

Abstract
Drug-induced liver injury (DILI) presents unique challenges for consumers, clinicians, and regulators. It is the most common cause of acute liver failure in the US. It is also one of the most common reasons for termination of new drugs during pre-clinical testing and withdrawal of new drugs post-marketing. DILI is generally divided into two forms: intrinsic and idiosyncratic. Many of the challenges with DILI are due in large part to poor understanding of the mechanisms of toxicity. Although useful models of intrinsic DILI are available, they are frequently misused. Modeling idiosyncratic DILI presents greater challenges, but promising new models have recently been developed. The purpose of this manuscript is to provide a critical review of the most popular animal models of DILI, and to discuss the future of DILI research.
Keywords: Acetaminophen, Carbon tetrachloride, intrinsic hepatotoxicity, idiosyncratic hepatotoxicity, immune tolerance
INTRODUCTION
Drug-induced liver injury (DILI) is a problem that affects consumers, clinicians, pharmaceutical companies, and regulators. For consumers, it is a marginal but ever-present risk. For clinicians, it is difficult to diagnose and can be difficult to treat. And for the pharmaceutical industry and regulators, it is an obstacle that must be avoided in order to bring a new product to the market and keep it there. DILI is the single most common cause of acute liver failure (ALF) in the US. It has been reported that DILI is responsible for approximately 60% of all ALF cases [1], but that is likely an underestimate; other studies have consistently demonstrated that 10-20% of ALF cases of indeterminate etiology are actually due to DILI [2–4]. DILI is also one of the most common reasons for post-marketing withdrawal of drugs and for new black box warnings throughout the world [5–7]
DILI is usually divided into two types that are commonly referred to as “intrinsic” and “idiosyncratic.” Although there is no universal definition of either, intrinsic DILI is typically said to be dose-dependent and predictable. The toxicity is attributed to chemical properties of the drug rather than some unique aspect of the drug consumer’s biology. On the other hand, idiosyncratic DILI (IDILI) is often described as non-dose-dependent (i.e. occurs at low doses), unpredictable, and rare (though the definition of “rare” also varies considerably). It is thought to be determined in large part by genetic variation. In reality, these definitions probably represent two ends of a spectrum. This is clear because the probability of IDILI increases with increasing daily dose [8–10] and the prevalence (and therefore predictability) of toxicity among users of IDILI-causing agents varies from drug to drug. Furthermore, there is some evidence that biological variation can also influence intrinsic hepatotoxicity [11–13].
The models used to study DILI are important. The challenges presented by DILI are due in part to our poor understanding of the mechanisms that drive it. Without knowledge of the mechanisms, it is difficult to develop ways to predict and avoid it. And in order to understand the causes, we need research models that can reproduce it. However, development of appropriate models has been onerous. It has been pointed out that adverse drug reactions that are idiosyncratic in humans are also idiosyncratic in mice [14], and even use of intrinsic hepatotoxicants in animals suffers from pitfalls that must be avoided. While novel in vitro models of DILI with promise for pre-clinical prediction of hepatotoxicity have been introduced, they lack important factors like a complete immune system and cross-talk with other organs. It seems unlikely that they will fully replace animals for research or drug development. In this manuscript, we review the major animal models of DILI and describe promising recent advances. The attributes of ideal DILI models are listed in Table 1, with examples of each.
Table 1.
Attributes of an ideal model of drug-induced liver injury.
| Attribute | Description | Example(s) |
|---|---|---|
| Clinical resemblance to human DILI | Doses, time course and/or pathology are similar | APAP overdose, Uetrecht-Pohl model, mitochondriopathy model |
| Mechanistic resemblance to human DILI | Pathophysiology is similar | APAP overdose, Uetrecht-Pohl model? |
| Experimental convenience | Animals develop injury in days to weeks | All current models |
MODELS OF INTRINSIC DILI
Animal models of intrinsic DILI are straightforward with regard to technique. In most cases, one can simply treat the animals with a large dose of the drug of interest to cause hepatotoxicity. However, the proper use of these models requires a basic understanding of the mechanisms of toxicity in each one. The most common models of intrinsic hepatotoxicity are listed in Table 2. By far, the two most common models in intrinsic DILI research are acetaminophen (APAP) and carbon tetrachloride (CCl4).
Table 2.
Major animal models of intrinsic drug-induced liver injury
| Drug | Favored Species | Typical Dose | Strengths | Weaknesses |
|---|---|---|---|---|
| Acetaminophen | Mouse | 200-600 mg/kg | Easy to use, clinically relevant | Potential interference with metabolism |
| CCl4 | Rat, Mouse | 1-2 mL/kg (10-20 mmol/kg) | Easy to use, can also model chronic DILI | Limited clinical relevance; potential interference with metabolism |
| Thioacetamide | Mouse, Rat | 100-300 mg/kg | Easy to use | Limited clinical relevance; potential interference with metabolism |
| Furosemide | Mouse | 200-500 mg/kg | Easy to use | Limited clinical relevance; potential interference with metabolism |
| Bromobenzene | Mouse, Rat | 0.5-1 mL/kg (5-10 mmol/kg) | Easy to use | Limited clinical relevance; potential interference with metabolism |
| Allyl alcohol | Mouse, Rat | 30-100 mg/kg | Easy to use | Limited clinical relevance; potential interference with metabolism |
Acetaminophen.
APAP is the most commonly used model of intrinsic DILI. It is also the most clinically relevant [15], as APAP overdose is the primary cause of acute liver failure in several countries [1]. As a result, the mechanisms of APAP hepatotoxicity have been well studied, though important gaps remain. The toxicity is initiated by conversion of APAP to an electrophile thought to be N-acetyl-p-benzoquinone imine (NAPQI) (Figure 1). That conversion is catalyzed by cytochrome P450 enzymes. NAPQI then binds to sulfhydryl groups on glutathione and proteins. Depletion of GSH makes the cells more susceptible to oxidative stress. The effects of protein binding are less clear, but there is mounting evidence that mitochondrial proteins are the critical targets [16,17]. For example, N-acetyl-m-aminophenol (AMAP), an isomer of APAP, does not bind to mitochondrial proteins nor cause toxicity in mouse hepatocytes, but does both in human hepatocytes [17]. The protein binding appears to cause inhibition of mitochondrial respiration during APAP toxicity [18] and mitochondrial oxidative stress develops [19–22]. It is thought that the initial oxidative stress activates redox-sensitive MAP kinases that converge on the c-Jun N-terminal kinases 1/2 (Jnk) [23–26], and phospho-Jnk then translocates to mitochondria where it exacerbates the oxidative stress by further inhibiting mitochondrial respiration [27–29]. Although recent data have cast doubt on the Jnk hypothesis [30], the weight of the evidence supports it [31]. Other kinases, such as the receptor interacting protein kinases (Ripk) 1 and 3 have also been demonstrated to be important [32–34]. However, the detailed mechanisms of the involvement of these kinases in APAP toxicity remain to be investigated; a recent study even questioned the relevance of Ripk 3 [35]. Eventually, loss of the mitochondrial membrane potential occurs [36], which causes swelling and rupture of mitochondrial membranes with release of endonucleases [37]. The endonucleases then translocate to the nucleus and cleave nuclear DNA [37,38]. The result is oncotic necrosis [16,39,40].
Figure 1. Mechanisms of Acetaminophen Hepatotoxicity.
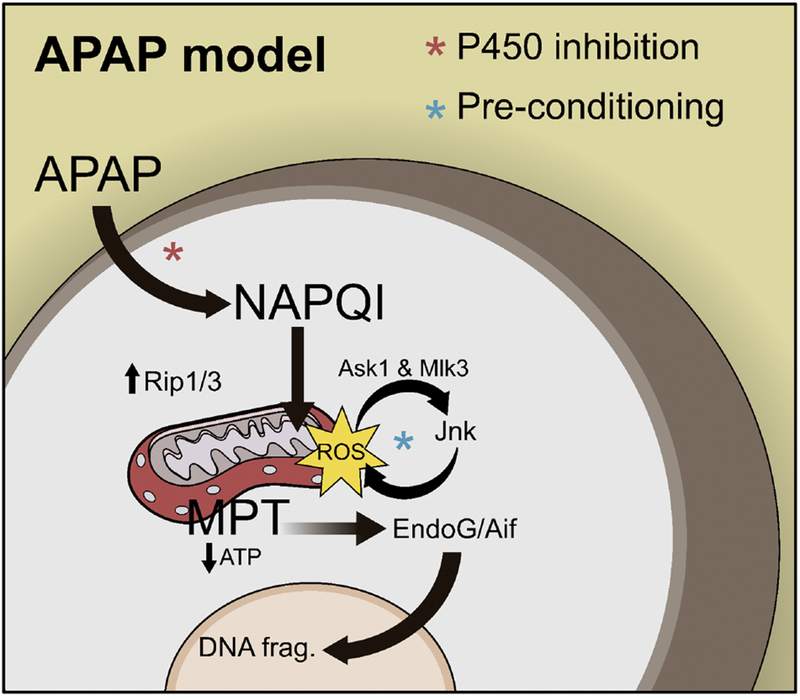
Acetaminophen (APAP) hepatotoxicity begins with formation of a reactive metabolite (NAPQI) that binds to proteins. Binding to mitochondrial proteins causes mitochondrial dysfunction and oxidative stress. The initial reactive oxygen species (ROS) activate kinases like the c-Jun N-terminal kinases 1/2 (Jnk), which then exacerbates the mitochondrial oxidative stress. Eventually, the mitochondrial permeability transition (MPT) occurs and mitochondrial polarization and ATP production are lost. The mitochondrial matrix swell and the outer membranes rupture releasing endonucleases (EndoG and AIF) that translocate to the nucleus and fragment nuclear DNA.
Mice are the preferred animal for studies of APAP overdose. Early studies involved multiple species, including rats [41,42], guinea pigs [41], cats [43] and hamsters [44]. However, it is now clear that mice are the best available model [45]. The doses of APAP that cause toxicity in mice and humans are similar; both species develop injury at doses ≥150 mg/kg, while the other species that have been tested are either much more resistant or much more susceptible [41,43,45]. More importantly, the mechanisms of toxicity appear to be the same in mice and humans [15]. There is strong evidence for glutathione depletion [40,46,47], protein binding [2,47,48], mitochondrial damage [16,17,40,47], oxidative stress [40], DNA fragmentation [16,49] and Jnk activation [47] in humans, like mice. On the other hand, rats have less mitochondrial protein binding than mice, despite receiving much higher doses [45], and there is no evidence of mitochondrial damage or oxidative stress in rats [45]. Conveniently, mice are generally easier to work with than other species, and numerous gene knock-out mice and transgenic substrains of mice are available for research purposes. One minor difference between humans and mice is the time course of liver injury; hepatotoxicity develops somewhat faster in mice, peaking around 12-24 h compared to 24-72 h in humans [16,40]
Typically, the mice are fasted for 12-16 h before treatment with 200-300 mg/kg APAP. The primary purpose of fasting is to reduce variation of hepatic GSH levels due to nutritional status; it ensures that all the animals have a similar baseline level of glutathione for glutathionylation and glycogen for glucuronidation. However, fasting is not required if higher doses of APAP (400-600 mg/kg) are given. Whether or not fasting the animals makes the model less clinically relevant is debatable. However, almost all research to date that has included a comparison of toxicity or regeneration mechanisms between mice and humans have used fasted mice and found that the mechanisms are similar between species, even in metabolomics experiments where one might expect fasting to alter the results [16,50–53]. One exception might be when studying autophagy. Fasting induces autophagy and may mask potential effects of pharmacological interventions. Thus, studies that showed the hepatoprotective effects of removing damaged mitochondria [54] or protein adducts [55] by autophagy used fed mice.
Several issues must be addressed when using the mouse model of APAP overdose. First, any intervention that reduces P450 expression, inhibits P450 activity, or otherwise reduces protein binding will also prevent or reduce the liver injury (Figure 1). Many drugs initially thought to protect against APAP through novel mechanisms actually inhibit APAP bioactivation [56–58]. Drug vehicles must also be considered. A classic example is dimethyl sulfoxide (DMSO), which is well known to inhibit Cyps [56,59–61]. To avoid inhibition of APAP bioactivation, interventions should be given at least 1.5 h after a dose of ≤ 300 mg/kg APAP. Anytime pre- or co-treatment is used, it is important to include vehicle controls, and to assess the effect of the drug(s) and vehicle(s) on either hepatic GSH depletion or protein binding.
A related issue is pre-conditioning. Any intervention that puts stress on hepatocytes prior to APAP treatment can induce expression of antioxidant genes that protect against toxicity (Figure 1). This phenomenon has been documented with both pharmacologic [62] and genetic interventions [63,64]. For example, liver-specific KO of autophagy genes causes chronic liver injury and repair that activates nuclear factor (erythroid-derived 2)-like 2 (Nrf2) [63]. The increased Nrf2 activity in the liver results in higher baseline GSH levels and thereby protects against APAP overdose [63]. Another example is the use of neutropenia-inducing antibodies 24 h before APAP treatment, which protects against APAP hepatotoxicity [52,65]. However, this protection is not caused by neutropenia as hypothesized but by causing a preconditioning effect due to the phagocytosis of the antibody-tagged, inactivated neutrophils by Kupffer cells, which triggers the extensive induction of defense genes including metallothionein [66]. Metallothionein protects against APAP overdose by assisting in the scavenging of NAPQI [67]. Thus, anytime an intervention or genotype protects against APAP hepatotoxicity, it is important to verify that it is not due to a pre-conditioning effect.
An additional consideration is the mouse strain used for the experiments. There is considerable variation in the extent of liver injury caused by APAP across strains [68]. Common inbred strains used in APAP research are C57Bl/6J, C57Bl/6N, and C3HeB/FeJ. Interestingly, C57Bl/6J mice carry a spontaneous mutation in the nicotinamide nucleotide transhydrogenase (Nnt) gene that results in loss of its activity. Normally, Nnt maintains high concentrations of NADPH in the mitochondrial matrix, which is important for several reactions including the reduction of glutathione disulfide (GSSG) back to the reduced form. However, under conditions when the mitochondrial electron transport chain is impaired, the enzyme works in the opposite direction, i.e. Nnt functions to generate more NADH for use in the electron transport chain, which leads to reduction of NADPH levels and consequently lower antioxidant capacity in the mitochondria [69]. In the case of APAP, this means that the push to feed more electrons into the transport chain leads to more reactive oxygen formation in combination with the impaired antioxidant defense. This explains why C57Bl/6N mice with functional Nnt are more susceptible to APAP toxicity than C57Bl/6J mice [70,71]. C3H/HeJ mice carry a spontaneous mutation in the toll-like receptor 4 (Tlr4) gene, which makes them less responsive to endotoxin. The C3H/HeJ mice are also less susceptible to APAP toxicity than other C3H substrains, which led one group to propose that Tlr4 is an important mediator of the injury [72]. Although there is no doubt that the extensive necrosis observed after APAP overdose leads to release of damage-associated molecular patterns, which are ligands for a variety of pattern recognition receptors and trigger the transcriptional activation of cytokines and chemokines in the liver [73–75], that interpretation of the data may be incorrect. This sterile formation of inflammatory mediators does cause the recruitment of neutrophils and monocyte-derived macrophages into the liver. However, the function of these inflammatory cells is controversial. There is experimental evidence both for and against a direct exaggerating effect of neutrophils and monocytes on APAP-induced liver injury [73–75], but the preponderance of evidence appears to support the idea that these inflammatory cells are recruited to remove necrotic cells and prepare the liver for recovery [74,75]. These animal data are supported by evidence that monocytes [76] and neutrophils [53] are pro-regenerative after APAP overdose in humans.
Carbon tetrachloride.
CCl4 is not a drug, but high doses (≥1 mL/kg) do cause reproducible acute liver injury that resembles intrinsic DILI. After APAP, CCl4 is probably the most common model of xenobiotic-induced acute liver injury. The effects of CCl4 on the liver are complex and there is debate over which are most important for its hepatotoxicity [77]. However, it is clear that the toxicity is dependent on the P450-catalyzed metabolism to the reactive metabolite trichloromethyl radical (CCl3·) (Figure 2). Early studies revealed that CCl4-induced necrosis is limited to the centrilobular area where P450 expression is greatest, that haloalkanes with stronger carbon-halogen bonds that are difficult to break are not hepatotoxic, that 14C from radiolabeled CCl4 accumulates in the endoplasmic reticulum where P450 activity is greatest, and that young rats with low P450 expression are resistant to CCl4-induced liver injury [78]. More direct approaches to test the involvement of P450s have since become available and also support that hypothesis [77,79]. Importantly, CCl3· can alkylate proteins, nucleic acids and lipids, altering their function. Major effects include reduced protein synthesis, steatosis, and altered calcium homeostasis [77]. CCl3· also reacts with molecular oxygen to form trichloromethylperoxy radical (CCl3OO·). In addition to binding to macromolecules like CCl3·, CCl3OO· can extract hydrogen from polyunsaturated fatty acids and thereby initiate the chain reaction known as lipid peroxidation (LPO). It appears that both alkylation and LPO are required for the hepatotoxicity of CCl4 [77]. Like APAP, acute CCl4 hepatotoxicity involves mitochondrial damage and mitochondrial DNA depletion in addition to LPO [79].
Figure 2. Mechanisms of Carbon Tetrachloride Hepatotoxicity.
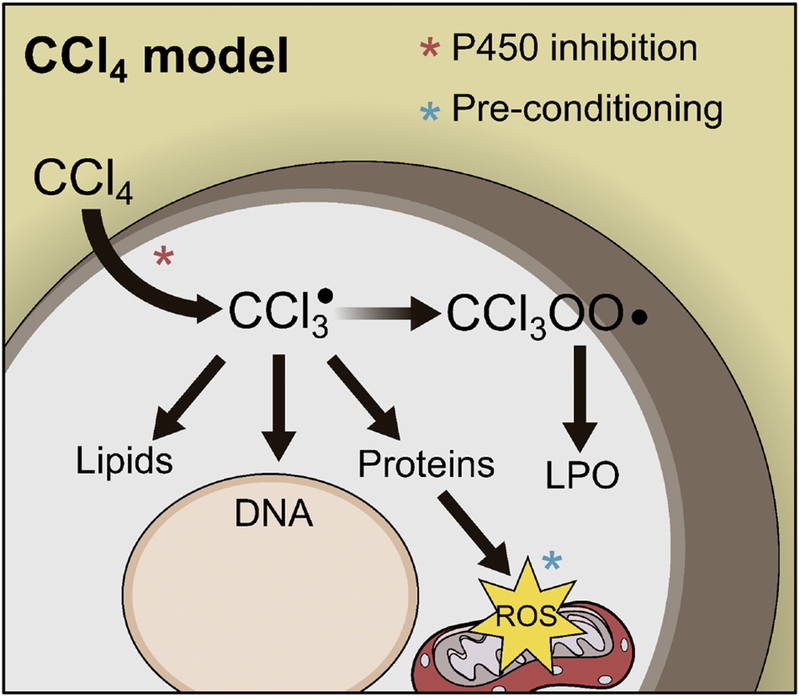
Carbon tetrachloride (CCl4) is converted to a radical (CCl3·) that binds to proteins, DNA and lipids. This causes mitochondrial damage and oxidative stress. CCl3· can also react with O2 to form CCl3OO·, which initiates the lipid peroxidation (LPO) chain reaction that damages cell membranes.
Rats are likely the best animals for studies of CCl4 hepatotoxicity when resemblance to humans is important. Although the rat is the least susceptible rodent species [80], there is evidence that metabolism of CCl4 is lower in both rats and humans than in other species [81] and it has been observed that the histopathology of CCl4 toxicity in rats is like that in humans [82]. Nevertheless, mice are often used for convenience and because of the availability of gene knock-out mice and transgenic substrains.
Many considerations for the APAP hepatotoxicity model of DILI are the same for CCl4. Again, any intervention that alters P450 expression or activity will likely affect the injury (Figure 2). The importance of P450-mediated bioactivation of CCl4 has been directly demonstrated using P450 inhibitors in multiple studies [79,83]. Thus, as for APAP, we recommend post-treatment when using any pharmacologic interventions. It has also been demonstrated that Nrf2 is activated in mouse liver tissue after acute CCl4 treatment [84] and that it protects against CCl4 [85]. Thus, it is likely that preconditioning effects that further upregulate antioxidant genes would protect (Figure 2). Finally, major differences between mouse sub-strains have also been reported for CCl4-induced liver injury. For example, C57Bl/6N mice develop more severe injury, but also recover much faster due to differences in the response of macrophages [86].
One application of CCl4 that sets it apart from APAP is that it can also be used to model chronic liver disease (CLD), such as alcohol associated liver diseases and fatty liver diseases. Lower doses (0.5 – 0.8 mL/kg) given twice per week over the course of 2-4 weeks will cause persistent liver injury with inflammation and fibrosis. Any intervention strategy, especially plant extracts with unknown composition, used in the chronic model of CCl4-induced toxicity has to be tested for its potential effects on P450-dependent metabolism. CCl4 is also often included as a second “hit” in two-hit models of CLD. Although most alcoholics and obese patients develop fatty liver, a minority progress to fibrosis or cirrhosis. The two-hit hypothesis of CLD is that fatty liver is merely the first hit, and one or more additional insults are required to drive disease progression [87]. Thus, CCl4 treatment is often mixed with other treatments as the second hit [87–90]. A promising novel model of non-alcoholic steatohepatitis (NASH) was recently developed using this approach. When C57Bl/6J mice were fed a high-fat Western diet combined with once per week exposure to a low dose of CCl4 (0.2 mL/kg) they developed steatosis and late-stage fibrosis by 12 weeks, with progression to hepatocellular carcinoma (HCC) by 24 weeks [90,91]. This appears to be the first mouse model to reproduce nearly all the histopathological and transcriptomic features of human NASH [90]
Other drugs and xenobiotics.
Thioacetamide (TAA) induces hepatotoxicity in mice and rats at doses ≥100 mg/kg. It is converted to the metabolites TAA S-oxide and S,S-dioxide by cytochrome P450 enzymes [92–94] and the S,S-dioxide initiates toxicity by binding to lipids and proteins. Few studies have directly addressed species differences in TAA hepatotoxicity, but values for serum ALT reported in the literature are typically much higher for mice than rats despite treatment with similar doses. Very few studies have investigated the pathophysiology of TAA hepatotoxicity, so not much is known about it. Other poorly studied model hepatotoxicants are furosemide, bromobenzene, and allyl alcohol. Similar to APAP, CCl4 and TAA, these drugs are converted to reactive metabolites that bind to proteins [95–98] or can induce lipid peroxidation [99] to cause hepatocyte necrosis. Finally, there are other xenobiotics that induce liver injury in rodents, but are not models of DILI per se. For example, the combination of endotoxin and an inhibitor of gene expression causes apoptotic death [100], which does not appear to be a common characteristic of intrinsic DILI [16,101].
Summary.
APAP is the archetype of intrinsic DILI, and APAP overdose in mice is by far the most common model. CCl4 is also popular, and has the advantage of being useful for studies of chronic DILI and fibrosis. However, consideration of the basic mechanisms of toxicity is important when using either APAP or CCl4. Reactive metabolite formation and protein binding are common features of almost all available animal models of intrinsic DILI. Some form of oxidative stress and mitochondrial damage are also involved in both the APAP and CCl4 models, and in several less common intrinsic DILI models [102–104]. Thus, intrinsic DILI appears to involve reactive metabolites, oxidative stress and mitochondrial damage in most cases.
MODELS OF IDIOSYNCRATIC DILI
The study of IDILI in animals poses greater technical challenges. Achieving an adverse reaction to a drug that is known to cause IDILI in humans typically requires a pre-treatment or genetic alteration designed to pre-dispose the animals to injury. Of course, any such pre-treatment has the potential to affect the clinical relevance of the model. The three major approaches that have been used so far involve either induction of inflammation, suppression of immune tolerance, or genetic manipulation of mitochondrial function. These models are summarized in Table 3.
Table 3.
Major animal models of idiosyncratic drug-induced liver injury
| Model | Strategy | Strengths | Weaknesses |
|---|---|---|---|
| Inflammagen model | LPS co-administration | Easy to use | Poor resemblance to human IDILI |
| Uetrecht-Pohl model | Compromised immune tolerance | Time course and pathology resemble human IDILI | No liver failure |
| Subclinical mitochondriopathy | Compromised ROS defense | Time course resembles human IDILI | Poor reproducibility |
Inflammagen model.
In the inflammagen model, animals are either pre-treated, co-treated, or post-treated with bacterial lipopolysaccharides (LPS) in order to induce inflammation. The model was based on observations made as far back as the 1940s, when it was discovered that pre-treatment with antibiotics reduces the hepatotoxicity of CCl4 [105]. Similar reports led to the hypothesis that IDILI is due in part to variation in exposure of humans to LPS [106]. That hypothesis has evolved to the idea that any randomly occurring inflammatory stimulus can precipitate IDILI. The first direct evidence to support that idea came from experiments with chlorpromazine (CPZ), a tricyclic antipsychotic. CPZ is now rarely used but was once a common cause of drug-induced liver disease. Therapeutic doses cause liver function impairment and elevated serum alkaline phosphatase (ALP) in approximately 40% of all users [107,108], and liver disease in approximately 1% [108]. However, extensive efforts for decades to develop an animal model of CPZ hepatotoxicity brought little success. Finally, Buchweitz et al. [109] were able to reproduce the clinical chemistry of human CPZ hepatotoxicity in rats by adding a 2 h pretreatment with LPS. Since then, the inflammagen model has been used to induce reproducible liver injury in rodents using relatively low doses of several other drugs that cause IDILI in humans including diclofenac [110], halothane [111], amiodarone [112], ranitidine [113] and trovafloxacin [114].
The human relevance of the inflammagen model is probably limited. One issue is that the timing of LPS treatment is important. Using this model, some drugs cause liver injury when LPS is administered as a pre-treatment [109,113], while others require very late post-treatment [112]. Such exact timing of increased LPS exposure seems unrealistic in humans. Uetrecht and Naisbitt [115] have described other major problems. First, the time from initiation of therapy to IDILI for a given drug is usually similar across patients. If exposure to LPS was a precipitating factor then one would expect that timing to be random because not all patients on a particular drug would encounter high levels of LPS after the same time period. Instead, the delay in injury is probably the time required to mount an adaptive immune response to a hapten or some other epitope created as a result of the drug [116]. Second, liver injury in the inflammagen model appears to be driven by an innate immune response, as it primarily involves neutrophils [113] The best example to illustrate that comes from APAP toxicity where the mechanism of APAP toxicity alone can be compared with the injury induced by LPS+APAP. Although an APAP dose close to the threshold of toxicity (150 mg/kg) can be aggravated with LPS pre-treatment [117], the time course of the injury is completely different. The delayed injury in the LPS+APAP experiment is consistent with a late neutrophil-mediated injury, which does not occur with APAP alone [118]. Thus, the LPS+drug toxicity is mainly a neutrophil-mediated toxicity, which is not typical for IDILI. In humans, IDILI is driven by lymphocytes. Third, sensitivity to toll-like receptor ligands including LPS falls precipitously after initial exposure, which likely limits their effects [9]. Finally, intestinal permeability is increased in patients with inflammatory bowel disease and they have elevated serum endotoxin values as a result [119], yet there is currently no evidence for increased risk of IDILI among patients with inflammatory diseases.
Suppression of immune tolerance (“Uetrecht-Pohl model”).
A recently developed approach to model IDILI in animals is to suppress immune tolerance in the liver (Figure 3). This approach is based on Temple’s corollary, which states that all drugs that cause serious IDILI in a few patients also cause much higher incidences of mild liver injury that spontaneously resolves despite continued treatment. The fact that most patients adapt to the initial mild injury strongly suggests that immune tolerance develops and prevents progression to liver failure. That led to the hypothesis that breakage of immune tolerance is necessary to develop a complete IDILI model. Consistent with that, Metushi et al. [120] observed that the idiosyncratic hepatotoxicant amodiaquine causes delayed-onset mild liver injury in female C57Bl/6J mice that spontaneously resolves, and that resolution occurred after an increase in programmed cell death 1 protein (Pd-1) positive T cells in the liver. Importantly, Pd-1 is critical for development of immune tolerance.
Figure 3. Importance of Immune Tolerance in Idiosyncratic DILI.
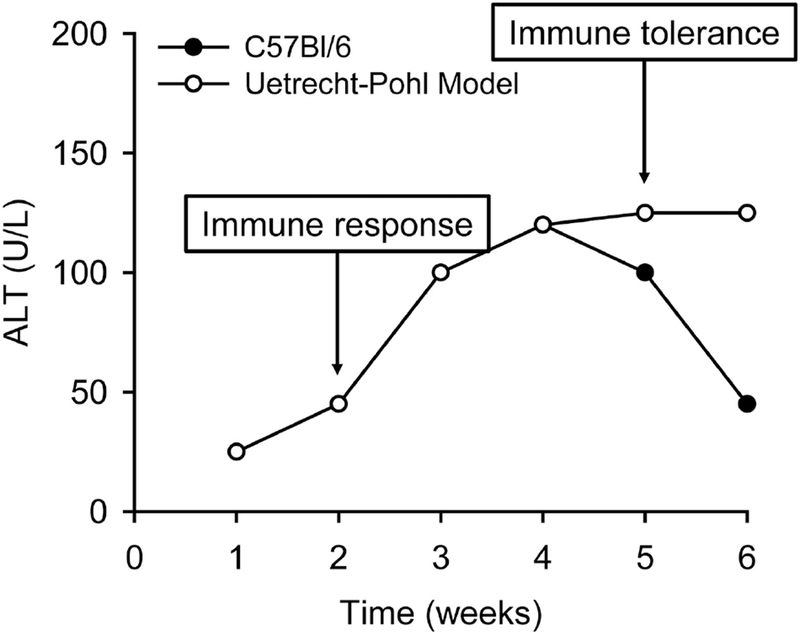
In WT C57Bl/6 mice, daily treatment with idiosyncratic hepatotoxicants causes delayed-onset transient liver injury. In the Uetrecht-Pohl model, various strategies are used to reduce regulation of the adaptive immune system and therefore prevent development of immune tolerance, and those mice experience ongoing liver injury. Results from that model have demonstrated that immune tolerance is a likely explanation for the transient nature of most IDILI in humans; loss of tolerance may explain why only some cases of IDILI are severe.
The authors later discovered that the injury appeared to be worse in Pd-1 KO (Pd-1−/−) mice [121], and that co-treatment of Pd-1−/− mice with an antibody against cytotoxic T-lymphocyte-associated protein 4 (CTLA4) on regulatory T cells prevented injury resolution [121]. At the same time, Chakraborty et al. [122] used a similar approach to delay resolution of halothane hepatotoxicity. Importantly, histology in both studies more closely resembled that of human IDILI than previous models [121,122]. Finally, a recent follow-up study demonstrated that the Pd-1−/−-anti-CTLA4 mouse model can also reproduce troglitazone and tolcapone IDILI, and can distinguish between hepatotoxic and non-hepatoxic drugs of the same class [14].
Despite the success of the Uetrecht-Pohl model to date, there are potential problems. First, it is a relatively new approach and requires further validation with other IDILI-causing drugs. Second, although there is evidence of sustained liver injury in this model, true liver failure has not yet been reported with the model. Finally, the strategies used to break immune tolerance in mice are extreme; it is unlikely to be so severe in humans, so other factors may need to be considered. Nevertheless, the available data are promising.
Subclinical mitochondriopathy.
Although there is widespread agreement that IDILI involves the adaptive immune system, it has been noted that associations with HLA variants are weak and cannot fully explain IDILI risk in patients [116]. As a result, it has been suggested that non-immune-mediated mechanisms may also be important. Interestingly, Ong et al. [123] reported that mice with partial deficiency of the mitochondrial superoxide dismutase 2 (Sod2+/−) developed mild liver injury as indicated by both serum ALT and histology after treatment with 30 mg/kg/day troglitazone for four weeks but WT mice did not. That result led to the hypothesis that some IDILI can be explained by increased susceptibility of patients with subclinical mitochondrial dysfunction. However, attempts to reproduce those results have had mixed success. Another group reported that the increase in ALT after chronic troglitazone treatment did not differ between WT and Sod2+/− mice [124]. In addition, attempts to mimic the troglitazone study with other drugs have resulted in only limited evidence for liver injury [125–127]. As a result, the Sod2+/− model has fallen out of favor. Associations between SOD2 variants and IDILI in humans have been discovered [128], but those variants have not been reported in more recent genome-wide association studies [129].
Summary.
Of the currently available animal models for IDILI, the Uetrecht-Pohl model appears to be the most similar to humans. Although early results look promising, challenges remain. There is still no complete model of serious IDILI with development of liver failure. Furthermore, although the Uetrecht-Pohl model suggests that loss of immune tolerance is likely an important part of the mechanisms of IDILI, we do not know what would cause that in humans. It is also possible that other, as yet unknown mechanisms play a role. An additional issue with all of the available models is that they have largely been designed with hepatocellular IDILI in mind. These models should also be validated for modeling of cholestatic and mixed IDILI.
DIFFERENCES IN DRUG METABOLISM BETWEEN ANIMALS AND HUMANS
Drug metabolism, and especially cytochrome P450-mediated metabolism, should be a major consideration when working with any model of DILI. It should be clear from our discussion of intrinsic DILI models that many depend upon metabolism to form a reactive metabolite. The prevailing view of IDILI is that it too depends upon reactive metabolites, which bind to proteins and create neoantigens that elicit an immune response (though other mechanisms have also been proposed) [116,130]. Importantly, there are dramatic species differences in metabolism. Although the common experimental models (mice, rats, dogs, and monkeys) all express homologs of the major P450s in humans, not all homologs have the same substrate specificity. In fact, CYP2E1 is the only human P450 that is functionally conserved across species [131]. Generally, mice bear the greatest resemblance in overall P450 function to humans among common research species, while rats are the most strikingly different [131]. Rats tend to have low P450 activity compared to other species, which may explain why they are less susceptible to some hepatotoxicants. Differences in expression or function of other enzymes can also be important. For example, dogs are poor acetylators because they lack N-acetyltransferase (Nat) genes [132] and the high incidence of idiosyncratic sulfonamide toxicity in dogs may be a result of that [133], while cats are poor glucuronidators and are highly susceptible to APAP hepatotoxicity because they lack functional Ugt1a6 [134]. Overall, it is critical to consider species differences in drug metabolism when using animal models of DILI.
CONCLUSIONS
DILI is a major clinical and regulatory problem. Many of the challenges presented by DILI are due to poor understanding of the mechanisms of toxicity caused by different drugs. Although reasonably good models of intrinsic DILI exist, they are often misused. A thorough understanding of what is known about the basic mechanisms of injury caused by intrinsic hepatotoxicants is necessary to ensure proper use. While IDILI is more difficult to reproduce in animals, the Uetrecht-Pohl model involving breakage of immune tolerance appears to be a major step forward. Overall, substantial progress is being made in our understanding of DILI using animal models, but considerable work remains to be done.
Supplementary Material
Highlights.
-
-
Drug-induced liver injury (DILI) is important clinically and during drug development
-
-
Intrinsic and idiosyncratic DILI are the two principal categories if DILI
-
-
Most relevant models for intrinsic DILI are acetaminophen overdose and CCL4
-
-
Idiosyncratic DILI models are less well developed but new ideas are being tested
-
-
Review provides critical discussion on validated and newly emerging models of DILI
ACKNOWLEDGEMENTS
This work was supported in part by the AASLD Foundation Pinnacle Research Award (MRM), start-up funds from the University of Arkansas for Medical Sciences (MRM), and National Institutes of Health (NIH) grants R01 DK102142 (HJ), P20 GM103549 (HJ) and P30 GM118247 (HJ). A portion of the start-up funds from UAMS were provided by NIH grant U54 TR001629 (PI: James, LP).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
REFERENCES
- 1.Lee WM. Etiologies of acute liver failure. Semin Liver Dis 2008;28:142–52. 10.1055/s-2008-1073114 [DOI] [PubMed] [Google Scholar]
- 2.Davern TJ, James LP, Hinson JA, Polson J, Larson AM, Fontana RJ, Lalani E, Munoz S, Shakil AO, Lee WM. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology 2006;130:687–94. 10.1053/j.gastro.2006.01.033 [DOI] [PubMed] [Google Scholar]
- 3.Ganger DR, Rule J, Rakela J, Bass N, Reuben A, Stravitz R, Sussman N, Larson AM, James L, Chiu C, Lee WM, Acute Liver Failure Study Group. Acute Liver Failure of Indeterminate Etiology: A Comprehensive Systematic Approach by An Expert Committee to Establish Causality. Am J Gastroenterol 2018; June 27. doi: 10.1038/s41395-018-0160-2. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.James LP, Alonso EM, Hynan LS, Hinson JA, Davern TJ, Lee WM, Squires RH, Pediatric Acute Liver Failure Study Group. Detection of Acetaminophen Protein Adducts in Children With Acute Liver Failure of Indeterminate Cause. Pediatrics 2006;118: e676–e681. 10.1542/peds.2006-0069 [DOI] [PubMed] [Google Scholar]
- 5.Issa AM, Phillips KA, Van Bebber S, Nidamarthy HG, Lasser KE, Haas JS, Alldredge BK, Wachter RM, Bates DW. Drug withdrawals in the United States: a systematic review of the evidence and analysis of trends. Curr Drug Saf 2007; 2:177–85. [DOI] [PubMed] [Google Scholar]
- 6.Onakpoya IJ, Heneghan CJ, Aronson JK. Post-marketing withdrawal of 462 medicinal products because of adverse drug reactions: a systematic review of the world literature. BMC Med 2016;14:10 10.1186/s12916-016-0553-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Solotke MT, Dhruva SS, Downing NS, Shah ND, Ross JS. New and incremental FDA black box warnings from 2008 to 2015. Expert Opin Drug Saf 2018;17:117–23. 10.1080/14740338.2018.1415323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lammert C, Einarsson S, Saha C, Niklasson A, Bjornsson E, Chalasani N. Relationship between daily dose of oral medications and idiosyncratic drug-induced liver injury: search for signals. Hepatology 2008;47:2003–9. 10.1002/hep.22272 [DOI] [PubMed] [Google Scholar]
- 9.Uetrecht J Prediction of a new drug’s potential to cause idiosyncratic reactions. Curr Opin Drug Discov Devel 2001;4:55–9. [PubMed] [Google Scholar]
- 10.Walgren JL, Mitchell MD, Thompson DC. Role of metabolism in drug-induced idiosyncratic hepatotoxicity. Crit. Rev Toxicol 2005; 35:325–61. 10.1080/10408440590935620 [DOI] [PubMed] [Google Scholar]
- 11.de Morais SM, Uetrecht JP, Wells PG. Decreased glucuronidation and increased bioactivation of acetaminophen in Gilbert’s syndrome. Gastroenterology 1992;102: 577–86. [DOI] [PubMed] [Google Scholar]
- 12.Rubin JB, Hameed B, Gottfried M, Lee WM, Sarkar M, Acute Liver Failure Study Group. Acetaminophen-induced Acute Liver Failure Is More Common and More Severe in Women. Clin Gastroenterol Hepatol 2018;16:936–46. 10.1016/j.cgh.2017.11.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watkins PB, Kaplowitz N, Slattery JT, Colonese CR, Colucci SV, Stewart PW, Harris SC. Aminotransferase Elevations in Healthy Adults Receiving 4 Grams of Acetaminophen Daily. JAMA 2006;296:87–93. 10.1001/jama.296.1.87 [DOI] [PubMed] [Google Scholar]
- 14.Mak A, Kato R, Weston K, Hayes A, Uetrecht J Editor’s Highlight: An Impaired Immune Tolerance Animal Model Distinguishes the Potential of Troglitazone/Pioglitazone and Tolcapone/Entacapone to Cause IDILI. Toxicol Sci 2018;161:412–20. 10.1093/toxsci/kfx219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jaeschke H, Xie Y, McGill MR. Acetaminophen-induced Liver Injury: from Animal Models to Humans. J Clin Transl Hepatol 2014;2:153–61. doi: 10.14218/JCTH.2014.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McGill MR, Sharpe MR, Williams CD, Taha M, Curry SC, Jaeschke H. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Invest 2012;122:1574–83. 10.1172/JCI59755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie Y, McGill MR, Du K, Dorko K, Kumer SC, Schmitt TM, Ding WX, Jaeschke H. Mitochondrial protein adducts formation and mitochondrial dysfunction during N-acetyl-m-aminophenol (AMAP)-induced hepatotoxicity in primary human hepatocytes. Toxicol Appl Pharmacol 2015b;289:213–22. 10.1016/j.taap.2015.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyers LL, Beierschmitt WP, Khairallah EA, Cohen SD. Acetaminophen-induced inhibition of hepatic mitochondrial respiration in mice. Toxicol Appl Pharmacol 1988;93:378–87. [DOI] [PubMed] [Google Scholar]
- 19.Cover C, Mansouri A, Knight TR, Bajt ML, Lemasters JJ, Pessayre D, Jaeschke H. Peroxynitrite-Induced Mitochondrial and Endonuclease-Mediated Nuclear DNA Damage in Acetaminophen Hepatotoxicity. J Pharmacol Exp Ther 2005;315:879–87. 10.1124/jpet.105.088898 [DOI] [PubMed] [Google Scholar]
- 20.Du K, Farhood A, Jaeschke H. Mitochondria-targeted antioxidant Mito-Tempo protects against acetaminophen hepatotoxicity. Arch Toxicol 2017;91:761–773. 10.1007/s00204-016-1692-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaeschke H Glutathione disulfide formation and oxidant stress during acetaminopheninduced hepatotoxicity in mice in vivo: the protective effect of allopurinol. J Pharmacol Exp Ther 1990;255:935–41. [PubMed] [Google Scholar]
- 22.Knight TR, Kurtz A, Bajt ML, Hinson JA, Jaeschke H. Vascular and hepatocellular peroxynitrite formation during acetaminophen toxicity: role of mitochondrial oxidant stress. Toxicol Sci 2001;62:212–20. [DOI] [PubMed] [Google Scholar]
- 23.Gunawan BK, Liu Z, Han D, Hanawa N, Gaarde WA, Kaplowitz N. c-Jun N-Terminal Kinase Plays a Major Role in Murine Acetaminophen Hepatotoxicity. Gastroenterology 2006;131:165–78. 10.1053/j.gastro.2006.03.045 [DOI] [PubMed] [Google Scholar]
- 24.Nakagawa H, Maeda S, Hikiba Y, Ohmae T, Shibata W, Yanai A, Sakamoto K, Ogura K, Noguchi T, Karin M, Ichijo H, Omata M. Deletion of apoptosis signal-regulating kinase 1 attenuates acetaminophen-induced liver injury by inhibiting c-Jun N-terminal kinase activation. Gastroenterology 2008;135:1311–21. 10.1053/j.gastro.2008.07.006 [DOI] [PubMed] [Google Scholar]
- 25.Sharma M, Gadang V, Jaeschke A. Critical Role for Mixed-Lineage Kinase 3 in Acetaminophen-Induced Hepatotoxicity. Mol Pharmacol 2012;82:1001–1007. 10.1124/mol.112.079863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie Y, Ramachandran A, Breckenridge DG, Liles JT, Lebofsky M, Farhood A, Jaeschke H. Inhibitor of apoptosis signal-regulating kinase 1 protects against acetaminopheninduced liver injury. Toxicol Appl Pharmacol 2015c;286:1–9. 10.1016/j.taap.2015.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, Kaplowitz N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J Biol Chem 2008;283:13565–77. 10.1074/jbc.M708916200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saito C, Lemasters JJ, Jaeschke H. c-Jun N-terminal kinase modulates oxidant stress and peroxynitrite formation independent of inducible nitric oxide synthase in acetaminophen hepatotoxicity. Toxicol Appl Pharmacol 2010;246:8–17. doi: 10.1016/j.taap.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Win S, Than TA, Fernandez-Checa JC, Kaplowitz N. JNK interaction with Sab mediates ER stress induced inhibition of mitochondrial respiration and cell death. Cell Death Dis 2014;5: e989–e989. 10.1038/cddis.2013.522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cubero FJ, Zoubek ME, Hu W, Peng J, Zhao G, Nevzorova YA, Al Masaoudi M, Bechmann LP, Boekschoten MV, Muller M, Preisinger C, Gassler N, Canbay AE, Luedde T, Davis RJ, Liedtke C, Trautwein C. Combined Activities of JNK1 and JNK2 in Hepatocytes Protect Against Toxic Liver Injury. Gastroenterology 2016;150: 968–81. 10.1053/j.gastro.2015.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Du K, Xie Y, McGill MR, Jaeschke H. Pathophysiological significance of c-jun N - terminal kinase in acetaminophen hepatotoxicity. Expert Opin Drug Metab Toxicol 2015;11: 1769–79. 10.1517/17425255.2015.1071353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li JX, Feng JM, Wang Y, Li XH, Chen XX, Su Y, Shen YY, Chen Y, Xiong B, Yang CH, Ding J, Miao ZH. The B-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis 2014;5:e1278 10.1038/cddis.2014.241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramachandran A, McGill MR, Xie Y, Ni HM, Ding WX, Jaeschke H. Receptor interacting protein kinase 3 is a critical early mediator of acetaminophen-induced hepatocyte necrosis in mice. Hepatology 2013;58:2099–108. 10.1002/hep.26547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang YF, He W, Zhang C, Liu XJ, Lu Y, Wang H, Zhang ZH, Chen X, Xu DX. Role of receptor interacting protein (RIP)1 on apoptosis-inducing factor-mediated necroptosis during acetaminophen-evoked acute liver failure in mice. Toxicol Lett 2014;225:445–53. 10.1016/j.toxlet.2014.01.005 [DOI] [PubMed] [Google Scholar]
- 35.Dara L, Johnson H, Suda J, Win S, Gaarde W, Han D, Kaplowitz N. Receptor interacting protein kinase 1 mediates murine acetaminophen toxicity independent of the necrosome and not through necroptosis. Hepatology 2015;62:1847–57. doi: 10.1002/hep.27939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kon K, Kim J-S, Jaeschke H, Lemasters JJ. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology 2004;40:1170–9. 10.1002/hep.20437 [DOI] [PubMed] [Google Scholar]
- 37.Bajt ML, Cover C, Lemasters JJ, Jaeschke H. Nuclear Translocation of Endonuclease G and Apoptosis-Inducing Factor during Acetaminophen-Induced Liver Cell Injury. Toxicol Sci 2006; 94:217–25. 10.1093/toxsci/kfl077 [DOI] [PubMed] [Google Scholar]
- 38.Bajt ML, Farhood A, Lemasters JJ, Jaeschke H. Mitochondrial Bax Translocation Accelerates DNA Fragmentation and Cell Necrosis in a Murine Model of Acetaminophen Hepatotoxicity. J Pharmacol Exp Ther 2008;324:8–14. 10.1124/jpet.107.129445 [DOI] [PubMed] [Google Scholar]
- 39.Gujral JS, Knight TR, Farhood A, Bajt ML, Jaeschke H. Mode of cell death after acetaminophen overdose in mice: apoptosis or oncotic necrosis? Toxicol Sci 2002;67:322–8. [DOI] [PubMed] [Google Scholar]
- 40.McGill MR, Yan HM, Ramachandran A, Murray GJ, Rollins DE, Jaeschke H. HepaRG cells: A human model to study mechanisms of acetaminophen hepatotoxicity. Hepatology 2011;53:974–82. 10.1002/hep.24132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boxill GC, Nash CB, Wheeler AG. Comparative pharmacological and toxicological evaluation of N-acetyl-p-aminophenol, salicylamide, and acetylsalicylic acid. J Am Pharm Assoc Am Pharm Assoc (Baltim) 1958;47:479–87. [DOI] [PubMed] [Google Scholar]
- 42.Boyd EM, Bereczky GM. Liver necrosis from paracetamol. Br J Pharmacol Chemother 1966;26:606–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eder H Chronic Toxicity Studies on Phenacetin, N-Acetyl-p-Aminophenol (NAPA) and Acetylsalicylic Acid on Cats. Acta Pharmacol Toxicol (Copenh) 1964;21:197–204. 10.1111/j.1600-0773.1964.tb01784.x [DOI] [PubMed] [Google Scholar]
- 44.Potter WZ, Thorgeirsson SS, Jollow DJ, Mitchell JR. Acetaminophen-Induced Hepatic Necrosis V. Correlation of Hepatic Necrosis, Covalent Binding and Glutathione Depletion in Hamsters. Pharmacology 1974;12:129–43. 10.1159/000136531 [DOI] [PubMed] [Google Scholar]
- 45.McGill MR, Williams CD, Xie Y, Ramachandran A, Jaeschke H. Acetaminophen-induced liver injury in rats and mice: comparison of protein adducts, mitochondrial dysfunction, and oxidative stress in the mechanism of toxicity. Toxicol Appl Pharmacol 2012;264:387–94. 10.1016/j.taap.2012.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davis M, Ideo G, Harrison NG, Williams R. Hepatic glutathione depletion and impaired bromosulphthalein clearance early after paracetamol overdose in man and the rat. Clin Sci Mol Med 1975;49:495–502. [DOI] [PubMed] [Google Scholar]
- 47.Xie Y, McGill MR, Dorko K, Kumer SC, Schmitt TM, Forster J, Jaeschke H. Mechanisms of acetaminophen-induced cell death in primary human hepatocytes. Toxicol Appl Pharmacol 2014;279:266–74. 10.1016/j.taap.2014.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.James LP, Farrar HC, Sullivan JE, Givens TG, Kearns GL, Wasserman GS, Walson PD, Hinson JA, Pumford NR, Pediatric Pharmacology Research Unit Network, NICHD. Measurement of acetaminophen-protein adducts in children and adolescents with acetaminophen overdoses. J Clin Pharmacol 2001;41:846–51. [DOI] [PubMed] [Google Scholar]
- 49.McGill MR, Staggs VS, Sharpe MR, Lee WM, Jaeschke H, Acute Liver Failure Study Group. Serum mitochondrial biomarkers and damage-associated molecular patterns are higher in acetaminophen overdose patients with poor outcome. Hepatology 2014;60:1336–45. 10.1002/hep.27265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bhattacharyya S, Yan K, Pence L, Simpson PM, Gill P, Letzig LG, Beger RD, Sullivan JE, Kearns GL, Reed MD, Marshall JD, Van Den Anker JN, James LP. Targeted liquid chromatography–mass spectrometry analysis of serum acylcarnitines in acetaminophen toxicity in children. Biomark Med 2014;8:147–59. 10.2217/bmm.13.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lutkewitte AJ, Schweitzer GG, Kennon-McGill S, Clemens MM, James LP, Jaeschke H, Finck BN, McGill MR.. Lipin deactivation after acetaminophen overdose causes phosphatidic acid accumulation in liver and plasma in mice and humans and enhances liver regeneration. Food Chem Toxicol 2018;115:273–83. 10.1016/j.fct.2018.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marques PE, Amaral SS, Pires DA, Nogueira LL, Soriani FM, Lima BHF, Lopes GAO, Russo RC, Ávila TV, Melgaço JG, Oliveira AG, Pinto MA, Lima CX, De Paula AM, Cara DC, Leite MF, Teixeira MM, Menezes GB. Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology 2012;56:1971–82. 10.1002/hep.25801 [DOI] [PubMed] [Google Scholar]
- 53.Williams CD, Bajt ML, Sharpe MR, McGill MR, Farhood A, Jaeschke H. Neutrophil activation during acetaminophen hepatotoxicity and repair in mice and humans. Toxicol Appl Pharmacol 2014;275:122–33. 10.1016/j.taap.2014.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 2012;55:222–32. doi: 10.1002/hep.24690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ni HM, McGill MR, Chao X, Du K, Williams JA, Xie Y, Jaeschke H, Ding WX. Removal of acetaminophen protein adducts by autophagy protects against acetaminophen-induced liver injury in mice. J Hepatol 2016;65:354–62. doi: 10.1016/j.jhep.2016.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Du K, McGill MR, Xie Y, Jaeschke H. Benzyl alcohol protects against acetaminophen hepatotoxicity by inhibiting cytochrome P450 enzymes but causes mitochondrial dysfunction and cell death at higher doses. Food Chem Toxicol 2015; 86:253–61. 10.1016/j.fct.2015.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Du K, Williams CD, McGill MR, Xie Y, Farhood A, Vinken M, Jaeschke H. The gap junction inhibitor 2-aminoethoxy-diphenyl-borate protects against acetaminophen hepatotoxicity by inhibiting cytochrome P450 enzymes and c-jun N-terminal kinase activation. Toxicol Appl Pharmacol 2013;273:484–91. 10.1016/j.taap.2013.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xie Y, Williams CD, McGill MR, Lebofsky M, Ramachandran A, Jaeschke H. Purinergic Receptor Antagonist A438079 Protects Against Acetaminophen-Induced Liver Injury by Inhibiting P450 Isoenzymes, Not by Inflammasome Activation. Toxicol Sci 2013;131:325–35. 10.1093/toxsci/kfs283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jaeschke H, Cover C, Bajt ML. Role of caspases in acetaminophen-induced liver injury. Life Sci 2006;78:1670–6. DOI: 10.1016/j.lfs.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 60.Kelava T, Cavar I, Culo F. Influence of small doses of various drug vehicles on acetaminophen-induced liver injury. Can J Physiol Pharmacol 2010;88:960–7. 10.1139/y10-065 [DOI] [PubMed] [Google Scholar]
- 61.Siegers CP. Antidotal effects of dimethyl sulphoxide against paracetamol-, bromobenzene-, and thioacetamide-induced hepatotoxicity. J Pharm Pharmacol 1978;30:375–7. [DOI] [PubMed] [Google Scholar]
- 62.Williams CD, McGill MR, Lebofsky M, Bajt ML, Jaeschke H. Protection against acetaminophen-induced liver injury by allopurinol is dependent on aldehyde oxidasemediated liver preconditioning. Toxicol Appl Pharmacol 2014;274:417–24. 10.1016/j.taap.2013.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ni HM, Boggess N, McGill MR, Lebofsky M, Borude P, Apte U, Jaeschke H, Ding WX. Liver-Specific Loss of Atg5 Causes Persistent Activation of Nrf2 and Protects Against Acetaminophen-Induced Liver Injury. Toxicol Sci 2012;127:438–50. 10.1093/toxsci/kfs133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Williams CD, McGill MR, Farhood A, Jaeschke H. Fas receptor-deficient lpr mice are protected against acetaminophen hepatotoxicity due to higher glutathione synthesis and enhanced detoxification of oxidant stress. Food Chem Toxicol 2013;58: 228–35. 10.1016/j.fct.2013.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu ZX, Han D, Gunawan B, Kaplowitz N. Neutrophil depletion protects against murine acetaminophen hepatotoxicity. Hepatology 2006;43:1220–30. DOI: 10.1002/hep.21175 [DOI] [PubMed] [Google Scholar]
- 66.Jaeschke H, Liu J. Neutrophil depletion protects against murine acetaminophen hepatotoxicity: another perspective. Hepatology 2007;45:1588–9. DOI: 10.1002/hep.21549 [DOI] [PubMed] [Google Scholar]
- 67.Saito C, Yan HM, Artigues A, Villar MT, Farhood A, Jaeschke H. Mechanism of protection by metallothionein against acetaminophen hepatotoxicity. Toxicol Appl Pharmacol 2010;242:182–90. doi: 10.1016/j.taap.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Harrill AH, Ross PK, Gatti DM, Threadgill DW, Rusyn I. Population-Based Discovery of Toxicogenomics Biomarkers for Hepatotoxicity Using a Laboratory Strain Diversity Panel. Toxicol Sci 2009;110:235–43. 10.1093/toxsci/kfp096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nickel AG, von Hardenberg A, Hohl M, Löffler JR, Kohlhaas M, Becker J, Reil JC, Kazakov A, Bonnekoh J, Stadelmaier M, Puhl SL, Wagner M, Bogeski I, Cortassa S, Kappl R, Pasieka B, Lafontaine M, Lancaster CR, Blacker TS, Hall AR, Duchen MR, Kästner L, Lipp P, Zeller T, Müller C, Knopp A, Laufs U, Böhm M, Hoth M, Maack C. Reversal of Mitochondrial Transhydrogenase Causes Oxidative Stress in Heart Failure. Cell Metab 2015;22:472–84. doi: 10.1016/j.cmet.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 70.Bourdi M, Davies JS, Pohl LR. Mispairing C57BL/6 Substrains of Genetically Engineered Mice and Wild-Type Controls Can Lead to Confounding Results as It Did in Studies of JNK2 in Acetaminophen and Concanavalin A Liver Injury. Chem Res Toxicol 2011;24:794–96. 10.1021/tx200143x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Duan L, Davis JS, Woolbright BL, Du K, Cahkraborty M, Weemhoff J, Jaeschke H, Bourdi M. Differential susceptibility to acetaminophen-induced liver injury in sub-strains of C57BL/6 mice: 6N versus 6J. Food Chem Toxicol 2016;98:107–18. 10.1016/j.fct.2016.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yohe HC, O’Hara KA, Hunt JA, Kitzmiller TJ, Wood SG, Bement JL, Bement WJ, Szakacs JG, Wrighton SA, Jacobs JM, Kostrubsky V, Sinclair PR, Sinclair JF. Involvement of Toll-like receptor 4 in acetaminophen hepatotoxicity. Am J Physiol Gastrointest Liver Physiol 2006;290:G1269–79. 10.1152/ajpgi.00239.2005 [DOI] [PubMed] [Google Scholar]
- 73.Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology 2012;143:1158–72. doi: 10.1053/j.gastro.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 74.Jaeschke H, Williams CD, Ramachandran A, Bajt ML. Acetaminophen hepatotoxicity and repair: the role of sterile inflammation and innate immunity. Liver Int 2012;32:8–20. doi: 10.1111/j.1478-3231.2011.02501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Woolbright B, Jaeschke H. The impact of sterile inflammation in acute liver injury. J Clin Transl Res 2017;3:170–88. 10.18053/jctres.03.2017S1.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Antoniades CG, Quaglia A, Taams LS, Mitry RR, Hussain M, Abeles R, Possamai LA, Bruce M, McPhail M, Starling C, Wagner B, Barnardo A, Pomplun S, Auzinger G, Bernal W, Heaton N, Vergani D, Thursz MR, Wendon J. Source and characterization of hepatic macrophages in acetaminophen-induced acute liver failure in humans. Hepatology 2012;56:735–46. doi: 10.1002/hep.25657. [DOI] [PubMed] [Google Scholar]
- 77.Weber LW, Boll M, Stampfl A. Hepatotoxicity and mechanism of action of haloalkanes: carbon tetrachloride as a toxicological model. Crit Rev Toxicol 2003;33:105–36. 10.1080/713611034 [DOI] [PubMed] [Google Scholar]
- 78.Slater TF. Necrogenic action of carbon tetrachloride in the rat: a speculative mechanism based on activation. Nature 1966;209: 36–40. [DOI] [PubMed] [Google Scholar]
- 79.Knockaert L, Berson A, Ribault C, Prost P-E, Fautrel A, Pajaud J, Lepage S, Lucas-Clerc C, Bégué J-M, Fromenty B, Robin M-A. Carbon tetrachloride-mediated lipid peroxidation induces early mitochondrial alterations in mouse liver. Lab Invest 2012;92: 396–410. 10.1038/labinvest.2011.193 [DOI] [PubMed] [Google Scholar]
- 80.Díaz Gómez MI, de Castro CR, D’Acosta N, de Fenos OM, de Ferreyra EC, Castro JA. Species differences in carbon tetrachloride-induced hepatotoxicity: the role of CCl4 activation and of lipid peroxidation. Toxicol Appl Pharmacol 1975;34:102–14. [DOI] [PubMed] [Google Scholar]
- 81.Thrall KD, Vucelick ME, Gies RA, Zangar RC, Weitz KK, Poet TS, Springer DL, Grant DM, Benson JM. Comparative metabolism of carbon tetrachloride in rats, mice, and hamsters using gas uptake and PBPK modeling. J Toxicol Environ Health A 2000;60:531–48. [DOI] [PubMed] [Google Scholar]
- 82.Teschke R Liver injury by carbon tetrachloride intoxication in 16 patients treated with forced ventilation to accelerate toxin removal via the lungs: A clinical report. Toxics 2018;6(2). pii: E25. doi: 10.3390/toxics6020025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Castro JA, De Ferreyra EC, De Castro CR, De Fenos OM, Sasame H, Gillette JR. Prevention of carbon tetrachloride-induced necrosis by inhibitors of drug metabolism--further studies on their mechanism of action. Biochem Pharmacol 1974;23:295–302. [DOI] [PubMed] [Google Scholar]
- 84.Randle LE, Goldring CEP, Benson CA, Metcalfe PN, Kitteringham NR, Park BK, Williams DP. Investigation of the effect of a panel of model hepatotoxins on the Nrf2-Keap1 defence response pathway in CD-1 mice. Toxicology 2008;243:249–60. 10.1016/j.tox.2007.10.011 [DOI] [PubMed] [Google Scholar]
- 85.Xu W, Hellerbrand C, Köhler UA, Bugnon P, Kan YW, Werner S, Beyer TA. The Nrf2 transcription factor protects from toxin-induced liver injury and fibrosis. Lab Investig 2008;88:1068–78. 10.1038/labinvest.2008.75 [DOI] [PubMed] [Google Scholar]
- 86.McCracken JM, Chalise P, Briley SM, Dennis KL, Jiang L, Duncan FE, Pritchard MT. C57BL/6 Substrains Exhibit Different Responses to Acute Carbon Tetrachloride Exposure: Implications for Work Involving Transgenic Mice. Gene Expr 2017;17:187–205. https://doi:10.3727/105221617X695050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tsukamoto H, Machida K, Dynnyk A, Mkrtchyan H. “Second Hit” Models of Alcoholic Liver Disease. Semin Liver Dis 2009;29:178–87. 10.1055/s-0029-1214373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chiang DJ, Roychowdhury S, Bush K, McMullen MR, Pisano S, Niese K, Olman MA, Pritchard MT, Nagy LE. Adenosine 2A receptor antagonist prevented and reversed liver fibrosis in a mouse model of ethanol-exacerbated liver fibrosis. PLoS One 2013;8:e69114 DOI: 10.1371/journal.pone.0069114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hall PD, Plummer JL, Ilsley AH, Cousins MJ. Hepatic fibrosis and cirrhosis after chronic administration of alcohol and “low-dose” carbon tetrachloride vapor in the rat. Hepatology 1991;13:815–9. [DOI] [PubMed] [Google Scholar]
- 90.Tsuchida T, Lee YA, Fujiwara N, Ybanez M, Allen B, Martins S, Fiel MI, Goossens N, Chou HI, Hoshida Y, Friedman SL. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J Hepatol 2018;69:385–95. 10.1016/j.jhep.2018.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kubota N, Kado S, Kano M, Masuoka N, Nagata Y, Kobayashi T, Miyazaki K, Ishikawa F. A high-fat diet and multiple administration of carbon tetrachloride induces liver injury and pathological features associated with non-alcoholic steatohepatitis in mice. Clin Exp Pharmacol Physiol 2013;40:422–30. 10.1111/1440-1681.12102 [DOI] [PubMed] [Google Scholar]
- 92.Hajovsky H, Hu G, Koen Y, Sarma D, Cui W, Moore DS, Staudinger JL, Hanzlik RP. Metabolism and toxicity of thioacetamide and thioacetamide S-oxide in rat hepatocytes. Chem Res Toxicol. 2012. September 17;25(9):1955–63. doi: 10.1021/tx3002719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hunter AL, Holscher MA, Neal RA. Thioacetamide-induced hepatic necrosis. I. Involvement of the mixed-function oxidase enzyme system. J Pharmacol Exp Ther 1977;200:439–48. [PubMed] [Google Scholar]
- 94.Kang JS, Wanibuchi H, Morimura K, Wongpoomchai R, Chusiri Y, Gonzalez FJ, Fukushima S. Role of CYP2E1 in thioacetamide-induced mouse hepatotoxicity. Toxicol Appl Pharmacol 2008;228:295–300. 10.1016/j.taap.2007.11.010 [DOI] [PubMed] [Google Scholar]
- 95.Belinsky SA, Bradford BU, Forman DT, Glassman EB, Felder MR, Thurman RG. Hepatotoxicity due to allyl alcohol in deermice depends on alcohol dehydrogenase. Hepatology 1985;5:1179–82. 10.1002/hep.1840050619 [DOI] [PubMed] [Google Scholar]
- 96.Brodie BB, Reid WD, Cho AK, Sipes G, Krishna G, Gillette JR. Possible mechanism of liver necrosis caused by aromatic organic compounds. Proc Natl Acad Sci USA 1971;68:160–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.McGill MR, Du K, Xie Y, Bajt ML, Ding WX, Jaeschke H. The role of the c-Jun Nterminal kinases 1/2 and receptor-interacting protein kinase 3 in furosemide-induced liver injury. Xenobiotica 2015;45:442–9. 10.3109/00498254.2014.986250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Williams DP, Antoine DJ, Butler PJ, Jones R, Randle L, Payne A, Howard M, Gardner I, Blagg J, Park BK. The Metabolism and Toxicity of Furosemide in the Wistar Rat and CD-1 Mouse: a Chemical and Biochemical Definition of the Toxicophore. J Pharmacol Exp Ther 2007;322:1208–20. 10.1124/jpet.107.125302 [DOI] [PubMed] [Google Scholar]
- 99.Jaeschke H, Kleinwaechter C, Wendel A. The role of acrolein in allyl alcohol-induced lipid peroxidation and liver cell damage in mice. Biochem Pharmacol 1987;36:51–7. [DOI] [PubMed] [Google Scholar]
- 100.Leist M, Gantner F, Bohlinger I, Tiegs G, Germann PG, Wendel A. Tumor necrosis factor-induced hepatocyte apoptosis precedes liver failure in experimental murine shock models. Am J Pathol 1995;146:1220–34. [PMC free article] [PubMed] [Google Scholar]
- 101.Bechmann LP, Marquitan G, Jochum C, Saner F, Gerken G, Canbay A. Apoptosis versus necrosis rate as a predictor in acute liver failure following acetaminophen intoxication compared with acute-on-chronic liver failure. Liver Int 2008;28:713–6. doi: 10.1111/j.1478-3231.2007.01566.x. [DOI] [PubMed] [Google Scholar]
- 102.Jaeschke H, McGill MR, Ramachandran A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: Lessons learned from acetaminophen hepatotoxicity. Drug Metab Rev 2012;44: 88–106. 10.3109/03602532.2011.602688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pessayre D, Fromenty B, Berson A, Robin MA, Lettéron P, Moreau R, Mansouri A. Central role of mitochondria in drug-induced liver injury. Drug Metab Rev 2012;44:34–87. 10.3109/03602532.2011.604086 [DOI] [PubMed] [Google Scholar]
- 104.Ramachandran A, Duan L, Akakpo JY, Jaeschke H. Mitochondrial dysfunction as a mechanism of drug-induced hepatotoxicity: current understanding and future perspectives. J Clin Transl Res, in press. 10.18053/jctres.04.201801.0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Leach BE, Forbes JC. Sulfonamide Drugs as Protective Agents Against Carbon Tetrachloride Poisoning. Exp Biol Med (Maywood) 1941;48:361–3. [Google Scholar]
- 106.Roth RA, Harkema JR, Pestka JP, Ganey P. Is exposure to bacterial endotoxin a determinant of susceptibility to intoxication from xenobiotic agents? Toxicol Appl Pharmacol 1997;147:300–11. 10.1006/taap.1997.8301 [DOI] [PubMed] [Google Scholar]
- 107.Dickes R, Schenker V, Deutsch L. Serial Liver-Function and Blood Studies in Patients Receiving Chlorpromazine. N Engl J Med 1957;256:1–7. 10.1056/NEJM195701032560101 [DOI] [PubMed] [Google Scholar]
- 108.Larrey D, Ripault M-P. Hepatotoxicity of psychotropic drugs and drugs of abuse, in: Kaplowitz N, DeLeve L (Eds.), Drug-Induced Liver Disease. Academic Press, Waltham, pp. 443–462, 2013. [Google Scholar]
- 109.Buchweitz JP, Ganey PE, Bursian SJ, Roth RA. Underlying endotoxemia augments toxic responses to chlorpromazine: is there a relationship to drug idiosyncrasy? J Pharmacol Exp Ther 2002;300:460–7. [DOI] [PubMed] [Google Scholar]
- 110.Deng X, Stachlewitz RF, Liguori MJ, Blomme EAG, Waring JF, Luyendyk JP, Maddox JF, Ganey PE, Roth RA. Modest Inflammation Enhances Diclofenac Hepatotoxicity in Rats: Role of Neutrophils and Bacterial Translocation. J Pharmacol Exp Ther 2006;319:1191–1199. 10.1124/jpet.106.110247 [DOI] [PubMed] [Google Scholar]
- 111.Dugan CM, MacDonald AE, Roth RA, Ganey PE. A mouse model of severe halothane hepatitis based on human risk factors. J Pharmacol Exp Ther 2010;333:364–72. 10.1124/jpet.109.164541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lu J, Jones AD, Harkema JR, Roth RA, Ganey PE. Amiodarone Exposure During Modest Inflammation Induces Idiosyncrasy-like Liver Injury in Rats: Role of Tumor Necrosis Factor-alpha. Toxicol Sci 2012;125:126–33. 10.1093/toxsci/kfr266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Luyendyk JP, Maddox JF, Cosma GN, Ganey PE, Cockerell GL, Roth RA. Ranitidine Treatment during a Modest Inflammatory Response Precipitates Idiosyncrasy-Like Liver Injury in Rats. J Pharmacol Exp Ther 2003;307: 9–16. 10.1124/jpet.103.054288 [DOI] [PubMed] [Google Scholar]
- 114.Shaw PJ, Hopfensperger MJ, Ganey PE, Roth RA. Lipopolysaccharide and trovafloxacin coexposure in mice causes idiosyncrasy-like liver injury dependent on tumor necrosis factor-alpha. Toxicol Sci 2007;100:259–66. 10.1093/toxsci/kfm218 [DOI] [PubMed] [Google Scholar]
- 115.Uetrecht J, Naisbitt DJ. Idiosyncratic Adverse Drug Reactions: Current Concepts Pharmacol Rev 2013;65:779–808. 10.1124/pr.113.007450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mosedale M, Watkins P. Drug-induced liver injury: Advances in mechanistic understanding that will inform risk management. Clin Pharmacol Ther 2017;101:469–80. 10.1002/cpt.564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Maddox JF, Amuzie CJ, Li M, Newport SW, Sparkenbaugh E, Cuff CF, Pestka JJ, Cantor GH, Roth RA, Ganey PE. Bacterial- and viral-induced inflammation increases sensitivity to acetaminophen hepatotoxicity. J Toxicol Environ Health A 2010;73:58–73. doi: 10.1080/15287390903249057. [DOI] [PubMed] [Google Scholar]
- 118.Williams CD, Bajt ML, Farhood A, Jaeschke H. Acetaminophen-induced hepatic neutrophil accumulation and inflammatory liver injury in CD18-deficient mice. Liver Int 2010;30:1280–92. doi: 10.1111/j.1478-3231.2010.02284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pastor Rojo O, López San Román A, Albéniz Arbizu E, de la Hera Martínez A, Ripoll Sevillano E, Albillos Martínez A. Serum lipopolysaccharide-binding protein in endotoxemic patients with inflammatory bowel disease. Inflamm Bowel Dis.2007;13(3):269–77. 10.1002/ibd.20019 [DOI] [PubMed] [Google Scholar]
- 120.Metushi IG, Cai P, Dervovic D, Liu F, Lobach A, Nakagawa T, Uetrecht J. Development of a novel mouse model of amodiaquine-induced liver injury with a delayed onset. J Immunotoxicol 2015;12:247–60. 10.3109/1547691X.2014.934977 [DOI] [PubMed] [Google Scholar]
- 121.Metushi IG, Hayes MA, Uetrecht J. Treatment of PD-1 −/− mice with amodiaquine and anti-CTLA4 leads to liver injury similar to idiosyncratic liver injury in patients. Hepatology 2015; 61:1332–42. 10.1002/hep.27549 [DOI] [PubMed] [Google Scholar]
- 122.Chakraborty M, Fullerton AM, Semple K, Chea LS, Proctor WR, Bourdi M, Kleiner DE, Zeng X, Ryan PM, Dagur PK, Berkson JD, Reilly TP, Pohl LR. Drug-induced allergic hepatitis develops in mice when myeloid-derived suppressor cells are depleted prior to halothane treatment. Hepatology 2015;62:546–57. 10.1002/hep.27764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ong MMK, Latchoumycandane C, Boelsterli UA. Troglitazone-Induced Hepatic Necrosis in an Animal Model of Silent Genetic Mitochondrial Abnormalities. Toxicol Sci 2007; 97:205–13. 10.1093/toxsci/kfl180 [DOI] [PubMed] [Google Scholar]
- 124.Fujimoto K, Kumagai K, Ito K, Arakawa S, Ando Y, Oda S, Yamoto T, Manabe S. Sensitivity of liver injury in heterozygous Sod2 knockout mice treated with troglitazone or acetaminophen. Toxicol Pathol 2009;37:193–200. 10.1177/0192623308329282 [DOI] [PubMed] [Google Scholar]
- 125.Hsiao CJ, Younis H, Boelsterli UA. Trovafloxacin, a fluoroquinolone antibiotic with hepatotoxic potential, causes mitochondrial peroxynitrite stress in a mouse model of underlying mitochondrial dysfunction. Chem Biol Interact 2010;188:204–13. 10.1016/j.cbi.2010.07.017 [DOI] [PubMed] [Google Scholar]
- 126.Kashimshetty R, Desai VG, Kale VM, Lee T, Moland CL, Branham WS, New LS, Chan ECY, Younis H, Boelsterli UA. Underlying mitochondrial dysfunction triggers flutamide-induced oxidative liver injury in a mouse model of idiosyncratic drug toxicity. Toxicol Appl Pharmacol 2009;238:150–9. 10.1016/j.taap.2009.05.007 [DOI] [PubMed] [Google Scholar]
- 127.Ong MMK, Wang AS, Leow KY, Khoo YM, Boelsterli UA. Nimesulide-induced hepatic mitochondrial injury in heterozygous Sod2+/− mice. Free Radic Biol Med 2006;40:420–9. 10.1016/j.freeradbiomed.2005.08.038 [DOI] [PubMed] [Google Scholar]
- 128.Lucena MI, García-Martín E, Andrade RJ, Martínez C, Stephens C, Ruiz JD, Ulzurrun E, Fernandez MC, Romero-Gomez M, Castiella A, Planas R, Durán JA, De Dios AM, Guarner C, Soriano G, Borraz Y, Agundez JAG. Mitochondrial superoxide dismutase and glutathione peroxidase in idiosyncratic drug-induced liver injury. Hepatology 2010;52:303–12. 10.1002/hep.23668 [DOI] [PubMed] [Google Scholar]
- 129.Nicoletti P, Aithal GP, Bjornsson ES, Andrade RJ, Sawle A, Arrese M, Barnhart HX, Bondon-Guitton E, Hayashi PH, Bessone F, Carvajal A, Cascorbi I, Cirulli ET, Chalasani N, Conforti A, Coulthard SA, Daly MJ, Day CP, Dillon JF, Fontana RJ, Grove JI, Hallberg P, Hernández N, Ibáñez L, Kullak-Ublick GA, Laitinen T, Larrey D, Lucena MI, Maitland-van der Zee AH, Martin JH, Molokhia M, Pirmohamed M, Powell EE, Qin S, Serrano J, Stephens C, Stolz A, Wadelius M, Watkins PB, Floratos A, Shen Y, Nelson MR, Urban TJ, Daly AK, International Drug-Induced Liver Injury Consortium, Drug-Induced Liver Injury Network Investigators, and International Serious Adverse Events Consortium. Association of Liver Injury From Specific Drugs, or Groups of Drugs, With Polymorphisms in HLA and Other Genes in a Genome-Wide Association Study. Gastroenterology 2017;152:1078–89. 10.1053/j.gastro.2016.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.McGill MR, Jaeschke H. Biomarkers of drug-induced liver injury: progress and utility in research, medicine, and regulation. Expert Rev Mol Diagn. 2018. doi: 10.1080/14737159.2018.1508998. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Turpeinen M, Ghiciuc C, Opritoui M, Tursas L, Pelkonen O, Pasanen M. Predictive value of animal models for human cytochrome P450 (CYP)-mediated metabolism: a comparative study in vitro. Xenobiotica. 2007;37:1367–77. 10.1080/00498250701658312 [DOI] [PubMed] [Google Scholar]
- 132.Trepanier LA, Ray K, Winand NJ, Spielberg SP, Cribb AE. Cytosolic arylamine Nacetyltransferase (NAT) deficiency in the dog and other canids due to an absence of NAT genes. Biochem Pharmacol. 1997;54:73–80. 10.1016/S0006-2952(97)00140-8 [DOI] [PubMed] [Google Scholar]
- 133.Trepanier LA. Idiosyncratic toxicity associated with potentiated sulfonamides in the dog. J Vet Pharmacol Ther. 2004;27:129–38. 10.1111/j.1365-2885.2004.00576.x [DOI] [PubMed] [Google Scholar]
- 134.Court MH, Greenblatt DJ. Molecular genetic basis for deficient acetaminophen glucuronidation by cats: UGT1A6 is a pseudogene, and evidence for reduced diversity of expressed hepatic UGT1A isoforms. Pharmacogenetics. 2000;10:355–69. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.