

Abstract
Lentiviral vector mobilization following HIV-1 infection of vector-transduced cells poses biosafety risks to vector-treated patients and their communities. The self-inactivating (SIN) vector design has reduced, however, not abolished mobilization of integrated vector genomes. Furthermore, an earlier study demonstrated the ability of the major product of reverse transcription, a circular SIN HIV-1 vector comprising a single-LTR to support production of high vector titers. Here, we demonstrate that configuring the internal vector expression cassette in opposite orientation to the LTRs abolishes mobilization of SIN vectors. This additional SIN mechanism is in part premised on induction of host PKR response to double-stranded RNAs comprised of mRNAs transcribed from cryptic transcription initiation sites around 3’SIN-LTR’s and the vector internal promoter. As anticipated, PKR response following transfection of opposite orientation vectors, negatively affects their titers. Importantly, shRNA-mediated knockdown of PKR rendered titers of SIN HIV-1 vectors comprising opposite orientation expression cassettes comparable to titers of conventional SIN vectors. High titer vectors carrying an expression cassette in opposite orientation to the LTRs efficiently delivered and maintained high levels of transgene expression in mouse livers. This study establishes opposite orientation expression cassettes as an additional PKR-dependent SIN mechanism that abolishes vector mobilization from integrated and episomal SIN lentiviral vectors.
Introduction
Recent successes of lentiviral vector-based gene replacement1, 2 and immunotherapy3, 4 at transmuting the pathologic course of human genetic and malignant proliferative diseases opened a new age in medicine. However, potential vector mobilization following infection of vector-transduced cells with HIV-1 poses biosafety risks to vector-treated patients and their communities. The development of self-inactivating (SIN) vectors significantly reduced, yet not abolished mobilization of integrated SIN vector genomes. The concept of SIN retroviral vectors is premised on deletion of the parental HIV-1 promoter and enhancer sequences from the 3’U3 in the vector cassette. The deleted 3’U3 is copied in the process of reverse-transcription to the 5’U3. Consequently, reverse-transcribed SIN vector genomes devoid of the parental enhancer promoter sequences should not support transcription of vector length mRNA. Although the first SIN vectors were developed by Yu et al as a means to enhance biosafety of ɣ-retroviral vectors 5 Shinya et al were the first to describe a SIN lentiviral vector.6 The first efficient SIN lentiviral vector system was developed and successfully employed by Miyoshi et al to transduce rat retina and brain tissues.7 However, later studies reported on residual SIN vector mobilization, due to initiation of aberrant transcription of vector length mRNA from SIN LTRs.8–10 This phenomenon was attributed to HIV-1 sequences within the vector’s packaging signal 9 and to host chromatin structures and regulatory elements in proximity to integrated vector genomes.8 The majority of lentiviral vector genomes shortly after transduction comprises episomal linear and circular (containing either one or two LTRs) DNAs. However, the ability of episomal lentiviral genomes to support vector mobilization has not been studied. Furthermore, in an earlier study, Ma et al employed a circular SIN vector genome comprising a single LTR to produce high titer lentiviral vectors.11 Concerned by these data, we sought to abolish residual vector mobilization from integrated and episomal vector genomes by incorporating an additional level of safety to the currently used SIN lentiviral vector system. We reasoned that any additional biosafety measure to eliminate vector mobilization should be mechanistically independent and thus, synergistic to the current SIN vector design in preventing vector mobilization. We theorized that configuring the vector internal expression cassette in opposite orientation (ECOO) to the SIN LTRs would minimize aberrant transcription from SIN LTRs (mediated by either the vector internal promoter11, parental HIV-1 sequences,9 or host regulatory elements8) and induce host Protein kinase-R (PKR) response to double stranded RNAs comprising LTR- and internal promoter-initiated transcripts. Indeed, here, we demonstrate dramatic reduction of vector mobilization from integrated and episomal lentiviral vector genomes comprising ECOO to the LTRs. Similar to earlier publications, lentiviral vectors carrying ECOO to the LTRs exhibited low titers12, 13. However, stable shRNA-mediated knockdown of PKR in vector producing cells rendered titers of the novel mobilization resistant vectors comparable to titers of their counterpart vectors, comprising expression cassettes in the same orientation to the LTRs. High titer mobilization resistant SIN vectors efficiently delivered and maintained firefly luciferase expression in mouse livers. Furthermore, we report here on efficient production of a lentiviral vector carrying an ECOO encoding a modified human β-globin protein under the regulation of the β-globin locus control region13. This study elucidates the mechanism of SIN vector mobilization, and establishes a highly efficient methodology of generating mobilization-resistant lentiviral vectors comprising expression cassettes in opposite orientation to the LTRs.
Methods and Materials
Cells
SODK014 and 293T cells were maintained in DMEM-High Glucose (Thermo Scientific, Waltham, MA) supplemented with 10% FBS (Atlantic biologicals, Miami, FL), 100 U/ml penicillin, 100 µg/ml streptomycin, and 250 ng/ml amphotericin B (Corning Cellgro, Manassas, VA). The SODK1 cell line 14 and all its derivatives were cultured in the pre-mentioned medium with the presence of 1 µg/ml Doxycycline (Dox) . HepG2 cells were cultured in MEM (Gibco, Gaithersburg, MD) medium with 10% FBS and antibiotics. shRNA’s targeting the hPKR and hβ-globin(HBB) genes in 293T cells were delivered by lentiviral vectors obtained from the TRC1 library. These include clones: TRCN0000196400 (pKR sh1), TRCN0000197012 (PKR sh2) and TRCN000002909 (HBB). Vector transduced 293T cells were selected with 1µg/ml puromycin (Invivogen, San Diego, CA) for two continuous weeks. PKR knockdown was confirmed by Western blot.
Plasmids
The lentiviral vector pTK485, pTK945, pTK979 and the packaging cassettes ΔNRF, pTK939 (Int-), VSV-G envelope, pMDL and the Rev plasmids, were described earlier 15–18. The Tat plasmid was a kind gift from Dr. Thomas Hope. All other lentiviral vector plasmids were generated by standard restriction enzyme digestion and ligation (New England Biolabs, Ipswich, MA). To simplify the cloning procedure, we synthesized a plasmid pTK1866 containing BamHI-cPPT-opposite orientated BGH - multiple cloning site (MCS) - WPRE- LTR-PmeI. BGH was flanked by BstZ17I, WPRE was flanked by ClaI to make future modification feasible. The expression cassette of CMV-GFP in pTK945 was generated by EcoRI/SpeI, blunted with DNA Polymerase Klenow Fragment, and inserted into HpaI located within MCS of pTK1866 with opposite orientation. The WPRE region was removed by ClaI digestion and re-ligation. The BGH in the aforementioned plasmid was removed by BstZ17I, and exchanged with SV40 Polyadenylation from pCI-neo by SmaI/FspI double digestion. The region (cPPT- SV40 op-GFP-CMV -WPRE-LTR) was digested by BamHI/ PmeI to substitute their counterparts in pTK945. To generate pTK1940 (GenBank accession MH297436), a ClaI fragment comprising the WPRE (nt 2908-3496 in pTK1940 and pTK1956) sequence was inserted in opposite orientation to the LTRs into a BsiWI site downstream to the SV40 polyA. pTK 1956 (GenBank accession MH297437) was created by replacing CMV-GFP expression cassette (XhoI/AscI) with hAAT-luciferase (AfeI/XhoI) from pTK979. All the unmatched overhung enzymatic digestion sites were blunted by DNA Polymerase I, Klenow Fragment, before ligation. All plasmids were confirmed by sequencing and restriction enzyme digestion.
Viral vector production and concentration
Lentiviral vector production via 3 plasmids transient transfection was described earlier. 16 For animal studies, viral vectors were concentrated by sucrose ultracentrifugation as described earlier. 19 Absence of replication competent retroviruses (RCR’s) was verified by three independent safety assays (GFP rescue assay, Tat transfer assay and Gag transfer assay) as described earlier. 14
Viral vector titration
Titers of vector transducing units (TU/ml) were determined by scoring GFP positive cell number under fluoresce microscope following serial dilutions on 293T cells. Titers of physical vector particles were determined by p24gag ELISA.20 Vector copy number (VCN) was determined by multiplex PCR using the ABI7300 realtime PCR system. NotI794 primer/prober set (F5’- taagaccaccgcacagca-3’; R5’- cacttctccaattgtccctca-3’; Roche Universal Probe Library (UPL) #25, 4686993001, Indianapolis, IN) was used for vector detection, and paired with human RNaseP primer/probe set (Thermo Scientific 4316844,) or mouse GAPDH (Roche 05190525001) as a reference gene.21
Western blot analysis
Naïve, PKR knocked down and β-Globin knocked down 293T cells were lysed in RIPA buffer. Protein samples were separated on 10% SDS–PAGE denaturing gels. Blots were detected with PKR antibody (1:1000, Cell signaling Technology, Cat# 3072S, Danvers, MA) or β-actin antibody (1: 1000,Santa Cruz, Cat# sc-1616, Dallas, TX) followed by a polyclonal goat anti-rabbit secondary Ab labeled with horseradish peroxidase (1:10,000; Pierce, Grand Island, NY ) treated with ECL (GE Health Amersham, Pittsburgh, PA), and imaged by the ChemiDoc MP Imaging System (BioRad, Hercules, CA).
FACS analysis
Target cells were harvested at 48 hours post-transduction with GFP expressing vectors. Harvested cells were washed with PBS, fixed with 2% paraformaldehyde for 15 minutes, and re-suspended in MACS buffer (2mM EDTA and 1%BSA in PBS). Fluorescence-activated cell sorter (FACS) analysis was performed with CyAn ADP (Becton–Dickinson). Percentage of GFP positive cell and mean fluorescence intensity (MFI) were analyzed with the FlowJo9.3.2 (Flowjo LLC, Ashland, OR) software.
Mobilization of integration deficient lentiviral vectors (IDLV) from the inducible packaging cell line SODK1
The inducible packaging cell line SODK114 was employed to evaluate IDLV mobilization. To induce vector production, SODk1 cells were washed with PBS (X3), and passaged onto poly-lysine (Sigma, St Loius, MO) pre-coated plates and cultured in doxycycline (Dox)-free media. At 5 days post-Dox withdrawal, 15 million cells were seeded onto 10cm plate for IDLV mobilization analysis. On day 6-post Dox withdrawal, 2×108 TU of IDLV were employed on the induced SODK1 cells in the presence of 5mM sodium butyrate. At twelve hours post-transduction, cells were washed and cultured with Dox-free fresh media containing 5mM sodium butyrate. Vector particles containing media were collected at 72 hours after addition of sodium butyrate. IDLV transduced SODK1 cells cultured in the presence of Dox (1µg/µl) and sodium butyrate (5mM) media served as controls. Mobilized vector particles were employed on 2-million naïve 293T target cells. To eliminate carrying over of non-mobilized IDLVs, titers of mobilized ICLVs (generated by the induced packaging cells) were determined after 5 passages of the above target cells in culture. Percent of GFP-positive cells and the number target cells at the time of transduction were used to calculate vector titers.
Mobilization of integration competent lentiviral vector (ICLV) from the inducible packaging cell line SODK1
SODK1 cells were transduced with ICLVs particles at an m.o.i. of 10. To dilute out episomal vector genomes, transduced SODk1 cells were passaged in culture (X5). To initiate vector production, transduced SODK1 cells were washed with PBS (X3), and passaged onto poly-lysine coated plates in Dox-free media. At 5 days post-Dox withdrawal, 15 million cells were seeded onto 10cm plate. Sodium butyrate (5mM) was added to culture media at day 6 post-Dox withdrawal. Vectors particles containing media were collected at 72 hours after addition of sodium butyrate. IDLV transduced SODK1 cells cultured in the presence of Dox (1µg/µl) and sodium butyrate (5mM) media served as controls. Titers (TU/ml) of mobilized vectors were determined by scoring GFP expression following serial dilutions on 293T cells.
Luciferase assay
Vector transduced HepG2 cells were lysed at 72 hr post-transduction and quantified with 1420 Multilabel Counter Victor 3 (PerkinElmer Waltham, MA) following manufacturers instruction of Luciferase Assay system (Promega Madison, WI). Relative Light Units (RLU) were further normalized with protein concentration and viral copy number (VCN per cell). Protein concentration was determined by the Braford method.
Animal studies
All animal cares and procedures were in accordance with the Guide for the Care and Use of Laboratory Animals and received prior approval by the UNC Animal Care and Usage Committee (Animal protocol 15-123.0). C57BL/6J female mice at age 5~6 week old were obtained from Jackson Lab. One week after animal arrival, 25 µg p24gag lentiviral vectors were administrated into each mouse via intraperitoneal injection. In vivo expression of vector-delivered firefly luciferase in live animals was determined at weeks 2, 4, 6 and 8 using IVIS Lumina optical system (PerkinElmer, Waltham, MA), as described previously. 21 Shortly, mice were injected with 250µl luciferin substrate (25mg/ml, Regis Technology, Grove, IL) via I.P. After 10 minutes incubation, mice were imaged for 5 minutes with IVIS imagine system.
Statistical analysis
Student’s T test in Excel software was used to determine the significance of in vitro assay of vector pTK 979 and pTK 1956.
Results
A circular SIN single-LTR vector cassette supports Tat independent production of high titer lentiviral vectors
Mobilization of HIV-1 based vectors following infection of vector-transduced cells with HIV and consequential spread of vector particles in vector-treated patients and their communities are major biosafety concerns. Using cell lines comprising integrated SIN vector genomes, several groups demonstrated significant reduction yet not complete elimination of lentiviral vector mobilization.8, 9 Although circular single-LTR vector genomes are a major product of lentiviral vector reverse transcription15, 22–24, their ability to mobilize packageable vector genomes has not been evaluated. Furthermore, in an earlier study, Ma et al., demonstrated that a DNA construct encoding a circular SIN single-LTR vector supported production of high vector titer 11. Since the precursor of the aforementioned shuttle SIN vector described by Ma et al., was isolated from lentiviral vector-transduced cells, we further studied the mechanism of SIN vector mobilization. Specifically, we characterized the role of HIV-1 Tat protein in mobilization of circular SIN single-LTR genomes pTK485 (Figure 1A and supplement figure 1), which are naturally generated in vector-transduced cells15, 22–24. To this end, vTK485 particles were produced by a transient four-plasmid transfection procedure, either in the presence or absence of the HIV-1 Tat protein. As shown in Figure 1B, production of high titer (3-4E+6 IU/ml) SIN vectors from a SIN single-LTR expression cassette (pTK485) was Tat-independent. These findings are in line with an earlier report by Poon et al.25 Vector mobilization from integrated SIN vector genomes was attributed to aberrant transcription initiation induced by either enhancer sequences located in the vector packaging signal or to host sequences in proximity to the vector integration site 8, 9. However, the high titers of SIN vectors generated from pTK485 indicated that a different or additional mechanism contributes to the highly efficient Tat-independent vector production from circular SIN vector genomes. Importantly, the process of vector circularization, locates the internal promoter upstream to the SIN LTR. Thus, we hypothesize that transcriptional activity induced by the internal promoter is the major contributor to episomal SIN vector mobilization. Furthermore, we theorized that positioning the internal expression cassette in opposite orientation to the LTR’s would significantly minimize internal promoter mediated mobilization of circularized episomal vectors.
Figure 1. A vector cassette comprising a single-SIN LTR supports production of high titer lentiviral vectors.
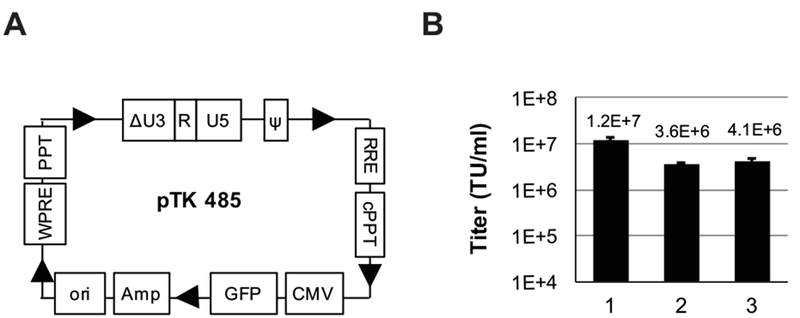
(A) Depiction of the single-SIN LTR vector pTK485 showing the green fluorescence protein (GFP) marker gene under the control of the cytomegalovirus immediate early promoter (CMV) promoter. The pUC Ori bacterial origin of replication (Ori) and the ampicillin resistance gene (Amp) in pTK485 facilitate transfer of reverse transcribed episomal vector genomes from vector transduced cells to bacteria.11 Additional cis-acting elements include the Rev-response element (RRE), the central polypurine tract (cPPT), the woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) and the 3’ polypurine tract (PPT). Arrows indicate the direction of transcription. (B) Titers of VSV-G pseudotyped pTK485 particles generated by transient transfection in 293T cells using different packaging cassettes. Lane 1 packaging by a second-generation packaging cassette (Delta-NRF) deleted of the HIV-1 vpr, nef and vif genes. Lanes 2 and 3; packaging by a third generation-packaging system comprising the gag-pol and rev gene expression cassettes (pMDL and pE2f-Rev, respectively) either without (lane 2) or with (lane 3) the HIV-1 Tat expression cassette (pSV-Tat). Transducing unit titers (TU/ml) were determined by scoring GFP positive cells following serial dilution on 293T cells at 72 hr post- transduction. Titers are represented as average of 3 independent experiment ± SD (Standard deviation).
Internal expression cassettes in opposite orientation to the LTR’s reduce production of lentiviral vector particles and vector titers
To reduce the likelihood of lentiviral vector mobilization, an internal expression cassette comprising the bovine growth hormone (BGH) polyadenylation (poly-A) site, a CMV promoter, and the green fluorescence protein (GFP) reporter gene was positioned in an opposite orientation to the SIN vector LTR’s (pTK1906 in Figure 2A). Production efficiency of pTK1906 by transient three-plasmid transfection was compared to production of its conventional counterpart vector (pTK945). As shown in figure 2B-C, positioning an internal expression cassette in opposite orientation to the LTRs dramatically reduced transducing- and physical- vector titers. Furthermore, the reduction in concentration of vector particles in culture media (as determined by p24gag ELISA) suggested that the opposite orientation expression cassette in the vector construct probably induced a host PKR response that effectively inhibited synthesis of viral structural proteins encoded by the co-transfected packaging cassettes.
Figure 2. Knockdown of PKR expression in vector producing cells minimizes the negative effects of opposite orientation expression cassettes on lentiviral vector titers.
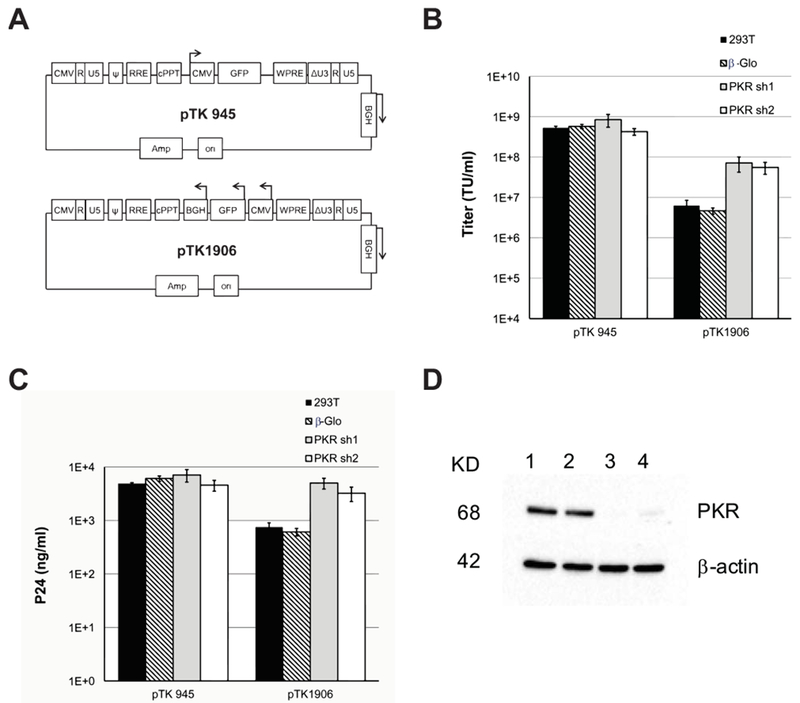
(A) Depiction of the SIN vectors pTK945 and pTK1906 carrying GFP expression cassettes in different orientations to the vector LTR’s. Arrows indicate the direction of transcription from the relevant internal promoters. (B) Bar-graph showing titers (TU/ml) of conventional and ECOO-comprising lentiviral vectors generated in naïve or PKR deficient cells. The conventional and ECOO-comprising vectors pTK945 and pTK1906 (respectively) were packaged by transient transfection in either naïve 293T cells (black bars) or following stable shRNA-mediated knockdown of PKR expression. Two different shRNA target clones (PKR sh1, PKR sh2) were employed to rule out shRNA off-target effects (PKR sh1-grey bar and PKR sh2-white bar). Vectors generated in 293T cells expressing β-globin-directed shRNA served as an additional negative control (diagonal lines). The vectors were generated in 3 independent experiments (n=3). Vector titers (TU/ml) were measured by scoring GFP expression following serial dilutions on 293T cells, and represented as average ± SD. (C) Concentration of physical vector particles in conditioned media was determined by p24gag ELISA. (D) Western blot analysis of PKR expression in the aforementioned four vector producing cell lines including naïve 293T cells (lane 1), cells expressing β-globin-directed shRNA (lane 2), cells expressing PKR-directed shRNA’s (PKR sh1-lane 3 and PKR sh2-lane 4). Probing with β-actin-directed antibody served as loading control.
PKR deficient packaging cells support efficient production of ECOO containing lentiviral vectors
To study the effects of the host PKR response on titers of ECOO comprising vectors, U6-shRNA expression cassettes carried by lentiviral vectors were employed to effectively knock down PKR expression in 293T cells (Figure 2D). Conventional and ECOO-comprising vectors were generated in either naïve or PKR deficient 293T cells. As shown in figure 2B-C, PKR knockdown in vector producing cells resulted in a ten-fold increase in titers of ECOO-comprising vectors, with minimal effect on titers of conventional vectors. Next, we sought to delineate the mechanisms that induce PKR response following transfection of ECOO comprising vectors.
The origin and position of poly-A sites in the vector cassette affect lentiviral vector titers
An internal poly-A site is required to facilitate efficient transgene expression from vectors comprising ECOO’s. Hence, we sought to characterize the effects of poly-A sites positioned upstream and in opposite orientation to conventional internal expression cassettes (in same orientation to the LTRs) on vector titers (Figure 3A). To this end, SIN vectors comprising either internal BGH (pTK1588) or SV40 (pTK1595) poly-A sites were generated in either naïve or PKR deficient 293T cells. Vector titers were determined and compared with titers of their conventional counterpart vector (pTK945). Unexpectedly, titer analysis (Figure 3B) indicated that the origin of the internal poly-A site had a major effect on vector titers. Specifically, regardless of PKR expression in vector producing cells, titers of SIN vectors comprising an internal SV40 poly-A site (pTK1595) were comparable to titers of their parental vector (pTK945) lacking an internal poly-A site. Per contra, when generated in naïve 293T cells, titers of a vector comprising an internal BGH poly-A site (pTK1588) were more than a 100-fold lower than titers of either control vectors or same vectors generated in PKR deficient cells. Since none of the above vectors contains an ECOO, we theorized that transcription through the parental LTR poly-A produced a BGH poly-A sequence located downstream to the 3’LTR. Consequently, the transcribed full-length vector mRNA’s comprised two complementary BGH poly-A sites, which generated an intra-molecular double stranded RNA structure and induced a host PKR response that negatively affected vector titers. To test the above notion, we developed two SIN vectors lacking the downstream BGH poly-A site (Figure 3A). The novel two vectors, either, comprising or lacking the internal BGH poly-A site (pTK1850 and pTK1848, respectively) were generated in either naïve or PKR deficient cells. As shown in figures 3C-D, both vectors (pTK1850 and pTK1848) exhibited comparable titers, which were only marginally lower than the titers of a conventional SIN vector (pTK945) containing a BGH poly-A site downstream to the 3’LTR. These findings suggested that in the absence of a functional ECOO the mere presence of an internal poly-A site induces only a minor reduction in vector titers.
Figure 3. Effects of internal polyadenylation (poly-A) sites and transcriptional activity of ECOO on vector titers.
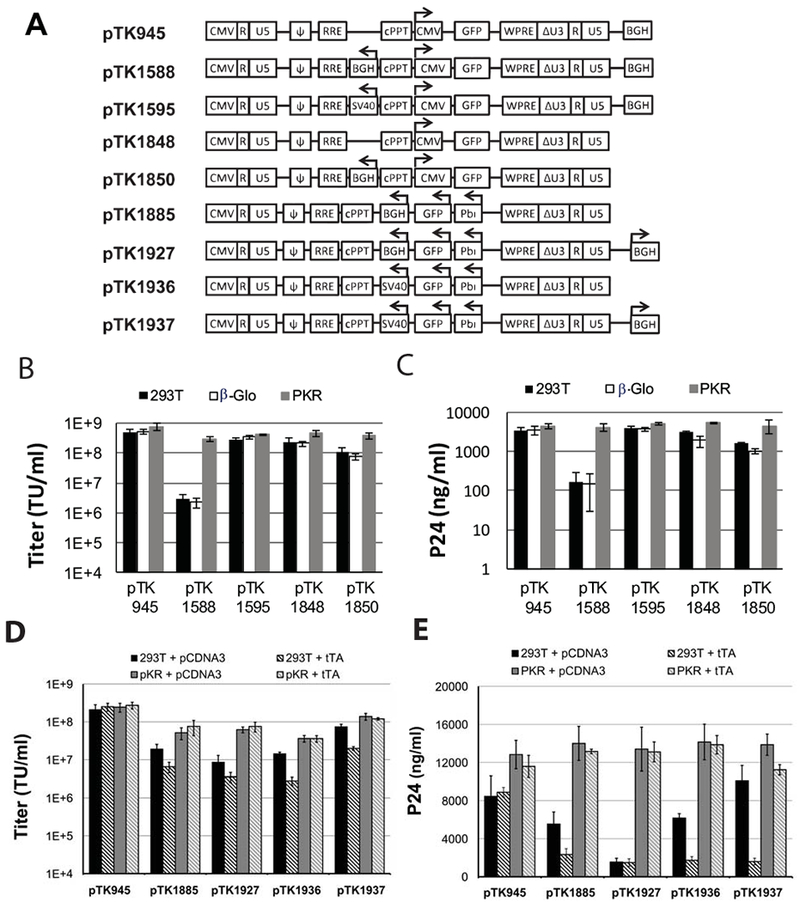
(A) Depiction of lentiviral vectors carrying poly-A signal sequences and internal promoters in different orientations. Arrows indicate the direction of transcription. BGH: bovine growth hormone poly-A signal; SV40: Simian virus 40 poly-A signal; pbi: tetracycline regulated promoter. (B) Bar graphs describing titers (TU/ml) and (C) physical particle concentration of the lentiviral vectors: pTK1588, pTK1595, pTK1848, pTK1850. Vectors were produced by transient transfection in 3 different 293T cell lines: naïve cells (black bars), β-globin knocked-down cells (white bars) and PKR knocked-down cells (grey bars). Titers of the conventional lentiviral vector pTK945 served as controls. Vectors were generated in 3 independent experiments (n=3), and represented as average ± SD. Titers (TU/ml) were determined by scoring GFP positive cells following serial dilutions on 293T cells. Concentrations of physical vector particles were determined by p24gag ELISA. (D-E) Bar-graphs describing vector titers and concentration of physical vector particles of lentiviral vectors carrying a tetracycline inducible promoter and either SV40 or BGH poly-A signals, including pTK1885, pTK1927, pTK1936, pTK1937 The vectors were generated in naïve (black and diagonal black lines bars) and PKR knockdown (grey and diagonal grey line bars) 293T cells, either in the presence (diagonal lines containing bars) or absence of the tetracycline-regulated trans-activator, tTA (black and grey bars). The conventional pTK945 vector served as control. Vectors were generated in 3 independent experiments (n=3), and titer was represented as average ± SD. (D) Vector titers were determined by scoring GFP positive cells following serial dilutions on SODK0 cells, which constitutively express tTA. (E) Concentrations of physical vector particles were determined by p24gag ELISA.
Transcription from ECOO reduces titers of SIN lentiviral vectors
Next, we sought to directly determine the effects of transcription from the ECOO at the time of vector production on vector titers. To this end, expression cassettes comprising an inducible tetracycline promoter and either a BGH or SV40 poly-A site were incorporated in opposite orientation to SIN vectors (Figure 3A: pTK1885, pTK1927 and pTK1936. pTK1937, respectively). Furthermore, to evaluate the combined effect of vector design and ECOO transcription on vector titers, a BGH poly-A site was incorporated downstream of the 3’LTR and compared to its unincorporated counterpart (Figure 3A: pTK1927, pTK1937 and pTK1885, pTK1936, respectively). Vector particles were generated by transient transfection in naïve and PKR deficient 293T cells either in the presence or absence of a tetracycline regulated trans activator (tTA). Vector titers were determined by scoring GFP expression following serial dilutions on 293- SODk0 cells, which constitutively express tTA.14 As shown in figures 3D-E, transcription from ECOO at the time of vector production (in the presence of tTA) significantly reduced vector titers generated in naïve 293T cells. The presence of two oppositely oriented BGH poly-A sites in pTK1927 further reduced its physical and transducing titers when generated in naïve 293T cells (Figure 3D-E). Per contra, transcription from ECOO and the presence of two BGH poly-sites in the vector cassettes had minimal effect on vector titers generated in PKR deficient 293T cells compared to the titers of a conventional SIN vector (pTK945). Note that transducing titers of vectors containing an internal SV40 poly-A site and a BGH poly-A site downstream to the 3’LTR (pTK1937) were higher than the titers of their counterpart vectors (pTK1936) that lack a BGH poly-A site downstream to the 3’LTR (Figure 3D). These data suggest that transcription from ECOO negatively affects vector titers generated in naïve 293T cells. Furthermore, the presence of a BGH poly-A site downstream to the 3’ LTR increases titers of conventional vectors, however, the presence of both an internal and an external (downstream to the 3’LTR) BGH poly-A sites significantly reduces titers of ECOO-comprising vectors.
Optimization of vector design to maximize titers of ECOO-comprising SIN vectors
To maximize titers of SIN vectors comprising ECOO, a series of vectors were developed to identify the optimal combination of poly-A sites within vector expression cassettes. (Figure 4A). Specifically, expression cassettes comprising either BGH (pTK1886 and pTK1906) or SV40 poly-A sites (pTK1907, 1909, 1908 and 1918) were cloned in opposite orientation to the LTR’s in vector constructs either containing or lacking a BGH poly-A site downstream to the 3’LTR. Note that the SV40 poly-A site was cloned either in the same (pTK1908 and pTK1918) or opposite (pTK1907 and pTK1909) orientation to the LTR’s. Vectors particles were generated in naïve and PKR deficient 293T cells. Concentration of physical vector particles and transducing vector titers were determined by p24gag ELISA and GFP expression scoring following serial dilutions on 293T cells, respectively. Levels of transgene expression in vector-transduced 293T cells were analyzed by FACScan analysis (supplemental figure 2). As shown in figures 4B-C, when generated in PKR deficient cells, the titers of pTK1909, a vector comprising an internal SV40 poly-A in opposite orientation to the LTRs and a BGH poly-A site downstream to the 3’LTR were comparable to the titers of a conventional SIN vector (pTK945), and more than 10-fold higher than the titers of the other ECOO vectors. These data suggest that in the absence of intra-molecular double stranded RNA structures (generated by two complementary BGH poly-A sequences), efficient shRNA-mediated knockdown of PKR facilitates production of high titer ECOO comprising vectors. The presence of a BGH poly-A site downstream to the 3’LTR further increases titers of vectors comprising an internal SV40 poly-A. Interestingly, internal SV40 poly-A sites positioned in opposite orientation to the internal CMV promoter (Figure 4A, pTK1908 and pTK1918) did not reduce transgene expression levels from ECOO (Figure 4D). Furthermore, transgene expression levels from all ECOO vectors were significantly lower than the level of expression from the conventional vector control, pTK945 (Figure 4D) indicating that addition of the woodchuck hepatitis virus post-transcriptional regulatory element (WPRE) to the internal expression cassette could improve the efficiency of ECOO comprising vectors.
Figure 4. Improved vector design enhances vector titers.
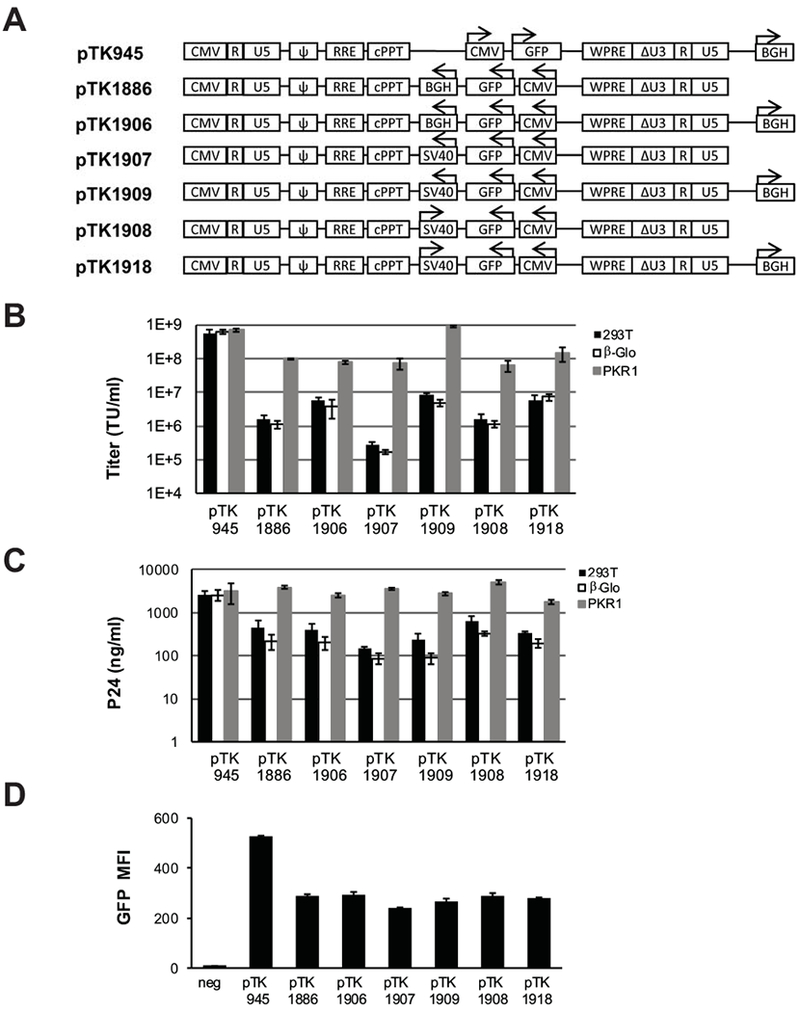
(A) Depiction of lentiviral vectors comprising SV40 and BGH poly-A sites in various positions and orientations to the LTR’s. Arrows indicate either the direction of transcription from the internal CMV promoter or the orientation of the parental SV40 poly-A signal. BGH: bovine growth hormone poly-A signal; SV40: Simian virus 40 poly-A signal; pbi: tetracycline regulated-promoter. (B-C) Bar graphs describing vector titers and concentrations of physical vector particles. Lentiviral vectors pTK1886, pTK1906, pTK1907, pTK1908, pTK1909, and pTK1918 were generated by transient transfection in 3 different 293T-based cell lines: naïve 293T cells (black bars), β-globin knockdown cells (white bars) and PKR1 knockdown cells (grey bars). The conventional pTK945 vector served as a positive control. Vectors were generated in 3 independent experiments (n=3), and represented as average ± SD. (B) Vector titers (TU/ml) were determined at 72hr post-transduction by scoring GFP positive cells following serial dilutions on 293T cells. (C) Concentration of physical vector particles at p24gag level was measured by ELISA, and represented as average ± SD. (D) FACscan analysis of transgene (GFP) expression levels. Vector transduced 293T cells were harvested for FACscan analysis at 72 hours post-transduction. Mean fluorescence intensity (MFI) was quantified with the FlowJo software. Error bars represent the standard deviation of 3 independent measures. To minimize the effect of multi-vector transduction events on transgene expression analysis, target cells were transduced at a relatively low m.o.i (<0.1). Consequently, a percent of GFP-positive cells was lower than 17.2%. Representative flow histograms are shown in supplemental figure 2.
Incorporation of the WPRE to ECOO’s increases levels of transgene expression
To enhance transgene expression from ECOO comprising vectors, a DNA sequence encoding the WPRE was positioned between the GFP reporter gene and the internal poly-A site (Figure 5A, pTK1940, GenBank accession MH297436). To address the concern that the lack of a WPRE sequence upstream to the 3’LTR would reduce vector titers, an additional WPRE sequence was incorporated between the 3’LTR and the BGH poly-A site (Figure 5A, pTK1954). Vector particles were generated either in naïve or PKR deficient 293T cells. Concentration of physical vector particles and vector titers were determined by p24gagELISA and by scoring GFP expression following serial dilutions on 293T cells (Figures 5B-C). Transgene (GFP) expression levels were determined by FACScan analysis. As shown in figures 5A and D, positioning a WPRE sequence between the transgene of interest (GFP) and the internal SV40 poly-A rendered transgene expression from ECOO (pTK1940 and pTK1954) comparable to the levels of expression obtained from a conventional vector control (pTK945). Interestingly, addition of a WPRE sequence between the 3’LTR and the BGH poly-A site (pTK1954) significantly reduced production of physical vector particles and vector titers generated in naïve 293T and to a lesser extent in PKR deficient cell lines cells (Figures 5C-D). Note that positioning a single WPRE sequence between the transgene of interest and the internal SV40 poly-A site (pTK1940) had no effect on production of physical vector particles nor on vector titers generated in PKR deficient cell lines.
Figure 5. Optimization of transgene expression from ECOO comprising vectors.
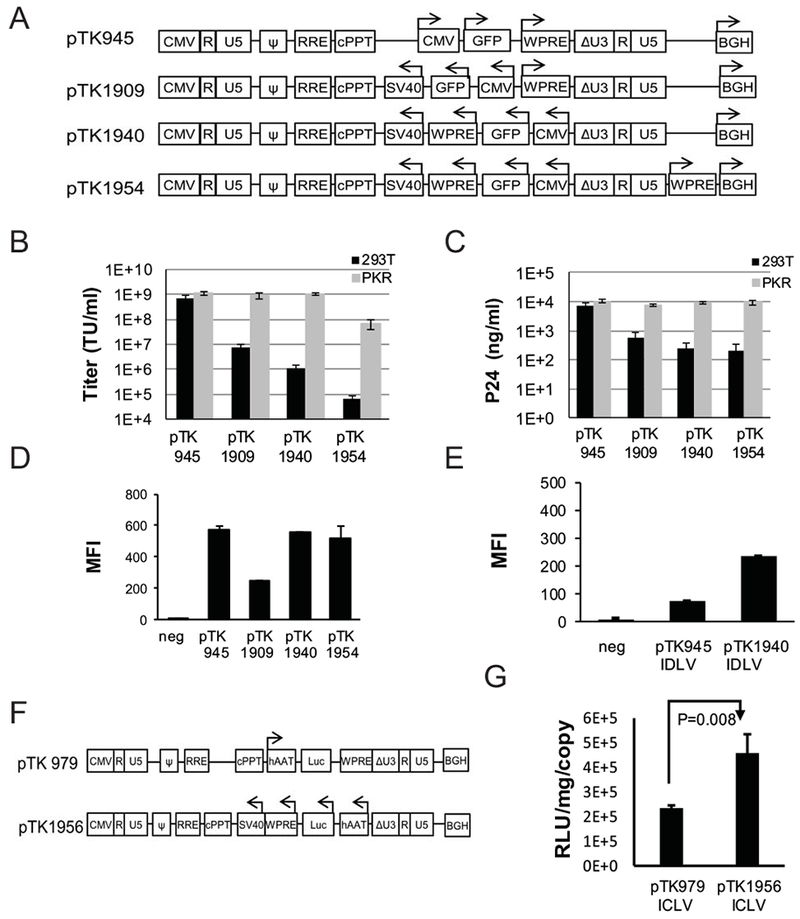
(A) Depiction of conventional and ECOO-comprising lentiviral vectors, pTK945, pTK1909, pTK1940 and pTK1954. Both vectors contain an internal WPRE (in opposite orientation to the LTR’s) between the transgene of interest (GFP) and the SV40 poly-A sequence. A second WPRE sequence in the same orientation to the LTR’s is positioned downstream to the 3’ LTR of pTK1954. Arrows indicate the direction of transcription from the internal CMV promoter or the orientation of the BGH poly-A and WPRE sequences positioned downstream to the 3’ LTR. (B-C) Bar graphs describing titers and concentrations of physical vector particles. Lentiviral vectors pTK1940 and pTK1954 were generated by transient transfection in either naïve or PKR deficient (PKR1 cells) 293T cells. The conventional vector, pTK945 and the WPRE-lacking vector pTK1909 served as positive and negative controls, respectively. (B) Transducing vector titers (TU/ml) were determined by scoring GFP-positive cells following dilutions on 293T cells. (C) Concentration of physical vector particles in conditioned media at 72h post transfection were determined by p24gag ELISA. (D) Bar graph describing FACscan analysis of GFP expression levels as MFI in 293T cells at 72hr post transduction with the ICLV’s pTK945, pTK1909, pTK1940 and pTK1954. The vectors were generated in PKR knockdown 293T cells. To minimize the effect of multi-vector transduction events on transgene expression analysis, target cells were transduced at a relatively low m.o.i (<0.1). Consequently, percentages of GFP-positive cells were lower than 19%. Representative flow histograms are shown in supplement figure 3. (E) Bar graph describing FACscan analysis of GFP expression levels as MFI in 293T cells at 72hr post transduction with the IDLV’s pTK945 and pTK1940. Naïve 293T served as a negative control. The vectors were generated in PKR knockdown 293T cells using an integrase deficient packaging cassette encoding the D64E integrase mutant. (F) Depiction of lentiviral vectors carrying firefly luciferase (Luc) expression cassettes driven by the liver specific, human alpha 1-antitrypsin (hAAT) promoter. Arrows indicate the direction of transcription from the internal hAAT promoter. (G) Bar graph describing efficiency of luciferase expression from the hAAT promoter at 72hr post transduction of HepG2 cells with conventional (pTK979) or ECOO-comprising vector pTK1956. Relative light units (RLU) were determined by luciferase assay and normalized to total protein concentration and vector copy number per cell (VCN) as determined by real time PCR. All data were represented as average ± SD of 3 independent experiments. P value (p=0.008) was determined by two tail student’s T test.
Superior transgene expression from integration deficient ECOO comprising lentiviral vectors
Intrigued by recent successful integration deficient lentiviral vectors (IDLV)-mediated gene delivery for vaccination and cancer immunotherapy in preclinical studies and human clinical trials, respectively4, 26, we sought to characterize the efficiency of transgene expression from integrase deficient ECOO comprising vectors. To this end, conventional and ECOO comprising vectors (pTK945 and pTK1940, respectively) were packaged into integrase-deficient vector particles and employed on 293T cells. Levels of transgene (GFP) expression were determined by FACScan analysis. As shown in figure 5E, transgene expression levels from ECOO comprising IDLV (pTK1940) were significantly higher than the levels of expression obtained from its conventional IDLV counterpart (pTK945).
Efficient transgene expression from a liver-specific promoter in ECOO comprising vectors
Several earlier studies demonstrated efficient liver transduction by lentiviral vectors carrying reporter and clinically relevant cDNAs 15, 21, 27. Hence, we sought to test the ability of ECOO comprising vectors to efficiently deliver and express a reporter gene under control of a liver specific promoter. To this end, an expression cassette comprising the liver specific promoter (hAAT), the firefly luciferase cDNA, the WPRE sequence and the SV40 poly-A was cloned in opposite orientation to the LTR’s (Figure 5F: pTK1956, GenBank accession MH297437). Vector particles of the above novel liver specific vector and its conventional counterpart (Figure 5F: pTK979) were generated by transient transfection14 and employed on the HepG2 cells. Levels of luciferase expression normalized to vector copy number were analyzed as described earlier. 21, 24 As shown in figure 5G, normalized expression levels were higher in HepG2 cells transduced with the ECOO comprising vectors than the normalized expression levels in similar cells transduced with a conventional vector.
Minimal mobilization of ECOO comprising vectors
To further characterize the suitability of ECOO comprising vectors for future clinical applications, we sought to compare their mobilization potential to that of conventional lentiviral vectors. ECOO comprising vectors (pTK1940) and their conventional SIN (pTK945) and non-SIN (pTK1087) counterparts (Figures 3A, 5A and 6A) were packaged by transient transfection into vector particles comprising either functional or mutant integrase (D64E) protein. Thus, generating integration competent and integration defective lentiviral vectors (ICLV and IDLV respectively). 15, 24, 27, 28 To specifically evaluate mobilization of episomal forms of the above vectors, IDLVs were employed on the tetracycline regulated, HIV-1 vector packaging cell line SODk1 14 either prior (in the presence of doxycycline) or after inducing (in the absence of doxycycline) production of VSV-G- pseudotyped vector particles . Titers of mobilized integrating vectors in conditioned media were determined by scoring GFP expression following serial dilutions on 293T cells. To eliminate potential carryover of the parental IDLV particles, the transduced reporter 293T cells were passaged 5 times prior to scoring GFP expression. As shown in figure 6B, titers of mobilized SIN IDLV (pTK945) were only mildly lower than the titers of mobilized non-SIN IDLV’s (pTK1087). These data suggest that vector mobilization from episomal vector genomes is independent of the parental HIV-1 promoter/enhancer sequences and is in line with the Tat independent production of a SIN single-LTR vector (Figure 1B). 11 Importantly, titers of mobilized episomal ECOO-comprising vectors (pTK1940) were more than fifty-fold lower. Mobilization efficiency of episomal SIN vector from a stable packaging cell line as described here was significantly higher than the level of mobilization of conventional integrated SIN vectors described earlier.8, 9 To characterize mobilization of integrated ECOO-comprising vectors, the above ICLVs (pTK1087, pTK945 and pTK1940) were employed on SODk1 cells. To dilute out episomal vector forms, transduced SODk1 cells were cultured in the presence of doxycycline for more than 2 weeks (5 passages) after which production of VSV-G-pseudotyped vector particles was induced by doxycycline withdrawal as described earlier 14, 29. Titers of integrating vector particles in conditioned media were determined by scoring GFP expression following serial dilutions on 293T cells. Vector copy number in SODk1 cells prior to doxycycline withdrawal was determined by qPCR. 30 As shown in figure 6C-D, titers of mobilized integrated conventional SIN vectors (pTK945) were several hundred fold lower than the titers of their non-SIN counterparts (pTK1087). These results are in line with the data presented earlier by Logan et al. and Hanawa et al. 8, 9 Most importantly, titers of mobilized ECOO comprising vectors were at the limit of detection (less than 10 TU/ml) and several hundred fold lower than the titers of mobilized conventional SIN vectors (pTK945). Furthermore, normalization of vector titers for vector copy number in SODk1 cells has not affected the observed differences in mobilization efficiency of conventional and ECOO-comprising vectors (Figure 6C). Intrigued by the low mobilization levels of ECOO-comprising SIN vectors (pTK1940), we sought to study the effects of positioning a lentiviral vector’s expression cassette in opposite orientation to the LTRs on mobilization of non-SIN vectors. To this end, a novel ECOO-comprising non-SIN vector pTK1976 was transduced on either naïve or PKR-deficient cells. In addition, conventional non-SIN and SIN vectors pTK1087 and pTK945 as well as the ECOO-comprising SIN vector pTK1940 were employed on naïve 293T cells and VCN was determined by qPCR. To characterize the level of vector mobilization, the above vector-transduced cells were transiently transfected with a VSV-G envelope and a lentiviral vector-packaging cassette. Titers of mobilized vectors in conditioned media were determined by scoring GFP expression following serial dilutions on 293T cells. As expected and shown in figures 6E-F, conventional non-SIN vector (pTK1087) exhibits the highest mobilization titers (prior and after normalization to VCN in vector producer cells). However, the mere positioning of the internal vector expression cassette in opposite orientation to the LTRs reduced VCN-normalized mobilization titers of non-SIN vectors (pTK1976) from 293T cells by more than 500–fold. These mobilization titers were only 5-fold higher than the mobilization titers of conventional SIN vectors (pTK945). Interestingly, VCN-normalized mobilization titers of pTK1976 from PKR-deficient 293T cells were more than 4-fold higher than the titers generated by naïve 293T cells. These data suggest the reduction in mobilization-titers of non-SIN ECOO-comprising vectors (pTK1976) is mediated by PKR-dependent and -independent mechanisms. Importantly, further analysis of vector mobilization-titers shows 96- and 18-fold reduction in vector mobilization titers following conversion of the parental LTR configuration in pTK1976 to a SIN one (similar to pTK1940-LTR) and after reverting the conventional orientation of the internal expression cassette in pTK945 (similar to pTK1940 vector design), respectively. Altogether, these findings indicate that three independent mechanisms (SIN vector design and the above two ECOO-related mechanisms) synergistically inhibit mobilization of ECOO-comprising integrated SIN vectors. Note that, under the above extreme conditions, mobilization of pTK1940 was not completely abolished. We speculate that some vector producing cells transcribe low levels of full length packagable mRNA, which do not elicit a robust PKR response. This facilitates production of low yet measurable levels of mobilized vector transducing units.
Figure 6. Minimal mobilization of ECOO comprising SIN vectors.
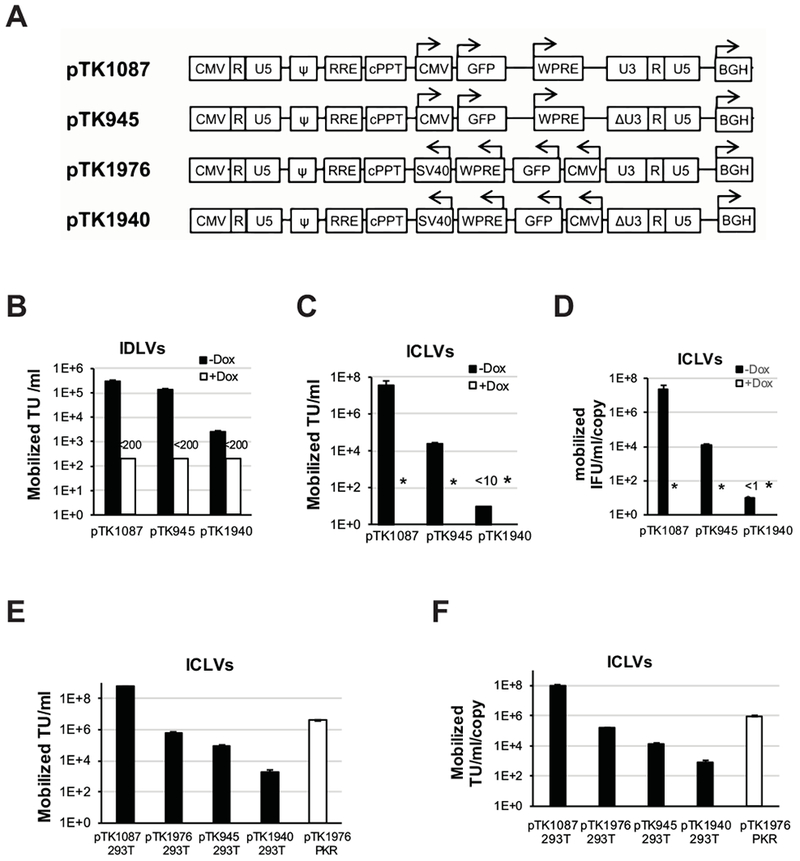
Mobilization efficiency of the SIN ECOO-comprising vector pTK1940 and its conventional SIN and non-SIN counterparts vectors, pTK945 and pTK1087, respectively. (A) Depiction of conventional and ECOO-comprising non-SIN/SIN vectors (pTK1087/pTK945 and pTK1976/pTk1940, respectively). The non-SIN U3 in the 3’LTR’s are indicated. Arrows indicate the direction of transcription from the internal CMV promoter and the orientation of the WPRE, SV40 and BGH poly-A signals. (B) Lentiviral vector mobilization from episomal vector genomes at 72h post IDLV transduction of the tetracycline regulated stable packaging cell line, SODk1, either in the presence of doxycycline or in the absence of doxycycline and the presence of sodium butyrate (uninduced or, induced respectively). Mobilized vector particles were employed on naïve 293T target cells. To dilute out carry over of non-mobilized IDLV’s, titers of mobilized ICLV’s (generated by the induced packaging cells) were determined following 5 passages of the above target cells in culture. Percentage of GFP-positive cells and the number target cells at the time of transduction were used to calculate vector titers. Black and white bars describe titers (TU/ml) of the above vectors mobilized from induced and uninduced SODk1 cells, respectively. Error bars represent standard deviation of 3 independent experiments. (C) Lentiviral vector mobilization from integrated vector genomes (pTK1087, pTK945 and pTK1940) in induced and uninduced SODk1 cells. Conditioned media were employed on 293T cells and vector titers were determined by scoring GFP positive cells following serial dilutions on 293T cells. Black and white bars describe titers (TU/ml) of the above vectors mobilized from induced and uninduced SODk1 cells, respectively. The results are average of three independent experiments. Asterisk indicates titers lower than detection level (<10TU/mL). (D) Normalized vector titers. To minimize bias of VCN in SODk1 on mobilized vector titers, calculated mobilization titers from ICLV-transduced SODk1 cells were normalized to VCN (vector producing SODk1 cells) as determined by real time PCR. Black and white bars describe normalized titers (TU/ml/copy) of the above vectors mobilized from induced and uninduced SODk1 cells, respectively. Titers were represented as average ± SD of 3 independent experiments. (E-F) Characterizing the combined effect of SIN-LTRs and the orientation of lentiviral vector internal expression cassettes on mobilization of integrated lentiviral vectors. PKR-deficient 293T cells transduced with integrating ECOO-comprising non-SIN vector (pTK1976) and naïve 293T cells transduced with either, conventional non-SIN and SIN vectors (pTK1087 and pTK945, respectively) or ECOO-comprising SIN and non-SIN vectors (pTK1976 and pTK1940, respectively) were analyzed for vector copy number by qPCR. The aforementioned vector-transduced 293T cell were transfected with a VSV-G envelope and the ΔNRF packaging cassettes. Mobilized vectors in conditioned media were collected at 72 hours post-transfection and tittered by scoring GFP expression following serial dilutions on 293T cells. (E) Bar graph describing titers of mobilized lentiviral vectors from integrated vector genomes in naïve (black bars) and PKR deficient (white bars) 293T cells. The results are average of three independent experiments. (F) Bar graph showing the above titers of mobilized vectors after normalization for VCN. The results are average of three independent experiments.
Superior hepatic gene delivery by ECOO comprising vectors
Minimal mobilization and efficient transgene expression render ECOO comprising vectors better suited to in vivo gene delivery applications than their conventional counterpart. Hence, we sought to evaluate the efficacy of ECOO-comprising vectors at hepatic gene delivery by direct comparison with their conventional counterparts. To this end, ECOO-comprising and conventional lentiviral vectors carrying the firefly luciferase under control of a liver specific (hAAT) promoter (Figure 5F: pTK1956 and pTK979, respectively) were packaged into VSV-G pseudotyped ICLVs. Concentrated vector particles were injected intraperitonealy to C57B6 mice (25µg p24gag/mouse). Luciferase expression in mouse livers was periodically imaged and quantified by the IVIS Lumina imagine system. As shown in figures 7A-B, transgene expression levels in mouse livers transduced with ECOO comprising vectors (pTK1956) were more than 5 fold higher than the levels of hepatic luciferase expression from conventional vectors (pTK979). These data underscore the superiority of the recently developed ECOO comprising vector as a means of in vivo gene delivery vehicle.
Figure 7. ECOO-comprising lentiviral vectors for hepatic gene delivery and ex-vivo applications.
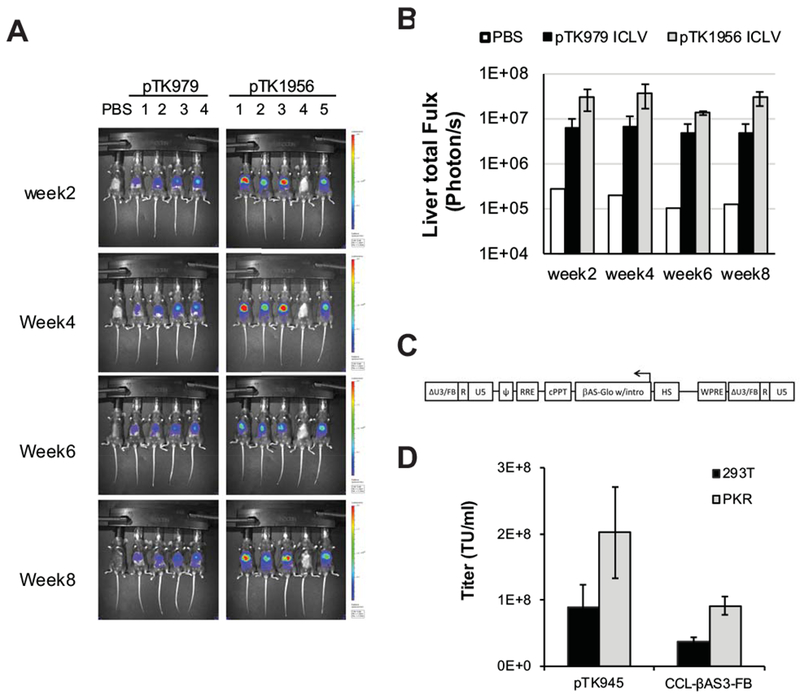
(A) In vivo imaging of lentiviral vectors-mediated hepatic luciferase expression. C57B6 mice were intraperitoneally injected with 1.5×109 TU of the ECOO-comprising vector pTK1956 (5 mice) and its conventional counterpart pTK979 (4 mice). As shown in figure 5F both vectors carry the firefly luciferase under the control of a liver specific promoter hAAT. A single mouse injected with PBS served as a negative control. Luciferase expression was imaged by the IVIS Lumina optical system at week 2, 4, 6, and 8 post-injection. (B) Bar graph describing average levels of luciferase expression in mouse livers (photon/sec) at weeks 2, 4, 6 and 8 after intraperitoneal administration of lentiviral vectors pTK979 (black bars, average ± SD, n=4) and pTK1956 (grey bars, n=4), and 1 mouse injected with PBS (white bar, n=1). Mouse #4 injected with pTK1956 was excluded from this analysis. (C) Depiction of The CCL-βAS3-FB LV provirus. The corresponding viral vector was developed and described earlier by Romero et al at in the laboratory of Dr. Donald B. Kohn at the University of California, Los Angeles (UCLA). The vector carries the HBBAS3 expression cassette in opposite orientation to the LTR’s (depicted as HBBAS3-Exp). It comprises the β-globin promoter, exons and introns of the β-globin gene as well as its 3’ and 5’ flanking regions including a poly-A site (between the globin coding sequence and the globin 3’ enhancer (Levasseur31 and Behringer 82 et al ). Also included is the β-globin mini-LCR with the hypersensitive sites 2-4. The SIN U3 regions contain a 77 bp mini-insulator (depicted as FB). (D) Bar graph describing titers of VSV-G pseudotyped the ECOO-comprising CCL-βAS3-FB and the conventional vector pTK945 generated in either naïve 293T cells (black bars) or PKR knockdown 293T cells (Grey bars). Error bars represent standard deviation of 2 independent experiments.
Efficient production of ECOO comprising vectors for clinically relevant ex-vivo applications
To further characterize the efficacy of the PKR-deficient packaging system at generating ECOO comprising vectors, we sought to evaluate production of a lentiviral vector, which was specifically developed for hematopoietic stem cell gene therapy in sickle cell disease patients. Romero et al. described the novel ECOO comprising vector, CCL-β AS3-FB, earlier (Figure 7C). 13 It encodes the modified human β-globin protein HbAS3 31 and contains the parental β-globin gene regulatory elements including its promoter, introns, poly-A site and the mini-locus control region (LCR) with hypersensitive sites (HS) 2-4. To support long-term and stable transgene expression, a 77bp insulator was incorporated in the vector’s SIN 3’U3. Similar to other lentiviral vectors, which efficiently delivered β-globin expression cassettes, the novel CCL-β AS3-FB exhibited low titers. 13, 32, 33 We conjectured that producing the CCL-β AS3-FB vector in PKR deficient cells would increase its titers. To test this hypothesis, CCL-β AS3-FB vector and a conventional vector (Figure 3A: pTK945) were generated by transient transfection in either naïve or PKR deficient 293T cells. Vector titers and concentration of physical vector particles in condition media were determine by qPCR on vector transduced 293T cells and by p24gag ELISA. As shown in figure 7D-E, titers of CCL-β AS3-FB vector generated in PKR deficient 293T cells were merely 2-3 fold higher than vector titers generated in naïve 293T cells and yet comparable to titers of the control vector pTK945 (up to 2×108 TU/ml). Altogether, the data presented here describe an efficient methodology of producing ECOO comprising lentiviral vectors, which are safer and more efficient than their conventional counterparts and thus better suited for clinical trials.
Discussion
Following numerous preclinical studies, years of promises, expectations, and early setbacks,34–37 recent accumulating successes of gene therapy clinical trials opened a new era in modern medicine. Currently, viral vectors serve as the method of choice to deliver therapeutic genetic cargos in gene therapy clinical trials. Lentiviral and AAV-based vectors have been used as a means to cure cancer as well as hereditary and acquired human diseases, most of which were not effectively treatable by conventional medicine.1, 3, 38–41 US FDA approvals of lentiviral and AAV vectors-based gene therapy to treat hematopoietic and lymphoid malignancies and a hereditary retinal disease 42–44 formally established a novel class of medications. Similar to conventional medications, the scope of disease indications for gene therapy protocols is dictated by risk/benefit considerations. Findings of earlier ɣ-retroviral and lentiviral vector-based preclinical studies and human clinical trials raised biosafety concerns regarding potential emergence of replication competent retroviruses (RCR) and integrating vector mediated insertional mutagenesis.34, 45–49. These biosafety concerns were significantly eased by a recent study summarizing biosafety test results of cancer and HIV-1 gene therapy clinical trials employing lenti/retro viral vectors-modified T cells.50 Overall, in these studies, biosafety assays of vector preparations, modified T-cells and treated patients could not detect RCR and demonstrated a long-term decrease in the vector modified T-cells population to less than 1%. These findings are highly encouraging, and yet additional studies are needed to characterize biosafety of ɣ-retroviral and lentiviral vector-based gene delivery in different clinical settings, especially those involving stem cell transduction and in vivo gene delivery. Lack of clinical and experimental reports on the adverse effects associated with vector mobilization reduced attention of regulators and investigators to this potential biosafety risk. Vector mobilization is defined as the undesired packaging and transfer of vector genomes from target cells, where they are performing a therapeutic objective, to other cell populations either within the host or to the community. Theoretically, vector mobilization can mediate several adverse processes including insertional mutagenesis, alteration in vector biodistribution, emergence of novel RCR’s, and induction of immune response to transgene products. Vector mobilization requires transcription of full-length vector genomes in a cell that concomitantly produces all viral proteins (structural and non-structural) needed to assemble productive vector particles. Genetic information encoding the above viral proteins could be provided by several mechanisms including inadvertent transferring of all or parts of the packaging cassettes to target cells, infection of target cells with parental wild type viruses, expression of endogenous retroviral genomes, or by a combination of these mechanisms. Human endogenous retroviruses (HERV), which invaded the human genome via germ cell infection in the last 2-40 million years, make ~8% of the human genome.51 Various genetic and epigenetic inactivation mechanisms rendered HERV non-infectious. However, human cell lines and tissues produce HERV proteins and noninfectious viral particles.52–55 Furthermore, infectious HERV viruses have been successfully reconstituted from human genomes.56, 57 The HERV-K envelope proteins, syncytin 1 and 2 are involved in human placenta formation and can pseudotype lentiviral vectors.52, 58 Thus, the receptors to syncytin-1 and −2 envelope proteins, which are synthesized in various human tissues (including germ cells), can potentially broaden biodistribution of syncytin-pseudotyped vectors.59–62
In contrast to AAV vector based therapies 63–65, the relatively small number of individuals that currently are or anticipated to be infected with HIV-1 reduces the likelihood of lentiviral vector mobilization. However, the necessity to avoid vector mobilization even in the presence of all HIV-1 proteins was the impetus to the development of SIN vectors, in which deletions of U3 sequences comprising the parental viral enhancer promoter inhibit transcription of full-length vector mRNA.5, 7, 66 Further characterization of SIN vector mobilization demonstrated reduced yet not completely abolished mobilization from integrated vector genomes. Vector mobilization in these studies was consequent upon cryptic transcription initiation of full-length vector mRNA, which was controlled by different transcription regulatory elements within the vector and in the host genome (determined by vector integration sites).8, 9 The fact that most of the cryptic transcription initiation sites reported by Hanawa et al. were downstream to the 5’R and could not produce full length packagable vector mRNA was in line with the relatively low titers (up to ~4×103 TU/ml) of SIN vectors mobilized from integrated vector genomes.8 Per contra, we show here that titers of SIN vectors mobilized from episomal vector genomes (Figures 1B and6B) are not significantly lower than the titers of their non-SIN counterparts. In theory, minimal if any host-genome effect on transcription of non-integrating vectors should render episomal SIN vector less mobilized than integrating vectors. However, vector circularization potentially positions various internal transcriptional regulatory elements upstream to the vector’s single or double LTR’s. We assert that this circularization event creates a new Tat independent promoter/enhancer constellation that renders the enhancer/promoter deletions of the SIN LTR ineffective at preventing transcription of full length vector mRNA. This notion is in line with an earlier study by Poon et al. showing Tat independent transcription from episomal HIV-1 genomes.67 The relatively high rate of vector mobilization from episomal vector genomes is in line with earlier studies describing significant differences in regulation of transgene expression from episomal and integrated lentiviral vector genomes.15, 28 Episomal lentiviral vectors comprise the majority of reverse transcription products 15, 22, 68 and thus serve as an efficient template for transcription of mobilizable, full-length mRNA’s. In dividing cells, episomal vector genomes lacking origin of replication are rapidly lost by dilution.68 Consequently, ex-vivo gene therapy protocols including gene delivery to stem cells and genetic engineering of T-cells carry lower risk of episomal vector mobilization.1, 40 Per contra, episomal vector genomes are highly stable in non-dividing cells; thus, in vivo administration of SIN lentiviral vector should raise biosafety concerns regarding potential vector mobilization.4
The development of the IDLV system successfully addressed the risk of insertional mutagenesis by reducing integration of vector genomes to less than 1%.24, 69 However, the risk of SIN vector mobilization is probably more significant following in vivo administration of IDLVs, considering more than 99% of their reverse transcribed genomes are episomes. Recently, 3’PPT-deleted vectors have been developed as a means to minimize illegitimate, integrase-independent integration of IDLV’s.24 The newly developed vectors have been successfully employed in immunotherapy clinical trials.4 3’PPT-deleted vectors generate mainly single-LTR circles, which are less likely than linear double-stranded vector genomes to recombine with the host genome, but are predicted to efficiently support vector mobilization.
To address the above biosafety concerns, we demonstrate here that positioning internal lentiviral vectors’ expression cassettes in opposite orientation to the LTRs, practically abolishes SIN vector mobilization. Premised on the fundamental differences in transcriptional regulation of gene expression from episomal and integrated vector genomes15, 67, we speculate that ECOO-associated reduction in vector mobilization from episomal and integrated vector genomes is mediated by different mechanisms. The relatively high levels of transgene expression from ECOO comprising IDLVs suggests that the negative effect of ECOO on episomal vector mobilization is probably secondary to silencing of cryptic transcription initiation sites in episomal vector’s SIN LTR and is not mediated by the host PKR response. Interestingly, three independent mechanisms reduce mobilization of integrated vectors. These include PKR-dependent and independent mechanisms that reduce mobilization of integrated non-SIN ECOO-comprising vectors and the SIN-LTR configuration that reduces mobilization of all integrated retroviral vectors. Consequently, ECOO-comprising vectors demonstrate the lowest level of mobilization, which renders them most suitable for a biosafety level-0 (BSL-0, the community), in which patients carrying large numbers of viral vectors genomes can potentially mobilize viral vectors and their derivatives to the community. Several research groups successfully employed ECOO-comprising vectors in hematopoietic stem cells (HSC)-based gene replacement therapy for different hemoglobinopathies.13, 31–33, 46, 70–73 To maintain long-term therapeutic level of transgene expression, most of the disease-tailored vectors carried human globin expression cassettes comprising transcriptional and post-transcriptional regulatory elements of the human β-globin gene including its’ promoter, mini-LCR, introns and poly-A site. However, difficulties in generating high vector titers limit the usage of these efficacious vectors in clinical trials.13, 32, 33 It is likely that the relatively large genetic payload of the above vectors negatively affect their titers. Since the parental globin promoter is not active in 293T cells, we observed merely a mild increase of ~2 fold in titers of the CCL-β AS3-FB vector in PKR deficient cells as compared with vector titers generated by naïve 293T cells. A similar increase in titers was observed for a conventional pTK945 vector. In theory these findings could be associated with low level of antisense mRNAs which could be transcribed from weak promoters embedded in the parental HIV-1 sequence and in the vector cassette and stabilized by the internal poly-A signal.74–77
Positioning the WPRE between the transgene of interest and the internal poly-A sequence significantly increased transgene expression from ECOO-comprising vectors without negatively affecting their titers. Unexpectedly, gene expression levels from ECOO-comprising vectors were comparable or higher than the levels of expression from their conventional counterparts. Interestingly, the molecular mechanism involved in this phenomenon, which was more distinct in IDLV’s has not been elucidated in this study. However, it is possible that positioning the internal promoter in opposite orientation to the LTR distanced it from negative regulatory elements in the vector’s 5’ untranslated region. This phenomenon is a significant advantage of ECOO-comprising vectors. Furthermore, in contrast to conventional vectors, ECOO-comprising vectors should facilitate delivery of intron-containing genes, which significantly increase transgene expression.78–81 Overall, the data presented here outline an efficient methodology of producing high titer ECOO-comprising vectors. The newly developed vector system is safer, more efficient at maintaining high levels of transgene expression and thus suites better than currently used lentiviral vectors for clinical trials.
Supplementary Material
ACKNOWLEDGMENTS
The following reagents were obtained through the National Institutes of Health (NIH) AIDS Research and Reference Reagent Program, Division of AIDS, the National Institute of Allergy and Infectious Diseases: the HIV-1 p24 monoclonal antibody (183-H12-5C) from Bruce Chesebro and Kathy Wehrly. The study was supported by UNC Flow Cytometry Core Facility, which is supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center. ‘The study was supported by NIH grants R01-HL128119 (to P.H., Y.B., T.S., M.H., B.Z., N.J.F., and T.K.), R01-DK058702 (to P.H., Y.B., T.S., M.H., B.Z., N.J.F., and T.K.). This study is dedicated to the U.S. Marine Corps and the Gold Star families. In memory of Henryk Goldszmit, the doctor who stayed.
Footnotes
CONFLICTS OF INTEREST
TK is an inventor of a PPT-deleted vector based technology owned by the University of North Carolina and licensed to a commercial entity. There is no conflicts of interest for all the other authors.
Reference:
- 1.Morgan RA, Gray D, Lomova A, Kohn DB. Hematopoietic Stem Cell Gene Therapy: Progress and Lessons Learned. Cell stem cell 2017; 21(5): 574–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Penati R, Fumagalli F, Calbi V, Bernardo ME, Aiuti A. Gene therapy for lysosomal storage disorders: recent advances for metachromatic leukodystrophy and mucopolysaccaridosis I. Journal of inherited metabolic disease 2017; 40(4): 543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oluwole OO, Davila ML. At The Bedside: Clinical review of chimeric antigen receptor (CAR) T cell therapy for B cell malignancies. Journal of leukocyte biology 2016; 100(6): 1265–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pollack SM, Lu H, Gnjatic S, Somaiah N, O’Malley RB, Jones RL et al. First-in-Human Treatment With a Dendritic Cell-targeting Lentiviral Vector-expressing NY-ESO-1, LV305, Induces Deep, Durable Response in Refractory Metastatic Synovial Sarcoma Patient. Journal of immunotherapy 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu SF, von Ruden T, Kantoff PW, Garber C, Seiberg M, Ruther U et al. Self-inactivating retroviral vectors designed for transfer of whole genes into mammalian cells. Proceedings of the National Academy of Sciences of the United States of America 1986; 83(10): 3194–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shinya E, et al. . A Safe HIV Vectors Packaging System Using the U3 Deficient LTR, . In: GENE THERAPY MEETING COLD SPRING HARBOR, 1994. p 150. [Google Scholar]
- 7.Miyoshi H, Blomer U, Takahashi M, Gage FH, Verma IM. Development of a self-inactivating lentivirus vector. Journal of virology 1998; 72(10): 8150–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanawa H, Persons DA, Nienhuis AW. Mobilization and mechanism of transcription of integrated self-inactivating lentiviral vectors. Journal of virology 2005; 79(13): 8410–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Logan AC, Haas DL, Kafri T, Kohn DB. Integrated self-inactivating lentiviral vectors produce full-length genomic transcripts competent for encapsidation and integration. Journal of virology 2004; 78(16): 8421–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lucke S, Grunwald T, Uberla K. Reduced mobilization of Rev-responsive element-deficient lentiviral vectors. Journal of virology 2005; 79(14): 9359–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma H, Kafri T. A single-LTR HIV-1 vector optimized for functional genomics applications. Molecular therapy : the journal of the American Society of Gene Therapy 2004; 10(1): 139–49. [DOI] [PubMed] [Google Scholar]
- 12.Liu YP, Vink MA, Westerink JT, Ramirez de Arellano E, Konstantinova P, Ter Brake O et al. Titers of lentiviral vectors encoding shRNAs and miRNAs are reduced by different mechanisms that require distinct repair strategies. Rna 2010; 16(7): 1328–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Romero Z, Urbinati F, Geiger S, Cooper AR, Wherley J, Kaufman ML et al. beta-globin gene transfer to human bone marrow for sickle cell disease. The Journal of clinical investigation 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kafri T, van Praag H, Ouyang L, Gage FH, Verma IM. A packaging cell line for lentivirus vectors. Journal of virology 1999; 73(1): 576–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bayer M, Kantor B, Cockrell A, Ma H, Zeithaml B, Li X et al. A large U3 deletion causes increased in vivo expression from a nonintegrating lentiviral vector. Molecular therapy : the journal of the American Society of Gene Therapy 2008; 16(12): 1968–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cockrell AS, van Praag H, Santistevan N, Ma H, Kafri T. The HIV-1 Rev/RRE system is required for HIV-1 5’ UTR cis elements to augment encapsidation of heterologous RNA into HIV-1 viral particles. Retrovirology 2011; 8: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D et al. A third-generation lentivirus vector with a conditional packaging system. Journal of virology 1998; 72(11): 8463–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu K, Ma H, McCown TJ, Verma IM, Kafri T. Generation of a stable cell line producing high-titer self-inactivating lentiviral vectors. Molecular therapy : the journal of the American Society of Gene Therapy 2001; 3(1): 97–104. [DOI] [PubMed] [Google Scholar]
- 19.Cockrell AS, Kafri T. Gene delivery by lentivirus vectors. Mol Biotechnol 2007; 36(3): 184–204. [DOI] [PubMed] [Google Scholar]
- 20.Cockrell AS, Ma H, Fu K, McCown TJ, Kafri T. A trans-lentiviral packaging cell line for high-titer conditional self-inactivating HIV-1 vectors. Molecular therapy : the journal of the American Society of Gene Therapy 2006; 14(2): 276–84. [DOI] [PubMed] [Google Scholar]
- 21.Suwanmanee T, Ferris MT, Hu P, Gui T, Montgomery SA, Pardo-Manuel de Villena F et al. Toward Personalized Gene Therapy: Characterizing the Host Genetic Control of Lentiviral-Vector-Mediated Hepatic Gene Delivery. Molecular therapy. Methods & clinical development 2017; 5: 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Butler SL, Hansen MS, Bushman FD. A quantitative assay for HIV DNA integration in vivo. Nature medicine 2001; 7(5): 631–4. [DOI] [PubMed] [Google Scholar]
- 23.Hazuda DJ, Felock P, Witmer M, Wolfe A, Stillmock K, Grobler JA et al. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science 2000; 287(5453): 646–50. [DOI] [PubMed] [Google Scholar]
- 24.Kantor B, Bayer M, Ma H, Samulski J, Li C, McCown T et al. Notable reduction in illegitimate integration mediated by a PPT-deleted, nonintegrating lentiviral vector. Molecular therapy : the journal of the American Society of Gene Therapy 2011; 19(3): 547–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poon B, Chen IS. Human immunodeficiency virus type 1 (HIV-1) Vpr enhances expression from unintegrated HIV-1 DNA. Journal of virology 2003; 77(7): 3962–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Negri D, Blasi M, LaBranche C, Parks R, Balachandran H, Lifton M et al. Immunization with an SIV-based IDLV Expressing HIV-1 Env 1086 Clade C Elicits Durable Humoral and Cellular Responses in Rhesus Macaques. Molecular therapy : the journal of the American Society of Gene Therapy 2016; 24(11): 2021–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suwanmanee T, Hu G, Gui T, Bartholomae CC, Kutschera I, von Kalle C et al. Integration-deficient lentiviral vectors expressing codon-optimized R338LhFIX restore normal hemostasis in hemophilia B mice. Molecular therapy : the journal of the American Society of Gene Therapy 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kantor B, Ma H, Webster-Cyriaque J, Monahan PE, Kafri T. Epigenetic activation of unintegrated HIV-1 genomes by gut-associated short chain fatty acids and its implications for HIV infection. Proceedings of the National Academy of Sciences of the United States of America 2009; 106(44): 18786–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu P, Li Y, Sands MS, McCown T, Kafri T. Generation of a stable packaging cell line producing high-titer PPT-deleted integration-deficient lentiviral vectors. Molecular therapy. Methods & clinical development 2015; 2: 15025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu P, Li Y, Nikolaishvili-Feinberg N, Scesa G, Bi Y, Pan D et al. Hematopoietic Stem cell transplantation and lentiviral vector-based gene therapy for Krabbe’s disease: Present convictions and future prospects. Journal of neuroscience research 2016; 94(11): 1152–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levasseur DN, Ryan TM, Pawlik KM, Townes TM. Correction of a mouse model of sickle cell disease: lentiviral/antisickling beta-globin gene transduction of unmobilized, purified hematopoietic stem cells. Blood 2003; 102(13): 4312–9. [DOI] [PubMed] [Google Scholar]
- 32.May C, Rivella S, Callegari J, Heller G, Gaensler KM, Luzzatto L et al. Therapeutic haemoglobin synthesis in beta-thalassaemic mice expressing lentivirus-encoded human beta-globin. Nature 2000; 406(6791): 82–6. [DOI] [PubMed] [Google Scholar]
- 33.Pawliuk R, Westerman KA, Fabry ME, Payen E, Tighe R, Bouhassira EE et al. Correction of sickle cell disease in transgenic mouse models by gene therapy. Science 2001; 294(5550): 2368–71. [DOI] [PubMed] [Google Scholar]
- 34.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003; 302(5644): 415–9. [DOI] [PubMed] [Google Scholar]
- 35.Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nature medicine 2006; 12(3): 342–7. [DOI] [PubMed] [Google Scholar]
- 36.Raper SE, Chirmule N, Lee FS, Wivel NA, Bagg A, Gao GP et al. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Molecular genetics and metabolism 2003; 80(1–2): 148–58. [DOI] [PubMed] [Google Scholar]
- 37.Somia N, Verma IM. Gene therapy: trials and tribulations. Nature reviews. Genetics 2000; 1(2): 91–9. [DOI] [PubMed] [Google Scholar]
- 38.Adair JE, Beard BC, Trobridge GD, Neff T, Rockhill JK, Silbergeld DL et al. Extended survival of glioblastoma patients after chemoprotective HSC gene therapy. Science translational medicine 2012; 4(133): 133ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Booth C, Gaspar HB, Thrasher AJ. Treating Immunodeficiency through HSC Gene Therapy. Trends in molecular medicine 2016; 22(4): 317–27. [DOI] [PubMed] [Google Scholar]
- 40.Frey NV, Porter DL. CAR T-cells merge into the fast lane of cancer care. American journal of hematology 2016; 91(1): 146–50. [DOI] [PubMed] [Google Scholar]
- 41.Keeler AM, ElMallah MK, Flotte TR. Gene Therapy 2017: Progress and Future Directions. Clinical and translational science 2017; 10(4): 242–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.FDA approves hereditary blindness gene therapy. Nature biotechnology 2018; 36(1): 6. [DOI] [PubMed] [Google Scholar]
- 43.Dyer O FDA panel recommends approval of first gene therapy in US. Bmj 2017; 358: j3443. [DOI] [PubMed] [Google Scholar]
- 44.Sheridan C First approval in sight for Novartis’ CAR-T therapy after panel vote. Nature biotechnology 2017; 35(8): 691–693. [DOI] [PubMed] [Google Scholar]
- 45.Braun CJ, Boztug K, Paruzynski A, Witzel M, Schwarzer A, Rothe M et al. Gene therapy for Wiskott-Aldrich syndrome--long-term efficacy and genotoxicity. Science translational medicine 2014; 6(227): 227ra33. [DOI] [PubMed] [Google Scholar]
- 46.Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature 2010; 467(7313): 318–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Donahue RE, Kessler SW, Bodine D, McDonagh K, Dunbar C, Goodman S et al. Helper virus induced T cell lymphoma in nonhuman primates after retroviral mediated gene transfer. The Journal of experimental medicine 1992; 176(4): 1125–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. The Journal of clinical investigation 2008; 118(9): 3143–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stein S, Ott MG, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nature medicine 2010; 16(2): 198–204. [DOI] [PubMed] [Google Scholar]
- 50.Marcucci KT, Jadlowsky JK, Hwang WT, Suhoski-Davis M, Gonzalez VE, Kulikovskaya I et al. Retroviral and Lentiviral Safety Analysis of Gene-Modified T Cell Products and Infused HIV and Oncology Patients. Molecular therapy : the journal of the American Society of Gene Therapy 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bannert N, Kurth R. Retroelements and the human genome: new perspectives on an old relation. Proceedings of the National Academy of Sciences of the United States of America 2004; 101 Suppl 2: 14572–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dupressoir A, Lavialle C, Heidmann T. From ancestral infectious retroviruses to bona fide cellular genes: role of the captured syncytins in placentation. Placenta 2012; 33(9): 663–71. [DOI] [PubMed] [Google Scholar]
- 53.Hohn O, Hanke K, Bannert N. HERV-K(HML-2), the Best Preserved Family of HERVs: Endogenization, Expression, and Implications in Health and Disease. Frontiers in oncology 2013; 3: 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lower R, Boller K, Hasenmaier B, Korbmacher C, Muller-Lantzsch N, Lower J et al. Identification of human endogenous retroviruses with complex mRNA expression and particle formation. Proceedings of the National Academy of Sciences of the United States of America 1993; 90(10): 4480–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nadeau MJ, Manghera M, Douville RN. Inside the Envelope: Endogenous Retrovirus-K Env as a Biomarker and Therapeutic Target. Frontiers in microbiology 2015; 6: 1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dewannieux M, Harper F, Richaud A, Letzelter C, Ribet D, Pierron G et al. Identification of an infectious progenitor for the multiple-copy HERV-K human endogenous retroelements. Genome research 2006; 16(12): 1548–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee YN, Bieniasz PD. Reconstitution of an infectious human endogenous retrovirus. PLoS pathogens 2007; 3(1): e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.An DS, Xie Y, Chen IS. Envelope gene of the human endogenous retrovirus HERV-W encodes a functional retrovirus envelope. Journal of virology 2001; 75(7): 3488–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bjerregaard B, Lemmen JG, Petersen MR, Ostrup E, Iversen LH, Almstrup K et al. Syncytin-1 and its receptor is present in human gametes. Journal of assisted reproduction and genetics 2014; 31(5): 533–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Esnault C, Priet S, Ribet D, Vernochet C, Bruls T, Lavialle C et al. A placenta-specific receptor for the fusogenic, endogenous retrovirus-derived, human syncytin-2. Proceedings of the National Academy of Sciences of the United States of America 2008; 105(45): 17532–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Green BJ, Lee CS, Rasko JE. Biodistribution of the RD114/mammalian type D retrovirus receptor, RDR. The journal of gene medicine 2004; 6(3): 249–59. [DOI] [PubMed] [Google Scholar]
- 62.Hummel J, Kammerer U, Muller N, Avota E, Schneider-Schaulies S. Human endogenous retrovirus envelope proteins target dendritic cells to suppress T-cell activation. European journal of immunology 2015; 45(6): 1748–59. [DOI] [PubMed] [Google Scholar]
- 63.Boutin S, Monteilhet V, Veron P, Leborgne C, Benveniste O, Montus MF et al. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Human gene therapy 2010; 21(6): 704–12. [DOI] [PubMed] [Google Scholar]
- 64.Halbert CL, Miller AD, McNamara S, Emerson J, Gibson RL, Ramsey B et al. Prevalence of neutralizing antibodies against adeno-associated virus (AAV) types 2, 5, and 6 in cystic fibrosis and normal populations: Implications for gene therapy using AAV vectors. Human gene therapy 2006; 17(4): 440–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hewitt FC, Li C, Gray SJ, Cockrell S, Washburn M, Samulski RJ. Reducing the risk of adeno-associated virus (AAV) vector mobilization with AAV type 5 vectors. Journal of virology 2009; 83(8): 3919–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zufferey R, Dull T, Mandel RJ, Bukovsky A, Quiroz D, Naldini L et al. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. Journal of virology 1998; 72(12): 9873–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Poon B, Chang MA, Chen IS. Vpr is required for efficient Nef expression from unintegrated human immunodeficiency virus type 1 DNA. Journal of virology 2007; 81(19): 10515–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Butler SL, Johnson EP, Bushman FD. Human immunodeficiency virus cDNA metabolism: notable stability of two-long terminal repeat circles. Journal of virology 2002; 76(8): 3739–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tareen SU, Kelley-Clarke B, Nicolai CJ, Cassiano LA, Nelson LT, Slough MM et al. Design of a novel integration-deficient lentivector technology that incorporates genetic and posttranslational elements to target human dendritic cells. Molecular therapy : the journal of the American Society of Gene Therapy 2014; 22(3): 575–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hanawa H, Hargrove PW, Kepes S, Srivastava DK, Nienhuis AW, Persons DA. Extended beta-globin locus control region elements promote consistent therapeutic expression of a gamma-globin lentiviral vector in murine beta-thalassemia. Blood 2004; 104(8): 2281–90. [DOI] [PubMed] [Google Scholar]
- 71.Miccio A, Cesari R, Lotti F, Rossi C, Sanvito F, Ponzoni M et al. In vivo selection of genetically modified erythroblastic progenitors leads to long-term correction of beta-thalassemia. Proceedings of the National Academy of Sciences of the United States of America 2008; 105(30): 10547–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pestina TI, Hargrove PW, Jay D, Gray JT, Boyd KM, Persons DA. Correction of murine sickle cell disease using gamma-globin lentiviral vectors to mediate high-level expression of fetal hemoglobin. Molecular therapy : the journal of the American Society of Gene Therapy 2009; 17(2): 245–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Puthenveetil G, Scholes J, Carbonell D, Qureshi N, Xia P, Zeng L et al. Successful correction of the human beta-thalassemia major phenotype using a lentiviral vector. Blood 2004; 104(12): 3445–53. [DOI] [PubMed] [Google Scholar]
- 74.Harwig A, Das AT, Berkhout B. HIV-1 RNAs: sense and antisense, large mRNAs and small siRNAs and miRNAs. Current opinion in HIV and AIDS 2015; 10(2): 103–9. [DOI] [PubMed] [Google Scholar]
- 75.Landry S, Halin M, Lefort S, Audet B, Vaquero C, Mesnard JM et al. Detection, characterization and regulation of antisense transcripts in HIV-1. Retrovirology 2007; 4: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Manghera M, Magnusson A, Douville RN. The sense behind retroviral anti-sense transcription. Virology journal 2017; 14(1): 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Michael NL, Vahey MT, d’Arcy L, Ehrenberg PK, Mosca JD, Rappaport J et al. Negative-strand RNA transcripts are produced in human immunodeficiency virus type 1-infected cells and patients by a novel promoter downregulated by Tat. Journal of virology 1994; 68(2): 979–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cooper AR, Lill GR, Gschweng EH, Kohn DB. Rescue of splicing-mediated intron loss maximizes expression in lentiviral vectors containing the human ubiquitin C promoter. Nucleic acids research 2015; 43(1): 682–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Le Hir H, Nott A, Moore MJ. How introns influence and enhance eukaryotic gene expression. Trends in biochemical sciences 2003; 28(4): 215–20. [DOI] [PubMed] [Google Scholar]
- 80.Nott A, Meislin SH, Moore MJ. A quantitative analysis of intron effects on mammalian gene expression. Rna 2003; 9(5): 607–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Valencia P, Dias AP, Reed R. Splicing promotes rapid and efficient mRNA export in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America 2008; 105(9): 3386–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Behringer RR, Hammer RE, Brinster RL, Palmiter RD, Townes TM. Two 3’ sequences direct adult erythroid-specific expression of human beta-globin genes in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America 1987; 84(20): 7056–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.