

Abstract
While the mechanisms underlying the functions of the complement system in the central nervous system (CNS) and systemically, namely opsonization, chemotaxis, membrane lysis, and regulation of inflammation are the same, the plethora of functions that complement orchestrates in the central nervous system (CNS) is complex. Strictly controlled expression of complement effector molecules, regulators and receptors across the gamut of life stages (embryogenesis, development and maturation, aging and disease) dictate fascinating contributions for this ancient system. Furthermore, it is becoming apparent that complement functions differ widely across distinct brain regions. This review provides a comprehensive overview of the newly identified roles for complement in the brain, including its roles in CNS development and function, during ageing and in the processes of neurodegeneration. The diversity and selectively of beneficial and detrimental activities of complement, while challenging, should lead to precision targeting of specific components to provide disease modifying treatments for devastating psychiatric and neurodegenerative disorders that are still without effective treatment.
Keywords: complement, C1q, C5a, synapse pruning, neurodevelopment, neurodegeneration
1. Introduction
The complement system is a well-known powerful effector mechanism of the immune system that normally contributes to protection from infection and resolution of injury [1]. While the underlying mechanisms of the canonical complement pathway functions of opsonization, chemotaxis, and membrane lysis, occur in the central nervous system (CNS), the precise role of complement in brain is complex, as several temporal and tissue region specific factors influence the presence and activities of the system. In addition, the source of complement components available can be the result of regulated synthesis within the local CNS tissue, or result from an influx from blood upon stroke or traumatic brain injury with destruction, or a transient increase in permeability, of the blood brain barrier (BBB), or both [2]. Effects can be due to the direct activation of complement by necrotic cells, cellular debris or misfolded protein (such as the fibrillar form of amyloid ß peptide in Alzheimer’s disease, as discussed below) or to penetrance of a compromised BBB (See, Alexander et al, in this issue) followed by complement activation as highlighted recently [3, 4]. However, the molecular events that lead from the activation of the initial complement cleavage to loss of brain function are not completely understood. Excessive activation and/or decreased regulation of the pathway may lead to direct neuronal damage. Alternatively, complement activation fragments can induce local activation of microglia/astrocytes that synergizes with other proinflammatory cascades, thus ultimately accelerating pathogenesis and neuronal dysfunction.
In the past 12-15 years, elegant studies have uncovered previously unknown beneficial roles for components of the complement cascade during normal embryogenesis and CNS development, including neuronal proliferation, differentiation and migration signals, and synaptic refinement (as reviewed in [5]). More recently, evidence for complement-dependent synapse elimination during critical neuronal maturation stages has emerged, which when aberrant, may contribute to neuropsychiatric and neurodevelopmental disorders [6]. In addition, excessive complement-mediated synapse pruning occurs in aging, Alzheimer’s disease models, and other disorders that correlate with cognitive or behavior impairments [7–10].
In yet another unexpected finding, C1q, in the absence of other complement components, induces gene expression critical for neuronal survival in vitro [11], and protection against oligomeric and fibrillar Aß-induced neuronal death [12], suggesting a beneficial homeostatic role for this component independent of the rest of the pathway. In addition, early components of complement participate in the non-phlogistic clearance of apoptotic cells and neuronal blebs [13]. All of these discoveries provide exciting potential for therapeutic intervention in multiple devastating neurological disorders which have, as of yet, no known effective treatments, but also demonstrate the need for careful targeting of the pathway to suppress detrimental consequences without inhibiting the beneficial roles this ancient and highly regulated system. This review focuses on functions of complement components synthesized and generated within the CNS itself, including the contributions of complement to normal brain development and homeostasis, and the consequences of malfunction or dysregulation in neuropsychiatric and neurodegenerative disorders.
2. Synthesis of Complement Proteins
The liver has long been recognized as the site of complement protein production both constitutively and in some cases (ex. C3, MBL) as inducible acute phase proteins (reviewed in [14]). However, over 35 years ago it was demonstrated that C1q, the initiating component of the classical complement cascade, could be synthesized in the absence of the C1 serine proteases C1r and C1s in the peripheral tissue myeloid cells [15]. The synthesis of most complement proteins is now recognized to be differentially induced in multiple cell types, including immune, endothelial, and epithelial cells, and pericytes (reviewed in [16]), and importantly, these factors can be induced variously, in CNS resident neurons, astrocytes, oligodendrocytes, and microglia during development, or with injury or aging (reviewed in [18]). In some cases, C1q, the recognition component of the classical complement pathway, is upregulated in the absence of concurrent upregulation of its associated serine proteases, C1r and C1s, ([19, 20] and reviewed in [18]) suggesting C1-independent C1q-mediated functions in the brain. An analysis of complement gene expression in human brains showed significant upregulation of complement component C1q mRNA and protein with aging relative to that of the younger brain [21, 22]. While C1q can be synthesized by some neurons during developmental critical periods [17], C1q protein is dramatically increased in the normal aging of mouse and human brain [23], and is predominantly synthesized in microglia [24]. C3 is highly upregulated in brain tissue with injury/inflammation but is predominately produced by astrocytes. All other complement proteins have been shown to be produced in brain as previously reviewed [18]. Thus, while in many cases of neurodegenerative diseases, such as lupus and multiple sclerosis, the blood brain barrier may be transiently compromised allowing entry of blood-derived complement, complement components are synthesized by resident brain cells, and thus are present even with an intact blood brain barrier in both human and mouse injured/aging brain [12, 19, 21, 25].
3. A role for complement proteins in early brain development
Recent studies have extended the realm of complement in neurodevelopment to the very early stages of brain formation. Early in CNS development, neural stem/progenitor cells (NPCs) rapidly proliferate to provide the substrate cells required for future neuronal and astrocyte lineages. Bolstered by prior reports for a role for complement anaphylatoxin receptors in adult neurogenesis [26], and evidence for C5aR1 in embryonic and induced pluripotent stem cell proliferation [27, 28], a recent study explored a role for C5aR1 in human and mouse embryonic neurogenesis [29]. Expression analysis demonstrated that C5aR1 was expressed on NPCs, in a highly polarized pattern on the apical (i.e. luminal) surface, which correlates with where stemness factors are localized. This restricted apical expression was also previously observed in neuroepithelial cells of the developing neural tube of both humans and mice at an earlier developmental stage [30, 31]. C5a-C5aR1 engagement in human and mouse NPCs resulted in p42/44 (ERK) phosphorylation, which was dependent on PKCζ, a known determinant of NPC polarity [32]. Accordingly, C5aR1 stimulation of human and mouse NPC cultures increased proliferation and maintained cell polarity. In mice in vivo, acute blockade of C5aR1 using a selective C5aR1 antagonist, PMX53 [33], during embryonic neurogenesis, reduced NPC proliferation in the ventricular zone, altering the balance between symmetric (proliferative) and asymmetric (neurogenic) division. This led to significant neurobehavioral alterations in adulthood that were linked to microstructural alterations in multiple regions of the brain.
Two further recent studies have extended the role of complement during early neurodevelopment to that of neuronal migration during embryonic cortical development [34, 35]. This process plays an important role in positioning cells into appropriate spatial relationships. Complement components, C3, MASP1 and MASP2 were identified as key effectors of this process, as shRNA knockdown of these factors resulted in impaired migration of neurons from the NPC proliferative zones, with many neurons arrested along the migratory tract, resulting in alterations in cortical layer thickness [34]. This phenotype could be rescued in part via exogenous administration of a selective C3aR agonist. In a separate study, knockdown or knockout (KO) of C1 inhibitor (C1-INH), a promiscuous complement protease inhibitor, also led to deficits in neuronal migration [35]. In this case, administration of a dual C3aR/C5aR agonist, but not a selective C3a agonist, completely restored the neuronal positioning defects induced by the lack of C1-INH. Interestingly, C1-INH knockdown or KO also resulted in deficits in embryonic NPC proliferation, whereas C3 KO and MASP-2 knockdown increased NPC proliferation, suggesting a complex role for complement in the regulation of NPC proliferation. It should also be noted that C1-INH additionally inhibits other serine proteases (particularly of the fibrinolytic, clotting, and kinin pathways), although the impact of this, if any, during brain development is unclear.
Overall, these studies demonstrate that upstream complement pathways and the downstream C5a-C5aR1 axis, plays key endogenous functional roles in embryonic brain development, maintaining NPC polarity and proliferation, and supporting neuronal migration. Pharmacological or genetic modulation of complement at several levels was found to have profound effects on normal mouse brain development. It is therefore interesting to speculate whether human neurodevelopment diseases such as schizophrenia, which have been shown to have links to upstream pathway complement factor mutations [6], may have aberrant downstream complement receptor (eg. C3aR/C5aR1) signaling leading to defects in neurogenesis and neuronal migration as a component of their pathophysiology, in addition to the postulated roles in synaptic refinement. This is set to be an exciting future avenue of complement research.
4. Complement in Developmental Synaptic Pruning
The classical complement cascade also plays a key role in the development and refinement of synapses during postnatal development, a period of remarkable plasticity. Synapse elimination is a normal developmental process in which subsets of synapses are eliminated while the remaining synapses are maintained and strengthened [36–38]. This process occurs during different critical periods, depending on the brain region and circuit, and has been extensively studied in the mammalian visual system. During early development, relay neurons in the dorsal lateral geniculate nucleus of the thalamus (dLGN) are multiply innervated and receive overlapping inputs from retinal neurons of the left and right eyes. During the first two postnatal weeks, overlapping retinal inputs are remodeled and pruned, resulting in the formation of eye-specific territories [39–42], in which most postsynaptic relay neurons become monocularly innervated. This process is driven by spontaneous retinal activity and dependent on competition between the two eyes [43–46]. Immunohistochemical analyses and high resolution imaging revealed that C1q and C3 are localized to subsets of synapses throughout postnatal brain and retina (47, 48), suggesting that complement proteins may be ’tagging’ weak or immature synapses for elimination in the healthy developing brain. Consistent with this hypothesis neuroanatomical tracings of retinogeniculate projections and electrophysiological recordings of dLGN neurons showed C1q and C3 Kos exhibit sustained defects in synaptic refinement and elimination, suggesting complement proteins cooperate with other pathways to regulate normal circuit development (Lee et al., 2014 ref 50). Although C1q and C3 KOs have defects in synaptic pruning, these mice still undergo a significant degree of synapse elimination suggesting that complement proteins cooperate with other pathways to regulate normal synaptic circuit development [50]. Several proteins associated with adaptive and innate immunity are expressed in the healthy brain and regulate circuit development and plasticity, including MHC Class1 (and interacting proteins) and neuronal pentraxins (51–54) that could interact, directly, or indirectly with complement to refine immature neural circuits.
Emerging research implicate microglia, the brain’s resident macrophages in complement-dependent and independent synaptic pruning in the developing brain [55–57]. Microglia are more phagocytic in the early postnatal brain in regions undergoing synaptic refinement, such as the visual thalamus and express several engulfment receptors, including complement receptor 3 (CR3) that recognize complement and other ‘eat me’ signals in the immune system. Electron microscopy and high resolution 3D reconstructions revealed that many microglia during this early postnatal period (postnatal day P5-8) contained fragments of presynaptic terminals in their lysosomes, and that microglia preferentially engulfed less-active synapses during activity –dependent synaptic competition in this system (36). In addition, mice deficient in C3, which is localized to synapses, or lack the C3b/iC3b receptor CR3, which is expressed on the surface of microglia, have impaired engulfment of synapses by microglia [36]. CR3 KO mice have more synapses and exuberant excitatory connectivity in multiple brain regions that phenocopy C1q and C3 KO mice [49, 58], indicating altered complement signaling early in development results in sustained defects in synaptic connectivity (Figure 1). It is not yet known how secreted complement proteins are recuited to specific (ie. less active) synapses, via complement-interacting proteins, or how microgia know which inputs not to prune. It is likely that there are mechanisms in the developing brain that protect appropriate axons and synapses from elimination, although little is known about complement inhibitors and ‘Don’t eat me’ signals in the healthy brain.
Figure 1. Microglia Prune Developing CNS Synapses in a complement-dependent manner.
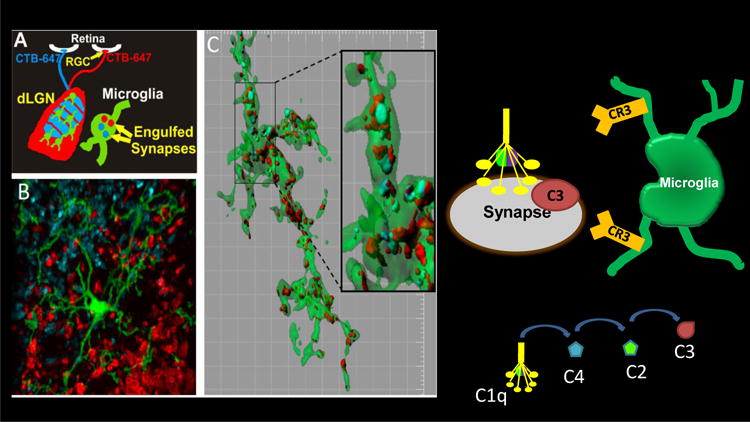
A Schematic of engulfment assay in which left and right eyes are injected with anterograde tracers, CTB-Alexa647 (blue) and CTB-Alexa488 (red), respectively B. Microglia (green) are visualized in the dLGN using a EGFP reporter mouse. C. Representative microglia (green) from the P5 dLGN surface rendered to visualize engulfed inputs (red and blue). [Grid line increments=5 μm] reveals presynaptic elements completely within microglia cytoplasm (green) (Schafer et al., Neuron 2012). Microglia engulfment of synapses is mediated, in part, by C3-CR3 phagocytic signaling as depicted on right.
Recent studies have demonstrated that microglia also engulf synaptic elements in the hippocampus, and visual cortex; however it is not yet known whether complement mediates synaptic remodeling in these regions during different developmental stages (Tremblay et al., 2010; Paolicelli et al., 2011). It is likely that different mechanisms instruct microglia to remodel specific pre and post-synaptic structures depending on local cues.
5. Functional Consequences of Aberrant Complement Activation During Development
What are the functional consequences of aberrant synaptic pruning? Alterations in complement mediated synaptic remodeling is hypothesized to contribute to synapse loss and cognitive dysfunction in neurodevelopmental and neurodegenerative disorders. Failure to prune could lead to excessive connectivity and neuronal excitability. Consistent with this idea, C1qKO mice exhibit enhanced excitatory synaptic connectivity and spontaneous absence seizures (Chu et al., 2010; ref 49). Conversely, inappropriate or excessive complement cascade activation during developmental critical periods could alter neural connectivity and result in aberrant synapse loss.
Irwin Feinberg proposed 30 years ago that altered pruning of cortical synapses might contribute to cognitive dysfunction and pathobiology of schizophrenia [59], however, this idea received little attention until recent genetic studies linked complement protein C4 (C4A) and other genes to schizophrenia [60]. It was recently discovered that schizophrenia’s association with the Major Histocompatibility Complex (MHC) locus arises, in part, from the existence of many common structural forms of the gene for complement component 4 (C4A). The C4 locus includes two C4 paralogues, C4A and C4B, each of which varies in copy number. Alleles that increase expression of C4, a protein necessary for complement activation and C3 deposition, are associated with the highest risk for schizophrenia [60]. Moreover, C4 protein decorates the synapses of human and mouse neurons and is necessary for developmental pruning in the mouse retinogeniculate system [60]. The mechanism by which C4A, but not C4B, underlies the increased risk of schizophrenia associated with C4A is not yet known. One hypothesis is that excessive activation of complement- or microglia-mediated pruning (i.e. due to increased expression of C4, or other mechanisms) during developmental critical periods leads to defects in synaptic connectivity that contribute to long-term deficits in cognition and behavior. Indeed, neuroanatomical and neuroimaging data studies suggest cortical thinning and loss of dendritic spines in some individuals with schizophrenia, but more evidence is needed to validate these findings. New animal models, including humanized mice and non-human primate models, could enable one to test this and other mechanisms nominated by emerging genetics. Intriguingly, alterations in microglia numbers and activation state have been observed in some individuals with autism [61–63]; however, it is not yet clear whether or how microglia and/or the complement cascade contributes to cognitive impairment in autism and other neurodevelopmental disorders.
6. Complement in Alzheimer’s disease and Dementias
Alzheimer’s disease (AD) is the most prevalent neurodegenerative disease of the elderly, with incidence doubling every 5 years after the age of 60, and ultimately affecting approximately 50% of people 85 and older. The devastating clinical ramification is loss of memory and cognitive ability ultimately leading to profound dementia. Evidence of neuroinflammation as a substantial component in the development of AD has been accumulating since the 1990′s [64, 65]. Given that age remains the most significant risk factor for sporadic AD, and that immune mediators of inflammation increase with aging and more so with AD [21, 66], immune activation in the brain is consistently identified as a potential pathogenic pathway and therapeutic target ([67, 68] and reviewed in [69, 70]). Indeed, Zhang, Gaiteri and colleagues have shown, using an unbiased integrative network-based approach, that immune- and microglia-specific networks including complement proteins involved in phagocytosis and inflammation are upregulated in human sporadic/late onset AD (LOAD). As mentioned above, expression of most complement components increases in brain during aging and more so in Alzheimer’s disease patients and animal models of Alzheimer’s disease, consistent with a role for complement in progression of the disease [21, 71]. There are at least three mechanisms by which the complement cascade may play a pathogenic role in pathogenesis of AD: 1. Excessive synaptic pruning triggered by changes at the synapse; 2. Synergistic activation of neuroinflammation; and 3. Deficits in CR1 mediated clearance in the vasculature. Since there is strong evidence for each of these mechanisms, reviewed below, it is plausible that all three mechanisms contribute to progression of the disease.
Following the discovery of complement-mediated synaptic pruning during development as discussed above [17, 36, 47], evidence of the involvement of early complement components in aberrant synapse elimination was described in mouse models of cognitive impairment [8–10]. Hong and colleagues, used super resolution microscopy to quantify synapses, defined as colocalization of pre- and post-synaptic proteins, and the early association of C1q or C3 with synaptic proteins in two different mouse models of AD. A striking decrease in synaptic density was seen in those AD models in brain areas most affected in AD, CA1, CA3 and dentate gyrus of the hippocampus, concomitant with an increased association of C1q or C3 with the post synaptic protein, PSD95 [10, 72]. Synaptic proteins were colocalized with microglia after injection of synaptotoxic oligomeric Aß into hippocampus of wild type mice but not CR3 knock out mice, all pointing to the clearance of iC3b tagged neuronal synapses via CR3, the complement receptor for iC3b [73]. Moreover, genetic and antibody-mediated inhibition of C1q, C3 rescued synapse loss and Aβ-mediated synaptic dysfunction, suggesting aberrant reactivation of a complement - mediated developmental pruning mechanisms mediates pathological synapse loss in AD.
Further investigations demonstrated that deficiency in C3 prevented the decrease in cognitive performance due to aging [74], as well as in the APP/PS1 AD mouse model [72]. In this later study, while amyloid accumulation was not diminished in the C3 KO, fewer microglia and astrocytes were colocalized with Aß plaques in the hippocampus. In addition, lower levels of the proinflammatory cytokines TNFα, IL-12 and gamma interferon (IFN-γ) were detected and a slight but statistically significant rescue of neuron number in the CA3 was detected in the C3 KO relative to the C3 sufficient APP/PS1 model, all suggesting that complement-mediated events are detrimental in these disease models [72]. An important technical and conceptual advance in these studies was the regional specificity revealed by super resolution microscopy and immunohistochemical analysis vs averaging expression/deposition of markers over the entire brain, consistent with regional differences seen in microglia more recently reported in normal mouse aging [75]. Similar extensive characterization of regionally selective complement-mediated synapse pruning and induction of inflammatory mediators have been reported in models of frontal temporal dementia associated with obsessive-compulsive disorder grooming behaviors [8] and West Nile virus infection associated with memory impairment [9].
In vitro studies have shown that fibrillar Aβ (fAß) can activate the classical complement pathway (in the absence of antibody) and the alternative pathway [76, 77]. Potential consequences include opsonization and engulfment of fAß, leukocyte recruitment (by anaphylatoxins C3a and C5a) and bystander lysis on neuronal cells by the membrane attack complex (MAC) (reviewed in [18]) (Figure 2). As stated above, synthesis of most complement factors has been shown to occur within the “injured” brain, including the AD brain, and it is well established that complement factors are found associated with fAß amyloid plaques [64, 78, 79], rather than with diffuse amyloid plaques lacking beta sheet [80], even early in the disease, as fibrillar plaques appear [81]. Complement components C1q, C3 and C4 colocalize with nearly 100% of the fibrillar amyloid plaques in humans with AD cognitive loss and in mouse models of AD, and is seen in other neurodegenerative diseases [8, 78, 79, 82, 83]. In addition, the complement membranolytic complex C5b-9 has been detected in the areas containing the fibrillar plaques in Alzheimer’s disease as well as on some NFTs (reviewed in [18]). Finally, activation of the complement cascade generates C5a, a fragment that recruits and activates microglia via a G-protein coupled receptor [84]. Observations of earlier appearance of thioflavine positive plaques, glial accumulation and hyperphosphorylation of tau in an AD model crossed to a strain of mice with elevated hemolytic (terminal pathway) complement activity [20], support the hypothesis that the activation of the complement pathway by fibrillar amyloid plaques contributes to a local complement-dependent acceleration of pathogenesis and neuronal dysfunction (Fig. 2). Human GWAS studies [85–87], as well as gene expression studies in mouse models of AD [88–90], are consistent with multiple, overlapping processes contributing to the ultimate synapse loss, neuronal degeneration and cognitive loss, but also suggest that microglia and immune response pathways involving inflammation and/or clearance mechanisms play dominant roles.
Fig. 2. C1q and C5a: Homeostasis and neuroinflammation in AD.
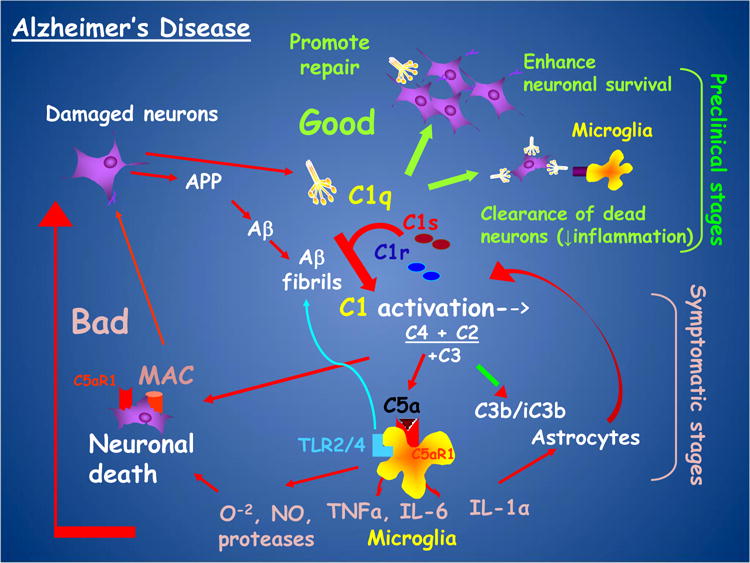
Proposed model of beneficial effects (green arrows) and detrimental effects (red arrows) of complement at different stages of AD progression.
Toll-like receptors (TLRs) are recognition components of the innate immune response that trigger protective inflammatory responses to the pathogens. A recent report demonstrated that LPS, the prototypical TLR4 inflammatory ligand, induced microglial secreted products, specifically TNFα, IL-1α and C1q, which resulted in the specific differentiation of a subset of astrocytes (A1) which consequently induced the production of an astrocyte secreted toxin that triggered apoptosis in neurons [91]. Fibrillar amyloid beta (fAβ) is also a TLR2 and TLR4 ligand inducing microglial inflammation (as reviewed in [92, 93]). Studies in the past decade have demonstrated that C5a-C5aR1 signaling synergizes with the TLR [94], in the activation of phagocytes in the periphery (reviewed in [95, 96]) exacerbating a proinflammatory response and producing detrimental levels of inflammatory cytokines (e.g. TNF-α, IL-1ß). Therefore, it is possible that proinflammatory functions of C5a and TLR in the CNS also synergize in inflammatory neurodegenerative diseases (Fig. 2). In vivo, the genetic deficiency of C5 has been shown to be one of a limited number of genetic differences associated with decreased amyloid deposition in DBA/2J mice versus C57Bl6 mice transgenic for the human APP gene [97], consistent with a contribution of C5a in the disease.
Treatment of two distinct mouse models of Alzheimer’s disease with PMX205, an antagonist of the receptor for the complement activation fragment C5a, led to less fibrillar amyloid accumulation, less activation of microglia and astrocytes, and a suppression of cognitive loss implicating a role for C5aR1 in disease progression [68]. Most recently, an additional AD mouse model, the “Arctic” mouse [98] that generates a human amyloid peptide that is highly resistant to degradation, was crossed to a C5ar1 null mouse [99]. The C5aR1 deficiency prevented spatial specific memory deficits [100] independent of changes in amyloid load (similar to the rescue of cognitive performance without a decrease in amyloid load in the C3 knock out AD model discussed above [72]). Importantly, Sholl analysis of neurons in the CA1 region of the hippocampus revealed a profound decrease in neuronal complexity temporally aligning with cognitive deficits in the C5aR1 sufficient AD model mice, whereas no such loss of neuritic branching was seen in the Arctic mice lacking C5aR1. Gene expression data from adult microglia isolated from brain at 4 different ages, demonstrate that a lack of C5aR1 in this AD model, prevents the polarization of microglia to a more inflammatory state seen with age while enhancing expression of genes involved in phagocytosis and lysosomal degradative enzymes [100]. This supports the postulated notion that amyloid plaques are not sufficient to cause the cognitive decline seen in AD/AD models, but rather the response to amyloid is key. Longitudinal analysis of mRNA and protein expression in microglia and astrocytes correlated with neuron function will further advance our understanding of the pivotal triggers of neurotoxicity, such as the recently proposed pathway in which activated microglia trigger astrocytes to become “A1” astrocytes that generate a neurotoxin [101]. How that program may be modulated by individual complement components, or by the concerted actions resulting from complement activation through C3 or C5 cleavage, remains to be seen.
Immunohistochemistry shows predominant staining of microglia when using well-characterized, specific antibodies to C5aR1 [102]. However, there is clear evidence for the expression of a receptor for C5a on neurons that is upregulated by the proinflammatory cytokine TNFα [103, 104]. In vitro C5a decreased primary neuron integrity (MAP2 staining), and the selective inhibition of C5aR1 using PMX53 blocked that C5a-mediated neuronal injury. Importantly, neurons from C5aR1KO mice were spared from C5a mediated toxicity, and PMX53 had no effect on the fAß-induced neuronal injury of these cells [103]. Furthermore, C5a, via neuronal C5aR1 receptor, contributes to neuronal apoptosis [105] and decreased cell viability in vitro [103, 106, 107], all suggesting that the protection in AD models by C5aR1 antagonism could be mediated via both direct effects on neurons and via microglia activation [103]. The activation of the upstream complement components involved in synaptic pruning could in some cases, such as an aging or in an injured environment, also result in the generation of C5a. As a result, it remains to be seen if C5a contributes to enhanced C1-, C3- and CR3-dependent synaptic pruning, and if so by what mechanism – by directly injuring neurons/synapses leading to “tagging” of synapses, by modulation of synaptic pruning or by induction of microglia/astrocyte induced inflammation and neurotoxicity, or some combination of these pathways.
CR1 is a cell-associated regulatory protein, which binds C3b (and weakly to C4b and C1q). Activation of either the classical or the alternative pathway by fAβ results in the covalent (via a thioester bridge) association of C3b with the amyloid fibers. In humans, erythrocyte CR1 plays a major role in the clearance of C3b-opsonized immune complexes via the mechanism called “immune adherence” in which C3b bound to immune complexes via its thioester bond, binds to erythrocyte CR1 that then transports the immune complexes to the liver and spleen for degradation and thus clearance. In a small study of 36 individuals, AD patients had significantly lower levels of C3b-opsonized Aβ bound to their erythrocytes than age-matched controls or mild cognitively impaired individuals consistent with a defect in peripheral amyloid clearance mechanisms [108]. Subsequently, polymorphisms in CR1 gene were identified as associated with AD risk [109]. While the initial risk associated SNPs were located in non-coding regions of the CR1 gene [109, 110], Keenan and colleagues identified a SNP that is within the coding region of CR1 [111] resulting in an amino acid change (S1610T) within the protein domain that has been attributed to C1q binding [112]. However, no significant differences in CR1 or sCR1 was detected in brain or plasma respectively, of individuals harboring either the rs4844609 or rs6656401 polymorphisms [113]. Additional studies by Rogers and colleagues provided evidence that CR1 on red blood cells could participate in CR1-mediated clearance of Aß from blood [114]. Furthermore, Aß capture by red blood cells (CR1 mediated) was enhanced in vitro by the presence of anti-Aß and complement (which would increase C3b association with fibrillar amyloid), and further enhanced by intravenously administration of anti-Aß in nonhuman primates [115]. Interestingly, the GWAS-identified variants show lower level/density of CR1 expression on red blood cells [116]. Given the multivalent nature of interaction of C3b on an activator such as fAß, as it binds to CR1 on a red cell for immune adherence and clearance, this characteristic low CR1 density may reduce the efficiency of peripheral clearance of amyloid ß, and thus lead to less efficient clearance from the blood and brain, impacting progression of AD.
While in humans CR1 and CR2 are coded for by distinct genes, mice express CR1 and CR2 proteins that result from differential splicing of a single Cr2 gene. In addition, CR1 in the mouse is not expressed on erythrocytes as in humans, although a transgenic mouse expressing CR1 in mouse erythrocytes has been reported [117]. Mice do have an additional gene (Crry), which functions similarly to CR1 regulating C3b amplification, but its structure and cell expression differs from the human, all of which makes mouse models of the role of red cell CR1 in AD more challenging. Clearly further studies are needed to determine the impact of CR1 function in AD.
7. The Role of Complement in Other Neurodegenerative Diseases
In addition to Alzheimer’s disease, there is mounting evidence for an involvement for complement proteins in other neurodegenerative diseases including Parkinson’s disease (PD), Huntington’s disease (HD) and amyotrophic lateral sclerosis (ALS). Increased microglial and astrocyte activation, correlating with neuroinflammation, is a common feature of these diseases, so it is not surprising that complement factors are suggested to potentially contribute to their pathogenesis [118]. Confirmation of complement activation in these conditions has come from both the identification of complement protein activation fragments in human patient samples, as well as in animal models of these diseases. There has also been attempts to identify the role of specific complement proteins in these diseases through the use of KO mice or pharmacological tools. Although the role of specific complement factors is, in many cases, still unknown, there is emerging evidence from clinical and animal studies for differing roles of upstream versus downstream (i.e. terminal) complement activation components in these diseases. A summary of the key findings for complement in neurodegenerative diseases is presented below.
7.1 Amyotrophic Lateral Sclerosis (ALS)
ALS is a motor neuron disease that leads to the progressive loss of motor neurons in both the motor cortex and spinal cord, leading to muscle paralysis and eventual death. In contrast to other neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease, ALS is generally diagnosed at an earlier age, and for the vast majority of patients, has a more rapid progression of pathology, with a median survival of 2-3 years from diagnosis [119]. There is only one approved medication in most countries for ALS, Riluzole, which only has a moderate effect on disease symptoms, and extends survival by a modest 2-3 months [120]. There is therefore great interest in ascertaining the molecular mechanism causing and driving ALS, to identify potential therapeutic targets. Neuroinflammation is a key feature of ALS, with evidence it may be a key driver of disease progression [121]. In line with this, inappropriate and uncontrolled complement activation has also been identified extensively in tissue samples from both ALS patients and in animal models (Kjældgaard, Garred and colleagues review in this same issue). For example, increased C3 and C4 activation products are found in post-mortem motor cortex and spinal cord tissue in ALS sufferers [122–124]. Additionally, complement activation fragments are present in the CSF and plasma of living patients, suggesting an ongoing complement activation process in affected individuals [125–127]. A key animal model of ALS utilizes various mutations of the SOD1 gene (eg. SOD1G93A), which are significant drivers of familial ALS [128]. These SOD1 mutant transgenic mice, which are widely used in ALS studies, develop similar behavioral and neuropathological symptoms of human ALS. SOD1 mutant mice have markedly elevated CNS and muscle expression of complement genes and proteins early in the disease process [129–132]. They also have increased microglial and astrocyte activation in the regions of motor neuron death in the CNS, which are associated with elevations in C5aR1 expression [130]. Similar findings of upregulated complement components and elevated microglial/macrophage C5aR1 expression in the CNS and muscle, are also observed in the TDP-43Q331K transgenic model of ALS [133].
Given the identification of dysregulated complement both in ALS patients, and in mouse ALS models, it is somewhat surprising that SOD1 mutant mice crossed with C1q-, C3-, or C4-KO mice, did not result in any significant alterations in survival or overall disease pathology [134, 135]. Only C4-deficient SOD1 mutant mice were observed to have significant reductions in activated macrophages within sciatic nerves [134]. The authors of these studies concluded that complement activation does not appear to play a significant role in this mouse model of ALS. The results contrasted somewhat an earlier study in SOD1G93A transgenic rats, where blockade of C5aR1 with the C5aR1 antagonist, PMX205, was shown to ameliorate motor deficits and extend survival, associated with reductions in gliosis [136]. Furthermore, C5aR1-KO mice crossed with SOD1G93A mutant mice, had extended survival in both genders [137]. More recently, therapeutic administration of PMX205 to SOD1 mutant mice also resulted in improved motor performance, and extended survival [29], suggesting that complement activation, at least at the level of C5a, is a driver of ALS pathology in mice and rats. The reasons for the apparently conflicting results between C1q, C3 and C4 KO and C5aR1 KO/antagonist studies, are not clear, but one suggestion is that in the absence of upstream complement proteins such as C3, there is downstream pathway compensation, potentially allowing for pathological terminal complement activation and C5a generation / C5aR1 engagement to occur [137]. Further studies, however, are required to precisely define the role of complement effector molecules in ALS, and their potential for therapeutic intervention in patients.
7.2 Parkinson’s disease
Parkinson’s disease (PD) is a progressive and terminal neurodegenerative disease that leads to loss of dopaminergic neurons within the basal ganglia, predominantly affecting motor function. Similar to the majority of neurodegenerative diseases, although there are multiple treatments to modulate disease symptoms, there are no treatments that can currently slow or halt the progression of the disease. There is therefore substantial interest in identifying whether immune proteins, such as complement, could be key disease drivers and would perhaps be amenable to therapeutic intervention. Evidence for complement involvement in PD began in the 1980′s where studies led by McGeer demonstrated activated complement products, C3d and C4d, within post-mortem brain of PD patients [138]. Many years later, iC3b and C9 were also demonstrated to be increased in the substantia nigra of PD patients [139], along with various alterations of complement factors within PD blood [140]. Furthermore, C5a was shown to synergize with IgG isolated from PD serum to mediate dopaminergic neuron death in rat mesencephalic neuron-glia cultures [141]. Despite these reports suggesting involvement of complement in PD pathology, in the MPTP toxin-induced PD model, mice lacking either C1q or C3, show no protection from nigrostriatal dopaminergic degeneration, perhaps indicating that complement does not play a role in the development of PD neuropathology [142, 143]. It should be noted however that a lack of C3 may not necessarily limit downstream complement activation (see above), and so the role of downstream complement effectors in PD pathology remains to be determined.
7.3 Huntington’s disease
HD is caused by an expansion of CAG triplet repeats in the HTT gene leading to aberrant huntingtin protein formation that accumulates within the striatum and cortex, leading to progressive neuronal death and multiple behavioral and motor symptoms. Similar to other neurodegenerative diseases, neuroinflammation is believed to be key in propagating and accelerating pathology, and is an attractive target for drug discovery. Continuing the theme from this section, complement activation products are also found in post-mortem brain tissue from HD subjects [123, 144]. Interestingly, it has also been shown that circulating blood immune cells isolated from HD patients, as well as immune cells and microglia in a transgenic HD model, have impaired migration responses to C5a, driven by defects in actin remodeling due to mutant huntingtin presence [145]. The consequences of this for HD pathology or immunity remain unclear. However, an early study using a toxin-based approach to induce acute striatal lesions demonstrated that therapeutic inhibition of C5aR1 was able to markedly inhibit neurodegeneration, and behavioral deficits associated with the model [146], suggesting that C5a may be a mediator of pathology in HD. Despite this, using a more relevant transgenic mouse model of HD (R6/2 mice), where the mutant human huntingtin gene is overexpressed, Larkin and Muchowski demonstrated that genetic deficiency in C3 (i.e. C3−/− × R6/2 mice), did not alter disease progression [147], reminiscent of prior findings in ALS [135] and PD [142] models. Whether specific therapeutic targeting of terminal complement factors, such as C5a-C5aR1 interaction, would be beneficial in transgenic HD mice models remains to be identified.
8. Therapeutic Implications
Specific antagonists of C5aR1 have been shown to inhibit peripheral acute C5a-induced inflammatory disorders as well as chronic debilitating disorders in animal models [148, 149] [150]). The C5aR1 specific antagonists PMX53 and PMX205, cyclic hexapeptides based on the C-terminal end of C5a that differ from each other only in substituting hydrocinnamate for acetylated phenylalanine, (reviewed in [151]), are orally active, although PMX205 has more favorable tissue distribution and CNS penetrance [152]. PMX205 and/or PMX53 have been shown to reduce disease activity in several animal models, including models of CNS disease such as brain trauma [153], ALS [154] and Huntington-like neurodegeneration [155]. Furthermore, PMX205, significantly and substantially limits pathology in 2 transgenic animal models of AD (Fig. 3) and trended toward a rescue of a behavioral deficit in aged transgenic mice [68]. These data suggest that specific targeting of C5aR1 using small, brain permeable antagonists such as PMX205 are promising for future clinical development.
Figure 3. C5aR1 Antagonist Suppresses AD-like Pathology in Mouse Models.
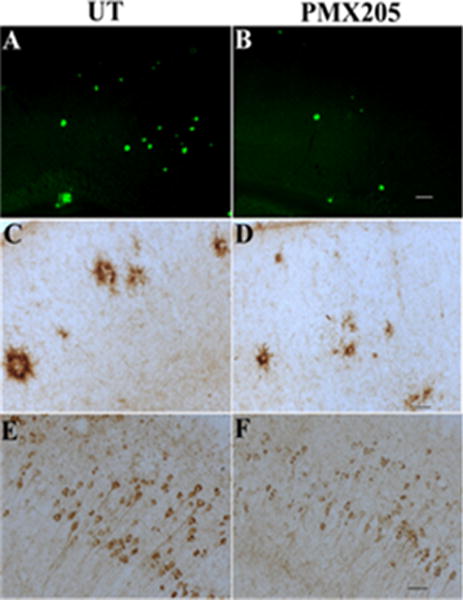
PMX205 substantially reduces thioflavine plaques (A,B), microglial CD45 (C,D) and hyperphosphorylated tau (AT100, E,F) (45-70%) in Tg2576 (A-D) and 3×Tg (E,F). Scale bar is 100um (A,B) or 50um (C-F). From Fonseca et al. (2009) J. Immunology 183:1375–1383. (currently #68)
The specificity of a complement inhibitor is an important consideration for future therapeutic translation for CNS disease. C5a also binds with equal affinity to a second 7-transmembrane receptor originally called C5L2, but now designated as C5aR2 [156, 157]. While several apparent controversies remain to be resolved (as discussed in [158, 159]), current evidence suggests that C5L2 may have both pro- or anti-inflammatory properties dependent upon the cell type, disease context and species [160–162]. Notably, the much utilized PMX series of compounds (ie. PMX53 and PMX205) have been shown to be specific for C5aR1, and do not cross-react with C5aR2 [163]. In addition, the protective effects of a specific genetic ablation of C5aR1 (C5aR1KO) in preventing behavioral deficits in AD transgenic mice [100], and survival in ALS transgenic mice [137], provides genetic target validation to support the pharmacologic data of PMX205.
Importantly for translation of complement inhibitors for CNS disease to the clinic, Eculizumab, a FDA-approved anti C5 monoclonal antibody that binds C5 preventing its cleavage and thus obviating the generation of the C5b-9 membrane attack complex (MAC) as well as C5a, has now been used in humans for specific autoimmune disorders [164]. Thus far, chronic suppression of the C5a/C5aR1 axis has not been harmful for extended clinical use in vaccinated adults. Furthermore, two of the C5aR1 antagonists, PMX53 and CCX168, have shown no detrimental toxicity in human phase Ia/IIb clinical trials of refractory rheumatoid arthritis [165] and ANCA-associated vasculitis [166] respectively. Various anti-C5a compounds have also completed Phase I and II trials with no reported adverse events (see [33] for review). One advantage of a selective non-antibody based C5aR1 antagonist for long term treatment of diseases such as AD is its potential for greater access to the brain (Kumar et al., 2018). In addition, while the antagonist blocks effects of C5a, the generation of C5b by the uninhibited cleavage of C5 enables the assembly of the bacteriolytic C5b-9 complex upon complement activation by pathogens that is critical in combating certain types of bacterial infections.
A method to mitigate or slow aberrant pruning could have translational relevance. Annexon Biosciences recently developed a functional C1q antibody that binds to the globular head domain of C1 and prevents activation of C1r/s and downstream classical complement cascade activation. Recent and emerging data show that this antibody enters the brain and decreases C3 synaptic deposition, synapse loss, and cognitive dysfunction in Alzheimer’s mouse models in which aberrant complement-mediated pruning is implicated (Hong et al., 2016). C1r/s inhibitors have been commercially developed and could also hold promise in future studies if brain penetrant compounds are developed. Given the broad funcions of complement in the brain and periphery, identification of complement interacting proteins at synapses could enable more specific inhibition of complement-dependent pruning in the brain.
9. Concluding Remarks
Application of modern advances in technology as well as creative thinking and well-designed experimentation has enabled a new understanding of the diverse roles of complement components in the brain. In addition to the now definitive synthesis of complement proteins in the brain itself, it is now established that complement activation fragments are critical during embryonic neurogenesis, with regulation of proliferation and differentiation of neural progenitor cells (C5a/C5aR1) and neuronal migration (C3a/C3aR). The discovery of a substantial role of the early classical complement components in sculpting neuronal synapses has created a new paradigm in the conceptual understanding of the role of these proteins in brain development and disease. These pathways can provide potential therapeutic targets to ameliorate or slow the progression of disease. C5aR1-specific antagonism and/or and genetic ablation of C5aR1 in mouse models of neurodegenerative disease, suggest a fundamental role for C5a, since both methods of inhibiting C5aR1 rescue behavior deficits. While further delineation of these processes is necessary, the results will undoubtedly instruct the pharmacological development of interventions for these quite diverse and currently intractable disorders of the nervous system. These disorders not only have a profound impact on quality of life of those affected and their families/communities, but also profoundly impact the health care systems worldwide, and thus disease-modifying treatments are desperately needed.
Highlights.
Complement has critical functional roles in embryonic brain development
C1, C4, C2, C3 and CR3 are critical in synaptic refinement during brain maturation
Complement activation contributes to excessive synaptic pruning in CNS disorders
Determination of the molecular triggers of complement involvement will direct the pharmacologic development of interventions for currently intractable disorders of the CNS.
Acknowledgments
This work was supported by the National Institutes of Health AG00536 (AJT), NIH RO1 NS084298 (BS), Cure Alzheimer’s Fund (BS) and a National Health and Medical Research Council (NHMRC) grant APP1082271 (TMW). TMW is supported NHMRC Career Development Fellowship APP1105420. The authors thank Dr L. Coulthard for his editorial suggestions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
BS is on the scientific advisory board of Annexon, Inc., a new company that will develop therapeutics for neurological diseases. The authors report no other conflict of interest.
References
- 1.Hajishengallis G, Reis ES, Mastellos DC, Ricklin D, Lambris JD. Novel mechanisms and functions of complement. Nat Immunol. 2017;18(12):1288–1298. doi: 10.1038/ni.3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Blasio D, Fumagalli S, Orsini F, Neglia L, Perego C, Ortolano F, Zanier ER, Picetti E, Locatelli M, Stocchetti N, et al. Human brain trauma severity is associated with lectin complement pathway activation. J Cereb Blood Flow Metab. 2018 doi: 10.1177/0271678X18758881. 271678X18758881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mader S, Brimberg L, Diamond B. The Role of Brain-Reactive Autoantibodies in Brain Pathology and Cognitive Impairment. Front Immunol. 2017;8:1101. doi: 10.3389/fimmu.2017.01101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alawieh A, Langley EF, Weber S, Adkins D, Tomlinson S. Identifying the role of complement in triggering neuroinflammation after traumatic brain injury. J Neurosci. 2018 doi: 10.1523/JNEUROSCI.2197-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coulthard LG, Hawksworth OA, Woodruff TM. Complement: The Emerging Architect of the Developing Brain. Trends Neurosci. 2018 doi: 10.1016/j.tins.2018.03.009. [DOI] [PubMed] [Google Scholar]
- 6.Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, Tooley K, Presumey J, Baum M, Van Doren V, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530(7589):177–183. doi: 10.1038/nature16549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sekar A, Bialas AR, de RH, Davis A, Hammond TR, Kamitaki N, Tooley K, Presumey J, Baum M, Van DV, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016 doi: 10.1038/nature16549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lui H, Zhang J, Makinson SR, Cahill MK, Kelley KW, Huang HY, Shang Y, Oldham MC, Martens LH, Gao F, et al. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell. 2016;165(4):921–935. doi: 10.1016/j.cell.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A, Yu J, Perez-Torres C, Frouin A, Wilton DK, et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature. 2016;534(7608):538–543. doi: 10.1038/nature18283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352:712–716. doi: 10.1126/science.aad8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benoit ME, Tenner AJ. Complement protein C1q-mediated neuroprotection is correlated with regulation of neuronal gene and microRNA expression. J Neurosci. 2011;31(9):3459–3469. doi: 10.1523/JNEUROSCI.3932-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benoit ME, Hernandez MX, Dinh ML, Benavente F, Vasquez O, Tenner AJ. C1q-induced LRP1B and GPR6 Proteins Expressed Early in Alzheimer Disease Mouse Models, Are Essential for the C1q-mediated Protection against Amyloid-beta Neurotoxicity. J BiolChem. 2013;288(1):654–665. doi: 10.1074/jbc.M112.400168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fraser DA, Pisalyaput K, Tenner AJ. C1q enhances microglial clearance of apoptotic neurons and neuronal blebs, and modulates subsequent inflammatory cytokine production. J Neurochem. 2010;112(3):733–743. doi: 10.1111/j.1471-4159.2009.06494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perlmutter DH, Colten HR. Molecular immunobiology of complement biosynthesis: a model of single-cell control of effector-inhibitor balance. Annu Rev Immunol. 1986;4:231–251. doi: 10.1146/annurev.iy.04.040186.001311. [DOI] [PubMed] [Google Scholar]
- 15.Bensa JC, Reboul A, Colomb MG. Biosynthesis in vitro of complement subcomponents C1q, C1s and C1 inhibitor by resting and stimulated human monocytes. Biochem J. 1983;216:385–392. doi: 10.1042/bj2160385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lubbers R, van Essen MF, van Kooten C, Trouw LA. Production of complement components by cells of the immune system. Clin Exp Immunol. 2017;188(2):183–194. doi: 10.1111/cei.12952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bialas AR, Stevens B. TGF-beta signaling regulates neuronal C1q expression and developmental synaptic refinement. NatNeurosci. 2013;16(12):1773–1782. doi: 10.1038/nn.3560. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Veerhuis R, Nielsen HM, Tenner AJ. Complement in the brain. Mol Immunol. 2011;48:1592–1603. doi: 10.1016/j.molimm.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lynch NJ, Willis CL, Nolan CC, Roscher S, Fowler MJ, Weihe E, Ray DE, Schwaeble WJ. Microglial activation and increased synthesis of complement component C1q precedes blood-brain barrier dysfunction in rats. Mol Immunol. 2004;40(10):709–716. doi: 10.1016/j.molimm.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 20.Fonseca MI, Chu SH, Berci AM, Benoit ME, Peters DG, Kimura Y, Tenner AJ. Contribution of complement activation pathways to neuropathology differs among mouse models of Alzheimer’s disease. J Neuroinflammation. 2011;8(1):4. doi: 10.1186/1742-2094-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cribbs DH, Berchtold NC, Perreau V, Coleman PD, Rogers J, Tenner AJ, Cotman CW. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: a microarray study. JNeuroinflammation. 2012;9(1):179. doi: 10.1186/1742-2094-9-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yasojima K, Schwab C, McGeer EG, McGeer PL. Up-regulated production and activation of the complement system in Alzheimer’s disease brain. AmJ Pathol. 1999;154(3):927–936. doi: 10.1016/S0002-9440(10)65340-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stephan AH, Madison DV, Mateos JM, Fraser DA, Lovelett EA, Coutellier L, Kim L, Tsai HH, Huang EJ, Rowitch DH, et al. A dramatic increase of C1q protein in the CNS during normal aging. J Neurosci. 2013;33(33):13460–13474. doi: 10.1523/JNEUROSCI.1333-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fonseca MI, Chu SH, Hernandez MX, Fang MJ, Modarresi L, Selvan P, MacGregor GR, Tenner AJ. Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J Neuroinflammation. 2017;14(1):48. doi: 10.1186/s12974-017-0814-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walker DG, McGeer PL. Complement gene expression in human brain: comparison between normal and Alzheimer disease cases. Brain ResMolBrain Res. 1992;14(1-2):109–116. doi: 10.1016/0169-328x(92)90017-6. [DOI] [PubMed] [Google Scholar]
- 26.Rahpeymai Y, Hietala MA, Wilhelmsson U, Fotheringham A, Davies I, Nilsson AK, Zwirner J, Wetsel RA, Gerard C, Pekny M, et al. Complement: a novel factor in basal and ischemia-induced neurogenesis. EMBO J. 2006;25(6):1364–1374. doi: 10.1038/sj.emboj.7601004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hawksworth OA, Coulthard LG, Taylor SM, Wolvetang EJ, Woodruff TM. Brief report: complement C5a promotes human embryonic stem cell pluripotency in the absence of FGF2. Stem Cells. 2014;32(12):3278–3284. doi: 10.1002/stem.1801. [DOI] [PubMed] [Google Scholar]
- 28.Hawksworth OA, Coulthard LG, Mantovani S, Woodruff TM. Complement in stem cells and development. Semin Immunol. 2018 doi: 10.1016/j.smim.2018.02.009. [DOI] [PubMed] [Google Scholar]
- 29.Coulthard LG, Hawksworth OA, Li R, Balachandran A, Lee JD, Sepehrband F, Kurniawan N, Jeanes A, Simmons DG, Wolvetang E, et al. Complement C5aR1 Signaling Promotes Polarization and Proliferation of Embryonic Neural Progenitor Cells through PKCzeta. J Neurosci. 2017;37(22):5395–5407. doi: 10.1523/JNEUROSCI.0525-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Denny KJ, Coulthard LG, Jeanes A, Lisgo S, Simmons DG, Callaway LK, Wlodarczyk B, Finnell RH, Woodruff TM, Taylor SM. C5a receptor signaling prevents folate deficiency-induced neural tube defects in mice. J Immunol. 2013;190(7):3493–3499. doi: 10.4049/jimmunol.1203072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeanes A, Coulthard LG, Mantovani S, Markham K, Woodruff TM. Co-ordinated expression of innate immune molecules during mouse neurulation. Mol Immunol. 2015;68(2 Pt A):253–260. doi: 10.1016/j.molimm.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 32.Gotz M, Huttner WB. The cell biology of neurogenesis. Nat Rev Mol Cell Biol. 2005;6(10):777–788. doi: 10.1038/nrm1739. [DOI] [PubMed] [Google Scholar]
- 33.Hawksworth OA, Li XX, Coulthard LG, Wolvetang EJ, Woodruff TM. New concepts on the therapeutic control of complement anaphylatoxin receptors. Mol Immunol. 2017;89:36–43. doi: 10.1016/j.molimm.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 34.Gorelik A, Sapir T, Haffner-Krausz R, Olender T, Woodruff TM, Reiner O. Developmental activities of the complement pathway in migrating neurons. Nat Commun. 2017;8:15096. doi: 10.1038/ncomms15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gorelik A, Sapir T, Woodruff TM, Reiner O. Serping1/C1 Inhibitor Affects Cortical Development in a Cell Autonomous and Non-cell Autonomous Manner. Front Cell Neurosci. 2017;11:169. doi: 10.3389/fncel.2017.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74(4):691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tremblay ME, Lowery RL, Majewska AK. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010;8(11):e1000527. doi: 10.1371/journal.pbio.1000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- 39.Hong YK, Chen C. Wiring and rewiring of the retinogeniculate synapse. Curr Opin Neurobiol. 2011;21(2):228–237. doi: 10.1016/j.conb.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guido W. Refinement of the retinogeniculate pathway. J Physiol. 2008;586(18):4357–4362. doi: 10.1113/jphysiol.2008.157115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jaubert-Miazza L, Green E, Lo FS, Bui K, Mills J, Guido W. Structural and functional composition of the developing retinogeniculate pathway in the mouse. Vis Neurosci. 2005;22(5):661–676. doi: 10.1017/S0952523805225154. [DOI] [PubMed] [Google Scholar]
- 42.Huberman AD, Feller MB, Chapman B. Mechanisms underlying development of visual maps and receptive fields. Annu Rev Neurosci. 2008;31:479–509. doi: 10.1146/annurev.neuro.31.060407.125533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sretavan DW, Shatz CJ. Prenatal development of retinal ganglion cell axons: segregation into eye-specific layers within the cat’s lateral geniculate nucleus. J Neurosci. 1986;6(1):234–251. doi: 10.1523/JNEUROSCI.06-01-00234.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Torborg CL, Feller MB. Spontaneous patterned retinal activity and the refinement of retinal projections. Progress in neurobiology. 2005;76(4):213–235. doi: 10.1016/j.pneurobio.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 45.Stellwagen D, Shatz C. An Instructive Role for Retinal Waves in the Development of Retinogeniculate Connectivity. Neuron. 2002;33(3):357–367. doi: 10.1016/s0896-6273(02)00577-9. [DOI] [PubMed] [Google Scholar]
- 46.Chen C, Regehr WG. Developmental remodeling of the retinogeniculate synapse. Neuron. 2000;28(3):955–966. doi: 10.1016/s0896-6273(00)00166-5. [DOI] [PubMed] [Google Scholar]
- 47.Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 48.Ahluwalia MS, Patton C, Stevens G, Tekautz T, Angelov L, Vogelbaum MA, Weil RJ, Chao S, Elson P, Suh JH, et al. Phase II trial of ritonavir/lopinavir in patients with progressive or recurrent high-grade gliomas. J Neurooncol. 2011;102(2):317–321. doi: 10.1007/s11060-010-0325-3. [DOI] [PubMed] [Google Scholar]
- 49.Chu Y, Jin X, Parada I, Pesic A, Stevens B, Barres B, Prince DA. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc Natl Acad Sci U S A. 2010;107(17):7975–7980. doi: 10.1073/pnas.0913449107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee H, Brott BK, Kirkby LA, Adelson JD, Cheng S, Feller MB, Datwani A, Shatz CJ. Synapse elimination and learning rules co-regulated by MHC class I H2-Db. Nature. 2014;509(7499):195–200. doi: 10.1038/nature13154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boulanger LM. Immune proteins in brain development and synaptic plasticity. Neuron. 2009;64(1):93–109. doi: 10.1016/j.neuron.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 52.Corriveau RA, Huh GS, Shatz CJ. Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron. 1998;21(3):505–520. doi: 10.1016/s0896-6273(00)80562-0. [DOI] [PubMed] [Google Scholar]
- 53.Shatz CJ. MHC class I: an unexpected role in neuronal plasticity. Neuron. 2009;64(1):40–45. doi: 10.1016/j.neuron.2009.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bjartmar L, Huberman AD, Ullian EM, Renteria RC, Liu X, Xu W, Prezioso J, Susman MW, Stellwagen D, Stokes CC, et al. Neuronal pentraxins mediate synaptic refinement in the developing visual system. J Neurosci. 2006;26(23):6269–6281. doi: 10.1523/JNEUROSCI.4212-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hong S, Dissing-Olesen L, Stevens B. New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol. 2016;36:128–134. doi: 10.1016/j.conb.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Neniskyte U, Gross CT. Errant gardeners: glial-cell-dependent synaptic pruning and neurodevelopmental disorders. Nat Rev Neurosci. 2017;18(11):658–670. doi: 10.1038/nrn.2017.110. [DOI] [PubMed] [Google Scholar]
- 57.Werneburg S, Feinberg PA, Johnson KM, Schafer DP. A microglia-cytokine axis to modulate synaptic connectivity and function. Curr Opin Neurobiol. 2017;47:138–145. doi: 10.1016/j.conb.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ma Y, Ramachandran A, Ford N, Parada I, Prince DA. Remodeling of dendrites and spines in the C1q knockout model of genetic epilepsy. Epilepsia. 2013;54(7):1232–1239. doi: 10.1111/epi.12195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Feinberg I. Schizophrenia: caused by a fault in programmed synaptic elimination during adolescence? Journal of psychiatric research. 1982;17(4):319–334. doi: 10.1016/0022-3956(82)90038-3. [DOI] [PubMed] [Google Scholar]
- 60.Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, Tooley K, Presumey J, Baum M, Van Doren V, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530(7589):177–183. doi: 10.1038/nature16549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morgan JT, Chana G, Pardo CA, Achim C, Semendeferi K, Buckwalter J, Courchesne E, Everall IP. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol Psychiatry. 2010;68(4):368–376. doi: 10.1016/j.biopsych.2010.05.024. [DOI] [PubMed] [Google Scholar]
- 62.Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005;57(1):67–81. doi: 10.1002/ana.20315. [DOI] [PubMed] [Google Scholar]
- 63.Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474(7351):380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, et al. Inflammation and Alzheimer’s disease. NeurobiolAging. 2000;21(3):383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lue LF, Rydel R, Brigham EF, Yang LB, Hampel H, Murphy GM, Jr, Brachova L, Yan SD, Walker DG, Shen Y, et al. Inflammatory repertoire of Alzheimer’s disease and nondemented elderly microglia in vitro. Glia. 2001;35(1):72–79. doi: 10.1002/glia.1072. [DOI] [PubMed] [Google Scholar]
- 66.Giunta B, Fernandez F, Nikolic WV, Obregon D, Rrapo E, Town T, Tan J. Inflammaging as a prodrome to Alzheimer’s disease. J Neuroinflammation. 2008;5:51. doi: 10.1186/1742-2094-5-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hu W, Ranaivo HR, Roy SM, Behanna HA, Wing LK, Munoz L, Guo L, Van Eldik LJ, Watterson DM. Development of a novel therapeutic suppressor of brain proinflammatory cytokine up-regulation that attenuates synaptic dysfunction and behavioral deficits. BioorgMed Chem Lett. 2007;17(2):414–418. doi: 10.1016/j.bmcl.2006.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fonseca MI, Ager RR, Chu SH, Yazan O, Sanderson SD, LaFerla FM, Taylor SM, Woodruff TM, Tenner AJ. Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. Journal of Immunology. 2009;183(2):1375–1383. doi: 10.4049/jimmunol.0901005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Heneka MT, Carson MJ, El KJ, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388–405. doi: 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wyss-Coray T, Rogers J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring HarbPerspectMed. 2012;2(1):a006346. doi: 10.1101/cshperspect.a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reichwald J, Danner S, Wiederhold KH, Staufenbiel M. Expression of complement system components during aging and amyloid deposition in APP transgenic mice. Journal of neuroinflammation. 2009;6:35. doi: 10.1186/1742-2094-6-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shi Q, Chowdhury S, Ma R, Le KX, Hong S, Caldarone BJ, Stevens B, Lemere CA. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci Transl Med. 2017;9(392) doi: 10.1126/scitranslmed.aaf6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352(6286):712–716. doi: 10.1126/science.aad8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shi Q, Colodner KJ, Matousek SB, Merry K, Hong S, Kenison JE, Frost JL, Le KX, Li S, Dodart JC, et al. Complement C3-Deficient Mice Fail to Display Age-Related Hippocampal Decline. JNeurosci. 2015;35(38):13029–13042. doi: 10.1523/JNEUROSCI.1698-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP, Freeman TC, Summers KM, McColl BW. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat Neurosci. 2016;19(3):504–516. doi: 10.1038/nn.4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jiang H, Burdick D, Glabe CG, Cotman CW, Tenner AJ. beta-Amyloid activates complement by binding to a specific region of the collagen-like domain of the C1q A chain. J Immunol. 1994;152(10):5050–5059. [PubMed] [Google Scholar]
- 77.Bradt BM, Kolb WP, Cooper NR. Complement-dependent proinflammatory properties of the Alzheimer’s disease beta-peptide. J Exp Med. 1998;188(3):431–438. doi: 10.1084/jem.188.3.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Loeffler DA, Camp DM, Bennett DA. Plaque complement activation and cognitive loss in Alzheimer’s disease. J Neuroinflammation. 2008;5(1):9. doi: 10.1186/1742-2094-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Afagh A, Cummings BJ, Cribbs DH, Cotman CW, Tenner AJ. Localization and cell association of C1q in Alzheimer’s disease brain. ExpNeurol. 1996;138(1):22–32. doi: 10.1006/exnr.1996.0043. [DOI] [PubMed] [Google Scholar]
- 80.Fonseca MI, Kawas CH, Troncoso JC, Tenner AJ. Neuronal localization of C1q in preclinical Alzheimer’s disease. NeurobiolDis. 2004;15(1):40–46. doi: 10.1016/j.nbd.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 81.Zanjani H, Finch CE, Kemper C, Atkinson J, McKeel D, Morris JC, Price JL. Complement activation in very early Alzheimer disease. Alzheimer Disease & Associated Disorders. 2005;19(2):55–66. doi: 10.1097/01.wad.0000165506.60370.94. [DOI] [PubMed] [Google Scholar]
- 82.Fan R, DeFilippis K, Van Nostrand WE. Induction of complement proteins in a mouse model for cerebral microvascular A beta deposition. JNeuroinflammation. 2007;4:22. doi: 10.1186/1742-2094-4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhou J, Fonseca MI, Pisalyaput K, Tenner AJ. Complement C3 and C4 expression in C1q sufficient and deficient mouse models of Alzheimer’s disease. JNeurochem. 2008;106(5):2080–2092. doi: 10.1111/j.1471-4159.2008.05558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yao J, Harvath L, Gilbert DL, Colton CA. Chemotaxis by a CNS macrophage, the microglia. JNeurosciRes. 1990;27(1):36–42. doi: 10.1002/jnr.490270106. [DOI] [PubMed] [Google Scholar]
- 85.Huang KL, Marcora E, Pimenova AA, Di Narzo AF, Kapoor M, Jin SC, Harari O, Bertelsen S, Fairfax BP, Czajkowski J, et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat Neurosci. 2017;20(8):1052–1061. doi: 10.1038/nn.4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, Kunkle BW, Boland A, Raybould R, Bis JC, et al. Rare coding variants in PLCG2 ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet. 2017 doi: 10.1038/ng.3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.International Genomics of Alzheimer’s Disease C. Convergent genetic and expression data implicate immunity in Alzheimer’s disease. Alzheimers Dement. 2015;11(6):658–671. doi: 10.1016/j.jalz.2014.05.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell. 2017;169(7):1276–1290 e1217. doi: 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 89.Spangenberg EE, Green KN. Inflammation in Alzheimer’s disease: Lessons learned from microglia-depletion models. Brain Behav Immun. 2017;61:1–11. doi: 10.1016/j.bbi.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Matcovitch-Natan O, Winter DR, Giladi A, Vargas Aguilar S, Spinrad A, Sarrazin S, Ben-Yehuda H, David E, Zelada Gonzalez F, Perrin P, et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science. 2016;353(6301):aad8670. doi: 10.1126/science.aad8670. [DOI] [PubMed] [Google Scholar]
- 91.Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017 doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cameron B, Landreth GE. Inflammation, microglia, and Alzheimer’s disease. NeurobiolDis. 2010;37(3):503–509. doi: 10.1016/j.nbd.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B, Rube CE, Walter J, Heneka MT, Hartmann T, et al. TLR2 is a primary receptor for Alzheimer’s amyloid beta peptide to trigger neuroinflammatory activation. Journal of Immunology. 2012;188(3):1098–1107. doi: 10.4049/jimmunol.1101121. [DOI] [PubMed] [Google Scholar]
- 94.Zhang X, Kimura Y, Fang C, Zhou L, Sfyroera G, Lambris JD, Wetsel RA, Miwa T, Song WC. Regulation of Toll-like receptor-mediated inflammatory response by complement in vivo. Blood. 2007;110(1):228–236. doi: 10.1182/blood-2006-12-063636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Song WC. Crosstalk between complement and toll-like receptors. ToxicolPathol. 2012;40(2):174–182. doi: 10.1177/0192623311428478. [DOI] [PubMed] [Google Scholar]
- 96.Hajishengallis G, Abe T, Maekawa T, Hajishengallis E, Lambris JD. Role of complement in host-microbe homeostasis of the periodontium. SeminImmunol. 2013;25(1):65–72. doi: 10.1016/j.smim.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ryman D, Gao Y, Lamb BT. Genetic loci modulating amyloid-beta levels in a mouse model of Alzheimer’s disease. NeurobiolAging. 2008;29(8):1190–1198. doi: 10.1016/j.neurobiolaging.2007.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cheng IH, Scearce-Levie K, Legleiter J, Palop JJ, Gerstein H, Bien-Ly N, Puolivali J, Lesne S, Ashe KH, Muchowski PJ, et al. Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem. 2007;282(33):23818–23828. doi: 10.1074/jbc.M701078200. [DOI] [PubMed] [Google Scholar]
- 99.Hollmann TJ, Mueller-Ortiz SL, Braun MC, Wetsel RA. Disruption of the C5a receptor gene increases resistance to acute Gram-negative bacteremia and endotoxic shock: Opposing roles of C3a and C5a. Molecular Immunology. 2008;45(7):1907–1915. doi: 10.1016/j.molimm.2007.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hernandez MX, Jiang S, Cole TA, Chu SH, Fonseca MI, Fang MJ, Hohsfield LA, Torres MD, Green KN, Wetsel RA, et al. Prevention of C5aR1 signaling delays microglial inflammatory polarization, favors clearance pathways and suppresses cognitive loss. Mol Neurodegener. 2017;12(1):66. doi: 10.1186/s13024-017-0210-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ager RR, Fonseca MI, Chu SH, Sanderson SD, Taylor SM, Woodruff TM, Tenner AJ. Microglial C5aR (CD88) expression correlates with amyloid-beta deposition in murine models of Alzheimer’s disease. J Neurochem. 2010;113(2):389–401. doi: 10.1111/j.1471-4159.2010.06595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hernandez MX, Namiranian P, Nguyen E, Fonseca MI, Tenner AJ. C5a Increases the Injury to Primary Neurons Elicited by Fibrillar Amyloid Beta. ASN Neuro. 2017;9(1):1759091416687871. doi: 10.1177/1759091416687871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pavlovski D, Thundyil J, Monk PN, Wetsel RA, Taylor SM, Woodruff TM. Generation of complement component C5a by ischemic neurons promotes neuronal apoptosis. FASEB J. 2012;26(9):3680–3690. doi: 10.1096/fj.11-202382. [DOI] [PubMed] [Google Scholar]
- 105.Farkas I, Takahashi M, Fukuda A, Yamamoto N, Akatsu H, Baranyi L, Tateyama H, Yamamoto T, Okada N, Okada H. Complement C5a receptor-mediated signaling may be involved in neurodegeneration in Alzheimer’s disease. Journal of Immunology. 2003;170(11):5764–5771. doi: 10.4049/jimmunol.170.11.5764. [DOI] [PubMed] [Google Scholar]
- 106.Humayun S, Gohar M, Volkening K, Moisse K, Leystra-Lantz C, Mepham J, McLean J, Strong MJ. The complement factor C5a receptor is upregulated in NFL−/− mouse motor neurons. J Neuroimmunol. 2009 doi: 10.1016/j.jneuroim.2009.01.028. [DOI] [PubMed] [Google Scholar]
- 107.Pavlovski D, Thundyil J, Monk PN, Wetsel RA, Taylor SM, Woodruff TM. Generation of complement component C5a by ischemic neurons promotes neuronal apoptosis. FASEB J. 2012;26(9):3680–3690. doi: 10.1096/fj.11-202382. [DOI] [PubMed] [Google Scholar]
- 108.Rogers J, Li R, Mastroeni D, Grover A, Leonard B, Ahern G, Cao P, Kolody H, Vedders L, Kolb WP, et al. Peripheral clearance of amyloid beta peptide by complement C3-dependent adherence to erythrocytes. Neurobiol Aging. 2006;27(12):1733–1739. doi: 10.1016/j.neurobiolaging.2005.09.043. [DOI] [PubMed] [Google Scholar]
- 109.Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 110.Brouwers N, Van CC, Engelborghs S, Lambert JC, Bettens K, Le BN, Pasquier F, Montoya AG, Peeters K, Mattheijssens M, et al. Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. MolPsychiatry. 2012;17(2):223–233. doi: 10.1038/mp.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Keenan BT, Shulman JM, Chibnik LB, Raj T, Tran D, Sabuncu MR, Allen AN, Corneveaux JJ, Hardy JA, Huentelman MJ, et al. A coding variant in CR1 interacts with APOE-{varepsilon}4 to influence cognitive decline. Hum Mol Genet. 2012 doi: 10.1093/hmg/dds054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Klickstein LB, Barbashov SF, Liu T, Jack RM, Nicholson-Weller A. Complement receptor type 1 (CR1, CD35) is a receptor for C1q. Immunity. 1997;7:345–355. doi: 10.1016/s1074-7613(00)80356-8. [DOI] [PubMed] [Google Scholar]
- 113.Fonseca MI, Chu S, Pierce AL, Brubaker WD, Hauhart RE, Mastroeni D, Clarke EV, Rogers J, Atkinson JP, Tenner AJ. Analysis of the Putative Role of CR1 in Alzheimer’s Disease: Genetic Association, Expression and Function. PLoSONE. 2016;11(2):e0149792. doi: 10.1371/journal.pone.0149792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Brubaker WD, Crane A, Johansson JU, Yen K, Garfinkel K, Mastroeni D, Asok P, Bradt B, Sabbagh M, Wallace TL, et al. Peripheral complement interactions with amyloid beta peptide: Erythrocyte clearance mechanisms. Alzheimers Dement. 2017 doi: 10.1016/j.jalz.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Crane A, Brubaker WD, Johansson JU, Trigunaite A, Ceballos J, Bradt B, Glavis-Bloom C, Wallace TL, Tenner AJ, Rogers J. Peripheral complement interactions with amyloid beta peptide in Alzheimer’s disease:2 Relationship to Abeta immunotherapy. Alzheimers Dement. 2017 doi: 10.1016/j.jalz.2017.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mahmoudi R, Kisserli A, Novella JL, Donvito B, Drame M, Reveil B, Duret V, Jolly D, Pham BN, Cohen JH. Alzheimer’s disease is associated with low density of the long CR1 isoform. NeurobiolAging. 2015;36(4):1766–1712. doi: 10.1016/j.neurobiolaging.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 117.Li J, Wang JP, Ghiran I, Cerny A, Szalai AJ, Briles DE, Finberg RW. Complement receptor 1 expression on mouse erythrocytes mediates clearance of Streptococcus pneumoniae by immune adherence. Infect Immun. 2010;78(7):3129–3135. doi: 10.1128/IAI.01263-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Brennan FH, Lee JD, Ruitenberg MJ, Woodruff TM. Therapeutic targeting of complement to modify disease course and improve outcomes in neurological conditions. Seminars in Immunology. 2016;28(3):292–308. doi: 10.1016/j.smim.2016.03.015. [DOI] [PubMed] [Google Scholar]
- 119.Pupillo E, Messina P, Logroscino G, Beghi E, Group S Long-term survival in amyotrophic lateral sclerosis: a population-based study. Ann Neurol. 2014;75(2):287–297. doi: 10.1002/ana.24096. [DOI] [PubMed] [Google Scholar]
- 120.Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, Shaw PJ, Simmons Z, van den Berg LH. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;3:17085. doi: 10.1038/nrdp.2017.85. [DOI] [PubMed] [Google Scholar]
- 121.Liu J, Wang F. Role of Neuroinflammation in Amyotrophic Lateral Sclerosis: Cellular Mechanisms and Therapeutic Implications. Front Immunol. 2017;8:1005. doi: 10.3389/fimmu.2017.01005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Donnenfeld H, Kascsak RJ, Bartfeld H. Deposits of IgG and C3 in the spinal cord and motor cortex of ALS patients. J Neuroimmunol. 1984;6(1):51–57. doi: 10.1016/0165-5728(84)90042-0. [DOI] [PubMed] [Google Scholar]
- 123.Yamada T, Akiyama H, McGeer PL. Complement-activated oligodendroglia: a new pathogenic entity identified by immunostaining with antibodies to human complement proteins C3d and C4d. Neurosci Lett. 1990;112(2-3):161–166. doi: 10.1016/0304-3940(90)90196-g. [DOI] [PubMed] [Google Scholar]
- 124.Sta M, Sylva-Steenland RM, Casula M, de Jong JM, Troost D, Aronica E, Baas F. Innate and adaptive immunity in amyotrophic lateral sclerosis: evidence of complement activation. Neurobiol Dis. 2011;42(3):211–220. doi: 10.1016/j.nbd.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 125.Annunziata P, Volpi N. High levels of C3c in the cerebrospinal fluid from amyotrophic lateral sclerosis patients. Acta Neurol Scand. 1985;72(1):61–64. doi: 10.1111/j.1600-0404.1985.tb01548.x. [DOI] [PubMed] [Google Scholar]
- 126.Mantovani S, Gordon R, Macmaw JK, Pfluger CM, Henderson RD, Noakes PG, McCombe PA, Woodruff TM. Elevation of the terminal complement activation products C5a and C5b-9 in ALS patient blood. J Neuroimmunol. 2014;276(1-2):213–218. doi: 10.1016/j.jneuroim.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 127.Xu Z, Lee A, Nouwens A, Henderson RD, McCombe PA. Mass spectrometry analysis of plasma from amyotrophic lateral sclerosis and control subjects. Amyotroph Lateral Scler Frontotemporal Degener. 2018:1–15. doi: 10.1080/21678421.2018.1433689. [DOI] [PubMed] [Google Scholar]
- 128.Woodruff TM, Costantini KJ, Taylor SM, Noakes PG. Role of complement in motor neuron disease: animal models and therapeutic potential of complement inhibitors. Adv Exp Med Biol. 2008;632:143–158. [PubMed] [Google Scholar]
- 129.Lobsiger CS, Boillee S, Cleveland DW. Toxicity from different SOD1 mutants dysregulates the complement system and the neuronal regenerative response in ALS motor neurons. Proc Natl Acad Sci U S A. 2007;104(18):7319–7326. doi: 10.1073/pnas.0702230104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lee JD, Kamaruzaman NA, Fung JN, Taylor SM, Turner BJ, Atkin JD, Woodruff TM, Noakes PG. Dysregulation of the complement cascade in the hSOD1G93A transgenic mouse model of amyotrophic lateral sclerosis. J Neuroinflammation. 2013;10:119. doi: 10.1186/1742-2094-10-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bahia El Idrissi N, Bosch S, Ramaglia V, Aronica E, Baas F, Troost D. Complement activation at the motor end-plates in amyotrophic lateral sclerosis. J Neuroinflammation. 2016;13(1):72. doi: 10.1186/s12974-016-0538-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wang HA, Lee JD, Lee KM, Woodruff TM, Noakes PG. Complement C5a-C5aR1 signalling drives skeletal muscle macrophage recruitment in the hSOD1(G93A) mouse model of amyotrophic lateral sclerosis. Skelet Muscle. 2017;7(1):10. doi: 10.1186/s13395-017-0128-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lee JD, Levin SC, Willis EF, Li R, Woodruff TM, Noakes PG. Complement components are upregulated and correlate with disease progression in the TDP-43Q331K mouse model of amyotrophic lateral sclerosis. J Neuroinflammation. 2018 doi: 10.1186/s12974-018-1217-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chiu IM, Phatnani H, Kuligowski M, Tapia JC, Carrasco MA, Zhang M, Maniatis T, Carroll MC. Activation of innate and humoral immunity in the peripheral nervous system of ALS transgenic mice. Proc Natl Acad Sci U S A. 2009;106(49):20960–20965. doi: 10.1073/pnas.0911405106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lobsiger CS, Boillee S, Pozniak C, Khan AM, McAlonis-Downes M, Lewcock JW, Cleveland DW. C1q induction and global complement pathway activation do not contribute to ALS toxicity in mutant SOD1 mice. Proc Natl Acad Sci U S A. 2013;110(46):E4385–4392. doi: 10.1073/pnas.1318309110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Woodruff TM, Costantini KJ, Crane JW, Atkin JD, Monk PN, Taylor SM, Noakes PG. The complement factor C5a contributes to pathology in a rat model of amyotrophic lateral sclerosis. J Immunol. 2008;181(12):8727–8734. doi: 10.4049/jimmunol.181.12.8727. [DOI] [PubMed] [Google Scholar]
- 137.Woodruff TM, Lee JD, Noakes PG. Role for terminal complement activation in amyotrophic lateral sclerosis disease progression. Proc Natl Acad Sci U S A. 2014;111(1):E3–4. doi: 10.1073/pnas.1321248111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yamada T, McGeer PL, McGeer EG. Lewy bodies in Parkinson’s disease are recognized by antibodies to complement proteins. Acta Neuropathol. 1992;84(1):100–104. doi: 10.1007/BF00427222. [DOI] [PubMed] [Google Scholar]
- 139.Loeffler DA, Camp DM, Conant SB. Complement activation in the Parkinson’s disease substantia nigra: an immunocytochemical study. J Neuroinflammation. 2006;3:29. doi: 10.1186/1742-2094-3-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Goldknopf IL, Sheta EA, Bryson J, Folsom B, Wilson C, Duty J, Yen AA, Appel SH. Complement C3c and related protein biomarkers in amyotrophic lateral sclerosis and Parkinson’s disease. Biochem Biophys Res Commun. 2006;342(4):1034–1039. doi: 10.1016/j.bbrc.2006.02.051. [DOI] [PubMed] [Google Scholar]
- 141.Wang XJ, Yan ZQ, Lu GQ, Stuart S, Chen SD. Parkinson disease IgG and C5a-induced synergistic dopaminergic neurotoxicity: role of microglia. Neurochem Int. 2007;50(1):39–50. doi: 10.1016/j.neuint.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 142.Liang Y, Li S, Guo Q, Zhang Y, Wen C, Zou Q, Su B. Complement 3-deficient mice are not protected against MPTP-induced dopaminergic neurotoxicity. Brain Res. 2007;1178:132–140. doi: 10.1016/j.brainres.2007.08.033. [DOI] [PubMed] [Google Scholar]
- 143.Depboylu C, Schorlemmer K, Klietz M, Oertel WH, Weihe E, Hoglinger GU, Schafer MK. Upregulation of microglial C1q expression has no effects on nigrostriatal dopaminergic injury in the MPTP mouse model of Parkinson disease. J Neuroimmunol. 2011;236(1-2):39–46. doi: 10.1016/j.jneuroim.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 144.Singhrao SK, Neal JW, Morgan BP, Gasque P. Increased complement biosynthesis by microglia and complement activation on neurons in Huntington’s disease. Exp Neurol. 1999;159(2):362–376. doi: 10.1006/exnr.1999.7170. [DOI] [PubMed] [Google Scholar]
- 145.Kwan W, Trager U, Davalos D, Chou A, Bouchard J, Andre R, Miller A, Weiss A, Giorgini F, Cheah C, et al. Mutant huntingtin impairs immune cell migration in Huntington disease. J Clin Invest. 2012;122(12):4737–4747. doi: 10.1172/JCI64484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Woodruff TM, Crane JW, Proctor LM, Buller KM, Shek AB, de Vos K, Pollitt S, Williams HM, Shiels IA, Monk PN, et al. Therapeutic activity of C5a receptor antagonists in a rat model of neurodegeneration. Faseb j. 2006;20(9):1407–1417. doi: 10.1096/fj.05-5814com. [DOI] [PubMed] [Google Scholar]
- 147.Larkin PB, Muchowski PJ. Genetic Deficiency of Complement Component 3 Does Not Alter Disease Progression in a Mouse Model of Huntington’s Disease. J Huntingtons Dis. 2012;1(1):107–118. doi: 10.3233/JHD-2012-120021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Qu H, Ricklin D, Lambris JD. Recent developments in low molecular weight complement inhibitors. Molecular Immunology. 2009;47(2-3):185–195. doi: 10.1016/j.molimm.2009.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Moriconi A, Cunha TM, Souza GR, Lopes AH, Cunha FQ, Carneiro VL, Pinto LG, Brandolini L, Aramini A, Bizzarri C, et al. Targeting the minor pocket of C5aR for the rational design of an oral allosteric inhibitor for inflammatory and neuropathic pain relief. ProcNatlAcadSciUSA. 2014;111(47):16937–16942. doi: 10.1073/pnas.1417365111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Xiao H, Dairaghi DJ, Powers JP, Ertl LS, Baumgart T, Wang Y, Seitz LC, Penfold ME, Gan L, Hu P, et al. C5a receptor (CD88) blockade protects against MPO-ANCA GN. Journal of the American Society of Nephrology. 2014;25(2):225–231. doi: 10.1681/ASN.2013020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kohl J. Drug evaluation: the C5a receptor antagonist PMX-53. CurrOpinMolTher. 2006;8(6):529–538. [PubMed] [Google Scholar]
- 152.Kumar V, Lee JD, Clark RJ, Woodruff TM. Development and validation of a LC-MS/MS assay for pharmacokinetic studies of complement C5a receptor antagonists PMX53 and PMX205 in mice. Scientific Reports. 2018 doi: 10.1038/s41598-018-26387-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Sewell DL, Nacewicz B, Liu F, Macvilay S, Erdei A, Lambris JD, Sandor M, Fabry Z. Complement C3 and C5 play critical roles in traumatic brain cryoinjury: blocking effects on neutrophil extravasation by C5a receptor antagonist. J Neuroimmunol. 2004;155(1-2):55–63. doi: 10.1016/j.jneuroim.2004.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Woodruff TM, Costantini KJ, Crane JW, Atkin JD, Monk PN, Taylor SM, Noakes PG. The complement factor C5a contributes to pathology in a rat model of amyotrophic lateral sclerosis. J Immunol. 2008;181(12):8727–8734. doi: 10.4049/jimmunol.181.12.8727. [DOI] [PubMed] [Google Scholar]
- 155.Woodruff TM, Crane JW, Proctor LM, Buller KM, Shek AB, de VK, Pollitt S, Williams HM, Shiels IA, Monk PN, et al. Therapeutic activity of C5a receptor antagonists in a rat model of neurodegeneration. FASEB J. 2006;20(9):1407–1417. doi: 10.1096/fj.05-5814com. [DOI] [PubMed] [Google Scholar]
- 156.Ohno M, Hirata T, Enomoto M, Araki T, Ishimaru H, Takahashi TA. A putative chemoattractant receptor, C5L2, is expressed in granulocyte and immature dendritic cells, but not in mature dendritic cells. Molecular Immunology. 2000;37(8):407–412. doi: 10.1016/s0161-5890(00)00067-5. [DOI] [PubMed] [Google Scholar]
- 157.Okinaga S, Slattery D, Humbles A, Zsengeller Z, Morteau O, Kinrade MB, Brodbeck RM, Krause JE, Choe HR, Gerard NP, et al. C5L2, a nonsignaling C5A binding protein. Biochemistry. 2003;42(31):9406–9415. doi: 10.1021/bi034489v. [DOI] [PubMed] [Google Scholar]
- 158.Ward PA. Functions of C5a receptors. Journal of Molecular Medicine. 2009;87(4):375–378. doi: 10.1007/s00109-009-0442-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Li R, Coulthard LG, Wu MC, Taylor SM, Woodruff TM. C5L2: a controversial receptor of complement anaphylatoxin, C5a. FASEB J. 2013;27(3):855–864. doi: 10.1096/fj.12-220509. [DOI] [PubMed] [Google Scholar]
- 160.Scola AM, Johswich KO, Morgan BP, Klos A, Monk PN. The human complement fragment receptor, C5L2, is a recycling decoy receptor. MolImmunol. 2009;46(6):1149–1162. doi: 10.1016/j.molimm.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lee H, Whitfeld PL, Mackay CR. Receptors for complement C5a. The importance of C5aR and the enigmatic role of C5L2. Immunol Cell Biol. 2008;86(2):153–160. doi: 10.1038/sj.icb.7100166. [DOI] [PubMed] [Google Scholar]
- 162.Croker DE, Halai R, Kaeslin G, Wende E, Fehlhaber B, Klos A, Monk PN, Cooper MA. C5a2 can modulate ERK1/2 signaling in macrophages via heteromer formation with C5a1 and beta-arrestin recruitment. Immunol Cell Biol. 2014;92(7):631–639. doi: 10.1038/icb.2014.32. [DOI] [PubMed] [Google Scholar]
- 163.Monk PN, Scola AM, Madala P, Fairlie DP. Function, structure and therapeutic potential of complement C5a receptors. Br J Pharmacol. 2007;152(4):429–448. doi: 10.1038/sj.bjp.0707332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sahin F, Ozkan MC, Mete NG, Yilmaz M, Oruc N, Gurgun A, Kayikcioglu M, Guler A, Gokcay F, Bilgir F, et al. Multidisciplinary clinical management of paroxysmal nocturnal hemoglobinuria. AmJBlood Res. 2015;5(1):1–9. [PMC free article] [PubMed] [Google Scholar]
- 165.Vergunst CE, Gerlag DM, Dinant H, Schulz L, Vinkenoog M, Smeets TJ, Sanders ME, Reedquist KA, Tak PP. Blocking the receptor for C5a in patients with rheumatoid arthritis does not reduce synovial inflammation. Rheumatology(Oxford) 2007;46(12):1773–1778. doi: 10.1093/rheumatology/kem222. [DOI] [PubMed] [Google Scholar]
- 166.Jayne DR, Bruchfeld AN, Harper L, Schaier M, Venning MC, Hamilton P, Burst V, Grundmann F, Jadoul M, Szombati I, et al. Randomized Trial of C5a Receptor Inhibitor Avacopan in ANCA-Associated Vasculitis. J Am Soc Nephrol. 2017 doi: 10.1681/ASN.2016111179. [DOI] [PMC free article] [PubMed] [Google Scholar]