the cross‐over design is low. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The hospital pharmacy provided the treatment and placebo for each participant and both were described as "indistinguishable," however the physician was aware of the treatment schedule because of the difficulty of blinding the attending physician to the treatment received. Unclear if this could have influenced results. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided. |
CHAMPS 2011.
| Methods | Randomised multicentre, phase II, double blind placebo‐controlled study. | |
| Participants | Eligible participants over the age of 5 years with HbSC and at least 1 vaso‐occlusive event in the previous 12 months (but none in the 4 weeks prior to study entry). 44 participants randomised; 11 to each treatment group (see 'Interventions'). Mean age: 13.6 years (range 5 ‐ 53 years), 43% females. |
|
| Interventions | Participants randomised to 1 of 4 arms in a factorial design: 1. hydroxyurea (20 mg/kg/day) and magnesium (0.6 mmol/kg/day in 2 doses); 2. hydroxyurea (20 mg/kg/day) and placebo; 3. placebo and magnesium (0.6 mmol/kg/day in 2 doses); 4. placebo and placebo. |
|
| Outcomes | Primary outcome: proportion of hyperdense red blood cells at 8 weeks. Secondary outcomes: central laboratory evaluations (including measurements of red cell density, HbF, red cell cation content, KCl co‐transport and Gardos channel activity, cell adhesion to endothelial cells and laminin, and erythrocyte membrane phosphatidyl serine (PS exposure)) at baseline (twice) and weeks 8, 16, 24, and 44. Participants were evaluated at 2 or 4 week intervals for 11 months (15 visits). |
|
| Notes | The study was not designed to measure efficacy and all analyses were considered exploratory. The study was terminated early due to low enrolment after 44 participants had been randomised (target 188). Due to factorial design, no data can be entered into analysis and results for hydroxyurea groups (groups 1 and 2) compared to no hydroxyurea groups are summarised narratively. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A sequential allocation algorithm (i.e. minimisation) was used due to small numbers within each strata (site and age group). |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 36 out of 44 participants completed 8 weeks and 22 out of 44 completed 44 weeks. Only those who completed the follow‐up time were included in analysis (8 weeks for primary endpoint and 44 weeks for secondary endpoints). This is not an ITT approach. |
| Selective reporting (reporting bias) | Low risk | Outcome defined in the methods section well described in the results section. Study was not designed to measure efficacy. |
| Other bias | Low risk | Study terminated early after only 44 of planned sample size of 188 were recruited. However, the study was not designed to measure efficacy and performed only exploratory analyses therefore the early termination is unlikely to have introduced bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | All tablets were 'over‐capsulated' to disguise appearance. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided. |
Jain 2012.
| Methods | Double‐blind randomised controlled study conducted in a tertiary hospital in Nagpur City, India. | |
| Participants | Indian children between the ages of 5 and 18 years with severe manifestations (defined as 3 or more blood transfusions or VOC requiring hospitalisation per year) despite high HbF. Exclusion criteria included seropositivity for HIV or chronic illness. 60 participants randomised; 30 to each treatment group. 53% females (16 females per group). Mean (SD) age ‐ hydroxyurea group: 12.73 (4.4) years, placebo group: 11.73 (4.08) years. |
|
| Interventions | Hydroxyurea (fixed dose 10 mg/mg/day) compared to placebo for 18 months. | |
| Outcomes | Primary outcome: frequency of VOC per participant per year. Secondary outcomes: frequency of blood transfusions, hospitalisations and HbF levels. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomised using randomisation tables. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants completed 18‐months follow‐up and were included in analysis. |
| Selective reporting (reporting bias) | Low risk | No protocol available, outcomes defined in the methods reported well in the results. |
| Other bias | Low risk | None identified. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants were blinded with placebo tablets of identical appearance. The clinician who assessed participants were not aware of treatment arm. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The laboratory technician and the clinician who assessed participants were not aware of treatment arm. |
MSH 1995.
| Methods | Randomised multicentre parallel, double‐blind placebo‐controlled study. Conducted in the USA and Canada across 21 sites. | |
| Participants | Adults over 18 years of age with HbSS genotype who had reported more than 3 'crises' to treating physician in the preceding 12 months and who had < 15% HbA. Exclusions included: HbA > 15%, pregnancy, opiate addiction, other potent anti‐sickling agents, cytopenia, CVA in the preceding 6 years, HIV antibody +, prior hydroxyurea therapy 152 randomised to hydroxyurea and 147 given placebo. |
|
| Interventions | Hydroxyurea starting at 15 mg/kg/day rising 12‐weekly by 5 mg/kg/day unless marrow depression (then cessation of drug until recovery and restarted at 2.5 mg/kg/day lower, i.e. MTD) compared to placebo for 2 years. | |
| Outcomes | Primary outcome: pain events ‐ attending hospital > 4 hours & parenteral opiate treatment. Secondary outcomes: 'crises' including ACS, CVA, priapism etc, daily pain charts, time to first and second crisis, FBC, F cells & Hb F%, dense cells. | |
| Notes | 93% follow‐up at 2 years, treatment stopped in 14 hydroxyurea and 6 placebo participants. Due to beneficial treatment effects, the study was stopped at a mean follow‐up of 21 months, before the planned 24 months of treatment had been completed for all participants. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Treatment assignments were made centrally using a computer programme to generate separate, randomised block assignment schedules for each clinic. |
| Allocation concealment (selection bias) | Unclear risk | There was no clear discussion on allocation concealment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Withdrawals from the study were well documented, all participants included in analysis in an ITT approach in the primary publications but later reported outcomes (such as quality of life) reported only for those who contributed data. |
| Selective reporting (reporting bias) | Low risk | All outcomes well defined in the methods and reported well in the results. |
| Other bias | Low risk | None identified. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Stated as double‐blind (physician and participant), where treatment was assigned in combinations of identically appearing capsules. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was no clear discussion on blinding of outcome assessors. |
SCATE 2015.
| Methods | Phase III randomised, partially masked (outcome assessors) multicentre study conducted in 3 centres in the USA, Jamaica and Brazil between May 2012 and August 2013. | |
| Participants | Children with SCA and conditional TCD ultrasound velocities (170 cm ‐ 199 cm per second). Exclusion criteria were prior abnormal TCD velocities or clinical stroke, red blood cell transfusion within 2 months of enrolment, concurrent use of another anti‐sickling medication or contraindication to hydroxyurea therapy (allergy, pregnancy, renal insufficiency). 22 participants randomised, 11 to each treatment group. Mean age (SD): hydroxyurea group: 6.2 (2.4) years, observation group: 6.6 (1.5) years. 64% females, 7 per group. 21 participants with HbSS and 1 with HbSβº‐thalassemia. |
|
| Interventions | Hydroxyurea (starting at 20 mg/kg/day escalated to a maximum of 35 mg/kg/day) compared to standard treatment (observation). | |
| Outcomes | Primary outcome: conversion to abnormal maximum abnormal TAMV. Secondary outcomes: changes in serial TCD velocities. Indicence of acute events including stroke. Health‐related quality of life (planned but not recorded, see 'Selective reporting' below). |
|
| Notes | The planned length of follow‐up was 30 months but the study was terminated early after 15 months of follow‐up due to slow participant accrual and the unlikelihood of meeting the trial recruitment target (100) and the primary endpoint. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was performed using an adaptive blocked algorithm. |
| Allocation concealment (selection bias) | Low risk | Central pharmacy distribution of allocations. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Numbers screened, randomised and number receiving randomised treatment reported. Analysis conducted using an ITT approach. |
| Selective reporting (reporting bias) | High risk | A planned outcome (health‐related quality of life) was not analysed or presented as the outcome was not sufficiently collected due to early study termination. |
| Other bias | High risk | Study terminated early after only 22 of planned sample size of 100 were recruited therefore study is likely to be statistically underpowered. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants and personnel could not be masked to treatment allocation by design (hydroxyurea compared to observation only). The primary outcome (conversion to abnormal TAMV) was objective and determined by masked outcome assessors so lack of blinding of participants and personnel is unlikely to have affected results. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | All TCD site examiners, central reviewers were masked and site clinicians were masked to participants TCD results. |
SWiTCH 2012.
| Methods | Phase III, multicentre, single masked (outcome assessors), non‐inferiority study conducted across 26 paediatric sites in the USA. | |
| Participants | Participants with previous clinical stroke, aged 7 to 17 years, PRBC transfusions for at least 18 months and transfusional iron overload. No specific exclusion criteria stated. 133 participants were randomised; 67 randomised to hydroxyurea treatment and 66 to standard treatment. Mean (SD) age at study enrolment in years: hydroxyurea group 13.0 (4.0) years, standard treatment group 13.3 (3.8) years. 132 out of 133 participants with HbSS genotype, 61 females (46%). |
|
| Interventions | Hydroxyurea starting at 20 mg/kg/day escalated to MTD and phlebotomy compared to standard treatment (transfusions and chelation) for 30 months. | |
| Outcomes | The primary outcome of the trial was a composite outcome. This involved occurrence of a secondary stroke and quantitative liver iron level change from baseline. Secondary outcomes included quality of life, non‐stroke neurological events, other SCD‐related events, growth and development, functional evaluations, neurocognitive evaluations, transfusion related complications, chelation related complications, hydroxyurea‐related complications, phlebotomy related complications, liver biopsy related complications and adverse and serious adverse events. |
|
| Notes | Numerous secondary outcomes were not reported in the main paper or any subsequent papers. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Study described as randomised, no further details given. |
| Allocation concealment (selection bias) | Unclear risk | No details were given on allocation concealment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Withdrawals from treatment reported, all individuals included in analysis in an ITT approach. |
| Selective reporting (reporting bias) | High risk | Many of the secondary outcomes (such as growth and development, functional evaluations, neurocognitive evaluations) have not yet been reported, If these results can be included at a later date then this judgement will be reconsidered. |
| Other bias | Low risk | None identified. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants and personnel could not be masked to treatment allocation by design (hydroxyurea compared to transfusion). The primary outcome (secondary stroke and quantitative liver iron level change from baseline) was objective and determined by masked outcome assessors so lack of blinding of participants and personnel is unlikely to have affected results. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The primary outcome (secondary stroke and quantitative liver iron level change from baseline) were determined by a group of treatment masked neurologists and neuroradiologists. |
TWiTCH 2016.
| Methods | Multicentre phase III randomised open‐label (partially masked) non‐inferiority study conducted at 26 paediatric hospitals and health centres in the USA and Canada. | |
| Participants | Children aged 4 ‐ 16 years with SCA and abnormal TCD ultrasound velocities (> 200 cm per second) if they had received 12 months of chronic transfusions. Exclusion criteria were documented clinical stroke, TIA or severe vasculopathy. 121 participants were randomised; 60 randomised to hydroxyurea treatment and 61 to standard treatment. Mean (SD) age at study enrolment in years: hydroxyurea group 9.5 (2.6) years, standard treatment group 9.7 (3.2) years. 119 out of 121 participants with HbSS genotype, 73 females (60%). |
|
| Interventions | Hydroxyurea starting at 20 mg/kg/day escalated to MTD and phlebotomy compared to standard treatment (transfusions and chelation) for 24 months. | |
| Outcomes | Primary outcome: maximum TCD time averaged mean velocity on the index side (i.e. the cerebral hemisphere with the higher mean arterial velocity at baseline assessment). Secondary outcomes: TCD velocity on the non‐index side, neurological events, new brain lesions, hepatic iron overload, SCD‐related events, treatment‐related complication (reported in this publication) neuropsychological status, quality of life and growth and (to be reported in future publications). |
|
| Notes | Study was terminated early at the first interim analysis when non‐inferiority was demonstrated, target sample size was met at early termination. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Centralised block randomisation with block size four, with stratification by site and balanced by baseline age and TCD velocity. |
| Allocation concealment (selection bias) | Low risk | Central randomisation and treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Numbers discontinuing the interventions stated, all participants included in analysis in an ITT analysis. |
| Selective reporting (reporting bias) | High risk | Outcomes of neuropsychological status, quality of life and growth were measured but results are not yet published. If these results can be included at a later date then this judgement will be reconsidered. |
| Other bias | Low risk | Study was terminated early at the first interim analysis when non‐inferiority was demonstrated, target sample size was met at early termination so the study is adequately powered to detect differences. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants and personnel could not be masked to treatment allocation by design (hydroxyurea compared to transfusion). The primary outcome (maximum TCD time averaged mean velocity) was objective and determined by masked outcome assessors so lack of blinding of participants and personnel is unlikely to have affected results. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | All TCD examinations were read centrally by observers blinded to treatment allocation and previous TCD results. |
ACS: acute chest syndrome ASS: acute splenic sequestration CVA: cerebro‐vascular accident Cytopenia: refers to either neutropenia or thrombocytopenia, anaemia is also a risk but was not reported FBC: full blood count HbA: adult haemoglobin HbF: fetal haemoglobin HbSβº: haemoglobin Sβºthalassaemia genotype HbSC: haemoglobin SC genotype HbSS: haemoglobin SS genotype ITT: intention‐to‐treat KCl: potassium chloride MTD: maximum tolerated dose PRBC: packed red blood cells SCA: sickle cell anaemia SCD: sickle cell disease SD: standard deviation TAMV: time averaged mean velocity TCD: transcranial doppler TIA: transient ischaemic attack VOC: vaso‐occlusive crisis
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Al‐Nood 2011 | This is not a randomised study. |
| De Montalembert 2006 | This study does not fulfil the inclusion criteria. The length of treatment was 8 days, the inclusion criteria state that treatment should be at least 1 month. |
| George 2013 | This does not make a randomised comparison of hydroxyurea and placebo or standard treatment (the randomised comparison is dosing schedules of hydroxyurea). |
| NCT00004492 | This does not make a randomised comparison of hydroxyurea and placebo or standard treatment. |
| NCT01848925 | This study does not fulfil the inclusion criteria. The length of treatment was 7 days, the inclusion criteria state that treatment should be at least 1 month. |
| NCT01960413 | This does not make a randomised comparison of hydroxyurea and placebo or standard treatment. |
| NCT02149537 | This is not an appropriate design to measure the effectiveness of hydroxyurea (cross‐over design of low‐dose hydroxyurea compared to no treatment to monitor those at increased risk of infection). |
| Pushi 2000 | This is not a randomised study. |
| Silva‐Pinto 2007 | This is not a randomised study. |
| Silva‐Pinto 2014 | This is not a randomised study. |
| Vichinsky 2013 | This does not make a randomised comparison of hydroxyurea and placebo or standard treatment. |
| Voskaridou 2005 | This is not a randomised study. |
Characteristics of studies awaiting assessment [ordered by study ID]
Anyanwu 2016.
| Methods | Prospective, randomised, placebo‐controlled, double‐blinded phase III trial (NOHARM trial) |
| Participants | Study participants will be recruited from the Mulago Hospital Sickle Cell Clinic (MHSCC) in Kampala, Uganda. Children aged 1 to 4 years with documented HbSS living in an area of meso‐endemic malaria transmission. |
| Interventions | Hydroxyurea 20 ± 2.5 mg/kg/day compared to placebo |
| Outcomes | Primary outcome: malaria incidence, defined as episodes of clinical malaria occurring over the 1‐year randomised study treatment period. Secondary outcomes: frequency of haematologic toxicities and AEs, relationships between hydroxyurea treatment and fetal haemoglobin, soluble intracellular adhesion molecule‐1, and nitric oxide levels, and between levels of these factors and risk of subsequent malaria. |
| Notes | Currently, only a protocol is available for the NOHARM study. We are unsure if this study meets the inclusion criteria of the review of 'Type of Participants' due to the study objectives around determining the incidence of malaria in SCA individuals. We will make an assessment of the eligibility of the population when study results are available. |
NCT02560935.
| Methods | Randomised, double‐blind, parallel group, dose‐controlled study (SPRING) |
| Participants | Participants with HbSS or HbSβº‐thalassemia, S variant with baseline haemoglobin less than 10 g/dL or other sickle cell syndromes apart from HbSC between the ages of 5 and 12 years, living in sub‐Saharan Africa, without prior overt stroke. Inclusion criteria for a 'non‐elevated TCD group' (participants who are not eligible to receive hydroxyurea therapy but are willing to be following for a minimum of 3 years). |
| Interventions | Hydroxyurea (moderate dose): 20 mg/kg/day (range 17.5 ‐ 26 mg/kg/day) for 24 months. Hydroxyurea (low dose): 10 mg/kg/day (range 7 ‐ 15 mg/kg/day) for 24 months. |
| Outcomes | Primary outcome: efficacy of moderate vs low‐dose hydroxyurea therapy for primary stroke prevention. Secondary outcome: incidence of all‐cause hospitalizations. Secondary outcome: long‐term safety of hydroxyurea therapy. |
| Notes | Unclear if this is an appropriate design (dose‐control) and population for the review ('non‐elevated TCD group'). Trial is ongoing (estimated completion date ‐ December 2021), we will make an assessment of the eligibility of the design and population when trial results are available. |
NCT02675790.
| Methods | Randomised, double‐blind, parallel group, dose‐controlled study (SPRINT). |
| Participants | Participants between the age of 1 and 18 years with SCA and a history of stroke up to 30 days before entry into the study, living in sub‐Saharan Africa. |
| Interventions | Hydroxyurea (moderate dose): 20 mg/kg/day (range 17.5 ‐ 26 mg/kg/day) for 24 months. Hydroxyurea (low dose): 10 mg/kg/day (range 7 ‐ 15 mg/kg/day) for 24 months. |
| Outcomes | Primary outcome: rate of clinical stroke recurrence. Secondary outcome: incidence of all‐cause hospitalisation. |
| Notes | Unclear if this is an appropriate design (dose‐control) and population for the review (participants can have already received hydroxyurea treatment). Study is ongoing (estimated completion date July 2019), we will make an assessment of the eligibility of the design and population when study results are available. |
AEs: adverse events HbSβº: haemoglobin Sβºthalassaemia genotype HbSC: haemoglobin SC genotype HbSS: haemoglobin SS genotype TCD: transcranial doppler
Characteristics of ongoing studies [ordered by study ID]
NCT01389024.
| Trial name or title | NCT01389024: Hydroxyurea to Prevent Brain Injury in Sickle Cell Disease (HUPrevent) |
| Methods | Randomised, double‐blind, parallel design, phase 2 study. |
| Participants | Participants with HbSS or HbSβº‐thalassemia aged between 9 and 48 months of age, with or without central nervous system complications. |
| Interventions | Hydroxyurea 20 mg/kg/day increased by 5 mg/kg every 8 weeks to maximum of 35 mg/kg/day; placebo (sucrose) 0.2 mL/kg/day increased to max of 0.35 mL/kg/day. |
| Outcomes | Primary outcome: central nervous system complications (a composite of abnormally elevated cerebral blood flow velocity as measured by TCD ultrasound, SCI, or stroke) Secondary outcome: proportion of participants with severe adverse events attributed to study procedures. |
| Starting date | October 2011 |
| Contact information | Johns Hopkins University Diane Weiss, BA (dweiss14@jhmi.edu) James F. Casella, MD (jcasella@jhmi.edu) |
| Notes | Estimated completion date October 2017 (final data collection date for primary outcome measure) |
HbSβº: haemoglobin Sβºthalassaemia genotype HbSC: Haemoglobin SC genotype HbSS: Haemoglobin SS genotype SCI: silent cerebral infarct TCD: transcranial doppler
Differences between protocol and review
The final secondary outcome (cost effectiveness of hydroxyurea) has been added at the 2005 update of this review but removed in the 2017 update of the review as economic methods which are outside the scope of Cochrane reviews are required to truly reflect cost‐effectiveness of an intervention.
The protocol stated that odds ratios would be used as the measure of treatment effect for dichotomous data, however on reflection, we felt it more appropriate to present risk ratios in preference to odds ratios (OR), as odds ratios give an inflated impression of the size of effect where event rates are high, as is the case of these studies.
2017 update: definition of primary outcome 'Pain Alteration' updated to better reflect the measures which are suitable for inclusion under this outcome.
2017 update: We changed the definition of the outcome: "Any reported adverse effects or toxicity of hydroxyurea recorded" to "Reported adverse effects or toxicity" as it is difficult to distinguish between adverse reactions (i.e. adverse events which are definitely drug related) and other adverse events which may be sickle or transfusion related. Where reported in study reports, we have separated reported adverse effects into drug‐related, sickle‐related, transfusion‐related etc.
In the 2017 update of the review, serious adverse events reported in included studies (whether treatment‐related or not) were included under the definition of primary outcome 'Life‐threatening illness.'
Contributions of authors
Original review (2001)
Sally Davies (SD) and Ade Olujohungbe (AO) conceived review and evaluated studies. SD drafted review.
2005 (minor) update
SD drafted review (with input from AO), but stepped down as lead author in September 2005 (remained as co‐author). Ashley Jones (AJ) became lead author in September 2005 and assisted in drafting the update and now acted as guarantor of the review.
2010 (minor) update
AJ drafted the update with input from SCD and AO. AJ acts as guarantor of the review.
2017 (major) update
SJN became lead author in 2016 and led the current update on screening, data extraction, risk of bias assessment, analysis, summary of findings table and drafting results and discussion. JH provided clinical interpretation of the review, drafted text of the discussion and commented on all sections of the review. AJ extracted and data and drafted text as well as commenting on later draft versions. SD stepped down as co‐author.
Sources of support
Internal sources
No sources of support supplied
External sources
-
National Institute for Health Research, UK.
This systematic review was supported by the National Institute for Health Research, via Cochrane infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group.
Declarations of interest
Sarah Nevitt: none known. Ashley Jones: none known. Jo Howard: no declarations in relation to the use of hydroxyurea. In general, has undertaken consultancy for Bluebird Bio; AesRx; and Global Blood Therapeutics. Her institution has the following grants: NIHR grant for the POMS trial (prevention of morbidity in SCD); and an NIH grant for natural history in stroke in SCD (pending).
New search for studies and content updated (conclusions changed)
References
References to studies included in this review
BABY HUG 2011 {published data only}
- Adams RJ, Barredo J, Bonds DR, Brown C, Casella J, Daner L, et al. TCD in infants: A report from the BABY HUG trial. Blood 2005;106(11). [Abstract no: 952] [Google Scholar]
- Adams RJ, Luden J, Miller S, Wang W, Rees R, Li D, et al. TCD in infants: a report from the Baby Hug study. 28th Annual Meeting of the National Sickle Cell Disease Program; 2005 Apr 9‐13; Cincinnati, Ohio. 2005:105.
- Alvarez O, Miller ST, Wang WC, Luo Z, McCarville MB, Schwartz GJ, et al. Effect of hydroxyurea treatment on renal function parameters: results from the multi‐center placebo‐controlled BABY HUG clinical trial for infants with sickle cell anemia. Pediatric Blood & Cancer 2012;59(4):668‐74. [CENTRAL: 848700; CRS: 5500125000000525; PUBMED: 22294512] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong FD, Elkin TD, Brown RC, Glass P, Rana S, Casella JF, et al. Developmental function in toddlers with sickle cell anemia. Pediatrics 2013;131(2):e406‐14. [CENTRAL: 853612; CRS: 5500125000000530; PUBMED: 23296434] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong FD, Elkin TD, Brown RC, Glass P, Rees RC, Wang WC, et al. Neurodevelopment in infants with sickle cell anemia: baseline data from the Baby HUG trial. Blood 2008;112(11):713. [Google Scholar]
- Armstrong FD, Rees RC, Li D, Bonner M, Elkin D, Strouse JJ, et al. Baseline developmental function by age for children in the pediatric hydroxyurea phase 3 clinical trial (Baby Hug). 28th Annual Meeting of the National Sickle Cell Disease Program; 2005 Apr 9‐13; Cincinnati, Ohio. 2005:137.
- Casella JF, Wang WC, Rogers ZR, Iyer RV, Rana S, Driscoll MC, et al. Progress of the multicenter trial of hydroxyurea in infants with sickle cell anemia (BABY HUG) and assessment of baseline splenic and renal function. Pediatric Academic Societies Annual Meeting. 2005; Vol. 57:1111.
- Kalpatthi R, Thompson B, Lu M, Wang WC, Patel N, Kutlar A, et al. Comparison of hematologic measurements between local and central laboratories: data from the BABY HUG trial. Clinical Biochemistry 2013;46(3):278‐81. [CENTRAL: 977455; CRS: 5500125000000527; PUBMED: 23123915] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebensburger JD, Miller ST, Howard TH, Casella JF, Brown RC, Lu M, et al. Influence of hemoglobin level on clinical findings in infants with sickle cell anemia; data from BABY HUG. 52nd ASH Meeting and Exposition; 2010 Dec 4‐7; Orlando. 2010. [Abstract no: 1631]
- Lebensburger JD, Miller ST, Howard TH, Casella JF, Brown RC, Lu M, et al. Influence of severity of anemia on clinical findings in infants with sickle cell anemia: analyses from the BABY HUG study. Pediatric Blood & Cancer 2012;59(4):675‐8. [CENTRAL: 854381; CRS: 5500125000000528; PUBMED: 22190441] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederman HM, Connolly MA, Kalpatthi R, Ware RE, Wang WC, Luchtman‐Jones L, et al. Immunologic effects of hydroxyurea in sickle cell anemia. Pediatrics 2014;134(4):686‐95. [CENTRAL: 1053679; CRS: 5500135000000943; PUBMED: 25180279] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederman HM, Connolly MA, Ware RE, Luchtman‐Jones L, Goldsmith JC. Effects of hydroxyurea (HU) on lymphocyte subsets and the immune response to pneumococcal, measles, mumps and rubella vaccination in the pediatric hydroxyurea phase III clinical trial ‐ BABY HUG ‐ (ClinicalTrials.gov Identifier: NCT00006400). Blood 2012;120(21). [Abstract no: 243; CENTRAL: 977456; CRS: 5500125000000532] [Google Scholar]
- McCarville MB, Luo Z, Huang X, Rees RC, Rogers ZR, Miller ST, et al. Abdominal ultrasound with scintigraphic and clinical correlates in infants with sickle cell anemia: baseline data from the BABY HUG trial. AJR American Journal of Roentgenology 2011;196(6):1399‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarville MB, Rees RC, Rogers ZR, Kalpatthi R, Miller ST, Wang WC, et al. Adbominal ultrasound findings in infants with sickle cell anemia; baseline data from the BABY HUG Trial. 3rd Annual Sickle Cell Disease Research and Educational Symposium and Annual Sickle Cell Disease Scientific Meeting; 2009 Feb 18‐20. 2009. [Abstract no: 212]
- McGann PT, Flanagan JM, Howard TA, Dertinger SD, He J, Kulharya AS, et al. Genotoxicity associated with hydroxyurea exposure in infants with sickle cell anemia: results from BABY‐HUG phase III clinical trial. 53rd ASH Annual Meeting and Exposition; 2011 Dec 10‐13; San Diego, California. 2011. [Abstract no: 8]
- McGann PT, Flanagan JM, Howard TA, Dertinger SD, He J, Kulharya AS, et al. Genotoxicity associated with hydroxyurea exposure in infants with sickle cell anemia: results from the BABY‐HUG Phase III Clinical Trial. Pediatric Blood & Cancer 2012;59(2):254‐7. [CENTRAL: 854422; CRS: 5500125000000524; PUBMED: 22012708] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller ST, Barredo J, Brown C, Bonds DR, Casella JF, Li D, et al. Renal concentrating ability in infants with sickle cell anemia; baseline data from Baby Hug, a multicenter trial. 29th Annual Meeting of the National Sickle Cell Disease Program; 2006 Apr 8‐12; Memphis, USA. 2006. [Abstract no: 141]
- Miller ST, Rey K, He J, Flanagan J, Fish BJ, Rogers ZR, et al. Massive accidental overdose of hydroxyurea in a young child with sickle cell anemia. Pediatric Blood & Cancer 2012;59(1):170‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller ST, Wang WC, Iyer R, Rana S, Lane P, Ware RE, et al. Urine concentrating ability in infants with sickle cell disease: baseline data from the phase III trial of hydroxyurea (BABY HUG). Pediatric Blood & Cancer 2010; Vol. 54, issue 2:265‐8. [DOI] [PMC free article] [PubMed]
- Miller ST, Wang WC, Iyer RV, Rana SR, Lane PA, Ware RE, et al. Urine concentrating ability in infants with sickle cell anemia: baseline data from the Baby HUG trial. Blood 2008;112(11):1413. [Google Scholar]
- Miller ST, Ware RE, Kutlar A, Alvarez OA, Iyer RV, Sarnaik SA, et al. Serum cystatin‐C levels in infants with sickle cell anemia: baseline data from the BABY HUG trial. Blood 2008;112(11):4791. [Google Scholar]
- Pavlakis SG, Rees RC, Huang X, Brown RC, Casella JF, Iyer RV, et al. Transcranial doppler ultrasonography (TCD) in infants with sickle cell anemia: baseline data from the BABY HUG trial. Pediatric Blood & Cancer 2010; Vol. 54, issue 2:256‐9. [DOI] [PMC free article] [PubMed]
- Rana S, Houston PE, Wang WC, Iyer RV, Goldsmith J, Casella JF, et al. Hydroxyurea and growth in young children with sickle cell disease. Pediatrics 2014;134(3):465‐472. Supplemental information. http://pediatrics.aappublications.org/content/134/3/465.supplemental. [CRS: 5500135000001451] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana S, Houston PE, Wang WC, Iyer RV, Goldsmith J, Casella JF, et al. Hydroxyurea and growth in young children with sickle cell disease. Pediatrics 2014;134(3):465‐72. [CRS: 5500135000000945; PUBMED: 25157002] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers ZR, Capparelli EV, Thompson B, Ware RE, Wang WC, Iyer RV, et al. Pharmacokinetics of hydroxyurea in young children with sickle cell anemia: a report from the Baby Hug trial. 29th Annual Meeting of the National Sickle Cell Disease Program; 2006 Apr 8‐12; Memphis, USA. 2006:157.
- Rogers ZR, Rees RC, Files B, Iyer RV, Shulkin BL, Shalaby‐Rana E, et al. Spleen function in infants with sickle cell anemia : baseline data from the BABY HUG trial. Blood 2008;112(11):1416. [Google Scholar]
- Rogers ZR, Rees RC, Files B, Iyer RV, Shulkin BL, Shalaby‐Rana E, et al. Spleen function in infants with sickle cell anemia: baseline data from the Baby Hug trial. 3rd Annual Sickle Cell Disease Research and Educational Symposium and Annual Sickle Cell Disease Scientific Meeting; 2009 Feb 18‐20. 2009. [Abstract no: 199]
- Rogers ZR, Rees RR, Wang WC, Li D, Iyer RV, Rana S, et al. Evaluation of splenic function in infants with sickle cell anemia in the Baby Hug trial. 28th Annual Meeting of the National Sickle Cell Disease Program; 2005 Apr 9‐13; Cincinnati, Ohio. 2005:106.
- Rogers ZR, Thompson B, Ware RE, Wang WC, Iyer RV, Miller ST, et al. Pharmacokinetics of hydroxyurea in young children with sickle cell anemia: a report from the BABY HUG trial. Blood. 2005, issue 11. [Abstract no:: 3184]
- Sheehan VA, Luo Z, Flanagan JM, Howard TA, Thompson BW, Wang WC, et al. Genetic modifiers of sickle cell anemia in the BABY HUG cohort: influence on laboratory and clinical phenotypes. American Journal of Hematology 2013;88(7):571‐6. [CENTRAL: 983421; CRS: 5500125000000710; PUBMED: 23606168] [DOI] [PubMed] [Google Scholar]
- Thompson BW, Miller ST, Rogers ZR, Rees RC, Ware RE, Waclawiw MA, et al. The pediatric hydroxyurea phase III clinical trial (BABY HUG): challenges of study design. Pediatric Blood & Cancer 2010; Vol. 54, issue 2:250‐5. [DOI] [PMC free article] [PubMed]
- Thompson BW, Wang WC, Miller ST, Rogers ZR, Ware RE, Thornburg CD, et al. The physiological and clinical effects of interrupting a treatment regimen of hydroxyurea in young children with sickle cell anemia (SCA). 53rd ASH Annual Meeting and Exposition; 2011 Dec 10‐13; San Diego. 2011. [Abstract no: 2134]
- Thornburg CD, Files BA, Luo Z, Miller ST, Kalpatthi R, Iyer R, et al. Impact of hydroxyurea on clinical events in the BABY HUG trial. Blood 2012;120(22):4304‐10; quiz 4448. [CENTRAL: 853818; CRS: 5500125000000526; PUBMED: 22915643] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornburg CD, Rogers ZR, Jeng MR, Rana SR, Iyer RV, Faughnan L, et al. Adherence to study medication and visits: data from the BABY HUG trial. Pediatric Blood & Cancer 2010; Vol. 54, issue 2:260‐4. [DOI] [PMC free article] [PubMed]
- Thornburg CD, Rogers ZR, Wang W, Jeng M, Rana SR, Iyer RV, et al. Study drug and visit adherence: data from the Baby HUG trial. Blood 2008;112(11):1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Luo Z, Alvarez O, Fixler J, Miller S, Ware RE, et al. Effects of hydroxyurea in asymptomatic infants with sickle cell anemia: Analysis F from the BABY HUG trial. American Journal of Hematology 2012;7:E20‐E21. [Google Scholar]
- Wang W, Rees RC, Miller ST, Brown RC, Casella JF, Iyer RV, et al. Transcranial doppler (TCD) ultrasonography in infants with sickle cell anemia: baseline data from the BABY HUG trial. Blood 2008;112(11):1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WC, Oyeku SO, Luo Z, Boulet SL, Miller ST, Casella JF, et al. Hydroxyurea is associated with lower costs of care of young children with sickle cell anemia. Pediatrics 2013;132(4):677‐83. [CENTRAL: 962768; CRS: 5500125000000529; PUBMED: 23999955] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WC, Ware RE, Miller ST, Iyer RV, Casella JF, Minniti CP, et al. Hydroxycarbamide in very young children with sickle‐cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet 2011;377(9778):1663‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WC, Yeku SO, Luo Z, Boulet SL, Miller ST, Fish B, et al. Costs associated with the care of very young children with sickle cell anemia (SCA): analysis from the BABY HUG study. 53rd ASH Annual Meeting and Exposition; 2011 Dec 10‐13; San Diego. 2011. [Abstract no: 171]
- Ware RE, Rees RC, Sarnaik SA, Iyer RV, Alvarez OA, Casella JF, et al. Renal function in infants with sickle cell anemia: baseline data from the BABY HUG trial. Blood 2008;112(11):1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware RE, Rees RC, Sarnaik SA, Iyer RV, Alvarez OA, Casella JF, et al. Renal function in infants with sickle cell anemia: baseline data from the BABY HUG trial. Journal of Pediatrics 2010; Vol. 156, issue 1:66‐70. [DOI] [PMC free article] [PubMed]
- Wynn L, Debenham E, Faughnan L, Martin B, Kelly T, Reed C, et al. Recruitment in the Baby Hug pediatric hydroxyurea phase 3 clinical trial. 35th Anniversary Convention of the National Sickle Cell Disease Program; 2007 Sep 17‐22; Washington DC, USA. 2007:245.
- Wynn L, Miller S, Faughnan L, Luo Z, Debenham E, Adix L, et al. Recruitment of infants with sickle cell anemia to a Phase III trial: data from the BABY HUG study. Contemporary Clinical Trials 2010; Vol. 31, issue 6:558‐63. [DOI] [PMC free article] [PubMed]
- Wynn LW, Faughnan L, Li D, Wang W, Martin B, Kelly T, et al. Recruitment of infants with sickle cell anemia to a phase III trials: data from the BABY HUG study. Blood 2008;112(11):1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Belgian Study 1996 {published data only}
- Ferster A, Vermylen C, Cornu G, Buyse M, Corazza F, Devalck C, et al. Hydroxyurea for treatment of severe sickle cell anemia: a pediatric clinical trial. Blood 1996;88(6):1960‐4. [PubMed] [Google Scholar]
CHAMPS 2011 {published data only}
- Wang W, Brugnara C, Snyder C, Wynn L, Rogers Z, Kalinyak K, et al. The effects of hydroxycarbamide and magnesium on haemoglobin SC disease: results of the multi‐centre CHAMPS trial. British Journal of Haematology 2011;152(6):771‐6. [CENTRAL: 801819; CRS: 5500125000000521; PUBMED: 21275961] [DOI] [PubMed] [Google Scholar]
- Wang WC, Snyder C, Brugnara C, Telen MJ, Steinberg MH, Wynn LW, et al. Effects of hydroxyurea (HU) and magnesium pidolate (Mg) in hemoglobin SC disease (HbSC): the "CHAMPS" trial. Blood 2009; Vol. 22. [Abstract no: 819]
Jain 2012 {published data only}
- Jain D. Low dose hydroxyurea in children severely affected with sickle cell disease: hospital based randomized controlled study. 4th Annual Sickle Cell Disease Research and Educational Symposium & Grant Writing Institute AND Annual Sickle Cell Disease Scientific Meeting; 2010 Feb 14‐19; Hollywood, Florida. 2010. [Abstract no: 076]
- Jain DL, Sarathi V, Desai S, Bhatnagar M, Lodha A. Low fixed‐dose hydroxyurea in severely affected Indian children with sickle cell disease. Hemoglobin 2012;36(4):323‐32. [CENTRAL: 879848; CRS: 5500125000000522; PUBMED: 22734586] [DOI] [PubMed] [Google Scholar]
MSH 1995 {published data only}
- Armstrong FD, Steinberg MH, Ballas SK, Ataga KI, Waclawiw MA, Kutlar A, et al. Development outcomes of offspring of adults treated with hydroxyurea in the multicenter study of hydroxyurea. Blood 2009, issue 22. [Abstract no: 1543]
- Ballas SK, Barton F, Castro O, Bellevue R, Investigators of the multicenter study of hydroxyurea in sickle cell anemia. Narcotic analgesia use among adult patients with sickle cell anemia. Blood 1995;86(10 Suppl 1):642a. [Google Scholar]
- Ballas SK, Barton F, Castro O, Koshy M, Bellevue R. Pattern of narcotic analgesic consumption among adult patients with sickle cell anemia. National Sickle Cell Disease Program 21st Annual Meeting; 1996 Mar. 1996:63.
- Ballas SK, Barton FB, Waclawiw MA, Swerdlow P, Eckman JR, Pegelow CH, et al. Hydroxyurea and sickle cell anemia: effect on quality of life. Health and Quality of Life Outcomes 2006;4:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballas SK, Bauserman RL, McCarthy WF, Castro OL, Smith WR, Waclawiw MA. Utilization of analgesics in the multicenter study of hydroxyurea in sickle cell anemia: Effect of sex,age, and geographical location. American Journal of Hematology 2010;85(8):613‐6. [DOI] [PubMed] [Google Scholar]
- Ballas SK, Bauserman RL, McCarthy WF, Castro OL, Smith WR, Waclawiw MA, IoftheMSofHinSCA. Hydroxyurea and acute painful crises in sickle cell anemia: effects on hospital length of stay and opioid utilization during hospitalization, outpatient acute care contacts, and at home. Journal of Pain and Symptom Management 2010;40(6):870‐82. [CENTRAL: 779191; CRS: 5500135000001680; PUBMED: 20864308] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballas SK, Bauserman RL, McCarthy WF, Waclawiw MA. The impact of hydroxyurea on career and employment of patients with sickle cell anemia. JAMA 2010; Vol. 102, issue 11:993‐9. [PubMed]
- Ballas SK, Bauserman RL, McCarthy WF, Waclawiw MA, Barton BA. Impact of hydroxyurea on employment among patients with sickle cell anemia. Blood 2009; Vol. 114, issue 22. [Abstract no: 2485]
- Ballas SK, Marcolina MJ, Dover GJ, Barton FB. Erythropoietic activity in patients with sickle cell anaemia before and after treatment with hydroxyurea. British Journal of Haematology 1999;105(2):491‐6. [PubMed] [Google Scholar]
- Ballas SK, Marcolina MJ, Investigators of the multicenter study of hydroxyurea in sickle cell anemia. In vivo RBC survival and ferrokinetic data in patients with sickle cell anemia before and after treatment with hydroxyurea. Blood 1995;86(10 Suppl 1):140a. [Google Scholar]
- Ballas SK, McCarthy WF, Bauseman RI, Castro OL, Swerdlow PS, Smith W, et al. Patterns of analgesic utilization in the multicenter study of hydroxyurea (MSH). Blood 2009; Vol. 114, issue 22. [Abstract no: 2577]
- Ballas SK, McCarthy WF, Bauserman RL, Castro OL, Waclawiw MA, Barton BA. Sickle cell genetic markers: geographic distribution and relation to pain outcomes in multicenter study of hydroxyurea in sickle cell anemia. Blood 2009; Vol. 114, issue 22. [Abstract no: 2582]
- Ballas SK, McCarthy WF, Bauserman RL, Valafar F, Waclawiw M, Barton BA, Kutlar A. Definition of the responder to hydroxyurea therapy: revisited. Blood 2009;114(22). [Abstract no: 1513] [Google Scholar]
- Ballas SK, McCarthy WF, Guo N, Brugnara C, Kling G, Bauserman R, et al. Early detection of responders to hydroxyurea therapy. 4th Annual Sickle Cell Disease Research and Educational Symposium & Grant Writing Institute AND Annual Sickle Cell Disease Scientific Meeting; 2010 Feb 14‐19; Hollywood, Florida. 2010; Vol. 26. [Abstract no: 030]
- Ballas SK, McCarthy WF, Guo N, DeCastro L, Bellevue R, Barton BA, et al. Exposure to hydroxyurea and pregnancy outcomes in patients with sickle cell anemia. Journal of the National Medical Association 2009;101(10):1046‐51. [CENTRAL: 730469; CRS: 5500050000000074; PUBMED: 19860305] [DOI] [PubMed] [Google Scholar]
- Barton F, Terrin M, Moore R, McMahon RP, Charache S. Ascertainment of the primary end point in the Multicenter Study of Hydroxyrea in Sickle Cell Anemia (MSH). The MSH Investigators. Controlled Clinical Trials 1996;17(2 Suppl):67S. [Google Scholar]
- Brandon AE, McCarthy WF, Barton FB, Terrin ML. Vital status determination of patients' lost to follow‐up in the multicenter study of hydroxyurea in sickle cell anemia (MSH) patients' follow‐up study. Clinical Trials 2004;2:209. [Google Scholar]
- Charache S. Effects of hydroxyurea therapy in patients with sickle cell anemia. Australian and New Zealand Journal of Medicine 1996;26:326. [Google Scholar]
- Charache S. Experimental therapy of sickle cell disease. Use of hydroxyurea. American Journal of Pediatric Hematology/Oncology 1994;16(1):62‐6. [PubMed] [Google Scholar]
- Charache S. Mechanism of action of hydroxyurea in the management of sickle cell anemia in adults. Seminars in Hematology 1997;34(3 Suppl 3):15‐21. [PubMed] [Google Scholar]
- Charache S. Preventing pain in sickle cell anemia (HB SS): baseline data from patients in a hydroxyurea trial. Blood 1993;82(10 Suppl):356a. [Google Scholar]
- Charache S, Barton FB, Moore RD, Terrin ML, Steinberg MH, Dover GJ, et al. Hydroxyurea and sickle cell anemia. Clinical utility of a myelosuppressive "switching" agent. Medicine 1996;75(6):300‐26. [DOI] [PubMed] [Google Scholar]
- Charache S, Terrin M, Moore RD, Dover GJ, Barton FB, Eckert SV, et al. Effect of hydroxyurea on the frequency of painful crisis in sickle cell anemia. New England Journal of Medicine 1995;332(20):1317‐22. [DOI] [PubMed] [Google Scholar]
- Charache S, Terrin ML, Moore RD, Dover GJ, McMahon RP, Barton FB, et al. Design of the multicenter study of hydroxyurea in sickle cell anemia. Controlled Clinical Trials 1995;16(6):432‐46. [DOI] [PubMed] [Google Scholar]
- Hackney AC, Heizer W, Hoffman E, Jones S, Strayhorn D, Orringer EP. Effect of hydroxyurea (HU) administration on the body weight, body composition and exercise performance of patients with sickle cell anemia. Blood 1995;86(10 Suppl 1):141a. [DOI] [PubMed] [Google Scholar]
- Hackney AC, Hezier W, Gulledge TP, Jones S, Strayhorn D, Busby M, et al. Effects of hydroxyurea administration on the body weight, body composition and exercise performance of patients with sickle‐cell anaemia. Clinical Science 1997;92(5):481‐6. [DOI] [PubMed] [Google Scholar]
- Handy C, Barton F, Moore R, McMahon R, Eckert S, Terrin M. Dose titration in the multicentre study of hydroxyurea in sickle cell anemia (MSH). Controlled Clinical Trials 1996;17(Suppl 2):92S. [Google Scholar]
- Heizer WD, Hackney AC, Busby M, Gulledge T, Jones S, Strayhorn G, et al. The composition and etiology of weight gain in sickle cell patients receiving hydroxyurea (HU): An ancillary study to the multicentre study of hydroxyurea (MSH). National Sickle Cell Disease Program 18th Annual Meeting; 1993 May. 1993:117a.
- Kutlar A, Barton F, Terrin M, Steinberg MH. Effect of hydroxyurea on hematologic and biochemical laboratory values in sickle cell disease: The MSH at 7‐8 years follow‐up. National Sickle Cell Disease Program 25th Annual Meeting; 2001 Apr. 2001. [Abstract no: #126]
- McCarthy WF, Bauserman RL, Barton BA, Guo N, Ballas SK, Smith W. Time series analysis of the pain diary data obtained during the multicenter study for hydroxyurea (MSH) clinical trial. Blood 2006;11. [Abstract no: 3807] [Google Scholar]
- McMahon RP, Waclawiw MA, Geller NL, Barton FB, Terrin ML, Bonds DR. An extension of stochastic curtailment for incompletely reported events: the multicenter study of hydroxyurea in sickle cell anemia (MSH). Controlled Clinical Trials 1997;18(5):420‐30. [DOI] [PubMed] [Google Scholar]
- Moore RD, Charache S, Terrin M, Barton FB, Ballas SK. Cost‐effectiveness of hydroxyurea in sickle cell anemia. National Sickle Cell Disease Program 23rd Meeting; 1999 Mar. 1999:210.
- Moore RD, Charache S, Terrin ML, Barton FB, Ballas SK, and the investigators of the MSH study of hydroxyurea in sickle cell anemia. Cost‐effectiveness of hydroxyurea in sickle cell anemia. American Journal of Hematology 2000;64(1):26‐31. [DOI] [PubMed] [Google Scholar]
- Orringer EP, Jones S, Strayhorn D, Hoffman E, Parker J, Greenberg C. The effect of hydroxyurea (HU) administration on circulating D‐dimer levels in patients with sickle cell anemia (HbSS). National Sickle Cell Disease Program 21st Meeting; 1996 Mar. 1996:131.
- Orringer EP, Jones S, Strayhorn D, Hoffman E, Parker J, Greenberg CS. The effect of hydroxyurea (HU) administration on circulating d‐dimer levels in patients with sickle cell anemia. Blood 1996;88(10 Suppl 1):496a. [Google Scholar]
- Smith WR, Ballas SK, McCarthy WF, Bauserman RL, Swerdlow PS, Steinberg MH. The association between hydroxyurea treatment and pain intensity, analgesic use, and utilization in ambulatory sickle cell anemia patients. Pain Medicine 2011;12(5):697‐705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WR, Bauseman RL, McCarthy WF, Barton BA, Ballas SK. Effect of geography and climate on pain frequency in patients enrolled in the multicenter study of hydroxyurea in sickle cell anemia. 3rd Annual Sickle Cell Disease Research and Educational Symposium AND Annual Sickle Cell Disease Scientific Meeting; 2009 Feb 18‐20. 2009. [Abstract no: 253]
- Steinberg MH. Determinants of fetal hemoglobin response to hydroxyurea. Seminars in Hematology 1997;3(Suppl 3):8‐14. [PubMed] [Google Scholar]
- Steinberg MH. Mortality at 3‐5 years: The multicenter study of hydroxyurea in sickle cell anemia (MSH). National Sickle Cell Disease Program Annual Meeting; 1997 Sep. 1997:68.
- Steinberg MH, Ballas S, Barton F, Terrin M, the MSH. Mortality at 4‐5 years: results from the multicenter study of hydroxyurea in sickle cell anemia (MSH). Blood 1997;90(10 Suppl 1 Pt 1):444a. [Google Scholar]
- Steinberg MH, Barton F, Castro O, Koshy M, Eckman J, Terrin M. Risks and benefits of hydroxyurea (HU) in adult sickle cell anaemia. Effects at 6‐ to 7‐ years. Blood 1999;94(10 Suppl 1 Pt 1):644a‐5a. [Google Scholar]
- Steinberg MH, Barton F, Castro O, Pegelow CH, Ballas SK, Kutlar A, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA 2003;289(13):1645‐51. [DOI] [PubMed] [Google Scholar]
- Steinberg MH, Barton F, Castro O, Ramirez G, Bellevue R, Terrin M, et al. Hydroxyurea (HU) is associated with reduced mortality in adults with sickle cell anemia. Blood 2000;96(11 Pt 1):485a. [Google Scholar]
- Steinberg MH, Castro O, Ballas SK, Barton F, Terrin M. The multicenter study of hydroxyurea in sickle cell anemia (MSH): mortality at 5‐6 years. Blood 1998;92(10 Suppl 1 Pt 1):496a. [Google Scholar]
- Steinberg MH, Lu ZH, Barton FB, Terrin ML, Charache S, Dover GJ. Fetal hemoglobin in sickle cell anemia: determinants of response to hydroxyurea. Blood 1997;89(3):1078‐8. [PubMed] [Google Scholar]
- Steinberg MH, Lu ZH, Barton M, Terrin S, Charache S, Dover G, et al. Fetal hemoglobin (Hb F) in sickle cell anemia (HbSS): Determinents of response to hydroxyurea (HU). Blood 1995;86(10 Suppl 1):418a. [PubMed] [Google Scholar]
- Steinberg MH, McCarthy WF, Castro O, Ballas SK, Armstrong FD, Smith W, et al. The risks and benefits of long‐term use of hydroxyurea in sickle cell anemia: A 17.5 year follow‐up. American Journal of Hematolology 2010; Vol. 85, issue 6:403‐8. [DOI] [PMC free article] [PubMed]
- Terrin ML, Barton FB, Bonds D, Ballas SK, Swerdlow P, Pegelow CH, et al. Effect of hydroxyurea on quality of life: 2‐year results from the multicenter study of hydroxyurea in sickle cell anemia. National Sickle Cell Disease Program 23rd Annual Meeting; 1999 Mar. 1999:161.
SCATE 2015 {published data only}
- Hankins JS, McCarville MB, Rankine‐Mullings A, Reid ME, Lobo CL, Moura PG, et al. Prevention of conversion to abnormal transcranial Doppler with hydroxyurea in sickle cell anemia: a phase III international randomized clinical trial. American Journal of Hematology 2015;90(12):1099‐105. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01531387. Sparing Conversion to Abnormal TCD (Transcranial Doppler) Elevation (SCATE). http://clinicaltrials.gov/show/NCT01531387.
SWiTCH 2012 {published data only}
- Alvarez O, Yovetich NA, Scott JP, Owen W, Miller ST, Schultz W, et al. Pain and other non‐neurological adverse events in children with sickle cell anemia and previous stroke who received hydroxyurea and phlebotomy or chronic transfusions and chelation: results from the SWiTCH clinical trial. American Journal of Hematology 2013;88(11):932‐8. [CENTRAL: 963136; CRS: 5500125000000520; PUBMED: 23861242] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aygun B, Mortier NA, Kesler K, Lockhart A, Schultz WH, Cohen AR, et al. Therapeutic phlebotomy is safe in children with sickle cell anaemia and can be effective treatment for transfusional iron overload. British Journal of Haematology 2015;169(2):262‐6. [CRS: 5500135000001418; PUBMED: 25612463] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aygun B, Mortier NA, Kesler K, Schultz WH, Alvarez OA, Rogers ZR, et al. Therapeutice phlebotomy in children with sickle cell anemia, stroke, and iron overload: the SWiTCH experience. 53rd ASH Annual Meeting and Exposition; 2011 Dec 10‐13; San Diego, California. 2011. [Abstract no: 1044]
- Kwiatkowski JL, Cohen AR, Garro J, Alvarez O, Nagasubramanian R, Sarnaik S, et al. Transfusional iron overload in children with sickle cell anemia on chronic transfusion therapy for secondary stroke prevention. American Journal of Hematology 2012;87(2):221‐3. [CENTRAL: 864015; CRS: 5500100000011226; PUBMED: 22120913] [DOI] [PubMed] [Google Scholar]
- NCT00122980. Stroke with transfusions changing to hydroxyurea. clinicaltrials.gov/show/NCT00122980 Date first received: 20 July 2005.
- Sheehan VA, Howard TA, Sabo A, Nagasaswamy U, Crosby JR, Davis B, et al. Genetic predictors of hemoglobin F response to hydroxyurea in sickle cell anemia. Blood 2012;120(21). [Abstract no: 241; CENTRAL: 977454; CRS: 5500125000000531] [Google Scholar]
- Ware RE, Helms RW. Stroke With Transfusions Changing to Hydroxyurea (SWiTCH). Blood 2012;119(17):3925‐32. [CENTRAL: 849022; CRS: 5500100000004120] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware RE, Helms RW. Stroke with transfusions changing to hydroxyurea (SWiTCH): a phase 3 randomised clinical trial for treatment of children with sickle cell anemia. 52nd ASH Meeting and Exposisiton; 2010 Dec 4‐7; Orlando, Florida. 2010. [Abstract no: 844]
- Ware RE, McMurray MA, Schultz WH, Alvarez OA, Aygun B, Cavalier ME, et al. Academic community standards for chronic transfusion therapy in children with sickle cell anemia and stroke. Blood 2006;108(11):Abst 1213. [Google Scholar]
- Ware RE, Schultz WH, Yovetich N, Mortier NA, Alvarez O, Hilliard L, et al. Stroke with transfusions changing to hydroxyurea (SWiTCH): A phase III randomized clinical trial for treatment of children with sickle cell anemia, stroke, and iron overload. Pediatric Blood & Cancer 2011;57(6):1011‐7. [CENTRAL: 806683; CRS: 5500100000004123] [DOI] [PMC free article] [PubMed] [Google Scholar]
TWiTCH 2016 {published data only}
- Aygun B, Wruck LM, Schultz WH, Mueller BU, Brown C, Luchtman‐Jones L, et al. Chronic transfusion practices for prevention of primary stroke in children with sickle cell anemia and abnormal TCD velocities. American Journal of Hematology 2012;87(4):428‐30. [DOI] [PubMed] [Google Scholar]
- Helton KJ, Roberts D, Schultz WH, Davis BR, Kalfa TA, Pressel SL, et al. Effects of chronic transfusion therapy on MRI and MRA in children with sickle cell anemia. Blood 2014;124(21):4052. [CENTRAL: 1261883; CRS: 5500135000001683] [Google Scholar]
- Imran H, Aygun B, Davis BR, Pressel SL, Herbert Schultz W, Jackson SM, et al. Effects of chronic transfusion therapy on transcranial doppler ultrasonography velocities in children with sickle cell anemia at risk for primary stroke: baseline findings from the Twitch trial. Blood 2014;124(21):87. [CENTRAL: 1261882; CRS: 5500135000001682] [Google Scholar]
- Ware RE, Davis BR, Schultz WH, Brown RC, Aygun B, Sarnaik S, et al. Hydroxycarbamide versus chronic transfusion for maintenance of transcranial doppler flow velocities in children with sickle cell anaemia—TCD With Transfusions Changing to Hydroxyurea (TWiTCH): a multicentre, open‐label, phase 3, non‐inferiority trial. Lancet 2016;387:661‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JC, Cohen A, Aygun B, Imran H, Luchtman‐Jones L, Thompson. Extrahepatic iron deposition in chronically transfused children with sickle cell anemia‐ baseline findings from the Twitch trial. Blood 2013;122(21):2238. [CENTRAL: 1261881; CRS: 5500135000001681] [Google Scholar]
- Wood JC, Cohen AR, Pressel SL, Aygun B, Imran H, Luchtman‐Jones L. Organ iron accumulation in chronically transfused children with sickle cell anaemia: baseline results from the TWiTCH trial. British Journal of Haematology 2016;172(1):122‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JC, Pressel S, Rogers ZR, Odame I, Kwiatkowski JL, Lee MT, et al. Liver iron concentration measurements by MRI in chronically transfused children with sickle cell anemia: baseline results from the TWiTCH trial. American Journal of Hematology 2015;90(9):806‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JC, Pressel S, Rogers ZR, Odame I, Kwiatkowski JL, Lee MT, et al. Liver iron concentration measurements by MRI in chronically transfused children with sickle cell anemia: Baseline results from the TWiTCH trial. American Journal of Hematology 2015;90(9):806‐10. [CENTRAL: 1090263; EMBASE: 2015311270; CRS: 5500050000000271] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Al‐Nood 2011 {published data only}
De Montalembert 2006 {published data only}
- Montalembert M, Bachir D, Hulin A, Gimeno L, Mogenet A, Bresson JL, et al. A phase 1 pharmacokinetics (PK) study of hydroxyurea (HU) 1,000 mg coated breakable tablets and 500mg capsules in pediatric and adult patients with sickle cell disease. Blood 2005;106(11):Abst 3194. [Abstract no: 3194] [Google Scholar]
- Montalembert M, Bachir D, Hulin A, Gimeno L, Mogenet A, Bresson JL, et al. Pharmacokinetics of hydroxyurea 1,000 mg coated breakable tablets and 500 mg capsules in pediatric and adult patients with sickle cell disease. Haematologica 2006;91(12):1685‐8. [PubMed] [Google Scholar]
George 2013 {published data only}
- George A, Aygun B, Mortier N, Sparreboom A, Ware R. A randomized controlled trial of a dose‐prediction equation to determine maximum tolerated dose of hydroxyurea in children with sickle cell anemia. Pediatric Blood & Cancer 2013;60 Suppl. [Abstract no: 588; CENTRAL: 1007869; CRS: 5500050000000224; EMBASE: 71047876] [Google Scholar]
NCT00004492 {published data only}
- NCT00004492. Phase I/II Randomized Study of Hydroxyurea With or Without Clotrimazole in Patients With Sickle Cell Anemia. https://clinicaltrials.gov/show/NCT00004492.
NCT01848925 {published data only}
- NCT01848925. A Phase I Open‐label, Randomized, Safety and Efficacy Study of SANGUINATE™ at Two Doses Levels Versus Hydroxyurea in Sickle Cell Disease (SCD) Patients. clinicaltrials.gov/show/NCT01848925 (date first received 23 April 2013).
NCT01960413 {published data only}
- NCT01960413. Phase 2 Study of Montelukast for the Treatment of Sickle Cell Anemia. https://clinicaltrials.gov/show/NCT01960413.
NCT02149537 {published data only}
- NCT02149537. Risk Stratification for Clinical Severity of Sickle Cell Disease in Nigeria and Assessment of Efficacy and Safety During Treatment With Hydroxyurea. https://clinicaltrials.gov/show/NCT02149537.
Pushi 2000 {published data only}
- Pushi A. Hydroxyurea ameliorates importantly the clinical course of sickle cell disease reducing the frequency of painful crises. Hematology Journal 2000;1(Suppl. 1):34, Abstract no: 130. [Google Scholar]
Silva‐Pinto 2007 {published data only}
- Silva‐Pinto AC, Carrara RC, Oliveira VC, Palma PV, Campos AD, Zago MA, et al. Hydroxyurea treatment of sickle cell diseases causes megaloblastic transformation of the bone marrow that is responsible for the increase of the mean corpuscular volume. Haematologica 2007;Suppl 1:298. [Google Scholar]
Silva‐Pinto 2014 {published data only}
- Silva‐Pinto AC, Dias‐Carlos C, Saldanha‐Araujo F, Ferreira FI, Palma PV, Araujo AG, et al. Hydroxycarbamide modulates components involved in the regulation of adenosine levels in blood cells from sickle‐cell anemia patients. Annals of Hematology 2014;93(9):1457‐65. [CENTRAL: 1000846; CRS: 5500131000000084; JID:: 9107334; PUBMED: 24696091] [DOI] [PubMed] [Google Scholar]
Vichinsky 2013 {published data only}
- Vichinsky E, Torres M, Glass J, Minniti CP, Barrette S, Habr D, et al. A randomized phase II study evaluating the efficacy and safety of deferasirox versus deferoxamine in patients with sickle cell disease (SCD0: 2‐year results including pharmacokinetics (PK) and safety of deferasirox with concomitant hydroxyurea therapy. Blood 2011;118(21). [Abstract no: 1082] [Google Scholar]
- Vichinsky E, Torres M, Minniti CP, Barrette S, Habr D, Zhang Y, et al. Efficacy and safety of deferasirox compared with deferoxamine in sickle cell disease: two‐year results including pharmacokinetics and concomitant hydroxyurea. American Journal of Hematology 2013;88(12):1068‐73. [CRS: 5500125000000523; PUBMED: 23946212] [DOI] [PubMed] [Google Scholar]
Voskaridou 2005 {published data only}
- Voskaridou E, Terpos E, Margeli A, Hantzi E, Meletis J, Papassotiriou I, et al. Renal dysfunction and osteodystrophy in patients with sickle cell thalassaemia under long‐term treatment with hydroxyurea. 10th Congress of the European Hematology Association; 2005 June 2‐5; Stockholm International Fairs, Sweden. 2005. [Abstract no: 0174]
References to studies awaiting assessment
Anyanwu 2016 {published data only}
- Anyanwu JN, Williams O, Sautter CL, Kasirye P, Hume H, Opoka RO, Latham T, Ndugwa C, Ware RE, John CC. Novel Use of Hydroxyurea in an African Region With Malaria: Protocol for a Randomized Controlled Clinical Trial. JMIR research protocols 2016;5(2):e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT02560935 {published data only}
- NCT02560935. Primary Prevention of Stroke in Children With SCD in Sub‐Saharan Africa II (SPRING). clinicaltrials.gov/show/NCT02560935 (first received 02 September 2015).
NCT02675790 {published data only}
- NCT02675790. Low Dose Hydroxyurea for Secondary Stroke Prevention in Children With Sickle Cell Disease in Sub‐Saharan Africa (SPRINT). clinicaltrials.gov/ct2/show/NCT02675790 (first received 08 January 2016).
References to ongoing studies
NCT01389024 {published data only}
- NCT01389024. Hydroxyurea to Prevent Brain Injury in Sickle Cell Disease (HUPrevent). clinicaltrials.gov/ct2/show/NCT01389024 (first received 30 June 2011).
Additional references
Aumont 2015
- Aumont C, Driss F, Lazure T, Picard V, Creidy R, Botton S, et al. Myelodysplastic syndrome with clonal cytogenetic abnormalities followed by fatal erythroid leukemia after 14 years of exposure to hydroxyurea for sickle cell anemia. American Journal of Hematology 2015;90(7):E131‐2. [DOI] [PubMed] [Google Scholar]
Ballas 1996
- Ballas SK, Barton F, Castro O, Koshy M, Bellevue R. Pattern of narcotic analgesic consumption among adult patients with sickle cell anemia. National Sickle Cell Disease Program 21st Annual Meeting; 1996 Mar. 1996:63.
Ballas 2006
- Ballas SK, Barton FB, Waclawiw MA, Swerdlow P, Eckman JR, Pegelow CH, et al. Hydroxyurea and sickle cell anemia: effect on quality of life. Health and Quality of Life Outcomes 2006;4:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ballas 2009
- Ballas SK, Guo N, DeCastro L, Bellevue R. Exposure to hydroxyurea and pregnancy outcomes in patients with sickle cell anemia. JAMA 2009;101(10):1046. [DOI] [PubMed] [Google Scholar]
Barton 1996
- Barton F, Terrin M, Moore R, McMahon RP, Charache S. Ascertainment of the primary end point in the Multicenter Study of Hydroxyrea in Sickle Cell Anemia (MSH). The MSH Investigators. Controlled Clinical Trials 1996;17:67S. [DOI] [PubMed] [Google Scholar]
Baz 2012
- Baz W, Najfeld V, Yotsuya M, Talwar J, Terjanian T, Forte F. Development of myelodysplastic syndrome and acute myeloid leukemia 15 years after hydroxyurea use in a patient with sickle cell anemia. Clinical Medicine Insights. Oncology 2012;6:149‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Berk 1995
- Berk PD, Wasserman LR, Fruchtman SM, Goldberg JD. Treatment of polycythemia vera: a summary of clinical trials conducted by the Polycythemia Vera Study Group. In: Wasserman LR BP, Berlin NI editor(s). Polycythemia Vera and the Myeloproliferation Disorders. Philadelphia: W B Saunders, 1995:166‐94. [Google Scholar]
Bernaudin 2016
- Bernaudin F, Verlhac S, Arnaud C, Kamdem A, Hau I, Leveillé E, et al. Long‐term treatment follow‐up of children with sickle cell disease monitored with abnormal transcranial Doppler velocities. Blood 2016;127(15):1814‐22. [10.1182/blood‐2015‐10‐675231. Epub 2016 Feb 5.] [DOI] [PubMed] [Google Scholar]
Berthaut 2008
- Berthaut I, Guignedoux G, Kirsch‐Noir F, Larouziere V, Ravel C, Bachir D, et al. Influence of sickle cell disease and treatment with hydroxyurea on sperm parameters and fertility of human males. Haematologica 2008;93(7):988‐93. [DOI] [PubMed] [Google Scholar]
Brousseau 2010
- Brousseau DC, A Panepinto J, Nimmer M, Hoffmann RG. The number of people with sickle‐cell disease in the United States: national and state estimates. American journal of hematology 2010 Jan 1;85(1):77‐8. [DOI] [PubMed] [Google Scholar]
Brozovic 1987
- Brozovic M, Davies SC. Management of sickle cell disease. Postgraduate Medical Journal 1987;63:605‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chakravorty 2015
- Chakravorty S, Williams TN. Sickle cell disease: a neglected chronic disease of increasing global health importance. Archives of Disease in Childhood 2015;100(1):48‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Charache 1995
- Charache S, Terrin TL, Moore RD, Dover GJ, Barton FB, Eckert SV, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anaemia. New England Journal of Medicine 1995;332:1317‐22. [DOI] [PubMed] [Google Scholar]
Davies 1997a
- Davies SC, Roberts‐Harewood M. Blood transfusion in sickle cell disease. Blood Reviews 1997;11:57‐71. [DOI] [PubMed] [Google Scholar]
Davies 1997b
- Davies SC, Oni L. The management of patients with sickle cell disease. British Medical Journal 1997;315:656‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JPT, Curtin F, Worthington HV, Vail A. Meta‐analysis involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Fruchtman 1994
- Fruchtman SM, Kaplan ME, Peterson P, Mack K, Berk PD, Wasserman LR. Acute leukemia (AL), hydroxyurea (HU), and polycythemia vera (PV): an analysis of risk from the Polycythemia Vera Study Group. Blood 1994;84(Suppl 1):518a. [Google Scholar]
Fuggle 1996
- Fuggle P, Shand PAX, Gill LJ, Davies SC. Pain, quality of life, and coping in sickle cell disease. Archives of Disease in Childhood 1996;75:199‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gardner 2016
- Gardner K, Douiri A, Drasar E, Allman M, Mwirigi A, Awogbade M, et al. Survival in adults with sickle cell disease in a high income setting. Blood 2016;128(10):1436‐1438. [DOI] [PubMed] [Google Scholar]
Gray 1991
- Gray A, Anionwu EN, Davies SC, Brozovic M. Mortality in sickle cell disease: the experience of a British centre. Journal of Clinical Pathology 1991;44:459‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Green 2014
- Green NS, Barral S. Emerging science of hydroxyurea therapy for pediatric sickle cell disease. Pediatric research 2013;75(1‐2):196‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
Grosse 2011
- Grosse SD, Odame I, Atrash HK, Amendah DD, Piel FB, Williams TN. Sickle cell disease in Africa: a neglected cause of early childhood mortality. American journal of preventive medicine 2011 Dec 31;41(6):S398‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Handy 1996
- Handy C, Barton F, Moore R, McMahon R, Eckert S, Terrin M. Dose titration in the Multicentre Study of Hydroxyurea in Sickle Cell Anemia (MSH). Controlled Clinical Trials 1996;17(Suppl 2):92S. [Google Scholar]
Hankins 2014
- Hankins JS, Aygun B, Nottage K, Thornburg C, Smeltzer MP, Ware RE, et al. From infancy to adolescence: fifteen years of continuous treatment with hydroxyurea in sickle cell anemia. Medicine (Baltimore) 2014;93(28):e215. [DOI: 10.1097/MD.0000000000000215] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hassell 2010
- Hassell KL. Population estimates of sickle cell disease in the US. American journal of preventive medicine 2010 Apr 30;38(4):S512‐21. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analysis. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Altman DG, Sterne JAC (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org..
Howard 2015
- Howard J, Telfer P. Sickle Cell Disease in Clinical Practice. London 2015: Springer‐Verlag, 2015. [Google Scholar]
Jones 2009
- Jones AP, Riley RD, Williamson PR, Whitehead A. Meta‐analysis of individual patient data versus aggregate data from longitudinal clinical trials. Clinical Trials 2009;6(1):16‐27. [DOI] [PubMed] [Google Scholar]
Lanzkron 2013
- Lanzkron S, Carroll CP, Haywood Jr C. Mortality Rates and Age at Death from Sickle Cell Disease: US. 1979‐2005. Public Health Reports 2013;128:110‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Luchtman‐Jones 2016
- Luchtman‐Jones L, Pressel S, Hilliard L, Brown RC, Smith MG, Thompson AA, et al. Effects of hydroxyurea treatment for patients with hemoglobin SC disease. American Journal of Hematology 2016;91(2):238‐42. [DOI] [PubMed] [Google Scholar]
Manci 2003
- Manci EA, Culberson DE, Yang YM, Gardner TM, Powell R, Haynes J Jr, et al. Causes of death in sickle cell disease: an autopsy study. British Journal of Haematology 2003;123(2):359‐65. [DOI] [PubMed] [Google Scholar]
McGann 2011
- McGann PT, Howard TA, Flanagan JM, Lahti JM, Ware RE. Chromosome damage and repair in children with sickle cell anaemia and long‐term hydroxycarbamide exposure. British Journal of Haematology 2011;154(1):134‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Najean 1997
- Najean Y, Rain JD. Treatment of polycythemia vera: the use of hydroxyurea and pipobroman in 292 patients under the age of 65 years. Blood 1997;90(9):3370‐7. [PubMed] [Google Scholar]
National Toxicology Program 2008
- National Toxicology Program. NTP‐CERHR monograph on the potential human reproductive and developmental effects of hydroxyurea. NTP CERHR MON 2008; Vol. Oct, issue 21:vii‐viii, v, ix‐III1. [NIH Publication no: 08‐5993] [PubMed]
NCEPOD 2008
- NCEPOD. ‘Sickle: A sickle crisis?’ A report of the national confidential enquiry into patient outcome and death. 2008. www.ncepod.org.uk.2008sc.html (accessed 19 September 2016).
NICE 2012
- National Insititute of Clinical Excellence (NICE). Sickle cell acute painful episode: management of an acute painful sickle cell episode in hospital. http://www.nice.org.uk/guidance/cg143 (accessed 06 March 2017) 2012. [PubMed]
NIH 2014
- Evidence Based Management of Sickle Cell Disease. Expert Panel Report. National Institute of Health. 2014. www.nhlbi.nih.gov/sites/www.nhlbi.nih.gov/files/sickle‐cell‐disease‐report%20020816.pdf (accessed 19 September 2016).
Perrine 1978
- Perrine RP, Pembrey ME, John P, Perrine S, Shoup F. Natural history of sickle cell anaemia in Saudi Arabs: a study of 270 subjects. Annals of Internal Medicine 1978;88:1‐6. [DOI] [PubMed] [Google Scholar]
Perronne 2002
- Perronne V, Roberts‐Harewood M, Bachir D, Roudot‐Thoraval F, Delord JM, Thuret I, et al. Patterns of mortality in sickle cell disease in adults in France and England. Haematology Journal 2002;3(1):56‐60. [DOI] [PubMed] [Google Scholar]
Piel 2013
- Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Dewi M, et al. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model‐based map and population estimates. Lancet 2013;381(9861):142‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Platt 1984
- Platt OS, Orkin SH, Dover GJ, Beardsley GP, Miller B, Nathan DG. Hydroxyurea increases fetal hemoglobin production in sickle cell anemia. Transactions of the Association of American Physicians 1984;97:268‐74. [PubMed] [Google Scholar]
Platt 1994
- Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, et al. Mortality in sickle cell disease ‐ life expectancy and risk factors for early death. New England Journal of Medicine 1994;330:1639‐43. [DOI] [PubMed] [Google Scholar]
Powars 1983
- Powars D, Overturf G, Turner E. Is there an increased risk of Haemophilus influenzae septicemia in sickle cell anaemia?. Pediatrics 1983;71:927‐31. [PubMed] [Google Scholar]
Quinn 2010
- Quinn CT, Rogers ZR, McCavit TL, Buchanan GR. Improved survival of children and adolescents with sickle cell disease. Blood 2010;115:3447‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rogers 1978
- Rogers DW, Clarke JM, Cupidore L, Ramlal A, Sparke BR, Serjeant GR. Early deaths in Jamaican children with sickle cell disease. British Medical Journal 1978;1:1515‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Steinberg 1999
- Steinberg MH. The Management of Sickle Cell Disease. New England Journal of Medicine 1999;340:1021‐30. [DOI] [PubMed] [Google Scholar]
Steinberg 2003
- Steinberg MH, Barton F, Castro O, Pegelow CH, Ballas SK, Kutlar A, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA 2003;289(13):1645‐51. [DOI] [PubMed] [Google Scholar]
Steinberg 2010
- Steinberg MH, McCarthy WF, Castro O, Ballas SK, Armstrong FD, Smith W, et al. The risks and benefits of long‐term use of hydroxyurea in sickle cell anemia: A 17.5 year follow‐up. American Journal of Hematolology 2010; Vol. 85, issue 6:403‐8. [DOI] [PMC free article] [PubMed]
Sterkers 1998
- Sterkers Y, Preudhomme C, Lai JL, Demory JL, Caulier MT, Wattel E, et al. Acute myeloid leukemia and myelodysplastic syndromes following essential thrombocythemia treated with hydroxyurea: high proportion of cases with 17p deletion. Blood 1998;91(2):616‐22. [PubMed] [Google Scholar]
STOP 2006
- Lee MT, Piomelli S, Granger S, et al. Stroke Prevention Trial in Sickle Cell Anemia (STOP): extended follow‐up and final results. Blood 2006;108(3):847‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Strouse 2008
- Strouse JJ, Lanzkron S, Beach MC, Haywood C, Park H, Witkop C, et al. Hydroxyurea for sickle cell disease: a systematic review for efficacy and toxicity in children. Pediatrics 2008;122(6):1332‐43. [DOI] [PubMed] [Google Scholar]
Telfer 2007
- Telfer P, Coen P, Chakravorty S, Wilkey O, Evans J, Newell H, et al. Clinical outcomes in children with sickle cell disease living in England: a neonatal cohort in East London.. Haematologica 2007;92(7):905‐12. [DOI] [PubMed] [Google Scholar]
Terrin 1999
- Terrin ML, Barton FB, Bonds D, Ballas SK, Swerdlow P, Pegelow CH, et al. Effect of hydroxyurea on quality of life: 2‐year results from the multicenter study of hydroxyurea in sickle cell anemia. National Sickle Cell Disease Program 23rd Annual Meeting; 1999 Mar. 1999:161.
Triadou 1994
- Triadou P, Maier‐Redelsperger M, Krishnamoorty R, Deschamps A, Casadevall N, Dunda O, et al. Fetal haemoglobin variations following hydroxyurea treatment in patients with cyanotic congenital heart disease. Nouvelle Revue Francaise d Hematologie 1994;36(5):367‐72. [PubMed] [Google Scholar]
van der Plas 2011
- Plas EM, van den Tweel XW, Geskus RB, Heijboer H, Biemond BJ, Peters M, et al. Mortality and causes of death in children with sickle cell disease in the Netherlands, before the introduction of neonatal screening. British Journal of Haematology 2011;155(1):106‐10. [DOI] [PubMed] [Google Scholar]
Veith 1985
- Veith R, Galanello R, Papayannopoulou T, Stamatoyannopoulos G. Stimulation of F‐cell production in patients with sickle cell anaemia treated with cytarabine or hydroxyurea. New England Journal of Medicine 1985;313:1571‐5. [DOI] [PubMed] [Google Scholar]
Voskaridou 2010
- Voskaridou E, Christoulas D, Bilalis A, Plata E, Varvagiannis K, Stamatopoulos G, et al. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: results of a 17‐year, single‐centre trial (LaSHS). Blood 2010;115:2354‐63. [DOI] [PubMed] [Google Scholar]
Ware 2006
- Ware RE, McMurray MA, Schultz WH, Alvarez OA, Aygun B, Cavalier ME, et al. Academic community standards for chronic transfusion therapy in children with sickle cell anemia and stroke. Blood 2006;108(11):Abst 1213. [Google Scholar]
Ware 2010
- Ware RE. How I use hydroxyurea to treat young patients with sickle cell anemia. Blood 2010;115:5300‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wierenga 2001
- Wierenga KJ, Hambleton IR, Lewis NA. Survival estimates for patients with homozygous sickle‐cell disease in Jamaica: a clinic‐based population study. Lancet 2001;357(9257):680‐3. [DOI] [PubMed] [Google Scholar]
Zimmerman 2004
- Zimmerman SA, Schultz WH, Davis JS, Pickens CV, Mortier NA, Howard TA, et al. Sustained long‐term hematologic efficacy of hydroxyurea at maximum tolerated dose in children with sickle cell disease. Blood 2004;103(6):2039‐45. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Jones 2001
- Jones AP, Davies SC, Olujohungbe A. Hydroxyurea for sickle cell disease. Cochrane Database of Systematic Reviews 2001, Issue 2. [DOI: 10.1002/14651858.CD002202] [DOI] [PubMed] [Google Scholar]