

Abstract
The age of personalized medicine continues to evolve within clinical oncology with the arsenal available to clinicians in a variety of malignancies expanding at an exponential rate. The development and advancement of molecular treatment modalities, including targeted therapy and immune checkpoint blockade, continue to flourish. Treatment with targeted therapy (BRAF, MEK, & other small molecule inhibitors) can be associated with swift disease control and high response rates, but limited durability when used as monotherapy. Conversely, treatment with immune checkpoint blockade monotherapy regimens (anti-CTLA-4 & anti-PD1/PDL1) tend to have lower response rates than those observed with BRAF-targeted therapy, though these treatments may offer long term durable disease control. With the advent of these forms of therapy, there was interest early on in empirically combining targeted therapy with immune checkpoint blockade with the hopes of preserving high response rates and adding durability, however there is now strong scientific rationale for combining these forms of therapy – and early evidence of synergy in pre-clinical models of melanoma. Clinical trials combining these strategies are ongoing, and mature data regarding response rates and durability are not yet available. Synergy may ultimately be apparent, however it has also become clear that complexities exist regarding toxicity when combining these therapies. Nonetheless, this increased appreciation of the complex interplay between oncogenic mutations and anti-tumor immunity has opened up tremendous opportunities for studying targeted agents and immunotherapy in combination, which extends far beyond melanoma to other solid tumors and also to hematologic malignancies.
Keywords: Melanoma, checkpoint blockade, targeted therapy, immunotherapy, clinical trials
Introduction
The last decade have seen a renaissance in clinical oncology. The development of highly specific, potent, and durable molecularly targeted and immune therapies continues to transform treatment for a variety of cancer types. One tumor type highlighting this renaissance in treatment is melanoma. Melanoma comprises <2% of all skin cancers, though it accounts for the majority of skin cancer-related deaths. Over 73,000 cases of malignant melanoma will be diagnosed in 2015 and its incidence has continued to rise for the past thirty years (1).
Until 2011, the only Food and drug administration (FDA)-approved treatments for metastatic melanoma were dacarbazine and high dose interleukin-2 (IL-2). However, within the past 5 years, ten new regimens have been FDA-approved for the treatment of stage IV melanoma (2–11), including molecularly-targeted agents as well as immunomodulatory agents (Figure 1), both as monotherapy as well as in combination. The improved survival associated with these regimens is groundbreaking (3, 12), and these agents are now being tested in earlier stage disease.
Figure 1. Timeline showing FDA approval of therapeutic regimens for melanoma.
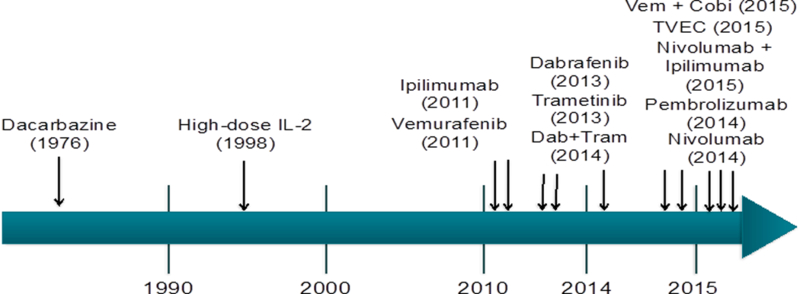
For 35 years only two systemic therapies existed for melanoma. The approval of ipilimumab in 2011 marked the beginning of an exponential approval of treatment regimens with 10 new regimens approved over the course of 5 years.
Despite these tremendous advances, these forms of therapy have limitations. Treatment with molecularly targeted therapy (such as with BRAF inhibitors or combined BRAF and MEK inhibitors) is associated with a high overall response rate but a limited duration of response due to the emergence of therapeutic resistance (Figure 2) (13). Conversely, treatment with immunotherapy is typically associated with a lower response rate than that observed with targeted therapy – however when responses are seen they tend to be more durable (Figure 2) (12). Given these limitations there was interest early on in combining targeted therapy and immunotherapy clinically to abrogate the weaknesses and capitalize on the strengths of each of these regimens, with the hopes of achieving high response rates with durable responses to therapy (Figure 2). In addition to this strong clinical rationale for combining these treatment modalities, there is now a strong scientific rationale to combine targeted therapy and immunotherapy for the treatment of cancer (14, 15). These concepts will be discussed herein, and limitations and promise of these combinations in other forms of cancer will also be discussed.
Figure 2. Response rate and duration of targeted and immunotherapy, as well as anticipated combination.
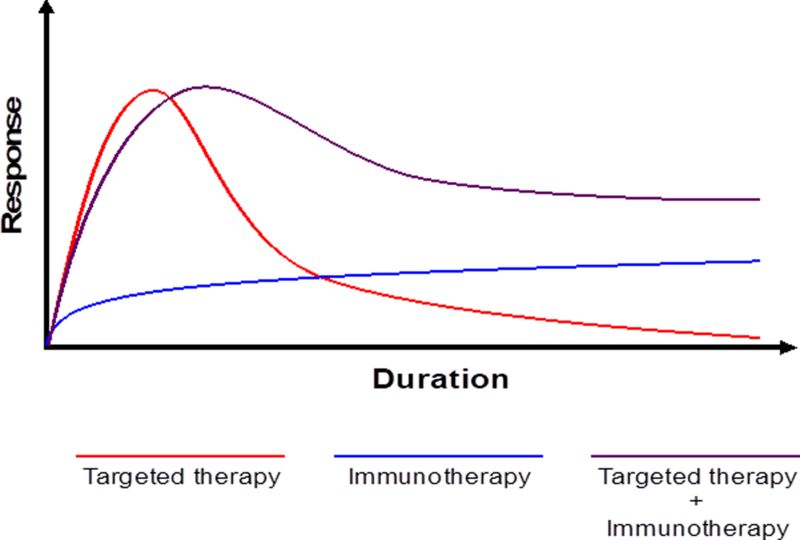
Targeted therapies with BRAF and MEK inhibitors provide rapid responses in the majority of treated patients, though these responses tend to be short-lived (red line). Treatment with checkpoint blockade benefits fewer patients, though those who do tend to do so durably (blue line). Preliminary evidence provides rationale for combining these forms of therapy and suggests their combination (purple line) may provide durable responses in a larger number of patients.
Summary of targeted therapy for melanoma
Within the last few decades, we have seen major advances in the treatment of cancer through a better understanding of genomic events contributing to carcinogenesis and mechanisms of therapeutic resistance. This is particularly evident in melanoma, with evidence of genomic alterations implicated in the process of melanomagenesis (16) and also in the setting of metastatic disease (17). Major oncogenic driver mutations have been identified in melanoma, including activating mutation in the BRAF oncogene (18, 19) with over half of metastases from cutaneous melanoma harboring activation point mutations at the V600 position. Activating mutations in the BRAF gene lead to constitutive signaling down the mitogen-activated protein kinase (MAPK) signaling pathway leading cellular proliferation and survival (18) as well as several other deleterious effects – including immune escape (Figure 3) (14).
Figure 3. Effects of BRAF mutations on tumor cell survival and the tumor microenvironment.
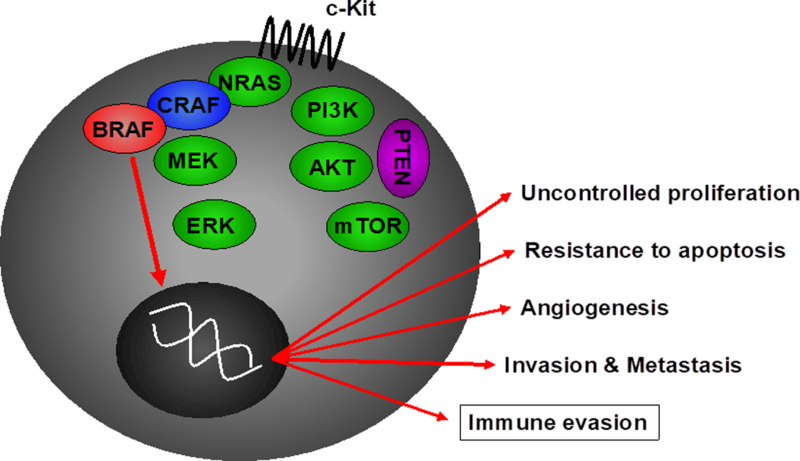
Oncogenic mutations in the BRAF gene lead to constitutive signaling down the MAPK pathway leading to multiple deleterious effects, including tumor cell proliferation, angiogenesis, invasion and metastasis, and resistance to apoptosis. Importantly, this can also lead to immune evasion, and immunogenicity may be increased through treatment with BRAF inhibitors.
Initial studies of pharmacologic agents targeting the BRAF oncogene (such as sorafenib) demonstrated a lack of specificity and limited clinical efficacy with significant toxicity (20). However, more specific agents were developed (such as Plexxikon 4032) that specifically target the V600 mutation, and these agents demonstrated significant promise in phase I studies (9, 10). These compounds were brought forward to phase III clinical trials, and demonstrated a higher response rate and significant improvement in overall survival in patients treated with BRAF inhibitor therapy (vemurafenib) compared to those treated with then standard of care dacarbazine (response rate of 48% vs. 5% and overall survival of 84% vs. 64% at 6 months, respectively) (21). Dabrafenib (GlaxoSmithKline), another agent targeting BRAFV600 mutant melanoma, demonstrated similarly high OR rates in a phase III trial (8) providing the basis for approval of each of these agents as monotherapy in 2011 and 2013, respectively (8).
Importantly, these forms of therapy require mutations in BRAF-V600, thus they are ineffective in half of patients with metastatic melanoma (i.e. in those whose tumors lack a BRAF mutation). In these patients, other targeted agents have been studied – including molecularly targeted therapy again MEK – a kinase downstream of RAF in the MAPK pathway. MEK inhibitors have demonstrated benefit as monotherapy in patients with BRAF-mutant metastatic melanoma (9), leading to the FDA approval of trametinib in 2013. These agents are now being tested as monotherapy and combinations (NCT01584648) in the treatment of BRAF wild type melanoma and also in other cancer types, however their efficacy is limited by a narrow therapeutic index with current agents (9, 10).
However despite the initially striking responses seen with BRAF-targeted therapy, therapeutic resistance and subsequent progression became the Achilles heel of BRAF targeted therapy, with a median time to progression of 6 months for BRAF inhibitor monotherapy (8, 21). Only a small fraction of patients experience a durable complete response to molecularly-targeted therapy (22), and efforts are underway to better understand which patients will achieve long-term benefit with BRAF-targeted therapy.
Numerous studies have elucidated acquired mechanisms of therapeutic resistance to BRAF-targeted therapy (23), and insights gained have led to treatment strategies to enhance responses to therapy. A prime example of this is the use of combined BRAF and MEK inhibition - based on the fact that a majority of BRAF-mutant melanomas demonstrate MAPK pathway reactivation at time of therapeutic resistance (23). This combination was tested in clinical trials in patients with BRAF-mutant melanoma, and results suggested that treatment with combined BRAF and MEK inhibition (dabrafenib plus trametinib vs. dabrafenib monotherapy) was associated with a higher overall response (76% versus 54%), and with a longer progression-free survival (9.4 vs. 5.8 months) (9) than with BRAF inhibitor monotherapy. In January 2014, the FDA granted accelerated approval to combined dabrafenib and trametinib for use in combination in patients with unresectable or metastatic BRAF-mutant melanoma. The following year, the combination of vemurafenib and cobimetinib was FDA approved (5).
Further strategies combining different molecularly-targeted agents focused on alternate signaling pathways (such as PI3K) are also in development (24), though mature data regarding the efficacy of these regimens is not available. Nonetheless it is becoming clear that combination strategies will be required in the majority of patients to combat de novo and acquired resistance. Importantly molecularly targeted agents (either as monotherapy or in combination) are also being used in multiple other tumor types (25), thus lessons learned through the experience with molecularly targeted therapy for melanoma (with regard to mechanisms of therapeutic resistance, immune effects of targeted agents, and strategies to overcome resistance) may be translatable to other tumor types.
Immune effects of targeted therapy
Though molecularly-targeted agents were developed to specifically inhibit tumor cell growth and proliferation via modulation of cell-autonomous pathways, studies by our group and others have demonstrated that these agents may also have an effect on other tumor cell intrinsic features (such as antigen processing and presentation) (19, 26, 27) and may also have profound effects on the tumor microenvironment – including on anti-tumor immunity (26).
We first began studying this nearly a decade ago, and demonstrated that MAPK pathway inhibition in melanoma cells was associated with enhanced melanoma antigen expression and reactivity to antigen-specific T lymphocytes (19). This phenomenon was observed in both BRAF-mutant melanoma cells (using selective BRAF inhibitors and / or MEK inhibitors) and also in BRAF-wild type cells (using MEK inhibitors). These studies provided the initial rationale for combining molecularly targeted therapy with immunotherapy in the treatment of melanoma, however also called the use of MEK inhibitors with immunotherapy into question – as we observed a significant decrease in T cell function when they were treated with MEK inhibitors in vitro (19). These in vitro studies were corroborated with in vivo data suggesting that treatment of patients with BRAF-targeted therapy in the setting of metastatic BRAF-mutant melanoma resulted in a much more favorable tumor microenvironment within 2 weeks of initiation of therapy – with increased melanoma antigen expression, increased CD8+ T cell infiltrate, decreased VEGF, and a decrease in immunosuppressive cytokines (26) – all suggesting immune mechanisms of response to BRAF-targeted therapy. Importantly, these changes were associated with enhanced cytotoxicity of CD8+ T cells with higher levels of granzyme B, and perforin, two crucial cytotoxic proteases involved in mediating T cell-induced tumor apoptosis. However simultaneously, there was an increase in PD-1 and Tim-3 expression on T lymphocytes and PD-L1 expression in the tumor microenvironment – suggesting an immune mechanism of resistance (28) – and providing the perfect rationale for combining BRAF-targeted therapy with immune checkpoint blockade.
It is important to note that the favorable immune responses to BRAF-targeted therapy appear to be early and transient (19, 26, 29), and are completely lost at time of progression – with lower levels of melanoma antigen expression and CD8+ T cells than seen before initiating therapy (26). Further work from our group assessing antigen specificity of these responses by T cell receptor sequencing suggests that treatment with BRAF-targeted therapy is associated with a more clonal T cell response in tumors, however differential responses are observed across patients (30). A large effort is now underway to better understand the complex interplay between host genomics and anti-tumor immunity in response to targeted therapy (31), though it is becoming increasingly clear that targeting oncogenic pathways can have a profound effect on anti-tumor immunity, and that significant genomic and immune mechanisms of resistance to these forms of therapy are at play.
One special consideration in understanding immune effects of targeted therapy involves the use of MEK inhibitors, as the MAPK pathway is known to be critical to T cell activation (32–34) and dendritic cell function. The potential for MEK inhibition to benefit patients is important, as this class of agents may be used on a broader patient population (in melanoma as well as in other malignancies). Though in vitro studies suggested that treatment with MEK inhibition leads to impaired T cell proliferation, cytokine secretion, and expansion of antigen-specific T cells, this appears to be heavily dependent on the timing of treatment as it relates to T cell activation (35), and subsequent studies in vivo have failed to demonstrate the same deleterious effects (36). However, additional studies need to be done and are currently underway. BRAF inhibitors do not appear to have any deleterious effects on T cell function, and may even potentiate T cell activation (37).
Together, the aforementioned studies provide a model of immune effects of targeted therapy with potential avenues of synergy with immune checkpoint blockade (Figure 4), however complexities may exist regarding optimal timing and sequence of therapy and need to be taking into consideration when contemplating combination strategies.
Figure 4. Effects of combined targeted and immunotherapy on the tumor microenvironment.
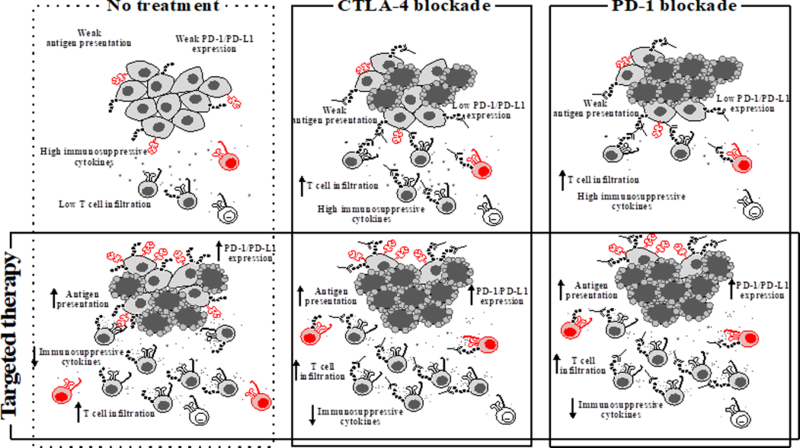
BRAF inhibition mediates numerous effects on the tumor microenvironment (TME), such as increases in melanocyte differentiation antigen expression, antigen presentation, T cell infiltration, granzyme B and perforin cytotoxicity, and PD-1/PD-L1 expression levels while decreasing levels of immunosuppressive cytokines in the TME. These changes provide rationale for combinations with checkpoint blockade immunotherapy through anti-CTLA-4 and anti-PD-1, as well as suggestion of an immune escape mechanism through PD-1/PD-L1 upregulation in the TME. Shown are the immune effects of targeted and immunotherapy as single regimens, as well as the effects when used in combination.
Summary of immunotherapy for melanoma (immune checkpoint blockade)
Immunotherapy as a whole can be broadly characterized as therapies that enhance the host’s cellular immune response to cancer. One of the first agents to be used in the treatment of metastatic melanoma was interleukin 2 (IL-2), which is a well-established T-cell stimulating molecule. The use of high dose IL-2 increases T-cell activation and proliferation in a broad, non-specific manner. Objective and impressively durable responses (OR) have been noted with IL-2 for metastatic melanoma as well as renal cell carcinoma (38–40), and although the complete response (CR) rate is low (6%), these CRs are often durable (beyond 20 years). However given its low overall response rate and high toxicity profile, this form of therapy is now infrequently used as monotherapy in the treatment of metastatic melanoma (41), and its use is reserved for combination with other strategies (such as in the treatment of melanoma using adoptive cell transfer) for its T cell stimulating properties.
A major breakthrough in the treatment of cancer has occurred in the past several years through the use of immune checkpoint blockade (ICB), a therapeutic strategy based on releasing inhibitory molecules that act upon the cytotoxic T-cell (Figure 5). Though these molecules are expressed as normal negative-feedback mechanisms to limit T cell autoreactivity (42), they may be aberrantly expressed in the setting of cancer, thereby facilitating immune escape (43). A deep mechanistic understanding of these molecules and their targeting has revolutionized the treatment of cancer over the past several years, and it is critical to have a deep mechanistic understanding of these to adequately conceptualize the design of combination strategies using these agents.
Figure 5. Costimulatory and inhibitory molecules and receptors.
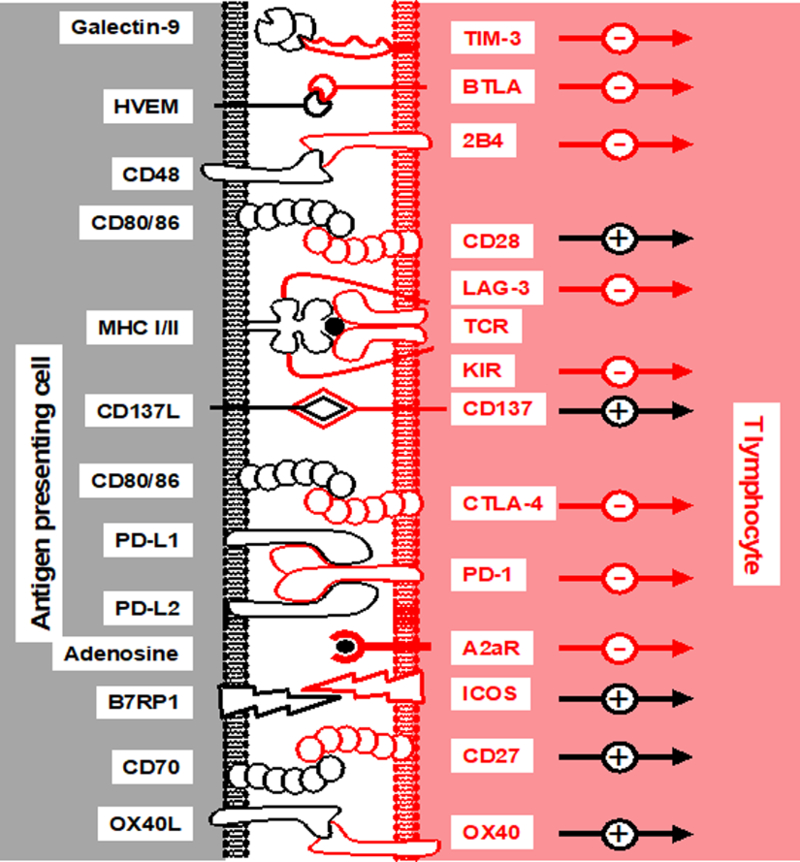
CD4 and CD8 T lymphocyte fate is dictated by interactions with costimulation and inhibitory molecules on the surface of antigen-presenting and tumor cells. Shown are these ligands and receptors, as well as the MHC-peptide/TCR interaction known as signal 1, as well as the impact of these interactions on T lymphocyte activation. +, activation; -, inhibition.
The first of these agents studied in the treatment of patients with stage IV melanoma was ipilimumab, a monoclonal antibody directed against cytotoxic T-lymphocyte antigen 4 (CTLA-4). CTLA-4 is a member of the immunoglobulin superfamily that regulates T-cell activation, which is expressed on the cell surface similarly to the T-cell co-stimulatory protein, CD28. CTLA-4 and CD28 competitively bind to B7–1 and B7–2 on antigen-presenting cells (APC) to determine T-cell fate, with CTLA-4 contributing an inhibitory signal (42). Work in the late 1990s by Allison et al., revealed that blockade of CTLA-4 binding could facilitate, and potentiate effective immune responses against tumor cells (44). Clinical trials in patients were subsequently conducted, and two large phase III trials showed a survival benefit in patients with metastatic melanoma treated with ipilimumab (3, 45). In a placebo-controlled trial, 676 patients were randomly assigned to ipilimumab plus a glycoprotein 100 (gp100) vaccine, ipilimumab alone, or gp100 alone. In these studies, overall survival (OS) was significantly higher in patients receiving ipilimumab than in those receiving gp100 alone. It was based on this survival benefit that Ipilimumab received FDA approval in 2011 for the treatment of stage IV melanoma.
The next major immune checkpoint axis to be studied was the programmed cell death protein-1 (PD-1) axis, which functions in the periphery to modulate T cell responses (46, 47). PD-1 interacts with 2 ligands that are members of the B7 like molecules (PD-L1 & PD-L2) (Figure 5) to dampen T cell responses, physiologically functioning to limit autoimmunity. The inhibitory effect of PD-1 is accomplished through a dual mechanism of promoting apoptosis in cytotoxic T-cells (programmed cell death, as the name implies) while simultaneously reducing apoptosis in regulatory T cells (Tregs) (48, 49). PD-1 protein is upregulated on activated T cells (46) with blockade of this molecule resulting in the cellular immune system’s anti-tumors activity by inhibiting this naturally occurring negative feedback mechanism.
The clinical use of PD-1 blocking antibodies has been tested in several clinical trials including in a phase I trial of 296 patients with either advanced melanoma or other solid tumors, which included non-small cell lung cancer, prostate cancer, renal cell carcinoma, and colorectal cancer (50). Treatment with a monoclonal antibody targeting PD-1 (nivolumab) was associated with a 28% response rate in patients with metastatic melanoma, with long-term responses longer than 1 year in 50% of responding patients. Anti-PD-1 based therapy was associated with a lower rate of grade 3 or 4 AEs compared with ipilimumab (51). Additional clinical trials have been performed (NCT01295827/NCT01704287), including a phase III trial with over 500 patients with metastatic melanoma demonstrating disease refractory to CTLA-4 blockade. In this trial (KEYNOTE-002) patients were randomized to treatment with pembrolizumab (2 mg/kg every three weeks), pembrolizumab (10 mg/kg every three weeks) or chemotherapy (carboplatin plus paclitaxel, paclitaxel alone, dacarbazine, or temozolomide per institutional standard) (52). Progression free survival (PFS) was the primary endpoint of the trial and was significantly improved with both pembrolizumab arms compared with chemotherapy, with the six-month progression-free rates at 34, 38, and 16 percent for pembrolizumab 2 mg/kg, pembrolizumab 10 mg/kg, and chemotherapy, respectively (52). Toxicity was limited in this trial, with the most common adverse events including fatigue, pruritus, and rash. Grade 3 immune related adverse events (IRAE) were seen in two patients treated with pembrolizumab 2 mg/kg (hepatitis, hypophysitis), and in eight patients given pembrolizumab 10 mg/kg (hepatitis, colitis, pneumonitis, and iritis or uveitis).
Given the differential mechanisms of action of CTLA-4 blockade and PD-1 blockade, their use together has been explored both pre-clinically (53) as well as in clinical trials (3, 4, 50), demonstrating enhanced anti-tumor activity in combination compared with monotherapy. However, the benefit comes at a cost associated with increased toxicity (2). The largest experiment combining anti-PD-1 and anti-CTLA-4 checkpoint inhibition comes from the CheckMate 067 phase III trial where over 900 treatment-naïve patients were randomly assigned to nivolumab (1 mg/kg every three weeks) plus ipilimumab (3 mg/kg every three weeks) for four doses followed by nivolumab (3 mg/kg every two weeks), nivolumab (3 mg/kg every two weeks), or ipilimumab (3 mg/kg every three weeks for four doses). The primary end points of this trial were PFS and OS. At 12 months (median follow-up) the median PFS with both the combination and nivolumab alone were superior to ipilimumab alone (11.5 versus 2.9 months and 6.9 versus 2.9 months, respectively). Not surprisingly, serious toxicities were more frequent with the combination than with monotherapy with either nivolumab or ipilimumab (2, 3, 11). Given these toxicities, additional combinations are being explored, including combinations with other novel checkpoint inhibitors and other modalities such as targeted therapy.
Combining Targeted Therapy and Immune Checkpoint Blockade: Rationale
Combination therapy in the cancer treatment is not a novel concept, as synergistic relationships have been described across multiple lines of therapy including chemotherapy, hormonal, and radiation therapies (54). Specifically in melanoma, developments in molecularly-targeted therapy and immune checkpoint blockade overlapped significantly from a temporal standpoint, and there was tremendous enthusiasm early on to empirically combine these treatment strategies in an effort to combat the limitations of each of these strategies and enhance responses to therapy (Figure 4). Over the past several years, there is now a growing scientific rationale to combine these strategies, and data supporting this concept will be discussed herein.
Preclinical studies of targeted therapy and checkpoint blockade immunotherapy
The earliest data supporting the combination of BRAF-targeted therapy and immune checkpoint blockade was published in 2010 (19), though there are now several hundred manuscripts in the literature exploring this concept (14, 15, 55, 56). Based on very strong data in humans regarding favorable effects of BRAF +/−MEK inhibitors on anti-tumor immunity and the tumor microenvironment (26, 57) as well as adaptive immune resistance to targeted therapy with an increase in immunomodulatory molecules such as PD-1 and PD-L1 (26), numerous murine studies have been performed to explore potential synergy of targeted therapy and immune checkpoint blockade in the treatment of melanoma.
One of the first of these studies published explored immune effects of targeted therapy, as well as potential synergy of targeted therapy and immune checkpoint blockade in a BRAF-mutant melanoma model (14). In these studies, CD8+ T cells were required for the response to BRAF-targeted therapy, and synergy was demonstrated when BRAF-targeted therapy was combined with immune checkpoint blockade against PD-1 (14). More recently, studies have explored BRAF and MEK inhibitors in combination with immune checkpoint inhibitors (15), and demonstrated synergy. Importantly, these studies also confirmed that combination therapy leads to increased homing of T cells to tumors, and also demonstrated that T cell function was not impaired by MEK inhibition in vivo (33). Additional studies are underway by numerous groups to better characterize the benefits and mechanisms of these combinations in mouse models.
As described previously, an important consideration in combining targeted therapy with immune checkpoint blockade is the effect of targeted agents on anti-tumor immunity. This has now been addressed in several studies (57), including one by Kakavand et al who sought to further study the effect of targeted therapy (BRAFi monotherapy vs BRAFi/MEKi) on PD-L1 expression and T-cell infiltrate in longitudinal tumor samples from patients with metastatic melanoma who were being treated with these agents. It was shown that PD-L1 expression highly correlated with TIL infiltrate, with BRAFi-treated patients similar increases in CD4 and CD8 T-cells compared to those receiving combined BRAF and MEK inhibition (29). The authors concluded that the addition of MEKi to BRAFi did not result in significant reduction in the immune infiltrate seen in early on treatment biopsies, and these findings are corroborated in other patient cohorts (26) as well as in well-designed murine studies (36). This again illustrates the potential advantage of performing clinical trials combining MAPK inhibition with immune checkpoint blockade (57).
Combining Targeted Therapy and Immune Checkpoint Blockade: Trials
Based on the observations regarding the immune effects of targeted therapy and compelling pre-clinical studies, there are currently several ongoing clinical trials (phase I/II) combining targeted therapy with either immune checkpoint blockade, radiation therapy, or cellular therapy (Table 1). To date, trials typically have used either a BRAF or immune checkpoint inhibitor backbone with or without MEK inhibition. Early data suggests potential synergy in these clinical trials (58), however mature data is not available and complexities exist regarding optimal timing, sequence and duration of therapy. Complexities also exist regarding toxicity of these combinations regimens, and will be discussed further below.
TABLE 1.
Clinical trials investigating combination targeted and immunotherapy.
| Targeted therapy + Checkpoint blockade | Targeted therapy + Cytokine | Targeted therapy + T cells | Targeted therapy + Radiation |
|---|---|---|---|
|
Dabrafenib ± Trametinib ± Ipilimumab (NCT01767454) |
Vemurafenib + High dose IL-2 (NCT01754376; NCT01683188) |
Vemurafenib + Tumor Infiltrating Lymphocytes (NCT00338377; NCT01585415; NCT01659151) |
Dabrafenib + Stereotactic Radiosurgery to the Brain (NCT01721603) |
|
Vemurafenib ± anti-PDL1 (NCT01656642) |
Vemurafenib + IL-2 (Infusional 96 hour) + IFN-γ (NCT01603212) |
Vemurafenib + Whole Brain Radiation or Radiosurgery to the Brain (NCT02145910) |
|
|
Dabrafenib ± Trametinib ± anti-PD1 (NCT02130466) |
Vemurafenib + Pegylated IFN-γ (NCT01959633) |
||
|
Trametinib ± Dabrafenib ± anti-PDL1 (NCT02027961) |
Vemurafenib + High dose IFNα-2b (NCT01943422) |
||
|
Anti-PDL1 + EGFR inhibitor (NCT024554933) |
|||
|
anti-PD1 ± Chemotherapy ± Ipilimumab ± Erlotinib ± Gefitinib (NCT02039674) |
|||
|
Ipilimumab + Imatinib (NCT01738139) |
|||
|
Vemurafenib + Ipilimumab (NCT01400451) |
|||
|
anti-PDL1 + Vemurafenib ± Cobimetinib (NCT01656642) |
One of the first trials exploring the use of combined BRAF-targeted therapy and immune checkpoint blockade began accruing patients in 2011 (NCT01400451). In this trial, patients were treated with a 4-week lead-in of BRAF inhibitor monotherapy (vemurafenib) and were then treated with anti-CTLA-4 blockade. Target accrual for this trial was 20, however the trial was closed early after 12 patients were accrued secondary to observed toxicity with the regimen (56). The dominant toxicity observed in this trial was hepatotoxicity with most patients experiencing grade 2–3 toxicity, and all toxicity being reversible either with temporary discontinuation of study drugs or with administration of corticosteroids (56). Though toxicity was admittedly not severe in this trial, it highlighted potential unexpected toxicities and the authors cautioned against the concurrent administration of vemurafenib and ipilimumab, particularly in light of the approval of both of these agents as monotherapy at time results of this trial were published. Importantly, analysis of CD8+ T cell infiltrate in longitudinal tumor samples from a patient on this trial demonstrated a low CD8+ T cell infiltrate at baseline with an increase in infiltrate at 2 weeks into BRAF-targeted therapy, however the infiltrate was lost at 4 weeks on therapy. Interestingly, the infiltrate was again seen with the initiation of immune checkpoint blockade, and persisted for several months (14).
A second trial exploring combined targeted therapy and CTLA-4 blockade was presented at the American Society of Clinical Oncology (ASCO) in 2014, and reported on 19 patients with BRAFV600E/K melanoma treated with dabrafenib (BRAFi) with or without a trametinib (MEKi) in combination with ipilimumab (NCT01767454) (59). The study had doublet and a triplet arms. The doublet arm had a run-in of dabrafenib (150 mg BID) for 2 weeks followed by received the aforementioned dabrafenib in addition to ipilimumab (3 mg/kg q3weeks for 4 doses) or dabrafenib at 100mg BID. In the triplet arms patients received dabrafenib and trametinib orally for 2 weeks (run-in), followed by ipilimumab (same dosing as doublet). The three planned triplet cohorts included dabrafenib 100 mg BID + trametinib 1 mg once daily + ipilimumab, dabrafenib 150 mg BID + trametinib 1 mg once daily+ ipilimumab, or dabrafenib 150 mg BID + trametinib 2 mg once daily + ipilimumab. Doublet patients appear to tolerate the regimen well. Although transaminitis was observed, no grade 3/4 hepatotoxicity or dose-limiting toxicities (DLTs) were observed. The most frequent adverse events (AEs; ≥2) were chills, fatigue, hand-foot syndrome, pyrexia, and maculopapular rash. The toxicity profile among the triplet cohorts was however noteworthy. Two cases of colitis associated with colonic perforation were noted in the first seven treated patients. Minor et al., reported that treatment with high dose steroids were required for both patients and one required emergent operative intervention (ileocecectomy) 41 days after the last dose of ipilimumab. Pathology of the resected bowel was consistent with ipilimumab-induced colitis (60). Further recruitment of patients in the triplet cohorts was suspended because of toxicity and the utility and safety of combining dabrafenib, tramenitinb, and ipilimumab remains in question (59).
In clinical trials single agent treatment with ipilimumab carries up to a 5% risk of significant colitis (3, 61). The triplet therapy (BRAFi/MEKi/α-CTLA-4) regimen appeared to in some manner augment this potentially life-threatening toxicity. Interestingly, single agent trametinib is fairly well tolerated and although gastrointestinal toxicity has been reported this is usually only Grade 1–2 and self-limiting (9, 10). Likewise double therapy with dabrafenib and trametinib is also usually well tolerated and infrequently associated with colitis, with no reports to date of colonic perforation (4, 22). The striking toxicity seen with the addition of ipilimumab to dabrafenib and trametinib reveals the caution that should be employed when evaluating new combinatorial strategies. It would seem these strategies are often more than simply a sum of their respective parts so to speak. The exact mechanism by which MEK inhibitors may add to the toxicity of ipilimumab is unclear and beyond the scope of this review, but likely related to previous mentioned effects on T-cell activation when combined with BRAF inhibition.
Several trials are now currently underway evaluating the combination of targeted therapy and either PD1 or PD-L1 blockade (NCT01656642, NCT02130466, & NCT02027961). At 1–2%, the incidence of grade 3–4 immune mediated colitis reported with PD-1 blockade appears than with ipilimumab making this a more attractive partner for the addition of targeted therapy (51). Current studies include a phase 1b open label study of atezolizumab (MPDL3280A), an engineered monoclonal PD-L1 antibody, in combination with vemurafenib (a BRAF inhibitor) and cobimetinib (a MEK inhibitor) in patients with previously untreated BRAFV600E or K metastatic melanoma (62). Early results by Hamid et al. presented at the Society of Melanoma Research (SMR) 2015 International Congress, showed that the combination resulted in an objective response rate of 76% (95% confidence interval [CI:] 50.1%–93.2%), including three complete responders (CR). Adverse events (AEs) were manageable and generally reversible with no reports of colonic perforation at the time of presentation (63).
Another combination targeted therapy and immune checkpoint blockade therapy trial was reported by Ribas et al. at ASCO 2015 (NCT02027961) (64). This trial is a phase I multicenter, open-label study, examined the feasibility of adding anti–PD-L1 immunotherapy (MEDI4736, durvalumab, a human IgG1 mAb blocking PD-L1 binding to PD-1 and CD80) to combined BRAF (dabrafenib) and MEK inhibitor (trametinib) therapy in patients with BRAF-mutant and BRAF wild-type metastatic melanoma. In this trial, patients were assigned to one of three distinct arms. In Arm A, patients with BRAF-mutant metastatic melanoma received therapy with MEDI4736 in combination with concurrent administration of dabrafenib and trametinib at approved doses until progressive disease (PD). In Arms B and C, patients with BRAF wild-type metastatic melanoma, received treatment with MEDI4736 and concurrently treatment with trametinib until PD (Arm B), or a 4 week lead-in with trametinib, followed by the addition of MEDI4736 and trametinib concurrently 2 weeks, then by MEDI4736 alone until PD (Arm C). This study allowed enrollment of patients who had progression on prior immunotherapy including anti-CTLA-4, anti-PD-1, or anti-PD-L1, but excluded patients who had received prior targeted therapy. The number of enrolled patients in Arms A, B, and C was 26, 20, and 19, respectively, with a median follow-up duration of 7.1, 6.8, and 3.7 months. Overall response rate for Arms A, B, and C was 69%, 21%, and 13%, respectively, and the corresponding disease control rate, including complete response, partial response, and stable disease (SD), was 100%, 79% and 80%, respectively. Of note, in up to 50 weeks of follow-up almost 90% of Arm A patients continued to derive clinical benefit (64). Overall toxicities were consistent with the expected AE profiles and previous experience of the individual agents. Their group also demonstrated the immune component of responses by identifying post-treatment markers of immune activation, which were strongest in patients with BRAF-mutant melanoma treated with BRAF and MEK inhibitors (64).
Importantly, targeted therapy combinations with immune checkpoint blockade are being explored in other solid tumors (NCT02039674). This is based on evidence that other oncogenic mutations can abrogate anti-tumor immunity, such as c-kit in gastrointestinal stromal tumors (GIST) (65) and that immune infiltrates in GIST are enhanced on treatment with imatinib (a c-kit inhibitor) (66). This provides the rationale for an ongoing clinical trial combining a c-kit inhibitor (imatinib) with immune checkpoint blockade (ipilimumab) in patients with c-kit mutant metastatic tumors (NCT01738139). Immunotherapy is now on the forefront of numerous clinical trials in multiple tumor types, including lung (NCT02352948, NCT02454933, NCT02453282), pancreatic (NCT00836407) (67), prostate (NCT00861614) (68), and hematologic malignancies (NCT01775631, NCT02036502, NCT02271945, and NCT02077959). Some of these trials have been designed with the concept of combining targeted therapy and immune checkpoint blockade.
Concluding Thoughts
There have been monumental advances in the treatment of cancer over the past decade through the use of immune checkpoint blockade and molecularly targeted therapy, with the approval of numerous agents as monotherapy as well as in combination. However, what is a current wealth of riches lends itself to complexity in terms of the development of appropriate clinical trials and standard of care regimens, and also with regard to proper sequence of different treatment regimens. Combination strategies will almost certainly be required in the majority of patients with metastatic disease, to combat therapeutic resistance and clonal evolution. Indeed there is a strong rationale to combine immune checkpoint blockade with strategies that may convert a “cold” tumor microenvironment to a “hot” one, and molecularly targeted therapy may fit the bill in this regard. Specifically, treatment with BRAF-targeted therapy may create a move favorable microenvironment, though adaptive resistance to therapy occurs early on – and is likely mediated by genetic, epigenetic, and immune events that help shape the response to therapy.
As we move forward in combining immune checkpoint blockade with other therapeutic regimens such as molecularly targeted therapy, it will be critical to understand the effects of each of the agents as monotherapy as well as in combination on the tumor cells themselves as well as the effects on other components of the microenvironment – such as stromal and immune cells. In the short term this is likely best accomplished via a comprehensive analysis of longitudinal tumor samples in the setting of therapy, though we clearly need to be doing parallel studies using blood-based assays and novel imaging techniques, as well as parallel studies in murine and other pre-clinical models. These insights will help us appreciate the appropriate timing and sequence of therapy, and will ultimately help us realize the power of personalized medicine – maximizing treatment responses while minimizing toxicity.
Footnotes
Disclosure of Potential Conflicts of Interest
J.A. Wargo has honoraria from speakers’ bureaus of Dava Oncology, Illumina and Bristol Myers Squibb and is an advisory board member for GlaxoSmithKline, Novartis and Roche/Genentech. No other potential conflicts of interest were disclosed.
References
- 1.NCI. SEER Statistical Fact Sheets: Melanoma of the Skin. 2015;
- 2.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013;369:122–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med 2015;372:2521–32. [DOI] [PubMed] [Google Scholar]
- 5.Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014;371:1867–76. [DOI] [PubMed] [Google Scholar]
- 6.Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med 2012;366:707–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol 2015;33:2780–8. [DOI] [PubMed] [Google Scholar]
- 8.Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012;380:358–65. [DOI] [PubMed] [Google Scholar]
- 9.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012;367:107–14. [DOI] [PubMed] [Google Scholar]
- 10.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 2012;367:1694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in Previously Untreated Melanoma without BRAF Mutation. N Engl J Med 2014. [DOI] [PubMed]
- 12.Prieto PA, Yang JC, Sherry RM, Hughes MS, Kammula US, White DE, et al. CTLA-4 blockade with ipilimumab: long-term follow-up of 177 patients with metastatic melanoma. Clin Cancer Res 2012;18:2039–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010;468:973–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cooper ZA, Juneja VR, Sage PT, Frederick DT, Piris A, Mitra D, et al. Response to BRAF inhibition in melanoma is enhanced when combined with immune checkpoint blockade. Cancer immunology research 2014;2:643–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu-Lieskovan S, Mok S, Homet Moreno B, Tsoi J, Robert L, Goedert L, et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Science translational medicine 2015;7:279ra41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shain AH, Yeh I, Kovalyshyn I, Sriharan A, Talevich E, Gagnon A, et al. The Genetic Evolution of Melanoma from Precursor Lesions. New England Journal of Medicine 2015;373:1926–36. [DOI] [PubMed] [Google Scholar]
- 17.Genomic Classification of Cutaneous Melanoma. Cell 2015;161:1681–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davies MA, Stemke-Hale K, Lin E, Tellez C, Deng W, Gopal YN, et al. Integrated Molecular and Clinical Analysis of AKT Activation in Metastatic Melanoma. Clin Cancer Res 2009;15:7538–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res 2010;70:5213–9. [DOI] [PubMed] [Google Scholar]
- 20.Amaravadi RK, Schuchter LM, McDermott DF, Kramer A, Giles L, Gramlich K, et al. Phase II Trial of Temozolomide and Sorafenib in Advanced Melanoma Patients with or without Brain Metastases. Clin Cancer Res 2009;15:7711–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. New England Journal of Medicine 2014;371:1877–88. [DOI] [PubMed] [Google Scholar]
- 23.Kwong LN, Boland GM, Frederick DT, Helms TL, Akid AT, Miller JP, et al. Co-clinical assessment identifies patterns of BRAF inhibitor resistance in melanoma. J Clin Invest 2015;125:1459–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nature reviews Cancer 2009;9:550–62. [DOI] [PubMed] [Google Scholar]
- 25.Corcoran RB, Atreya CE, Falchook GS, Kwak EL, Ryan DP, Bendell JC, et al. Combined BRAF and MEK Inhibition With Dabrafenib and Trametinib in BRAF V600-Mutant Colorectal Cancer. J Clin Oncol 2015;33:4023–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res 2013;19:1225–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bradley SD, Chen Z, Melendez B, Talukder A, Khalili JS, Rodriguez-Cruz T, et al. BRAFV600E Co-opts a Conserved MHC Class I Internalization Pathway to Diminish Antigen Presentation and CD8+ T-cell Recognition of Melanoma. Cancer immunology research 2015;3:602–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Science translational medicine 2012;4:127ra37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res 2012;18:1386–94. [DOI] [PubMed] [Google Scholar]
- 30.Cooper ZA, Frederick DT, Juneja VR, Sullivan RJ, Lawrence DP, Piris A, et al. BRAF inhibition is associated with increased clonality in tumor-infiltrating lymphocytes. Oncoimmunology 2013;2:e26615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hugo W, Shi H, Sun L, Piva M, Song C, Kong X, et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell 2015;162:1271–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koya RC, Mok S, Otte N, Blacketor KJ, Comin-Anduix B, Tumeh PC, et al. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer Res 2012;72:3928–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vella LJ, Andrews MC, Pasam A, Woods K, Behren A, Cebon JS. The kinase inhibitors dabrafenib and trametinib affect isolated immune cell populations. Oncoimmunology 2014;3:e946367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vella LJ, Pasam A, Dimopoulos N, Andrews M, Knights A, Puaux AL, et al. MEK inhibition, alone or in combination with BRAF inhibition, affects multiple functions of isolated normal human lymphocytes and dendritic cells. Cancer immunology research 2014;2:351–60. [DOI] [PubMed] [Google Scholar]
- 35.Shindo T, Kim TK, Benjamin CL, Wieder ED, Levy RB, Komanduri KV. MEK inhibitors selectively suppress alloreactivity and graft-versus-host disease in a memory stage-dependent manner. Blood 2013;121:4617–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu L, Mayes PA, Eastman S, Shi H, Yadavilli S, Zhang T, et al. The BRAF and MEK Inhibitors Dabrafenib and Trametinib: Effects on Immune Function and in Combination with Immunomodulatory Antibodies Targeting PD-1, PD-L1, and CTLA-4. Clin Cancer Res 2015;21:1639–51. [DOI] [PubMed] [Google Scholar]
- 37.Comin-Anduix B, Chodon T, Sazegar H, Matsunaga D, Mock S, Jalil J, et al. The oncogenic BRAF kinase inhibitor PLX4032/RG7204 does not affect the viability or function of human lymphocytes across a wide range of concentrations. Clin Cancer Res 2010;16:6040–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol 1999;17:2105–16. [DOI] [PubMed] [Google Scholar]
- 39.Atkins MB, Kunkel L, Sznol M, Rosenberg SA. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: long-term survival update. The cancer journal from Scientific American 2000;6 Suppl 1:S11–4. [PubMed] [Google Scholar]
- 40.Klapper JA, Downey SG, Smith FO, Yang JC, Hughes MS, Kammula US, et al. High-dose interleukin-2 for the treatment of metastatic renal cell carcinoma : a retrospective analysis of response and survival in patients treated in the surgery branch at the National Cancer Institute between 1986 and 2006. Cancer 2008;113:293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amaria R, Reuben A, Cooper ZA, Wargo JA. Update on use of aldesleukin for treatment of high-risk metastatic melanoma. ImmunoTargets and Therapy 2015;4:79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 1995;182:459–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nature reviews Cancer 2012;12:252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science (New York, NY) 1996;271:1734–6. [DOI] [PubMed] [Google Scholar]
- 45.Maio M, Grob JJ, Aamdal S, Bondarenko I, Robert C, Thomas L, et al. Five-year survival rates for treatment-naive patients with advanced melanoma who received ipilimumab plus dacarbazine in a phase III trial. J Clin Oncol 2015;33:1191–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 2000;192:1027–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol 2007;8:239–45. [DOI] [PubMed] [Google Scholar]
- 48.Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev 2010;236:219–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fife BT, Pauken KE. The role of the PD-1 pathway in autoimmunity and peripheral tolerance. Ann N Y Acad Sci 2011;1217:45–59. [DOI] [PubMed] [Google Scholar]
- 50.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014;32:1020–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ribas A, Puzanov I, Dummer R, Schadendorf D, Hamid O, Robert C, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. The Lancet Oncology 2015;16:908–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proceedings of the National Academy of Sciences 2010;107:4275–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001;344:783–92. [DOI] [PubMed] [Google Scholar]
- 55.Cooper ZA, Frederick DT, Ahmed Z, Wargo JA. Combining checkpoint inhibitors and BRAF-targeted agents against metastatic melanoma. Oncoimmunology 2013;2:e24320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med 2013;368:1365–6. [DOI] [PubMed] [Google Scholar]
- 57.Kakavand H, Wilmott JS, Menzies AM, Vilain R, Haydu LE, Yearley JH, et al. PD-L1 Expression and Tumor-Infiltrating Lymphocytes Define Different Subsets of MAPK Inhibitor-Treated Melanoma Patients. Clin Cancer Res 2015;21:3140–8. [DOI] [PubMed] [Google Scholar]
- 58.Cooper ZA, Reuben A, Amaria RN, Wargo JA. Evidence of synergy with combined BRAF-targeted therapy and immune checkpoint blockade for metastatic melanoma. Oncoimmunology 2014;3:e954956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Puzanov IC MK; Linette GP; Patel SP; Luke JJ; Sosman JA; Wolchok JD; Hamid O; Minor DR; Orford KW; Hug BA; Ma B; Matthys GM; Hoos A Phase 1 study of the BRAF inhibitor dabrafenib (D) with or without the MEK inhibitor trametinib (T) in combination with ipilimumab (Ipi) for V600E/K mutation positive unresectable or metastatic melanoma (MM). 50th Annual Meeting of the American Society of Clinical Oncology (ASCO); 2014; Chicago, IL: Journal of Clinical Oncology. [Google Scholar]
- 60.Minor DR, Puzanov I, Callahan MK, Hug BA, Hoos A. Severe gastrointestinal toxicity with administration of trametinib in combination with dabrafenib and ipilimumab. Pigment cell & melanoma research 2015;28:611–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wolchok JD, Neyns B, Linette G, Negrier S, Lutzky J, Thomas L, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. The Lancet Oncology 2010;11:155–64. [DOI] [PubMed] [Google Scholar]
- 62.Omid Hamid JAS, Lawrence Donald P., Sullivan Ryan J., Ibrahim Nageatte, Kluger Harriet M., Boasberg Peter D., Flaherty Keith, Hwu Patrick, Ballinger Marcus, Mokatrin Ahmad, Kowanetz Marcin, Chen Daniel S., Stephen Hodi F. Clinical activity, safety, and biomarkers of MPDL3280A, an engineered PD-L1 antibody in patients with locally advanced or metastatic melanoma. 2013 ASCO Annual Meeting; 2013.
- 63.Hamid O Preliminary clinical safety, tolerability and activity of atezolizumab (anti-PDl1) combined with Zelboraf in BRAFv600 metastatic melanoma. Society for Melanoma Research; 2015; San Francisco, CA. [Google Scholar]
- 64.Antoni Ribas MB, Lutzky Jose, Lawrence Donald P., Robert Caroline, Miller Wilson, Linette Gerald P., Ascierto Paolo Antonio, Kuzel Timothy, Algazi Alain Patrick, Postow Michael Andrew, Nathan Paul D., Curti Brendan D., Robbins Paul B., Li Xia, Blake-Haskins John A., Gordon Michael S.. Phase I study combining anti-PD-L1 (MEDI4736) with BRAF (dabrafenib) and/or MEK (trametinib) inhibitors in advanced melanoma. 2015. ASCO Annual Meeting
- 65.McLean SR, Gana-Weisz M, Hartzoulakis B, Frow R, Whelan J, Selwood D, et al. Imatinib binding and cKIT inhibition is abrogated by the cKIT kinase domain I missense mutation Val654Ala. Molecular cancer therapeutics 2005;4:2008–15. [DOI] [PubMed] [Google Scholar]
- 66.Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med 2011;17:1094–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Le DT, Lutz E, Uram JN, Sugar EA, Onners B, Solt S, et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. Journal of immunotherapy (Hagerstown, Md : 1997) 2013;36:382–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, van den Eertwegh AJ, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184–043): a multicentre, randomised, double-blind, phase 3 trial. The Lancet Oncology 2014;15:700–12. [DOI] [PMC free article] [PubMed] [Google Scholar]