

Abstract
Background and Purpose:
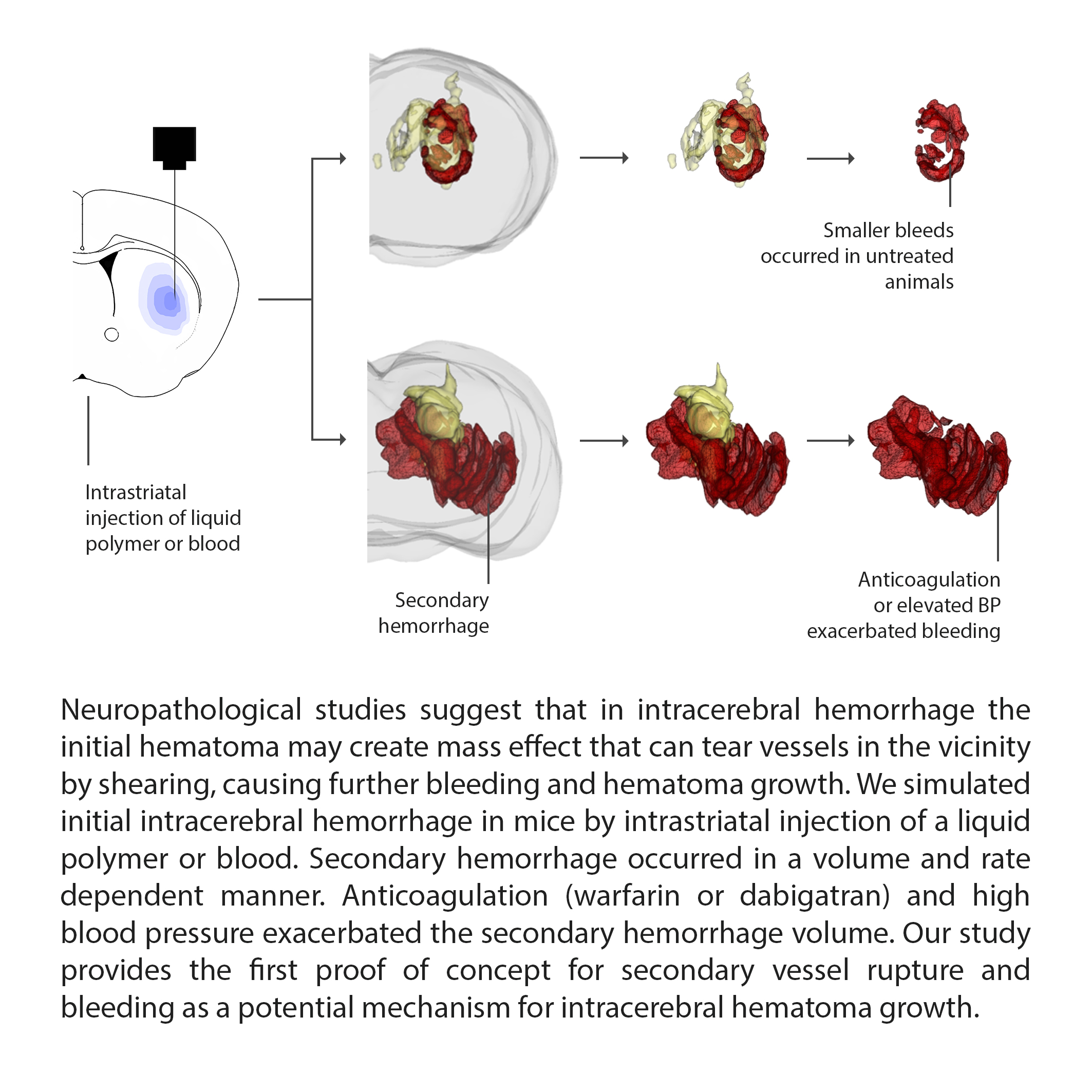
Mechanisms contributing to acute hematoma growth in intracerebral hemorrhage are not well understood. Neuropathological studies suggest that the initial hematoma may create mass effect that can tear vessels in the vicinity by shearing, causing further bleeding and hematoma growth.
Methods:
To test this in mice, we simulated initial intracerebral hemorrhage by intrastriatal injection of a liquid polymer that coagulates upon contact with tissue, and measured the presence and volume of bleeding secondary to the mass effect using Hemoglobin-ELISA 15 minutes after injection.
Results:
Secondary hemorrhage occurred in a volume (4, 7.5 or 15 μl of polymer) and rate (0.05, 0.5 or 5 μl/sec) dependent manner. Anticoagulation (warfarin or dabigatran) exacerbated the secondary hemorrhage volume. In a second model of hematoma expansion, we confirmed that intrastriatal whole blood injection (15 μl, 0.5 μl/sec) also caused secondary bleeding, using acute Evans blue extravasation as a surrogate. Anticoagulation once again exacerbated secondary hemorrhage after intrastriatal whole blood injection. Secondary hemorrhage directly and significantly correlated with arterial blood pressures in both non-anticoagulated and anticoagulated mice, when modulated by phenylephrine or labetalol.
Conclusions:
Our study provides the first proof of concept for secondary vessel rupture and bleeding as a potential mechanism for intracerebral hematoma growth.
Keywords: Intracerebral hemorrhage, hematoma expansion, pathophysiology, blood pressure, anticoagulation
Introduction
Intracerebral hemorrhage (ICH) is a deadly disease with short-term mortality of 40% and 6-month dependency rate of 80%.1, 2 A third of ICH patients develop significant hematoma expansion, and this number is doubled in anticoagulated patients.3 Hematoma expansion worsens outcomes, which provides a therapeutic opportunity to intervene. However, mechanisms contributing to hematoma expansion are poorly understood. Presumably, ICH starts with the rupture of a single vessel, giving rise to an initial hematoma. In 1971, Fisher introduced the idea that the mass effect of the initial hematoma may tear surrounding arteries or arterioles by shearing, leading to secondary bleeding and hematoma expansion, like an avalanche.4 He proposed that at least parts of the final hematoma volume are explained by this theory.
Additional human data support this theory. For example, the spot sign (contrast extravasation within an ICH) on CT angiograms, especially when multiple (30–50% of cases with spot signs), is suggestive of bleeding from secondary ruptures.5–8 Simultaneous extravasation from multiple vessels has been reported during cerebral angiography after ICH.9 In a case where ICH occurred while the patient was in the MRI-scanner, the developing hematoma changed its pace of growth in the different axes over time, implicating cascades of local vessel bursts.10 Consistent with this, the density and irregular shape of ICH has been predictive of growth in large cohorts as well.11 While such observations are suggestive, the theory has never been examined in an experimental setting. To test the “avalanche” hypothesis of secondary bleeding after ICH, we developed a new experimental model in mice and modified the established autologous blood injection model, and examined the influence of the volume and the speed of growth of the initial hematoma, anticoagulation, and blood pressure, on secondary hemorrhage. Our data provide proof-of-concept, and identify coagulation status and blood pressure as potential determinants of secondary arterial rupture and bleeding.
Material and Methods
Animals, study design, rigor and statistics
Our manuscript adheres to the AHA Journals’ implementation of the Transparency and Openness Promotion (TOP) Guidelines. The authors declare that all supporting data are available within the article and its online supplementary files. All experiments were performed under protocols approved by the Massachusetts General Hospital Institutional Animal Care and Use Committees following the NIH Guide for Use and Care of Laboratory Animals. For the entire study, male CD-1 mice aged 12 to 16 weeks were used. Mice were randomly assigned to study groups. Experiments were performed in a blinded fashion where feasible. Secondary ICH after liquid polymer injection was studied in 3 cohorts (n=80 total) to examine the effects of primary ICH volume, primary ICH growth rate, and anticoagulation. Secondary ICH after direct blood injection was studied in 5 cohorts (n=44 total) to examine the effects of blood pressure and anticoagulation. Data are presented as whisker-box plots (whisker, full range; box, interquartile range; horizontal line, median; +, mean), or mean ± standard deviation. Statistical tests (Graphpad Prism 7) for each dataset are indicated in figure legends.
Intrastriatal liquid polymer injection and measurement of extravascular tissue hemoglobin
To obtain the initial proof-of-concept, we sought to mimic primary intrastriatal hemorrhage by injecting a space occupying, non-blood liquid that gradually solidified after contact with tissue, and can be easily distinguished from secondary bleeding. As a substance with biomechanical properties similar to blood, we used Onyx18, which is a non-adhesive liquid embolic agent used in clinical practice for the embolization of brain arteriovenous malformations. It is comprised of 6% ethylene vinyl alcohol copolymer dissolved in dimethyl sulfoxide (DMSO) and tantalum powder. Upon injection, Onyx18 precipitates into a coherent solid (for more information see www.ev3.net) that makes it suitable to simulate ICH. Onyx18 has a viscosity comparable to blood, hardens from the periphery to the inside, and allows slow but sustained injection to yield ICH-like mass effect with shapes similar to those routinely observed in the clinical setting. Before usage liquid polymer was shaken for 20 minutes according to the manufacturer’s manual. Mice were anesthetized using isoflurane (1.5–2% in 30%O2/70%N2O) and allowed to breathe spontaneously. A small burr hole was drilled 2.5 mm lateral, 0.5 mm anterior to bregma. A 28G syringe was flushed with DMSO before usage, analogous to the preparation of the catheter system in clinical practice, and loaded with liquid polymer. The needle was slowly lowered to a depth of 3.5 mm into the striatum, and the polymer was injected at various volumes and rates using an infusion pump (Figure 1). After injection, the needle was removed and mice were allowed to wake up in their cages. There was no backflow as the polymer gradually solidified upon contact with tissue. Fifteen minutes after onyx18 injection, mice were transcardially perfused with 60 ml of phosphate-buffered saline (PBS) under deep (5%) isoflurane anesthesia to clear intravascular blood, which was confirmed grossly after brain removal by absence of blood in pial vessels. Brains were removed and cut into 1 mm coronal slices by means of a mouse brain matrix, and solidified polymer removed from brain tissue. Tissue between 3 mm anterior and 3 mm posterior to the needle track was divided into left and right hemisphere, homogenized in cell lysate buffer and centrifuged at 14,000 rpm for 20 minutes. To determine the protein concentration Bio-Rad Protein Assay was used. A Mouse Hemoglobin ELISA (abcam®) was performed for ipsilateral hemispheres thereafter.
Figure 1: Secondary hemorrhage after intrastriatal liquid polymer injection.

(A) Graphic representation of polymer injection into the right striatum (left panel) and gross coronal section cut surface showing the black solidified polymer (15 μl) surrounded by secondary bleeding in a non-anticoagulated and an anticoagulated animal. Sham (i.e. needle insertion only) did not cause detectable secondary hemorrhage. Sections were used for three-dimensional reconstruction (Figure 1B; Supplemental Movie 1).
(B) Three-dimensional reconstruction of serial coronal sections through the polymer in representative non-anticoagulated (left panel) and anticoagulated (right panel) animals (polymer in yellow, blood in red; see Supplemental Movie 1). Extension of polymer into the ventricle is evident, just as primary ICH often does.
(C) Increasing volumes of polymer injection (4, 7.5 or 15 μl) at a fixed rate of 0.5 μl/sec caused progressively larger volumes of secondary hemorrhage that appeared to be bimodal, as measured by hemoglobin ELISA (p=0.0001; Kruskal-Wallis non-parametric one-way ANOVA followed by Dunn’s multiple comparisons test; *p<0.05 vs. 4 μl; #p<0.05 vs. sham). Increasing speeds of polymer injection (0.05, 0.5, 5 μl/sec) caused progressively larger volumes of secondary hemorrhage that once again appeared to be bimodal, as measured by hemoglobin ELISA (p=0.0011; Kruskal-Wallis non-parametric one-way ANOVA followed by Dunn’s multiple comparisons test; *p<0.05 vs. other injection rates).
(D) Warfarin or dabigatran anticoagulation significantly increased secondary hemorrhage compared with non-anticoagulated animals. The bimodal distribution appeared to be accentuated by anticoagulation; approximately one third of the anticoagulated cohort had hemorrhage levels that were 15-fold higher than the rest (*p=0.0326; χ2 for hemoglobin >700 or <700 ng/mg).
Intrastriatal blood injection and measurement of Evans blue leakage
To directly simulate ICH in a separate cohort, we injected arterial blood into the striatum. Femoral artery was catheterized under isoflurane anesthesia (1.5–2% in 30%O2/70%N2O) in spontaneously breathing mice. Rectal temperature was monitored and maintained at 37°C via a heating pad (TC-1000 Temperature Controller, CWE, Ardmore, PA, USA). Blood for injection was drawn from the heart of deeply anesthetized (5% isoflurane) donor mice of the same strain and transferred to tubes containing 10 μL EDTA. In order to measure secondary hemorrhage, 200 μl of Evans blue (2% in normal saline) was injected through a femoral artery catheter 10 minutes before intrastriatal blood injection. Mice were then placed in a stereotaxic frame. A burr hole was drilled on the right side at 2.5 mm lateral and 0.5 mm anterior from bregma. Thereafter, a 28 G x1/2 syringe was filled with donor blood, the needle slowly lowered 3.5 mm deep from cortical surface into the striatum, and 15 μl was injected using an infusion pump at a rate of 0.5 μl/sec. The needle was left in place for two minutes and then slowly removed. Sham operated animals received no injection after needle insertion. Fifteen minutes after intracerebral blood injection or sham, mice were deeply anesthetized (5% isoflurane) and perfused transcardially with 60 ml PBS to wash out the remaining blood and Evans blue from the circulation. Three animals died shortly before the 15-minute time point for tissue harvesting (1 each in warfarin, phenylephrine and labetalol groups). These mice were processed as usual and included in the analyses, but since less time has passed after the primary bleed, the volume of secondary bleeds in these mice may have been slightly underestimated. Brains were removed and cut into 1 mm slices using a mouse brain matrix. 6 slices around the needle track were identified and divided into left and right hemispheres. Samples were stored at −80 °C until homogenization in RIPA buffer at room temperature and centrifugation at 3,000 rcf for 10 min. The supernatant was read (ex/em: 585 nm/680 nm) using a fluorescence plate reader (SpectraMax M5, Molecular Devices, CA, USA). Calculations were based on standards obtained by supernatant of naïve brain tissue spiked with known amounts of Evans Blue (2×105 ng/ml). The amount of Evans blue was quantified according to a linear standard curve and is expressed as ng Evans blue per mg brain tissue.
Blood pressure monitoring and manipulation
Blood pressure (BP) was monitored throughout the intrastriatal blood injection protocol via the femoral artery catheter (MacLab, ADInstruments, Colorado Springs, MO, USA). To elevate the BP, selective α1-adrenergic agonist phenylephrine (10 mg/kg in 120 μl saline) was injected intraperitoneally after the intraarterial Evans blue injection. The striatal blood injection was performed when BP peaked, typically within 5 minutes. To lower the BP, α1 and β1/β2 adrenergic blocker labetalol (7.5 mg/kg in 180 μl saline) was administered through the femoral artery catheter over 5 seconds after the onset of striatal blood injection.
Anticoagulation
Warfarin (2.5 mg/kg) was administered via drinking water for 30 hours following a previously established protocol.12 Briefly, a 5 mg Coumadin tablet (crystalline warfarin sodium, Bristol Myers Squibb, New York City, NY, USA) was dissolved in 375 ml of tap water. No other water was supplied. The international normalized ratio (INR) values were 5.6±2.2, consistent with previous reports.12–14 Control mice received regular tap water. Dabigatran (110 mg, Pradaxa, Boehringer Ingelheim, Ingelheim Germany) was dissolved in tap water at a concentration of 20 mg/ml,15 and administered via orogastric gavage at a dose of 75 mg/kg in 3.75 ml/kg volume every 8 hours for 3 doses.13, 15, 16
Results
Liquid polymer injection created a well-circumscribed solid mass that was limited to the striatum. Multiple foci of secondary hemorrhage were visible mainly around the perimeter of the polymer, as evidence of secondary vessel rupture (Figure 1A). This was best demonstrated by three-dimensional reconstructions of serial cryosections (Figure 1B, Supplemental Movie 1). Sham operated mice occasionally showed minimal secondary hemorrhage within the needle track. When quantified by hemoglobin ELISA, the secondary bleeding volume depended on the injected polymer volume (p=0.0001; Figure 1C). In contrast to secondary bleeding caused by 7.5 and 15 μl polymer injection, bleeding after 4 μl polymer did not differ from sham group suggesting a threshold effect. Secondary hemorrhage volume was also related to the speed of polymer injection, lending support to the avalanche theory (p=0.0011; Figure 1C). Anticoagulation with warfarin or dabigatran dramatically increased the secondary hemorrhage in a third of the cohort (p=0.0123; Figure 1D, Supplemental Movie 1B). Secondary hemorrhage volumes did not differ between warfarin and dabigatran, although the sample size was not powered to test this. As an important control, 50% or 100% DMSO injections yielded much lower tissue hemoglobin content compared to liquid polymer injections, and comparable to needle insertion alone, suggesting that DMSO in the polymer does not affect the natural course of secondary bleeding (Supplemental Figure I).
Intrastriatal blood injection resulted in grossly and histologically visible Evans blue leakage predominantly around the primary injection site (Figure 2A), with a spatial distribution similar to blood after liquid polymer injection as described above. Since Evans blue was quantified only 15 minutes after the primary ICH, extravasation could only be explained by secondary bleeding, rather than blood-brain barrier opening (e.g. transcytosis), which doesn’t occur until several hours after the onset of ICH.17 We next examined the impact of mean arterial BP and anticoagulation on secondary hemorrhage in this model. Intrastriatal blood injection caused an acute increase in BP by approximately 40 mmHg, which persisted until sacrifice 15 minutes later (Figure 2B). Treatment with phenylephrine elevated the resting BP by about 60 mmHg, which further increased by another 25 mmHg after intrastriatal blood injection. Treatment with labetalol diminished the BP surge caused by intrastriatal blood injection, and brought the post-injection BP back to pre-injection levels. Anticoagulation per se did not directly influence the BP. Altogether, these interventions created three tiers of BP after ICH, alone or in combination with anticoagulation (Figure 2B).
Figure 2: Secondary hemorrhage after intrastriatal blood injection.

(A) Coronal cut through the primary hematoma (15 μl) and secondary hemorrhage marked by grossly visible Evans blue around the periphery after intrastriatal blood injection (left panel), and cryosections from the same brain showing DAB staining (middle panel) and Evans blue fluorescence (right panel) (image size 920 × 1800 μm). Similar images were obtained using 70kDa FITC-dextran as a surrogate for secondary bleeding (not shown).
(B) The time course of mean arterial BP is shown at 10 minutes before blood injection (−10 min), just before Phenylephrine (Phe) administration (−5 to −8 min), at baseline immediately before blood injection (−0.5 min), peak BP after blood injection (2 to 3 min), and 5, 10 and 15 min after blood injection. Labetalol (Labet) was administered right after the completion of intrastriatal blood injection, with or without warfarin (Warf) anticoagulation. These interventions created three tiers of BP (upper tier: ICH+Phe and ICH+Warf+Phe; middle tier: ICH and ICH+Warf; lower tier: ICH+Warf+Labet). The average time of Phe administration in the relevant groups, and the time when peak BP is reached after intrastriatal blood injection in all groups are shown with their standard deviations as horizontal error bars.
(C) Hemispheric Evans blue fluorescence as a surrogate for secondary bleeding in six experimental groups (p<0.0001 ipsilateral vs. contralateral hemisphere and among treatments; two-way ANOVA for repeated measures followed by Newman-Keuls multiple comparisons test; *p<0.05 vs. contralateral; †p<0.05 vs. ipsilateral sham; #p<0.05 vs. ipsilateral ICH, ICH+Phe and ICH+Warf+Labet.
(D) Secondary hemorrhage was directly correlated with BP in both non-anticoagulated (circles) and anticoagulated (triangles) cohorts, shown and analyzed separately along with p values, best fit and 95% confidence intervals (linear regression). The slopes of two fits did not significantly differ (p=0.7169).
All ICH groups had larger secondary hemorrhage than sham controls within the ipsilateral hemisphere (p<0.0001; Figure 2C). Tissue Evans blue concentrations were significantly higher within the ipsilateral compared with contralateral hemisphere in all ICH groups, but not sham controls (p<0.0001; Figure 2C). As with the liquid polymer method, anticoagulation markedly augmented secondary hemorrhage. Elevating the BP with phenylephrine did not further increase secondary hemorrhage, suggesting a ceiling effect. However, lowering the BP by labetalol significantly reduced secondary hemorrhage in anticoagulated animals. Secondary hemorrhage significantly correlated with BP in both non-anticoagulated and anticoagulated cohorts (Figure 2D).
Discussion
Here, we provide the first experimental evidence supporting Fisher’s avalanche theory of secondary hematoma expansion after intracerebral hemorrhage 4 through mechanical shear or tear of other vessels in the vicinity. As pathological evidence of secondary vessel rupture, multiple blood spots were seen around the initial hematoma. Using two independent but complementary experimental models, we show that the volume of secondary bleeding strongly depended on the volume and speed of growth of the initial hematoma. Anticoagulation with warfarin or dabigatran markedly enhanced secondary bleeding. Lastly, the volume of secondary bleeding correlated with BP and could be ameliorated using antihypertensive treatment in the setting of anticoagulation.
Review of the scatterplots of secondary hemorrhage volume suggests they occur in a bimodal distribution, with reasonably distinct groups of small vs. large secondary hemorrhages particularly in the presence of anticoagulation (Figures 1C–D, 2C). The bimodal distribution of secondary hemorrhage volume suggests that it does not necessarily occur in every ICH, perhaps critically dependent on the presence of a vulnerable artery or arteriole in the vicinity of the growing mass. Consistent with this, in cases of small human ICH Fisher has observed only one ruptured vessel on serial sections.18 Our experimental results also parallel the bimodal distribution of hemorrhage volumes measured in 46 lobar hemorrhage patients, which demonstrated reasonably distinct groups of “microbleeds” and “macrobleeds.”19 Anticoagulation presumably enhances the hematoma expansion by increasing the likelihood of multiple secondary microbleeds that grow or coalesce into macrobleeds, which may cause additional vessels to rupture creating an avalanche or domino effect. Additional determinants of secondary hemorrhage are likely to be the location and physical properties of initial hematoma formation, coagulation status and the presence, size and structural vulnerability of nearby arteries and arterioles for mechanical tear. For example, increased hemorrhage volumes after intracerebral blood injection in diabetic rats as compared to non-diabetic controls may be explained by impaired coagulation or higher vulnerability of diabetic arteries to shear forces.20 Besides the mechanical disruption of blood vessels by expanding hematoma (i.e. the avalanche theory), other factors might also contribute to secondary bleeding and hematoma expansion, such as plasma kallikrein in the hyperacute stage 20, 21, and upregulation and activation and of proteolytic enzymes such as MMP-2 and MMP-9 in later stages.
In an attempt to estimate the actual volume of secondary hemorrhages we performed calculations using the following parameters: (a) ~72 ml/kg blood volume, 35 g body weight, and 150 mg hemoglobin/ml circulating blood in normal CD1 mice 22, and (b) 1.6 mg/ml circulating blood Evans blue concentrations after systemic administration based on the dosage. For side by side comparisons, we used data from 15 μl liquid polymer or homologous blood injection experiments at 0.5 μl/sec injection rate in non-anticoagulated mice. In liquid polymer experiments, the measured total extravasated tissue hemoglobin content corresponded to approximately 0.2±0.1 μl of secondary hemorrhage (as seen in Fig 1B, non-anticoagulated). In homologous blood injection experiments, the measured total extravasated tissue Evans blue content corresponded to 1.7±1.0 μl of secondary hemorrhage (~10% of primary hematoma). In comparison, anticoagulated mice developed much larger secondary hemorrhages. For example, liquid polymer injection in warfarin-treated animals (Fig 1D, blue symbols) yielded 0.9±1.3 μl of secondary hemorrhage, while homologous blood injection in warfarin-treated animals (Fig 2C, red bars) yielded 2.8±1.6 μl of secondary hemorrhage (~20% of primary hematoma). In none of the animals the calculated secondary bleeding volume exceeded the primary injection volume of 15 μl. Of course, these calculated numbers are only approximations, as there are many potential levels of inaccuracies in measurements and estimations. However, they at least provide a rough scale of the secondary bleeding in this model. These calculations showed that secondary bleeds were in fact smaller than the primary hematoma volumes in all cases. Therefore, some apparently very large volumes of secondary bleeds on tissue sections in anticoagulated mice were likely due to diffusion of blood into the tissue to some extent, making the hematomas look larger than the actual amount of blood. The calculations also showed that homologous blood injection yielded larger secondary bleeding than liquid polymer, perhaps because of the solid mass nature of the latter causing pressure hemostasis. Importantly, we do not intend to suggest that the estimations of secondary bleeding in this experimental model accurately reflect the clinical setting. But they provide the proof of concept to support “the avalanche theory”.
Our data for the first time suggest that dabigatran augments hemorrhage to a similar extent as warfarin in a model of ICH, which contradicts prior reports in another ICH model and in traumatic brain injury.15, 16 While data from human studies also suggest that dabigatran is favorable in comparison to warfarin in terms of a reduced rate of occurrence of ICH, number of fatal outcomes was reported to be similar between groups.23, 24 The results of our study emphasize that hematoma expansion might be aggravated in a subset of patients.
A recent meta-analysis of large clinical studies suggested that intensive BP-lowering is associated with a lower risk of significant ICH expansion,25 although this has not convincingly improved outcomes in randomized clinical trials.26–28 In our experiments higher BP was associated with larger secondary bleeding and the secondary bleeding was markedly reduced using labetalol in anticoagulated animals. Since hematoma expansion usually occurs early after symptom onset, treatment would be expected to be more effective in the earliest phases.10 Yet, clinical trials typically enroll patients in a broader time window, and it is possible that the absence of benefit from antihypertensive treatment in some trials may be due to late treatment.25, 29 Moreover, the efficacy of antihypertensive treatment is likely underestimated because clinical trials often excluded patients at the highest risk for hematoma expansion, such as those with large initial hematoma volumes or who are anticoagulated,25 who might benefit most from aggressive BP lowering.
Elevated BP is common after ICH,30 but it is unclear whether it is the cause or the consequence of hematoma expansion.31 In the blood injection model, we observed a marked increase in BP as a direct response to hematoma formation in otherwise normotensive animals, presumably caused by elevated intracranial pressure (Cushing-Kocher response). In clinical practice the onset of this immediate rise in MAP is difficult to detect since patients arrive later at the hospital. Our results add to the evidence that BP is a determinant of hematoma expansion. Intracerebral hemorrhage frequently causes an immediate rise in intracranial pressure, which again may lead to increased BP and subsequently larger hematomas. In our model we observed larger secondary bleeding volumes with faster hematoma growth. This may be explained partly by more pronounced pressure and shearing forces but also by a more sudden rise in intracranial pressure.
Our study was designed to obtain proof of concept, and as such, it has limitations. First, we focused on the hyperacute stage (i.e. first 15 minutes), and later time points of assessment might yield larger secondary hemorrhages. Future work will examine the same animal models longitudinally using neuroimaging to better define the natural history of secondary hemorrhage. Second, we used a non-blood liquid polymer to mimic acute hematoma expansion in the first part of our study, and used Evans blue as a surrogate for hemorrhage due to vessel rupture in the second part. While the liquid polymer has a similar viscosity as blood it is impossible to completely mimic the rheological properties of blood which consists of cells and plasma, and as such physical forces leading to mechanical vascular damage by the polymer might be different. Although Evans blue can leak through the blood-brain barrier, this does not take place until hours after ICH. Therefore, Evans blue detected around the hematoma 15 minutes after the primary ICH likely reflected secondary disruption of vessel integrity. Future work could use fluorescent-labeled circulating red blood cells to directly visualize and quantify secondary hemorrhage. Third, we used EDTA for the blood injection model to prevent clotting before injection. We cannot exclude that small amounts of EDTA entered into the brain tissue and artificially exaggerated bleeding from the adjacent vessels. Fourth, we exclusively studied young non-diseased mice, whereas ICH typically occurs in old patients, whose vessels are often remodeled due to chronic hypertension, or have small vessel disease such as amyloid angiopathy. Such vessels might be more brittle and prone to break than the elastic vessel walls of younger brains, and therefore, may further accentuate secondary bleeding. Future studies will need to better define the conditions predisposing to or protecting against secondary tears contributing to acute hematoma expansion.
In summary, we provide initial proof of concept suggesting that the injection of blood or a substance with properties similar to blood causes secondary vessel rupture and bleeding depending on the rate and the amount of injection. Vulnerability to secondary bleeding is enhanced in the presence of anticoagulation and high BP. Using this novel model, future work will investigate the biological mechanisms of hematoma expansion in detail (e.g. effects of aging, sex and comorbidities) and test therapeutic strategies. A better understanding of the pathophysiology of secondary hemorrhage might translate into new treatments limiting hematoma growth in the acute phase by preventing vessel rupture.
Supplementary Material
{kind=link}
Disclosures:
MB: Supported by a fellowship from the Boehringer Ingelheim Fonds; CA: NIH (NS055104, NS061505), the Fondation Leducq, the Heitman Foundation, and the Ellison Foundation; SMG: NIH (NS096730); BJB: NIH (NS096730).
References
- 1.Jakubovic R, Aviv RI. Intracerebral hemorrhage: Toward physiological imaging of hemorrhage risk in acute and chronic bleeding. Frontiers in neurology. 2012;3:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flaherty ML, Haverbusch M, Sekar P, Kissela B, Kleindorfer D, Moomaw CJ, et al. Long-term mortality after intracerebral hemorrhage. Neurology. 2006;66:1182–1186 [DOI] [PubMed] [Google Scholar]
- 3.Parry-Jones AR, Di Napoli M, Goldstein JN, Schreuder FH, Tetri S, Tatlisumak T, et al. Reversal strategies for vitamin k antagonists in acute intracerebral hemorrhage. Annals of neurology. 2015; 78(1): 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fisher CM. Pathological observations in hypertensive cerebral hemorrhage. Journal of neuropathology and experimental neurology. 1971;30:536–550 [DOI] [PubMed] [Google Scholar]
- 5.Boulouis G, Dumas A, Betensky RA, Brouwers HB, Fotiadis P, Vashkevich A, et al. Anatomic pattern of intracerebral hemorrhage expansion: Relation to ct angiography spot sign and hematoma center. Stroke; a journal of cerebral circulation. 2014;45:1154–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brouwers HB, Falcone GJ, McNamara KA, Ayres AM, Oleinik A, Schwab K, et al. Cta spot sign predicts hematoma expansion in patients with delayed presentation after intracerebral hemorrhage. Neurocritical care. 2012;17:421–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Romero JM, Heit JJ, Delgado Almandoz JE, Goldstein JN, Lu J, Halpern E, et al. Spot sign score predicts rapid bleeding in spontaneous intracerebral hemorrhage. Emergency radiology. 2012;19:195–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morotti A, Brouwers HB, Romero JM, Jessel MJ, Vashkevich A, Schwab K, et al. Intensive blood pressure reduction and spot sign in intracerebral hemorrhage: A secondary analysis of a randomized clinical trial. JAMA Neurol. 2017;74(8):950–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Komiyama M, Yasui T, Tamura K, Nagata Y, Fu Y, Yagura H. Simultaneous bleeding from multiple lenticulostriate arteries in hypertensive intracerebral haemorrhage. Neuroradiology. 1995;37:129–130 [DOI] [PubMed] [Google Scholar]
- 10.Edlow BL, Bove RM, Viswanathan A, Greenberg SM, Silverman SB. The pattern and pace of hyperacute hemorrhage expansion. Neurocritical care. 2012;17:250–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barras CD, Tress BM, Christensen S, MacGregor L, Collins M, Desmond PM, et al. Density and shape as ct predictors of intracerebral hemorrhage growth. Stroke; a journal of cerebral circulation. 2009;40:1325–1331 [DOI] [PubMed] [Google Scholar]
- 12.Foerch C, Arai K, Jin G, Park KP, Pallast S, van Leyen K, et al. Experimental model of warfarin-associated intracerebral hemorrhage. Stroke. 2008;39:3397–3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Won SY, Schlunk F, Dinkel J, Karatas H, Leung W, Hayakawa K, et al. Imaging of contrast medium extravasation in anticoagulation-associated intracerebral hemorrhage with dual-energy computed tomography. Stroke; a journal of cerebral circulation. 2013;44:2883–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schlunk F, Van Cott EM, Hayakawa K, Pfeilschifter W, Lo EH, Foerch C. Recombinant activated coagulation factor vii and prothrombin complex concentrates are equally effective in reducing hematoma volume in experimental warfarin-associated intracerebral hemorrhage. Stroke; a journal of cerebral circulation. 2012;43:246–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lauer A, Cianchetti FA, Van Cott EM, Schlunk F, Schulz E, Pfeilschifter W, et al. Anticoagulation with the oral direct thrombin inhibitor dabigatran does not enlarge hematoma volume in experimental intracerebral hemorrhage. Circulation. 2011;124:1654–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schaefer JH, Leung W, Wu L, Van Cott EM, Lok J, Whalen M, et al. Translational insights into traumatic brain injury occurring during dabigatran or warfarin anticoagulation. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2014;34:870–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006;5:53–63 [DOI] [PubMed] [Google Scholar]
- 18.Fisher CM. Hypertensive cerebral hemorrhage. Demonstration of the source of bleeding. J Neuropathol Exp Neurol. 2003;62:104–107 [DOI] [PubMed] [Google Scholar]
- 19.Greenberg SM, Nandigam RN, Delgado P, Betensky RA, Rosand J, Viswanathan A, et al. Microbleeds versus macrobleeds: Evidence for distinct entities. Stroke. 2009;40:2382–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu J, Gao BB, Clermont AC, Blair P, Chilcote TJ, Sinha S, et al. Hyperglycemia-induced cerebral hematoma expansion is mediated by plasma kallikrein. Nature medicine. 2011;17:206–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmaier AH, McCrae KR. The plasma kallikrein-kinin system: Its evolution from contact activation. J Thromb Haemost. 2007;5:2323–2329 [DOI] [PubMed] [Google Scholar]
- 22.Diehl KH, Hull R, Morton D, Pfister R, Rabemampianina Y, Smith D, et al. A good practice guide to the administration of substances and removal of blood, including routes and volumes. J Appl Toxicol. 2001;21:15–23 [DOI] [PubMed] [Google Scholar]
- 23.Grysiewicz R, Gorelick PB. Incidence, mortality, and risk factors for oral anticoagulant-associated intracranial hemorrhage in patients with atrial fibrillation. Journal of stroke and cerebrovascular diseases : the official journal of National Stroke Association. 2014;23:2479–2488 [DOI] [PubMed] [Google Scholar]
- 24.Hart RG, Diener HC, Yang S, Connolly SJ, Wallentin L, Reilly PA, et al. Intracranial hemorrhage in atrial fibrillation patients during anticoagulation with warfarin or dabigatran: The re-ly trial. Stroke; a journal of cerebral circulation. 2012;43:1511–1517 [DOI] [PubMed] [Google Scholar]
- 25.Boulouis G, Morotti A, Goldstein JN, Charidimou A. Intensive blood pressure lowering in patients with acute intracerebral haemorrhage: Clinical outcomes and haemorrhage expansion. Systematic review and meta-analysis of randomised trials. J Neurol Neurosurg Psychiatry. 2017;88:339–345 [DOI] [PubMed] [Google Scholar]
- 26.Anderson CS, Heeley E, Huang Y, Wang J, Stapf C, Delcourt C, et al. Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage. The New England journal of medicine. 2013;368:2355–2365 [DOI] [PubMed] [Google Scholar]
- 27.Goldstein J, Brouwers H, Romero J, McNamara K, Schwab K, Greenberg S, et al. Score-it: The spot sign score in restricting ich growth horizontal line an atach-ii ancillary study. Journal of vascular and interventional neurology. 2012;5:20–25 [PMC free article] [PubMed] [Google Scholar]
- 28.Mayer SA. Recombinant activated factor vii for acute intracerebral hemorrhage. Stroke; a journal of cerebral circulation. 2007;38:763–767 [DOI] [PubMed] [Google Scholar]
- 29.Anderson CS, Chalmers J, Stapf C. Blood-pressure lowering in acute intracerebral hemorrhage. The New England journal of medicine. 2013;369:1274–1275 [DOI] [PubMed] [Google Scholar]
- 30.Schlunk F, Chang Y, Ayres A, Battey T, Vashkevich A, Raffeld M, et al. Blood pressure burden and outcome in warfarin-related intracerebral hemorrhage. International journal of stroke : official journal of the International Stroke Society. 2016;11:898–909 [DOI] [PubMed] [Google Scholar]
- 31.Shah QA, Ezzeddine MA, Qureshi AI. Acute hypertension in intracerebral hemorrhage: Pathophysiology and treatment. Journal of the neurological sciences. 2007;261:74–79 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.