

Abstract
Background
Despite the availability of effective drug therapies that reduce low‐density lipoprotein (LDL)‐cholesterol (LDL‐C), cardiovascular disease (CVD) remains an important cause of mortality and morbidity. Therefore, additional LDL‐C reduction may be warranted, especially for patients who are unresponsive to, or unable to take, existing LDL‐C‐reducing therapies. By inhibiting the proprotein convertase subtilisin/kexin type 9 (PCSK9) enzyme, monoclonal antibodies (PCSK9 inhibitors) may further reduce LDL‐C, potentially reducing CVD risk as well.
Objectives
Primary
To quantify short‐term (24 weeks), medium‐term (one year), and long‐term (five years) effects of PCSK9 inhibitors on lipid parameters and on the incidence of CVD.
Secondary
To quantify the safety of PCSK9 inhibitors, with specific focus on the incidence of type 2 diabetes, cognitive function, and cancer. Additionally, to determine if specific patient subgroups were more or less likely to benefit from the use of PCSK9 inhibitors.
Search methods
We identified studies by systematically searching the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE, Embase, and Web of Science. We also searched Clinicaltrials.gov and the International Clinical Trials Registry Platform and screened the reference lists of included studies. We identified the studies included in this review through electronic literature searches conducted up to May 2016, and added three large trials published in March 2017.
Selection criteria
All parallel‐group and factorial randomised controlled trials (RCTs) with a follow‐up time of at least 24 weeks were eligible.
Data collection and analysis
Two review authors independently reviewed and extracted data. When data were available, we calculated pooled effect estimates.
Main results
We included 20 studies with data on 67,237 participants (median age 61 years; range 52 to 64 years). Twelve trials randomised participants to alirocumab, three trials to bococizumab, one to RG7652, and four to evolocumab. Owing to the small number of trials using agents other than alirocumab, we did not differentiate between types of PCSK9 inhibitors used. We compared PCSK9 inhibitors with placebo (thirteen RCTs), ezetimibe (two RCTs) or ezetimibe and statins (five RCTs).
Compared with placebo, PCSK9 inhibitors decreased LDL‐C by 53.86% (95% confidence interval (CI) 58.64 to 49.08; eight studies; 4782 participants; GRADE: moderate) at 24 weeks; compared with ezetimibe, PCSK9 inhibitors decreased LDL‐C by 30.20% (95% CI 34.18 to 26.23; two studies; 823 participants; GRADE: moderate), and compared with ezetimibe and statins, PCSK9 inhibitors decreased LDL‐C by 39.20% (95% CI 56.15 to 22.26; five studies; 5376 participants; GRADE: moderate).
Compared with placebo, PCSK9 inhibitors decreased the risk of CVD events, with a risk difference (RD) of 0.91% (odds ratio (OR) of 0.86, 95% CI 0.80 to 0.92; eight studies; 59,294 participants; GRADE: moderate). Compared with ezetimibe and statins, PCSK9 inhibitors appeared to have a stronger protective effect on CVD risk, although with considerable uncertainty (RD 1.06%, OR 0.45, 95% CI 0.27 to 0.75; three studies; 4770 participants; GRADE: very low). No data were available for the ezetimibe only comparison. Compared with placebo, PCSK9 probably had little or no effect on mortality (RD 0.03%, OR 1.02, 95% CI 0.91 to 1.14; 12 studies; 60,684 participants; GRADE: moderate). Compared with placebo, PCSK9 inhibitors increased the risk of any adverse events (RD 1.54%, OR 1.08, 95% CI 1.04 to 1.12; 13 studies; 54,204 participants; GRADE: low). Similar effects were observed for the comparison of ezetimibe and statins: RD 3.70%, OR 1.18, 95% CI 1.05 to 1.34; four studies; 5376 participants; GRADE: low. Clinical event data were unavailable for the ezetimibe only comparison.
Authors' conclusions
Over short‐term to medium‐term follow‐up, PCSK9 inhibitors reduced LDL‐C. Studies with medium‐term follow‐up time (longest median follow‐up recorded was 26 months) reported that PCSK9 inhibitors (compared with placebo) decreased CVD risk but may have increased the risk of any adverse events (driven by SPIRE‐1 and ‐2 trials). Available evidence suggests that PCSK9 inhibitor use probably leads to little or no difference in mortality. Evidence on relative efficacy and safety when PCSK9 inhibitors were compared with active treatments was of low to very low quality (GRADE); follow‐up times were short and events were few. Large trials with longer follow‐up are needed to evaluate PCSK9 inhibitors versus active treatments as well as placebo. Owing to the predominant inclusion of high‐risk patients in these studies, applicability of results to primary prevention is limited. Finally, estimated risk differences indicate that PCSK9 inhibitors only modestly change absolute risks (often to less than 1%).
Keywords: Humans; Middle Aged; Antibodies, Monoclonal; Antibodies, Monoclonal/therapeutic use; Antibodies, Monoclonal, Humanized; Antibodies, Monoclonal, Humanized/therapeutic use; Cardiovascular Diseases; Cardiovascular Diseases/prevention & control; Cause of Death; Cholesterol, LDL; Cholesterol, LDL/blood; Cholinergic Antagonists; Cholinergic Antagonists/therapeutic use; Ezetimibe; Ezetimibe/therapeutic use; Hydroxymethylglutaryl‐CoA Reductase Inhibitors; Hydroxymethylglutaryl‐CoA Reductase Inhibitors/therapeutic use; Primary Prevention; Primary Prevention/methods; Proprotein Convertase 9; Proprotein Convertase 9/antagonists & inhibitors; Randomized Controlled Trials as Topic; Secondary Prevention; Secondary Prevention/methods; Time Factors
PCSK9 inhibitors for prevention of cardiovascular disease
Research question
Describe the effectiveness and safety of PCSK9 inhibitors for cardiovascular disease prevention.
Background
Despite the availability of effective drug therapies (statins or ezetimibe) that reduce low‐density (LDL) cholesterol (LDL‐C), cardiovascular disease (CVD) remains an important cause of mortality and morbidity. Additional LDL‐C reduction may therefore be warranted, especially for patients who are unresponsive to, or are unable to use, existing LDL‐C reducing therapies. PCSK‐9 inhibition produced by monoclonal antibodies (PCSK9 inhibitors) may further reduce LDL‐C levels and CVD risk.
Study characteristics
Review authors identified 20 studies that evaluated the effects of PCSK9 inhibitors in participants at high risk of CVD; studies were conducted in outpatient clinic settings. Review authors identified the studies included in this review through electronic literature searches conducted up to May 2016, and added three large trials published in March 2017.
Key results
PCSK9 inhibitors constitute a class of drugs that decrease LDL‐C and therefore may decrease the incidence of CVD. We examined the results of 20 studies, which showed beneficial effects on blood cholesterol concentrations of PCSK9 inhibitors at both six months and one year of follow‐up. Although the magnitude of this beneficial effect differed between studies, all showed beneficial effects. In comparisons of PCSK9 inhibitors versus no PCSK9 inhibitors, current evidence suggests that PCSK9 inhibitors decrease CVD incidence without affecting the incidence of all‐cause mortality. In comparisons of PCSK9 inhibitors versus alternative (more established) treatments such as statins or ezetimibe, high‐quality evidence is lacking. Differences in risk between people treated with and without PCKS9 inhibitors suggest the absolute treatment benefit will likely be modest (e.g. < 1% change in risk).
Quality of evidence
Most of the included randomised controlled trials (RCTs) were designed to explore biomarker associations; however, as all trials were industry funded, GRADE assessment revealed that the quality of the evidence was moderate. For associations with clinical endpoints (mortality and CVD), the quality of the evidence was moderate (placebo comparison) to very low (ezetimibe and statin comparisons).
Summary of findings
Summary of findings for the main comparison.
Summary of findings for PCSK9 compared with placebo
| PCSK9 inhibitors compared with placebo in addition to statin and/or ezetimibe background care | |||||||
| Patient or population: people at high risk of cardiovascular disease (history of CVD or high LDL‐C despite treatment) Setting: outpatient care settings Intervention: PCSK9 monoclonal antibodies Comparison: placebo | |||||||
| Outcomes | Ilustrative comparative risk or mean (95% CI) | Relative effect (95% CI) | Mean difference (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
|
Assumed risk or mean biomarker using placebo* |
Corresponding risk or mean using PCSK9 inhibition† |
||||||
| LDL‐C reduction (LDL‐C) Follow‐up: 6 months | Mean LDL‐C reduction was ‐6.12 mean percentage change form baseline | Mean LDL‐C reduction in the intervention group was ‐59.98 (‐64.76 to ‐55.19) percentage change form baseline | ‐53.86% (‐58.64 to ‐49.08) in percentage reduction from baseline | 4782 (8 RCTs) | ⊕⊕⊕⊝ MODERATEa | Negative is beneficial | |
| Cardiovascular disease (CVD) Follow‐up: 6 months to 36 months |
Cardiovascular disease risk was 64 per 1000 participants | Cardiovascular disease risk in the intervention group was 55 (51 lower to 59 lower) per 1000 participants | OR 0.86 (0.80 to 0.92) | 59294 (8 RCTs) | ⊕⊕⊕⊝ MODERATEb | Below 1 is beneficial | |
| All‐cause mortality (mortality) Follow‐up: 6 months to 36 months |
All‐cause mortality risk was 18 per 1000 participants | All‐cause mortality risk in the intervention group was 18 (16 lower to 20 higher) per 1000 participants | OR 1.02 (0.91 to 1.14) | 60684 (12 RCTs) | ⊕⊕⊕⊝ MODERATEb | Below 1 is beneficial | |
| Any adverse events (adverse events) Follow‐up: 6 months to 36 months |
Risk of any adverse events was 692 per 1000 participants | Risk of any adverse events in the intervention group was 707 (700 higher to 715 higher) per 1000 participants | OR 1.08 (1.04 to 1.12) | 61038 (13 RCTs) | ⊕⊕⊝⊝ LOWb,c | Below 1 is beneficial | |
| CI: confidence interval | |||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to the estimate of effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of effect but may be substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | |||||||
aUnclear randomisation processes and high risk of other biases. Downgrading one level because of "limitations in the design and implementation of available studies suggesting high likelihood of bias"
bResults predominantly determined by 3 large RCTs with a relatively short median follow‐up of 7 months (SPIRE‐1), 12 months (SPIRE‐2), and 26 months (FOURIER). SPIRE‐1/2 trials terminated prematurely owing to an unanticipated drug‐antibody response. Downgrading one level because of "indirectness of evidence"
cEffect was driven by the discontinued SPIRE trials. Downgrading one level because of "limitations in the design and implementation of available studies suggesting high likelihood of bias"
*Assumed risks or mean LDL‐C was based on the comparison arms of included trials
†Corresponding risk or mean was based on the risk difference reported in Table 7 or the mean difference in LDL‐C
Summary of findings 2.
Summary of findings for PCSK9 compared with ezetimibe
| PCSK9 Inhibitors compared to ezetimibe. | |||||||
| Patient or population: people at high risk of cardiovascular disease (history of CVD or high LDL‐C despite treatment) Setting: outpatient care settings Intervention: PCSK9 monoclonal antibodies Comparison: ezetimibe | |||||||
| Outcomes | Ilustrative comparative risk or mean (95% CI) | Relative effect (95%CI) | Mean difference (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
|
Assumed risk or mean biomarker using ezetimibe* |
Corresponding risk or mean using PCSK9 inhibition† |
||||||
| LDL‐C reduction (LDL‐C) Follow up: 6 months | Mean LDL‐C reduction was ‐6.12 mean percentage change form baseline | Mean LDL‐C reduction in the intervention group was ‐36.32 (‐40.29 to ‐32.34) percentage change from baseline | ‐30.20% (‐34.18 to ‐26.23) in percentage reduction from baseline | 823 (2 RCTs) | ⊕⊕⊕⊝ MODERATEa | Negative is beneficial | |
| Cardiovascular disease (CVD) | Cardiovascular disease risk was 64 per 1000 participants | Data unavailable | |||||
| All‐cause mortality (mortality) | All‐cause mortality risk was 6 per 1000 participants | Data unavailable | |||||
| Any adverse events (adverse events) | Risk of any adverse events was 692 per 1000 participants | Data unavailable | |||||
| CI: confidence interval | |||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to the estimate of effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of effect but may be substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | |||||||
aHigh risk of other biases. Downgrading one level because of "limitations in the design and implementation of available studies suggesting high likelihood of bias"
*Assumed risks or mean LDL‐C was based on the comparison arms of included trials
†Corresponding risk or mean was based on the risk difference reported in Table 7 or the mean difference in LDL‐C.
Summary of findings 3.
Summary of findings for PCSK9 compared with ezetimibe and statins
| PCSK9 inhibitors compared with ezetimibe and statins | |||||||
| Patient or population: people at high risk of cardiovascular disease (history of CVD or high LDL‐C despite treatment) Setting: outpatient care settings Intervention: PCSK9 monoclonal antibodies Comparison: ezetimibe and statins | |||||||
| Outcomes | Ilustrative comparative risk or mean (95% CI) | Relative effect (95%CI) | Mean difference (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
|
Assumed risk or mean biomarker with ezetimibe and statins* |
Corresponding risk or mean with PCSK9 inhibition† |
||||||
| LDL‐C reduction (LDL‐C) Follow‐up: 6 months | Mean LDL‐C reduction was ‐6.12 mean percentage change form baseline | Mean LDL‐C reduction in the intervention group was ‐45.32 (‐62.27 to ‐28.37) percentage change form baseline | ‐39.20% (‐56.15 to ‐22.26) in percentage reduction from baseline | 5376 (5 RCTs) | ⊕⊕⊕⊝ MODERATEa | Negative is beneficial | |
| Cardiovascular disease (CVD) Follow‐up: 6 months to 11 months |
Cardiovascular disease risk was 64 per 1000 participants | Cardiovascular disease risk in the intervention group was 53 (47 lower to 60 lower) per 1000 participants | OR 0.45 (0.27 to 0.75 | 4770 (3 RCTs) | ⊕⊝⊝⊝ VERY LOWa,b,c | Below 1 is beneficial | |
| All‐cause mortality (mortality) | All‐cause mortality risk was 6 per 1000 participants | Data unavailable | |||||
| Any adverse events (adverse events) Follow‐up: 6 months to 11 months |
Risk of any adverse events was 692 per 1000 participants | Risk of any adverse events in the intervention group was 729 (703 lower to 755 higher) per 1000 participants | OR 1.18 (1.05 to 1.34) | 5376 (5 RCTs) | ⊕⊕⊝⊝ LOWa,b | Below 1 is beneficial | |
| CI: confidence interval | |||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to the estimate of effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of effect but may be substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | |||||||
aMost data were based on OSLER‐1 and/or OSLER‐2, which were open‐label studies. Downgrading one level because of "limitations in the design and implementation of available studies suggesting high likelihood of bias"
bITT results were often unavailable; instead data were extracted on the basis of an as treated sample. Downgrading one level because of "limitations in the design and implementation of available studies suggesting high likelihood of bias"
cNumber of events was low. Downgrading one level because of " Imprecision of results"
*Assumed risks or mean LDL‐C was based on comparison arms of included trials
†Corresponding risk or mean was based on the risk difference reported in Table 7 or the mean difference in LDL‐C
Background
Description of the condition
Cardiovascular disease (CVD), including fatal and non‐fatal cardiac and vascular diseases, remains a major cause of mortality and morbidity both in the United Kingdom (UK) and globally (Capewell 2008; Kreatsoulas 2010; Krishnamurthi 2013; Moran 2014; Murray 2012; Roger 2011; WHO 2008). Cardiovascular disease imposes a serious personal, financial, and societal burden with estimated direct costs of GBP 14,300,000,000 (i.e. 20% of National Health Service (NHS) funding), indirect costs of GBP 16,200,000,000, and an attributed mortality percentage of 35% in the UK (Capewell 2008). This burden is especially high in patients with familial hypercholesterolaemia (FH) who have loss of function mutation, which affects 1 in 250 individuals of European descent (Benn 2012; Knowles 2014; Nordestgaard 2013). These mutations prevent removal of circulating low‐density lipoprotein cholesterol (LDL‐C), which is one of the most important modifiable risk factors for CVD (Grundy 2004), both in patients with FH and in the general population. Autosomal dominant FH is caused by heterozygous mutations in the low‐density lipoprotein receptor (LDLR: OMIM #143890) (Sudhof 1985), apolipoprotein B (APOB; OMIM #144010) ‐ the major constituent apoprotein of LDL‐C (Garcia 2001; Innerarity 1987; Nordestgaard 2013), or the gene for proprotein convertase subtilisin/kexin type 9 (PCSK9; #603776) (Abifadel 2003). A rare autosomal recessive form of FH is caused by mutations in the gene for the low‐density lipoprotein receptor adaptor protein 1 (LDRRAP1; OMIM #603813). Patients with FH have higher risk of premature coronary heart disease that can be reduced with statin treatment. Polygenic elevation in LDL‐C concentration, which is associated with higher risk of coronary heart disease (CHD), is caused by additive effects of common, largely independently inherited polymorphisms located in more than 50 loci throughout the genome (Willer 2013).
Description of the intervention
Interventions of proven efficacy in reducing cardiovascular events through lowering of LDL‐C include statin drugs targeting 3‐hydroxy‐3‐methyl‐glutaryl‐CoA (HMG‐CoA) reductase and ezetimibe targeting the Niemann‐Pick C1‐like 1 intestinal cholesterol transporter protein (Cannon 2015; CTT 2005a; CTT 2005b; CTT 2012). Cardiovascular risk is reduced but not abolished among patients receiving these medications, suggesting that additional LDL‐C reduction via alternative pathways may result in further reduction in CVD events, especially among patients who have an inadequate response to, or are intolerant of, statins or ezetimibe (Mancini 2011; Marks 2003).
A new pharmacological target for further reduction of LDL‐C is the proprotein convertase subtilisin/kexin type 9 (PCSK9) enzyme. Monoclonal antibodies (mAbs) against the PCSK9 enzyme (PCSK9 inhibitors) are currently being evaluated in phase 3 trials. PCSK9 inhibitors are administered subcutaneously every two or four weeks. Reported mean half‐life times for subcutaneous administration have been six to seven days, with minimal differences due to administration site (abdomen or upper arm) and LDL‐C reaching its lowest level at 15 days (Lunven 2014). The impact, if any, of environmental factors or comedications on PCSK9 mAb efficacy is still mostly unknown (Lunven 2014).
How the intervention might work
PCSK9 is synthesised and secreted by hepatocytes and binds to the LDL‐C receptor (LDLR) on the hepatocyte surface, promoting internalisation and degradation. Reduction in surface LDLR reduces uptake of LDL particles and increases LDL‐C concentration in the blood (Cohen 2005; Cohen 2006). Therefore, inhibitors of PCSK9 are expected to lower LDL‐C. Moreover, inhibition of PCSK9 may further enhance the lipid‐lowering effects of statins, which are thought to be limited by a statin‐induced increase in PCSK9 expression (Catapano 2013).
PCSK9 inhibitors bind to the PCSK9 enzyme with high affinity, disrupting its ability to bind with LDLR. By preventing PCSK9 from binding to LDLR, inhibitors against PCSK9 maintain surface LDLR expression with the aim of reducing LDL‐C serum concentration. This is supported by the finding that variations in the PCSK9 gene are associated with long‐term elevations in LDL‐C and higher risk of CHD (Benn 2010; Chasman 2012). Alternatively, loss of function mutations in PCKS9 that lower LDL‐C levels have also been associated with decreased CHD risk (Cohen 2006). This provides evidence in favour of the PCSK9 enzyme as a valid therapeutic target for prevention of CVD.
Why it is important to do this review
Statins are widely prescribed to reduce LDL‐C levels and CVD risk in patients at increased risk. Patients taking statins reduce their risk of CVD by around 20% to 25% for every 1 mmol/L decrease in LDL‐C (CTT 2005a; CTT 2012), which may be further reduced by taking ezetimibe (Cannon 2015). Given the strong and positive associations, without clear threshold, between LDL‐C and CVD as described in prospective studies (CTT 2005a; CTT 2012), it is expected that further reduction in LDL‐C may lead to further prevention of CVD events. This could be especially important for patients not tolerating statins, those with very high levels of LDL‐C, and those at high cardiovascular risk. Previously, a narrative review of phase 1 and 2 trials found that PCSK9 inhibitors were generally well tolerated (Catapano 2013); however, information on the medium‐term to long‐term safety and efficacy of these drugs has not yet been reviewed. Research on statins seems to suggest the following unintended (safety) endpoints: type 2 diabetes (T2DM), possible weight gain (Sattar 2010; Swerdlow 2014), liver inflammation, and rarely myositis (Collins 2016). It is uncertain if reducing LDL‐C via a different mechanism might be associated with the same or a different set of adverse events. Although a recent meta‐analysis (Navarese 2015), which included the ODYSSEY Long Term trial, showed that PCSK9 inhibitors indeed reduced LDL‐C and cardiovascular‐related mortality, this finding was based mostly on short‐term studies (< 24 weeks) and excluded larger trials with longer follow‐up, such as OSLER‐1 and OSLER‐2 randomised controlled trials (RCTs) and recently published phase 3 trials (FOURIER and SPIRE‐1 and SPIRE‐2) (Sabatine 2015). Furthermore, with recent Food and Drug Administration (FDA) and European Medicines Agency (EMA) approvals of alirocumab (Praluent) and evolocumab (Repatha), these drugs have become available to (selected) patients, and (remaining) questions on long‐term efficacy and safety have become increasingly important to answer. Specifically, the EMA has approved Praluent and Repatha for patients with primary hypercholesterolaemia, and the FDA has approved both drugs for patients with heterozygous familial hypercholesterolaemia or a history of clinical atherosclerotic cardiovascular disease. These recommendations have found their way into the 2016 European Society of Cardiology (ESC)/European Atherosclerosis Society (EAS) Guidelines for the Management of Dyslipidaemias, which recommend consideration of a PCSK9 inhibitor for pharmacological treatment of hypercholesterolaemia "in patients at very high‐risk, with persistent high LDL‐C despite treatment with maximal tolerated statin dose, in combination with ezetimibe or in patients with statin intolerance". The same guidelines recommend that "treatment with a PCSK9 antibody should be considered in FH patients with CVD or at very high‐risk for CHD" (Catapano 2016). Recently, Pfizer discontinued the development of bococizumab, citing lack of long‐term efficacy due to increased immunogenicity over time (Pfizer 2017). Consequently, we considered it timely to conduct a systematic review of RCTs to quantify the long‐term efficacy and safety of inhibitors of PCSK9 for CVD prevention. For this review, CVD is defined as a composite of fatal and non‐fatal cardiac and vascular diseases, including stroke.
Objectives
Primary
To quantify short‐term (24 weeks), medium‐term (one year), and long‐term (five years) effects of PCSK9 inhibitors on lipid parameters and on the incidence of CVD.
Secondary
To quantify the safety of PCSK9 inhibitors, with specific focus on the incidence of type 2 diabetes, cognitive function, and cancer. Additionally, to determine if specific patient subgroups are more or less likely to benefit from the use of PCSK9 inhibitors.
Methods
Criteria for considering studies for this review
Types of studies
We included parallel‐group and factorial RCTs with follow‐up time of at least 24 weeks. Cluster RCTs, cross‐over trials, and non‐randomised studies were ineligible for this review, and we excluded them during title and abstract screening; we note a single cross‐over trial that we have excluded for this reason (Nissen 2016). RCTs were eligible if they were reported as full‐text articles or were published as abstracts, or if they were available only as unpublished data.
Types of participants
RCTs were eligible if they included adults 18 years of age or older, with or without a prior history of CVD. Participants could have normal lipid levels or hypercholesterolaemia. We applied no restriction on comorbidities.
Types of interventions
We included trials if they randomised participants to a PCSK9 inhibitor and to placebo, statins, or ezetimibe, or a combination of these.
Types of outcome measures
Primary outcomes
Lipid parameters (total cholesterol, LDL‐C, high‐density lipoprotein cholesterol (HDL‐C), triglycerides, apolipoprotein A1, apolipoprotein B and lipoprotein(a)): mean difference (MD) in mean percentage change from baseline or difference at the end of follow‐up
Composite endpoint of CVD, defined as urgent coronary revascularisation, unstable angina pectoris, non‐fatal and fatal myocardial infarction, non‐fatal and fatal stroke, and CHD death
Secondary outcomes
All‐cause mortality
Any adverse events, including type 2 diabetes (T2DM) and cancer
Cognitive function as standardised mean difference (SMD), as mean percentage change from baseline, or as difference between treatment arms at the end of follow‐up
Fasting glucose and glycosylated haemoglobin (HbA1c) as mean percentage change from baseline or as difference at the end of follow‐up
Quality of life as SMD, as mean percentage change from baseline, or as difference at the end of follow‐up
Search methods for identification of studies
Electronic searches
We identified trials through systematic searches (Lefebvre 2011) of the following databases.
Cochrane Central Register of Controlled Trials (CENTRAL; 2016, Issue 4 of 12) in the Cochrane Library.
MEDLINE (Ovid, 1946 to April week 4 2016).
Embase (Ovid, 1980 to week 19 2016).
Web of Science Core Collection (Thomson Reuters, 1970 to 8 May 2016).
Please see Appendix 1 for the search strategies used. We applied the sensitivity‐maximising version of the Cochrane RCT filter (Lefebvre 2011) to MEDLINE and adaptations of it to Embase and Web of Science. We limited searches to records from 2005, as PCSK9 was discovered as a potential target in 2003 (Farnier 2014; Seidah 2003), hence we excluded papers published before 2005. We imposed no language restrictions.
We identified the studies included in this review through electronic literature searches conducted up to May 2016. Through these searches, we identified several ongoing studies, and during the latter stages of finalising the review, we became aware of the publication of three of them in March 2017. We decided to incorporate data from those studies into this version of the review because of their size and impact on review findings.
Additionally, we searched ClinicalTrials.gov (www.ClinicalTrials.gov) and the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP) Search Portal (apps.who.int/trialsearch/) for relevant RCTs on 18 September 2016.
Searching other resources
We searched the following websites for unpublished studies on 19 September 2016.
Food and Drug Administration (FDA) website (http://www.fda.gov/).
Pharmaceutical company websites (e.g. Regeneron ‐ http://www.regeneron.com/; Sanofi ‐ http://en.sanofi.com/).
ProQuest dissertations and theses (PQDT; http://www.proquest.com/products‐services/pqdt.html).
Additionally, we screened reference lists of included studies for relevant RCTs.
Data collection and analysis
Selection of studies
Two review authors (AFS and LSP) independently screened search results by title and abstract, and subsequently the full text, for potentially relevant studies. A third review author (JPC) resolved disagreements. We distilled multiple reports on a single RCT into a single entry. We have provided a PRISMA (Preferred Reporting Items for Systematic Reviews and Meta‐Analyses) flow diagram, as well as details of studies excluded after full text assessment (see Characteristics of excluded studies).
Data extraction and management
Two review authors (AFS and LSP) independently extracted data and resolved differences by returning to the original publication and, if needed, by consulting a third review author (JPC). When appropriate, we extracted data on numbers of events versus no events, means, standard deviations, crude point estimates, or standard error estimates. For continuous endpoints, we extracted data on change from baseline or on differences between study arms at completion of follow‐up. When possible, we focused on estimates adjusted for baseline measurements (Vickers 2001). When reported, we extracted results from an intention‐to‐treat (ITT) analysis. For adverse events, we tried to extract results for per‐protocol or as‐treated populations. When available, we used the study protocol, appendices and design papers as additional sources of information. We combined full‐text screening, data extraction, and data entry using a Microsoft Access database (available from AFS).
Assessment of risk of bias in included studies
We assessed risk of bias using the Cochrane risk of bias tool (Higgins 2011a) on the basis of the following items.
Random sequence generation (selection bias).
Allocation (selection bias).
Blinding of participants and personnel (performance bias).
Blinding of outcome assessment (detection bias).
Incomplete outcome data (attrition bias).
Selective reporting (reporting bias).
Other potential sources of bias.
We graded individual items as having "low", "unclear", or "high" risk of bias. We presented "risk of bias" per study and for the outcome LDL‐C (which can be seen more generally as risk of bias for biomarker outcomes).
Assessment of bias in conducting the systematic review
We conducted this Cochrane review according to the published protocol (Schmidt 2015) and reported deviations from it in the Differences between protocol and review section.
Measures of treatment effect
We reported results as mean differences (MDs) for continuous outcomes and as odds ratios (ORs) and risk differences (RDs) for binary endpoints. In the manuscript, we focus on MDs and ORs; however, we include estimates of RDs of meta‐analysed treatment effects because of their relevance for individual patients (Newcombe 2014); given that ORs and RDs represent the same data, we provide only forest plots for OR estimates and report pooled RD (and OR) estimates in Table 4 and Table 5. We calculated confidence intervals (CIs) using the Wald method, assuming a standard normal distribution, or a t‐distribution when appropriate.
Table 1.
Summary results ‐ clinical events analyses as odds ratios
|
Number of studies |
Number of events in the PCSK9 arm |
Number of participants in the PCSK9 arm |
Number of events in the comparison arm |
Number of participants in the comparison arm |
Fixed‐effect (95% CI) |
Between‐study heterogeneity P value |
|
| Placebo comparison | |||||||
| All‐cause mortality | 12 | 580 | 31358 | 558 | 29326 | 1.02 (0.91 to 1.14) | 0.159 |
| Any cardiovascular event | 8 | 1790 | 30355 | 2009 | 28939 | 0.86 (0.80 to 0.92) | 0.803 |
| Any myocardial infarction | 10 | 686 | 30610 | 869 | 29038 | 0.77 (0.69 to 0.85) | 0.674 |
| Any stroke | 8 | 265 | 29828 | 340 | 28672 | 0.76 (0.65 to 0.89) | 0.185 |
| Any adverse event | 13 | 22593 | 31611 | 20435 | 29427 | 1.08 (1.04 to 1.12) | 0.38 |
| Myalgia | 12 | 1249 | 31428 | 1094 | 29363 | 1.07 (0.99 to 1.16) | 0.873 |
| Influenza | 6 | 191 | 2923 | 82 | 1477 | 1.19 (0.91 to 1.55) | 1 |
| Hypertension | 8 | 110 | 3436 | 60 | 1593 | 0.86 (0.62 to 1.18) | 0.741 |
| Cancer | 5 | 83 | 2851 | 46 | 1442 | 0.91 (0.63 to 1.31) | 0.964 |
| Type 2 diabetes | 7 | 956 | 17535 | 911 | 16681 | 1.04 (0.95 to 1.14) | 0.983 |
| Elevated creatinine | 8 | 319 | 30399 | 309 | 28933 | 0.85 (0.73 to 0.99) | 0.419 |
| Neurological events | 5 | 289 | 16036 | 242 | 14919 | 1.04 (0.88 to 1.24) | 0.759 |
| Ezetimibe and statin comparison | |||||||
| All‐cause mortality | |||||||
| Any cardiovascular event | 3 | 29 | 3079 | 33 | 1691 | 0.45 (0.27 to 0.75) | 0.712 |
| Any myocardial infarction | |||||||
| Any stroke | |||||||
| Any adverse event | 5 | 2290 | 3309 | 1347 | 2067 | 1.18 (1.05 to 1.34) | 0.478 |
| Myalgia | 5 | 127 | 3309 | 81 | 2067 | 1.09 (0.81 to 1.48) | 0.715 |
| Influenza | 4 | 113 | 3183 | 51 | 1942 | 1.28 (0.91 to 1.80) | 0.45 |
| Hypertension | 3 | 6 | 207 | 12 | 453 | 1.10 (0.41 to 2.96) | 0.893 |
| Cancer | |||||||
| Type 2 diabetes | 4 | 35 | 3183 | 22 | 1942 | 1.10 (0.63 to 1.93) | 0.057 |
| Elevated creatinine | 5 | 20 | 3183 | 29 | 1942 | 0.51 (0.28 to 0.92) | 0.969 |
| Neurological events | 2 | 5 | 207 | 9 | 453 | 1.22 (0.40 to 3.69) | 1 |
Table 2.
Summary results ‐ clinical events analyses as risk differences
|
Number of studies |
Number of events in the PCSK9 arm |
Number of participants in the PCSK9 arm |
Number of events in the comparison arm |
Number of participants in the comparison arm |
Fixed‐effect (95% CI) |
Between‐study heterogeneity P value |
|
| Placebo comparison | |||||||
| All‐cause mortality | 12 | 580 | 31358 | 558 | 29326 | 0.000 (‐0.002 to 0.002) | 0.781 |
| Any cardiovascular event | 8 | 1790 | 30355 | 2009 | 28939 | ‐0.009 (‐0.013 to ‐0.005) | 0.005 |
| Any myocardial infarction | 10 | 686 | 30610 | 869 | 29038 | ‐0.007 (‐0.009 to ‐0.004) | < 0.001 |
| Any stroke | 8 | 265 | 29828 | 340 | 28672 | ‐0.003 (‐0.004 to ‐0.001) | 0.409 |
| Any adverse event | 13 | 22593 | 31611 | 20435 | 29427 | 0.015 (0.008 to 0.023) | < 0.001 |
| Myalgia | 12 | 1249 | 31428 | 1094 | 29363 | 0.002 (‐0.001 to 0.006) | 0.979 |
| Influenza | 6 | 191 | 2923 | 82 | 1477 | 0.010 (‐0.005 to 0.025) | 0.513 |
| Hypertension | 8 | 110 | 3436 | 60 | 1593 | ‐0.005 (‐0.016 to 0.006) | 1 |
| Cancer | 5 | 83 | 2851 | 46 | 1442 | ‐0.003 (‐0.013 to 0.008) | 0.892 |
| Type 2 diabetes | 7 | 956 | 17535 | 911 | 16681 | 0.002 (‐0.03 to 0.07) | 0.73 |
| Elevated creatinine | 8 | 319 | 30399 | 309 | 28933 | ‐0.002 (‐0.003 to ‐0.000) | < 0.001 |
| Neurological events | 5 | 289 | 16036 | 242 | 14919 | 0.001 (‐0.002 to 0.004) | 0.923 |
| Ezetimibe and statin comparison | |||||||
| All‐cause mortality | |||||||
| Any cardiovascular event | 3 | 29 | 3079 | 33 | 1691 | ‐0.011 (‐0.017 to ‐0.004) | 1 |
| Any myocardial infarction | |||||||
| Any stroke | |||||||
| Any adverse event | 5 | 2290 | 3309 | 1347 | 2067 | 0.037 (0.011 to 0.063) | 0.862 |
| Myalgia | 5 | 127 | 3309 | 81 | 2067 | 0.003 (‐0.007 to 0.014) | 0.901 |
| Influenza | 4 | 113 | 3183 | 51 | 1942 | 0.009 (‐0.002 to 0.019) | 1 |
| Hypertension | 3 | 6 | 207 | 12 | 453 | 0.002 (‐0.020 to 0.025) | 0.922 |
| Cancer | |||||||
| Type 2 diabetes | 4 | 35 | 3183 | 22 | 1942 | 0.001 (‐0.007 to 0.008) | 0.027 |
| Elevated creatinine | 5 | 20 | 3183 | 29 | 1942 | ‐0.006 (‐0.012 to ‐0.000) | 0.984 |
| Neurological events | 2 | 5 | 207 | 9 | 453 | 0.004 (‐0.019 to 0.028) | 1 |
Unit of analysis issues
This Cochrane review focused exclusively on parallel‐group designed RCTs, hence we had no unit of analysis issues.
Dealing with missing data
We contacted trial authors to request missing data.
Assessment of heterogeneity
We measured between‐study heterogeneity by using the I² statistic with a one‐sided confidence interval (with a z‐value of ‐1.96) and tested it using a Q test. For binary endpoints, we measured between‐study heterogeneity by using Tau² and tested it using a likelihood ratio test.
Originally, we intended to refrain from meta‐analysis if heterogeneity was greater than 50%. Although we observed a large amount of heterogeneity in the biomarker estimates, we nevertheless meta‐analysed the data. We made this decision because we believed that between‐study differences in treatment effects did not preclude a clinically relevant combination of data (see results and discussion).
Assessment of reporting biases
Fewer than 10 trials reported on the same comparator groups (see data synthesis and results), hence we did not assess reporting bias.
Data synthesis
Before meta‐analysing results, we grouped trials together on the basis of comparator treatment(s) received, including placebo, ezetimibe, and ezetimibe or statin. Trials comparing PCSK9 mAbs against statins only, or comparing mAbs types, were unavailable.
We combined study‐specific estimates in R (R Development Core Team 2014) and combined continuous data using the inverse variance method for fixed‐effect meta‐analysis. For binary data, we reconstructed individual participant data on the basis of cell counts, and we combined results using generalised linear models (GLMs) with a random intercept for study (Bradburn 2007; Sweeting 2004). For continuous data, we reported both fixed‐effect and random‐effects estimates, and for binary endpoints, we reported only fixed‐effect estimates, because owing to data sparseness, random‐effects models were unreliable.
In the case of multiple treatment or comparator arms, we pooled estimates across arms to facilitate a comparison between inhibitors and comparison therapy. Alternatively, we could have compared results from a single intervention arm versus multiple comparator groups (or vice versa), but this would have resulted in correlated effect estimates with erroneously small P values (i.e. increased type 1 errors).
'Summary of findings' table
We created 'Summary of findings' tables (using the GRADE approach to assess the quality of evidence; Grade Working Group 2004) for each comparison group separately and (on the basis of the protocol) included outcomes, LDL‐C, CVD events, adverse events, and mortality. We calculated risk under the intervention using estimated mean differences or risk differences; we included odds ratios in the table but did not use them to calculate (reduced) risk under treatment. Given that all studies provided participants with a combination of statins or ezetimibe, we estimated the mean or risk under the comparison treatment using the entire sample of trials. We changed column names to reflect this approach.
Subgroup analysis and investigation of heterogeneity
We assessed potential sources of between‐study heterogeneity in PCSK9 inhibitor effects on LDL‐C (at six months) using the following subgroup analyses: gender, age, history of CHD, diabetes at baseline, baseline LDL‐C level, and familial or non‐familial hypercholesterolaemia. We calculated interaction effects within study (Altman 2003) and meta‐analysed them, preventing bias due to study‐specific factors (Schmidt 2014b). We explored these subgroup analyses separately for RCTs comparing PCSK9 mAbs against placebo or against ezetimibe. Study authors did not report subgroup effects in sufficient detail for RCTs comparing PCSK9 mAbs against ezetimibe or statin for inclusion in the analysis.
Additionally (owing to the limited number of RCTs, only for trials using a placebo comparator), we employed meta‐regression (weighted for inverse variance weights; Thompson 2002) to explore whether between‐study heterogeneity was related to baseline characteristics described before, with the addition of ethnicity and proportion of missing LDL‐C measurements.
Sensitivity analysis
We attempted the following sensitivity analyses.
We stratified trials by allocated dose of PCSK9 mAb. Owing to the limited number of trials, we did this only for the placebo comparison and the endpoints of LDL‐C and apolipoprotein B at six months.
We intended to explore the influence of perceived risk of bias by grouping studies that had a low perceived risk of bias on all items (see Characteristics of included studies) and comparing six‐month LDL‐C reduction in studies that did not have low risk of bias on all items (higher‐risk group). However, none of the trials had low risk of bias on all items, hence we did not perform this sensitivity analysis.
We also intended to explore differences due to type of PCSK9 mAb, but we had already explored this by stratifying doses for placebo trials. For remaining comparison groups, RCTs were too few for meaningful exploration of this.
Please note that in our published protocol, we originally set out to perform these sensitivity analyses for CVD and mortality as well; however, owing to the limited number of events, we were not able to perform these analyses. Simillarly, we aimed to explore the impact of missing data by stratifying RCTs on missing 0% to 5%, 6% to 10%, and more than 10% of LDL‐C, CVD, or mortality data. However, owing to data sparseness, we did this only for the LDL‐C endpoint and used a meta‐regression analysis instead.
Reaching conclusions
We based our conclusions on findings derived from quantitative synthesis of included studies.
Results
Description of studies
Results of the search
The search yielded 1066 hits, which we supplemented by 11 additional records obtained by cross‐referencing trial registry sites and other sources (see Figure 1 for a flow diagram). After screening titles and abstracts, we retrieved 42 full‐text articles and excluded 25 of these. We included 19 references describing 20 studies. Most studies had multiple publications (e.g. conference abstracts) that we distilled into a single entry. For the ODYSSEY trials, we extracted additional information from a recent FDA report (FDA 2015).
Figure 1.
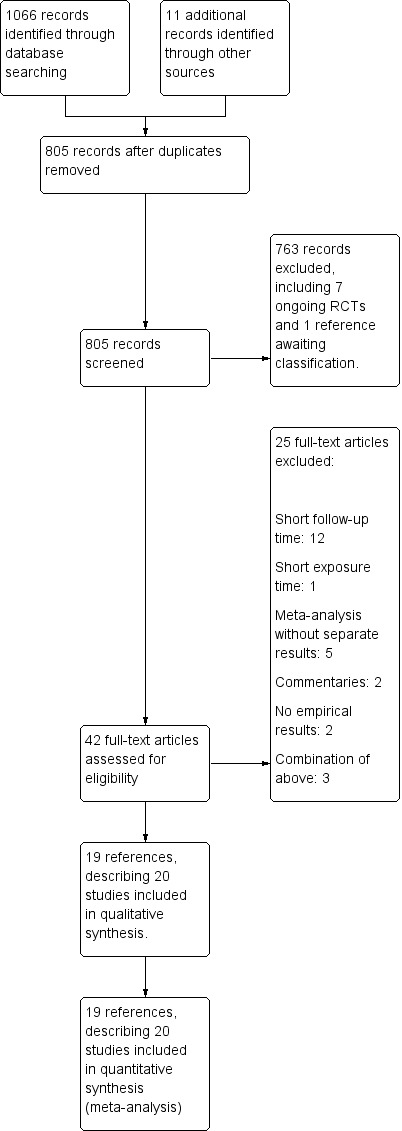
Study flow diagram.
Included studies
PCSK9 inhibitors; settings and participants
Investigators collected a combined sample of 68,341 participants in these 20 trials; of these, 1104 participants were included twice ‐ once in OSLER‐1, and once in the meta‐analysis of OSLER‐1 and OSLER‐2 (OLSER‐2 was unavailable separately). Of 67,237 unique participants, 20,210 were women (30%; of 67,130 participants for whom gender was reported; see Characteristics of included studies), 6984 did not have a history of CVD (11%; of 61,382 participants with reported CVD history), 2513 had FH (7%; of 33,707 with reported FH status), 25,536 had a T2DM diagnosis at baseline (39%; of 65,740 participants with recorded T2DM status). We note that the three FH studies focused exclusively on participants with FH (self‐identified). Caucasians were the predominant ethnic group included in these studies (86%). All trials included participants treated in outpatient care settings.
All included studies were industry‐sponsored, multi‐centre trials; most focused on alirocumab (REGN727, SAR256553), three explored bococizumab (RM316, PF‐04950615; Ballantyne 2015;SPIRE 1/2), one examined RG7652 (Equator), and four studied evolocumab (AMG145). The evolocumab trials (Descartes; OSLER‐1; OSLER 1/2) are closely related in the sense that, after completing the Descartes study, participants were offered enrolment in the OSLER‐2 study. The OSLER‐2 has been published only in combination with OSLER‐1 (which similarly limited enrolment to participants who first completed a 12‐week "parent" trial). Given that the OSLER‐2 trial has not been published separately, we included meta‐analysis results of OSLER‐1 and OSLER‐2, but we also used OLSER‐1 data for outcomes not reported in the meta‐analysis of OLSER‐1 and OSLER‐2 trials.
Comparison group
Investigators in 13 trials randomised participants to placebo or PCSK9 inhibitors (Ballantyne 2015; Descartes; Equator; FOURIER; ODYSSEY CHOICE I; ODYSSEY CHOICE II; ODYSSEY COMBO I; ODYSSEY FH I; ODYSSEY FH II; ODYSSEY HIGH FH; ODYSSEY Long Term; SPIRE 1/2, with SPIRE1/2 counted as two studies) as add‐on to background therapy, which could consist of ezetimibe, statins, and other interventions (see Characteristics of included studies). They randomised participants enrolled in ODYSSEY COMBO II and ODYSSEY MONO to alirocumab or ezetimibe. Finally, the remaining five studies (OSLER‐1; OSLER 1/2; ODYSSEY ALTERNATIVE; ODYSSEY OPTIONS I; ODYSSEY OPTIONS II) compared participants receiving a PCSK9 inhibitor with those receiving ezetimibe or statins, or usual care involving both ezetimibe and statins. Note that the OPTIONS I and OPTIONS II trials compared alirocumab with ezetimibe and atorvastatin, atorvastatin, or rosuvastatin. As described in the Methods section, to prevent erroneously small P values (due to use of the same alirocumab arm twice), we combined multiple arms of comparison groups and estimated effects of alirocumab versus ezetimibe and statin.
Researchers administered PCSK9 inhibitors every two weeks, every four weeks, or every eight weeks; for the sake of comparison, we calculated the two weeks' equivalence dosage (see Characteristics of included studies), which ranged from 50 mg to 210 mg every two weeks. In most studies (except ODYSSEY FH II, ODYSSEY HIGH FH, DESCARTES, OSLER‐1, and ODYSSEY LONG TERM), participants received different dosages of PCSK9, often depending on a predefined up‐titration criterion such as LDL‐C reduction or history of CVD; to account for these within‐study differences in dosage by stratified analyses (see methods and results), we grouped studies (when needed) by using a dosage range instead of a single dosage.
Excluded studies
We excluded 25 trials (Characteristics of excluded studies), predominantly owing to follow‐up time less than 24 weeks (see main objectives), or because trials described a meta‐analysis while providing little to no detail on individual studies (which were already included separately). Besides these excluded trials, we identified seven ongoing trials (Characteristics of ongoing studies) that fit our inclusion criteria; of these, two trials (ODYSSEY Outcomes; TAUSSIG) focus on long‐term effects on clinical outcomes, and one describes the six SPIRE biomarker trials and is pending classification.
Risk of bias in included studies
We have provided per study risk of bias with rationale in the Characteristics of included studies table. All studies described used a randomised trial design; we have discussed risk of bias for biomarker endpoints in the following sections and have summarised this information in Figure 2 and Figure 3, See section on "Detection and attrition bias of the association with clinical endpoints" for risk of bias reflecting clinical endpoints.
Figure 2.
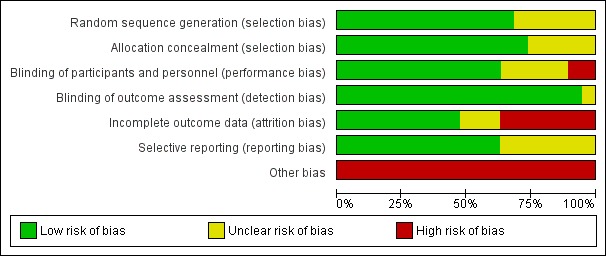
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Figure 3.
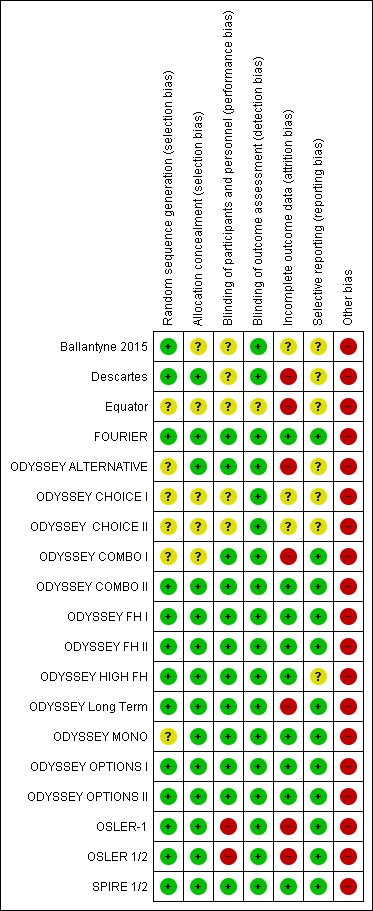
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Six trials (Equator; ODYSSEY ALTERNATIVE; ODYSSEY CHOICE I; ODYSSEY CHOICE II; ODYSSEY COMBO I; ODYSSEY MONO) did not provide sufficient detail on how randomisation was achieved (unclear risk of bias). The remaining studies typically used a voice‐based or Internet‐based centralised response system, and we perceived them to have low risk of bias.
We ensured allocation concealment by using centralised allocation and in some cases permuted blocks. Five RCTs (Ballantyne 2015; Equator; ODYSSEY CHOICE I; ODYSSEY CHOICE II; ODYSSEY COMBO I) did not sufficiently report on this item, and we perceived them as having unclear risk of bias (we contacted study authors but they did not respond).
Blinding
Owing to the open‐label design, the OSLER‐1 and OSLER‐2 studies are at high risk of performance bias. It seems plausible that knowledge of drug exposure may influence choices on lifestyle or additional clinical care, which may distort difference in biomarkers and clinical events across treatment arms.
Most studies assessed biomarkers in a central laboratory, making detection bias unlikely; one possible exception is the Equator study, which did not describe how biomarkers were assessed.
Incomplete outcome data
Loss due to follow‐up (attrition bias) was typically low (arbitrarily defined as < 5%), except in the Descartes, Equator, ODYSSEY ALTERNATIVE, ODYSSEY COMBO I, ODYSSEY Long Term, and OSLER‐1 trials, and in meta‐analysis of OSLER 1/2. Most studies used advanced analytics, such as mixed‐effects models or (multiple) imputations, to ameliorate loss due to follow‐up (even if this was minor) and to ensure the ITT analysis. However, information on both performance of these methods and appropriateness of assumptions underlying these methods was missing.
Selective reporting
We compared endpoints described in study protocols and on clinicaltrials.gov versus endpoints reported in the primary publication, and general found good agreement. We assigned seven trials (contributing 2901 participants) an unclear risk of bias grade because the full publication was unavailable, hence we could not fully compare results.
Other potential sources of bias
In accordance with guidance provided by Cochrane (Lundh 2017), we assigned high risk of bias to all industry‐funded trials.
Detection and attrition bias of association with clinical endpoints
Most studies reported clinical endpoints based on the safety sample, typically defined as the sample that received at least one dose of the allocated study drug, and not the sample randomised. Especially worrisome were the Descartes, Equator, ODYSSEY ALTERNATIVE, ODYSSEY COMBO I, ODYSSEY Long Term, and OSLER‐1 trials, which, as described, had considerable attrition. Positive exceptions were the SPIRE‐1, SPIRE‐2, and FOURIER trials, which were specifically designed to explore clinical endpoints, used the ITT sample, and report small numbers of participants lost to follow‐up. Although potential lack of blinding seems unlikely to bias biomarker measurements, it may pose a considerable source of bias for detection of clinical endpoints. Of particular concern are the OSLER‐1 and OSLER‐2 studies, which were open‐label trials (high risk of bias); however, other studies did not always adequately explain how clinical endpoints were detected and how detection bias was prevented (unclear risk of bias; see Characteristics of included studies).
Effects of interventions
See: Table 1; Table 2; Table 3
See 'Summary of findings' tables for the following.
PCSK9 mAb against placebo (Table 1).
PCSK9 mAb against ezetimibe and statins (Table 2).
PCSK9 mAb against ezetimibe (Table 3).
Biomarker effects in comparison of PCSK9 mAb against placebo at six months
At six months follow‐up, the effect of PCSK9 inhibitors on LDL‐C compared with placebo was noted as ‐53.86% (95% CI ‐58.64 to ‐49.08; eight studies; 4782 participants; GRADE: moderate) reduction from baseline (Figure 4) (see Table 6 and Appendix 2 for remaining forest plots). Review authors observed similar reductions for triglycerides (‐11.39%, 95% CI ‐17.04 to ‐5.74); total cholesterol (‐31.41%, 95% CI ‐43.65 to ‐19.16); apolipoprotein B (‐41.93%, 95% CI ‐49.76 to ‐34.10); lipoprotein(a) (‐19.80%, 95% CI ‐25.43 to ‐14.17); and non‐HDL‐C (‐47.17%, 95% CI ‐53.92 to ‐40.42) (see Table 6). Treatment effect estimates on HDL‐C and apolipoprotein A1 at six months were as follows: 6.00, 95% CI 4.31 to 7.69; and 3.50%, 95% CI 2.37 to 4.64, respectively. Findings of two studies reveal that the association with HbA1c, as absolute change from baseline, was 0.01% (95% CI ‐0.06 to 0.08).
Table 3.
Summary results ‐ biomarker analyses at 6 months
|
Number of studies |
Number of PCSK9 participants |
Number of comparator arm participants |
Fixed‐effect (95% CI) |
Random‐effects (95% CI) |
Between‐study heterogeneity P value |
|
| Placebo comparison | ||||||
| LDL‐C % change | 8 | 3255 | 1527 | ‐57.62 (‐59.37 to ‐55.87) | ‐53.86 (‐58.64 to ‐49.08) | < 0.001 |
| HDL‐C % change | 5 | 2324 | 1175 | 5.48 (4.37 to 6.59) | 6.00 (4.31 to 7.69) | 0.19 |
| Triglycerides % change | 5 | 2324 | 1175 | ‐14.62 (‐16.74 to ‐12.50) | ‐11.39 (‐17.04 to ‐5.74) | < 0.001 |
| Total cholesterol % change | 2 | 1762 | 895 | ‐35.79 (‐37.36 to ‐34.23) | ‐31.41 (‐43.65 to ‐19.16) | < 0.001 |
| Apolipoprotein A1 % change | 3 | 2043 | 1033 | 3.49 (2.38 to 4.60) | 3.50 (2.37 to 4.64) | 0.36 |
| Apolipoprotein B % change | 6 | 2507 | 1239 | ‐47.79 (‐49.51 to ‐46.08) | ‐41.93 (‐49.76 to ‐34.10) | < 0.001 |
| Lipoprotein(a) % change | 4 | 2252 | 1140 | ‐22.43 (‐24.30 to ‐20.56) | ‐19.80 (‐25.43 to ‐14.17) | < 0.001 |
| Non‐HDL‐C % change | 4 | 2252 | 1140 | ‐50.03 (‐51.73 to ‐48.33) | ‐47.17 (‐53.92 to ‐40.42) | < 0.001 |
| HbA1c absolute change | 2 | 490 | 245 | 0.00 (‐0.04 to 0.05) | 0.01 (‐0.06 to 0.08) | 0.151 |
| Ezetimibe and statin comparison | ||||||
| LDL‐C % change | 5 | 3309 | 2067 | ‐52.17 (‐53.91 to ‐50.43) | ‐39.20 (‐56.15 to ‐22.26) | < 0.001 |
| HDL‐C % change | 3 | 333 | 578 | 7.53 (5.54 to 9.51) | 6.42 (1.31 to 11.52) | 0.002 |
| Triglycerides % change | 2 | 229 | 327 | ‐3.47 (‐8.26 to 1.32) | ‐3.47 (‐8.26 to 1.32) | 0.46 |
| Total cholesterol % change | ||||||
| Apolipoprotein A1 % change | ||||||
| Apolipoprotein B % change | 3 | 333 | 578 | ‐26.86 (‐29.50 to ‐24.22) | ‐26.72 (‐30.26 to ‐23.19) | 0.169 |
| Lipoprotein(a) % change | 2 | 207 | 453 | ‐19.51 (‐24.48 to ‐14.53) | ‐19.51 (‐24.48 to ‐14.53) | 0.60 |
| Non‐HDL‐C % change | 2 | 207 | 453 | ‐28.19 (‐32.79 to ‐23.59) | ‐28.19 (‐32.79 to ‐23.59) | 0.65 |
| HbA1c absolute change | ||||||
| Ezetimibe comparison | ||||||
| LDL‐C % change | 2 | 531 | 292 | ‐30.20 (‐34.18 to ‐26.23) | ‐30.20 (‐34.18 to ‐26.23) | 0.71 |
| HDL‐C % change | 2 | 531 | 292 | 7.40 (5.11 to 9.70) | 7.01 (3.70 to 10.32) | 0.22 |
| Triglycerides % change | 2 | 531 | 292 | ‐0.43 (‐4.90 to 4.03) | ‐0.43 (‐4.90 to 4.03) | 0.89 |
| Total cholesterol % change | 2 | 531 | 292 | ‐15.51 (‐18.18 to ‐12.83) | ‐15.84 (‐19.37 to ‐12.30) | 0.24 |
| Apolipoprotein A1 % change | 2 | 531 | 292 | 6.13 (4.34 to 7.91) | 6.13 (4.34 to 7.91) | 0.68 |
| Apolipoprotein B % change | 2 | 531 | 292 | ‐23.18 (‐26.28 to ‐20.08) | ‐23.18 (‐26.28 to ‐20.08) | 0.37 |
| Lipoprotein(a) % change | 2 | 531 | 292 | ‐18.70 (‐23.03 to ‐14.37) | ‐13.69 (‐30.60 to 3.21) | 0.003 |
| Non‐HDL‐C % change | 2 | 531 | 292 | ‐23.45 (‐27.07 to ‐19.83) | ‐23.45 (‐27.07 to ‐19.83) | 0.57 |
| HbA1c absolute change | ||||||
Figure 4.

Association of PCSK9 inhibitors compared with placebo with mean percentage change from baseline in LDL‐C at six months.
Biomarker effects in comparison of PCSK9 mAb against ezetimibe and statins at six months
Compared with those given ezetimibe and statins, participants receiving PCSK9 inhibitors showed a reduction (percentage change from baseline) of ‐39.20% in LDL‐C (95% CI ‐56.15 to ‐22.26; five studies; 5376 participants; GRADE: moderate) (Figure 5); ‐3.47% (95% CI ‐8.26 to 1.32) in triglycerides; ‐26.72% (95% CI ‐30.26 to ‐23.19) in apolipoprotein B; ‐19.51% (95% CI ‐24.48 to ‐14.53) in lipoprotein(a); ‐28.19% (95% CI ‐32.79 to ‐23.59) in non‐HDL‐C, and 6.42% (95% CI 1.31 to 11.52) in HDL‐C (see Table 6 and Appendix 2 for remaining forest plots). No information was available on total cholesterol, apolipoprotein A1, or HbA1c.
Figure 5.

Association of PCSK9 inhibitors compared with ezetimibe and statins with mean percentage change from baseline in LDL‐C at six months.
Biomarker effects in comparison of PCSK9 mAb against ezetimibe at six months
Two trials (ODYSSEY COMBO II; ODYSSEY MONO) evaluated PCSK9 mAb against ezetimibe and reported the following effects (percentage change from baseline) on biomarkers: ‐30.20% (95% CI ‐34.18 to ‐26.23; two studies; 823 participants; GRADE: moderate) for LDL‐C (Figure 6); ‐0.43% (95% CI ‐4.90 to 4.03) for triglycerides; ‐15.84% (95% CI ‐19.37 to ‐12.30) for total cholesterol; ‐13.69% (95% CI ‐30.60 to 3.21) for lipoprotein(a); ‐23.18% (95% CI ‐26.28 to ‐20.08) for apolipoprotein B; ‐23.45% (95% CI ‐27.07 to ‐19.83) for non‐HDL‐C; 7.01% (95% CI 3.70 to 10.32) for HDL‐C; and 6.13% (95% CI 4.34 to 7.91) for apolipoprotein A1. Information on HbA1c was unavailable.
Figure 6.

Association of PCSK9 inhibitors compared with ezetimibe with mean percentage change from baseline in LDL‐C at six months.
Biomarker effects of PCSK9 mAb after one year
At one year, effect estimates of PCSK9 inhibitors versus placebo were available for six trials (Descartes; FOURIER; ODYSSEY COMBO I; ODYSSEY Long Term; SPIRE 1/2, with SPIRE1/2 counted as two studies) and generally showed similar effect estimates as for six months: ‐52.87% (95% CI ‐60.03 to ‐45.72) for LDL‐C; ‐28.47% (95% CI ‐38.85 to ‐18.10) for total cholesterol; ‐12.53% (95% CI ‐15.45 to ‐9.61) for triglycerides; ‐43.51% (95% CI ‐48.88 to ‐38.13) for apolipoprotein B; 3.00% (95% CI 1.31 to 4.69) for apolipoprotein A1; ‐43.46% (95% CI ‐57.45 to ‐29.47) for non‐HDL‐C; and 6.06% (95% CI 4.30 to 7.82) for HDL‐C. Associations with glucose and HbA1c were 1.80 mg/dL (95% CI 0.61 to 2.99) and 0.02% (95% CI ‐0.01 to 0.05). Results for other biomarkers were unavailable.
The meta‐analysis (OSLER 1/2) provided estimates at one year for PCSK9 mAbs compared with ezetimibe and statins, again reporting similar effect estimates as before (see Table 7 and Appendix 2 for remaining forest plots). Studies comparing PCSK9 inhibitors against ezetimibe did not follow participants up to one year.
Table 4.
Summary results ‐ biomarker analyses at 1 year
|
Number of studies |
Number of PCSK9 participants |
Number of comparator arm participants |
Fixed‐effect (95% CI) |
Random‐effects (95% CI) |
Between‐study heterogeneity P value |
|
| Placebo comparison | ||||||
| LDL‐C % change | 6 | 29865 | 28694 | ‐52.80 (‐53.46 to 52.14) | ‐52.87 (‐60.03 to ‐45.72) | < 0.001 |
| HDL‐C % change | 4 | 14528 | 14127 | 5.55 (5.07 to 6.03) | 6.06 (4.30 to 7.82) | 0.102 |
| Triglycerides % change | 3 | 14319 | 14020 | ‐12.53 (‐15.45 to ‐9.61) | ‐12.53 (‐15.45 to ‐9.61) | 0.679 |
| Total cholesterol % change | 2 | 808 | 409 | ‐31.33 (‐33.80 to ‐28.86) | ‐28.47 (‐38.85 to ‐18.10) | < 0.001 |
| Apolipoprotein A1 % change | 1 | 599 | 302 | 3.00 (1.31 to 4.69) | ||
| Apolipoprotein B % change | 4 | 14528 | 14127 | ‐47.18 (‐48.29 to ‐48.29) | ‐43.51 (‐48.88 to ‐38.13) | < 0.001 |
| Lipoprotein(a) % change | ||||||
| Non‐HDL‐C % change | 2 | 808 | 409 | ‐47.16 (‐50.77 to ‐43.55) | ‐43.46 (‐57.45 to ‐29.47) | 0.001 |
| Glucose (mg/dL) absolute change* | 2 | 13720 | 13718 | 1.80 (0.61 to 2.99) | ||
| HbA1c absolute change* | 2 | 13720 | 13718 | 0.02 (‐0.01 to 0.05) | ||
| Ezetimibe and statin comparison | ||||||
| LDL‐C % change | 1 | 2976 | 1489 | ‐58.40 (‐60.40 to ‐56.40) | ||
| HDL‐C % change | 1 | 736 | 368 | 5.40 (3.09 to 7.71) | ||
| Triglycerides % change | 1 | 736 | 368 | ‐10.00 (‐13.59 to ‐6.41) | ||
| Total cholesterol % change | ||||||
| Apolipoprotein A1 % change | 1 | 736 | 368 | 4.30 (2.61 to 5.99) | ||
| Apolipoprotein B % change | 1 | 736 | 368 | ‐38.80 (‐41.18 to ‐36.42) | ||
| Lipoprotein(a) % change | 1 | 736 | 368 | ‐20.80 (‐23.95 to ‐17.65) | ||
| Non‐HDL‐C % change | 1 | 736 | 368 | ‐44.00 (‐46.77 to ‐41.23) | ||
| Glucose (mg/dL) absolute change* | ||||||
| HbA1c absolute change | ||||||
*On the basis of the combined analysis of SPIRE‐1 and SPIRE‐2, study‐specific estimates were unavailable, hence no random‐effects or between‐study heterogeneity estimates could be calculated
Exploration of between‐study heterogeneity
Generally, between‐study heterogeneity (measured as I²) in treatment response was high. To explore this, we performed the following subgroup analyses on LDL‐C and apolipoprotein B.
Grouping studies with similar PCSK9 dosages (Included studies) compared with placebo at six months follow‐up resulted in mean percentage changes in LDL‐C of ‐54.37% (95% CI ‐59.14 to ‐49.60) for bi‐weekly 75 to 150 mg mAbs; ‐51.95% (95% CI ‐63.73 to ‐40.17) for bi‐weekly 150 mg mAbs; and ‐54.00% (95% CI ‐77.46 to ‐30.54) for bi‐weekly 50 to 200 mg mAbs compared with the overall effect in all RCTs combined of ‐53.86% (95% CI ‐58.64 to ‐49.08) (see Figure 7). Mean percentage changes in apolipoprotein B were ‐40.99% (95% CI ‐50.78 to ‐31.20) for biweekly 75 to 150 mg mAbs; ‐41.74% (95% CI ‐55.22 to ‐28.26) for biweekly 150 mg mAbs; and ‐45.50% (95% CI ‐65.27 to ‐25.73) for biweekly 50 to 200 mg mAbs compared with the overall effect in all RCTs combined of ‐41.93% (95% CI ‐49.76 to ‐34.10) (see Figure 8). Between‐study heterogeneity persisted despite grouping of RCTs administering similar dosages and reporting no clear dose‐response effect (increasing effectiveness).
Figure 7.

Sensitivity analyses grouping RCTs by PCSK9 dose compared with placebo on 6 months LDL‐C mean percentage change from baseline.
Figure 8.

Sensitivity analyses grouping RCTs by PCSK9 dose compared with placebo on 6 months apolipoprotein B mean percentage change from baseline.
To further explore sources of between‐study heterogeneity, we meta‐analysed reported subgroup effect estimates on PCSK9 mAbs compared with placebo on six months mean percentage change in LDL‐C (Figure 9). These analyses suggested that some between‐study heterogeneity may be explained by more pronounced effects in participants who were 65 years of age or younger, had a body mass index (BMI) of 30 or greater, or had a history of T2DM. High baseline levels of LDL‐C and total PCSK9 seemed to be related to treatment response but were available for only a single trial (ODYSSEY Long Term). We performed similar analyses for trials comparing PCSK9 inhibitors versus ezetimibe, but with a maximum sample size of two RCTs, results were imprecise (Figure 10). Finally, using meta‐regression (Figure 11), we found that the proportion of Caucasians and the proportion of participants for whom follow‐up LDL‐C measurements were missing were related, and effects of PCSK9 inhibitors on mean percentage change in LDL‐C were increased. However, these estimates became non‐significant after correction for unexplained between‐study heterogeneity based on a random‐effects model.
Figure 9.

Subgroup and interaction effects of six months mean percentage change in LDL‐C for PCSK9 trials using a placebo comparison arm.
Figure 10.

Subgroup and interaction effects of six months mean percentage change in LDL‐C for PCSK9 trials using a ezetimibe comparison arm.
Figure 11.

Meta‐regression of PCSK9 mAbs compared with placebo at six months mean percentage change in LDL‐C. The long dashed line represents the fixed effect, the long‐short dashed line random effects, circle diameter is proportionate to the inverse of the variance (i.e. equal to study precision).
PCSK9 effects on clinical endpoints in comparison with placebo
Owing to the fact that original publications did not report treatment effect estimates with clinical endpoints over time, results on clinical endpoints (summarised in Table 4 and Table 5) are based on the maximum follow‐up available.
Odds ratio estimates of PCSK9 inhibitors compared with placebo with intended effects were as follows: OR 1.02 (95% CI 0.91 to 1.14; 12 studies; 60,684 participants; GRADE: moderate) for all‐cause mortality; OR 0.86 (95% CI 0.80 to 0.92; eight studies; 59,294 participants; GRADE: moderate) for any CVD event (Figure 12); OR 0.77 (95% CI 0.69 to 0.85) for any myocardial infarction (MI); and OR 0.76 (95% CI 0.65 to 0.89) for any stroke. Treatment effect estimates of unintended effects were as follows: OR 1.08 (95% CI 1.04 to 1.12; 13 studies; 61,038 participants; GRADE: low) for any adverse events (Figure 13); OR 1.07 (95% CI 0.99 to 1.16) for myalgia; OR 1.19 (95% CI 0.91 to 1.55) for influenza; OR 0.86 (95% CI 0.62 to 1.18) for hypertension; OR 0.91 (95% CI 0.63 to 1.31) for any cancer diagnosis; OR 1.04 (95% CI 0.95 to 1.14) for T2DM; OR 0.85 (95% CI 0.73 to 0.99) for elevated creatinine; and OR 1.04 (95% CI 0.88 to 1.24) for neurological events. Exclusion of terminated SPIRE‐1/2 ‐ bococizumab ‐ trials from any adverse events and myalgia meta‐analyses resulted in attenuated effect estimates of OR 1.01 (95% CI 0.96 to 1.06) and OR 1.17 (95% CI 0.87 to 1.56). Evaluation of these treatment effect estimates on the RD scale revealed that the effect of PCSK9 inhibitors on the risk of an event was typically modest, with changes in risk often less than 1% (see Table 5).
Figure 12.

Association of PCSK9 inhibitors compared with placebo with the incidence of any CVD.
Figure 13.

Association of PCSK9 inhibitors compared with placebo with the incidence of any adverse events.
PCSK9 effects on clinical endpoints in comparison with ezetimibe and statins
Odds ratio estimates of PCSK9 inhibitors compared with ezetimibe and statins with intended effects were as follows: OR 0.45 (95% CI 0.27 to 0.75; three studies; 4770 participants; GRADE: very low) for any CVD event (Figure 14 data on all‐cause mortality and any MI were unavailable. Treatment effect estimates with unintended effects were as follows: OR 1.18 (95% CI 1.05 to 1.34; five studies; 5376 participants; GRADE: low) for any adverse events (Figure 15); OR 1.09 (95% CI 0.81 to 1.48) for myalgia; OR 1.28 (95% CI 0.91 to 1.80) for influenza; OR 1.10 (95% CI 0.41 to 2.96) for hypertension; OR 1.10 (95% CI 0.63 to 1.93) for T2DM, OR 0.51 (95% CI 0.28 to 0.92) for elevated creatinine; and OR 1.22 (95% CI 0.40 to 3.69) for neurological events. Data for any stroke and for cancer were unavailable.
Figure 14.

Association of PCSK9 inhibitors compared with ezetimibe and statins with the incidence of any CVD.
Figure 15.

Association of PCSK9 inhibitors compared with ezetimibe and statins with the incidence of any adverse events.
Evaluation of these estimates on the RD scale revealed that effects of PCSK9 inhibitors on risks of an event were typically modest; changes in risk often were less than 1% (see Table 5).
Outcomes and comparisons without data
See respective sections for details on missing outcome data that were unavailable for some comparisons. Clinical outcome data were insufficiently available to perform a meta‐analysis for comparison with ezetimibe. Data on quality of life were unavailable for all studies. Although the substudy of the FOURIER ‐ EBBINGHAUB ‐ presented little or no effect on cognitive function, these data had not been published in sufficient detail to be included here. Regardless of the publication status of the EBBINGHAUB trial, data on cognitive function were not published by other trials, hence we decided (post hoc) to extract data on neurological events.
Discussion
Summary of main results
In this systematic review and meta‐analysis, we showed that randomised trials evaluating PCSK9 inhibitors (primarily against placebo) had a beneficial profile in terms of cardiovascular risk factors that most likely explain their protective effects on cardiovascular events.
In terms of cardiovascular biomarkers, treatment with PCSK9 inhibitors was characterised by a decrease in low‐density lipoprotein cholesterol (LDL‐C), apolipoprotein B, non‐high‐density lipoprotein cholesterol (HDL‐C), triglycerides, and lipoprotein(a), and a modest increase in HDL‐C and apolipoprotein A1. Investigators reported some differences in biomarker response depending on the use of placebo or active comparisons.
Although we observed high between‐study heterogeneity for biomarker outcomes, most study authors agreed on direction of effect and deemed that differences in magnitude were similar enough to provide clinically relevant treatment effect estimates. We did not observe a dose‐response effect of PCSK9 inhibitors on LDL‐C or apolipoprotein B when comparing trials with similar PCSK9 monoclonal antibody (mAb) dosages. A dose‐response effect may have been due to the crude categorisation used by review authors and/or to grouping of studies by different comparator drugs or by other differences in study‐specific factors.
Trials published to date comparing PCSK9 inhibitors against placebo showed potentially little to no effect on all‐cause mortality; nevertheless, PCSK9 inhibitors showed protective effects on cardiovascular disease (CVD) events, myocardial infarction (MI), and any stroke. Treatment with PCSK9 inhibitors was associated with a modest increase in the risk of any adverse events (odds ratio (OR) 1.08, 95% confidence interval (CI) 1.04 to 1.12), largely driven by the SPIRE‐1 and SPIRE‐2 trials, which used an agent that was discontinued owing to immunogenicity. When looking at specific adverse events (extracted in this systematic review), we found that compared with placebo, PCSK9 inhibitors did not show significant association with type 2 diabetes (T2DM), cancers, or neurocognitive events, possibly as the result of limited follow‐up duration. It is important to note that recent phase 3 trials (FOURIER, SPIRE‐1, and SPIRE‐2) did not report on cancer. Regarding minor adverse events, PCSK9 inhibitors showed potentially increased risk of myalgia and influenza, with the former becoming non‐significant after the SPIRE‐1/2 trials were excluded. Study authors reported that they observed a protective effect with PCSK9 inhibitors, which decreased the risk of elevated creatinine (compared with placebo and active treatments). Trials comparing PCSK9 inhibitors against ezetimibe and statins described a more pronounced protective effect on CVD risk when compared with placebo; this discrepancy is likely related to the lower quality of evidence. Researchers provided no data on all‐cause mortality, stroke, or MI. Information on clinical endpoints was unavailable for the ezetimibe only comparison.
Estimation of the same associations on a risk difference scale (Table 7) revealed that PCSK9 inhibitors only modestly changed the outcome risk, often with less than 1% change in risk.
Overall completeness and applicability of evidence
Given selection criteria and study designs reported by published trials evaluating PCSK9 inhibitors, we consider it important to highlight situations that may limit the applicability of existing evidence.
First, most of the evidence was obtained from people with established atherosclerotic CVD or at high risk of cardiovascular events; therefore evidence regarding the use of PCSK9 inhibitors for primary prevention remains controversial. Second, information on clinical endpoints for the placebo comparison was based on the large sample size in the FOURIER and SPIRE‐1 and SPIRE‐2 trials. Although these trials were large, median follow‐up was less than three years, hence information on long‐term efficacy and safety is absent. For the other comparisons, follow‐up was shorter and events were fewer, prohibiting any strong recommendations at this time. Third, information on the safety of PCSK9 inhibitors did not reveal an increase in risk of cancer or T2DM. However, the largest trials to date (FOURIER and SPIRE‐1 and SPIRE‐2) did not provide cancer data, and again, follow‐up time was very modest, leaving questions on long‐term effectiveness and risk unanswered. Three recent genetic studies with large sample size and long‐term follow‐up showed that variation in the PCSK9 locus was associated with increased glucose and T2DM (Ference 2016; Lotta 2016; Schmidt 2017). Lack of significant association with T2DM may be due to the relatively small number of T2DM events collected to date (< 2000) as a comparison; the association of statins with T2DM was discovered only after more than 4000 events were reported (Swerdlow 2014).
Quality of the evidence
Although all available data were derived from industry‐sponsored randomised controlled trials (RCTs), most trials seemed to have low risk of bias. Exceptions are the open‐label OSLER trials, which were at high risk of performance bias. Another important potential source of bias was attrition bias, whereby some RCTs included missing observations for more than 5% of enrolled participants. Most trials tried to minimise this bias by using advanced analytics that explicitly (multiple imputation) or implicitly (mixed‐effects models) imputed these missing observations, thus ensuring that all comparison were made on an intention‐to‐treat (ITT) basis. The appropriateness of these models (and their underlying assumptions) was not reported, hence these imputation algorithms may have failed to correct for potential attribution bias. For the placebo comparison, however, the large number of participants in the FOURIER and SPIRE‐1/2 trials had very low attrition rates and generally were perceived to have low risk of bias.
The quality of evidence was high for the biomarker endpoints in comparison with placebo or ezetimibe. For the comparison of PCSK9 mAbs against ezetimibe and statins, we graded quality as moderate owing to inclusion of the open‐label OSLER trials. Despite the GRADE (Grade Working Group 2004) recommendation to downgrade evidence associated with high between‐study heterogeneity, we decided against this approach because most studies (i.e. LDL‐C outcomes) reported the same direction of effects. Heterogeneity reflected a difference in magnitude ‐ not in direction of effect (confirmed by clinical experts JPC and ADH). Furthermore, use of random‐effects models resulted in point estimates and confidence intervals that are free of bias (Thompson 1999), even in the presence of heterogeneity. Although we believe that this between‐study heterogeneity should not be reflected in our GRADE score, it does reflect a potential need for personalised medicine (Schmidt 2016).
For intended effect and clinical outcomes (i.e. CVD, all‐cause mortality, and MI) with PCSK9 inhibitors compared with placebo, we graded the quality of the evidence as moderate. Results were derived from three trials with large sample sizes (FOURIER, SPIRE‐1 and ‐2), two of which used the terminated bococizumab drug. Furthermore, median follow‐up was less than three years, hence long‐term effectiveness and safety remain uncertain, potentially influencing the absence of an effect on all‐cause mortality or other outcomes with longer lag time. We graded the quality of the evidence as very low in the PCSK9 mAb‐to‐ezetimibe and statin comparison, again owing to inclusion of the open‐label OSLER trials. Bias due to unblinded allocation may explain the likely overly large effect of PCSK9 inhibitors against ezetimibe and statins on CVD events (OR 0.45, 95 CI% 0.27 to 0.75) versus PCSK9 mAb against placebo (OR 0.86, 95% CI 0.0.80 to 0.92).
Given the reported antibody drug response, inclusion of the discontinued bococizumab trials may seem controversial. However, owing to the limited large sample size of trials with modestly long follow‐up, we decided to include these data. Side effects may differ between PCSK9 inhibitors, for example, the potential myalgia effect in the placebo comparison seemed more pronounced in the SPIRE‐1 and SPIRE‐2 trials than in the FOURIER trials (evolocumab). Owing to the limited number of adequately sampled trials, we could not perform formal analyses.
Potential biases in the review process
The meta‐analysis presented may show some weaknesses. First, meta‐analyses explored a large number of endpoints, increasing the probability of a false positive finding. We did not correct for multiple testing because we sought to inform ongoing trials, which can act as an independent and final arbiter. Second, despite our best efforts, we may have failed to identify certain PCSK9 inhibitor trials. Given that we are unaware of the results of any unidentified RCTs, this seems unlikely to bias our results but will obviously reduce sample size. Third, although we set out to report effect estimates with clinical endpoints, similar to biomarker endpoints, at six months, one year, and five years of follow‐up, we found that this was impossible owing to the limited sample size and the fact that the original RCT did not present data in sufficient detail. Fourth, we did not present data by type of PCSK9 inhibitor because of the limited sample size and the focus of trials on alirocumab, making such an analysis uninformative at the moment. Fifth, effect estimates of PCSK9 compared with ezetimibe and statin may be further biased by the limited number of events influencing both point estimates and confidence intervals (Bradburn 2007; Sweeting 2004). We tried to deal with this potential source of bias by re‐creating individual participant data (based on reported cell frequencies) and by estimating a combined effect by using a mixed‐effect model with random intercept (and slope) for study indicator. Nevertheless, we found that random‐effects models (mixed‐effect model with random intercept and slope) often did not converge, hence we did not report these estimates. Given the large sample size included in FOURIER and SPIRE‐1 and SPIRE‐2 trials for the placebo comparison, sparse data are less of an issue for effect estimates on major CVD events but remain inconclusive for rarer CVD and non‐CVD events such as haemorrhagic stroke, cancers, and T2DM. Furthermore, although the FOURIER and SPIRE trials collected data on a large number of participants, investigators provided relatively short follow‐up times, leaving open the question of long‐term efficacy and safety.
Agreements and disagreements with other studies or reviews
We are aware of two previous systematic reviews and meta‐analyses on PCSK9 inhibitors (Navarese 2015; Zhang 2015); both included a large number of RCTs with short follow‐up of 12 weeks, which we excluded here, as well as several longer‐term follow‐up studies that we included.
The meta‐analysis of Zhang 2015 revealed a protective effect on mortality of alirocumab versus placebo (OR 0.43, 95% CI 0.19 to 0.96) and of alirocumab versus ezetimibe (OR 0.48, 95% CI 0.16 to 1.45); these effects are different from the more neutral effect that we observed (OR 1.02, 95% CI 0.91 to 1.14). This difference may have occurred because Zhang 2015 relied predominantly on short‐term follow‐up studies, limiting the number of events per study, and this likely biased effect estimates.
Similar to this review, Zhang found a protective effect of alirocumab on elevated creatine kinase versus placebo (OR 0.72, 95% CI 0.52 to 1.01) and versus ezetimibe (OR 0.75, 95% CI 0.46 to 1.24). Review authors found a similar protective effect of elevated creatine kinase for evolucumab (vs placebo or ezetimibe), as well as protective effects of elevated alanine aminotransferase and aspartate aminotransferase (no information was reported on mortality for evolucumab). Contrary to our meta‐analyses, Zhang 2015 found a non‐significant decrease in influenza for both alirocumab and evolucumab. Results may be concordant with a null effect, as in both Zhang's review and ours, these associations did not reach significance at an alpha of 0.05. Alternatively, Zhang included trials with only a few weeks of follow‐up, potentially excluding the annual influenza season, and the shorter duration of exposure conveys less risk.
Navarese 2015 reported a protective effect of PCSK9 inhibitors (vs all types of comparators) for all‐cause mortality (OR 0.45, 95% CI 0.23 to 0.86) and a decreased incidence of increased serum creatine kinase levels (OR 0.72, 95% CI 0.54 to 0.96), as well as protective effects for cardiovascular mortality and MI (OR 0.50, 95% CI 0.23 to 1.10; OR 0.49, 95% CI 0.26 to 0.93, respectively). As with the Zhang 2015 study, results were based on many short‐term trials with very few events per study, hence the caveats described before continue to hold.
Finally, we are aware that a recent network meta‐analysis (Lipinski 2016) indirectly comparing mAb versus placebo showed an OR for neurocognitive adverse events of 2.34 (95% CI 1.11 to 4.93). This association was not observed in the current meta‐analyses, which directly compared PCSK9 inhibitors versus placebo and therefore were less susceptible to bias.
Authors' conclusions
Moderate‐quality evidence shows that PCSK9 inhibitors decrease LDL‐C and related lipid biomarkers on a short‐term (24 weeks) and medium‐term (one year) basis (GRADE: moderate). When compared against placebo, PCSK9 inhibitors reduce risks of CVD, MI, and any stroke (GRADE: moderate); however, owing to limited follow‐up (< 3 years) and few adequately sampled trials (three with large samples), information on long‐term safety and efficacy is lacking.
Effects of PCSK9 inhibitors compared with statin and ezetimibe were of lower quality (GRADE: low to very low), mainly because the number of events per RCT was limited. Additionally, some trials had perceived high risk of bias as the result of incomplete follow‐up, and others were not adequately blinded (OSLER studies). Both comparisons revealed an increase in any adverse events (GRADE: low), which, in the placebo compassion, was driven by SPIRE‐1 and SPIRE‐2 results. Evidence found to date shows no effect on type 2 diabetes and cancers, but the SPIRE‐1 and SPIRE‐2 trials reported an increase in glucose. Additionaly, we observed an unexpected decrease in the incidence of elevated creatine in the PCSK9 inhibitor arm (placebo and statins and ezetimibe groups). PCSK9 inhibitor effects on mortality were not recorded for the ezetimibe and statin comparison, and were potentially neutral for the placebo comparison; the latter may be related to the modest follow‐up time mentioned. Observed high heterogeneity in biomarker response suggests that personalised PCSK9 treatment regimens may be needed to optimise patient benefit.
Besides exploring the long‐term effects of PCSK9 inhibition on CVD‐related endpoints, especially when compared against active comparisons such as ezetimibe and statins, ongoing research should explore potential safety issues. Given the magnitude of the between‐study heterogeneity discussed, future studies should explore (the need for) personalised medicine algorithms to ensure that patients benefit optimally. Currenlty, no data have been obtained on the comparison of PCSK9 inhibitors themselves; ideally, these should be explored by a factorial RCT (instead of between RCTs on the basis of network meta‐analysis).
Acknowledgements
None provided.
Appendices
Appendix 1. Search strategies
MEDLINE search strategy
1. exp antibodies, monoclonal/ 2. monoclonal antibod*.tw. 3. MAB*.tw. 4. evolocumab.tw. 5. amg 145.tw. 6. amg145.tw. 7. alirocumab.tw. 8. regn 727.tw. 9. regn727.tw. 10. sar 236553.tw. 11. sar236553.tw. 12. 1D05‐IgG2.tw. 13. LGT209.tw. 14. RG7652.tw. 15. Bococizumab.tw. 16. "pf 04950615".tw. 17. pf04950615.tw. 18. rn 316.tw. 19. rn316.tw. 20. or/1‐19 21. exp Proprotein Convertases/ 22. proprotein convertase*.tw. 23. pro‐protein convertase*.tw. 24. pcsk9.tw. 25. serine proteinase*.tw. 26. or/21‐25 27. exp Cardiovascular Diseases/ 28. cardio*.tw. 29. cardia*.tw. 30. heart*.tw. 31. coronary*.tw. 32. angina*.tw. 33. ventric*.tw. 34. myocard*.tw. 35. pericard*.tw. 36. isch?em*.tw. 37. emboli*.tw. 38. arrhythmi*.tw. 39. thrombo*.tw. 40. atrial fibrillat*.tw. 41. tachycardi*.tw. 42. endocardi*.tw. 43. (sick adj sinus).tw. 44. exp Stroke/ 45. (stroke or stokes).tw. 46. cerebrovasc*.tw. 47. cerebral vascular.tw. 48. apoplexy.tw. 49. (brain adj2 accident*).tw. 50. ((brain* or cerebral or lacunar) adj2 infarct*).tw. 51. exp Hyperlipidemias/ 52. hyperlipid*.tw. 53. hyperlip?emia*.tw. 54. hypercholesterol*.tw. 55. hypercholester?emia*.tw. 56. hyperlipoprotein?emia*.tw. 57. hypertriglycerid?emia*.tw. 58. exp Arteriosclerosis/ 59. exp Cholesterol/ 60. cholesterol.tw. 61. "coronary risk factor* ".tw. 62. exp Cognition/ 63. exp dementia/ 64. cognitive function*.tw. 65. dementia.tw. 66. alzheimer*.tw. 67. or/27‐66 68. 20 and 26 and 67 69. randomized controlled trial.pt. 70. controlled clinical trial.pt. 71. randomized.ab. 72. placebo.ab. 73. drug therapy.fs. 74. randomly.ab. 75. trial.ab. 76. groups.ab. 77. 69 or 70 or 71 or 72 or 73 or 74 or 75 or 76 78. exp animals/ not humans.sh. 79. 77 not 78 80. 68 and 79 81. limit 80 to yr="2005 ‐Current"
CENTRAL search strategy
#1 MeSH descriptor: [Antibodies, Monoclonal] explode all trees
#2 monoclonal next antibod*
#3 MAB*
#4 evolocumab
#5 "amg 145" or amg145
#6 alirocumab
#7 "regn 727" or regn727 or "sar 236553" or sar236553 or 1D05‐IgG2 or LGT209 or RG7652
#8 Bococizumab
#9 "pf 04950615" or pf04950615 or "rn 316" or rn316
#10 #1 or #2 or #3 or #4 or #5 or #6 or #7 or #8 or #9
#11 MeSH descriptor: [Proprotein Convertases] explode all trees
#12 proprotein next convertase*
#13 pro‐protein next convertase*
#14 pcsk9
#15 serine next proteinase*
#16 #11 or #12 or #13 or #14 or #15
#17 MeSH descriptor: [Cardiovascular Diseases] explode all trees
#18 cardio*
#19 cardia*
#20 heart*
#21 coronary*
#22 angina*
#23 ventric*
#24 myocard*
#25 pericard*
#26 isch?em*
#27 emboli*
#28 arrhythmi*
#29 thrombo*
#30 atrial next fibrillat*
#31 tachycardi*
#32 endocardi*
#33 (sick next sinus)
#34 MeSH descriptor: [Stroke] explode all trees
#35 (stroke or stokes)
#36 cerebrovasc*
#37 cerebral next vascular
#38 apoplexy
#39 (brain near/2 accident*)
#40 ((brain* or cerebral or lacunar) near/2 infarct*)
#41 MeSH descriptor: [Hyperlipidemias] explode all trees
#42 hyperlipid*
#43 hyperlip?emia*
#44 hypercholesterol*
#45 hypercholester?emia*
#46 hyperlipoprotein?emia*
#47 hypertriglycerid?emia*
#48 MeSH descriptor: [Arteriosclerosis] explode all trees
#49 MeSH descriptor: [Cholesterol] explode all trees
#50 cholesterol
#51 "coronary risk factor*"
#52 MeSH descriptor: [Cognition] explode all trees
#53 MeSH descriptor: [Dementia] explode all trees
#54 cognitive next function*
#55 dementia
#56 alzheimer*
#57 #17 or #18 or #19 or #20 or #21 or #22 or #23 or #24 or #25 or #26 or #27 or #28 or #29 or #30 or #31 or #32 or #33 or #34 or #35 or #36 or #37 or #38 or #39 or #40 or #41 or #42 or #43 or #44 or #45 or #46 or #47 or #48 or #49 or #50 or #51 or #52 or #53 or #54 or #55 or #56
#58 #10 and #16 and #57 Publication Year from 2005 to 2014
Embase search strategy
1. exp monoclonal antibody/
2. monoclonal antibod*.tw.
3. MAB*.tw.
4. evolocumab.tw.
5. amg 145.tw.
6. amg145.tw.
7. alirocumab.tw.
8. regn 727.tw.
9. regn727.tw.
10. sar 236553.tw.
11. sar236553.tw.
12. 1D05‐IgG2.tw.
13. LGT209.tw.
14. RG7652.tw.
15. Bococizumab.tw.
16. "pf 04950615".tw.
17. pf04950615.tw.
18. rn 316.tw.
19. rn316.tw.
20. or/1‐19
21. exp serine proteinase/
22. proprotein convertase*.tw.
23. pro‐protein convertase*.tw.
24. serine proteinase*.tw.
25. pcsk9.tw.
26. or/21‐25
27. exp cardiovascular disease/
28. cardio*.tw.
29. cardia*.tw.
30. heart*.tw.
31. coronary*.tw.
32. angina*.tw.
33. ventric*.tw.
34. myocard*.tw.
35. pericard*.tw.
36. isch?em*.tw.
37. emboli*.tw.
38. arrhythmi*.tw.
39. thrombo*.tw.
40. atrial fibrillat*.tw.
41. tachycardi*.tw.
42. endocardi*.tw.
43. (sick adj sinus).tw.
44. exp cerebrovascular disease/
45. (stroke or stokes).tw.
46. cerebrovasc*.tw.
47. cerebral vascular.tw.
48. apoplexy.tw.
49. (brain adj2 accident*).tw.
50. ((brain* or cerebral or lacunar) adj2 infarct*).tw.
51. exp hyperlipidemia/
52. hyperlipid*.tw.
53. hyperlip?emia*.tw.
54. hypercholesterol*.tw.
55. hypercholester?emia*.tw.
56. hyperlipoprotein?emia*.tw.
57. hypertriglycerid?emia*.tw.
58. exp Arteriosclerosis/
59. exp Cholesterol/
60. cholesterol.tw.
61. "coronary risk factor*".tw.
62. exp cognition/
63. exp dementia/
64. cognitive function*.tw.
65. dementia.tw.
66. alzheimer*.tw.
67. or/27‐66
68. 20 and 26 and 67
69. random$.tw.
70. factorial$.tw.
71. crossover$.tw.
72. cross over$.tw.
73. cross‐over$.tw.
74. placebo$.tw.
75. (doubl$ adj blind$).tw.
76. (singl$ adj blind$).tw.
77. assign$.tw.
78. allocat$.tw.
79. volunteer$.tw.
80. crossover procedure/
81. double blind procedure/
82. randomized controlled trial/
83. single blind procedure/
84. 69 or 70 or 71 or 72 or 73 or 74 or 75 or 76 or 77 or 78 or 79 or 80 or 81 or 82 or 83
85. (animal/ or nonhuman/) not human/
86. 84 not 85
87. 68 and 86
88. limit 87 to embase
89. limit 88 to yr="2005 ‐Current"
Web of Science search strategy
# 12 #11 AND #10
# 11 TS=((random* or blind* or allocat* or assign* or trial* or placebo* or crossover* or cross‐over*))
# 10 #9 AND #8 AND #7
# 9 TS=("proprotein convertase*" or "pro‐protein convertase*" or pcsk9 or "serine proteinase*")
# 8 TS=("monoclonal antibod*" or MAB* or evolocumab or "amg 145" or amg145 or alirocumab or "regn 727" or regn727 or "sar 236553" or sar236553 or 1D05‐IgG2 or LGT209 or RG7652 or Bococizumab or "pf 04950615" or pf04950615 or "rn 316" or rn316)
# 7 #6 OR #5 OR #4 OR #3 OR #2 OR #1
# 6 TS=("cognitive function*" or dementia or alzheimer*)
# 5 TS=(cardio* OR cardia* OR heart* OR coronary* OR angina* OR ventric* OR myocard*)
# 4 TS=(pericard* OR isch?em* OR emboli* OR arrhythmi* OR thrombo*)
# 3 TS=("atrial fibrillat*" OR tachycardi* OR endocardi*)
# 2 TS=(stroke OR stokes OR cerebrovasc* OR cerebral OR apoplexy OR (brain SAME accident*) OR (brain SAME infarct*))
# 1 TS=(hyperlipid* OR hyperlip?emia* OR hypercholesterol* OR hypercholester?emia* OR hyperlipoprotein?emia* OR hypertriglycerid?emia*)
Appendix 2. Biomarker forest plots
Figure 16; Figure 17; Figure 18; Figure 19; Figure 20; Figure 21; Figure 22; Figure 23; Figure 24; Figure 25; Figure 26; Figure 27; Figure 28; Figure 29; Figure 30; Figure 31; Figure 32; Figure 33; Figure 34; Figure 35; Figure 36; Figure 37; Figure 38; Figure 39; Figure 40; Figure 41; Figure 42; Figure 43; Figure 44; Figure 45; Figure 46; Figure 47; Figure 48; Figure 49
Figure 16.

Association of PCSK9 inhibitors compared with placebo with mean percentage change from baseline in HDL‐C at six months.
Figure 17.

Association of PCSK9 inhibitors compared with placebo with mean percentage change from baseline in triglycerides at six months.
Figure 18.

Association of PCSK9 inhibitors compared with placebo with mean percentage change from baseline in total cholesterol at six months.
Figure 19.

Association of PCSK9 inhibitors compared with placebo with mean percentage change from baseline in apolipoprotein A1 at six months.
Figure 20.

Association of PCSK9 inhibitors compared with placebo with mean percentage change from baseline in apolipoprotein B at six months.
Figure 21.

Association of PCSK9 inhibitors compared with placebo with mean percentage change from baseline in lipoprotein(a) at six months.
Figure 22.

Association of PCSK9 inhibitors compared with placebo with mean percentage change from baseline in non‐HDL‐C at six months.
Figure 23.

Association of PCSK9 inhibitors compared with placebo with mean absolute change from baseline in HbA1c at six months.
Figure 24.

Association of PCSK9 inhibitors compared with ezetimibe and statins with mean percentage change from baseline in HDL‐C at six months.
Figure 25.

Association of PCSK9 inhibitors compared with ezetimibe and statins with mean percentage change from baseline in triglycerides at six months.
Figure 26.

Association of PCSK9 inhibitors compared with ezetimibe and statins with mean percentage change from baseline in apolipoprotein B at six months.
Figure 27.

Association of PCSK9 inhibitors compared with ezetimibe and statins with mean percentage change from baseline in lipoprotein(a) at six months.
Figure 28.

Association of PCSK9 inhibitors compared with ezetimibe and statins with mean percentage change from baseline in non‐HDL‐C at six months.
Figure 29.

Association of PCSK9 inhibitors compared with ezetimibe with mean percentage change from baseline in HDL‐C at six months.
Figure 30.

Association of PCSK9 inhibitors compared with ezetimibe with mean percentage change from baseline in triglycerides at six months.
Figure 31.

Association of PCSK9 inhibitors compared with ezetimibe with mean percentage change from baseline in total cholesterol at six months.
Figure 32.

Association of PCSK9 inhibitors compared with ezetimibe with mean percentage change from baseline in apolipoprotein A1 at six months.
Figure 33.

Association of PCSK9 inhibitors compared with ezetimibe with mean percentage change from baseline in apolipoprotein B at six months.
Figure 34.

Association of PCSK9 inhibitors compared with ezetimibe with mean percentage change from baseline in lipoprotein(a) at six months.
Figure 35.

Association of PCSK9 inhibitors compared with ezetimibe with mean percentage change from baseline in non‐HDL‐C at six months.
Figure 36.

Association of PCSK9 inhibitors compared with placebo with mean percentage change from baseline in LDL‐C at 12 months.
Figure 37.

Association of PCSK9 inhibitors compared with placebo with mean percentage change from baseline in HDL‐C at 12 months.
Figure 38.

Association of PCSK9 inhibitors compared with placebo with mean percentage change from baseline in triglycerides at 12 months.
Figure 39.

Association of PCSK9 inhibitors compared with placebo with mean percentage change from baseline in total cholesterol at 12 months.
Figure 40.

Association of PCSK9 inhibitors compared with placebo with mean percentage change from baseline in apolipoprotein A1 at 12 months.
Figure 41.

Association of PCSK9 inhibitors compared with placebo with mean percentage change from baseline in apolipoprotein B at 12 months.
Figure 42.

Association of PCSK9 inhibitors compared with placebo with mean percentage change from baseline in non‐HDL‐C at 12 months.
Figure 43.

Association of PCSK9 inhibitors compared with ezetimibe and statins with mean percentage change from baseline in LDL‐C at 12 months.
Figure 44.

Association of PCSK9 inhibitors compared with ezetimibe and statins with mean percentage change from baseline in HDL‐C at 12 months.
Figure 45.

Association of PCSK9 inhibitors compared with ezetimibe and statins with mean percentage change from baseline in triglycerides at 12 months.
Figure 46.

Association of PCSK9 inhibitors compared with ezetimibe and statins with mean percentage change from baseline in apolipoprotein A1 at 12 months.
Figure 47.

Association of PCSK9 inhibitors compared with ezetimibe and statins with mean percentage change from baseline in apolipoprotein B at 12 months.
Figure 48.

Association of PCSK9 inhibitors compared with ezetimibe and statins with mean percentage change from baseline in lipoprotein(a) at 12 months.
Figure 49.

Association of PCSK9 inhibitors compared with ezetimibe and statins with mean percentage change from baseline in non‐HDL‐C at 12 months.
Appendix 3. Clinical endpoint forest plots
Figure 50; Figure 51; Figure 52; Figure 53; Figure 54; Figure 55; Figure 56; Figure 57; Figure 58; Figure 59; Figure 60; Figure 61; Figure 62; Figure 63; Figure 64; Figure 65
Figure 50.

Association of PCSK9 inhibitors compared with placebo with the incidence of all‐cause mortality.
Figure 51.

Association of PCSK9 inhibitors compared with placebo with the incidence of any MI.
Figure 52.

Association of PCSK9 inhibitors compared with placebo with the incidence of any stroke.
Figure 53.

Association of PCSK9 inhibitors compared with placebo with the incidence of myalgia.
Figure 54.

Association of PCSK9 inhibitors compared with placebo with the incidence of influenza.
Figure 55.

Association of PCSK9 inhibitors compared with placebo with the incidence of hypertension.
Figure 56.

Association of PCSK9 inhibitors compared with placebo with the incidence of any cancer diagnosis.
Figure 57.

Association of PCSK9 inhibitors compared with placebo with the incidence of type 2 diabetes.
Figure 58.

Association of PCSK9 inhibitors compared with placebo with the incidence of elevated creatine.
Figure 59.

Association of PCSK9 inhibitors compared with placebo with the incidence of neurological events.
Figure 60.

Association of PCSK9 inhibitors compared with ezetimibe and statins with the incidence of myalgia.
Figure 61.

Association of PCSK9 inhibitors compared with ezetimibe and statins with the incidence of influenza.
Figure 62.

Association of PCSK9 inhibitors compared with ezetimibe and statins with the incidence of hypertension.
Figure 63.

Association of PCSK9 inhibitors compared with ezetimibe and statins with the incidence of type 2 diabetes.
Figure 64.

Association of PCSK9 inhibitors compared with ezetimibe and statins with the incidence of elevated creatinine.
Figure 65.

Association of PCSK9 inhibitors compared with ezetimibe and statins with the incidence neurological events
Data and analyses
This review has no analyses.
Differences between protocol and review
We note the following deviations from the protocol.
We intended to present a 'Risk of bias' figure depicting risk of bias per item, weighted for how much an individual RCT contributed to the overall effect estimate of PCSK9 inhibitors on LDL‐C. However, some studies did not report on LDL‐C at all, or did not report it at the same time point, making it impossible to present such a figure.
Owing to the small number of events off all‐cause mortality and the CVD endpoints, we decided against using the usual inverse variance method of pooling, which may result in biased estimates. Instead, we pooled clinical events by reconstructing individual participant data based on cell frequencies, and analysed these data using a mixed‐effect generalised linear regression model (Bradburn 2007; Sweeting 2004) with a random intercept (fixed‐effect).
We meta‐analysed biomarker results despite considerable heterogeneity in continuous endpoints, this contrary to the protocol statement that no meta‐analysis would be performed if heterogeneity would be larger than 50%. We decided to combine results because estimates were universally on one side of the neutral effect.
Owing to the small number of events, we performed all subgroup analyses for LDL‐C instead of CVD. Similarly, subgroups explored were slightly different from those described in the protocol as the result of available data.
We intended to extract data for continuous endpoints as mean percentage change from baseline, or as the difference at the end of follow‐up. However, the latter was unavailable in most studies, and we focused on the former.
Instead of data on cognitive function, we decided (post hoc) to extract data on neurological events.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
| Methods |
Type of RCT: 5:2 parallel‐group, double‐blind dose‐ranging RCTs Settings: outpatient care Duration: 24 weeks Start and stop dates: 07/2012 and 05/2013 |
|
| Participants |
Number of participants: 354 Number lost to follow‐up: NA Women: 182 (51%) Age (SD), years: 59 (11) History of CVD: NA Participants with FH: NA Participants with hypercholesterolaemia on stable statin therapy with fasting LDL‐C of 80 mg/dL or more and triglycerides of 400 mg/dL or less |
|
| Interventions |
Background therapy: statin therapy Randomised therapy: bococizumab (RN316) vs placebo Bococizumab dose: Participants were offered 50 mg, 100 mg, 150 mg once every 2 weeks, or 200 mg, 300 mg every 4 weeks, resulting in a dosage range of 50 mg to 150 mg every 2 weeks Intervention was continued for 24 weeks with dose reduction at day 43 (14‐week regimen) or at day 57 (28‐week regimen) |
|
| Outcomes | Adverse events | |
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation by interactive voice‐response system |
| Allocation concealment (selection bias) | Unclear risk | Unclear how the interactive voice system was implemented |
| Blinding of participants and personnel (performance bias) LDL‐C | Unclear risk | Although paper and appendix describe the study as double‐blind, it is unclear how this was maintained and who was blinded. However, no LDL‐C measurement was available at/near any of the predefined time points, making this less important |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Although paper and appendix describe the study as double‐blind, it is unclear how this was maintained and who was blinded. Any lack of blinding of participants and personnel seems unlikely to bias LDL‐C assessment, which was performed in independent laboratories. On the other hand, outcomes such as adverse events may be biased owing to detection bias |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not reported; mixed‐effects models, including baseline measurement, were used for continuous outcomes |
| Selective reporting (reporting bias) | Unclear risk | Study protocol was unavailable |
| Other bias | High risk | Funded by Pfizer |
| Methods |
Type of RCT: 2:1 parallel‐group, double‐blind RCT with stratified randomisation Settings: outpatient care Duration: 52 weeks Start and stop dates: 01/2012 and 11/2013 |
|
| Participants |
Number of participants: 905 (901 with baseline data) Number lost to follow‐up: 134 Women: 471 (52%) Age (SD), years: 56 (11) History of CVD: 136 (15%) Participants with FH: NA Participants with fasting LDL‐C of 75 mg/dL or more and fasting triglyceride level of 400 mg/dL |
|
| Interventions |
Background therapy: standard of care, which consisted of diet only, daily atorvastatin 10 mg, 80 mg, or 80 mg + 10 mg ezetimibe Randomised therapy: evolocumab every 4 weeks vs placebo Evolocumab dose: 48 weeks of 420 mg each 4 weeks. Two‐week equivalent dose of 210 mg |
|
| Outcomes | CVD, lipids, any adverse events, all‐cause mortality, glucose, HbA1c (change from baseline) | |
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation performed centrally using an interactive voice‐response system |
| Allocation concealment (selection bias) | Low risk | Randomisation performed centrally using an interactive system |
| Blinding of participants and personnel (performance bias) LDL‐C | Unclear risk | Although paper and appendix describe the study as double‐blind, it is unclear how this was maintained and who was blinded. Lack of blinding will likely cause a change in adherence and/or participant choices regarding SOC/lifestyle, which may influences outcomes |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Although paper and appendix describe the study as double‐blind, it is unclear how this was maintained and who was blinded. However, any lack of blinding of participants and personnel seems unlikely to bias LDL‐C assessment, which was performed in independent laboratories. Outcomes such as adverse events may be biased owing to detection bias |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 4 participants were randomised but were not included in the ITT (small number, good). However, at 2 weeks of follow‐up, the number of available patients had decreased by about 15% (number of missing measurements 44 (14.57%) in comparison arm, and 90 (15.03%) in intervention arm). In some of these cases, missing patients are likely due to different enrolment times, limiting follow‐up; however, reported numbers of discontinued participants were similarly high: 73 in the evolocumab arm and 28 in the placebo arm. Missing LDL‐C data were imputed used linear mixed models, including baseline measurements. Other missing lipid measurements were imputed using a last observation carried forward approach and were analysed by ANCOVA |
| Selective reporting (reporting bias) | Unclear risk | Study protocol was unavailable |
| Other bias | High risk | Funded by Amgen |
| Methods |
Type of RCT: 1:3 parallel‐group, double‐blind dose‐ranging RCT Settings: outpatient care Duration: 24 weeks Start and stop dates: NA |
|
| Participants |
Number of participants: 248 (247 with baseline data) Number lost to follow‐up: 20 Women: 107 (43%) Age (SD), years: 64 (8) History of CVD: 129 (52%) Participants with FH: NA Participants with established CHD or CHD equivalent risk (not defined further) |
|
| Interventions |
Background therapy: standard of care, potentially including statin therapy Randomised therapy: 24 weeks of RG7652 (MPSK3169A) every 4, 8, or 12 weeks vs placebo RUG7652 dose: 5 dosage regimens were administered: 200 mg every 8 weeks, 400 mg every 8 weeks, 800 mg every 12 weeks, 400 mg every 4 weeks, 800 mg every 8 weeks, resulting in a 2‐week equivalent dose of 50 mg to 200 mg |
|
| Outcomes | Lipids, any adverse events, CVD, all‐cause mortality | |
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Only an abstract/poster was available |
| Allocation concealment (selection bias) | Unclear risk | Only an abstract/poster was available |
| Blinding of participants and personnel (performance bias) LDL‐C | Unclear risk | Only an abstract/poster was available |
| Blinding of outcome assessment (detection bias) LDL‐C | Unclear risk | Only an abstract/poster was available. Unknown if a central laboratory was used |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 1 participant was excluded from modified ITT population, and 19 participants (7.66%) did not complete the study |
| Selective reporting (reporting bias) | Unclear risk | Full paper has not yet been published |
| Other bias | High risk | Funded by F. Hoffman‐La Roche Ltd |
| Methods |
Type of RCT: 1:1 parallel‐group, double‐blind RCT Settings: outpatient care Duration: 157 weeks (36 months) Start and stop dates: 02/2013; 11/2016 |
|
| Participants |
Number of participants: 27,564 (39 did not receive treatment) Number lost to follow‐up: 1558 participants had observed LDL‐C measurements at 36 months, 1375 completed follow‐up time of 36 months for the primary endpoint of CVD Women: 6769 (25%) Age (SD), years: 63 (9) History of CVD: 27,564 (100%), not reported but inferred on the basis of inclusion criteria Participants with FH: NA Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Background therapy: statin therapy. Randomized therapy: evolocumab compared to placebo. RUG7652 dose: 140 mg/2w or to 420 mg/4w of evolocumab. Resulting in a two week equivalent dose of 140mg‐210mg. |
|
| Outcomes | LDL‐C, any adverse events, CVD, all‐cause mortality, T2DM | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central computerized system |
| Allocation concealment (selection bias) | Low risk | Central allocation |
| Blinding of participants and personnel (performance bias) LDL‐C | Low risk | Both were blinded |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Central laboratory and blinded adjudication |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 27564 patients were randomized of whom 39 did not receive any treatment. The number of participants available reduced considerably over time to only 1375 subjects remaining at study end. However, as reported loss to follow‐up was only 0.1% and the decrease in number reflects different enrolment times. |
| Selective reporting (reporting bias) | Low risk | Reports on most endpoints |
| Other bias | High risk | Amgen |
| Methods |
Type of RCT: 1:2 parallel group, double‐blind RCT. Settings: outpatient care. Duration: 24 weeks Start and stop dates: 12/2013; 06/2017 |
|
| Participants |
Number of participants: 233 Number lost to follow‐up: NA Women: 103 (44%) Age(SD): 63 (10) History of CVD: NA FH participants: 29 (12%) Participants with primary hypercholesterolaemia (heFH or non‐FH) with high CV risk with muscle related statin intolerance, or moderate CV risk without muscle related statin intolerance. |
|
| Interventions |
Background therapy: ezetimibe, fenofibrate or diet alone. Randomized therapy: alirocumab versus placebo. Alirocumab dose: 24 weeks of 75 mg alirocumab every 2 weeks or 150 mg Alirocumab every 4 weeks. At 12 weeks participants could switch to 150 mg every 2 weeks. Resulting in a two week equivalent dose of 75‐150mg. |
|
| Outcomes | lipids, any adverse events, all‐cause mortality. | |
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported. |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) LDL‐C | Unclear risk | Described as double‐blind however no details are provided on who was blinded. However, taking account of the other Odyssey trials seems likely both patients and personal were blinded. |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | No details are provided. However, LDL‐C and other biomarkers are unlikely biased by any lack of blinded assessment. Furthermore, all other Odyssey trails implemented blinded assessment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No details are provided on missing data. |
| Selective reporting (reporting bias) | Unclear risk | The full paper has not yet been published. |
| Other bias | High risk | Funded by Sanofi and Regeneron. |
| Methods |
Type of RCT: 1:1 parallel‐group RCT, with stratification for CVD history Settings: outpatient care Duration: 24 weeks Start and stop dates: 09/2012 and 09/2016 |
|
| Participants |
Number of participants: 251 (excluding 63 participants in an atorvastatin rechallenge arm) Number lost to follow‐up: 80 Women: 114 (45%) Age (SD), years: 63 (10) History of CVD: 115 (46%) FH participants: 38 (15%) Participants with primary hypercholesterolaemia and moderate, high, or very high CV risk, who are intolerant to statins 377 participants with a history of statin intolerance, and of moderate, high, or very high CV risk. Moderate CV risk defined as SCORE risk of 1% or more but lower than 5%; high risk defined as score risk of 5% or more, or moderate chronic kidney disease, diabetes without target organ damage heFH; very high risk defined as history of documented CHD, ischaemic stroke, peripheral artery disease, TIA, abdominal aortic aneurysm, or carotid artery stent procedure, or carotid endarterectomy or carotid artery stent procedure, or renal artery stenosis or renal artery stent procedure or diabetes with target organ damage |
|
| Interventions |
Background therapy: National Cholesterol Education Program Adult Treatment Panel III therapeutic lifestyle changes diet. Participants were allowed to continue to use bile acid, nicotinic acid, fenofibrate, or mega‐3 acid Randomised therapy: alirocumab and placebo vs daily 10 mg ezetimibe or 20 mg atorvastatin and placebo Alirocumab dose: 24 weeks 75 mg alirocumab every 2 weeks, with uptitration of alirocumab to 150 mg every 2 weeks at week 12. Resulting in a 2‐week equivalent dose of 75 mg to 150 mg |
|
| Outcomes | MACE, lipids, any adverse events, all‐cause mortality | |
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Low risk | Permuted‐block design and central allocation |
| Blinding of participants and personnel (performance bias) LDL‐C | Low risk | Placebo‐controlled, patients self‐administered. Unclear if staff was also blinded. Any potential unblinding of staff would be unlikely to result in bias in association with biomarkers |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters assessed at central blinded laboratory |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 36 (28.6%) participants in the alirocumab arm had missing lipid measurements compared with 44 (36.1%) in the ezetimibe arm. Potenially, these "missing" participants simply did not make the required follow‐up time (24 weeks) owing to late enrolment; without specific description of the reason for these lower numbers, some concern is warranted |
| Selective reporting (reporting bias) | Unclear risk | Full paper has not yet been published |
| Other bias | High risk | Funded by Sanofi and Regeneron |
| Methods |
Type of RCT: 1:2 parallel‐group, double‐blind, stratified RCT Settings: outpatient care. Duration: 24 weeks Start and stop dates: 10/2013 and 05/2015 |
|
| Participants |
Number of participants: 803 Number lost to follow‐up: NA Women: 341 (42%) Age (SD), years: 60 (10) History of CVD: NA Participants with FH: 45 (6%) Participants with poorly controlled hypercholesterolaemia and moderate CV risk with or without muscle‐related statin intolerance, or with high CV risk receiving maximally tolerated dose. No definition of poorly controlled or moderate/high CV risk was provided |
|
| Interventions |
Background therapy: statin therapy. Randomized therapy: alirocumab vs placebo. At 12 weeks, participants could switch to 150 mg every 2 weeks Alirocumab dose: 48 weeks 75 mg alirocumab every 2 weeks or 300 mg alirocumab every 4 weeks. Resulting in a 2‐week equivalent dose of 75 mg to 150 mg. Treatment was allocated stratified on statin use or not |
|
| Outcomes | Lipids, any adverse events, all‐cause mortality | |
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) LDL‐C | Unclear risk | Described as double‐blind, but no details provided on who was blinded. However, taking account of the other Odyssey trials, seems likely both participants and personal were blinded |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | No details are provided. However, LDL‐C and other biomarkers are unlikely biased by any lack of blinded assessment. Furthermore, all other Odyssey trials implemented blinded assessment using a central laboratory |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No details of missing data are provided |
| Selective reporting (reporting bias) | Unclear risk | Full paper has not yet been published |
| Other bias | High risk | Funded by Sanofi and Regeneron |
| Methods |
Type of RCT: 1:2 parallel‐group, double‐blind, stratified RCT Settings: outpatient care Duration: 52 weeks Start and stop dates: 07/2012 and 04/2014 |
|
| Participants |
Number of participants: 316 Number lost to follow‐up: 30 Women: 108 (34%) Age (SD), years: 63 (9) History of CVD: 247 (78%) FH participants: 0 Participants with hypercholesterolaemia (LDL‐C ≥ 70 mg/dL) and established CVD or LDL‐C of 100 mg/dL and CHD risk equivalents (e.g. chronic kidney disease) and on a maximally tolerated dose of statin, with possible addition of other lipid‐lowering therapies |
|
| Interventions |
Background therapy: both add‐on to maximal tolerated dose of statin Randomised therapy: alirocumab vs placebo Alirocumab dose: 104 weeks of 75 mg alirocumab every 2 weeks, with uptitration of alirocumab to 150 mg every 2 weeks at week 12. resulting in a 2‐week equivalent dose of 75 mg to 150 mg |
|
| Outcomes | CVD, lipids, any adverse events, all‐cause mortality | |
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Does not mention randomisation but presumably similar as COMBO II: using an interactive voice‐response system |
| Allocation concealment (selection bias) | Unclear risk | Does not describe this |
| Blinding of participants and personnel (performance bias) LDL‐C | Low risk | Both were blinded |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Central laboratory |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 20 (9.57%) participants in the alirocumab arm had missing lipid measurements compared with 10 (9.34%) in the comparator arm. Potenially, these "missing" participants simply did not make the required follow‐up time (24 weeks) owing to late enrolment; however, without specific description of the reasons for these lower numbers, some concern is warranted |
| Selective reporting (reporting bias) | Low risk | Reports on protocol‐defined endpoints |
| Other bias | High risk | Funded by Sanofi and Regeneron |
| Methods |
Type of RCT: 2:1 parallel‐group, double‐blind, stratified, permuted‐block RCT Settings: outpatient care Duration: 104 weeks Start and stop dates: 08/2012 and 07/2015 |
|
| Participants |
Number of participants: 720 Number lost to follow‐up: 13 Women: 190 (26%) Age (SD), years: 62 (9) History of CVD: 649 (90%) FH participants: 0 Participants with hypercholesterolaemia (not defined) and established CHD or CHD risk equivalents (Ischaemic stroke, peripheral artery disease, moderate chronic kidney disease, or diabetes mellitus plus 2 or more additional risk factors) and on a maximally tolerated dose of statin, without addition of other lipid‐lowering therapies |
|
| Interventions |
Background therapy: add‐on to maximal tolerated dose of statin Randomised therapy: alirocumab and ezetimibe placebo vs 10 mg daily of ezetimibe and placebo Alirocumab: 104 weeks of 75 mg alirocumab every 2 weeks, with uptitration of alirocumab to 150 mg every 2 weeks at week 12, resulting in a 2‐week equivalent dose of 75 mg to 150 mg |
|
| Outcomes | CVD, lipids, any adverse events, all‐cause mortality | |
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Using an interactive voice‐response system |
| Allocation concealment (selection bias) | Low risk | Permuted blocks |
| Blinding of participants and personnel (performance bias) LDL‐C | Low risk | Both were blinded |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Central laboratory |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 12 (2.51%) participants in the alirocumab arm had missing lipid measurements compared with 1 (0.41) in the comparator arm. Additionally, mixed‐effects (ANCOVA) models were used |
| Selective reporting (reporting bias) | Low risk | Reports on protocol‐defined endpoints |
| Other bias | High risk | Funded by Sanofi and Regeneron |
| Methods |
Type of RCT: 2:1 parallel‐group, double‐blind, stratified RCT Settings: outpatient care Duration: 78 weeks Start and stop dates: 07/2012; 12/2014 |
|
| Participants |
Number of participants: 486 Number lost to follow‐up: 1 Women: 212 (44%) Age (SD), years: 52 (13) History of CVD: 225 (46%) Participants with FH: 485 (100%) Participants with heterozygous familial hypercholesterolaemia on a maximally tolerated dose of statin with LDL‐C of 70 mg/dL or higher or 100 mg/dL or higher, depending on CV risk |
|
| Interventions |
Background therapy: add‐on to maximal tolerated dose of statin and possible addition of other lipid‐lowering therapies Randomized therapy: alirocumab vs placebo Alirocumab dose: 78 weeks of 75 mg alirocumab every 2 weeks, with possible uptitration of alirocumab to 150 mg every 2 weeks at week 12. Resulting in a 2‐week equivalent dose of 75 mg to 150 mg |
|
| Outcomes | CVD, lipids, any adverse events, all‐cause mortality | |
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Centralised interactive voice‐response system or interactive Web‐response system |
| Allocation concealment (selection bias) | Low risk | Central allocation |
| Blinding of participants and personnel (performance bias) LDL‐C | Low risk | Both were blinded |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Central laboratory |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 1 (0.31%) participant in the alirocumab arm had missing lipid measurements compared with 0 in the comparator arm. Additionally, mixed‐effects (ANCOVA) models were used |
| Selective reporting (reporting bias) | Low risk | Reports on protocol‐defined endpoints |
| Other bias | High risk | Funded by Sanofi and Regeneron |
| Methods |
Type of RCT: 2:1 parallel‐group, double‐blind, stratified RCT Settings: outpatient care Duration: 52 weeks Start and stop dates: 12/2012 and 01/2015 |
|
| Participants |
Number of participants: 249 Number lost to follow‐up: 2 Women: 118 (47%) Age (SD), years: 53.2 (17.2) History of CVD: 89 (36%) Participants with FH: 249 (100%) Participants with heFH not adequately controlled with a maximally tolerated daily dose of statin with or without the other LMT, at a stable dose before the screening visit |
|
| Interventions |
Background therapy: add‐on to maximal tolerated dose of statin and possible addition of other lipid‐lowering therapies Randomised therapy: alirocumab vs placebo Alirocumab dose: 78 weeks 75 mg alirocumab every 2 weeks, with possible uptitration of alirocumab to 150 mg every 2 weeks at week 12. Resulting in a 2‐week equivalent dose of 75 mg to 150 mg |
|
| Outcomes | CVD, lipids, any adverse events, all‐cause mortality | |
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Centralised interactive voice‐response system or interactive Web‐response system |
| Allocation concealment (selection bias) | Low risk | Central allocation |
| Blinding of participants and personnel (performance bias) LDL‐C | Low risk | Both were blinded |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Central laboratory |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 1 (0.60%) portion of the alirocumab arm had missing lipid measurements compared with 1 (1.22%) participant in the comparator arm. Additionally, mixed‐effects (ANCOVA) models were used |
| Selective reporting (reporting bias) | Low risk | Reports on protocol‐defined endpoints |
| Other bias | High risk | Funded by Sanofi and Regeneron |
| Methods |
Type of RCT: 2:1 parallel‐group, double‐blind, stratified RCT Settings: outpatient care Duration: 78 weeks Start and stop dates: 12/2012 and 01/2015 |
|
| Participants |
Number of participants: 107 Number lost to follow‐up: 1 Women: NA Age (SD), years: NA History of CVD: 64 (60%) Participants with FH: 107 (100%) Participants with heterozygous familial hypercholesterolaemia on a maximally tolerated dose of statin with LDL‐C ≥ 160 mg/dL |
|
| Interventions |
Background therapy: both add‐on to maximal tolerated dose of statin and possible addition of other lipid‐lowering therapies Randomized therapy: alirocumab vs placebo Alirocumab dose: 78 weeks of 150 mg alirocumab every 2 weeks |
|
| Outcomes | CVD, lipids, any adverse events, all‐cause mortality | |
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Centralised interactive voice‐response system or interactive Web‐response system |
| Allocation concealment (selection bias) | Low risk | Central allocation |
| Blinding of participants and personnel (performance bias) LDL‐C | Low risk | Both were blinded |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Central laboratory |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 1 (1.38%) participant in the alirocumab arm had missing lipid measurements compared with 0 in the comparator arm. Additionally, mixed‐effects (ANCOVA) models were used |
| Selective reporting (reporting bias) | Unclear risk | Full paper has not yet been published |
| Other bias | High risk | Funded by Sanofi and Regeneron |
| Methods |
Type of RCT: 2:1 parallel‐group, double‐blind RCT with stratified randomisation Settings: outpatient care Duration: 78 weeks Start and stop dates: 01/2012 and 11/2014 |
|
| Participants |
Number of participants: 2341 Number lost to follow‐up: 247 Women: 884 (38%) Age (SD), years: 63 (11) History of CVD: 1607 (68%) Participants with FH: 415 (18%) Participants with heterozygous familial hypercholesterolaemia or established coronary heart disease or coronary heart disease risk equivalent |
|
| Interventions |
Background therapy: standard of care Randomized therapy: alirocumab vs placebo for 78 weeks Alirocumab dose: 150 mg alirocumab every 2 weeks |
|
| Outcomes | CVD, lipids, any adverse events, all‐cause mortality | |
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central computer‐generated allocation system |
| Allocation concealment (selection bias) | Low risk | Central computer‐generated allocation system |
| Blinding of participants and personnel (performance bias) LDL‐C | Low risk | Participants and investigators were blinded with placebo identically packaged as alirocumab |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Biomarkers were assessed at a central laboratory blinded for allocation. Clinical endpoints and adverse advents were similarly assessed in a blinded fashion |
| Incomplete outcome data (attrition bias) All outcomes | High risk | ITT analysis excludes participants (167 (10.8%) in the intervention arm and 80 (10.1%) in the control arm) who missed LDL‐C measurements during first 24 weeks. In total, 437 alirocumab patients did not complete study follow‐up compared with 193 placebo participants. Categorical outcomes were analysed using an available case analysis. Missing biomarker values were imputed using mixed models or multiple imputations |
| Selective reporting (reporting bias) | Low risk | Reports on protocol‐defined endpoints |
| Other bias | High risk | Funded by Sanofi and Regeneron |
| Methods |
Type of RCT: 1:1 1:1 parallel‐group, double‐blind RCT Settings: outpatient care Duration: 24 weeks Start and stop dates: 07/2012 and 07/2013 |
|
| Participants |
Number of participants: 103 Number lost to follow‐up: 0 Women: 48 (47%) Age (SD), years: 60 (5) History of CVD: 103 (100%) Participants with FH: 0 Participants with 10‐year risk of fatal CV events between 1% and < 5% |
|
| Interventions |
Background therapy: National Cholesterol Education Program Adult Treatment Panel III therapeutic lifestyle changes diet Randomized therapy: alirocumab and placebo ezetimibe daily vs 10 mg ezetimibe daily plus alirocumab biweekly placebo Alirocumab dose: 24 weeks 75 mg alirocumab every 2 weeks, at 12 weeks LDL‐C dependent uptitration of alirocumab occurred to 150 mg biweekly. Resulting in a 2‐week equivalent dose of 75 mg to 150 mg |
|
| Outcomes | CVD, lipids, any adverse events | |
| Notes | · LDL‐C was calculated using the Friedewald formula · NCT01644474 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Low risk | Permuted‐block design |
| Blinding of participants and personnel (performance bias) LDL‐C | Low risk | Participants were blinded for treatment allocation and self‐administered treatments |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Central laboratory |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants were available at 24 weeks of follow‐up |
| Selective reporting (reporting bias) | Low risk | Reports on protocol‐defined endpoints |
| Other bias | High risk | Funded by Sanofi and Regeneron |
| Methods |
Type of RCT: 2:1 parallel‐group, double‐blind, stratified, permuted‐block designed RCT Settings: outpatient care Duration: 24 weeks Start and stop dates: NA |
|
| Participants |
Number of participants: 355 Number lost to follow‐up: 10 Women: 124 (35%) Age (SD), years: 63 (10) History of CVD: 200 (56%) FH participants: 31 (9%) Participants with history of CVD and LDL‐C levels ≥ 70 mg/dL, or CVD risk factors and LDL‐C ≥ 100 mg/dL |
|
| Interventions |
Background therapy: 24 weeks 20 or 40 mg of baseline atorvastatin and National Cholesterol Education Program Adult Treatment Panel III Randomised therapy: alirocumab versus 10 mg ezetimibe per day, or 20 or 40 mg atorvastatin, or for atorvastatin 40 mg regimen only, switch to rosuvastatin 40 mg Alirocumab dose: 75 mg alirocumab every 2 weeks, with uptitration of alirocumab to 150 mg at week 12. Resulting in a 2‐week equivalent dose of 75 mg to 150 mg Resulting in 7 groups
All blinded with placebo alirocumab and over‐encapsulated tables for ezetimibe, atorvastatin, and rosuvastatin |
|
| Outcomes | CVD, lipids, any adverse events, all‐cause mortality | |
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Centralised interactive voice‐response system or interactive Web‐response system |
| Allocation concealment (selection bias) | Low risk | Permuted‐block design and central allocation |
| Blinding of participants and personnel (performance bias) LDL‐C | Low risk | Both were blinded |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Central laboratory |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 4 (3.85%) participants in the alirocumab arm had missing lipids measurements compared with 6 (2.39%) in the comparator arm. Additionally, mixed‐effects (ANCOVA) models were used |
| Selective reporting (reporting bias) | Low risk | Reports on protocol‐defined endpoints |
| Other bias | High risk | Funded by Sanofi and Regeneron |
| Methods |
Type of RCT: double‐blind, placebo‐controlled, parallel‐group RCT Settings: outpatient care Duration: 24 weeks Start and stop dates: NA |
|
| Participants |
Number of participants: 305 Number lost to follow‐up: 7 Women: 118 (39%) Age (SD), years: 61 (10) History of CVD: 177 (58%) Participants with FH: 41 (13%) Participants with a history of CVD and LDL‐C levels ≥ 70 mg/dL, or CVD risk factors and LDL‐C ≥ 100 mg/dL |
|
| Interventions |
Background therapy: Patients received 24 weeks 10 or 20 mg of baseline rosuvastatin and National Cholesterol Education Program Adult Treatment Panel III Randomised therapy: alirocumab vs add‐on 10 mg ezetimibe per day, or additional 10 or 20 mg of rosuvastatin Alirocumab dose: add‐on of 75 mg alirocumab every 2 weeks, with uptitration of alirocumab to 150 mg at week 12. Resulting in a 2‐week equivalent dose of 75 mg to 150 mg Resulting in 6 groups
All blinded with placebo alirocumab and over‐encapsulated tables for ezetimibe, rosuvastatin |
|
| Outcomes | CVD, lipids, any adverse events, all‐cause mortality | |
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Centralised interactive voice‐response system or interactive Web‐response system |
| Allocation concealment (selection bias) | Low risk | Permuted‐block design and central allocation |
| Blinding of participants and personnel (performance bias) LDL‐C | Low risk | Both were blinded |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Central laboratory |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 2 (1.94%) participants in the alirocumab arm had missing lipid measurements compared with 5 (2.48%) in the comparator arms. Additionally, mixed‐effects (ANCOVA) models were used |
| Selective reporting (reporting bias) | Low risk | Reports on protocol‐defined endpoints |
| Other bias | High risk | Funded by Sanofi and Regeneron |
| Methods |
Type of RCT: meta‐analysis of OSLER‐1 and OSLER‐2 RCTs Settings: outpatient care Duration: 52 weeks/48 weeks Start and stop dates: NA |
|
| Participants |
Number of participants: 4465 Number lost to follow‐up: 738 Women: 2210 (49%) Age (SD), years: 58 (11) History of CVD: NA Participants with FH: NA Participants with and without a history of CVD or familial hypercholesterolaemia; all were previously enrolled in phase 2 to 3 PCSK9 inhibitor trials and completed these without serious adverse events |
|
| Interventions |
Background therapy: standard of care (including statins and/or ezetimibe). Randomised therapy: evolocumab vs standard of care only, for 52/48 weeks Evolocumab dose: 420 mg evolocumab every 4 weeks (OSLER–1, OSLER‐2), or 140 mg every 2 weeks (OSLER–2), resulting in 2‐week equivalent dose of 140 mg to 210 mg |
|
| Outcomes | CVD, LDL‐C, any adverse events, all‐cause mortality | |
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was performed centrally with the use of an interactive voice‐response or Web‐response system |
| Allocation concealment (selection bias) | Low risk | Central allocation |
| Blinding of participants and personnel (performance bias) LDL‐C | High risk | No blinding; lack of blinding will likely cause a change in adherence and/or in a participant's choices regarding SOC/lifestyle that may influences outcomes |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Central laboratory. Outcomes directly assessed by study personnel, such as adverse events, may be biased owing to detection bias |
| Incomplete outcome data (attrition bias) All outcomes | High risk | At week 48, 270 (18.13%) SOC participants were unavailable, and 468 (15.72%) in the intervention arm were unavailable. Portion of these "unavailable" participants were due to differences in enrolment dates limiting follow‐up, but with a reported percentage of 7.2%, a considerable proportion of participants were genuinely lost to follow‐up. No mention of how missing data were handled |
| Selective reporting (reporting bias) | Low risk | Reports on protocol‐defined endpoints |
| Other bias | High risk | Analyses such as the Wilcoxon test, or Cox proportional hazards model without stratification for centre, ignore clustering of participants by studies or by study centres Funded by Amgen |
| Methods |
Type of RCT: 1:2 parallel‐group, open‐label stratified trial Settings: outpatient care Duration: 52 weeks Start and stop dates: NA |
|
| Participants |
Number of participants: 1104 Number lost to follow‐up: 169 Women: 610 (55%) Age (SD), years: 56 (12) History of CVD: 210 (19%) FH participants: 414 (38%) Participants with and without a history of CVD or familial hypercholesterolaemia; all were previously enrolled in phase 2 PCSK9 inhibitor trials and completed these trials without serious adverse events |
|
| Interventions |
Background therapy: standard of care (SOC) Randomized therapy: evolocumab vs standard of care for 52 weeks Evolocumab dose: 420 mg evolocumab every 4 weeks, resulting in a 2‐week equivalent dose of 210 mg |
|
| Outcomes | CVD, lipids, any adverse events, all‐cause mortality | |
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was performed centrally with the use of an interactive voice‐response or Web‐response system |
| Allocation concealment (selection bias) | Low risk | Central allocation |
| Blinding of participants and personnel (performance bias) LDL‐C | High risk | No blinding. Lack of blinding will likely cause a change in adherence and/or in participants regarding SOC/lifestyle that may influences outcomes |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Central laboratory. Besides lipids, outcomes such as adverse events may be biased owing to detection bias |
| Incomplete outcome data (attrition bias) All outcomes | High risk | At week 52, 73/368 = 19.83% of SOC dropped out, and 96/736 = 13.04% of intervention arm dropped out. No mention of how missing data were handled |
| Selective reporting (reporting bias) | Low risk | Reports on protocol‐defined endpoints |
| Other bias | High risk | Funded by Amgen |
| Methods |
Type of RCT: 1:1 parallel‐group RCTs, double‐blind, permuted‐block design stratified by geographic region Settings: outpatient care Duration: 143 weeks Start and stop dates: both 10/2013 and 01/2017 |
|
| Participants |
Number of participants: 27438 (39 participants did not receive treatment) Number lost to follow‐up: At week 52, the number of participants available for biomarker measurements could be as low as 7814. For clinical endpoints, only 11 participants made it to the end of follow‐up (143 weeks), but this is likely to happen with participants starting at different times and early termination of trials due to an antidrug‐antibody response Women: 8111 (30%) Age (SD), years: 63 (9) History of CVD: 23198 (85%) Participants with FH: 1072 (4%) Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Background therapy: statins and/or ezetimibe Randomized therapy: bococizumab compared with placebo Evolocumab dose: 150 mg/2w downtitration to 75 mg/2w |
|
| Outcomes | CVD, lipids, any adverse events, all‐cause mortality | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Interactive Response Technology (IRT) System (Interactive Web Response (IWR)/Interactive Voice Response (IVR) system) |
| Allocation concealment (selection bias) | Low risk | Permuted blocks |
| Blinding of participants and personnel (performance bias) LDL‐C | Low risk | Both were blinded |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Central laboratory and blinded adjudicated clinical outcomes |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number of available participants at 52 weeks could be as low as 7814, depending on the biomarker measured in a total of 27,438 randomised participants. For clinical endpoints, only 11 participants made it to the end of follow‐up (143 weeks). Both of these issues are related to early termination of these trials and participants enrolling at different moments in time; actual loss to follow‐up was 0.9% |
| Selective reporting (reporting bias) | Low risk | Reports on most endpoints |
| Other bias | High risk | Pfizer funded |
ANCOVA: analysis of covariance
CABG: coronary artery bypass graft
CHD: coronary heart disease
CV: cardiovascular
CVD: cardiovascular disease
FH: familial hypercholesterolaemia
HbA1c: glycosylated haemoglobin
HDL‐C: high‐density lipoprotein cholesterol
heFH: heterozygous familial hypercholesterolaemia
ITT: intention‐to‐treat
LDL‐C: low‐density lipoprotein cholesterol
LMT: lipid modifying treatments
MACE: major adverse cardiac events
MDRD: Modification of Diet in Renal Disease
NYHA: New York Heart Association
PCI: percutaneous coronary intervention
RCT: randomised controlled trial
SD: standard deviation
SOC: standard of care
TIA: transient ischaemic attack
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Baruch 2013 | Follow‐up time too short |
| Cho 2014 | Follow‐up time too short |
| Desai 2014 | Follow‐up time too short |
| Dias 2012 | Follow‐up time too short |
| Dufour 2012 | Meta‐analysis without separate results |
| Gaudet 2012 | Meta‐analysis of 3 studies without separate results |
| Gaudet 2013 | Meta‐analysis of 3 studies without separate results |
| Gumbiner 2012 | Follow‐up time too short |
| Hopkins 2013 | Follow‐up time too short |
| Jones 2015 | Meta‐analysis of 4 studies without separate results |
| Kastelein 2015 | Follow‐up time too short |
| Kawashiri 2012 | No randomisation to PCSK9 inhibitor |
| Mabuchi 2015 | No empirical results |
| Maxwell 2012 | No empirical results |
| Mearns 2014 | No empirical results |
| Pordy 2013 | Dose‐response modelling |
| Raal 2014 | Follow‐up time too short |
| Raal 2014a | Meta‐analysis without separate results |
| Shaywitz 2012 | Follow‐up time too short |
| Stawowy 2014 | Follow‐up time too short |
| Stein 2012 | This reference published on a subset of the data included in OSLER‐1 |
| Stein 2013 | Follow‐up time too short |
| Swergold 2010 | Follow‐up time too short |
| Swergold 2011 | Follow‐up time too short |
| Wan 2013 | Follow‐up time too short |
Characteristics of studies awaiting assessment [ordered by study ID]
| Methods | Six parallel, multi‐national lipid‐lowering trials |
| Participants | 4300 patients with hyperlipidaemia |
| Interventions | 150 mg bococizumab or placebo subcutaneously every 2 weeks |
| Outcomes | Lipids, any adverse events, clinical endpoints |
| Notes | Given the short follow‐up time, the focus on biomarkers, and the fact that drug development has been terminated, incorporation of these trials will have limited impact |
Characteristics of ongoing studies [ordered by study ID]
| Trial name or title | ANITSCHKOW |
| Methods | Parallel randomised controlled trials |
| Participants | People 50 to 80 years of age with baseline Lp(a) ≥ 50 mg/dL and LDL‐C ≥ 100 mg/dL |
| Interventions | Evolocumab compared with placebo with background statin therapy for all |
| Outcomes | Number of participants with treatment‐related adverse events as assessed by Common Terminology Criteria for Adverse Events version 4 |
| Starting date | April 2016 |
| Contact information | |
| Notes | Amgen |
| Trial name or title | EBBINGHAUS |
| Methods | Parallel randomised controlled trials |
| Participants | People 40 to 85 years of age Inclusion criteria
Exclusion criteria
|
| Interventions | Evolocumab compared with statin therapy in combination with placebo |
| Outcomes | Mean change from baseline over time in spatial working memory (SWM) index of executive function |
| Starting date | July 2014 |
| Contact information | |
| Notes | Amgen, substudy of FOURIER |
| Trial name or title | HAUSER‐RCT |
| Methods | Parallel randomised controlled trials |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Evolocumab compared with placebo |
| Outcomes | Percentage change from baseline in low‐density lipoprotein cholesterol levels |
| Starting date | February 2015 |
| Contact information | |
| Notes | Amgen |
| Trial name or title | A Double Blind, Randomized, Placebo Controlled, Multicenter Study to Evaluate Safety, Tolerability, and Efficacy on LDL‐C of Evolocumab (AMG 145) in Subjects With HIV and With Hyperlipidemia and/or Mixed Dyslipidemia |
| Methods | Parallel randomised controlled trials |
| Participants | Human immunodeficiency virus (HIV)‐positive individuals with hyperlipidaemia or mixed dyslipidaemia (time frame: week 24) |
| Interventions | Evolocumab compared with placebo |
| Outcomes | Percent change from baseline in low‐density lipoprotein cholesterol (LDL‐C) |
| Starting date | June 2016 |
| Contact information | |
| Notes | Amgen |
| Trial name or title | ODYSSEY DM‐Dyslipidemia |
| Methods | Open‐label parallel randomised controlled trials |
| Participants | Inclusion criteria
Exclusion criteria
The above information is not intended to contain all considerations relevant to a patient's potential participation in a clinical trial |
| Interventions |
Drug: placebo Drug: statins
Interventions: drug: placebo Drug: statins Drug: ezetimibe Drug: fenofibrate Drug: nicotinic acid Drug: omega‐3 fatty acids |
| Outcomes | Percent change in non‐HDL‐C in the intent‐to‐treat (ITT) population |
| Starting date | December 2015 |
| Contact information | |
| Notes | Sanofi |
| Trial name or title | ODYSSEY Outcomes |
| Methods | Parallel randomised controlled trials |
| Participants | Inclusion criteria
Exclusion criteria
The above information is not intended to contain all considerations relevant to a patient's potential participation in a clinical trial |
| Interventions | Alirocumab compared with placebo |
| Outcomes | Time from randomisation to first occurrence of one of the following clinical events: CHD death, any non‐fatal MI, fatal and non‐fatal ischaemic stroke, unstable angina requiring hospitalisation |
| Starting date | August 2012 |
| Contact information | |
| Notes | Sanofi |
| Trial name or title | TAUSSIG |
| Methods | Open‐label parallel randomised controlled trial |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | 1 monthly dose of evolocumab compared with 2 monthly dosages of evolocumab |
| Outcomes | Incidence of treatment‐emergent adverse events |
| Starting date | June 2015 |
| Contact information | |
| Notes | Amgen |
Contributions of authors
AFS drafted the protocol and the full review and conducted all analyses. AFS and LSP screened hits and extracted data. LSP, JTW, JPO, AH, and JPC provided guidance during critical revision of the manuscript.
Sources of support
Internal sources
-
Springboard fellowship, Other.
AF Schmidt is funded by a UCL springboard fellowship.
External sources
-
Biomedical Research Centre, UK.
This project was supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre.
Declarations of interest
Amand F Schmidt: none known.
Lucy S Pearce: none known.
John T Wilkins: none known.
John P Overington: none known.
Aroon Hingorani is a member of the organisation committee of The Genetics of Subsequent Coronary Heart Disease Consortium and the Heart failure Molecular Epidemiology for Therapeutic Targets Consortium (HERMES) each comprising over 20 member cohorts. A number of Pharma companies have provided direct and in kind support for these initiatives, but AH is not a direct recipient of any of these funds.
Juan P Casas: none known.
New
References
References to studies included in this review
- Ballantyne CM, Neutel J, Cropp A, Duggan W, Wang EQ, Plowchalk D, et al. Results of bococizumab, a monoclonal antibody against proprotein convertase subtilisin/kexin type 9, from a randomized, placebo‐controlled, dose‐ranging study in statin‐treated subjects with hypercholesterolemia. American Journal of Cardiology 2015;115(9):1212‐21. [DOI] [PubMed] [Google Scholar]
- Blom DJ, Hala T, Bolognese M, Lillestol MJ, Toth PD, Burgess L, et al. A 52‐week placebo‐controlled trial of evolocumab in hyperlipidemia. New England Journal of Medicine 2014;370:1809‐19. [DOI] [PubMed] [Google Scholar]
- Tingley W, Mosesova S, Baruch A, Davis JD, Budha N, Vilimovskij A, et al. Effects of RG7652, a monoclonal antibody against proprotein convertase Subtilisin/Kexin type 9, on LDL cholesterol in patients with coronary heart disease or high risk: results from the EQUATOR study. European Heart Journal. 2014:371. [Google Scholar]
- Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. New England Journal of Medicine 2017;online:1‐10. [DOI] [PubMed] [Google Scholar]
- Moriarty PM, Thompson PD, Cannon CP, Guyton JR, Bergeron J, Zieve FJ, et al. Efficacy and safety of alirocumab vs ezetimibe in statin‐intolerant patients, with a statin rechallenge arm: the ODYSSEY ALTERNATIVE randomized trial. Journal of Clinical Lipidology 2015;9(6):758‐69. [DOI] [PubMed] [Google Scholar]
- Stroes E, Guyton JR, Farnier M, Rader D, Moriarty PM, Bergeron J, et al. Efficacy and safety of 150 mg and 300 mg every 3 weeks in patients with poorly controlled hypercholesterolemia: the ODYSSEY CHOICE I and CHOICE II studies. Journal of the American College of Cardiology. 2015; Vol. abstract:Exhibit 99.1. [Google Scholar]
- Stroes E, Guyton JR, Farnier M, Rader D, Moriarty PM, Bergeron J, et al. Efficacy and safety of 150 mg and 300 mg every 3 weeks in patients with poorly controlled hypercholesterolemia: the ODYSSEY CHOICE I and CHOICE II studies. Journal of the American College of Cardiology. 2015; Vol. abstract:Exhibit 99.1. [Google Scholar]
- Kereiakes DJ, Robinson JG, Cannon CP, Lorenzato C, Pordy R, Chaudhari U, et al. Efficacy and safety of the proprotein convertase subtilisin/kexin type 9 inhibitor alirocumab among high cardiovascular risk patients on maximally tolerated statin therapy: The ODYSSEY COMBO I study. American Heart Journal 2015;169(6):906. [DOI] [PubMed] [Google Scholar]
- Cannon CP, Cariou B, Blom D, McKenney JM, Lorenzato C, Pordy R, et al. Efficacy and safety of alirocumab in high cardiovascular risk patients with inadequately controlled hypercholesterolaemia on maximally tolerated doses of statins: the ODYSSEY COMBO II randomized controlled trial. European Heart Journal 2015;36(19):1186‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastelein JJ, Ginsberg HN, Langslet G, Hovingh GK, Ceska R, Dufour R, et al. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. European Heart Journal 2015;36(43):2996‐3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastelein JJ, Ginsberg HN, Langslet G, Hovingh GK, Ceska R, Dufour R, et al. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. European Heart Journal 2015;36(43):2996‐3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg HN, Rader DJ, Raal F, Guyton JR, Lorenzato C, Pordy R, et al. ODYSSEY HIGH FH: Efficacy and safety of alirocumab in patients with severe heterozygous familial hypercholesterolemia. Conference abstract. Published online 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JG, Farnier M, Krempf M, Bergeron J, Luc G, Averna M, et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. New England Journal of Medicine 2015;372(16):1489‐99. [DOI] [PubMed] [Google Scholar]
- Roth EM, Taskinen MR, Ginsberg HN, Kastelein JJP, Colhoun HM, Robinson JG, et al. Monotherapy with the PCSK9 inhibitor alirocumab versus ezetimibe in patients with hypercholesterolemia: results of a 24 week, double‐blind, randomized Phase 3 trial. International Journal of Cardiology 2014;176:55‐61. [DOI] [PubMed] [Google Scholar]
- Bays H, Gaudet D, Weiss R, Ruiz JL, Watts GF, Gouni‐Berthold I, et al. Alirocumab as add‐on to atorvastatin versus other lipid treatment strategies: ODYSSEY OPTIONS I randomized trial. Journal of Clinical Endocrinology and Metabolism 2015;100(8):3140‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnier M, Jones P, Severance R, Averna M, Steinhagen‐Thiessen E, Colhoun HM, et al. Efficacy and safety of adding alirocumab to rosuvastatin versus adding ezetimibe or doubling the rosuvastatin dose in high cardiovascular‐risk patients: the ODYSSEY OPTIONS II randomized trial. Atherosclerorsis 2016;244:138‐46. [DOI] [PubMed] [Google Scholar]
- Koren MJ, Giugliano RP, Raal FJ, Sullivan D, Bolognese M, Langslet G, et al. Efficacy and safety of longer‐term administration of evolocumab (AMG 145) in patients with hypercholesterolemia: 52‐week results from the Open‐Label Study of Long‐Term Evaluation Against LDL‐C (OSLER) randomized trial. Circulation 2014;129:234‐43. [DOI] [PubMed] [Google Scholar]
- Sabatine MS, Giugliano RP, Wiviott SD, Raal FJ, Blom DJ, Robinson J, et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. New England Journal of Medicine 2015;372(16):1500‐9. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Revking J, Amarenco P, Brunell R, Curto M, Civeria F, et al. Cardiovascular efficacy and safety of bococizumab in high‐risk patients. New England Journal of Medicine 2017;online:1‐13. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
- Baruch A, Peng K, Leabman M, Budha N, Luca D, Cowan KJ, et al. Effect of RG7652, a mAb against PCSK9, on apolipoprotein B, oxidized LDL, lipoprotein(A) and lipoprotein‐associated phospholipase a2 in healthy individuals with elevated LDL‐C. Circulation. 2013; Vol. 22. [Google Scholar]
- Cho L, Rocco M, Colquhoun D, Sullivan D, Rosenson RS, Dent R, et al. Clinical profile of statin intolerance in the phase 3 gauss‐2 study. Canadian Journal of Cardiology. 2014:S79. [DOI] [PubMed] [Google Scholar]
- Desai NR, Giugliano RP, Zhou J, Kohli P, Somaratne R, Hoffman E, et al. AMG 145, a monoclonal antibody against PCSK9, facilitates achievement of National Cholesterol Education Program‐Adult Treatment Panel III low‐density lipoprotein cholesterol goals among high‐risk patients: an analysis from the LAPLACE‐TIMI 57 trial (LDL‐C assessment with PCSK9 monoclonal antibody inhibition combined with statin thErapy‐thrombolysis in myocardial infarction 57). Journal of the American College of Cardiology 2014;63:430‐3. [DOI] [PubMed] [Google Scholar]
- Dias CS, Shaywitz AJ, Wasserman SM, Smith BP, Gao B, Stolman DS, et al. Effects of AMG 145 on low‐density lipoprotein cholesterol levels: results from 2 randomized, double‐blind, placebo‐controlled, ascending‐dose phase 1 studies in healthy volunteers and hypercholesterolemic subjects on statins. Journal of the American College of Cardiology 2012;60:1888‐98. [DOI] [PubMed] [Google Scholar]
- Dufour R, Moriarty PM, Genestin E, Sasiela WJ, Du Y, Ferrand AC, et al. Effect of REGN727/SAR236553 anti‐proprotein convertase subtilisin/kexin type 9 fully human monoclonal antibody in patients with elevated triglycerides/low high‐density lipoprotein cholesterol: data from three phase 2 studies (NCT:01266876; 01288469; 01288443). Circulation. 2012. [Google Scholar]
- Gaudet D, Kereiakes D, McKenney J, Roth E, Hanotin C, Gipe D, et al. Effect of SAR236553/REGN727 fully human monoclonal anti‐proprotein convertase subtilisin/kexin type 9 antibody on plasma lipoprotein(a) concentrations: pooled analysis from three phase 2 studies (NCT:01266876; 01288469; 01288443). Circulation. 2012. [Google Scholar]
- Gaudet D, Kereiakes D, McKenney J, Roth E, Hanotin C, Gipe D, et al. Alirocumab, a fully human monoclonal antibody to PCSK9, reduces high plasma lp(a) concentration: pooled analysis of 352 patients from phase 2. Journal of Clinical Lipidology 2013;7(3):283‐4. [Google Scholar]
- Gumbiner B, Udata C, Joh T, Liang H, Wan H, Shelton D, et al. The effects of multiple dose administration of RN316 (PF‐04950619), a humanized IgG2a monoclonal antibody binding proprotein convertase snbtilisin kexin type 9, in hypercholesterolemic subjects. Circulation. 2012; Vol. 126, issue 21. [Google Scholar]
- Hopkins PN, Swergold GD, Mellis S, Bruckert E, Luc G, Mendoza J, et al. A randomized placebo‐phase clinical trial with the monoclonal antibody alirocumab demonstrates reductions in low‐density lipoprotein cholesterol in patients with proprotein convertase subtilisin/kexin type 9 gain‐of‐function mutations. Circulation. 2013. [Google Scholar]
- Jones PH, Bays H, Chaudhari U, Pordy R, Lorenzato C, Miller K, et al. Pooled safety and adverse events in nine randomized, placebo‐controlled, phase 2 and 3 clinical trials of alirocumab. Journal of the American College of Cardiology. 2015:A1363. [Google Scholar]
- Kastelein J, Nissen S, Rader D, Krueger K, Wang MD. Safety and efficacy of LY 3015014, a new monoclonal antibody to proprotein convertase subtilisin/kexin type 9 (PCSK 9) with an inherently longer duration of action, in patients with primary hypercholesterolemia: a randomized, placebo‐controlled, dose‐ranging, phase 2 study. Journal of the American College of Cardiology. 2015:A1591. [Google Scholar]
- Kawashiri MA, Nohara A, Noguchi T, Tada H, Nakanishi C, Mori M, et al. Efficacy and safety of coadministration of rosuvastatin, ezetimibe, and colestimide in heterozygous familial hypercholesterolemia. American Journal of Cardiology 2012;109:364‐9. [DOI] [PubMed] [Google Scholar]
- Mabuchi H, Nohara A. Therapy: PCSK9 inhibitors for treating familial hypercholesterolaemia. Nature Reviews Endocrinology 2015;11(1):8‐9. [DOI] [PubMed] [Google Scholar]
- Maxwell KN, Breslow JL. Antibodies to PCSK9: a superior way to lower LDL cholesterol?. Circulation Research 2012;111:274‐7. [DOI] [PubMed] [Google Scholar]
- Mearns BM. Dyslipidaemia: 1‐year results from OSLER trial of anti‐PCSK9 monoclonal antibody evolocumab. Nature Reviews Cardiology 2014;11:63. [DOI] [PubMed] [Google Scholar]
- Pordy R, Lecorps G, Bessac L, Sasiela WJ, Ginsberg H. Alirocumab, a fully human monoclonal antibody to proprotein convertase subtilisin/kexin type 9: therapeutic dosing in phase 3 studies. Journal of Clinical Lipidology 2013;7(3):279. [Google Scholar]
- Raal F, Nelson P, Langslet G, Basart DCG, Civeira F, Lopez‐Miranda J, et al. Safety, tolerability, and efficacy of long‐term administration of monthly AMG 145 in subjects with heterozygous familial hypercholesterolemia. Global Heart. 2014; Vol. 1:e139. [Google Scholar]
- Raal F, Giugliano RP, Sabatine MS, Koren MJ, Blom D, Honarpour N, et al. Long‐term reduction in lipoprotein(A) with the PCSK9 monoclonal antibody evolocumab (AMG 145): a pooled analysis of 3278 patients in phase 2, 3, and open label extension studies. Circulation. 2014. [DOI] [PubMed] [Google Scholar]
- Shaywitz AJ, Dias C, Smith B, Gao B, Gibbs J, Emery M, et al. AMG 145, a fully human monoclonal antibody against PCSK9, reduces LDL‐C in healthy volunteers and patients on stable doses of statins+. Journal of Clinical Lipidology. 2012; Vol. 6, issue 3:286‐7. [Google Scholar]
- Stawowy P, Just IA, Kaschina E. Inhibition of PCSK9: a novel approach for the treatment of dyslipidemia. Coronary Artery Disease 2014;25:353‐9. [DOI] [PubMed] [Google Scholar]
- Stein EA, Mellis S, Yancopoulos GD, Stahl N, Logan D, Smith WB, et al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. New England Journal of Medicine 2012;366:1108‐18. [DOI] [PubMed] [Google Scholar]
- Stein EA, Somaratne R, Schou MB, Civeira F, Sullivan D, Watts GF, et al. Efficacy and tolerability of long‐term treatment with AMG 145 in patients with statin intolerance. Circulation. 2013. [Google Scholar]
- Swergold G, Biedermann S, Renard R, Nadler D, Wu R, Mellis S. Safety, lipid, and lipoprotein effects of REGN727/SAR236553, a fully‐human proprotein convertase subtilisin kexin 9 (PCSK9) monoclonal antibody administered intravenously to healthy volunteers. Circulation 2010;122:2. [Google Scholar]
- Swergold G, Smith W, Mellis S, Logan D, Webb C, Wu R, et al. Inhibition of proprotein convertase subtilisin/kexin type 9 with a monoclonal antibody REGN727/SAR236553, effectively reduces low‐density‐lipoprotein cholesterol, as mono or add‐on therapy in heterozygous familial and non familial hypercholesterolemia. Circulation. 2011; Vol. 124:2. [Google Scholar]
- Wan H, Gumbiner B, Joh T, Udata C, Forgues P, Garzone PD. Effects of RN316 (pf‐04950615), a humanized igg2a monoclonal antibody binding proprotein convertase subtilisin kexin type 9, on lipoprotein particles in hypercholesterolemic subjects. Journal of the American College of Cardiology. 2013:E1387. [Google Scholar]
References to studies awaiting assessment
- Ridker PM, Tardif JC, Amarenco P, DUggan W, Glyn RJ, Jukema WJ, et al. Lipid‐reduction variability and antidrug‐antibody formation with bococizumab. New England Journal of Medicine 2017;online first:1‐10. [DOI] [PubMed] [Google Scholar]
References to ongoing studies
- NCT02729025. Effects of Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) Inhibition on Arterial Wall Inflamation Study in Patients With Elevated Lipoprotein(a) (Lp(a)). (ANITSCHKOW). https://clinicaltrials.gov/ct2/show/NCT02729025 (first received: March 8, 2016).
- NCT02207634. Evaluating PCSK9 Binding antiBody Influence oN coGnitive HeAlth in High cardiovascUlar Risk Subjects (EBBINGHAUS). https://clinicaltrials.gov/ct2/show/NCT02207634 (first received July 31, 2014).
- NCT02392559. Trial Assessing Efficacy, Safety and Tolerability of PCSK9 Inhibition in Paediatric Subjects With Genetic LDL Disorders (HAUSER‐RCT). https://clinicaltrials.gov/ct2/show/NCT02392559 (first received February 25, 2015).
- NCT02833844. Safety, Tolerability & Efficacy on LDL‐C of Evolocumab in Subjects With HIV & Hyperlipidemia/Mixed Dyslipidemia. https://clinicaltrials.gov/ct2/show/NCT02833844 (first received June 13, 2016).
- NCT02642159. Efficacy and Safety of Alirocumab Versus Usual Care on Top of Maximally Tolerated Statin Therapy in Patients With Type 2 Diabetes and Mixed Dyslipidemia (ODYSSEY DM‐Dyslipidemia). https://clinicaltrials.gov/ct2/show/NCT02642159 (first received December 24, 2015).
- NCT01663402. ODYSSEY Outcomes: Evaluation of Cardiovascular Outcomes After an Acute Coronary Syndrome During Treatment With Alirocumab. https://clinicaltrials.gov/ct2/show/NCT01663402 (first received August 8, 2012).
- NCT01624142. Trial Assessing Long Term USe of PCSK9 Inhibition in Subjects With Genetic LDL Disorders (TAUSSIG). https://clinicaltrials.gov/ct2/show/NCT01624142 (first received June 5, 2012).
Additional references
- Abifadel M, Varret M, Rabès JP, Allard D, Ouguerram K, Devillers M, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nature Genetics 2013;34(2):154‐6. [DOI] [PubMed] [Google Scholar]
- Altman DG, Bland JM. Interaction revisited: the difference between two estimates. BMJ 2003;326(7382):219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benn M, Nordestgaard BG, Grande P, Schnohr P, Tybjaerg‐Hansen A. PCSK9 R46L, low‐density lipoprotein cholesterol levels, and risk of ischemic heart disease: 3 independent studies and meta‐analyses. Journal of the American College of Cardiology 2010;55(22):2833‐42. [DOI] [PubMed] [Google Scholar]
- Benn M, Watts GF, Tybjaerg‐Hansen A, Nordestgaard BG. Familial hypercholesterolemia in the Danish general population: prevalence, coronary artery disease, and cholesterol‐lowering medication. Journal of Clinical Endocrinology and Metabolism 2012;97(11):3956‐64. [DOI] [PubMed] [Google Scholar]
- Bradburn MJ, Deeks JJ, Berlin JA, Localio AR. Much ado about nothing: a comparison of the performance of meta‐analytical methods with rare events. Statistics in Medicine 2007;26(1):53‐77. [DOI] [PubMed] [Google Scholar]
- Cannon CP, Blazing MA, Giugliano RP, McCagg A, White JA, Theroux P, et al. Ezetimibe added to statin therapy after acute coronary syndromes. New England Journal of Medicine 2015;372(25):2387‐97. [DOI] [PubMed] [Google Scholar]
- Capewell S, Allender S, Critchley J, Lloyd‐Williams F, O'Flaherty M, Rayner M, et al. Modelling the UK burden of cardiovascular disease to 2020: a research report for the Cardio & Vascular Coalition and the British Heart Foundation. British Heart Foundation, 2008. http://www.healthimpact.org.uk/Home/Links (accessed 01 February 2015).
- Catapano AL, Papadopoulos N. The safety of therapeutic monoclonal antibodies: implications for cardiovascular disease and targeting the PCSK9 pathway. Atherosclerosis 2013;228(1):18‐28. [DOI] [PubMed] [Google Scholar]
- Catapano AL, Graham I, Backer G, Wiklund O, Chapman MJ, Drexel H, et al. 2016 ESC/EAS Guidelines for the Management of Dyslipidaemias. European Heart Journal 2016;Ahead of print:1‐72. [Google Scholar]
- Chasman DI, Giulianini F, MacFadyen J, Barratt BJ, Nyberg F, Ridker PM. Genetic determinants of statin‐induced low‐density lipoprotein cholesterol reduction: the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) trial. Circulation. Cardiovascular Genetics. 2012;5(2):257‐64. [DOI] [PubMed] [Google Scholar]
- Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nature Genetics 2005;37(2):161‐5. [DOI] [PubMed] [Google Scholar]
- Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. New England Journal of Medicine 2006;354(12):1264‐72. [DOI] [PubMed] [Google Scholar]
- Collins R, Reith C, Emberson J, Armitage J, Baigent C, et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet 2016;Ahead of print:1‐30. [DOI] [PubMed] [Google Scholar]
- Cholesterol Treatment Trialists' (CTT) Collaborators. Efficacy and safety of cholesterol‐lowering treatment: prospective meta‐analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005;366(9493):1267‐78. [DOI] [PubMed] [Google Scholar]
- Cholesterol Treatment Trialists' (CTT) Collaboration, Fulcher J, O'Connell R, Voysey M, Emberson J, Blackwell L, et al. Efficacy and safety of LDL‐lowering therapy among men and women: meta‐analysis of individual data from 174 000 participants in 27 randomised trials. Lancet 2005;385(9976):1397‐405. [DOI] [PubMed] [Google Scholar]
- Cholesterol Treatment Trialists' (CTT) Collaborators, Mihaylova B, Emberson J, Blackwell L, Keech A, Simes J, et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta‐analysis of individual data from 27 randomised trials. Lancet 2012;380(9841):581‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnier M. PCSK9: From discovery to therapeutic applications. Archives of Cardiovascular Disease 2014;107(1):58‐66. [DOI] [PubMed] [Google Scholar]
- Food and Drug Administration Center for Drug Evaluation and Research. Briefing document Praluent (alirocumab) injection. The Endocrinologic and Metabolic Drugs Advisory Committee Meeting 09‐06‐2015; Vol. BLA 125559.
- Ference BA, Robinson JG, Brook RD, Catapano AL, Chapman J, Neff DR, et al. Variation in PCSK9 and HMGCR and risk of cardiovascular disease and diabetes. New England Journal of Medicine 2016;22:2144‐53. [DOI] [PubMed] [Google Scholar]
- Garcia CK, Wilund K, Arca M, Zuliani G, Fellin R, Maioli M, et al. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science 2001;292(5520):1934‐8. [DOI] [PubMed] [Google Scholar]
- GRADE Working Group. Grading quality of evidence and strength of recommendations. BMJ 2004;328(1):1490‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy SM, Cleeman JI, Merz CN, Brewer HB Jr, Clark LT, Hunninghake DB, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III Guidelines. Journal of the American College of Cardiology 2004;44(3):720‐32. [DOI] [PubMed] [Google Scholar]
- Higgins JPT, Altman DG, Sterne JAC. Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. www.cochrane‐handbook.org.
- Innerarity TL, Weisgraber KH, Arnold KS, Mahley RW, Krauss RM, Vega GL, et al. Familial defective apolipoprotein B‐100: low density lipoproteins with abnormal receptor binding. Proceedings of the National Academy of Sciences 1987;84:6919‐6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles JW, O'Brein EC, Greendale K, Wilemon K, Genest J, Sperling LS, et al. Reducing the burden of disease and death from familial hypercholesterolemia: a call to action. American Heart Journal 2014;168(6):807‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreatsoulas C, Anand SS. The impact of social determinants on cardiovascular disease. Canadian Journal of Cardiology 2010;26(Suppl C):8C‐13C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthi RV, Feigin VL, Forouzanfar MH, Mensah GA, Connor M, Bennett DA, et al. Global and regional burden of first‐ever ischaemic and haemorrhagic stroke during 1990‐2010: findings from the Global Burden of Disease Study 2010. Lancet Global Health 2013;1(5):e259‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. www.cochrane‐handbook.org. [Google Scholar]
- Lipinsky MJ, Benedetto U, Escarcega RO, Biondi‐Zoccai G, Lhermusier T, Baker NC, et al. The impact of proprotein convertase subtilisin‐kexin type 9 serine protease inhibitors on lipid levels and outcomes in patients with primary hypercholesterolaemia: a network meta‐analysis. European Heart Journal 2016;37(6):536‐45. [DOI] [PubMed] [Google Scholar]
- Lotta LA, Sharp SJ, Burgess S, Perry JR, Stewart ID, Willems SM, et al. Association between low‐density lipoprotein cholesterol–lowering genetic variants and risk of type 2 diabetes. JAMA 2016;13:1383‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundh A, Lexchin J, Mintzes B, Schroll JB, Bero L. Industry sponsorship and research outcome. Cochrane Database of Systematic Reviews 2017, Issue 2. [DOI: 10.1002/14651858.MR000033.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunven C, Paehler T, Poitiers F, Brunet A, Rey J, Hanotin C, et al. A randomized study of the relative pharmacokinetics, pharmacodynamics, and safety of alirocumab, a fully human monoclonal antibody to PCSK9, after single subcutaneous administration at three different injection sites in healthy subjects. Cardiovascascular Therapy 2014;32(6):297‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini GBJ, Baker S, Bergeron J, Fitchett D, Frohlich J, Genest J, et al. Diagnosis, prevention, and management of statin adverse effects and intolerance: proceedings of a Canadian Working Group Consensus Conference. Canadian Journal of Cardiology 2011;27(5):635‐62. [DOI] [PubMed] [Google Scholar]
- Marks D, Thorogood M, Neil HAW, Humpries SE. A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemia. Atherosclerosis 2003;168(1):1‐14. [DOI] [PubMed] [Google Scholar]
- Moran AE, Forouzanfar MH, Roth GA, Mensah GA, Ezzati M, Flaxman A, et al. The global burden of ischemic heart disease in 1990 and 2010: the Global Burden of Disease 2010 study. Circulation 2014;129(14):1493‐501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray CJ, Vos T, Lozano R, Naghavi M, Flaxman AD, Michaud C, et al. Disability‐adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990‐2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012;380(9859):2197‐223. [DOI] [PubMed] [Google Scholar]
- Navarese EP, Kołodziejczak M, Schulze V, Gurbel PA, Tantry U, et al. Effects of proprotein convertase subtilisin/kexin type 9 antibodies in adults with hypercholesterolemia: a systematic review and meta‐analysis. Annals of Internal Medicine2015; Vol. 163, issue 1:40‐51. [DOI: 10.7326/M14-2957] [DOI] [PubMed]
- Newcombe RG, Bender R. Implementing GRADE: calculating the risk difference from the baseline risk and the relative risk. Evidence‐Based Medicine 2014;19(1):6‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen SE, Stroes E, Dent‐Acosta RE, Rosenson RS, Lehman SJ, Sattar N, et al. Efficacy and tolerability of evolocumab vs ezetimibe in patients with muscle‐related statin intolerance: the GAUSS‐3 randomized clinical trial. JAMA 2016;315(15):1580‐90. [DOI] [PubMed] [Google Scholar]
- Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, et al. Familial hypercholesterolaemia is underdiagnosedand undertreated in the general population:guidance for clinicians to prevent coronary heart disease. European Heart Journal 2013;34(45):3478‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfizer. Pfizer discontinues global development of bococizumab, its investigational PCSK9 inhibitor. http://www.pfizer.com/news/press‐release/press‐release‐detail/pfizer_discontinues_global_development_of_bococizumab_its_investigational_pcsk9_inhibitor (accessed 24 April 2017).
- R Development Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing, 2014.
- Roger VL, Go AS, Lloyd‐Jones DM, Adams RJ, Berry JD, Brown TM, et al. Heart disease and stroke statistics ‐ 2011 update: a report from the American Heart Association. Circulation 2011;123(4):e18‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatine MS, Giugliano RP, Wiviott SD, Raal FJ, Blom DJ, Robinson J, et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. New England Journal of Medicine 2015;372(16):1500‐9. [DOI] [PubMed] [Google Scholar]
- Sattar N, Preiss D, Murray HM, Welsh P, Buckley BM, Craen AJ, et al. Statins and risk of incident diabetes: a collaborative meta‐analysis of randomised statin trials. Lancet 2010;375(9716):735‐42. [DOI] [PubMed] [Google Scholar]
- Schmidt AF, Hoes AW, Groenwold RH. Comments on 'the use of propensity scores and observational data to estimate randomized controlled trial generalizability bias' by Taylor R. Pressler and Eloise E. Kaizar, Statistics in Medicine 2013. Statistics in Medicine 2014;33(3):536‐7. [DOI] [PubMed] [Google Scholar]
- Schmidt AF, Pearce LS, Wilkins JP, Overington JP, Hingorani A, Casas JP. PCSK9 monoclonal antibodies for the primary and secondary prevention of cardiovascular disease. Cochrane Database of Systematic Reviews 2015, Issue 6. [DOI: 10.1002/14651858.CD011748] [DOI] [Google Scholar]
- Schmidt AF, Klungel OH, Nielen M, Boer A, Groenwold RHH, Hoes AW. Tailoring treatments using treatment effect modification. Pharmacoepidemiology and Drug Safety 2016;25(4):355‐62. [DOI] [PubMed] [Google Scholar]
- Schmidt AF, Swerdlow DI, Holmes MV, Patel RS, Fairhurst‐Hunter Z, Lyall DM, et al. PCSK9 genetic variants and risk of type 2 diabetes: a mendelian randomisation study. The Lancet Diabetes & Endocrinology 2017;5:97‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Jasmin SB, Stifani S, et al. The secretory proprotein convertase neural apoptosis‐regulated convertase 1 (NARC‐1): liver regeneration and neuronal differentiation. Proceedings of the National Academy of Sciences of the United States of America 2003;100(3):928‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, Goldstein JL, Brown MS, Russell DW. The LDL receptor gene: a mosaic of exons shared with different proteins. Science 1985;228(4701):815‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeting MJ, Sutton AJ, Lambert, PC. What to add to nothing? Use and avoidance of continuity corrections in meta‐analysis of sparse data. Statistics in Medicine 2004;23(1):1351‐75. [DOI] [PubMed] [Google Scholar]
- Swerdlow DI, Preiss D, Kuchenbaecker KB, Holmes MV, Engmann JE, Shah T, et al. HMG‐coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. Lancet 2014;385(9965):351‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SG, Sharp SJ. Explaining heterogeneity in meta‐analysis: a comparison of methods. Statistics in Medicine 1999;18(20):2693‐708. [DOI] [PubMed] [Google Scholar]
- Thompson SG, Higgins JP. How should meta‐regression analyses be undertaken and interpreted?. Statistics in Medicine 2002;21(11):1559‐73. [DOI] [PubMed] [Google Scholar]
- Vickers AJ, Altman DG. Statistics notes: analysing controlled trials with baseline and follow up measurements. BMJ 2001;323(7321):1123‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. The global burden of disease: 2004 update. http://www.who.int/healthinfo/global_burden_disease/2004_report_update/en/ (accessed 01 February 2015).
- Global Lipids Genetics Consortium, Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, et al. Discovery and refinement of loci associated with lipid levels. Nature Genetics 2013;45(11):1274‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XL, Zhu QQ, Zhu L, Chen JZ, Chen QH, Li QN, et al. Safety and efficacy of anti‐PCSK9 antibodies: a meta‐analysis of 25 randomized, controlled trials. BMC Medicine 2015;13(123):1‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]