

Abstract
Background
Uveitis is a group of intraocular inflammatory diseases whose primary treatment involves immunosuppression. Although corticosteroids (CSs) remain the mainstay therapy, sirolimus is among the recently studied immunomodulatory drugs for treating noninfectious uveitis (NIU).
Objective
The aim of this review was to assess and summarize the updated evidence on the outcomes of treatment with sirolimus for NIU.
Materials and methods
Two reviewers conducted a systematic search on November 5, 2018, of electronic databases (EMBASE, MEDLINE, and The Cochrane Library) and clinical trial registers having no restrictions on language or publication date. The primary outcome was uveitis activity as measured by vitreous haze (VH), while the secondary outcomes included central macular thickness (CMT), best-corrected visual acuity (BCVA), CS-sparing effect, IOP elevation, and other adverse events. A meta-analysis was conducted on selected studies with appropriate clinical and methodological homogeneity.
Results
Seven studies were included and reviewed. Four randomized clinical trials were eligible for meta-analysis: SAVE 2013, One-year outcomes of the SAVE study, SAVE 2 2016, SAKURA 2016. The pooled proportions of inflammation control (VH improvement) were 38% (95% CI 16.19%–62.66%) during a 6-month follow-up and 49.97% (95% CI 16.19%–83.03%) during a 6- to 12-month follow-up with the latter showing a significantly higher response rate (p=0.0472). BCVA improvement was 62.2% (95% CI 33.17%–87.11%) during a 6-month follow-up and 56.86% (95% CI 20.91%–89.05%) during a 6- to 12-month follow-up with no significant difference between the two (p=0.3705). Increased IOP remained at 7.11% (95% CI 3.46%–12.68%) for both a 6-month follow-up and up to a 12-month follow-up duration. The CS-sparing effect of sirolimus was also well demonstrated. A reduction in CMT was observed, and only minor drug-related adverse events were reported in all the studies reviewed.
Conclusion
This review provided evidence that sirolimus is a promising treatment option for controlling inflammatory activity, improving visual acuity, and sparing CS use with minor adverse events for NIU.
Keywords: non-infectious uveitis, immunosuppression, corticosteroid-sparing, sirolimus, mTOR inhibitors, uveitis treatment, vitreous haze
Video abstract
Introduction
Uveitis is a group of intraocular inflammatory diseases1 with significant clinical, socioeconomic, and quality-of-life impact.2–4 As the third leading cause of preventable blindness in the world (fifth in the US), uveitis should engender greater efforts toward the development of sight-saving treatments.5,6 Potentially blinding complications include cataract, glaucoma or increased IOP, and macular edema (ME).7 Uveitis is responsible for about 5%–20% of visual loss in the USA and Europe and as high as 25% in developing countries.8 Up to 90% of cases affected belong to the working class (20–60 years of age), with nearly half occurring at the third to fourth decade of life.8
Corticosteroids (CSs) have always been the mainstay of treatment for most forms of noninfectious uveitis (NIU). In fact, the Standardization of Uveitis Nomenclature (SUN) Working Group guidelines recommend its use as the first-line therapy for active disease.9,10 Topical CS preparations are usually given for cases of anterior uveitis, while periocular, intravitreal (IVT), or systemic preparations are available for intermediate uveitis, posterior uveitis, and panuveitis. However, certain limitations with CS therapy arise in cases of refractory uveitis and in patients who develop CS-related complications such as hypertension, diabetes, cataract formation, IOP elevation, or glaucoma due to prolonged use.11–15 Thus, uveitis treatment guidelines recommend that chronic use of systemic CSs should be no more than 10 mg/day prednisone-equivalent dose to reduce the incidence of potentially serious adverse events,16 but even adverse outcomes can still occur with a dose as low as 7.5 mg/day.17 In such situations, other forms of immunosuppressive therapy may be warranted.10
Immunomodulatory therapy (IMT) is an alternative for controlling the inflammatory process and serves as adjunctive therapy that helps in reducing CS use. Although CSs are usually required to control acute inflammation, IMT agents are available to downregulate chronic inflammation and prevent recurrences.18 A number of drug classes belong to this group of agents, and choosing which among these drugs to use depends not only on drug efficacy but also on tolerability.19 Such an agent that demonstrates both efficacious steroid-sparing IMT and favorable side effect profile is therefore needed.20
Sirolimus is a recently studied and developed IMT agent for treating noninfectious intermediate uveitis, posterior uveitis, and panuveitis.21 Sirolimus, also known as rapamycin, is a bacteria-derived IMT agent that suppresses T cell proliferation by inhibiting the expression of IL-2, IL-4, and IL-15. This is mediated by the binding of the immunophilin FKBP-12 and, therefore, preventing it from binding and activating the mTOR.22,23 This may have a beneficial effect in the context of uveitis, as the immune dysfunction in NIU is thought to be primarily T cell mediated.24,25
The US Food and Drug Administration has approved the systemic use of sirolimus in preventing kidney transplant rejection as well as a sirolimuseluting coronary stent for enhancing coronary luminal diameter in patients with ischemic heart disease. Currently, the therapeutic use of sirolimus in ophthalmology is still under investigation.22,23
The systemic use of sirolimus is associated with cytotoxic, especially hematological, adverse effects (AEs) that can limit its use in treating patients with uveitis.21 However, local preparations of the drug that can be administered by subconjunctival (SCJ) or IVT injections have been developed and proven suitable based on preclinical studies.26 Clinical trials are now focused on determining the optimal effective dose of sirolimus in such preparations.27–29
The aim of this article was to systematically review the current evidence concerning the therapeutic use of sirolimus for patients with NIU and to perform a meta-analysis on the available data.
Materials and methods
This systematic review and meta-analysis were conducted by using the methods described by the Cochrane Collaboration for systematic reviews of interventions and in concordance with the PRISMA guidelines for systematic reviews and meta-analyses.30,31 The details of the systematic review protocol were submitted for registration on the International Prospective Register of Systematic Reviews (PROSPERO) database (registration number: CRD42018115715).
The aim of this meta-analysis was to assess the effectiveness and treatment outcomes of the mTOR inhibitor sirolimus (rapamycin) for the treatment of NIU. This was achieved by conducting a statistical study of clinical trials, which evaluated or described the use of sirolimus in cases of NIU.
Eligibility criteria
The following criteria were used in the selection of studies for review:
NIU-related studies with inflammatory activity grading defined based on the SUN working group criteria
Patients with refractory uveitis receiving a chronic course of immunosuppressive therapy and systemic steroid treatment
Sirolimus as the intervention competed with or without any comparator (eg, another pharmacological agent or placebo)
Studies with a median follow-up duration of at least 3 months.
Outcome measurements
The primary outcome assessed is the clinical activity of uveitis by vitreous haze (VH) score after giving sirolimus in different doses and intervals. The following secondary outcomes were recorded and assessed: best-corrected visual acuity (BCVA) in ETDRS letters, clinical measures of uveitis activity (anterior chamber cells, presence of active inflammatory vascular and chorioretinal lesions, and central macular thickness [CMT]), concurrent requirement of CSs and other immunomodulatory treatment to control uveitis, time to treatment failure defined as the time up to a worsening (two-grade increase) in the SUN cell activity score, and occurrence of adverse events.
Searches
The literature search for review was conducted using the electronic databases of published studies namely: Cochrane Library (which comprises a portal to EMBASE and PubMed), PMC (which includes MEDLINE), and registers of clinical trials of ClinicalTrials.gov with the terms “sirolimus” or “rapamycin” matched with “uveitis”. No restrictions were made on uveitis such as using the words “anterior”, “posterior”, “infectious”, “non-infectious” to expand the number of hits in the literature to be reviewed. No restrictions were made on language, year of publication, and study types included to deliver more evidence related to the effectiveness of sirolimus in cases of uveitis. The search strategy for CENTRAL, PMC, and ClinicalTrials.gov is summarized in Tables S2–S4.
Selection of studies
Two reviewers (Cabahug and Sapno) independently screened the titles and abstracts of the searched studies and determined their relevance to this meta-analysis. Full-text articles were retrieved and assessed for eligibility. Evaluations of methodological quality and risk of bias were made using the Cochrane risk of bias tool and a modified Jadad scale.32–35 The Jadad scale assesses three primary quality factors: randomization, blinding, and reported withdrawals. The modified augmented version will also assess other methodological factors such as exclusion criteria, the intervention used, the control used, and data reporting providing a total quality score of 10 (Table 1).34,35
Table 1.
The modified Jadad questionnaire for assessing the risk of bias
| Modified Jadad Questionnaire |
|---|
| 1) “Was the study described as randomized?” |
| 2) “Was the randomization protocol detailed and appropriate?” |
| 3) “Was the study described as double-blind?” |
| 4) “Was the blinding process detailed and appropriate?” |
| 5) “Did the study have a control group?” |
| 6) “Was the control detailed and appropriate?” |
| 7) “Was there an adequate exclusion criterion?” |
| 8) “Was the intervention used at a therapeutic dose?” |
| 9) “Was there a description of withdrawals and dropouts?” |
| 10) “Were the data clearly and adequately reported?” |
| Yes. 1 point; No. 0 point; (total score of 10) |
Key information gathered from the selected articles was listed in a standard form containing relevant details such as study design type, number of patients, type of intervention, dosage, follow-up duration, and outcome measurements. A PRISMA flow diagram was used to illustrate the details of the selection process including reasons for the exclusion of articles.31
Data synthesis and analysis
All collected data from the selected studies were summarized and tabulated for possible statistical treatment. Meta-analysis was undertaken, where appropriate clinical and methodological homogeneity exists.
Meta-analysis on single proportions was utilized in order to identify the pooled response rate of reduction in VH, BCVA improvement, IOP elevation, CMT, CS-sparing effect, and safety outcomes whenever possible. MedCalc uses a Freeman–Tukey transformation (arcsine square root transformation; Freeman and Tukey, 1950)36 to calculate the weighted summary proportion under the fixed- and random-effects model (DerSimonian and Laird, 1986)37.
Results
Selection of studies
A systematic search of multiple electronic databases yielded a total of 277 possible relevant articles; of these, 256 were excluded after screening for the titles and abstracts. After a full-text evaluation of the remaining 21 articles, 15 relevant studies specific to our research questions were included. Eight potentially eligible articles were then removed for failing to meet the inclusion criteria and for having data that could not be synthesized together. Ultimately, seven studies were included for our systematic review, but only four randomized controlled trials (RCTs) out of seven of these studies were eligible for the meta-analysis. The details of the systematic selection process are shown in Figure 1.
Figure 1.

PRISMA flowchart.
Characteristics of included studies
A total of seven studies were included for our systematic review, but only four studies were included in our risk of bias assessment and meta-analysis. The SAVE 201328 trial and its follow-up study, Ibrahim et al,21 were Phase I, open-label, randomized clinical trials conducted in one clinical center in USA. SAVE 2 201629 was a prospective randomized, Phase II, open-label interventional clinical trial conducted at four clinical centers also in USA. Lastly, SAKURA 2016,27 the largest clinical trial of sirolimus to date, was a Phase III, randomized, double-masked multinational study conducted in the European Union, India, Israel, Japan, Latin America, and USA. Although all the abovementioned studies were provided by a study drug from Santen, Inc. (Santen Pharmaceutical Co., Ltd., Osaka, Japan), only SAVE 2 201629 and SAKURA 201627 were industry sponsored. Table 2 summarizes all the characteristics of the four studies including the risk of bias assessment.
Table 2.
Main characteristics of the included studies
| SAVE 201328 | ||||||
| Methods | Open-label RCT Patients grouped into three disease categories with 1:1 randomization into one of two treatment arms. The eye with more advanced disease was selected as the study eye in patients with bilateral uveitis. If both eyes were affected equally, the study eye was selected before randomization at the investigator’s discretion. If the fellow eye was contraindicated, proven to be ineffective or rejected by the patient, the injection of sirolimus was administered to the fellow eye at the discretion of the investigator at the same dose and route of administration of the study eye, but at least 14 days apart from the injection of the study eye (Page 13) |
|||||
| Participants | Country: USA Number of people randomized: 30 Average age: 47 years (range 29–65 years) Sex: 50% male, 50% female Inclusion criteria: • ≥18 years of age • BCVA of 20/400 or better in both eyes • Noninfectious, active uveitis defined as having at least 1+ VH and/or at least 1+ vitreous cell count based on the SUN scale • Receiving no other treatments or with sufficient inflammation requiring systemic treatment such as prednisone ≥10 mg/day and/or at least one other systemic immunosuppressants • Inactive disease using the SUN scale, defined as having 0.5+ VH or less and a grade of 0.5+ vitreous cell count or less and are receiving prednisone <10 mg/day and/or at least one other systemic immunosuppressants • Having posterior uveitis, intermediate uveitis, or panuveitis (posterior component must be greater than the anterior component Exclusion criteria: • Patients with bilateral uveitis who are receiving systemic IMTs for the treatment of the fellow eye and cannot be controlled with standard local therapies alone • Presence of significant ocular disease that could compromise vision in the study eye • Ocular procedures such as the following: capsulotomy within 30 days prior to day 0; vitreoretinal surgery or scleral buckling, IVT injections or posterior subtenon steroids, and intraocular surgery within 90 days prior to day 0; ocular surgery anticipated within the first 180 days following day 0 • IOP ≥25 mmHg (glaucoma patients maintained on no more than two topical medications with IOP <25 mmHg are allowed) • Inadequate pupillary dilation • Media opacity that would limit clinical visualization and evaluation • Presence of the following: any form of ocular malignancy, herpetic infection in the study eye or adnexa, active or inactive toxoplasmosis in either eye • Non-ocular exclusion criteria: allergy or hypersensitivity to sirolimus or fluorescein dye, immunosuppressive therapy within 30 days of day 0, receiving strong inducers of CYP3A4 and P-gp, any recent infection within 30 days of baseline, immunocompromised patients, history of CMV infection or clinical evidence of active CMV infection at baseline, malignancy in remission for <5 years prior to study, history of other diseases that contraindicate the use of an investigational drug, females who are pregnant or lactating and females of childbearing potential who are not using adequate contraceptive precautions, sexually active males with partners of child-bearing potential who are not using adequate contraceptive precautions |
|||||
| Interventions | IVT sirolimus 352 μg (n=15) SCJ sirolimus 1,320 μg (n=15) Sirolimus was frozen in 2.0 mL vials as 0.5 mL sterile injection solution. A 30 G needle was used to deliver IVT injections on a Hamilton glass syringe and on a tuberculin syringe to deliver SCJ injections. Injections (SCJ or IVT) were given on days 0, 60, and 120. Follow-up after injection of the study eye was planned on the 14th and 30th day (±2 days). The SAVE study’s primary end point was set at month 6. Patients were monitored up to 12 months (Pages 12–13) |
|||||
| Outcomes | Main outcomes were as follows: 1. Bioactivity: The primary analysis of bioactivity was carried out at month 6 and evaluated by assessing the proportion of patients who achieved a complete or partial response in the study eye. Another end point, the secondary end point, was defined as the ability of sirolimus to reduce or prevent uveitis in the study eye. This is expressed by the frequency of inflammation during the first 6-month study period 2. Ocular tolerability: Adverse events related to sirolimus, both systemic and ocular, were monitored and recorded. Follow-up after injection of the study eye was planned on the 14th and 30th day (±2 days) (Page 13) |
|||||
| Notes | Date study conducted: Not reported Funding: Not reported Conflict of interest: Quan Dong Nguyen works for Santen, Inc. as part of the Scientific Advisory Board. Two authors (Joel Naor and Naveed Shams) are employed under Santen, Inc. The remaining authors have no conflicts of interest Trial registration: NCT00908466 |
|||||
| Risk of bias | ||||||
| Bias | Author’s judgment | Support for judgment | ||||
| Random sequence generation (selection bias) | Unclear | Not reported | ||||
| Allocation concealment (selection bias) | Unclear | Not reported | ||||
| Blinding of participants and personnel (performance bias) | High | Unmasked study. This is a proof-of-concept, open-label, randomized clinical trial | ||||
| Blinding of outcome assessment (detection bias) | Low | The Wilmer Eye Institute’s RIRRC was the coordinating, data management and reading center for the SAVE study. Its readers were masked in treatment groups (Page 13) |
||||
| Incomplete outcome data (attrition bias) | Low | Before the primary end point at month 6, two subjects, one from each study group and both from category 2, left the study. Bioactivity data collected from both subjects were not transmitted to month 6; since there was a significant amount of missing information to allow an appropriate assessment, data from these two subjects were removed when results were compared at month 6 to baseline. However, adverse events from both subjects were included in the safety results analysis (Pages 2–3) |
||||
| Selective reporting (reporting bias) | Low | Outcomes and events that happened during the 6-month study were reported in the SAVE study (additional file) | ||||
| Other bias | Low | None | ||||
| Overall risk of bias | Low | Majority of items show low risk | ||||
| Ibrahim et al21 | ||||||
| Methods | Open-label RCT Patients grouped into three disease categories with 1:1 randomization into one of two treatment arms. The eye with more advanced disease was selected as the study eye in patients with bilateral uveitis. If both eyes were affected equally, the study eye was selected before randomization at the investigator’s discretion. If the fellow eye was contraindicated, proven to be ineffective, or rejected by the patient, the injection of sirolimus was administered to the fellow eye at the discretion of the investigator at the same dose and route of administration of the study eye, but at least 14 days apart from the injection of the study eye (Page 2) |
|||||
| Participants | Country: USA Number of people randomized: 30 Average age: 47 years (range 29–65 years) Sex: 50% male, 50% female Inclusion criteria: Not reported but may be found in additional file (Table S1) of SAVE study28 Exclusion criteria: Not reported but may be found in additional file (Table S1) of SAVE study28 |
|||||
| Interventions | IVT sirolimus 352 μg (n=15) SCJ sirolimus 1,320 μg (n=15) Each study eye received mandatory baseline treatment for months 2 and 4. During the follow-up period of 6–12 months, eyes included were eligible to receive sirolimus at the same dose and route as required and at intervals of no <2 months between injections. (Page 2) A detailed description was provided in the SAVE study |
|||||
| Outcomes | Main outcome measures were bioactivity and ocular tolerability. Response to treatment for patients with active disease at baseline was measured using the SUN working group criteria, defined as reduction in VH by at least two steps when compared to baseline or reduction of one step to no haze. This was measured at 6 and 12 months However, in inactive patients (category 3), the efficacy or success of sirolimus was measured by the number of subjects who maintained quiescent uveitis during the entire 6-month period while lowering or discontinuing the previously taken CS treatment. Secondary outcome measures were as follows: change in BCVA as measured by ETDRS charts from baseline and change in macular thickness measured by SD-OCT. In patients with intermediate uveitis, posterior uveitis, and panuveitis, the safety and tolerability of SCJ and IVT injections of sirolimus were assessed by evaluating the frequency and severity of systemic and ocular adverse events and their relationship to the study drug. (Pages 2–3) Follow-up schedule was reported in the SAVE study and was extended from 6 months to 1 year |
|||||
| Notes | Date study conducted: Not reported Funding: Grants are from Santen, Inc. and the Research to Prevent Blindness to The Johns Hopkins University and the University of Nebraska Medical Center Conflict of interest: QDN works for Santen, XOMA, Inc., and AbbVie, Inc. as part of the Scientific Advisory Board; leads the Study Steering Committees SAKURA, EyeGuard, and VISUAL studies. The remaining authors have no conflicts of interest. Trial registration: NCT00908466 |
|||||
| Risk of bias | ||||||
| Bias | Author’s judgment | Support for judgment | ||||
| Random sequence generation (selection bias) | Unclear | Not reported | ||||
| Allocation concealment (selection bias) | Unclear | Not reported | ||||
| Blinding of participants and personnel (performance bias) | High | Unmasked study. This is a proof-of-concept, open-label, randomized clinical trial | ||||
| Blinding of outcome assessment (detection bias) | Unclear | This was a follow-up study of the SAVE trial and although blinding of outcome assessment was mentioned in the original article, this was not reported for in this study | ||||
| Incomplete outcome data (attrition bias) | Low | The primary 6-month report showed the baseline characteristics. Before the end point of month 12, six patients left the study. Two out of six patients left the study before the primary end point at month 6 and the reasons for their dropout were detailed in the previous report. Both patients did not complete the three mandatory injections and therefore bioactivity data from both patients were not included in the analysis of month 6 or 12 end points. Twenty-four patients completed the secondary end point of the study at month 12 with an 80% completion. Patients who did not have a visit for month 12, but visited the study at month 6 and beyond, were included in the analysis for month 12 (Page 3) |
||||
| Selective reporting (reporting bias) | Low | All outcomes mentioned on trial registration were reported | ||||
| Other bias | Low | None | ||||
| Overall risk of bias | Unclear | An equal number of low and unclear risk | ||||
| SAVE 2 201629 | ||||||
| Methods | Open-label RCT Randomization of the patients was stratified by two disease categories and was randomized into one of the two treatment arms in a ratio of 1:1. The eye with more advanced disease was selected as the study eye in patients with bilateral uveitis. If both eyes were affected equally, the study eye was selected before randomization at the investigator’s discretion. Patients with unilateral uveitis, however, if develop uveitis on the fellow eye at any point during the study period, will be treated with the same approach as that of study subjects with bilateral uveitis (Page 2) |
|||||
| Participants | Country: USA Number of people randomized: 24 Average age: 49 years (range 34–64 years) Sex: 29% male, 71% female Inclusion criteria: • Age ≥18 years • Diagnosed with active, noninfectious intermediate uveitis, posterior uveitis, or panuveitis (active uveitis was defined as having ≥1+ VH) • Treatment naïve (disease category 1) or patients with sufficient inflammation requiring systemic treatment such as prednisone ≥10 mg/day (or equivalent dose of another CS) and/or at least one systemic IMT other than CSs (disease category 2) • BCVA of ≥20/400 (approximately 20 ETDRS letters, Snellen equivalent) in both the study and fellow eye Exclusion criteria: • Patients requiring systemic IMT for bilateral disease • Presence of concomitant ocular conditions such as diabetic retinopathy and macular degeneration • Treatment with IVT injections (including but not limited to anti-VEGF) 60 days prior to the baseline • Intraocular surgery including vitreoretinal surgery within 90 days of baseline • IOP ≥25 mmHg in the study eye (glaucoma patients maintained on ≤2 topical medications with IOP <25 mmHg were allowed to participate) • Presence of active or inactive ocular infections (including herpes and toxoplasmosis) and periocular infections in either eye • Non-ocular exclusion criteria: patients with allergy to sirolimus, those receiving strong enzyme inducers/inhibitors of CYP34A and P-gp (eg, rifampin and ketoconazole), history of immunodeficiency, malignancy, metabolic dysfunction, pregnancy, or lactation |
|||||
| Interventions | IVT sirolimus • Group 1: A 20 μL of 2% (22 μg/μL sirolimus) to make a 440 μg (n=11) on sterile vials was given on days 0, 30, 60, 90, 120, and 150 • Group 2: A 20 μL of 4% (44 μg/μL sirolimus) to make an 880 μg (n=13) on sterile vials was given every 2 months on days 0, 60, and 120 A specially designed plastic syringe for the SAVE 2 study was provided by the drug company, which has one specific mark at 20 μL to allow the investigators/injectors to give exactly 20 μL on each study eye (Page 2) |
|||||
| Outcomes | Main outcomes: 1. Complete or partial response in the study eye 2. Reduction or prevention of recurrences in the study eye 3. Change in BCVA, CMT, VHZ, VCC, and anterior chamber cells compared to baseline Follow-up visits scheduled at 30, 60, 90, 120 days (6 months) |
|||||
| Notes | Date study conducted: Not reported Funding: Support is partly from an unrestricted grant from RPB and by an unrestricted educational grant from Santen, Inc. Sirolimus was provided by the drug manufacturer. Conflict of interest: QDN leads the SAKURA Study Steering Committee and is part of the Santen Scientific Advisory Board. Trial registration: NCT01280669 |
|||||
| Risk of bias | ||||||
| Bias | Author’s judgment | Support for judgment | ||||
| Random sequence generation (selection bias) | Unclear | Not reported | ||||
| Allocation concealment (selection bias) | Unclear | Not reported | ||||
| Blinding of participants and personnel (performance bias) | High | Unmasked study. This is a multicenter, open-label, randomized clinical trial | ||||
| Blinding of outcome assessment (detection bias) | High | Data with regard to BCVA (ETDRS letters read at 4 m), CMT, VHZ, vitreous cells, amount of anterior cells, daily dose of prednisone (or other CSs) were recorded and summarized on respective treatment group and disease category within treatment groups on every visit (Page 3) |
||||
| Incomplete outcome data (attrition bias) | Low | Out of the 24 patients enrolled in the study, at month 6, three patients did not complete the primary end point visit. After month 3 (group 1, category 2), one patient was lost to follow up and a second patient (group 2, category 2) chose a different therapeutic option, leaving the study at month 4. The third patient needed rescue treatment at month 4 (group 1, category 1) due to increase in inflammation Since all subjects completed their visit to month 3, data were still included on month 6. In the final analysis, 11 patients were included in group 1 (seven of which were category 1 patients) and 13 patients in group 2 (six of which were category 1 patients) (Page 3) | ||||
| Selective reporting (reporting bias) | Low | All outcomes mentioned on trial registration were reported Adverse events, both ocular and non-ocular, were analyzed in total and as subsets based on severity and relationship to sirolimus | ||||
| Other bias | Low | None | ||||
| Overall risk of bias | Low | Although some items could not be fully assessed, we believe that randomization and allocation concealment should be adequate in this multicenter trial aiming at drug registration, as per regulatory requirement | ||||
| SAKURA 201627 | ||||||
| Methods | Double-masked RCT Randomization of the patients at baseline in a ratio of 1:1:1 to IVT sirolimus after a screening period of 30 days. The eye with greater VH score was selected to be the study eye in patients with bilateral disease. However, in patients with equal VH score on both eyes, right eye was chosen as the study eye (Page 2) |
|||||
| Participants | Countries: European Union, India, Israel, Japan, Latin America, and USA Number of people randomized: 347 (348 eyes) Average age: 46 years (range 18–83 years) Sex: 59.9% were female (208) Inclusion criteria: • Age ≥18 years • Diagnosed with active, noninfectious intermediate uveitis, posterior uveitis, or panuveitis (active uveitis was defined as having ≥1+ VH) • BCVA of ≥20/400 (≥19 ETDRS letters, Snellen equivalent) in the study eye and ≥20/200 in the fellow eye • Anterior component of uveitis had to be less than the posterior component if present Exclusion criteria: • Active infectious uveitis • Primary diagnosis of anterior uveitis • Uncontrolled glaucoma • Ocular procedures such as the use of IVT injections, posterior subtenon CSs 90 days before baseline, and vitrectomy in the study eye |
|||||
| Interventions | IVT sirolimus administered via a 20 mL injection on days 1, 60, and 120: – 44 μg (n=114), also the comparator – 440 μg (n=117) – 880 μg (n=117) (Page 2) |
|||||
| Outcomes | Primary outcome: percentage of subjects with VH 0 response at month 5 without the use of rescue therapy Secondary outcomes: • VH 0 or 0.5+ response rate • The proportion of subjects with a VH score of 0 or a ≥2 U improvement from baseline • CS-tapering success rate • Changes in BCVA • CRT • NEI VFQ-25 score |
|||||
| Notes | Date study conducted: May 31, 2011, to March 31, 2013 Funding: Sponsored by Santen, Inc. Conflicts of interest of all authors were disclosed. Trial registration: NCT01358266 |
|||||
| Risk of bias | ||||||
| Bias | Author’s judgment | Support for judgment | ||||
| Random sequence generation (selection bias) | Low | A unique randomization number was assigned to each subject. In order to randomize eligible subjects, a permuted block randomization stratified by predefined geographical area and baseline VH score of the study eye (1.5+, 2+, 3+, or 4+) was used. Also, the ivRS/iwRS was applied to generate a separate permuted-block randomization scheme with a fixed block for each stratum (Supplementary materials27) | ||||
| Allocation concealment (selection bias) | Low | |||||
| Blinding of participants and personnel (performance bias) | Low | During the double-masked treatment period, all study sponsors (with the exception of clinical supplies), vendors, contract research staff, clinical researchers, site staff, and study subjects were masked. The study drug, which is formulatedas a clear, nondispersive, nonaqueous sirolimus solution in a vehicle composed of polyethylene glycol 400 and ethanol, was provided to site staff in numbered vials assigned by the randomization system to each subject (Supplementary materials27) | ||||
| Blinding of outcome assessment (detection bias) | Low | During the double-masked treatment period, all study sponsors (with the exception of clinical supplies), vendors, contract research staff, clinical researchers, site staff, and study subjects were masked (Supplementary materials27) | ||||
| Incomplete outcome data (attrition bias) | Low | Two randomized but not treated subjects with screen failure were excluded from the security population. Inadvertently, one subject was registered twice and randomized to receive a dose of 880 mg each time, with two different subject identification numbers assigned to a different study eye. The security population excluded two randomized but not treated subjects with screen failure. One subject was inadvertently registered twice and randomized to receive a dose of 880 mg each time, with two different identification numbers assigned to a different study eye (Page 2416) | ||||
| Selective reporting (reporting bias) | Low | Efficacy and safety outcomes were assessed. Ocular adverse events and general disorders were reported | ||||
| Other bias | Low | None | ||||
| Overall risk of bias | Low | Low risk of bias for most items | ||||
Abbreviations: BCVA, best-corrected visual acuity; CMT, central macular thickness; CMV, cytomegalovirus; CRT, central retinal thickness; CS, corticosteroid; ETDRS, Early Treatment of Diabetic Retinopathy Study; IMT, immunomodulatory therapy; IVRS, interactive voice response system; IVT, intravitreal; IWRS, interactive web response system; RCT, randomized controlled trial; RIRRC, Retinal Imaging Research and Reading Center; RPB, research to prevent blindness; SCJ, subconjunctival; SUN, Standardization of Uveitis Nomenclature; VEGF, vascular endothelial growth factor; VH, vitreous haze; SD-OCT, spectral domain-ocular coherence tomography; VHZ, vitreous haze; VCC, vitreous cell count.
Types of participants
All patients included in the RCT studies were 18 years old and above, clinically diagnosed with NIU (posterior, intermediate, or panuveitis), and assessed using the SUN classification activity score for VH, with sufficient inflammation requiring systemic treatment and with a BCVA of 20/400 or better.
Types of interventions
Sirolimus was administered by either IVT or SCJ injections. IVT doses studied were 44 μg, 352 μg, 440 μg, and 880 μg. Only one study evaluated a 1,320 μg dose of sirolimus given subconjunctivally.
Types of outcomes
The primary outcome of all studies was uveitis activity as measured by VH. Secondary outcomes measured were BCVA, CMT, IOP, and CS-sparing effect as measured by CS dose. Safety outcomes and adverse events were also noted.
Characteristics of studies excluded from the meta-analysis
Sen et al,38 Vigil et al,39 and Shanmuganathan et al40 also studied sirolimus for treating patients with uveitis. The limited time of follow-up and differences in study design precluded their data for undergoing meta-analysis. All three studies, however, supported the potential efficacy of sirolimus. Main characteristics of the studies and level of evidence based on the Scottish Intercollegiate Guidelines Network (SIGN)41 criteria are summarized in Table 3.
Table 3.
Main characteristics of studies excluded from the meta-analysis
| Study | Study design | Participants | Intervention | Outcome/results | Evidence strength |
|---|---|---|---|---|---|
| Sen et al38 USA | Phase I, prospective nonrandomized study | Five patients with chronic active anterior uveitis | Single 1,320 μg SCJ sirolimus injection in the study eye Study duration: 4 months |
Primary outcome: Two-step reduction in AC inflammation (according to SUN criteria) within 4 weeks after treatment Results: Three out of five (60%) patients met the primary outcome All patients reported no serious adverse events |
Level 3 |
| Vigil et al39 USA | Prospective, randomized, open-label trial (QoL assessment of the SAVE trial) | 30 patients with noninfectious intermediate uveitis, posterior uveitis, and panuveitis | Two treatment groups Group 1: IVT injections of 352 μg of sirolimus Group 2: SCJ injections of 1,320 μg of sirolimus in the study eye on days 0, 60, and 120 Study duration: 12 months |
Primary outcome: Vision-related QoL measured by the NEI VFQ-39 Results: 26 out of 30 (86.7%) patients completed the VFQ-39 at baseline and at month 6, while 23 out of 30 (76.7%) patients completed up to month 12 Composite scores showed significant improvement in all patients Better tolerability was reported with IVT sirolimus injections |
Level 1+ |
| Shanmuganathan et al40 UK | Prospective, nonrandomized, open-label trial | Eight patients with severe NIU | Systemic sirolimus started at 4 mg daily (increased in 2 mg increments depending on the disease activity and trough blood levels) | Primary outcomes: BCVA gain (>2 lines), control of uveitis activity (inflammatory cell count), CS dose reduction, and symptomatic relief Results: Five out of eight (62.5%) patients responded well to sirolimus treatment Three out of eight (37.5%) had treatment failure in controlling uveitis or had intolerable side effects |
Level 3 |
Abbreviations: BCVA, best-corrected visual acuity; CS, corticosteroid; IVT, intravitreal; NEI-VFQ-39, National Eye Institute 39-Question Visual Function Questionnaire; NIU, noninfectious uveitis; QoL, quality of life; SCJ, subconjunctival; SUN, Standardization of Uveitis Nomenclature; AC, anterior chamber.
Risk of bias of included studies
Two reviewers (Cabahug and Sapno) independently assessed the risk of bias among the included studies. To assess the methodological quality, the “modified Jadad scale” (Table 4) as well as the “Cochrane risk of bias assessment tool” (Figure 2) was used. The mean Jadad score was 8.0 (high quality). Each study was assessed for the presence of any selection bias, performance bias, detection bias, attrition bias, selective reporting, and other potential sources of bias (Figure 3). Three studies had a low overall risk of bias with SAKURA 2016 having the lowest risk, while Ibrahim et al21 had an unclear risk of bias. Collectively, the overall risk of bias of all the selected studies was 75% low risk of bias and 25% unclear risk of bias (Figure 2).
Table 4.
Modified Jadad scores of the four included studies
| SAVE28 2013 | Ibrahim et al21 | SAVE 229 2016 | SAKURA27 2016 | |
|---|---|---|---|---|
| “Was the study described as randomized?” | Y | Y | Y | Y |
| “Was the randomization protocol detailed and appropriate?” | Y | Y | Y | Y |
| “Was the study described as double-blind?” | N | N | N | Y |
| “Was the blinding process detailed and appropriate?” | N | N | N | Y |
| “Did the study have a control group?” | Y | Y | Y | Y |
| “Was the control detailed and appropriate?” | N | N | Y | Y |
| “Was there an adequate exclusion criterion?” | Y | Y | Y | Y |
| “Was the intervention used at a therapeutic dose?” | Y | Y | Y | Y |
| “Was there a description of withdrawals and dropouts?” | Y | Y | Y | Y |
| “Were the data clearly and adequately reported?” | Y | Y | Y | Y |
| Score (total =10) | 7 | 7 | 8 | 10 |
Note: Ibrahim et al21 is a follow-up study of the SAVE trial.
Abbreviation: RCT, randomized controlled trial.
Figure 2.
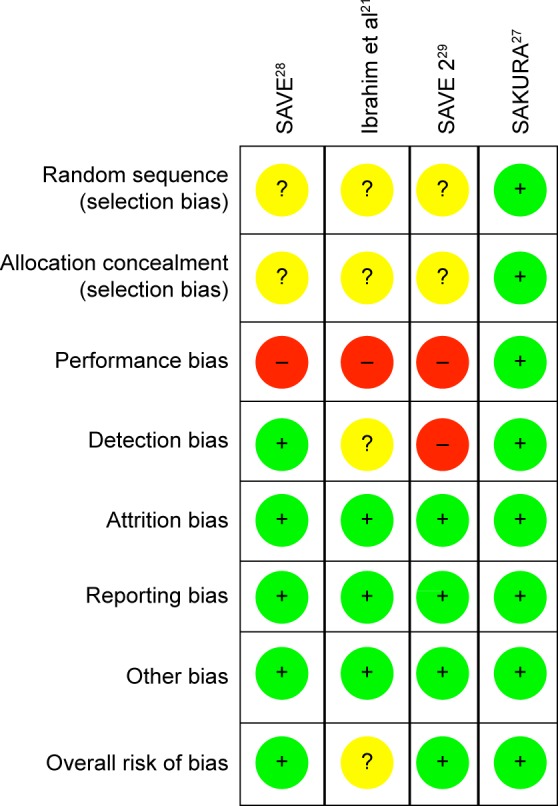
Risk of bias summary: (+) low risk, (?) unclear risk, (−) high risk.
Figure 3.
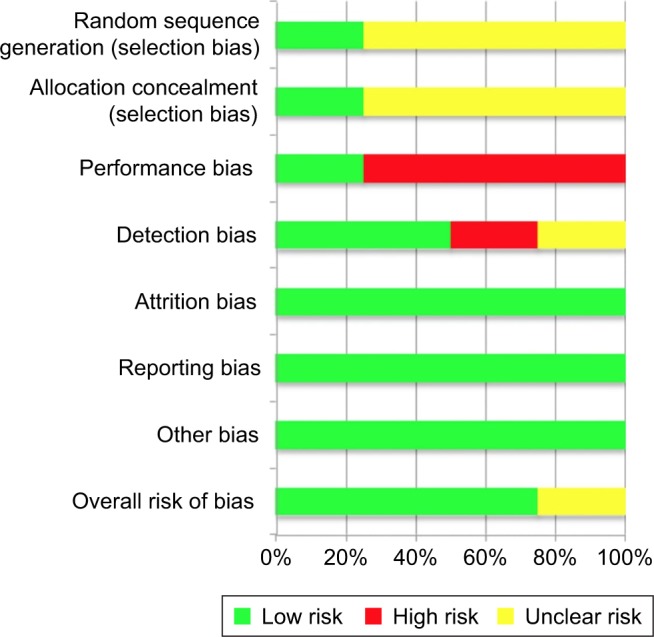
Risk of bias graph.
Studies awaiting assessment
The SAKURA 2 trial, which was also sponsored by Santen Inc., was recently completed with supposedly mixed results compared to the original SAKURA trial. However, data were still unavailable. The authors have attempted to contact the representatives of the company requesting their official results.42
The LUMINA trial (ClinicalTrials.gov Identifier: NCT03711929) is the latest Phase III trial yet to be commenced by Santen Inc. The study design is similar to the SAKURA trial except that this study included a sham treatment arm. The expected completion date is April 17, 2021.43
Control of uveitis activity
Figure 4 shows a meta-analysis on percentage improvement in VH with a follow-up duration of 6 months, where the test of heterogeneity of 74.95% I2 (p=0.0185) suggests that a random-effects model is preferred. The resulting overall pooled proportion of percentage improvement in VH is 38%, with 95% CI of 16.19–62.66. The biggest weight given is on SAKURA 201627 (82.73). Forest plot, where the diamond marker did not intersect 0, suggests that the 38% pooled prevalence is significantly higher than 0.
Figure 4.

The pooled proportion of percentage improvement in VH with a follow-up duration of 6 months.
Abbreviation: VH, vitreous haze.
Figure 5 shows a meta-analysis on percentage improvement in VH with a follow-up duration of 6–12 months, where the test of heterogeneity of 88.03% I2 (p=0.0002) suggests that a random-effects model is preferred. The resulting overall pooled proportion of percentage improvement in VH is 49.97%, with 95% CI of 16.93–83.03. The biggest weight given is on SAKURA 201627 (37.63). Forest plot, where the diamond marker did not intersect 0, suggests that the 49.97% pooled prevalence is significantly higher than 0.
Figure 5.

The pooled proportion of percentage improvement in VH with a follow-up duration of 6–12 months.
Abbreviation: VH, vitreous haze.
The two different meta-analysis runs were compared, specifically, the pooled response rate of 38% and 49.97%, and the resulting p-value of 0.0472 indicates that the run where the 1-year duration was included has significantly higher response rate as compared to the run where all studies are only up to 6 months.
Visual acuity
Figure 6 shows a meta-analysis on percentage improvement in BCVA with a follow-up duration of 6 months, where the test of heterogeneity of 83.75% I2 (p=0.0021) suggests that a random-effects model is preferred. The resulting overall pooled proportion of percentage improvement in BCVA is 62.2%, with 95% CI of 33.17–87.11. The biggest weight given is on SAKURA 201627 (39.1). Forest plot, where the diamond marker did not intersect 0, suggests that the 62.6% pooled prevalence is significantly higher than 0.
Figure 6.

The pooled proportion of percentage improvement in BCVA with a follow-up duration of 6 months.
Abbreviation: BCVA, best-corrected visual acuity.
Figure 7 shows a meta-analysis on percentage improvement in BCVA with a follow-up duration of 6–12 months, where the test of heterogeneity of 90.22% I2 (p=0.0001) suggests that a random-effects model is preferred. The resulting overall pooled proportion of percentage improvement in BCVA is 56.86%, with 95% CI of 20.91–89.05. The biggest weight given is on SAKURA 201627 (36.75). Forest plot, where the diamond marker did not intersect 0, suggests that the 56.86% pooled prevalence is significantly higher than 0.
Figure 7.

The pooled proportion of percentage improvement in BCVA with a follow-up duration of 6–12 months.
Abbreviation: BCVA, best-corrected visual acuity.
The two different meta-analysis runs about BCVA improvement were compared, specifically, the pooled BCVA improvement of 62.2% and 56.86%, and a resulting p-value of 0.3705 indicates that the two runs resulted in same pooled BCVA improvement indicating that no significant difference exists.
IOP
Figure 8 shows a meta-analysis on percent increase in IOP with a follow-up duration of 6 months, where the test of heterogeneity of 0.00% I2 (p=0.6545) suggests that a fixed-effects model is preferred. The resulting overall pooled proportion of percent increase in IOP is 7.1%, with 95% CI of 3.46–12.68. The biggest weight given is on SAKURA 201627 (80.1). Forest plot, where the diamond marker did not intersect 0, suggests that the 7.1% pooled prevalence is significantly higher than 0.
Figure 8.

The pooled proportion of percentage increase in IOP with a follow-up duration of 6 months.
SAVE 201328 and Ibrahim et al21 reported the same number of events for percent increase in IOP indicating no change after 1 year. Thus, a meta-analysis on percent increase in IOP with a follow-up duration of 6–12 months was not performed.
Other outcomes
Outcomes where data were not reported similarly precluded meta-analysis; hence, a narrative synthesis is reported in the following sections.
CS-sparing effect
SAVE 201328 and Ibrahim et al21 reported that 11 subjects (IVT group n=6, SCJ group 2 n=5) with active uveitis who were receiving prednisone ≥10 mg/day and/or at least one systemic IMT (category 2) successfully tapered their CS dose to <10 mg/day, with a median dose reduced up to 8 mg/day and 2 mg/day at month 6 and month 12, respectively, from a 20 mg/day median dose at baseline. Subjects with inactive disease at baseline (category 3) also showed a reduction with a median dose to 3 mg/day and 2 mg/day at months 6 and 12, respectively, from a median dose of 9 mg/day at baseline. CS was stopped completely in five patients from category 2. However, it was impossible to reduce CS dose in one patient and ended the study with a 20 mg/day dose.
In SAVE 2 2016,29 dose reduction was reported in nine out of ten patients who received ≥10 mg daily of systemic CS treatment at baseline to <5 mg daily after 6 months of treatment (440 μg n=4 [100%]; 880 μg n=5 [83.3%]) as a result of improvement in VH.
SAKURA 201627 also reported a dose reduction in 69 patients treated with CS at baseline. CS dosage was ≤5 mg at month 5 in 48 patients (69.6%) and eventually discontinued in all except for one patient (47/69 [68.1%]). Each dose group (44 μg n=14/22 [63.6%]; 440 μg n=20/26 [76.9%]; 880 μg n=14/21 [66.7%]) reached this end point in similar proportions. However, comparing the proportion of patients who met both the primary end point and the CS-tapering end point, the 440 μg dose group had significantly higher number of such patients than the 44 μg dose group (P=0.011).
CMT
SAVE 201328 and Ibrahim et al21 reported that, at baseline, 37% (n=11) of subjects had ME (IVT n=7, SCJ n=4), defined as having an average CMT of >315 μm. In patients without ME (n=17), the average thickness was 272 μm (27), 265 μm (29), and 269 μm (35) at baseline, month 6, and month 12, respectively, with no changes from baseline in any patient during the follow-up at month 6 or 12. In patients who had ME, the baseline average (SD) CMT was 505 μm (156). CMT in the IVT group increased to 615 μm (168) at month 6 and 616 μm (165) at month 12, with a mean change of 105 μm and 106 μm at months 6 and 12, respectively, from an average of 510 μm (194) at baseline. The SCJ group showed a reduction in CMT with a mean change of 30 μm and 47 μm, at months 6 and 12, respectively, to 451 μm (114) at month 6 and to 434 μm (122) at month 12 from 481 μm (131) at baseline. No statistical difference was present between the changes in CMT from baseline at either month 6 or 12 (P=0.169 and 0.182 at month 6 and 12, respectively).
SAVE 2 201629 reported that patients with baseline ME receiving the 440 μg dose (n=7) had a decrease in CMT with a mean change of 42 mm from baseline at month 6, while those receiving 880 μg (n=6) had an increase in CMT with a mean change of 82 mm from baseline. Patients without ME at baseline (n=11) did not have any significant change in mean CMT at month 6.
Lastly, SAKURA 201627 also showed that CMT decreased by 50 mm or more in nearly 50% of all patients with baseline ME (CMT >300 mm, n=99) in each of the three treatment groups (44 μg, 45.5%; 440 μg, 55.2%; 880 μg, 48.6%) with no significant differences between the three groups (P=0.061).
Safety outcomes/AEs
SAVE 201328 and Ibrahim et al21 reported mild-to-moderate ocular and systemic AEs with both IVT and SCJ injections of sirolimus, and no systemic AEs were related to treatment. Vitreous floaters were the common complaint in the IVT group, while in the SCJ group conjunctival hyperemia and chemosis were among the common complaints reported.
SAVE 2 201629 also reported mild-to-moderate non-ocular AEs (eg, sinus infection), but all were treatment unrelated. Ocular AEs (eg, ocular pain) were reported in both treatment groups (440 μg, 36.4%; 880 μg, 46.2%), while more serious events tended to be reported in the 880 μg group (eg, anterior uveitis and cataract) not commonly seen in the 440 μg group. Fourteen patients who received bilateral IVT sirolimus injections also tolerated treatment well. Systemic AEs (eg, sinus infection) were reported in three patients (21.4%), while local AEs (eg, floaters) were reported in two patients (14.3%) suggesting that bilateral IVT injections of sirolimus may be given safely on the same day. The SAVE 2 study also suggested that both monthly 440 μg and every other monthly 880 μg IVT sirolimus injections provide sufficient control of ocular inflammation in most cases of NIU, while the monthly injections were noted to be more beneficial in reducing ME.
Lastly, SAKURA 201627 demonstrated that treatment-related ocular serious AEs with sirolimus were uncommon, but the severity tended to be dose dependent. Sterile endophthalmitis was reported in 0%, 0.9%, and 3.4% of patients in the 44 μg, 440 μg, and 880 μg dose groups, respectively. Similarly, cataract progression was seen in 0.9%, 0.9%, and 2.6% of patients in the 44 μg, 440 μg, and 880 μg dose groups, respectively. Two patients, one of each from the 44 μg and 440 μg dose groups, had glaucoma. The mean IOP in all three treatment groups remained nearly constant (≤1.1 mmHg difference) from baseline up to month 5. All of the above-mentioned findings suggest that IVT sirolimus may not be associated with AEs commonly reported in previous studies involving intraocular CS treatment and that non-ocular AEs occurred with low frequency unrelated to treatment (eg, headache in 4.6% of all patients). Finally, SAKURA 201627 suggested that a 440 μg IVT sirolimus injection was the optimal dose in having a better benefit-to-risk profile for treating most cases of NIU leading to improvement in ocular inflammation with a low incidence of AEs commonly associated with intraocular CS treatment in patients with active disease.
Discussion
Summary of evidence
The data provided in this review and meta-analysis summarize the available evidence for the use of sirolimus in the treatment of NIU. Currently, the potential use of sirolimus in such cases is limited by the paucity of RCTs available. We have presented a pooled proportion of the outcomes studied with sirolimus treatment at 6 months and up to 1-year follow-up. It should be noted that there was only one study evaluating the 1-year outcomes.28
The pooled proportion of VH improvement with a 6-month follow-up was 38%, but this increased to nearly 50% when 1-year follow-up study data were included. This suggests a trend to further improvement in uveitis control when longer follow-ups are included which was not usually evaluated with current trials available. The pooled proportion of BCVA improvement was relatively the same after 6 months (62.2%) and 1 year (56.86%). An improvement in visual acuity can probably be appreciated early in the course of treatment, although it is still not clear whether good visual acuity and its duration can be maintained. Improvement in visual acuity follows inflammation control as seen by the reduction in VH in these patients.
CSs are still the mainstay of treatment in uveitis, used both as rescue therapy for acute disease and as long-term therapy in chronic or even refractory cases.12,44 CSs have many desirable features, including speed of onset and efficacy in both local and systemic forms. In the MUST trial, disease inactivity was reported in 88% of eyes randomized to the fluocinolone acetonide implant and in 71% of eyes randomized to systemic CS usage.12,44 They do however have an unfavorable side effect profile, with dose- and duration-dependent adverse systemic effects. Alternative IMT has been developed to address these concerns. These “steroid-sparing agents” have improved tolerability while maintaining similar inflammation controls. Unfortunately, these agents may be associated with different, but equally severe, side effects, and in many cases lack both the speed of onset and efficacy of CS. Nevertheless, among these agents, sirolimus remains a promising option as a potential CS-sparing IMT.
The pooled proportion of IOP elevation after both 6 months and 1 year of treatment with sirolimus was roughly 7%. This relatively low rate can be causally attributed to the overall CS-sparing effect of sirolimus. As summarized in our results, the majority of patients successfully tapered their CS dose to as low as ≤5–10 mg/day.
Blair et al44 also reported a comprehensive review of the role of mTOR inhibitors for treating NIU, including evidence on everolimus. Although a meta-analysis was not conducted, their conclusions from the data they gathered also supported our results. Published evidence thus to date suggests that sirolimus (and everolimus) appears to be well tolerated especially when locally administered and may prove useful in the management of NIU.44,45
In general, therapeutic agents administered locally provide a more rapid onset of action with fewer systemic side effects, and recent trials mainly focused on such preparations of sirolimus. In effect, the data from our meta-analysis will only apply to IVT and perhaps SCJ administrations of sirolimus. Overall, it appears that this is the most optimal and favorable route for the drug when considered for ocular conditions.
Limitations
The strength of evidence provided in this review is limited by the small number of clinical trials evaluating sirolimus. The trials available used an active control or a dose–response control. Although the existence of an effect can be readily documented, measuring the drug’s absolute effect size is not possible unless a placebo-controlled design is available. RCTs with sham treatment arms are therefore recommended. It should also be noted that there are various possible etiologies for uveitis which may be difficult to account with present clinical trial designs. The wide range of percentage improvement in VH may be attributable to this factor. Lastly, our review also did not include unpublished studies which would result in publication bias. Nevertheless, we are confident that the evidence we provide is enough to overcome this small fraction. Although there seems to be an increasing trend of evidence supporting the efficacy of sirolimus, the remaining gaps may be addressed once further evidence from ongoing or future high-quality clinical trials are available.
Conclusion
The efficacy of sirolimus in treating NIU was positively determined in a meta-analysis done on three RCTs and established that potent control of uveitis activity as a result of the reduction in VH was well demonstrated up to 12-month follow-up duration. Sirolimus treatment also resulted in moderate visual acuity improvement and had relatively low adverse events with good CS-sparing effect. However, data from ongoing and future large-scale clinical trials of appropriate design are needed to further strengthen the evidence provided.
Supplementary materials
Supplementary appendix
Intravitreal Sirolimus in Active Noninfectious Uveitis of the Posterior Segment: The Multinational, Randomized, Phase 3 SAKURA Study 1
Nguyen QD, Merrill PT, Clark WL, Banker AS, Fardeau C, Franco P, LeHoang P, Ohno S, Rathinam SR, Thurau S, Abraham A, Wilson L, Yang Y, Shams N; for the SAKURA Study Group
Table S1.
Inclusion and exclusion criteria1
| Inclusion criteria | |
|---|---|
|
| |
| 1. ≥18 years of age | |
| 2. Having diagnosis of uveitis determined by the investigator to be noninfectious based on the patient’s medical history, history of present illness, ocular examination, review of systems, physical examination, and any relevant, pertinent laboratory evaluations | |
| 3. Patients with active uveitis, defined as having at least 1+ VH and/or at least 1+ vitreous cell count (SUN scale), and | |
| 4. are receiving no other treatment; or | |
| 5. are receiving prednisone ≥10 mg/day and/or at least one other systemic immunosuppressants | |
| 6. Patients with inactive disease, defined as having 0.5+ VH or less and a grade of 0.5+ vitreous cell count or less (SUN scale), and are receiving prednisone <10 mg/day and/or at least one other systemic immunosuppressants | |
| 7. Having posterior uveitis, intermediate uveitis, or panuveitis; for panuveitis, if an anterior component is present, it must be less than the posterior component | |
| 8. Sufficient inflammation to require systemic treatment | |
| 9. BCVA of 20/400 or better in both eyes | |
|
| |
| Exclusion criteria | |
|
| |
| Non-ocular | Ocular |
|
| |
| 1. Allergy or hypersensitivity to sirolimus or fluorescein dye 2. Immunosuppressive therapy within 30 days of day 0 3. Patients who are receiving strong inducers of CYP3A4 and P-gp and have any recent infection within 30 days of baseline 4. Immunocompromised patients 5. History of CMV infection or clinical evidence of active CMV infection at baseline 6. Malignancy in remission for <5 years prior to study 7. History of other diseases, metabolic dysfunction, physical examination finding, or clinical laboratory finding giving reasonable suspicion of a disease condition that contraindicates the use of an investigational drug might affect the interpretation of the results of the study or renders the patient at high risk for treatment complications 8. Females who are pregnant or lactating and females of child-bearing potential who are not using adequate contraceptive precautions 9. Sexually active males with partners of child-bearing potential who are not using adequate contraceptive precautions |
1. Patients with bilateral uveitis who are receiving systemic IMT for the treatment of the fellow eye and cannot be controlled with standard local therapies alone 2. Any significant ocular disease that could compromise vision in the study eye 3. Any IVT injections or posterior subtenon’s steroids within 90 days prior to day 0 4. Intraocular surgery within 90 days prior to day 0 5. Capsulotomy within 30 days prior to day 0 6. History of vitreoretinal surgery or scleral buckling within 90 days prior to day 0 7. Any ocular surgery anticipated within the first 180 days following day 0 8. IOP ≥25 mmHg (glaucoma patients maintained on no more than two topical medications with IOP <25 mmHg are allowed) 9. Pupillary dilation inadequate for quality stereoscopic fundus photography 10. Media opacity that would limit clinical visualization, IVFA, or OCT evaluation 11. Presence of any form of ocular malignancy History of herpetic infection in the study eye or adnexa 12. Presence of known active or inactive toxoplasmosis in either eye 13. Ocular or periocular infection in either eye |
Abbreviations: BCVA, best-corrected visual acuity; CMV, cytomegalovirus; IMT, immunomodulatory therapy; IVFA, intravenous fluorescein angiography; IVT, intravitreal; OCT, optical coherence tomography; SUN, Standardization of Uveitis Nomenclature; VH, vitreous haze.
Table S2.
CENTRAL search strategy
| Date Run: | 11/05/2018 05:15:28 | |
|---|---|---|
| ID | Search | Hits |
| #1 | MeSH descriptor: [Uveitis] explode all trees | 537 |
| #2 | Uveitic | 110 |
| #3 | #1 OR #2 | 615 |
| #4 | Sirolimus | 2,970 |
| #5 | Rapamycin | 1,821 |
| #6 | mTOR inhibitor | 534 |
| #7 | #4 OR #5 OR #6 | 3,947 |
| #8 | #3 AND #7 | 7 |
| #9 | (sirolimus): ti,ab,kw AND (uveitis): ti,ab,kw | |
| (Word variations have been searched) | 23 | |
| #10 | #8 OR #9 | 23 hits |
Table S3.
PMC search strategy
| Date Run: 11/06/2018 09:22:29 | |
|---|---|
| (“uveitis”[MeSH Terms] OR “uveitis”[All Fields]) AND | 247 hits |
| (“sirolimus”[MeSH Terms] OR “sirolimus”[All Fields]) | |
| URL: https://www.ncbi.nlm.nih.gov/pmc/?term=(%22uveitis%22%5BMeSH+Terms%5D+OR+%22uveitis%22%5BAll+Fields%5D)+AND+(%22sirolimus%22%5BMeSH+Terms%5D+OR+%22sirolimus%22%5BAll+Fields%5D)&cmd=DetailsSearch |
Table S4.
ClinicalTrials.gov search strategy
| Date Run: 11/7/2018 07:15:18 | |
|---|---|
| CONDITION or DISEASE: Uveitis | 7 hits |
| OTHER TERMS: Sirolimus | |
| (Uveitis AND Sirolimus) |
Reference
- 1.Nguyen QD, Ibrahim MA, Watters A, et al. Ocular tolerability and efficacy of intravitreal and subconjunctival injections of sirolimus in patients with non-infectious uveitis: primary 6-month results of the SAVE study. J Ophthalmic Inflamm Infect. 2013;3(1):32. doi: 10.1186/1869-5760-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Randomization and Masking
Randomization occurred after subjects met all eligibility requirements at screening and Day 1. Each subject was assigned a unique randomization number. A permuted-block randomization stratified by pre-defined geographic region and baseline vitreous haze score of the study eye (1.5+, 2+, 3+, or 4+) was employed to randomize eligible subjects, and a separate permuted-block randomization scheme with a fixed block was generated for each stratum by an Interactive Voice Response System (IVRS)/Interactive Web Response System (IWRS). A computer algorithm for random number generation produced the treatment assignments.
All study sponsor personnel (with the exception of clinical supplies personnel), vendors, contract research organization staff, clinical investigators, site staff, and study subjects were masked to the treatment assignments during the Double-Masked Treatment Period. The study drug, which is formulated as a clear, nondispersive, non-aqueous solution of sirolimus in a vehicle composed of polyethylene glycol 400 and ethanol, was provided to site staff in numbered vials assigned to each subject by the randomization system. Each vial contained 20 μL of sirolimus at a concentration of 2.2 μg/μL (44 μg), 22 μg/μL (440 μg), or 44 μg/μL (880 μg). Study sites received the drug vials frozen and stored them in secure, locked, dark, temperature-controlled freezers with restricted access until the time of use. A sterile, single-use 250-μL syringe custom-marked at 20 μL was provided separately for intravitreal injection use and filled on site. The clinical investigator was required to use the syringe provided and administer sirolimus according to intravitreal injection guidelines.
Changes to Study Design
This study was originally planned as two studies to be conducted in separate predefined geographic regions (Region 1: United States and Latin America; Region 2: European Union and Asia) under a single protocol. Given that NIU of the posterior segment is an orphan disease, enrollment in each study was slower than anticipated. The sponsor, in consultation with the US FDA, restructured the studies to include the 347 subjects randomized up to March 31, 2013, in a single study, regardless of geographic region. The 347 subjects randomized up to that date comprise the population of the present study (SAKURA Study 1), with subjects enrolling from April 1, 2013, randomized into a second study (SAKURA Study 2).
Under the original plan to conduct two studies defined by geographic region, a sample size of 213 subjects (71 per treatment group) subjects per study was calculated as necessary to provide sufficient statistical power to assess the primary endpoint. Revised calculations based on the new study population defined by date of enrollment indicated that 348 subjects (116 subjects per group) would provide >80% power to detect a difference of 16% in response rate between sirolimus 44 μg (assuming aresponse rate of 8%) and sirolimus 440 μg or 880 μg (assuming a response rate of 24%) by Fisher’s exact test (two-sided, Bonferroni corrected α=0.025).
Acknowledgments
Reginald Arimado was a hired biostatistician who assisted us in conducting the meta-analysis and provided the necessary software to perform the task. No funding from any sponsor or external source was received.
Abbreviations
- AE
adverse effects
- BCVA
best-corrected visual acuity
- CMT
central macular thickness
- CS
corticosteroid
- ETDRS
Early Treatment of Diabetic Retinopathy Study
- IMT
immunomodulatory therapy
- IVT
intravitreal
- ME
macular edema
- MUST
Multicenter Uveitis Steroid Treatment
- NIU
noninfectious uveitis
- PROSPERO
Prospective Register of Systematic Reviews
- RCT
randomized controlled trial
- SAKURA
Sirolimus study assessing double-masked uveitis treatment
- SAVE
sirolimus as a therapeutic approach for uveitis
- SCJ
subconjunctival
- SIGN
Scottish Intercollegiate Guidelines Network
- SUN
Standardization of Uveitis Nomenclature
- VH
vitreous haze
Footnotes
Disclosure
Harvey S Uy has received research funding from Santen, Inc. for an unrelated study. The other authors report no conflicts of interest in this work.
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
References
- 1.Krishna U, Ajanaku D, Denniston A, Gkika T. Uveitis: a sight-threatening disease which can impact all systems. Postgrad Med J. 2017;93(1106):766–773. doi: 10.1136/postgradmedj-2017-134891. [DOI] [PubMed] [Google Scholar]
- 2.Fabiani C, Vitale A, Orland I, et al. Impact of uveitis on quality of life: a prospective study from a tertiary referral rheumatology-ophthalmology collaborative uveitis center in Italy. Isr Med Assoc J. 2017;19:478–483. [PubMed] [Google Scholar]
- 3.Thorne J, Skup M, Tundia N, et al. Direct and indirect resource use, healthcare costs and work force absence in patients with non-infectious intermediate, posterior or panuveitis. Acta Ophthalmol. 2016;94(5):e331–e339. doi: 10.1111/aos.12987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chu D, Johnson S, Mallya U, Davis M, Sorg R, Duh M. Healthcare costs and utilization for privately insured patients treated for non-infectious uveitis in the USA. J Ophthalmic Inflamm Infect. 2013;3(1):64. doi: 10.1186/1869-5760-3-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.The Ocular Immunology and Uveitis Foundation Glossary of terms, incidence and prevalence of Uveitis. [Accessed November 10, 2018]. Uveitis.org | OIUF. Published April 10, 2017 Available from: http://www.uveitis.org/patients/education/glossary.
- 6.NIH study shows two treatments for uveitis equally effective National Eye Institute. [Accessed November 5, 2018]. Published August 17, 2011 Available from: https://nei.nih.gov/news/statements/uveitis.
- 7.Jones N. The Manchester Uveitis Clinic: the first 3000 patients, 2: uveitis manifestations, complications, medical and surgical management. Ocul Immunol Inflamm. 2014;23(2):127–134. doi: 10.3109/09273948.2014.968671. [DOI] [PubMed] [Google Scholar]
- 8.de Smet M, Taylor S, Bodaghi B, et al. Understanding uveitis: the impact of research on visual outcomes. Prog Retin Eye Res. 2011;30(6):452–470. doi: 10.1016/j.preteyeres.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 9.Jabs DA, Nussenblatt RB, Rosenbaum JT. Standardization of uveitis nomenclature (SUN) working group. Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol. 2005;140(3):509–516. doi: 10.1016/j.ajo.2005.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenbaum J, Pasadhika S. Update on the use of systemic biologic agents in the treatment of noninfectious uveitis. Biologics Targets Ther. 2014;8:67–81. doi: 10.2147/BTT.S41477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yap YC, Papathomas T, Kamal A. Results of intravitreal dexamethasone implant 0.7 mg (Ozurdex®) in noninfectious posterior uveitis. Int J Ophthalmol. 2015;8:835–838. doi: 10.3980/j.issn.2222-3959.2015.04.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kempen J, Altaweel M, Holbrook J, et al. Randomized comparison of systemic anti-inflammatory therapy versus fluocinolone acetonide implant for intermediate, posterior, and panuveitis: the multicenter uveitis steroid treatment trial. Ophthalmol. 2011;118(10):1916–1926. doi: 10.1016/j.ophtha.2011.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lowder C, Belfort R, Jr, Lightman S, et al. Dexamethasone intravitreal implant for noninfectious intermediate or posterior uveitis. Arch Ophthalmol. 2011;129(5):545. doi: 10.1001/archophthalmol.2010.339. [DOI] [PubMed] [Google Scholar]
- 14.Jaffe G, Martin D, Callanan D, Pearson P, Levy B, Comstock T. Fluocinolone acetonide implant (Retisert) for noninfectious posterior uveitis. Ophthalmol. 2006;113(6):1020–1027. doi: 10.1016/j.ophtha.2006.02.021. [DOI] [PubMed] [Google Scholar]
- 15.You C, Sahawneh H, Ma L, Kubaisi B, Schmidt A, Foster S. A review and update on orphan drugs for the treatment of noninfectious uveitis. Clin Ophthalmol. 2017;11:257–265. doi: 10.2147/OPTH.S121734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jabs D, Rosenbaum J, Foster C, et al. Guidelines for the use of immunosuppressive drugs in patients with ocular inflammatory disorders: recommendations of an expert panel. Am J Ophthalmol. 2000;130(4):492–513. doi: 10.1016/s0002-9394(00)00659-0. [DOI] [PubMed] [Google Scholar]
- 17.Nguyen Q, Hatef E, Kayen B, et al. A cross-sectional study of the current treatment patterns in noninfectious uveitis among specialists in the United States. Ophthalmol. 2011;118(1):184–190. doi: 10.1016/j.ophtha.2010.03.029. [DOI] [PubMed] [Google Scholar]
- 18.Pujari S, Kempen J, Newcomb C, et al. Cyclophosphamide for ocular inflammatory diseases. Ophthalmol. 2010;117(2):356–365. doi: 10.1016/j.ophtha.2009.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Durrani K, Zakka FR, Ahmed M, Memon M, Siddique SS, Foster CS. Systemic therapy with conventional and novel immunomodulatory agents for ocular inflammatory disease. Surv Ophthalmol. 2011;56(6):474–510. doi: 10.1016/j.survophthal.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 20.Denniston A, Barry R, Nguyen QD, Lee R, Murray P. Pharmacotherapy for uveitis: current management and emerging therapy. Clin Ophthalmol. 20141891 doi: 10.2147/OPTH. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ibrahim MA, Sepah YJ, Watters A, et al. One-year outcomes of the SAVE study: sirolimus as a therapeutic approach for UVEitis. Transl Vis Sci Technol. 2015;4(2):4. doi: 10.1167/tvst.4.2.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Napoli KL, Taylor PJ. From beach to bedside: history of the development of sirolimus. Ther Drug Monit. 2001;23:559–586. doi: 10.1097/00007691-200110000-00012. [DOI] [PubMed] [Google Scholar]
- 23.Sehgal SN. Rapamune® (RAPA, rapamycin, sirolimus): mechanism of action immunosuppressive effect results from blockade of signal transduction and inhibition of cell cycle progression. Clin Biochem. 1998;31(5):335–340. doi: 10.1016/s0009-9120(98)00045-9. [DOI] [PubMed] [Google Scholar]
- 24.Curnow SJ, Scheel-Toellner D, Jenkinson W, et al. Inhibition of T cell apoptosis in the aqueous humor of patients with uveitis by IL-6/soluble IL-6 receptor trans-signaling. J Immunol. 2004;173(8):5290–5297. doi: 10.4049/jimmunol.173.8.5290. [DOI] [PubMed] [Google Scholar]
- 25.Agarwal A, Rajagopalan N, Hassan M, et al. Sirolimus for retinal and uveitic diseases. Dev Ophthalmol Retinal Pharmacotherapeutics. 2016;55:276–281. doi: 10.1159/000438951. [DOI] [PubMed] [Google Scholar]
- 26.Dugel PU, Blumenkranz MS, Haller JA, et al. A randomized, dose-escalation study of subconjunctival and intravitreal injections of sirolimus in patients with diabetic macular edema. Ophthalmol. 2012;119(1):124–131. doi: 10.1016/j.ophtha.2011.07.034. [DOI] [PubMed] [Google Scholar]
- 27.Nguyen QD, Merrill PT, Clark WL, et al. Intravitreal sirolimus for noninfectious uveitis: a phase III Sirolimus study Assessing double-masKed Uveitis TReAtment (SAKURA) Ophthalmol. 2016;123(11):2413–2423. doi: 10.1016/j.ophtha.2016.07.029. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen QD, Ibrahim MA, Watters A, et al. Ocular tolerability and efficacy of intravitreal and subconjunctival injections of sirolimus in patients with non-infectious uveitis: primary 6-month results of the SAVE study. J Ophthalmic Inflamm Infect. 2013;3(1):32. doi: 10.1186/1869-5760-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen QD, Sadiq MA, Soliman MK, et al. The effect of different dosing schedules of intravitreal sirolimus, a mammalian target of rapamycin (mTOR) inhibitor, in the treatment of non-infectious uveitis (An American Ophthalmological Society Thesis) Trans Am Ophthalmol Soc. 2016;114:T3, 1–14. [PMC free article] [PubMed] [Google Scholar]
- 30.Higgins JPT, Green S. Cochrane handbook for systematic reviews of interventions. Cochrane Handbook for Systematic Reviews of Interventions. [Accessed November 10, 2018]. Published March 2011 Available from: http://handbook-5-1.cochrane.org/
- 31.Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Int J Surg. 2010;8(5):336–341. doi: 10.1016/j.ijsu.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 32.Higgins JPT, Altman DG, Gotzsche PC, et al. The Cochrane Collaboration tool for assessing risk of bias in randomised trials. BMJ. 2011;343(2):d5928. doi: 10.1136/bmj.d5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berger V, Alperson S. A general framework for the evaluation of clinical trial quality. Rev Recent Clin Trials. 2009;4(2):79–88. doi: 10.2174/157488709788186021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jadad AR, Moore R, Carroll D, et al. Assessing the quality of reports of randomized clinical trials: is blinding necessary? Control Clin Trials. 1996;17(1):1–12. doi: 10.1016/0197-2456(95)00134-4. [DOI] [PubMed] [Google Scholar]
- 35.Sarris J, Byrne GJ. A systematic review of insomnia and complementary medicine. Sleep Med Rev. 2011;15(2):99–106. doi: 10.1016/j.smrv.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 36.Freeman MF, Tukey JW. Transformations Related to the Angular and the Square Root. The Annals of Mathematical Statistics. 1950;21(4):607–611. doi: 10.1214/aoms/1177729756. [DOI] [Google Scholar]
- 37.Dersimonian R, Laird N. Meta-analysis in clinical trials. Controlled Clinical Trials. 1986;7(3):177–188. doi: 10.1016/0197-2456(86)90046-2. [DOI] [PubMed] [Google Scholar]
- 38.Sen HN, Larson TA, Meleth AD, Smith WM, Nussenblatt RB. Subconjunctival sirolimus for the treatment of chronic active anterior uveitis: results of a pilot trial. Am J Ophthalmol. 2012;153(6):1038–1042. doi: 10.1016/j.ajo.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vigil EM, Sepah YJ, Watters AL, et al. Assessment of changes in quality of life among patients in the SAVE study – sirolimus as therapeutic approach to uVEitis: a randomized study to assess the safety and bioactivity of intravitreal and subconjunctival injections of sirolimus in patients with non-infectious uveitis. J Ophthalmic Inflamm Infect. 2015;5(1):13. doi: 10.1186/s12348-015-0044-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shanmuganathan VA, Casely EM, Raj D, et al. The efficacy of sirolimus in the treatment of patients with refractory uveitis. Br J Ophthalmol. 2005;89(6):666–669. doi: 10.1136/bjo.2004.048199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harbour R, Miller J. A new system for grading recommendations in evidence based guidelines. BMJ. 2001;323(7308):334–336. doi: 10.1136/bmj.323.7308.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Santen Pharmaceutical Co., Ltd Santen announces phase III SAKURA program topline results in patients with non-infectious Uveitis of the posterior segment PR Newswire: news distribution, targeting and monitoring Published November 28, 2016 Available from: https://www.prnewswire.com/news-releases/santen-announces-phase-iii-sakura-program-topline-results-in-patients-with-non-infectious-uveitis-of-the-posterior-segment-300368781.htmlAccessed November 10, 2018a
- 43.Santen Pharmaceutical Co A phase III study assessing the efficacy and safety of intravitreal injections of 440 ug DE-109 for the treatment of active, non-infectious uveitis of the posterior segment of the eye – full text view. Search of: Spain – list results – Clinicaltrials.gov. [Accessed November 10, 2018]. Available from: https://clinicaltrials.gov/ct2/show/NCT03711929.
- 44.Blair J, Barry R, Moore DJ, Denniston AK. A comprehensive review of mTOR-inhibiting pharmacotherapy for the treatment of non-infectious uveitis. Curr Pharm Des. 2017;23:20. doi: 10.2174/1381612823666170111125550. [DOI] [PubMed] [Google Scholar]
- 45.Nguyen QD, Merrill PT, Sepah YJ, et al. Intravitreal sirolimus for the treatment of noninfectious uveitis. Ophthalmol. 2018;125(12):1984–1993. doi: 10.1016/j.ophtha.2018.06.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.
Inclusion and exclusion criteria1
| Inclusion criteria | |
|---|---|
|
| |
| 1. ≥18 years of age | |
| 2. Having diagnosis of uveitis determined by the investigator to be noninfectious based on the patient’s medical history, history of present illness, ocular examination, review of systems, physical examination, and any relevant, pertinent laboratory evaluations | |
| 3. Patients with active uveitis, defined as having at least 1+ VH and/or at least 1+ vitreous cell count (SUN scale), and | |
| 4. are receiving no other treatment; or | |
| 5. are receiving prednisone ≥10 mg/day and/or at least one other systemic immunosuppressants | |
| 6. Patients with inactive disease, defined as having 0.5+ VH or less and a grade of 0.5+ vitreous cell count or less (SUN scale), and are receiving prednisone <10 mg/day and/or at least one other systemic immunosuppressants | |
| 7. Having posterior uveitis, intermediate uveitis, or panuveitis; for panuveitis, if an anterior component is present, it must be less than the posterior component | |
| 8. Sufficient inflammation to require systemic treatment | |
| 9. BCVA of 20/400 or better in both eyes | |
|
| |
| Exclusion criteria | |
|
| |
| Non-ocular | Ocular |
|
| |
| 1. Allergy or hypersensitivity to sirolimus or fluorescein dye 2. Immunosuppressive therapy within 30 days of day 0 3. Patients who are receiving strong inducers of CYP3A4 and P-gp and have any recent infection within 30 days of baseline 4. Immunocompromised patients 5. History of CMV infection or clinical evidence of active CMV infection at baseline 6. Malignancy in remission for <5 years prior to study 7. History of other diseases, metabolic dysfunction, physical examination finding, or clinical laboratory finding giving reasonable suspicion of a disease condition that contraindicates the use of an investigational drug might affect the interpretation of the results of the study or renders the patient at high risk for treatment complications 8. Females who are pregnant or lactating and females of child-bearing potential who are not using adequate contraceptive precautions 9. Sexually active males with partners of child-bearing potential who are not using adequate contraceptive precautions |
1. Patients with bilateral uveitis who are receiving systemic IMT for the treatment of the fellow eye and cannot be controlled with standard local therapies alone 2. Any significant ocular disease that could compromise vision in the study eye 3. Any IVT injections or posterior subtenon’s steroids within 90 days prior to day 0 4. Intraocular surgery within 90 days prior to day 0 5. Capsulotomy within 30 days prior to day 0 6. History of vitreoretinal surgery or scleral buckling within 90 days prior to day 0 7. Any ocular surgery anticipated within the first 180 days following day 0 8. IOP ≥25 mmHg (glaucoma patients maintained on no more than two topical medications with IOP <25 mmHg are allowed) 9. Pupillary dilation inadequate for quality stereoscopic fundus photography 10. Media opacity that would limit clinical visualization, IVFA, or OCT evaluation 11. Presence of any form of ocular malignancy History of herpetic infection in the study eye or adnexa 12. Presence of known active or inactive toxoplasmosis in either eye 13. Ocular or periocular infection in either eye |
Abbreviations: BCVA, best-corrected visual acuity; CMV, cytomegalovirus; IMT, immunomodulatory therapy; IVFA, intravenous fluorescein angiography; IVT, intravitreal; OCT, optical coherence tomography; SUN, Standardization of Uveitis Nomenclature; VH, vitreous haze.
Table S2.
CENTRAL search strategy
| Date Run: | 11/05/2018 05:15:28 | |
|---|---|---|
| ID | Search | Hits |
| #1 | MeSH descriptor: [Uveitis] explode all trees | 537 |
| #2 | Uveitic | 110 |
| #3 | #1 OR #2 | 615 |
| #4 | Sirolimus | 2,970 |
| #5 | Rapamycin | 1,821 |
| #6 | mTOR inhibitor | 534 |
| #7 | #4 OR #5 OR #6 | 3,947 |
| #8 | #3 AND #7 | 7 |
| #9 | (sirolimus): ti,ab,kw AND (uveitis): ti,ab,kw | |
| (Word variations have been searched) | 23 | |
| #10 | #8 OR #9 | 23 hits |
Table S3.
PMC search strategy
| Date Run: 11/06/2018 09:22:29 | |
|---|---|
| (“uveitis”[MeSH Terms] OR “uveitis”[All Fields]) AND | 247 hits |
| (“sirolimus”[MeSH Terms] OR “sirolimus”[All Fields]) | |
| URL: https://www.ncbi.nlm.nih.gov/pmc/?term=(%22uveitis%22%5BMeSH+Terms%5D+OR+%22uveitis%22%5BAll+Fields%5D)+AND+(%22sirolimus%22%5BMeSH+Terms%5D+OR+%22sirolimus%22%5BAll+Fields%5D)&cmd=DetailsSearch |
Table S4.
ClinicalTrials.gov search strategy
| Date Run: 11/7/2018 07:15:18 | |
|---|---|
| CONDITION or DISEASE: Uveitis | 7 hits |
| OTHER TERMS: Sirolimus | |
| (Uveitis AND Sirolimus) |