

Abstract
Evidence has emerged that the failing heart increases utilization of ketone bodies. We sought to determine whether this fuel shift is adaptive. Mice rendered incapable of oxidizing the ketone body 3-hydroxybutyrate (3OHB) in the heart exhibited worsened heart failure in response to fasting or a pressure overload/ischemic insult compared with WT controls. Increased delivery of 3OHB ameliorated pathologic cardiac remodeling and dysfunction in mice and in a canine pacing model of progressive heart failure. 3OHB was shown to enhance bioenergetic thermodynamics of isolated mitochondria in the context of limiting levels of fatty acids. These results indicate that the heart utilizes 3OHB as a metabolic stress defense and suggest that strategies aimed at increasing ketone delivery to the heart could prove useful in the treatment of heart failure.
Keywords: Cardiology, Metabolism
Keywords: Heart failure, Mitochondria, Mouse models
Strategies aimed at increasing cardiac ketone body delivery show promise as a new heart failure therapy in pre-clinical models.
Introduction
As an incessantly working pump, the adult mammalian heart has very high energy demands that are met by a specialized mitochondrial system. Fatty acids serve as the chief fuel for mitochondrial ATP production in the normal adult heart (1, 2). Glucose is also an important cardiac energy substrate, allowing for adaptive fuel utilization shifts during development and in accordance with diverse nutritional and physiological conditions. The mitochondrial fuel utilization machinery of the adult heart is programmed for fuel utilization flexibility during the perinatal and postnatal developmental periods (2). Specifically, the fetal heart has relatively low mitochondrial oxidative capacity and relies largely on glucose and lactate as fuels, with fatty acids serving as a minor energy substrate (3, 4). At birth, a well-defined transcriptional circuitry triggers mitochondrial biogenesis, followed by induction of genes involved in mitochondrial fatty acid oxidation (FAO) during the postnatal period (2, 5–9). The corresponding developmental maturation renders the heart a fuel omnivore, relying on both fatty acids and glucose.
The heart changes fuel utilization preferences in chronic pathophysiological circumstances that lead to heart failure (HF). For example, during development of cardiac hypertrophy and HF caused by chronic hypertension or recurrent ischemic insult, cardiac myocyte mitochondria undergo a reprogramming resulting in a reduction in capacity for mitochondrial FAO with increased reliance on glucose oxidation and glycolysis (10–13). Significant evidence suggests that this metabolic reprogramming leads to bioenergetic derangements that contribute to the pathogenesis of HF. For example, human genetic defects in FAO can cause childhood cardiomyopathy in conditions that increase reliance of the heart on fatty acids as a fuel, such as with prolonged fasting (14). In addition, magnetic resonance spectroscopy measurements of humans with cardiac hypertrophy and HF have shown that reduced myocardial phosphocreatine (PCr)/ATP ratio correlates with worsened outcomes (15–19).
Recently, we and others discovered an unrecognized fuel metabolic shift in the hypertrophied and failing heart (20, 21). Unbiased metabolomic profiles in well-defined mouse models of HF and in the failing human heart were indicative of increased ketone body oxidation. Proteomic studies identified increased levels of D-β-hydroxybutyrate dehydrogenase (BDH1) in the failing heart (20). BDH1 is a mitochondrial enzyme that catalyzes the first step in the oxidation of 3-hydroxybutyrate (3OHB), a ketone body that is synthesized mainly in the liver. 13C-NMR spectroscopy studies confirmed that the hypertrophied mouse heart utilizes 3OHB to a greater extent than the normal heart (20), and in vivo measurements confirmed a higher ketone body uptake by canine failing hearts (22). Taken together, the results of these recent studies suggest that, in the context of reduced capacity to oxidize fatty acids as a fuel, the heart reprograms to increase utilization of ketone bodies generated in the liver. However, the role of this fuel switch as adaptive, maladaptive, or of no consequence to the pathogenesis of HF is unknown.
In this study, we sought to assess the importance of ketone body utilization in the stressed and diseased heart. Our results indicate that the heart increases utilization of 3OHB as an adaptive metabolic response during periods of nutritional and chronic hemodynamic stress. Moreover, strategies aimed at increasing ketone delivery to the heart reduced pathologic cardiac remodeling and dysfunction in mice and in a large-animal model of progressive HF. Studies conducted in isolated mouse mitochondria provided evidence that 3OHB provides ancillary substrate and enhances respiratory efficiency. These results raise the intriguing possibility that therapeutic strategies aimed at increasing myocardial 3OHB utilization could prevent or reverse the energy metabolic derangements and fuel starvation that contributes to the syndrome of HF.
Results
Generation of mice incapable of oxidizing 3OHB in the heart.
We first sought to define the cardiac phenotype of mice that are unable to oxidize 3OHB. To this end, cardiac-specific BDH1-deficient (csBDH1–/–) mice were generated by crossing mice harboring loxP sites within the Bdh1 gene with a line expressing Cre recombinase downstream of the cardiac-specific α myosin heavy chain promoter (αMHC-Cre mice) (23) to create mice with the final genotype of Bdh1fl/flMHC-Cre+/WT (Bdh1fl/flCre+) (Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.124079DS1). csBDH1–/– mice appeared grossly normal, including no significant difference in body weight compared with Cre– littermate controls (Supplemental Figure 1B). Assessment of offspring ratios from breeding pairs of Bdh1fl/flCre+ × Bdh1fl/flCre– revealed a small but finite deviation from Mendelian inheritance patterns at weaning, suggesting incomplete penetrance of a perinatal lethal phenotype (Supplemental Table 1). Thereafter, no difference in survival between the csBDH1–/– and control mice was observed. Levels of circulating 3OHB were similar in csBDH1–/– and littermate controls (Supplemental Figure 1C). csBDH1–/– mice did not exhibit a cardiac phenotype under basal conditions. Specifically, there were no abnormalities in ventricular mass or left ventricular (LV) chamber size or function, as determined by echocardiographic analysis (Supplemental Figure 1D and data not shown).
The myocardial substrate utilization profile of the csBDH1–/– mouse heart was determined by 13C-NMR spectroscopy, which allows for assessment of the relative contribution of specific labeled substrates for acetyl-CoA formation and entry into the tricarboxylic acid (TCA) cycle. Hearts isolated from csBDH1–/– and control groups were perfused in the Langendorff mode with 0.5 mM D-3-hydroxy-[2,4-13C2]butyrate ([2,4-13C2]3OHB) and unlabeled 0.4 mM palmitate or with 0.4 mM [U-13C] palmitate and unlabeled 0.5 mM D-3-hydroxybutyrate, both in the presence of unlabeled 1 mM lactate and 5 mM glucose. Functional performance was similar between the groups (rate-pressure product for controls = 41,716 ± 2,314 mmHg × bpm; csBDH1–/– = 42,826 ± 3050 mmHg × bpm). The fractional contribution (Fc) of [2,4-13C2]3OHB was virtually undetectable in the csBDH1–/– hearts compared with approximately 0.4 for the control group (Figure 1A). As expected, 13C-palmitate accounted for the majority of Fc to acetyl-CoA production (~0.6) for control hearts. Palmitate utilization was modestly but significantly elevated in csBDH1–/– hearts compared with controls, consistent with a compensatory increase in the context of an inability to oxidize 3OHB (Figure 1B). These results confirm a complete deficit of 3OHB oxidation in the hearts of csBDH1–/– mice.
Figure 1. csBDH1–/– mouse hearts are incapable of oxidizing D-3-hydroxybutyrate.

(A) (Left) The fractional enrichment of acetyl-CoA (Fc), representing oxidation of 13C-labeled D-3-hydroxybutyrate into the TCA cycle, is shown in Bdh1fl/flCre– (control) and csBDH1–/– isolated perfused mouse hearts (12- to 16-week-old male littermates) (n = 6). (Right) Representative in vitro NMR spectra displaying 13C labeling of glutamate at the 4- and 3-carbon (glu C-4 and glu C-3) positions in tissue extract from the hearts of control mice (top) and csBDH1–/– mouse (bottom) is shown. The latter has complete absence of signal (1% natural abundance). (B) Fc for 13C-labeled palmitate perfused isolated mouse hearts is shown (n = 5–6) (12- to 16-week-old male littermates). (C) Levels of myocardial 3-hydroxybutyrate (3OHB) per wet weight (ww) measured in control and csBDH1–/– male mice 8–10 weeks after 4-hour fast (n = 5). Bars represent mean ± SEM; *P < 0.05 control vs. csBDH1–/– using unpaired, 2-tailed Mann-Whitney test.
To assess the uptake and fate of 3OHB in csBDH1–/– hearts, quantitative mass spectrometry–based measurements were performed. Levels of 3OHB were significantly elevated in the csBDH1–/– myocardium of fed mice (Figure 1C). The observation that 13C-palmitate oxidation and 3OHB levels are increased in the BDH1–/– heart indicates that the normal adult mouse heart is capable of oxidizing ketone bodies as a minor fuel, even in nonstressed conditions.
BDH1 is necessary to maintain cardiac function in the context of a nutritional stress.
Tissues that rely on glucose as a chief fuel source, including many regions in the brain, shift to ketone oxidation as an ancillary fuel source during periods of fasting and starvation (24). Less is known about the importance of ketone body oxidation in the heart during states of nutritional stress, given that this organ — in contrast to the brain — is capable of high-capacity FAO (1, 25). The csBDH1–/– mice afforded us the opportunity to assess the necessity of 3OHB as a fuel source in the heart in the context of nutritional deprivation. Accordingly, csBDH1–/– and littermate control mice were subjected to a 24-hour fast. There were no significant differences in the fed or fasting levels of circulating 3OHB or glucose between groups (Supplemental Figure 2A). To assess the cardiac functional response to prolonged fasting, echocardiographic studies were conducted at the conclusion of the fasting period. The fasted csBDH1–/– mice exhibited significant alterations in LV function compared with fasted Bdh1fl/flCre– controls, as reflected by percent LV fractional shortening (FS) and longitudinal strain rate, together with an increase in LV internal diameter and volumes (Supplemental Figure 2B and Supplemental Table 2). These results strongly suggest that a shift to myocardial 3OHB oxidation serves as an adaptive alternative fuel source to maintain cardiac function in response to prolonged fasting and possibly other nutritional stress states.
Inability to oxidize 3OHB in the heart results in worsened pathologic cardiac remodeling in the context of pressure overload/ischemic stress.
Recent studies have shown that the hypertrophied and failing heart oxidize ketone bodies coincident with an induction in the expression of myocardial ketolytic enzymes BDH1 and succinyl-CoA-3-oxaloacid CoA transferase (SCOT) (20, 21, 26). An important question is whether this shift in myocardial fuel utilization is adaptive, maladaptive, or inconsequential to the disease process. As an initial step to address this question, csBDH1–/– and Bdh1fl/flCre– littermate control mice were subjected to an established mouse surgical HF model involving pressure overload via transverse aortic constriction (TAC) combined with a small apical myocardial infarction (TAC/MI) that results in predictable, progressive, pathological LV dilatation and contractile dysfunction over a 4-week period (20, 27, 28). Following surgery, mortality rates were no different between the 2 groups. Echocardiography performed 4 weeks after TAC/MI surgery demonstrated that csBDH1–/– mice had developed more severe LV dysfunction and pathological remodeling compared with the control group. Specifically, LV ejection fraction (LVEF) was significantly lower in the csBDH1–/– mice (Figure 2A and Supplemental Table 3). In addition, csBDH1–/– mice exhibited greater LV end-diastolic volume (EDV) and end-systolic volume (ESV) compared with controls (Figure 2B and Supplemental Table 3).
Figure 2. Inability to oxidize 3OHB in the heart results in worsened pathologic cardiac remodeling in the context of pressure overload/ischemic stress.

Echocardiographic data collected 4 weeks following sham or TAC/MI surgeries in Bdh1fl/flCre– (control) and csBDH1–/– male mice aged 8–11 weeks at time of surgery. (A) EF graphed as percent, (B) EDV (left), and ESV (right) graphed as volume (µl). Bars represent mean ± SEM (Sham, n = 5; TAC/MI, n = 8–9); *P < 0.05 TAC/MI control vs. TAC/MI csBDH1–/–, using unpaired, 2-tailed t test. EF, ejection fraction; TAC/MI, transverse aortic constriction with myocardial infarction; EDV, end-diastolic volume; ESV, end-systolic volume, csBDH1–/–, cardiac-specific β-hydroxybutyrate dehydrogenase–deficient.
Molecular signatures of cardiac remodeling were also indicative of worsened LV remodeling in csBDH1–/– mice after TAC/MI intervention. Induction of natriuretic peptide A (Nppa) was significantly greater in LV of csBDH1–/– mice compared with the control group (Supplemental Figure 3A). Similarly, LV expression of genes encoding ATPase sarcoplasmic/ER Ca2+ transporting 2 (Atp2a2), cardiac troponin I (Tnni3), and genes involved in fatty acid utilization — which are well-known to be downregulated in the failing heart — were reduced to a greater extent in the csBDH1–/– mice (Supplemental Figure 3, B and C). Lastly, as we have recently shown, cardiac expression of Bdh1 was induced in hearts of the control mice following TAC/MI (20). Bdh1 was not induced by TAC/MI in csBDH1–/– mice (Supplemental Figure 3D), indicating that the increased myocardial expression of Bdh1 observed in HF is specific to cardiac myocytes.
Increased delivery of ketone bodies to the heart ameliorates pathological cardiac remodeling and dysfunction.
We next sought to determine whether increasing levels of circulating ketones would alter cardiac remodeling in mice following TAC/MI. To this end, WT mice were fed normal chow or a ketogenic diet (KD) starting 1 week before TAC/MI surgery and for the 4-week postsurgical period (Supplemental Figure 4A). The KD was confirmed to induce significant ketonemia prior to surgery (mean fed blood 3OHB levels with standard chow = 0.5611 ± 0.036 mM; KD group = 1.213 ± 0.1802 mM; P < 0.0001), and circulating 3OHB levels remained elevated at 4 weeks after surgery (Supplemental Figure 4B). Following TAC/MI surgery, no significant difference in mortality rates was observed between the chow and KD groups (data not shown). In addition, there was no significant difference in LVEF between the groups (Figure 3A and Supplemental Table 4). However, several pathologic LV remodeling endpoints were improved in the KD group, as evidenced by assessment of LV volumes. Specifically, LVEDV and LVESV were both significantly reduced in the TAC/MI KD group compared with controls (Figure 3B and Supplemental Table 4).
Figure 3. Increased delivery of ketone bodies to the heart supplied by a ketogenic diet ameliorates pathological cardiac remodeling.

Results of echocardiography 4 weeks after TAC/MI surgery on C57BL/6N mice fed standard chow (Chow) vs. ketogenic diet (KD) starting 1 week prior to surgery (total of 5 weeks on diet). (A) LVEF graphed as percent, (B) EDV (left) and ESV (right) graphed as volume (µl). Bars represent mean ± SEM (n = 8–15), *P < 0.05 Chow vs. KD or #P < 0.05 Sham vs. TAC/MI using 1-way ANOVA with Tukey’s post hoc test to correct for multiple comparisons. EF, ejection fraction; TAC/MI, transverse aortic constriction with myocardial infarction; EDV, end-diastolic volume; ESV, end-systolic volume.
A KD, while increasing circulating levels of ketone bodies, introduces other significant dietary variables, including a marked enrichment in long-chain fatty acids together with reduced carbohydrate and amino acid composition. We sought to address the direct impact of chronically increasing 3OHB delivery to the heart. To this end, studies were conducted in the canine tachypacing model of dilated cardiomyopathy that recapitulates, with high reproducibility, many of the functional, structural, biochemical, and molecular alterations that occur in human congestive HF (29). Importantly, this large-animal model allows chronic delivery of 3OHB via an indwelling catheter in the right ventricle together with in vivo measurements of hemodynamics and myocardial substrate metabolism. The graded tachypacing protocol results in severe and predictable cardiac decompensation over a period of approximately 4 weeks (30). Starting on day 13 of pacing, when cardiac function was still in the compensated stage, 3OHB was infused continuously into the right ventricle until the end of the protocol (day 29 of pacing), resulting in a steady-state arterial plasma concentration of approximately 2.5-fold higher than that measured in an untreated historical control HF group used for comparison in this study (Supplemental Figure 5A). The infusion did not alter arterial pH and did not affect food or water intake, as also indicated by the absence of significant changes in body weight compared with the HF control group at the end of the study (data not shown). The impact of chronic ketone body administration on cardiac function and LV remodeling was dramatic. With 3OHB infusion, heart rate and cardiac output remained at prepacing levels, and the progressive pacing-induced diminution in LVEF was markedly blunted (Figure 4A and Supplemental Table 5). The 3OHB infusion completely prevented the rise in LV end-diastolic pressure (LVEDP) and attenuated the increase in LV end-diastolic diameter (LVEDD) (Figure 4A and Supplemental Table 5). The latter effect indicated reduced chamber remodeling, as confirmed by the preservation of cardiac weight relative to control nonpaced hearts, while pacing alone led to a significant increase in cardiac mass (data not shown). The characteristic reduction in dP/dtmax and dP/dtmin was not influenced by 3OHB under basal conditions (Supplemental Table 5), but dP/dtmax was increased by 3OHB in response to a dobutamine challenge (Figure 4B). In addition, although mean arterial pressure was unchanged by the treatment, total peripheral resistance and effective arterial elastance (Ea), 2 indices of afterload, were lower in the 3OHB-infused animals (Figure 4A and Supplemental Table 5). Taken together, these results demonstrate a remarkable protective effect of 3OHB infusion on pathologic cardiac remodeling and suggest that the beneficial effects involve direct actions on the myocyte, as well as afterload reduction.
Figure 4. Chronically increased delivery of ketone bodies to the heart by direct infusion ameliorates pathological cardiac remodeling.

(A) Results of echocardiography and hemodynamic monitoring at weeks 0, 2, and 4 of tachypacing in dogs without (HF) or with infusion of 3-hydroxybuytrate (HF + 3OHB). Note that the “2 week” echo was performed on day 14 for the HF dogs, and — for the HF+3OHB group — the echo was performed on days 12–13 immediately before starting 3OHB infusion. The set of data for HF was randomly selected from a historical pool. (B) β-Adrenergic response to dobutamine infused after 4 weeks of cardiac pacing. Dots and bars represent mean ± SEM (n = 7–8 per group), *P < 0.05 vs. baseline or #P < 0.05 HF vs. HF + 3OHB at the corresponding time point using 2-way ANOVA followed by Student-Newman test for multiple comparisons. HR, heart rate; EF, ejection fraction; CO, cardiac output; LVEDP, left ventricular end-diastolic pressure; LVEDD, left ventricular end-diastolic diameter; TPR, total peripheral resistance; Ea, effective arterial elastance; dP/dtmax, maximal first derivative of left ventricular pressure.
The impact of 3OHB on cardiac substrate oxidation rates was assessed at the conclusion of the pacing protocol by infusing [9,10-3H]-oleate and [U-14C]-glucose isotopic tracers and measuring coronary blood flow. In addition, arteriovenous (A-V) differences of ketone bodies and lactate across the heart were multiplied by coronary flow to calculate net cardiac uptake. As expected, the uptake of ketone bodies was increased with HF compared with historical non–cardiac-paced controls and further with 3OHB infusion (Figure 5A). The infusion of 3OHB did not significantly alter the arterial levels of free fatty acid (FFA) and glucose, while it caused a slight, albeit significant, reduction in circulating lactate (Supplemental Figure 5A). Consistent with previous studies in this model (22), palmitate oxidation rates decreased and glucose oxidation rates increased in the HF group compared with controls in the absence of 3OHB treatment (Figure 5, B and C). In the failing heart, myocardial glucose uptake and oxidation rates were markedly suppressed by 3OHB infusion, whereas FFA uptake and oxidation was not significantly changed (Figure 5, B–E). 3OHB infusion reduced net lactate uptake, which could reflect reduced consumption and/or increased production by the myocyte (Figure 5F).
Figure 5. Impact of 3OHB infusion on cardiac substrate utilization.

Cardiac substrate utilization measured at the last day of the cardiac pacing protocol in dogs without (HF) or with infusion of 3-hydroxybuytrate (HF + 3OHB) compared with nonpaced controls. The set of data for HF and nonpaced controls was randomly selected from historical pools. (A) Cardiac uptake of ketone bodies. (B and C) Myocardial oxidation of (B) FFA and (C) glucose. (D and E) Cardiac uptake of (D) glucose and (E) FFA. (F) Cardiac uptake of lactate. (G) Myocardial oxygen consumption (MVO2). Bars represent mean ± SEM (n = 6 for all groups), *P < 0.05 vs. Control; #P < 0.05 HF vs. HF + 3OHB by 1-way ANOVA followed by Student-Newman test for multiple comparisons.
Notably, the reduction in myocardial O2 consumption (MVO2) occurring in the HF group was prevented by 3OHB infusion, consistent with increased work (Figure 5G). Since pressure-volume or diameter loops were not measured in this study, we could not precisely calculate cardiac efficiency. However, stroke volume was determined by echocardiography, and — by approximating the height of the pressure-volume loop as equal to mean arterial pressure minus half of the LVEDP — we calculated a close estimate of the pressure-volume loop area. This was then divided by MVO2 to determine myocardial mechanical energetic efficiency (31). These calculations indicated that myocardial mechanical efficiency was approximately 30% higher in dogs infused with 3OHB than nontreated dogs (79,850.70 ± 5,549.48 vs. 59,770.58 ± 6,171.35 mmHg × beat/100 g, P < 0.05).
Interestingly, the impact of 3OHB on myocardial substrate utilization exhibited a different pattern in nonpaced control hearts. Specifically, 3OHB infusion resulted in decreased palmitate uptake and oxidation rates, whereas glucose uptake and oxidation rates remained unchanged and net lactate uptake was reduced (Supplemental Figure 5B). In addition, chronic 3OHB infusion decreased MVO2 in the nonpaced control animals (Supplemental Figure 5B). Taken together, the substrate utilization results suggest that ketones compete with the main myocardial substrates in a pattern that is influenced by the existence of cardiac dysfunction.
3OHB augments respiratory efficiency in isolated heart mitochondria.
Infusion of 3OHB resulted in a marked improvement in cardiac function and remodeling in the tachypacing model, together with evidence of increased myocardial uptake of the ketone body. These observations suggest that, in the context of reduced capacity for oxidizing the main substrates (fatty acids and glucose), 3OHB improved cardiac bioenergetics, thereby augmenting contractile function. One potential mechanism for the observed effect on energetics is that 3OHB provides a substrate for generating additional acetyl-CoA (32, 33), leading to increased production of NADH to drive energy transfer and ATP production (32, 33). We sought to discern the energetic value of 3OHB using a recently developed bioenergetics assay platform in which comprehensive assessment of mitochondrial respiratory flux is conducted on freshly isolated mouse heart mitochondria (34). The platform features a modified version of the creatine kinase (CK) energetic clamp technique (25, 35, 36), which permits dynamic control of the extramitochondrial concentration ratio of ATP/ADP — and, thus, the energy of ATP hydrolysis (ΔGATP) — within a near-physiologic range. In simple terms, the technique evaluates how well a given population of mitochondria exposed to a specific mixture of carbon fuels responds to an energetic challenge. The transition from a high to low ATP/ADP ratio mimics an increase in energy demand, akin to a transition between rest and exercise, and thereby serves as an in vitro stress test. Analysis of the linear relationship between energy demand (ATP/ADP, ΔGATP) and steady-state oxygen flux (JO2) allows for an estimation of respiratory conductance (i.e., reciprocal of resistance), wherein a steeper slope indicates greater sensitivity and improved kinetics. Both the absolute rates of oxygen consumption and respiratory sensitivity (slope) depend on energy gradients and fluxes controlled by 3 principal regulatory nodes (Supplemental Figure 6): (a) the dehydrogenase enzymes, (b) the electron transport system (ETS), and (c) ATP synthesis and transport, which together mediate the transfer of energy from that available in carbon substrates to electron potential energy (ΔGredox), to the proton motive force (PMF, ΔGH+), to the free energy of ΔGATP. Thus, to gain insight into the free energies that drive the transduction process, the dynamic JO2 assays are combined with parallel assessments of mitochondrial membrane potential (ΔΨ), the primary contributor to the PMF, and NAD(P)H/NAD+ redox state.
The assay system was first used to evaluate 3OHB-supported energy transduction in the context of Km concentrations of the anaplerotic substrates, pyruvate and malate (P/M), in the absence of fatty acid (Supplemental Figure 6). Addition of 2 mM 3OHB caused a modest but significant increase in respiratory sensitivity (slope of JO2 vs. ΔGATP), while affording a more polarized (more negative) ΔΨ and a more reduced NAD(P)H/NAD+ redox state (e.g., a higher NAD[P]H/NAD+ ratio). Ketone-mediated enhancements in JO2 and energy transfer were even more impressive when added to Km concentrations of either P/αKG or αKG alone (Supplemental Figure 6). Notably, in all 3 cases, the relationship between JO2 and ΔΨ shifted rightward, such that mitochondria exposed to 3OHB were able to maintain a greater (more negative) ΔΨ for any given rate of oxygen consumption (Supplemental Figure 6).
We next sought to examine the impact of 3OHB on mitochondria fueled with a more physiological mixture of substrates, including P/M and fatty acids. Here, octanoyl-carnitine (OC) concentrations ranging from 10–100 μM were used to mimic impaired versus robust flux through the β-oxidation pathway, as occurs in failing hearts versus healthy hearts, respectively. Notably, increasing provision of OC resulted in greater maximal JO2 (Figure 6A), along with a more polarized ΔΨ (Figure 6B) and a more reduced NAD(P)H/NAD+ redox state (Figure 6C). Regardless of the OC concentration, addition of 3OHB had little impact on absolute JO2 and respiratory sensitivity but again produced a more negative ΔΨ, a more reduced NAD(P)H/NAD+ redox state, and a clear rightward shift in ΔΨ versus JO2, suggesting improved respiratory efficiency (Figure 6D), particularly at the 2 lower OC doses.
Figure 6. 3OHB augments respiratory efficiency in isolated heart mitochondria.

Freshly isolated mitochondria from heart ventricles of C57BL/6N mice were utilized to assess the impact of 3-hydroxybutyrate (3OHB) on the relationship between (A) oxygen consumption rate (JO2) (B) mitochondrial membrane potential (ΔΨ) in millivolts (mV), and (C) NAD(P)H/NAD(P)+ redox state versus the estimated Gibbs energy of ATP hydrolysis (ΔGATP). Mitochondria were fueled with pyruvate + malate (P/M, 110 μM each) and increasing concentrations of L-octanoylcarnitine (OC; 10, 50, or 100 μM) in the absence (purple) or presence (red) of 2 mM 3OHB. (D) Mitochondrial respiratory efficiency was evaluated by plotting JO2 against ΔΨ in the presence of P/M + 10–100 μM OC ± 3OHB. Dotted lines separate the submaximal and maximal portions of JO2 vs. ΔGATP. Triangle denotes the changing concentrations of ATP relative to ADP (ATP/ADP), resulting in a reciprocal change in energy demand. Data are mean ± SEM (n = 5). Measurements made at submaximal JO2 (A–C) were analyzed by 2-way ANOVA (†main effect of ketone; ‡ketone, ΔGATP interaction; P < 0.05), whereas those representing maximal JO2 (ΔGATP = –12.95) were analyzed via t test (*P < 0.05).
To assess the necessity of 3OHB oxidation for the observed effects, identical experiments were repeated in heart mitochondria isolated from csBDH1–/– mice compared with Bdh1fl/flCre–controls. The findings demonstrate that the efficiency profile observed in the presence of 3OHB depends on BDH1 activity. Specifically, the 3OHB-mediated effects on ΔΨ, respiratory efficiency, and NADH redox were not observed in BDH1–/– mitochondria (Figure 7 and Supplemental Figure 8). Moreover, the S-enantiomer of 3OHB (S-3OHB, which is not a substrate for BDH1) did not confer the improvement in the respiratory efficiency profile observed with the R-enatiomer (R-3OHB) (Figure 7). The necessity of BDH1 for the effects of 3OHB on NADH generation and JO2 are further shown in Supplemental Figure 7. Lastly, the use of acetoacetate (which bypasses the BDH1 step) produced a blunted response (Figure 7). In summary, while R-3OHB did not fully compensate for a limitation in FAO, this specific ketone clearly improved bioenergetic parameters when flux through the FAO pathway was limited. The energetic profile of 3OHB-supported respiration is consistent with increased flux through NADH generating dehydrogenase enzymes and increased redox driving forces across the ETS, which in turn supports a more polarized ΔΨ for any given rate of oxygen consumption. These results suggest that ketones can defend against pathologic remodeling and HF not only by averting a severe acetyl-CoA deficit, but also by enhancing mitochondrial respiratory thermodynamics, including maintenance of a stronger PMF.
Figure 7. The effects of 3OHB on mitochondrial respiratory efficiency depend on BDH1 flux.

(A–C) Freshly isolated mitochondria from heart ventricles of BDH1fl/fl (controls), csBDH1–/–, or WT C57BL/6N mice were used to assess the impact of 3OHB on the relationship between (A) oxygen consumption rate (JO2), (B) membrane potential (ΔΨ ) in millivolts (mV), and (C) NAD(P)H/NAD(P)+ redox state versus the estimated Gibbs energy of ATP hydrolysis (ΔGATP). Mitochondria were fueled with pyruvate + malate (110 μM each) and 50 μM L-octanoylcarnitine in the absence (purple) or presence of 2 mM of the (R, red) or (S, green) enantiomer of 3OHB, or acetoacetate (AcAc, gray). (D) Mitochondrial respiratory efficiency was evaluated by plotting JO2 against ΔΨ. Dotted lines separate the submaximal and maximal portions of JO2 vs. ΔGATP. Triangle denotes the changing concentrations of ATP relative to ADP (ATP:ADP), resulting in a reciprocal change in energy demand. Data represent mean ± SEM (n = 4–6). Comparisons between BDH1fl/fl versus csBDH1–/– mitochondria were analyzed by 3-way ANOVA followed by Tukey HSD (#ketone/genotype interaction; §ketone effect; P< 0.05 and family-wise error rate [FWER] < 0.05) using measurements (A–C) made at submaximal JO2. Energy fluxes representing maximal JO2 (ΔGATP = –12.95) were analyzed by 2-way ANOVA and Tukey HSD (*FWER < 0.05, relative to vehicle control). Comparison of R and S enantiomers were analyzed by 2-way ANOVA (submaximal JO2) and 1-way ANOVA (maximal JO2) each followed by Tukey HSD (§effect of R at submaximal JO2; ¤effect of S at maximal JO2; FWER < 0.05). Effects of AcAc were analyzed by 2-way ANOVA at submaximal JO2 (†main effect of ketone; ‡ketone/ΔGATP interaction, P < 0.05) and t test at maximal JO2 (*P < 0.05).
Discussion
During the development of HF, myocardial fuel metabolism is reprogrammed, resulting in reduced capacity to oxidize fatty acids, the chief myocardial fuel (10, 12, 37). Recently, we and others found that the hypertrophied and failing mouse heart utilizes ketone bodies at increased rates (20, 21). These results, together with observations that circulating ketone bodies are increased in humans and experimental dogs with HF (22, 38, 39), suggest that the failing heart oxidizes ketone bodies as an ancillary fuel in the context of reduced capacity to burn fatty acids, the chief substrate of the normal heart. However, the role of chronic ketone utilization as adaptive or maladaptive in the failing heart is unknown. In this study, we provide evidence that this fuel switch comprises an adaptive stress response based on the following lines of evidence. First, mice with cardiac deficiency of BDH1 cope poorly with nutritional deprivation and chronic pressure overload stress. Secondly, strategies aimed at increasing availability of ketone bodies ameliorated pathologic cardiac remodeling in both murine and large-animal models of HF. Lastly, in the context of reduced capacity to oxidize fatty acids and glucose, such as occurs in the failing heart, 3OHB enhanced bioenergetics in isolated mitochondria.
Our stable isotope labeling protocol confirmed that the mitochondrial enzyme BDH1 is necessary for myocardial 3OHB oxidation in mouse hearts. Accordingly, the related cytosolic short-chain fatty acid dehydrogenase, BDH2, does not likely contribute significantly to 3OHB oxidation, raising a question as to its function. Our results also indicate that the normal mouse heart oxidizes 3OHB as a minor fuel, as evidenced by the increase in myocardial 3OHB levels and enhanced palmitate oxidation rates in the BDH1–/– heart. Interestingly, we found that BDH1 is necessary to maintain cardiac function in the context of the nutritional stress imposed by prolonged fasting. Specifically, the csBDH1–/– mice developed modest but significant fasting-induced reductions in LV function and increases in LV volume following a 24-hour fast. It is well known that the brain shifts to oxidation of ketone bodies as a fuel in the context of nutritional deprivation. The cardiac response of csBDH1–/– mice to fasting suggests that this adaptive shift to reliance on ketone bodies as a fuel is also operative in the heart. Hepatic production of ketone bodies 3OHB and acetoacetate is markedly increased in the prolonged fasting state. Notably, the csBDH1–/– mice should still be able to oxidize acetoacetate in the heart via the reaction catalyzed by 3-oxoacid CoA-transferase 1 (Oxct1) (40). Accordingly, our results suggest that acetoacetate cannot fully substitute for 3OHB to meet the demands of the heart in the fasting state.
The observation that csBDH1–/– mice develop worsened cardiac remodeling compared with controls following TAC/MI supports the conclusion that a shift to myocardial 3OHB oxidation in the hypertrophied and failing heart is adaptive. This conclusion is also supported by the previous observation that Oxct1-deficient mice also exhibit modest cardiac dysfunction with prolonged pressure overload (41). It should be noted that the csBDH1–/– mice exhibited greater LV dysfunction and remodeling than Oxct1-deficient mice in the context of a HF intervention. This could relate to the more aggressive HF model used here (TAC/MI) compared with TAC alone in the study with Oxct1–/– mice (41). However, we cannot exclude other explanations for this phenotypic difference, such as the impact on redox state or metabolite accumulation profile related to the specific enzymatic action of BDH1. Regardless, the results of the Oxct1–/– and csBDH1–/– mice, together with the observed increase in 3OHB oxidation in the failing heart that occurs in the context of reduced utilization of fatty acids, suggest that ketone bodies are being used as an alternate oxidative fuel in order to maintain adequate ATP generation. This result is further supported by the recent observation that transgenic mice overexpressing BDH1 exhibit less pathologic remodeling in response to TAC (42).
The response of csBDH1–/– mice to TAC/MI prompted us to explore the impact of enhancing myocardial ketone delivery on the pathologic remodeling process that leads to HF. Administration of a KD, prior to and after the TAC/MI procedure in mice, reduced LV dilatation. However, KD also introduces major changes in nutrient composition. To avoid these potentially confounding factors and directly assess the effects of increasing ketone body delivery, 3OHB was infused in a well-established canine pacing model of HF (29), starting during the phase of compensated cardiac dysfunction. The results were striking. 3OHB infusion completely prevented changes in LVEDP, preserved baseline heart rate, and markedly reduced pacing-induced diminutions in cardiac output and EF, thus halting the progression toward congestive HF. The chronic infusion of 3OHB also reduced LV remodeling, as evidenced by the less pronounced LV dilatation and lack of cardiac hypertrophy. Moreover, the enhanced dP/dtmax during β-adrenergic stimulation in 3OHB-treated dogs indicates a better-preserved myocyte contractile reserve. It should also be noted that our in vivo findings raise the possibility that 3OHB infusion also influences cardiac output and remodeling by lowering afterload, as evidenced by reduced peripheral resistance and effective Ea. Such an effect could be related to a direct vasodilatory action exerted by 3OHB on resistance vessels, a secondary response to an increase in cardiac output, and/or decreased sympathetic tone. Future studies aimed at the extracardiac effects of 3OHB should prove interesting.
The capacity for FAO is reduced in the failing heart, setting the stage for a compensatory increase in glucose utilization, though increased glycolysis has been shown to outpace the increases in downstream pyruvate oxidation due to PDH inhibition and elevated pyruvate carboxylation via malic enzyme 1 (43). These collective fuel metabolic disturbances set the stage for a mitochondrial fuel (acetyl-CoA) input deficit. The myocardial substrate utilization studies following the pacing protocol indicate that the exogenous 3OHB served as an additional fuel for the failing canine heart. This conclusion is supported by the observations that cardiac 3OHB uptake was increased, together with an increase in oxygen consumption. The changes in myocardial substrate oxidation observed with 3OHB infusion are also consistent with the conclusion that 3OHB provides an additional source of acetyl-CoA to the TCA cycle. The observed decrease in glucose oxidation rates likely reflects the well-known impact of increased acetyl-CoA levels on pyruvate dehydrogenase activity, otherwise known as the Randle effect (44). Interestingly, the effect of 3OHB on substrate utilization in the normal (nonpaced) canine heart was different than that of the failing heart. In the normal heart, 3OHB infusion competed with FAO rather than glucose oxidation. This likely reflects the high rates of β-oxidation and derivative acetyl-CoA production in the normal heart. The substrate competition results in the normal canine heart shown here differ from those of a recent report that described a reduction on cardiac glucose uptake with acute infusion of 3OHB in humans without HF (45). The basis for this discrepancy is unclear but could relate to species differences or the impact of acute versus chronic infusion of 3OHB. It should also be noted that, in the study by Gormsen et al., glucose uptake — rather than glucose oxidation — was measured.
The bioenergetic effects of 3OHB oxidation were evaluated in freshly isolated mouse heart mitochondria using a recently developed assay platform that permits comprehensive assessment of respiratory fluxes and determinants of energy transfer (34). Importantly, this approach provides for a highly tractable model system wherein substrate supply and energetic backpressure (ΔGATP) can be precisely controlled. In the context of physiologic energy demands and limiting amounts of fatty acids and other substrates (as occurs in the failing heart), provision of 3OHB to intact mitochondria enhanced respiratory kinetics and thermodynamics. Our results support the conclusion that oxidation of 3OHB provides an alternate and uniquely efficient source of acetyl-CoA in the context of reduced oxidation of fatty acids and pyruvate in the failing heart. Accordingly, increased generation of acetyl-CoA provides substrate for the TCA cycle, leading to increased production of NADH. NADH is also produced by the Bdh1-mediated oxidative reaction converting 3OHB to acetoacetate. We propose that the increased flow of reducing equivalents at Complex I of the ETC augments and protects the proton driving force and ΔGATP (Supplemental Figure 9). Notably, the findings here align with the results of previous work conducted in healthy dogs and in rats, showing that provision of 3OHB improves cardiac respiratory efficiency and proton driving force (32, 33, 46). In these studies, the investigators surmised that the 3OHB oxidation reaction augments ΔGredox and promotes electron flux through Complex I of the ETS, while limiting flux through FAD-linked enzymes. In theory, the resulting improvement in redox driving forces across the ETS should support a more polarized ΔΨ for a given rate of oxygen consumption — the exact phenotype observed in our in vitro assays. This observation is noteworthy because, in working hearts, the energy harnessed in the steady-state PMF (Δp) determines the extent to which the ATP synthase complex can displace the ATP/ADP ratio from equilibrium, which dictates the free energy of ΔGATP and, thus, the power available for cardiac work.
Taken together, our results support the conclusion that ketone bodies provide an additional myocardial fuel source when fatty acids and glucose cannot fully satisfy energy demands such as occurs in HF or in the context of prolonged fasting. Accordingly, increasing the availability of 3OHB exerts cardioprotective effects in pathophysiologic circumstances that lead to HF by providing an ancillary fuel source and improving mitochondrial energetics. It should be noted that our data do not exclude additional cardioprotective actions of 3OHB beyond energetics. For example, our results raise the possibility that 3OHB can exert beneficial extracardiac effects, including reducing peripheral resistance, although we have not determined if this is a primary or secondary effect. In addition, 3OHB has been shown to convey antiinflammatory functions and exert epigenomic effects, including inhibition of histone deacetylases (HDACs), which have been implicated in pathologic cardiac hypertrophy (47, 48). Taken together, our results strongly suggest that increasing delivery of 3OHB to the heart should be considered as a potentially novel ancillary therapeutic approach to HF.
Methods
Mouse studies
General.
Mouse studies were performed on male mice 6–12 weeks of age (see below for further detail). Mice were housed in a facility with 12-hour light/dark cycles. Mice were provided ad libitum access to water and either normal chow (16.4% protein, 4.0% fat, and 48.5% carbohydrates; Harlan Teklad) or KD (20% protein, 80% fat, and 0.1% carbohydrate; Research Diets Inc., catalog D03022101) as specified. In certain circumstances, singly housed mice were fasted for 24 hours with ad libitum access to water. For experiments involving isolated cardiac mitochondria, all mice were on a C57BL/6NJ (BL6N) background, male, and aged 16–30 weeks. Prior to tissue removal, animals were anesthetized with pentobarbital (Nembutal, Oak Pharmaceuticals, 100 mg/kg). TAC/MI cardiac surgeries were utilized to induce HF, as described (27, 49). Ultrasound examination of the cardiovascular system was noninvasively performed using a Vevo2100 Ultrasound System (VisualSonics Inc.), as described previously (49). More details on TAC, TAC/MI, and echocardiography can be found in Supplemental Methods.
Generation of csBDH1–/– mice
To generate cardiac-specific BDH1-KO mice, targeted ES cells were purchased from EUCOMM (Bdh1tm1a[EUCOMM]Wtsi) and injected into C57BL/6J blastocysts (Charles River Laboratories). The allele contains flippase recognition target (FRT) sites flanking the lacZ and neo cassettes (gene trap), as well as Loxp sites flanking exons 3 and 4 (Supplemental Figure 1A). Founder mice were crossed to C57BL/6J to confirm germline transmission, followed by crossing to the flippase (FLP) mouse (The Jackson Laboratory, 009086) to remove the gene trap. Subsequent mating was performed to remove the FLP allele and create the Bdh1flox mice. The Bdh1flox mice were backcrossed twice with WT BL6N mice to obtain Bdh1flox mice on a BL6N (Nnt WT) genetic background. Finally, to create the cardiac-specific BDH1–/– mice, a Cre recombinase strategy was employed using the αMHC promoter (Supplemental Figure 1A) (23). Breeding pairs were established with Bdh1flox BL6N mice and hemizygous αMHC-Cre BL6N mice. The litters generated from final breeding pairs included mice with floxed Bdh1 alleles and either αMHC-Cre+ or αMHC-Cre– genotype.
For genotyping, ear punch samples were collected from pups at 4 weeks of age (after weaning). DNA was extracted from ear punch samples by adding 100 μl of 25 mM NaOH (Thermo Fisher Scientific) and 0.2 mM EDTA (Thermo Fisher Scientific, pH 12), and by heating the samples at 95°C for 20 minutes. Subsequent addition of 100 μl of 40 mM Tris (Thermo Fisher Scientific, pH 5) neutralized the samples. Samples were used immediately following extraction or stored at 4°C. Standard PCR protocols using the following primers (manufacturered by IDT) were utilized to amplify the DNA region of interest. PCR products were analyzed using standard gel electrophoresis. Bdh1, meltine temperature (Tm) = 58°C, (forward) 5′-TGCAGGAATCAGTGCTCTCTCCTAGCA-3′, (reverse) 5′-GGTGTCAGGGCTGAAGGATG-3′; Cre, Tm = 60°C, (forward) 5′-CCGGTGAACGTGCAAAACAGGCTCTA-3′, (reverse) 5′-CTTCCAGGGCGCGAGTTGATAGC-3′; Nnt, Tm = 60°C, (WT, forward) 5′- GGGCATAGGAAGCAAATACCAAGTTG-3′, (mutant, forward) 5′- GTGGAATTCCGCTGAGAGAACTCTT-3′, (common, reverse) 5′- GTAGGGCCAACTGTTTCTGCATGA-3′.
Canine studies
Surgical instrumentation.
Thirteen male mongrel dogs (aged 9–12 months; 21–27 kg) were chronically instrumented with a solid state pressure gauge inserted in the left ventricle, 3 fluid-filled catheters (1 inserted in the descending thoracic aorta, 1 in the right ventricle, and 1 in the left atrium), a Doppler flow probe placed around the left circumflex coronary artery, and 2 pacing leads attached to the LV epicardial surface, as previously described (22, 30). The dogs wore cotton/nylon jackets to protect the exteriorized wires and catheters. Continuous intravascular infusions were performed by connecting external pumps (50 ml capacity), held in the pockets of the jacket, to the right ventricular catheter or, in case of occlusion, to the left atrial one. All the experiments were performed in conscious, nonsedated dogs recumbent on the right side on the laboratory table.
General protocol.
The study included 4 groups of dogs, characterized and named as follows. (a) HF + 3OHB group: 8 dogs underwent cardiac pacing with an external pacemaker at the rate of 210 beats/min for 3 weeks and at 240 beats/min for an additional week to induce HF, as previously described (22, 30). Starting at 12–13 days of pacing, sodium D-3OHB dissolved in sterile water was continuously infused first at the dose of 2.5 μmol/kg/min and progressively increased until day 16, when the dose of 5 μmol/kg/min was reached and maintained constant until day 29 of pacing. The total infusion volume was approximately 55 ml/day, a negligible amount in dogs of this size, if compared with their total blood volume of 2.5–3 liters. (b) Control + 3OHB group: in 5 dogs, the heart was not paced and was 3OHB infused in a similar fashion for 14 days. (c) HF group: the control HF group not infused with 3OHB was composed of 8 dogs randomly drawn from a larger historical pool of 15 dogs previously studied by us for a recent study on cardiac metabolism (22). (d) Control group: a non–heart-paced control group was obtained by randomly drawing 6 dogs from a larger historical pool of 10 dogs utilized for the same study mentioned above. The control group was necessary as a term of comparison for the study on cardiac metabolism, since in HF and HF + 3OHB groups radioisotopes could be infused only once; hence, baseline metabolism could not be measured before starting the pacing protocol, as for functional measurements. Dogs were euthanized after performing the final functional and metabolic measurements. The end-point for the HF group was based on the time when LVEDP reached 25 mmHg, which — in our model — typically occurs after approximately 4 weeks of pacing (30); therefore, dogs of the HF + 3OHB group were euthanized at 29 days (14–15 days of 3OHB infusion), regardless of their stage of cardiac dysfunction. Dogs of the control + 3OHB group were euthanized after 14 days of 3OHB infusion. After euthanasia, the hearts were extracted and weighed.
Recorded and calculated functional parameters.
Hemodynamics and cardiac morphofunctional parameters were measured, respectively, by connecting the chronically implanted probes to recorders or by echocardiography. In the HF group, measurements were taken at baseline, before cardiac pacing, and then every week. In the HF + 3OHB group, intermediate measurements were not taken at 14 days, but at 13 days — immediately before starting the 3OHB infusion. In the control + 3OHB group, measurements were taken at baseline and after 14 days of 3OHB infusion. Directly measured hemodynamic parameters were heart rate; LV end-diastolic, aortic systolic, and diastolic pressures; and coronary blood flow. Calculated parameters were mean aortic pressure, mean blood flow in the left circumflex coronary artery, and the first derivative of LV pressure (dP/dt). dP/dtmax is an index of contractility, and dP/dtmin is an index of diastolic relaxation rate during the respective isovolumic phases. Data for each time point were obtained by averaging measures over 1 respiratory cycle. Hemodynamic measurements were also taken, after 29 days of cardiac pacing, during β-adrenergic stimulation with dobutamine infusion at 5, 10, and 15 μg × kg−1 × min−1, with 5 minutes for each dose (50). Echocardiographic measurements were performed based on the right parasternal short axis and long axis, as well as apical 4-chamber and 2-chamber views of at least 5 consecutive cardiac cycles at a frame rate of 40.70 frames per second (fps). EF was calculated using the Simpson formula on the apical views. Cardiac output was calculated by multiplying stroke volume by heart rate. Total peripheral resistance, an index of mean afterload, was calculated by dividing mean arterial pressure by cardiac output. Effective Ea, a more integrated index of both mean and pulsatile afterload (51), was calculated by dividing mean arterial pressure by stroke volume.
Cardiac oxygen and energy substrate consumption.
Cardiac metabolism was assessed on the last day of the protocol. A catheter was inserted into the coronary sinus, through peripheral veins, under X-ray fluoroscopic guidance. Blood partial pressure of O2 (PO2) and O2 concentrations were determined in paired arterial and coronary sinus blood samples, and coronary blood flow was measured to calculate MVO2. The isotopic tracers [9,10-3H]-oleate (0.7 μCi/min) and [U-14C]-glucose (20 μCi as a bolus, followed by 0.3 μCi/min) were infused through a peripheral vein to track the metabolic fate of FFA and glucose, respectively, which are used by cardiac muscle as a source of energy (52). After 40 minutes of tracer infusion, baseline hemodynamics were recorded, and paired blood samples were withdrawn from aorta and coronary sinus. The concentrations of total and radiolabeled FFA and glucose,14CO2, 3H2O, lactic acid, and ketone bodies were determined in arterial and coronary sinus blood samples, and the rate of cardiac substrate uptake and oxidation were calculated using coronary blood flow, according to well-established methods (22, 52). All the calculated values of MVO2 and cardiac substrate uptake and oxidation were normalized by heart rate and heart weight.
RNA analyses
Aliquots of pulverized tissue were placed into Precellys homogenization tubes on ice. QIAzol Lysis Reagent (Qiagen, 700 μl) was added to each sample. Samples were then placed into a prechilled Precellys Homogenizer. The homogenization protocol used 3 cycles of 20 seconds at 6,800 rpm agitation, followed by a 10-second pause. The remainder of the RNA isolation followed the manufacturer’s instructions (Qiagen miRNeasy). Isolated RNA was diluted to 0.1 μg/μl for use in cDNA synthesis. Quantitative PCR (qPCR) was performed as described previously (20) using the appropriate primers (Supplemental Table 2).
Immunoblotting analyses
Western immunoblotting was performed with either heart or liver tissue lysate prepared as previously described (53). Protein concentrations were quantified with the Micro BCA Protein Assay Kit (Thermo Fisher Scientific). Primary antibodies for BDH1 (Proteintech, catalog 15417-1-AP) and SDHA (Invitrogen, catalog 459200) were diluted 1:5,000 in 1:1 Odyssey Blocking Buffer (LI-COR Biosciences) and 0.1% Tween in Tris-buffered saline (TBS-T, MilliporeSigma) and incubated overnight at 4°C. Secondary antibodies IRDye 800CW donkey anti–mouse or anti–rabbit IgG (H + L) (LI-COR Biosciences, catalogs 926-32212 and 926-32211) were added to blot at 1:7,500 dilution in 1:1 Odyssey Blocking Buffer and TBS-T. Odyssey Image Studio software (LI-COR Biosciences) was used for quantification.
Metabolite measurements
Myocardial 3OHB measurements were performed by the Metabolomics Core at Sanford Burnham Prebys Medical Discovery Institute in Lake Nona using previously described methods (20, 28). This was followed by the derivatization of organic acids, as described (49). More details can be found in Supplemental Methods.
Blood samples were taken between 8–10 a.m. from the tail vein for determination of β-hydroxybutyrate concentrations using a Precision Xtra Ketone Meter according to manufacturer’s instructions (Abbott). Plasma glucose and FFAs were measured using kits from Cayman Chemical (catalogs 10009582 and 700310, respectively), according to the manufacturer’s instructions.
NMR-based substrate oxidation measurements
Mice were heparinized (100 U, Mckesson Medical) by i.p. injection and anesthetized with 85 mg/kg ketamine (Hospira) and 12 mg/kg xylazine (Akorn Inc.). Following sacrifice, hearts isolated from csBDH1–/–, and control groups were perfused in the Langendorff mode with a fluid-filled intraventricular balloon for hemodynamic monitoring and to apply an external load at a 5-mm diastolic pressure. Perfusate contained either 0.5 mM D-3-hydroxy-[2,4-13C2] butyrate ([2,4-13C2]3OHB) (Isotec, MilliporeSigma) and unlabeled 0.4 mM palmitate/BSA (MilliporeSigma) or 0.4 mM [U-13C] palmitate/BSA (Cambridge Isotope Laboratories Inc., MilliporeSigma) and unlabeled 0.5 mM D-3-hydroxybutyrate (MilliporeSigma), both in the presence of unlabeled 1 mM lactate (MilliporeSigma) and 10 mM glucose (MilliporeSigma). Following each perfusion, hearts were snap frozen with liquid nitrogen–cooled tongs. NMR spectroscopy of tissue extracts was used to quantify the fractional enrichment of acetyl-CoA (Fc) from glutamate isotopomers produced from labeled substrate oxidation — ketone or palmitate, as specified in text (54, 55).
Mitochondrial isolation
For experiments involving isolated cardiac mitochondria, all mice were on a BL6N background, male, and aged 16–24 weeks. Prior to tissue removal, animals were anesthetized with pentobarbital (Nembutal, Oak Pharmaceuticals, 100 mg/kg).
Mitochondria were isolated from murine hearts by differential centrifugation. The following buffers were utilized for all isolations: Buffer A, phosphate buffered saline (pH 7.4) with EDTA (10 mM); Buffer B, MOPS (50 mM, pH 7.1), KCl (100 mM), EGTA (1 mM), and MgSO4 (5 mM); Buffer C, Buffer B, supplemented with BSA (2 g/l, MilliporeSigma, A3803). Heart tissue was excised and immediately placed in ice-cold Buffer A. The atria and aorta were trimmed away. Tissues were minced, incubated 5 minutes on ice in Buffer A supplemented with 0.05% (wt/vol) trypsin (MilliporeSigma, T4799), and centrifuged at 200 g for 5 minutes at 4°C, and the supernatant was discarded to remove trypsin. Tissue pellets were suspended in Buffer C and homogenized with a Potter-Elvehjem tissue grinder with polytetrafluoroethylene (PTFE) pestle. After centrifugation at 500 g for 5 minutes at 4°C, the supernatant from this step was centrifuged at 10,000 g for 10 minutes at 4°C to pellet mitochondria. Mitochondrial pellets were gently washed in 1.4 ml of Buffer B, transferred to a 1.7 ml microcentrifuge tube, and centrifuged at 10,000 g for 5 minutes at 4°C. The supernatant was aspirated and the mitochondrial pellet resuspended in 130 μl of Buffer B. Mitochondrial protein concentration was determined using the bicinchoninic acid (BCA) assay and subsequently diluted to a working concentration of 10 mg/ml in Buffer B. Functional assays involving isolated mitochondria were performed in Buffer D: potassium-MES (105 mM, pH 7.2), KCl (30 mM), KH2PO4 (10 mM), MgCl2 (5 mM), EGTA (1 mM), BSA (2.5 g/L).
Substrates
The following substrates were used for the experiments described above: Potassium Pyruvate (Combi-blocks, QA-1116, prepared fresh daily); L-Malic Acid (MilliporeSigma, M1000), from 1M stock solutions (pH 7 with KOH) stored at –20°C; α-Ketoglutaric acid potassium salt (MilliporeSigma, K2000, prepared fresh daily); L-Octanoylcarnitine (MilliporeSigma, 50892) from 50 mM stock solutions stored at –20°C; (R)-3-Hydroxybutyric acid (MilliporeSigma, 54920, prepared fresh daily), (S)-3-Hydroxybutyric acid (MilliporeSigma, 54925, prepared fresh daily); and Acetoacetate (MilliporeSigma, A8509, prepared fresh daily).
Creatine kinase clamp
Mitochondrial respiration, ΔΨ, and NAD(P)H/NAD(P)+ redox states were measured using a modified version of the creatine (Cr) energetic clamp technique (35, 56). In this assay, the free energy of ATP hydrolysis (ΔG’ATP) can be calculated based on known amounts of Cr, PCr, and ATP, in combination with excess amounts of CK and the equilibrium constant for the CK reaction (i.e., K’CK). Calculation of ΔG’ATP was performed according to the following formula:
Equation 1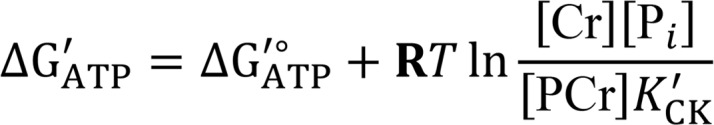
where ΔG’°ATP is the standard apparent transformed Gibbs energy (under a specified pH, ionic strength, free magnesium, and pressure), R is the gas constant (8.3145 J × K–1 × mol–1), and T is temperature in Kelvin (310.15 K), and K’CK is the apparent equilibrium constant for the CK reaction. Given that experiments were performed via sequential additions of PCr, both the ΔG’°ATP and K’CK were determined at each titration step based on the changes in buffer ionic strength and free magnesium, as previously described (57, 58). Calculation of ΔG’ATP at each titration point was performed using the recently published online tool (https://dmpio.github.io/bioenergetic-calculators/).
Mitochondrial respiratory control
High-resolution O2 consumption measurements were conducted using the Oroboros Oxygraph-2K (Oroboros Instruments) at 37°C in a 2-ml reaction volume. Buffer for all assays was Buffer D, supplemented with creatine (Cr, 5 mM; MilliporeSigma, C3630), PCr (1.5 mM; MilliporeSigma, P1937), and CK (20 U/ml; MilliporeSigma, CK-RO). Isolated mitochondria were added to the assay buffer (0.025 mg/ml), followed by the addition of respiratory substrates, then ATP (5 mM; MilliporeSigma, A9062). Then, sequential additions of PCr were added to produce concentrations of 3, 6, 9, 12, and 15 mM.
ΔΨ and NAD(P)H/NAD(P)+ redox
Fluorescent determination of ΔΨ and NAD(P)H/NAD(P)+ were carried out simultaneously via a QuantaMaster Spectrofluorometer (QM-400; Horiba Scientific). The ΔΨ was determined using the ratiometric fluorescent dye tetramethylrhodamine, methyl ester (TMRM) (59). Briefly, the excitation/emission intensities at 572/590 nm and 551/590 nm were recorded, and the 572/551 nm ratio was subsequently converted to millivolts using a KCl standard curve in the presence of valinomycin. The excitation/emission parameters for NAD(P)H were 340/450 nm. All experiments were carried out at 37°C in a 0.2-ml reaction volume with constant stirring. All assays were performed in Buffer D, supplemented with creatine (Cr, 5 mM; MilliporeSigma, C3630), PCr (1.5 mM; MilliporeSigma, P1937), ATP (5 mM; MilliporeSigma, A9062), CK (20 U/ml; MilliporeSigma, CK-RO), and TMRM (0.2 μM; Thermo Fisher Scientific, T668). Isolated mitochondria were added to the assay buffer (0.1 mg/ml), followed by the addition of respiratory substrates, and then sequential PCr additions to produce concentrations of 3, 6, 9, 12, and 15 mM. Next, potassium cyanide (4 mM) was added to generate a state of 100% reduction within the NAD(P)H/NAD(P)+ redox couple. Finally, alamethicin (12.5 μg/ml; AG Scientific, A-1286) was added to generate a state of 0% reduction. The NAD(P)H/NAD(P)+ throughout the experiment was expressed as a percent reduction based on the following formula: (F – F0%)(F100% – F0%)–1
BDH1 activity
Rates of NADH production were determined as previously described (34). More detail can be found in Supplemental Methods.
Statistics
GraphPad Prism 7.0 was used for all statistical analyses except χ2 test. Significance was defined as P < 0.05. Statistical tests were chosen based on the number of compared variables and the size of samples (60). For comparisons of 2 groups with 1 variable: if at least 1 comparison group n < 5, an F-test was applied to determine if variance between groups differed significantly. If variance did not differ significantly, a 2-tailed unpaired t test was applied. If F-test results showed significant difference in variance, a 2-tailed unpaired t test with Welch’s correction was applied (61). For experiments with a small sample size (such as Figure 1, at least 1 group less than n = 8), a 2-tailed Mann-Whitney U test was used (62). When both groups had n = 8 or greater, a D’Agostino-Pearson omnibus normality test was used to determine if values followed a Gaussian distribution (63). If values were determined to be normally distributed, an F-test was applied to determine if variance between groups differed significantly. Depending on these results, either a 2-tailed t test or 2-tailed t test with Welch’s correction was applied as described above. If values were determined to come from a nonnormal distribution, a 2-tailed unpaired Mann-Whitney U test was applied. Comparisons of more than 2 groups were analyzed with 1- or 2-way ANOVA (normal distribution or n < 5 in at least 1 group) with post hoc analysis as described in individual legends (64). For determination of Mendelian inheritance patterns, χ2 test was applied using Excel (65). The χ2 test was conducted using 1 degree of freedom with χ2 table set at 3.841. Statistical analysis for the canine studies was performed by using commercially available software (SigmaStat 4.0). Data were found normally distributed; therefore, changes were compared by t tests and paired t tests or 1-way and 2-way ANOVA followed by Student–Newman post hoc test, depending on the number of groups and time points involved in the comparisons. Data are presented as mean ± SEM. For all the statistical analyses, significance was accepted at P < 0.05.
For experiments using isolated cardiac mitochondria, statistical analyses were performed using the python libraries pandas (0.23.4) and statsmodels (0.9.0). For JO2, ΔΨ, and NAD(P)H/NAD(P)+ redox potential plotted against ΔGATP, an ANOVA (2-way, ketone × ΔGATP; or 3-way, ketone × ΔGATP × genotype) was performed with a significant threshold (α) of P ≤ 0.05, followed by Tukey’s multiple comparisons test. These analyses excluded measurements made at the lowest ΔGATP, which represents an extreme, nonphysiological energetic condition positioned outside the linear range of JO2. Maximal JO2 and corresponding energetics recorded upon addition of 1.5 mM PCr at ΔGATP of –12.95 kCal/mol (approximately state 3 respiration) were analyzed separately by 2-tailed t test. Data are presented as mean ± SEM, and plots were generated using the python library matplotlib (3.0.2).
Study approval
All experiments performed with animals were conducted with protocols approved by IACUCs at SBP-LN, Duke University, and Temple University.
Author contributions
Conceptualization was contributed by JLH, RBV, DMM, FAR, and DPK; methodology was contributed by JLH, MTD, CK, JCP, CP, EDL, and PAC; investigation was contributed by JLH, MTD, CK, JCP, TRM, CP, EDL, and PAC; formal analysis was contributed by JLH, MTD, CK, JCP, RBV, EDL, and PAC; writing the original draft was contributed by JLH and DPK; review and editing were contributed by JLH, RBV, EDL, PAC, DMM, FAR, and DPK; funding acquisition was contributed by EDL, DMM, FAR, and DPK; and supervision was contributed by DPK.
Supplementary Material
Acknowledgments
We thank Teresa Leone for scientific discussion and help with manuscript preparation. We would like to acknowledge the Cardiometabolic Phenotyping and Metabolomics Cores at SBP-LN. We thank Rosa Rosario and the SBP-LN Vivarium staff for assistance with the animal studies and husbandry, Ling Lai for helpful scientific discussions and expert assistance in acquiring data for Figure 1, and Kapil Kapoor for expert assistance with the echocardiographic studies. This work was supported by NIH grants HL058493 (DPK), HL128349 (DPK and DMM), HL129120 (FAR), and HL132525 (EDL).
Version 1. 01/22/2019
In-Press Preview
Version 2. 02/21/2019
Electronic publication
Footnotes
Conflict of interest: DPK serves on the Pfizer Cardiovascular, Metabolic, and Endocrine Diseases Therapeutic Area (CVMET) Scientific Advisory Panel. FAR received funds for research unrelated to this work from Amgen.
License: Copyright 2019, American Society for Clinical Investigation.
Reference information: JCI Insight. 2019;4(4):e124079. https://doi.org/10.1172/jci.insight.124079.
Contributor Information
Julie L. Horton, Email: juliehortonphd@gmail.com.
Michael T. Davidson, Email: davidson.michael@duke.edu.
Clara Kurishima, Email: clarakurishima@gmail.com.
Rick B. Vega, Email: richard.vega@flhosp.org.
Jeffery C. Powers, Email: jeff.powers@temple.edu.
Timothy R. Matsuura, Email: Timothy.Matsuura@pennmedicine.upenn.edu.
Christopher Petucci, Email: Christopher.Petucci@pennmedicine.upenn.edu.
E. Douglas Lewandowski, Email: doug.lewandowski@osumc.edu.
Peter A. Crawford, Email: crawforp@umn.edu.
Deborah M. Muoio, Email: debbie.muoio@duke.edu.
Fabio A. Recchia, Email: tue68462@temple.edu.
Daniel P. Kelly, Email: dankelly@upenn.edu.
References
- 1.LaNoue K, Nicklas WJ, Williamson JR. Control of citric acid cycle activity in rat heart mitochondria. J Biol Chem. 1970;245(1):102–111. [PubMed] [Google Scholar]
- 2.Dorn GW, Vega RB, Kelly DP. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015;29(19):1981–1991. doi: 10.1101/gad.269894.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Griffin JL, O’Donnell JM, White LT, Hajjar RJ, Lewandowski ED. Postnatal expression and activity of the mitochondrial 2-oxoglutarate-malate carrier in intact hearts. Am J Physiol, Cell Physiol. 2000;279(6):C1704–C1709. doi: 10.1152/ajpcell.2000.279.6.C1704. [DOI] [PubMed] [Google Scholar]
- 4.Lopaschuk GD, Spafford MA. Energy substrate utilization by isolated working hearts from newborn rabbits. Am J Physiol. 1990;258(5 Pt 2):H1274–H1280. doi: 10.1152/ajpheart.1990.258.5.H1274. [DOI] [PubMed] [Google Scholar]
- 5.Martin OJ, et al. A role for peroxisome proliferator-activated receptor γ coactivator-1 in the control of mitochondrial dynamics during postnatal cardiac growth. Circ Res. 2014;114(4):626–636. doi: 10.1161/CIRCRESAHA.114.302562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106(7):847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gulick T, Cresci S, Caira T, Moore DD, Kelly DP. The peroxisome proliferator-activated receptor regulates mitochondrial fatty acid oxidative enzyme gene expression. Proc Natl Acad Sci USA. 1994;91(23):11012–11016. doi: 10.1073/pnas.91.23.11012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huss JM, Kopp RP, Kelly DP. Peroxisome proliferator-activated receptor coactivator-1alpha (PGC-1alpha) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-alpha and -gamma. Identification of novel leucine-rich interaction motif within PGC-1alpha. J Biol Chem. 2002;277(43):40265–40274. doi: 10.1074/jbc.M206324200. [DOI] [PubMed] [Google Scholar]
- 9.Scarpulla RC. Nuclear control of respiratory chain expression by nuclear respiratory factors and PGC-1-related coactivator. Ann N Y Acad Sci. 2008;1147:321–334. doi: 10.1196/annals.1427.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taegtmeyer H, Overturf ML. Effects of moderate hypertension on cardiac function and metabolism in the rabbit. Hypertension. 1988;11(5):416–426. doi: 10.1161/01.HYP.11.5.416. [DOI] [PubMed] [Google Scholar]
- 11.Sack MN, Rader TA, Park S, Bastin J, McCune SA, Kelly DP. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation. 1996;94(11):2837–2842. doi: 10.1161/01.CIR.94.11.2837. [DOI] [PubMed] [Google Scholar]
- 12.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85(3):1093–1129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 13.Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90(1):207–258. doi: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- 14.Kelly DP, Strauss AW. Inherited cardiomyopathies. N Engl J Med. 1994;330(13):913–919. doi: 10.1056/NEJM199403313301308. [DOI] [PubMed] [Google Scholar]
- 15.Ingwall JS, et al. The creatine kinase system in normal and diseased human myocardium. N Engl J Med. 1985;313(17):1050–1054. doi: 10.1056/NEJM198510243131704. [DOI] [PubMed] [Google Scholar]
- 16.Neubauer S, et al. Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation. 1997;96(7):2190–2196. doi: 10.1161/01.CIR.96.7.2190. [DOI] [PubMed] [Google Scholar]
- 17.Weiss RG, Gerstenblith G, Bottomley PA. ATP flux through creatine kinase in the normal, stressed, and failing human heart. Proc Natl Acad Sci USA. 2005;102(3):808–813. doi: 10.1073/pnas.0408962102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neubauer S. The failing heart--an engine out of fuel. N Engl J Med. 2007;356(11):1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 19.Bottomley PA, et al. Metabolic rates of ATP transfer through creatine kinase (CK Flux) predict clinical heart failure events and death. Sci Transl Med. 2013;5(215):215re3. doi: 10.1126/scitranslmed.3007328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aubert G, et al. The Failing Heart Relies on Ketone Bodies as a Fuel. Circulation. 2016;133(8):698–705. doi: 10.1161/CIRCULATIONAHA.115.017355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bedi KC, et al. Evidence for Intramyocardial Disruption of Lipid Metabolism and Increased Myocardial Ketone Utilization in Advanced Human Heart Failure. Circulation. 2016;133(8):706–716. doi: 10.1161/CIRCULATIONAHA.115.017545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seki M, et al. Acute and Chronic Increases of Circulating FSTL1 Normalize Energy Substrate Metabolism in Pacing-Induced Heart Failure. Circ Heart Fail. 2018;11(1):e004486. doi: 10.1161/CIRCHEARTFAILURE.117.004486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA, Schneider MD. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J Clin Invest. 1997;100(1):169–179. doi: 10.1172/JCI119509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Owen OE, Morgan AP, Kemp HG, Sullivan JM, Herrera MG, Cahill GF. Brain metabolism during fasting. J Clin Invest. 1967;46(10):1589–1595. doi: 10.1172/JCI105650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taegtmeyer H, Hems R, Krebs HA. Utilization of energy-providing substrates in the isolated working rat heart. Biochem J. 1980;186(3):701–711. doi: 10.1042/bj1860701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huynh K. Heart failure: Ketone bodies as fuel in heart failure. Nat Rev Cardiol. 2016;13(3):122–123. doi: 10.1038/nrcardio.2016.22. [DOI] [PubMed] [Google Scholar]
- 27.Weinheimer CJ, Lai L, Kelly DP, Kovacs A. Novel mouse model of left ventricular pressure overload and infarction causing predictable ventricular remodelling and progression to heart failure. Clin Exp Pharmacol Physiol. 2015;42(1):33–40. doi: 10.1111/1440-1681.12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horton JL, et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight. 2016;2(1):e84897. doi: 10.1172/jci.insight.84897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dixon JA, Spinale FG. Large animal models of heart failure: a critical link in the translation of basic science to clinical practice. Circ Heart Fail. 2009;2(3):262–271. doi: 10.1161/CIRCHEARTFAILURE.108.814459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woitek F, et al. Intracoronary Cytoprotective Gene Therapy: A Study of VEGF-B167 in a Pre-Clinical Animal Model of Dilated Cardiomyopathy. J Am Coll Cardiol. 2015;66(2):139–153. doi: 10.1016/j.jacc.2015.04.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saavedra WF, et al. Imbalance between xanthine oxidase and nitric oxide synthase signaling pathways underlies mechanoenergetic uncoupling in the failing heart. Circ Res. 2002;90(3):297–304. doi: 10.1161/hh0302.104531. [DOI] [PubMed] [Google Scholar]
- 32.Kim DK, Heineman FW, Balaban RS. Effects of beta-hydroxybutyrate on oxidative metabolism and phosphorylation potential in canine heart in vivo. Am J Physiol. 1991;260(6 Pt 2):H1767–H1773. doi: 10.1152/ajpheart.1991.260.6.H1767. [DOI] [PubMed] [Google Scholar]
- 33.Veech RL. The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot Essent Fatty Acids. 2004;70(3):309–319. doi: 10.1016/j.plefa.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 34.Fisher-Wellman KH, Davidson MT, Narowski TM, Lin CT, Koves TR, Muoio DM. Mitochondrial Diagnostics: A Multiplexed Assay Platform for Comprehensive Assessment of Mitochondrial Energy Fluxes. Cell Rep. 2018;24(13):3593–3606.e10. doi: 10.1016/j.celrep.2018.08.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Messer JI, Jackman MR, Willis WT. Pyruvate and citric acid cycle carbon requirements in isolated skeletal muscle mitochondria. Am J Physiol, Cell Physiol. 2004;286(3):C565–C572. doi: 10.1152/ajpcell.00146.2003. [DOI] [PubMed] [Google Scholar]
- 36.Glancy B, Barstow T, Willis WT. Linear relation between time constant of oxygen uptake kinetics, total creatine, and mitochondrial content in vitro. Am J Physiol, Cell Physiol. 2008;294(1):C79–C87. doi: 10.1152/ajpcell.00138.2007. [DOI] [PubMed] [Google Scholar]
- 37.Lopaschuk GD, Jaswal JS. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J Cardiovasc Pharmacol. 2010;56(2):130–140. doi: 10.1097/FJC.0b013e3181e74a14. [DOI] [PubMed] [Google Scholar]
- 38.Yokokawa T, et al. A case of acute decompensated heart failure evaluated by series of exhaled acetone concentrations as noninvasive biomarker of heart failure severity. Int J Cardiol. 2016;204:112–113. doi: 10.1016/j.ijcard.2015.11.173. [DOI] [PubMed] [Google Scholar]
- 39.Puchalska P, Crawford PA. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017;25(2):262–284. doi: 10.1016/j.cmet.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cotter DG, Schugar RC, Crawford PA. Ketone body metabolism and cardiovascular disease. Am J Physiol Heart Circ Physiol. 2013;304(8):H1060–H1076. doi: 10.1152/ajpheart.00646.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schugar RC, Moll AR, André d’Avignon D, Weinheimer CJ, Kovacs A, Crawford PA. Cardiomyocyte-specific deficiency of ketone body metabolism promotes accelerated pathological remodeling. Mol Metab. 2014;3(7):754–769. doi: 10.1016/j.molmet.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uchihashi M, et al. Cardiac-Specific Bdh1 Overexpression Ameliorates Oxidative Stress and Cardiac Remodeling in Pressure Overload-Induced Heart Failure. Circ Heart Fail. 2017;10(12):e004417. doi: 10.1161/CIRCHEARTFAILURE.117.004417. [DOI] [PubMed] [Google Scholar]
- 43.Lahey R, et al. Enhanced Redox State and Efficiency of Glucose Oxidation With miR Based Suppression of Maladaptive NADPH-Dependent Malic Enzyme 1 Expression in Hypertrophied Hearts. Circ Res. 2018;122(6):836–845. doi: 10.1161/CIRCRESAHA.118.312660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1(7285):785–789. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 45.Gormsen LC, et al. Ketone Body Infusion With 3-Hydroxybutyrate Reduces Myocardial Glucose Uptake and Increases Blood Flow in Humans: A Positron Emission Tomography Study. J Am Heart Assoc. 2017;6(3):e005066. doi: 10.1161/JAHA.116.005066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sato K, et al. Insulin, ketone bodies, and mitochondrial energy transduction. FASEB J. 1995;9(8):651–658. doi: 10.1096/fasebj.9.8.7768357. [DOI] [PubMed] [Google Scholar]
- 47.Shimazu T, et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339(6116):211–214. doi: 10.1126/science.1227166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Youm YH, et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. 2015;21(3):263–269. doi: 10.1038/nm.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lai L, et al. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: a multisystems approach. Circ Heart Fail. 2014;7(6):1022–1031. doi: 10.1161/CIRCHEARTFAILURE.114.001469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qanud K, et al. Reverse changes in cardiac substrate oxidation in dogs recovering from heart failure. Am J Physiol Heart Circ Physiol. 2008;295(5):H2098–H2105. doi: 10.1152/ajpheart.00471.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kelly RP, et al. Effective arterial elastance as index of arterial vascular load in humans. Circulation. 1992;86(2):513–521. doi: 10.1161/01.CIR.86.2.513. [DOI] [PubMed] [Google Scholar]
- 52.Taegtmeyer H, et al. Assessing Cardiac Metabolism: A Scientific Statement From the American Heart Association. Circ Res. 2016;118(10):1659–1701. doi: 10.1161/RES.0000000000000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cresci S, Wright LD, Spratt JA, Briggs FN, Kelly DP. Activation of a novel metabolic gene regulatory pathway by chronic stimulation of skeletal muscle. Am J Physiol. 1996;270(5 Pt 1):C1413–C1420. doi: 10.1152/ajpcell.1996.270.5.C1413. [DOI] [PubMed] [Google Scholar]
- 54.Malloy CR, Sherry AD, Jeffrey FM. Evaluation of carbon flux and substrate selection through alternate pathways involving the citric acid cycle of the heart by 13C NMR spectroscopy. J Biol Chem. 1988;263(15):6964–6971. [PubMed] [Google Scholar]
- 55.Carley AN, Taglieri DM, Bi J, Solaro RJ, Lewandowski ED. Metabolic efficiency promotes protection from pressure overload in hearts expressing slow skeletal troponin I. Circ Heart Fail. 2015;8(1):119–127. doi: 10.1161/CIRCHEARTFAILURE.114.001496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Glancy B, Willis WT, Chess DJ, Balaban RS. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry. 2013;52(16):2793–2809. doi: 10.1021/bi3015983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Golding EM, Teague WE, Dobson GP. Adjustment of K’ to varying pH and pMg for the creatine kinase, adenylate kinase and ATP hydrolysis equilibria permitting quantitative bioenergetic assessment. J Exp Biol. 1995;198(Pt 8):1775–1782. doi: 10.1242/jeb.198.8.1775. [DOI] [PubMed] [Google Scholar]
- 58.Teague WE, Golding EM, Dobson GP. Adjustment of K’ for the creatine kinase, adenylate kinase and ATP hydrolysis equilibria to varying temperature and ionic strength. J Exp Biol. 1996;199(Pt 2):509–512. doi: 10.1242/jeb.199.2.509. [DOI] [PubMed] [Google Scholar]
- 59.Scaduto RC, Grotyohann LW. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys J. 1999;76(1 Pt 1):469–477. doi: 10.1016/S0006-3495(99)77214-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. de Winter JCF. Using the Student’s t-test with extremely small sample sizes. Practical Assessment, Research & Evaluation. 2013;18(10): https://pareonline.net/pdf/v18n10.pdf.
- 61.Sawilowsky SS. Fermat, Schubert, Einstein, and Behrens-Fisher: The Probable Difference Between Two Means. Journal of Modern Applied Statistical Methods. 2002;1(2):462–472. [Google Scholar]
- 62. Sheskin DJ. Handbook of Parametric and Nonparametric Statistical Procedures: Fourth Edition. Boca Raton, FL: Chapman & Hall/CRC; 2007. [Google Scholar]
- 63. D’Agostino RB. Goodness-of-Fit Techniques. Boca Raton, FL: CRC Press Taylor & Francis Book; 1986. [Google Scholar]
- 64. Zar JH. Biostatistical Analysis. 5th ed. London, England: Pearson College Division; 2010. [Google Scholar]
- 65.Montoliu L. Mendel: a simple excel workbook to compare the observed and expected distributions of genotypes/phenotypes in transgenic and knockout mouse crosses involving up to three unlinked loci by means of a χ2 test. Transgenic Res. 2012;21(3):677–681. doi: 10.1007/s11248-011-9544-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.