

Abstract
Single-cell technologies that can quantify features of individual cells within a tumor are critical for treatment strategies aiming to target cancer cells while sparing or activating beneficial cells. Given that key players in protein networks are often the primary targets of precision oncology strategies, it is imperative to transcend the nucleic acid message and read cellular actions in human solid tumors. Here, we review the advantages of multiplex, single-cell mass cytometry in tissue and solid tumor investigations. Mass cytometry can quantitatively probe nearly any cellular feature or target. In discussing the ability of mass cytometry to reveal and characterize a broad spectrum of cell types, identify rare cells, and study functional behavior through protein signaling networks in millions of individual cells from a tumor, this review surveys publications of scientific advances in solid tumor biology made with the aid of mass cytometry. Advances discussed include functional identification of rare tumor and tumor-infiltrating immune cells and dissection of cellular mechanisms of immunotherapy in solid tumors and the periphery. The review concludes by highlighting ways to incorporate single-cell mass cytometry in solid tumor precision oncology efforts and rapidly developing cytometry techniques for quantifying cell location and sequenced nucleic acids.
Keywords: immune cell, immunotherapy, mass cytometry, proteomics, signaling, single cell
THE NEED FOR SINGLE-CELL PROTEIN ANALYSES IN CANCER
Currently, genome-based oncology drives cancer research to better prognosticate and inform treatment of individual patients. For example, in glioblastoma, genomic efforts have identified mutations in the IDH1/2 genes and epigenetic silencing of the MGMT DNA-repair gene as important predictors of survival [1]. Genomic studies of glioblastoma have also revealed genetic alterations in potentially targetable receptor tyrosine kinase (RTK) pathways in nearly two-thirds of patients [2]. Leveraging these discoveries, several genome-based precision oncology clinical trials have been performed in solid tumors. In these, up to a third of the patients with advanced solid tumors who received a targeted therapy matched to their genomic alteration had desirable outcomes [3–7]. However, progress still needs to be made. For example, molecularly targeted therapy for multiple advanced solid tumors based on large-scale genomic profiling did not improve outcomes compared to standard-of-care therapy in the first multi-institutional, randomized controlled trial SHIVA [8], and trials developed based on mutations in RTK pathways in glioblastomas also have not improved survival [9–12]. Although other clinical trials are underway [13], these initial results highlight progress and challenges in genomic oncology and invite a complementary understanding of post-translational cellular functions that is expected to significantly enhance therapies of solid tumors.
Cellular diversity in tumors hinders genomics-guided targeted molecular therapy from achieving widespread success [13, 14]. Virtually all forms of solid cancers are currently diagnosed histopathologically and contextualized with a grade and stage that estimates the tumor’s malignant potential and extent of burden in an individual. However, there is a growing movement to enhance this qualitative classification approach based on quantitative, single-cell tools. In both blood cancers and solid tumors, single-cell approaches have revealed striking clinical and cellular differences within groups considered to be distinct entities according to pathological classification schemes. Cell biology and clinical outcome can be correlated in ways that do not align with traditional pathological classification; features including phospho-protein signaling responses [15–19] or presence of tumor-infiltrating immune cells [20] can quantitatively stratify patient survival. In light of this, precision oncology must now measure and incorporate information from diverse tumor cell types in molecular targeted therapies.
Single-cell approaches stand ready for this challenge, as most cellular features can now be quantified [21], and cells in tumors can be readily resolved and deeply characterized. These approaches can uncover subpopulations of cells that may be responsible for a tumor’s adaptive potential and therapy resistance. Although single-cell genomic approaches can provide critical insights into the genetic, epigenetic, and transcriptional bases for cancer drug resistance [22, 23], genetic biomarkers by themselves do not comprehensively inform effective therapies [13]. For example, an integrative proteomics effort in prostate cancer using high-throughput mass spectrometry revealed that proteomic changes are not reliably predicted by gene copy number, DNA methylation, and RNA expression [24]. In fact, alternations in pathways, including metabolic shifts in the tricarboxylic acid cycle, not discovered by RNA expression were revealed through a proteomic approach [24]. Many of the resistance mechanisms known are also mediated by post-translational changes in proteins within individual cells [25]. Thus, an integrated approach that combines proteomic and genomic information from millions of individual tumor cells may be able to guide combination targeted therapies and effectively anticipate resistance mechanisms (Fig. 1). Such an approach has the potential to offer a greater understanding of the regulation of cancer cell identity, which is determined by a dynamic interplay of nucleic acids and proteins, for therapeutic perturbations [26]. Currently, clinical applications of protein based single-cell analyses are lacking in solid tumors and must be integrated with single-cell genomic approaches for the most comprehensive, targeted approach to precision oncology in solid tumors. Here, we review the utility of mass cytometry for single-cell quantitative protein analyses in solid tumors and discuss its potential to reveal single-cell biology for precision oncology approaches.
Figure 1. Single-cell technologies quantify different cellular features and targets.
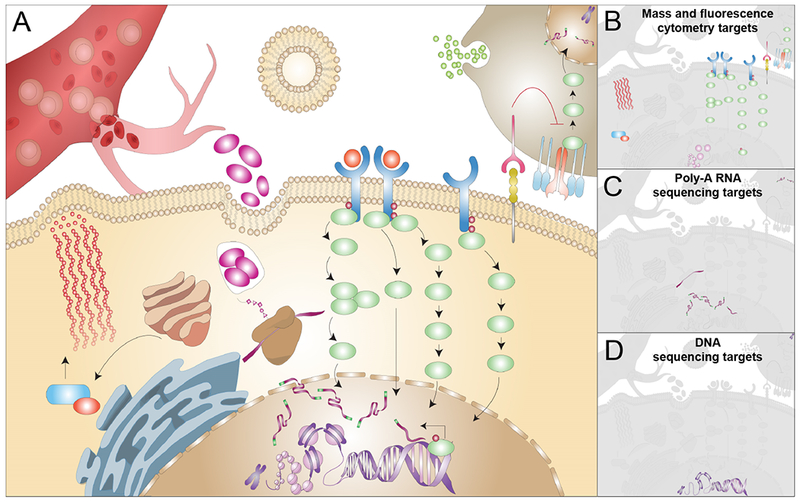
(A) A cancer cell situated in its tumor microenvironment is depicted as surrounded by an immune cell and a blood vessel. The dynamic functions each tumor cell executes result from both intrinsic programs and the diverse, contrasting signals these cells encounter across the tumor microenviroment. In order to understand tumor cell behavior, single-cell technologies commonly probe one of the three major classes of biomolecules responsible for carrying out cellular functions. Examples of common single-cell molecular features that can be quantified include (B) various protein states by mass and fluorescence cytometry, (C) mRNA, and (D) DNA by single-cell RNA and DNA sequencing technologies. Note that platforms may be adapted to capture more than one molecular class. By itself, each approach typically provides information on only a fraction of cellular functions.
ADVANTAGES OF MASS CYTOMETRY FOR SINGLE-CELL PROTEIN ANALYSES
Technical overview, throughput, and cost
Cytometry by inductively coupled plasma time-of-flight mass spectrometry (mass cytometry; also referred to using the Fluidigm instrument name, CyTOF®, Fluidigm Corporation, South San Francisco, CA, USA) is a single-cell method that enables highly multiplexed and quantitative measurements of proteins and their modifications. Sample preparation and protein detection are similar to the more widely used fluorescence flow cytometry with a few important differences (Table 1): (a) Instead of fluorophores, antibodies used to detect proteins are typically tagged with unique, stable, heavy-metal isotopes (e.g., lanthanides) not found in biological samples; (b) in traditional flow cytometry, cells may be identified in part by their intrinsic light scatter characteristics; whereas, in mass cytometry, the cells are stained with a cell marker, such as rhodium- or iridium-conjugated DNA intercalator [27] or antibodies against common proteins, such as total histone H3 [28], CD298 [29], or CD45 [30]; (c) cells are typically fixed prior to their analysis with mass cytometry and are consumed, in contrast to fluorescence-activated cell sorting, as each cell is vaporized into a cloud of ionized single atoms in the mass cytometer. Typically, atoms with a mass of < 75 Da are filtered by a quadrupole, leaving metals in a certain range of charge and mass chosen to sensitively detect the mass tag reporters and minimize unwanted signals. The ionized atoms (all with a similar charge) are accelerated by an electrical potential towards a detector. The ion ‘time-of-flight’ depends on its mass and is recorded during the acceleration. ‘Events’, such as cells or beads, are parsed and a sum of detected ions is recorded for each chosen measurement channel (corresponding to a unique mass). Thus, ion counts per cell provide a quantification of a probe, such as a metal-labeled antibody. Because of the sample introduction design in the original generation of mass cytometers, up to 60% of the loaded cells could be lost [31, 32], and the time required to detect all ions in a single cell ion cloud while maintaining single cell resolution limits the rate of single cells analyzed to 1000–2000 cells·s−1 (or ~ 3.6–7.2 million cells in an hour) [33]. In practice, 500 cells or events per second is typical as it reduces signal overlap between events in sequence. However, the ability to label cells in a given sample with metals not used by the 40 or more metal-conjugated antibodies in a panel allows for barcoding capabilities that greatly increase the number of samples that can be simultaneously analyzed. Typically, up to 20 samples can be run together in a single tube [34]. Such an approach enables simultaneous quantitative analyses and efficient resource utilization by reducing time and reagent costs. Several reference protocols are available on how to perform single-cell mass cytometry assays [35–37], including in solid tissues and tumors [38]. General computational methods for analysis and interpretation of mass cytometry data have been recently reviewed [31, 39–41].
Table 1.
Comparison of common single cell flow cytometry technologies by probe type
| Mass cytometry | Fluorescence cytometry | Single cell poly-A RNA-seq | |
|---|---|---|---|
| Detection method | Time-of-flight of ionized heavy elemental isotopes | Emitted light and light scatter | Nucleic acid sequence |
| Example probes | Metal-conjugated antibodies, metal-conjugated small molecules | Fluorochrome-conjugated antibodies, fluorescent molecules | Poly-A targeted oligonucleotides, antibody-conjugated oligonucleotides [144] |
| Example cellular features and targets measured | Proteins, phospho-proteins, chromatin modifications [124], RNA transcripts [131–133], platinum drug uptake [128, 129], cell cycle status and DNA synthesis [130], cell size [125], apoptosis [48], and viability [27] | Proteins, phospho-proteins, chromatin modifications [145], RNA transcripts [146], fluorescent drug uptake [147], metabolism and redox state [148, 149], cell cycle status and DNA synthesis [150], cell size and granularity, apoptosis [151] and viability [152] | Poly-adenylated RNA transcripts, CITE-seq probes [144] |
| Minimum cells per sample needed at start of protocol | 50 000 cells | 50 000 cells | 200 cells [153]a |
| Cell capture rate | 30–60% [31, 32] | > 95% | 5–65% [154] |
| Target capture | > 95% | > 95% | 10–40% [154–156] |
| Example of analyzed cell events per study | 2 000 000 cells per study | 20 000 000 cells per study | 5000 cells per study [157] |
| Resolution, arcsinh (max / error)b | ~6-9 e.g., arcsinh(12,000/5) | ~5-9 e.g., arcsinh(262, 144/150), arcsinh(4,194,300/6,000)c | ~2-3 e.g., arcsinh(10/1) |
| Cell throughput | Up to 104 cells·s–1 [33] | Up to 105 cells·s–1 [31] | Up to 104 cells·s–1 [155] |
| Features/available channels | 50/200 channels | 30/64 channelsc | ~ 30 000/N/A |
| Degree of crosstalk between parameters | 3% (range: 0–8.6%) [55] | 19.5% (4.9–51.1%) [56] | N/A |
| Accessibility | Major research institutions | > 20 features: major research institutions; ~ 4 color common | Major research institutions |
| Total cost | $0.004-$0.01/cell [31] | < $0.001/cell | $0.05-$3.00/cell [158] |
Number calculated based on 10 assumed number of cell types, rarest cell type makes up 10%, and at least 10 cells are desired per cell type; see reference for a sample size calculator.
Max is an example maximum range of the instrument scale; error is the typical error for that scale.
Includes capabilities of novel instruments such as Cytek™ Aurora (Cytek Biosciences), ZE5™ (Bio-Rad), and FACSymphony™ (BD)
For common mass cytometry assays, the total cost per cell is estimated to be $0.01/cell (United States Dollar, USD) [42] to $0.004/cell USD in our experience [43], including personnel time, antibodies, and instrument time, which is currently significantly lower than single-cell RNA sequencing [42, 44] (Table 1). Although one report estimated the cost of mass cytometry to be about 1000 fold higher (~ $35/cell USD [45]), this is likely a gross overestimation as a routine mass cytometry pilot experiment on 12 samples might cost ~ $3500, not $35 000 000 USD.
Multiplexing capability
Isotopic discrimination of more than 100 elemental masses confers multiplexing capability to mass cytometry. In conventional fluorescence flow cytometry, the ability to increase parameterization can be limited by the spectral overlap of fluorophores. Given the mass cytometer’s precise isotopic discrimination of elemental isotopes, the current practical limitation to measurement channels is the availability of stable, non-biological, elemental isotopes that fall into a common mass range for detection and are above the background caused by biological elements and the argon carrier gas used. Presently, there are 37 channels for commercial mass-tagged antibodies provided by Fluidigm (South San Francisco, CA, USA). The use of rhodium for viability and iridium for cell marking brings the feature list to 39 different parameters that can be routinely measured in each of the typically millions of single cells in a single experiment [42]. Although a large majority of the elemental isotopes used to conjugate antibodies are lanthanides, other metals such as indium, yttrium, and bismuth can also be used. See protocol [46] for conjugating antibodies with these heavy-metals. Beyond this, users can employ quantum dot labeled antibodies to gain additional channels [47], iodine for cell cycle tracking [48], and palladium [49] and platinum [50] for cellular barcoding, allowing advanced users to exceed 50 measurement channels per cell. Of note, several advanced fluorescence flow cytometers which have emerged as potential competitors of mass cytometry have a multiplexing capability of up to 50 parameters (Table 1). These include FACSymphony™ (BD Biosciences, San Jose, CA, USA) and ZE5™ (Bio-Rad Laboratories, Inc., Hercules, CA, USA), which enhance conventional detection technology, and Cytek™ Aurora (Cytek Biosciences, Fremont, CA, USA) and SP6800 (Sony Biotechnology, San Jose, CA, USA), which detect and deconvolute a known ‘spectral signature’ for relative quantification of fluorescent probes.
Sensitivity, contamination, and noise
Detection in mass cytometry is relatively free from reporter instability (such as fluorophore quenching) and sensitivity variability. The elemental mass tags used are detected with relatively comparable sensitivity, differing by only 2–4 fold across the mass spectrum presently employed, as opposed to fluorophores, whose sensitivities differ by 10–50 fold [42, 51]. Currently, 300–1000 copies of a single immuno-targeted protein can be detected in a single cell, which is slightly lower than the sensitivities of the more efficient fluorophores [52]. This limit is placed by the number of isotope atoms present on a single antibody molecule (currently ~ 100–140 atoms per antibody) [52] and likely to be overcome with technologies that increase the atom carrying capacity [53, 54].
Isotopic discrimination in mass cytometry results in data which are less significantly contaminated with issues of signal overlap or spillover into another reporter channel. The main causes of detection channel crosstalk are (a) the instrument’s intrinsic abundance sensitivity (related to small time-of-flight differences of highly abundant probe ions, usually < 4%); (b) isotopic impurities in preparations of mass isotope tags (presence of undesired, naturally occurring isotopes, usually < 4%); and, (c) ion oxidation resulting in a species 16 Da more massive (usually < 2%). However, compensation or correction for unintended crosstalk between measurement channels is not required in mass cytometry. Because an instrument’s abundance sensitivity and isotopic impurities are quantitatively known and consistent, for most masses the total crosstalk in a full, properly designed panel is minimal [34]. For example, the average crosstalk for 35 channels in mass cytometry was reported to be 2.7% (range: 0–8.6%] [55]. Nonetheless, a non-negative least squares approach has been proposed to compensate for these sources of crosstalk [55]. The lack of crosstalk in mass cytometry contrasts with fluorescence cytometry, especially in panels employing more than 20 fluorescent probes and traditional approaches that do not employ spectral deconvolution. In a cutting-edge multiplexed fluorescence cytometry experiment, the average crosstalk for 27 channels was reported to be 19.5% (range: 4.9–51.1%] [56] (Table 1). Although fluorescence crosstalk can be partially corrected, crosstalk above ~ 10% can negatively affect the ability of computational tools to distinguish populations. By detecting and deciphering the spectral signature, novel flow cytometers such as the Aurora and SP6800 are designed to circumvent spillover and use the distinct spectral pattern of each dye to improve resolution per channel in high-dimensional panels.
Mass cytometry data are relatively free from background noise, due to negligible abundance of the heavy metals used in biological samples, and from auto-fluorescence seen in fluorescence flow cytometry of native cells or due to fluorescing exogenous agents in a treated sample [51]. This allows for high resolution of molecules that are in low abundance and therefore could be obscured by background noise on other platforms. Furthermore, minimal technical noise is introduced into mass cytometry data compared to, for example, single-cell RNA sequencing. Although this platform lacks crosstalk issues, adjustment for technical noise and signal dropout bias are critical for reliable analyses [43, 57]. For example, a statistical model of recent single-cell RNA sequencing platforms used external RNA spike-ins to predict that up to 80% of stochastic allele-specific expression can be attributed to technical noise [58], and noted that up to 95% of measurements can be zeroes [43].
Mass cytometry data can be reliably comparable across conditions and time using metal-embedded internal bead standards spiked-in with samples [59] or by spiking patient samples with CD45-barcoded reference peripheral blood mononuclear cells (PBMCs) to better normalize experimental variability and differential antibody staining [60]. Low technical noise and adjustment of internal bead intensities make normalization of mass cytometry data feasible, reduce batch effects, and facilitate accurate comparisons between samples over time measured on the same instrument [59] as well as between instruments [61]. This reproducibility of mass cytometry measurements across instruments and laboratories invites collaborative science and enables critical comparisons. Feasibility of multicenter mass cytometry assays was recently demonstrated across six international centers, which were consistent in the performance of mass cytometry assays on healthy control human PBMCs [62]. The suitability of mass cytometry for large patient cohort studies or multicenter studies is a major advantage over single-cell technologies such as RNA sequencing [22, 63].
A large set of recently developed computational tools [64–70] (and the basic analysis approaches well summarized in references [31, 39–41]) allow optimal characterization of rare, new, or unexpected subpopulations in the high-dimensional datasets generated by a multiplexed mass cytometry assay which could otherwise remain concealed or sub-optimally characterized with conventional exploration using biaxial gating. For example, Levine et al. [18] used mass cytometry to detect rare subpopulations of cells in pediatric acute myeloid leukemia samples constituting as low as 0.06% of a sample. Impressively, this limit was pushed by a recent study that mapped the human dendritic cell lineage, where certain lineages, such as the pre-dendritic cell, comprise ~ 0.001% of PBMCs [71]. Furthermore, rare subsets can be discovered using small samples. For example, multiple lineages of immune cells were detected in healthy human skin biopsy samples of 1 × 106 cells [72]. Detection of plasmacytoid dendritic cells (~ 0.2% in PBMCs) was possible in a sample with as few as 3000 PBMCs [72]. This depth makes mass cytometry very suitable to study cellular diversity in small samples compared to single-cell RNA sequencing, which requires assaying a greater number of single cells [22] (the largest number of single cells analyzed by single-cell RNA sequencing to date falls short of half a million [73]). Assuming the presence of only binary phenotypic markers (i.e. present or absent, ignoring levels of expression), mass cytometry is theoretically capable of distinctly determining cell types on the order of billions [47]. The results and discoveries made through mass cytometry can also be adopted to other platforms, such as fluorescence flow cytometry.
CHALLENGES UNIQUE TO THE STUDY OF SOLID TUMORS
Mass cytometry is not yet as widely used to study solid tissues and tumors despite its new prominence in the study of ‘liquid’ tissues or tissues that readily disaggregate into single-cell suspensions (i.e. hematopoietic and lymphoid tissues) [34, 38]. For mass cytometry to become a more prominent choice of investigational method for the study of solid tissues and tumors at least two obstacles must be overcome: access to tumor specimens and generation of viable single-cell suspensions.
A common obstacle for the study of solid tumors, and especially for precision oncology guided treatment, is access to tumor samples for research. This requires tumor biopsies to be collected at critical timepoints in patients’ clinical course. Unlike hematological malignancies which can be sampled in an outpatient setting at the desired clinical timepoints, solid tumor biopsies often entail major operations associated with significantly greater risks. Therefore, multiple operations to guide biomarker discovery, post-therapy response, and cellular or molecular surveillance of solid tumors are rarely performed. However, based on our experience, we offer two workarounds: (a) Pursue collaborations between clinicians and investigators to increase access to the tumor samples. The regular institutional interdisciplinary oncology conferences (often called tumor boards) and patient-centered conferences are excellent forums for clinicians and researchers to express their desire for collaborating in precision oncology efforts and to cross-pollinate ideas. Initiating or joining a multicenter effort is another possible avenue. (b) If access to fresh tissue is not feasible, using clinical archives of embedded human tissues is another solution. Formalin-fixed, paraffin-embedded (FFPE) tissues can be disaggregated into single cells for analysis using mass cytometry in a technique called FFPE-DISSECT [74, 75], or they can be used directly for another novel technique, imaging mass cytometry (IMC) [76] (discussed below).
Solid tumor samples require disaggregation into single-cell suspensions prior to analysis by mass cytometry. This process is critical; and if done without sufficient care, phenotypic markers or cell subsets may be lost and cellular biology may not accurately reflect the biology intended to be studied. Samples may also be sensitive to handling and storage conditions [77]. We and others recently introduced a standardized procedure for preparing viable single-cell suspensions from solid tumors (gliomas, melanoma, lung cancer [28, 38] and ovarian tumors [78]) for mass cytometry that maintains cellular diversity. While these protocols serve as important starting points, ideal disaggregation conditions may vary for other solid tissues or tumors. Therefore, we recommend carefully designed, well-controlled investigations to design and optimize a protocol for disaggregation and storage of the desired tumor samples into single-cell suspensions.
MASS CYTOMETRY TO REVEAL CANCER BIOLOGY BEYOND GENOMICS
How genomic alternations could lead to neoplastic growth and therapy resistance in cells requires shifting experimental focus to include proteins. They are the executors of the altered genetic code that link it to biological functions and lend functional identity to cancer cells. These proteins occupy a large portion of the cellular landscape and are presently beyond the investigative reach of single-cell genomic sequencing technologies (Fig. 1). Here, we review recent studies that exemplify the suitability of mass cytometry (a) to unveil tumor and immune microenvironment composition and identify novel immunotherapy targets and biomarkers of therapy response; and (b) to understand signaling networks within individual cancer cells.
Mass cytometry identifies rare functional immune cell subsets in the tumor microenvironment
Tumors can exploit the programmed cell death protein-1 (PD-1)/programmed cell death-ligand 1 (PD-L1) and cytotoxic T lymphocyte-associated protein-4 (CTLA-4) pathways to evade immune surveillance [79]. Immunotherapy targeting these pathways (‘immune checkpoint inhibitors’) has recently become an innovative addition to the armamentarium of cancer therapies [80]. However, its success is not uniform across all forms of cancers, and more importantly, primary and acquired resistance to immunotherapy limit its revolutionary therapeutic potential [79]. Understanding the immune cell make-up in tumors and the basis for variable immunotherapy response and resistance is necessary. Mass cytometry can be used to generate initial maps of the immune landscape as well as identify tumor-specific and systemic responses to immunotherapy.
An initial, general overview of solid tumor immune cell maps (generated in clear cell renal cell carcinoma [63, 81], lung cancer [82, 83], colorectal adenocarcinoma [83], hepatocellular carcinoma [84, 85], glioma [86, 87], malignant pleural mesothelioma [88], and melanoma [89]) convey a tumor-specific immune cell landscape dominated by T cells and monocytes/macrophages with small changes in other cell subtypes between tumor and normal tissues. However, granular examinations of these maps revealed rare and/or dysfunctional tumor-infiltrated immune cell subsets that were significantly associated with clinical outcomes. For example, an abundance of Treg cells, known to suppress anti-tumor immunity [90], and CD8+ T cells were noted in an ‘exhausted’ state, denoted by their high PD-1 expression among other markers indicating persistent antigen stimulation [91]. Furthermore, a subset of the tumor-infiltrated CD8+ T cells which lack CD39 expression were noted in lung and colorectal tumors to be simply bystander cells [83]. They were not specific for tumor antigens and lacked hallmarks of ‘exhaustion’ [83]. Quantification of the proportion of CD39+CD8+ T cells in a tumor may inform immunotherapy response. The tumor-infiltrated natural killer cells had impaired cytolytic activity in several different cancer types [82, 84, 86]. CD141+dendritic cells, which are known to potentiate anti-tumor immunity by presenting tumor antigens to CD8+ T cells [82, 92], were noted to be depleted in lung cancer. Regulatory T cells and CD8+ resident memory T cells were more enriched, had greater PD-1 expression, and were functionally more immunosuppressive in Hepatitis B virus-related hepatocellular carcinoma than in non-virus-related hepatocellular carcinoma [85]. Subsets of tumor-infiltrative CD14+ macrophages in hepatocellular carcinoma with an ‘exhausted-like’ phenotype (i.e., expressing PD-L1) were also discovered [84]. In a murine model of prostate cancer, polymorphonuclear myeloid-derived suppressor cells (CD11b+Gr1+) were the most prominent tumor-infiltrated immune cells and served as therapeutic targets for their critical role in tumor progression [93]. Infiltrating CD38+ myeloid-derived suppressor cells in renal cancer were found to correlate with immunosuppression [63].
With the aid of mass cytometry, insights into tumor-specific and systemic effects of immunotherapy have also been obtained. Analyses of tumor-infiltrating T cell responses to anti-PD-1 immunotherapy in murine solid tumor models [94, 95], human melanoma [94], and human liver cancer [84] revealed expansion of the ‘exhausted’-like CD8+ T cells. Although anti-CTLA-4 immunotherapy evoked a similar response, it also distinctly resulted in expansion of ICOS+ TH1-like CD4+ T cells, suggesting distinct cellular mechanisms of these two immunotherapies [94]. An even deeper analysis in a murine sarcoma model revealed immunotherapy induces tumor-infiltrated neoantigen (tumor-specific mutant antigen)-specific CD8+ T cell expansion selectively in the tumor but not in the peripheral compartments [95]. This illuminated a basis for the increased benefit of immunotherapy in solid tumors with increased mutational burden [95, 96].
Mass cytometry analysis of the peripheral compartment also led to several insights into the effects of immunotherapy in solid tumors. Systemic immune network analyses in murine models of triple-negative breast cancer and melanoma patients revealed that maintenance of systemic immunity is required for effective anti-tumor immunity following immunotherapy, even for local (intratumoral) therapy, which results in post-therapy upregulation of PD-L1 known to mediate immunosuppression in distal tumors [97]. A key finding in this study was the identification of a specific subset of an activated, effector memory CD4+ TH1 cell that conferred immunoprotection against tumors [97]. However, the relationship between the presence of circulating effector memory CD4+ T cells and anti-PD1 immunotherapy response in melanoma patients is debated: one found a lower frequency of these cells with response [98], another found increase of these cells correlated with response [99], and yet another claimed no correlation with response [100]. T cells in melanoma patients who responded to anti-PD1 immunotherapy demonstrated an upregulation of CTLA-4 and CD69 [98]. CD8+ T cells specifically upregulated CD62L, CD69, CCR4, and granzyme B and were enriched in the tumors, suggesting their stimulated migratory capacity [98]. Cells resembling CD8+ central memory T cells also showed a proliferative burst [98]. Interestingly, in recurrent glioblastoma patients from a recent clinical trial who did not respond to anti-PD-1 immunotherapy, immunotherapy drove the peripheral Treg cells to become more ‘exhausted’-like and similar to the tumor-infiltrated Treg cells [87, 101].
Overall, mass cytometry was instrumental in the above detailed identifications of rare tumor-infiltrating and peripheral immune cells to uncover functional responses to immunotherapy. These studies leveraged its multiplexing capability using carefully designed panels of over 20–30 markers that functionally characterized cell subpopulations and used its power to detect and characterize rare cell subtypes to quantitively expose a wide range of phenotypic diversity of tumor-infiltrated immune cells. Most importantly, these studies indicate that mass cytometry can potentially be clinically employed to predict therapy response, especially to immunotherapy.
Mass cytometry phenotypically characterizes cellular diversity of tumors
Although thus far mass cytometry has been most frequently applied to understand the immune compartment of the tumor, recent efforts [78, 102] have used it to reveal cellular heterogeneity of the tumor itself. For example, the cellular heterogeneity of high-grade serous ovarian cancer was recently exposed with mass cytometry [78]. Although individual tumor samples varied in their cellular composition, mass cytometry revealed two phenotypically important ovarian cancer cell subsets. Whereas most cells expressed E-cadherin or vimentin in a mutually exclusive manner, a snapshot of epithelial-to-mesenchymal transition was captured in some cells that co-expressed these markers in addition to several putative cancer stem cell markers, signaling proteins associated with stem-ness, and a metastasis-promoting protein. Another specific cell subtype (Vimentin+/c-MYC+/HE4+) was identified whose frequency in tumor samples correlated with carboplatin treatment failure and tumor relapse.
Mass cytometry enables single-cell signaling network mapping
Tumor growth, adaptation, and survival is dependent on cellular signaling [103], which can be independent of genetics. Therefore, solely targeting the the proteins of genetic alterations may not yield the best possible outcomes. For example, although mutant-specific BRAF inhibitors have demonstrated clinical effectiveness in melanoma [104, 105], understanding changes in the RAS/RAF/MEK/ERK signaling pathway has led to the emergence of combined BRAF and MEK inhibition therapy, which is superior [13, 106]. By quantifying basal and activated protein signal responses to stimulations, mass cytometry enables single-cell signaling network mapping to identify protein-based signals that mediate tumor survival and therapy resistance in various cell subsets. Adaptive signals can emerge at any signaling node along the signaling pathway or shift reliance onto other signaling pathways altogether. Cancer cells are known to block the normal proteolytic shedding of cell surface growth receptors to encourage signaling bypass when treated with kinase inhibition [107]. In melanoma patients, for example, progression-free survival with BRAF/MEK inhibitors was noted to correlate with shedding of RTKs detected in blood plasma, but not RTK mRNA expression [107]. Adaptive rewiring in protein signaling networks mediating therapy resistance was also observed in glioblastoma in response to mTOR kinase inhibition therapy. In fact, this genetically independent rewiring was detected within days and observed to promote downstream mTOR signaling via ERK and SRC [108]. Combination therapy with an mTOR kinase inhibitor and either an ERK or SRC inhibitor resulted in the most effective tumor control in a murine model [108].
Capturing signaling states by measuring protein phosphorylation in various cell subsets can also define or reveal their phenotype. For example, detection of WNT/β-catenin and STAT3 signaling in a CD133+ cell line derived from a salivary adenoid cystic carcinoma by mass cytometry helped characterize these cells as cancer stem cell-like [109]. We recently identified novel tumor cell subsets based on basal signaling profile in glioblastoma [102]. Differential signaling responses to tumor necrosis factor–α captured using mass cytometry helped identify various cell types within the modular cytoarchitecture of a normal colonic epithelium [75]. These cells utilized shared regulatory mechanisms between signaling pathways. However, cells in colon cancer had reduced differentiation and loss of signaling and regulation of key kinases and protein effectors [75]. Furthermore, the cancer epithelium lacked the modular organization of signaling pathways in the cells.
Mass cytometry is well-suited to map protein-based signaling networks in cancer. Its multiplexing capabilities together with the recently developed computational tools [64–70] provide for a much deeper and a systems level of understanding of signaling networks than traditionally possible. Its use is growing to study signaling in many solid tumor cell lines (adenoid cystic carcinoma [109], melanoma [110], head and neck squamous cell carcinoma [111], and neuroblastoma [112]) as well as primary samples of solid tumors (colon cancer [75], high-grade serous ovarian tumors [78], head and neck squamous cell carcinoma [111], and glioblastoma [102]).
MASS CYTOMETRY SUITS PRECISION ONCOLOGY PRACTICES
Although presently clinical use of a mass cytometer is not approved by the United States Food and Drug Administration, mass cytometry has a strong potential for clinical utility, especially in areas where fluorescence-based flow cytometry is already in clinical use. The ability of samples to be analyzed quickly suits the often fast-paced clinical environment. For example, a tumor sample can be analyzed by mass cytometry within <6 h of its receipt [28]. More importantly, the above studies highlight the power of mass cytometry to unmask a large diversity of tumor and infiltrating immune cells and cell subset-specific signaling networks to provide a high-resolution mechanistic understanding of cellular and molecular responses to therapy.
Mass cytometry can be used in comprehensively profiling immune signatures for monitoring chemotherapy responses [113–115] and, given its capability to detect rare cell subsets, to identify circulating tumor cells in peripheral blood and monitor tumor burden. It can further be used to clinically monitor the extent of therapy response. For example, it helped reveal that BRAF and MEK inhibitors effectively diminished Nestin-expressing tumor cells in BRAFV600Mut melanoma patients [116]. Those cells that persisted post-treatment expressed SOX2 or SOX10 [116]. The kinetics of the change in Nestin-expressing cells suggested that the signaling inhibitors used to treat the cancer were also directly altering the identity of the cancerous cells. The percentage of Nestinlow melanoma cells can now be quantified as a metric of therapy response.
Mass cytometry may also be used clinically to predict therapy response. In renal cancer, tumor immune signatures correlated with progression-free survival [63]. This signature may now be clinically integrated to enhance prognostication. Taken further, mass cytometry aided in the development of a clinical predictive model for anti-PD1 immunotherapy response using peripheral CD14+CD16−HLA-DRhigh monocytes in melanoma patients [98]. The model was applied to another set of melanoma patients with predictive success [98]. Another model was also generated using mass cytometry to predict a sustained response to yttrium-90 radioembolisation of hepatocellular carcinoma based on pre-treatment single-cell immune profiles of the patients’ PBMCs [117]. Hence, mass cytometry can serve as an important tool in precision oncology approaches that can be clinically integrated to develop and apply predictive signatures from high-dimensional data. Others have broadened its clinical application to predict recovery of patients undergoing surgery [118–120] or who suffered trauma [121], to predict vaccine response [122], and to stratify mortality in elderly patients [123].
BEYOND PROTEINS: INNOVATIVE APPLICATIONS OF MASS CYTOMETRY
Probing cell states and nucleic acids
The capabilities of mass cytometry extend beyond quantifying proteins and their post-translational states to provide understanding of various other cellular processes. For example, antibody-based probes can be designed to profile global epigenetic changes by probing chromatin modifications in single cells [124]. Non-antibody reagents can be utilized to quantitatively assess cell membrane properties (such as cell size [125] and integrity/cell viability [126]), metabolic states such as hypoxia [127], uptake of chemotherapy agents containing heavy metals [128, 129], DNA (total content [27], nucleotide incorporation and cell cycle analyses [130]), and RNA [131] (Table 1). Further, to obtain a more comprehensive understanding of cell behavior two novel mass cytometry-based methods have been developed that allow simultaneous detection of proteins and mRNA transcripts in single cells [132, 133]. One utilizes a proximity ligation assay for RNA [132] and the other utilizes metal-tagged in situ mRNA hybridization [131, 133].
Mapping spatial organization in tissues and solid tumors
Imaging mass cytometry [134] and multiplex ion beam imaging (MIBI) [135] are two recently developed, targeted imaging methods of exploring spatial organization in tumor samples. They overcome limitations of conventional immunofluorescence and immunohistochemistry imaging by offering quantitative multiplexed imaging without spectral overlap. These platforms are conceptually similar: tissue sections of interest are stained with antibodies tagged with isotopically pure elemental probes. In IMC, a pulsed laser coupled to a mass cytometer sequentially ablates microsections of the tissue ~ 1 μm in diameter in a linear fashion. The ablated tissue microsections vaporize and are subjected to mass cytometry analysis for detection of antibody-specific mass tags. By rastering across a spatially indexed microsection, an image of the mass tag abundances in different tissue areas can be progressively built. In MIBI, an oxygen primary ion beam rasters across microsections and releases the antibody tagged heavy-metal isotopes as secondary ions which are detected using a mass spectrometer. Currently, MIBI is able to render a slightly higher resolution of 200–300 nm, although its image processing speed is slower than IMC (~ 500 μm2 by MIBI [135] vs. 1.5 mm2 by IMC [136] in 2 h). Importantly, both of these techniques can be applied archived clinical FFPE tissue sections (for protocol refer to [76]). For example, quantitative measurements of immune and non-immune tumor compartments were recently shown to be feasible in FFPE samples of breast and lung cancer using IMC [137].
Relatively few studies have employed MIBI as it is not yet widely available, and the first reports of IMC are just emerging as it became commercially available in 2017. These reports have used human FFPE breast cancer samples to image 19–32 (IMC) [134, 138, 139] and 10–36 (MIBI) [135, 140] different proteins. Using IMC, high levels of phosphorylated S6 protein were noted in stromal cells, which were at times situated adjacent to the tumor margin, indicating signaling activity likely induced by tumor cells [134]. In some samples, expression of carbonic anhydrase IX, a reporter for hypoxia, gradually increased towards the tumor core. Using novel computational tools to analyze spatial networks of cells in IMC data, distinct tumor cell subsets were revealed to be associated with tumor-associated macrophages (such as phospho-S6-expressing proliferative and carbonic anhydrase IX-expressing hypoxic tumor cells) and different ‘niches’ or neighborhoods with macrophages were identified [138]. The landscape of these niches changed with tumor grade [138]. IMC was also used to quantify spatial distribution of the platinum-based chemotherapeutic agent cisplatin within pancreatic cancer. Interestingly, platinum was found extensively bound to collagen fibers in tumors and normal tissue [136, 141]. In addition to proteins and drugs, mRNA can also be spatially visualized with mass cytometry [133]. A recent study performed simultaneous multiplexed imaging of mRNA (using mass-tagged in situ mRNA hybridization) and proteins using IMC to spatially characterize cells expressing CXCL10, a T cell chemoattractant, in FFPE breast cancer samples [139]. Detecting as little as six mRNA copies per cell, the study also demonstrated that at a single-cell level correlations between mRNA and protein levels for two genes were generally weak, sample-specific, and gene-specific [139]. Although fewer studies utilizing MIBI are presently available compared to IMC, a recent study used MIBI to perform multiplexed imaging of 36 proteins in 41 triple-negative breast cancer [140]. The study evaluated the spatial organization of immune cells and identified three patterns: cold (minimal immune infiltration), mixed pattern of tumor and immune cells, and compartmentalized organization with distinct regions of predominantly tumor or immune cells. Interestingly, expression of immuno-regulatory markers correlated with these spatial organizations. For example, in the mixed pattern, PD1 was expressed by CD4+ T cells while tumor cells expressed PD-L1 and indoleamine-2,3-dioxygenase (IDO). In the compartmentalized organization, which was associated with a greater patient survival compared to mixed, immune cells expressed PD-L1 and IDO and CD8+ T cells expressed PD1.
Using both antibody-based and non-antibody reagents, these preliminary studies demonstrate the tremendous potential of IMC and MIBI in revealing spatial organization of cells, localization of rare cellular subsets, cell morphology (size and shape), cell proliferation (DNA nucleotide incorporation and cell cycle analysis), and gene expression. Prior imaging studies have suggested that tumors can form morphologically and functionally distinct niches that harbor cancer stem cells and maintain niche-specific communicative signals with the microenvironment to enhance growth and evade immune surveillance. Glioblastoma is one example where three specialized tumor niches are widely recognized: the perivascular niche, where cancer stem cells are found along the vasculature [142]; the vascular-invasive niche, where tumor cells use normal blood vessels to invade surrounding brain parenchyma; and the hypoxic niche, which contains necrotic areas surrounded by hypoxia-dependent tumor cells [143]. All of these structures are potentially uniquely affected by therapy. IMC and MIBI have the potential to offer insights into the interplay between cancer cells and microenvironment constituents specific to these niches, including the cell-specific effects of chemotherapy and mechanisms underlying failed (for example, anti-angiogenic therapy in glioblastoma) and successful targeted treatments.
CONCLUSION
Relying on traditional approaches that analyze sample aggregates or unidimensional molecular information in single cells limits our potential to comprehensively understand cancer. In addition to genomic elements, proteins must also be measured in single cells of tumor samples. Mass cytometry is well suited for this need. It offers highly multiplexed and relative unbiased quantitative protein measurements to understand the phenotypic, functional, and spatial diversity of the cells in tissues and solid tumors. Together, a proteogenomic approach with single-cell genomics and mass cytometry has a greater potential to offer insights into the cancer cell identity in order to make precision oncology approaches more effective.
ACKNOWLEDGEMENTS
The authors thank the members of the Ihrie and Irish labs at Vanderbilt for helpful discussions.
FUNDING
Michael David Greene Brain Cancer Fund; US Department of Defense. Grant Number: W81XWH-16-1-0171; Research Update in Neuroscience for Neurosurgeons Award, Society of Neurological Surgeons, United States (A.M.M.); Vanderbilt-Ingram Cancer Center; Southeastern Brain Tumor Foundation; National Cancer Institute. Grant Numbers: F32 CA224962 (A.M.M.), R00 CA143231; National Institute of Neurological Disorders and Stroke. Grant Number: R01 NS096238
Abbreviations
- CTLA-4
cytotoxic T lymphocyte-associated protein-4
- FFPE
formalin fixed, paraffin embedded
- IDO
indoleamine-2,3-dioxygenase
- IMC
imaging mass cytometry
- MIBI
multiplex ion beam imaging
- PBMCs
peripheral blood mononuclear cells
- PD-1
programmed cell death protein-1
- PD-L1
programmed cell death-ligand 1
- RTK
receptor tyrosine kinase
Footnotes
CONFLICTS OF INTEREST
JMI is co-founder and a board member of Cytobank Inc. and received research support from Incyte Corp, Janssen, and Pharmacyclics.
REFERENCES
- 1.Alexander BM & Cloughesy TF (2017) Adult glioblastoma. J Clin Oncol 35, 2402–2409. [DOI] [PubMed] [Google Scholar]
- 2.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH et al. (2013) The somatic genomic landscape of glioblastoma. Cell 155, 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Massard C, Michiels S, Ferte C, Le Deley MC, Lacroix L, Hollebecque A, Verlingue L, Ileana E, Rosellini S, Ammari S et al. (2017) High-throughput genomics and clinical outcome in hard-to-treat advanced cancers: results of the MOSCATO 01 trial. Cancer Discov 7, 586–595. [DOI] [PubMed] [Google Scholar]
- 4.Wheler JJ, Janku F, Naing A, Li Y, Stephen B, Zinner R, Subbiah V, Fu S, Karp D, Falchook GS et al. (2016) Cancer therapy directed by comprehensive genomic profiling: a single center study. Can Res 76, 3690–3701. [DOI] [PubMed] [Google Scholar]
- 5.Stockley TL, Oza AM, Berman HK, Leighl NB, Knox JJ, Shepherd FA, Chen EX, Krzyzanowska MK, Dhani N, Joshua AM et al. (2016) Molecular profiling of advanced solid tumors and patient outcomes with genotype-matched clinical trials: the Princess Margaret IMPACT/COMPACT trial. Genome Med 8, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsimberidou AM, Iskander NG, Hong DS, Wheler JJ, Falchook GS, Fu S, Piha-Paul S, Naing A, Janku F, Luthra R et al. (2012) Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center initiative. Clin Cancer Res 18, 6373–6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Von Hoff DD, Stephenson JJ Jr, Rosen P, Loesch DM, Borad MJ, Anthony S, Jameson G, Brown S, Cantafio N, Richards DA et al. (2010) Pilot study using molecular profiling of patients’ tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol 28, 4877–4883. [DOI] [PubMed] [Google Scholar]
- 8.Le Tourneau C, Delord JP, Goncalves A, Gavoille C, Dubot C, Isambert N, Campone M, Tredan O, Massiani MA, Mauborgne C et al. (2015) Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol 16, 1324–1334. [DOI] [PubMed] [Google Scholar]
- 9.De Witt Hamer PC (2010) Small molecule kinase inhibitors in glioblastoma: a systematic review of clinical studies. Neuro Oncol 12, 304–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reardon DA, Wen PY & Mellinghoff IK (2014) Targeted molecular therapies against epidermal growth factor receptor: past experiences and challenges. Neuro Oncol 16(Suppl 8), viii7–viii13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bastien JI, McNeill KA & Fine HA (2015) Molecular characterizations of glioblastoma, targeted therapy, and clinical results to date. Cancer 121, 502–516. [DOI] [PubMed] [Google Scholar]
- 12.Sim HW, Morgan ER & Mason WP (2018) Contemporary management of high-grade gliomas. CNS Oncol 7, 51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Senft D, Leiserson MDM, Ruppin E & Ronai ZA (2017) Precision oncology: the road ahead. Trends Mol Med 23, 874–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tannock IF & Hickman JA (2016) Limits to personalized cancer medicine. N Engl J Med 375, 1289–1294. [DOI] [PubMed] [Google Scholar]
- 15.Irish JM, Myklebust JH, Alizadeh AA, Houot R, Sharman JP, Czerwinski DK, Nolan GP & Levy R (2010) B-cell signaling networks reveal a negative prognostic human lymphoma cell subset that emerges during tumor progression. Proc Natl Acad Sci USA 107, 12747–12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Myklebust JH, Brody J, Kohrt HE, Kolstad A, Czerwinski DK, Walchli S, Green MR, Troen G, Liestol K, Beiske K et al. (2017) Distinct patterns of B-cell receptor signaling in non-Hodgkin lymphomas identified by single-cell profiling. Blood 129, 759–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Irish JM, Hovland R, Krutzik PO, Perez OD, Bruserud O, Gjertsen BT & Nolan GP (2004) Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell 118, 217–228. [DOI] [PubMed] [Google Scholar]
- 18.Levine JH, Simonds EF, Bendall SC, Davis KL, Amir AD, Tadmor MD, Litvin O, Fienberg HG, Jager A, Zunder ER et al. (2015) Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell 162, 184–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Good Z, Sarno J, Jager A, Samusik N, Aghaeepour N, Simonds EF, White L, Lacayo NJ, Fantl WJ, Fazio G et al. (2018) Single-cell developmental classification of B cell precursor acute lymphoblastic leukemia at diagnosis reveals predictors of relapse. Nat Med 24, 474–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barnes TA & Amir E (2017) HYPE or HOPE: the prognostic value of infiltrating immune cells in cancer. Br J Cancer 117, 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Irish JM & Doxie DB (2014) High-dimensional single-cell cancer biology. Curr Top Microbiol Immunol 377, 1–21. 10.1007/82_2014_367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parker NR, Hudson AL, Khong P, Parkinson JF, Dwight T, Ikin RJ, Zhu Y, Cheng ZJ, Vafaee F, Chen J et al. (2016) Intratumoral heterogeneity identified at the epigenetic, genetic and transcriptional level in glioblastoma. Sci Rep 6, 22477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shalek AK & Benson M (2017) Single-cell analyses to tailor treatments. Sci Transl Med 9, pii: eaan4730 10.1126/scitranslmed.aan4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Latonen L, Afyounian E, Jylha A, Nattinen J, Aapola U, Annala M, Kivinummi KK, Tammela TTL, Beuerman RW, Uusitalo H et al. (2018) Integrative proteomics in prostate cancer uncovers robustness against genomic and transcriptomic aberrations during disease progression. Nat Commun 9, 1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holohan C, Van Schaeybroeck S, Longley DB & Johnston PG (2013) Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 13, 714–726. [DOI] [PubMed] [Google Scholar]
- 26.Macaulay IC, Ponting CP & Voet T (2017) Single-cell multiomics: multiple measurements from single cells. Trends Genet 155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ornatsky OI, Lou X, Nitz M, Schafer S, Sheldrick WS, Baranov VI, Bandura DR & Tanner SD (2008) Study of cell antigens and intracellular DNA by identification of element-containing labels and metallointercalators using inductively coupled plasma mass spectrometry. Anal Chem 80, 2539–2547. [DOI] [PubMed] [Google Scholar]
- 28.Leelatian N, Doxie DB, Greenplate AR, Sinnaeve J, Ihrie RA & Irish JM (2017) Preparing viable single cells from human tissue and tumors for cytomic analysis. Curr Protoc Mol Biol 118, 25C.1.1–25C.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hartmann FJ, Simonds EF & Bendall SC (2018) A universal live cell barcoding-platform for multiplexed human single cell analysis. Sci Rep 8, 10770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mei HE, Leipold MD, Schulz AR, Chester C & Maecker HT (2015) Barcoding of live human peripheral blood mononuclear cells for multiplexed mass cytometry. J Immunol 194, 2022–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matos TR, Liu H & Ritz J (2017) Research techniques made simple: mass cytometry analysis tools for decrypting the complexity of biological systems. J Invest Dermatol 137, e43–e51. [DOI] [PubMed] [Google Scholar]
- 32.Leipold MD, Newell EW & Maecker HT (2015) Multiparameter phenotyping of human PBMCs using mass cytometry. Methods Mol Biol 1343, 81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Di Palma S & Bodenmiller B (2015) Unraveling cell populations in tumors by single-cell mass cytometry. Curr Opin Biotechnol 31, 122–129. [DOI] [PubMed] [Google Scholar]
- 34.Behbehani GK (2017) Applications of mass cytometry in clinical medicine: the promise and perils of clinical CyTOF. Clin Lab Med 37, 945–964. [DOI] [PubMed] [Google Scholar]
- 35.Leelatian N, Diggins KE & Irish JM (2015) Characterizing phenotypes and signaling networks of single human cells by mass cytometry. Methods Mol Biol 1346, 99–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bandyopadhyay S, Fisher DAC, Malkova O & Oh ST (2017) Analysis of signaling networks at the single-cell level using mass cytometry. Methods Mol Biol 1636, 371–392. [DOI] [PubMed] [Google Scholar]
- 37.Brodie TM & Tosevski V (2017) High-dimensional single-cell analysis with mass cytometry. Curr Protoc Immunol 118, 5.11.1–5.11.25. [DOI] [PubMed] [Google Scholar]
- 38.Leelatian N, Doxie DB, Greenplate AR, Mobley BC, Lehman JM, Sinnaeve J, Kauffmann RM, Werkhaven JA, Mistry AM, Weaver KD et al. (2017) Single cell analysis of human tissues and solid tumors with mass cytometry. Cytometry B Clin Cytom 92, 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Diggins KE, Ferrell PB Jr & Irish JM (2015) Methods for discovery and characterization of cell subsets in high dimensional mass cytometry data. Methods 82, 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Diggins KE, Greenplate AR, Leelatian N, Wogsland CE & Irish JM (2017) Characterizing cell subsets using marker enrichment modeling. Nat Methods 14, 275–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tirosh I & Suva ML (2018) Dissecting human gliomas by single-cell RNA sequencing. Neuro Oncol 20, 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spitzer MH & Nolan GP (2016) Mass cytometry: single cells, many features. Cell 165, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang B, Zhu J, Pierson E, Ramazzotti D & Batzoglou S (2017) Visualization and analysis of single-cell RNA-seq data by kernel-based similarity learning. Nat Methods 14, 414–416. [DOI] [PubMed] [Google Scholar]
- 44.Chattopadhyay PK, Gierahn TM, Roederer M & Love JC (2014) Single-cell technologies for monitoring immune systems. Nat Immunol 15, 128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Papalexi E & Satija R (2018) Single-cell RNA sequencing to explore immune cell heterogeneity. Nat Rev Immunol 18, 35–45. [DOI] [PubMed] [Google Scholar]
- 46.Han G, Spitzer MH, Bendall SC, Fantl WJ & Nolan GP (2018) Metal-isotope-tagged monoclonal antibodies for high-dimensional mass cytometry. Nat Protoc 13, 2121–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bendall SC, Nolan GP, Roederer M & Chattopadhyay PK (2012) A deep profiler’s guide to cytometry. Trends Immunol 33, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Behbehani GK, Bendall SC, Clutter MR, Fantl WJ & Nolan GP (2012) Single-cell mass cytometry adapted to measurements of the cell cycle. Cytometry A 81, 552–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zunder ER, Finck R, Behbehani GK, Amir AD, Krishnaswamy S, Gonzalez VD, Lorang CG, Bjornson Z, Spitzer MH, Bodenmiller B et al. (2015) Palladium-based mass tag cell barcoding with a doublet-filtering scheme and single-cell deconvolution algorithm. Nat Protoc 10, 316–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mei HE, Leipold MD & Maecker HT (2016) Platinum-conjugated antibodies for application in mass cytometry. Cytometry A 89, 292–300. [DOI] [PubMed] [Google Scholar]
- 51.Huang L, Michael SA, Chen Y & Wu H (2017) Current advances in highly multiplexed antibody-based single-cell proteomic measurements. Chem Asian J 12, 1680–1691. [DOI] [PubMed] [Google Scholar]
- 52.Bjornson ZB, Nolan GP & Fantl WJ (2013) Single-cell mass cytometry for analysis of immune system functional states. Curr Opin Immunol 25, 484–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Illy N, Majonis D, Herrera I, Ornatsky O & Winnik MA (2012) Metal-chelating polymers by anionic ring-opening polymerization and their use in quantitative mass cytometry. Biomacromol 13, 2359–2369. [DOI] [PubMed] [Google Scholar]
- 54.Majonis D, Herrera I, Ornatsky O, Schulze M, Lou X, Soleimani M, Nitz M & Winnik MA (2010) Synthesis of a functional metal-chelating polymer and steps toward quantitative mass cytometry bioassays. Anal Chem 82, 8961–8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chevrier S, Crowell HL, Zanotelli VRT, Engler S, Robinson MD & Bodenmiller B (2018) Compensation of signal spillover in suspension and imaging mass cytometry. Cell Syst 6, 612–620.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mair F & Prlic M (2018) OMIP-044: 28-color immunophenotyping of the human dendritic cell compartment. Cytometry A 93, 402–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kolodziejczyk AA, Kim JK, Svensson V, Marioni JC & Teichmann SA (2015) The technology and biology of single-cell RNA sequencing. Mol Cell 58, 610–620. [DOI] [PubMed] [Google Scholar]
- 58.Kim JK, Kolodziejczyk AA, Ilicic T, Teichmann SA & Marioni JC (2015) Characterizing noise structure in single-cell RNA-seq distinguishes genuine from technical stochastic allelic expression. Nat Commun 6, 8687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Finck R, Simonds EF, Jager A, Krishnaswamy S, Sachs K, Fantl W, Pe’er D, Nolan GP & Bendall SC (2013) Normalization of mass cytometry data with bead standards. Cytometry A 83, 483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kleinsteuber K, Corleis B, Rashidi N, Nchinda N, Lisanti A, Cho JL, Medoff BD, Kwon D & Walker BD (2016) Standardization and quality control for high-dimensional mass cytometry studies of human samples. Cytometry A 89, 903–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tricot S, Meyrand M, Sammicheli C, Elhmouzi-Younes J, Corneau A, Bertholet S, Malissen M, Le Grand R, Nuti S, Luche H et al. (2015) Evaluating the efficiency of isotope transmission for improved panel design and a comparison of the detection sensitivities of mass cytometer instruments. Cytometry A 87, 357–368. [DOI] [PubMed] [Google Scholar]
- 62.Leipold MD, Obermoser G, Fenwick C, Kleinstuber K, Rashidi N, McNevin JP, Nau AN, Wagar LE, Rozot V, Davis MM et al. (2018) Comparison of CyTOF assays across sites: results of a six-center pilot study. J Immunol Methods 453, 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chevrier S, Levine JH, Zanotelli VRT, Silina K, Schulz D, Bacac M, Ries CH, Ailles L, Jewett MAS, Moch H et al. (2017) An immune atlas of clear cell renal cell carcinoma. Cell 169, 736–749.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Amir AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP & Pe’er D (2013) viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol 31, 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krishnaswamy S, Spitzer MH, Mingueneau M, Bendall SC, Litvin O, Stone E, Pe’er D & Nolan GP (2014) Systems biology. Conditional density-based analysis of T cell signaling in single-cell data. Science 346, 1250689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bruggner RV, Bodenmiller B, Dill DL, Tibshirani RJ & Nolan GP (2014) Automated identification of stratifying signatures in cellular subpopulations. Proc Natl Acad Sci USA 111, E2770–E2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Spitzer MH, Gherardini PF, Fragiadakis GK, Bhattacharya N, Yuan RT, Hotson AN, Finck R, Carmi Y, Zunder ER, Fantl WJ et al. (2015) Immunology. An interactive reference framework for modeling a dynamic immune system. Science 349, 1259425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zunder ER, Lujan E, Goltsev Y, Wernig M & Nolan GP (2015) A continuous molecular roadmap to iPSC reprogramming through progression analysis of single-cell mass cytometry. Cell Stem Cell 16, 323–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bendall SC, Davis KL, Amir AD, Tadmor MD, Simonds EF, Chen TJ, Shenfeld DK, Nolan GP & Pe’er D (2014) Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell 157, 714–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Samusik N, Good Z, Spitzer MH, Davis KL & Nolan GP (2016) Automated mapping of phenotype space with single-cell data. Nat Methods 13, 493–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.See P, Dutertre CA, Chen J, Gunther P, McGovern N, Irac SE, Gunawan M, Beyer M, Handler K, Duan K et al. (2017) Mapping the human DC lineage through the integration of high-dimensional techniques. Science 356, pii: eaag3009. 10.1126/science.aag3009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yao Y, Liu R, Shin MS, Trentalange M, Allore H, Nassar A, Kang I, Pober JS & Montgomery RR (2014) CyTOF supports efficient detection of immune cell subsets from small samples. J Immunol Methods 415, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rozenblatt-Rosen O, Stubbington MJT, Regev A & Teichmann SA (2017) The human cell atlas: from vision to reality. Nature 550, 451–453. [DOI] [PubMed] [Google Scholar]
- 74.Simmons AJ, Banerjee A, McKinley ET, Scurrah CR, Herring CA, Gewin LS, Masuzaki R, Karp SJ, Franklin JL, Gerdes MJ et al. (2015) Cytometry-based single-cell analysis of intact epithelial signaling reveals MAPK activation divergent from TNF-alpha-induced apoptosis in vivo. Mol Syst Biol 11, 835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Simmons AJ, Scurrah CR, McKinley ET, Herring CA, Irish JM, Washington MK, Coffey RJ & Lau KS (2016) Impaired coordination between signaling pathways is revealed in human colorectal cancer using single-cell mass cytometry of archival tissue blocks. Sci Signal 9, rs11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chang Q, Ornatsky O & Hedley D (2017) Staining of frozen and formalin-fixed, paraffin-embedded tissues with metal-labeled antibodies for imaging mass cytometry analysis. Curr Protoc Cytom 82, 12.47.1–12.47.8. [DOI] [PubMed] [Google Scholar]
- 77.Kadic E, Moniz RJ, Huo Y, Chi A & Kariv I (2017) Effect of cryopreservation on delineation of immune cell subpopulations in tumor specimens as determinated by multiparametric single cell mass cytometry analysis. BMC Immunol 18, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gonzalez VD, Samusik N, Chen TJ, Savig ES, Aghaeepour N, Quigley DA, Huang YW, Giangarra V, Borowsky AD, Hubbard NE et al. (2018) Commonly occurring cell subsets in high-grade serous ovarian tumors identified by single-cell mass cytometry. Cell Rep 22, 1875–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Syn NL, Teng MWL, Mok TSK & Soo RA (2017) De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol 18, e731–e741. [DOI] [PubMed] [Google Scholar]
- 80.Boussiotis VA (2016) Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med 375, 1767–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Siska PJ, Beckermann KE, Mason FM, Andrejeva G, Greenplate AR, Sendor AB, Chiang YJ, Corona AL, Gemta LF, Vincent BG et al. (2017) Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight 2, pii: 93411. 10.1172/jci.insight.93411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, Bigenwald C, Remark R, Sweeney R, Becker CD, Levine JH et al. (2017) Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell 169, 750–765.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, Yeong JPS, Nahar R, Zhang T, Kared H et al. (2018) Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557, 575–579. [DOI] [PubMed] [Google Scholar]
- 84.Chew V, Lai L, Pan L, Lim CJ, Li J, Ong R, Chua C, Leong JY, Lim KH, Toh HC et al. (2017) Delineation of an immunosuppressive gradient in hepatocellular carcinoma using high-dimensional proteomic and transcriptomic analyses. Proc Natl Acad Sci USA 114, E5900–E5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lim CJ, Lee YH, Pan L, Lai L, Chua C, Wasser M, Lim TKH, Yeong J, Toh HC, Lee SY et al. (2018) Multidimensional analyses reveal distinct immune microenvironment in hepatitis B virus-related hepatocellular carcinoma. Gut, pii: gutjnl-2018-316510. 10.1136/gutjnl-2018-316510 [DOI] [PubMed] [Google Scholar]
- 86.Vasquez JC, Huttner A, Zhang L, Marks A, Chan A, Baehring JM, Kahle KT & Dhodapkar KM (2017) SOX2 immunity and tissue resident memory in children and young adults with glioma. J Neurooncol 134, 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lowther DE, Goods BA, Lucca LE, Lerner BA, Raddassi K, van Dijk D, Hernandez AL, Duan X, Gunel M, Coric V et al. (2016) PD-1 marks dysfunctional regulatory T cells in malignant gliomas. JCI Insight 1, pii: e85935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee HS, Jang HJ, Choi JM, Zhang J, de Rosen VL, Wheeler TM, Lee JS, Tu T, Jindra PT, Kerman RH et al. (2018) Comprehensive immunoproteogenomic analyses of malignant pleural mesothelioma. JCI Insight 3, pii: 98575. 10.1172/jci.insight.98575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Boddupalli CS, Bar N, Kadaveru K, Krauthammer M, Pornputtapong N, Mai Z, Ariyan S, Narayan D, Kluger H, Deng Y et al. (2016) Interlesional diversity of T cell receptors in melanoma with immune checkpoints enriched in tissue-resident memory T cells. JCI Insight 1, e88955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Palucka AK & Coussens LM (2016) The basis of oncoimmunology. Cell 164, 1233–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wherry EJ & Kurachi M (2015) Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 15, 486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, Kaisho T, Bogunovic D, Bhardwaj N & Krummel MF (2016) Critical role for CD103(+)/CD141(+) dendritic cells bearing CCR7 for Tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell 30, 324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang G, Lu X, Dey P, Deng P, Wu CC, Jiang S, Fang Z, Zhao K, Konaparthi R, Hua S et al. (2016) Targeting YAP-dependent MDSC infiltration impairs tumor progression. Cancer Discov 6, 80–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, Sharma P, Wang J, Wargo JA, Pe’er D et al. (2017) Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell 170, 1120–1133.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fehlings M, Simoni Y, Penny HL, Becht E, Loh CY, Gubin MM, Ward JP, Wong SC, Schreiber RD & Newell EW (2017) Checkpoint blockade immunotherapy reshapes the high-dimensional phenotypic heterogeneity of murine intratumoural neoantigen-specific CD8(+) T cells. Nat Commun 8, 562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yarchoan M, Hopkins A & Jaffee EM (2017) Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med 377, 2500–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, Gherardini PF, Prestwood TR, Chabon J, Bendall SC et al. (2017) Systemic immunity is required for effective cancer immunotherapy. Cell 168, 487–502.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Krieg C, Nowicka M, Guglietta S, Schindler S, Hartmann FJ, Weber LM, Dummer R, Robinson MD, Levesque MP & Becher B (2018) High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat Med 24, 144–153. [DOI] [PubMed] [Google Scholar]
- 99.Takeuchi Y, Tanemura A, Tada Y, Katayama I, Kumanogoh A & Nishikawa H (2018) Clinical response to PD-1 blockade correlates with a sub-fraction of peripheral central memory CD4+ T cells in patients with malignant melanoma. Int Immunol 30, 13–22. [DOI] [PubMed] [Google Scholar]
- 100.Subrahmanyam PB, Dong Z, Gusenleitner D, Giobbie-Hurder A, Severgnini M, Zhou J, Manos M, Eastman LM, Maecker HT & Hodi FS (2018) Distinct predictive biomarker candidates for response to anti-CTLA-4 and anti-PD-1 immunotherapy in melanoma patients. J Immunother Cancer 6, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Reardon DA, Omuro A, Brandes AA, Rieger J, Wick A, Sepulveda J, Phuphanich S, de Souza P, Ahluwalia MS, Lim M et al. (2017) OS10.3 randomized phase 3 study evaluating the efficacy and safety of nivolumab vs bevacizumab in patients with recurrent glioblastoma: CheckMate 143. Neuro Oncol 19, iii21. [Google Scholar]
- 102.Leelatian N, Sinnaeve J, Mobley BC, Mistry AM, Liu D, Weaver KD, Thompson RC, Chambless LB, Ihrie RA & Irish JM (2017) Abstract 364: mass cytometry of human glioblastoma characterizes more than 99 percent of cells and reveals intratumoral cell subsets defined by contrasting signaling network profiles. Can Res 77, 364. [Google Scholar]
- 103.Irish JM, Kotecha N & Nolan GP (2006) Mapping normal and cancer cell signalling networks: towards single-cell proteomics. Nat Rev Cancer 6, 146–155. [DOI] [PubMed] [Google Scholar]
- 104.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M et al. (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364, 2507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH Jr, Kaempgen E et al. (2012) Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 380, 358–365. [DOI] [PubMed] [Google Scholar]
- 106.Johnson DB, Pollack MH & Sosman JA (2016) Emerging targeted therapies for melanoma. Expert Opin Emerg Drugs 21, 195–207. [DOI] [PubMed] [Google Scholar]
- 107.Miller MA, Oudin MJ, Sullivan RJ, Wang SJ, Meyer AS, Im H, Frederick DT, Tadros J, Griffith LG, Lee H et al. (2016) Reduced proteolytic shedding of receptor tyrosine kinases is a post-translational mechanism of kinase inhibitor resistance. Cancer Discov 6, 382–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wei W, Shin YS, Xue M, Matsutani T, Masui K, Yang H, Ikegami S, Gu Y, Herrmann K, Johnson D et al. (2016) Single-cell phosphoproteomics resolves adaptive signaling dynamics and informs targeted combination therapy in glioblastoma. Cancer Cell 29, 563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Panaccione A, Zhang Y, Ryan M, Moskaluk CA, Anderson KS, Yarbrough WG & Ivanov SV (2017) MYB fusions and CD markers as tools for authentication and purification of cancer stem cells from salivary adenoid cystic carcinoma. Stem Cell Res 21, 160–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fernandez M, Sutterluty-Fall H, Schwarzler C, Lemeille S, Boehncke WH & Merat R (2017) Overexpression of the human antigen R suppresses the immediate paradoxical proliferation of melanoma cell subpopulations in response to suboptimal BRAF inhibition. Cancer Med 6, 1652–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Brodie TM, Tosevski V & Medova M (2018) OMIP-045: characterizing human head and neck tumors and cancer cell lines with mass cytometry. Cytometry A 93, 406–410. [DOI] [PubMed] [Google Scholar]
- 112.Nolo R, Herbrich S, Rao A, Zweidler-McKay P, Kannan S & Gopalakrishnan V (2017) Targeting P-selectin blocks neuroblastoma growth. Oncotarget 8, 86657–86670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Baumgart S, Peddinghaus A, Schulte-Wrede U, Mei HE & Grutzkau A (2017) OMIP-034: comprehensive immune phenotyping of human peripheral leukocytes by mass cytometry for monitoring immunomodulatory therapies. Cytometry A 91, 34–38. [DOI] [PubMed] [Google Scholar]
- 114.Wistuba-Hamprecht K, Martens A, Weide B, Teng KW, Zelba H, Guffart E, Chen J, Garbe C, Newell EW, Larbi A et al. (2017) Establishing high dimensional immune signatures from peripheral blood via mass cytometry in a discovery cohort of stage IV melanoma patients. J Immunol 198, 927–936. [DOI] [PubMed] [Google Scholar]
- 115.Hiniker SM, Reddy SA, Maecker HT, Subrahmanyam PB, Rosenberg-Hasson Y, Swetter SM, Saha S, Shura L & Knox SJ (2016) A prospective clinical trial combining radiation therapy with systemic immunotherapy in metastatic melanoma. Int J Radiat Oncol Biol Phys 96, 578–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Doxie DB, Greenplate AR, Gandelman JS, Diggins KE, Roe CE, Dahlman KB, Sosman JA, Kelley MC & Irish JM (2018) BRAF and MEK inhibitor therapy eliminates Nestin-expressing melanoma cells in human tumors. Pigment Cell Melanoma Res 31, 708–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chew V, Lee YH, Pan L, Nasir NJM, Lim CJ, Chua C, Lai L, Hazirah SN, Lim TKH, Goh BKP et al. (2018) Immune activation underlies a sustained clinical response to Yttrium-90 radioembolisation in hepatocellular carcinoma. Gut, pii: gutjnl-2017-315485. 10.1136/gutjnl-2017-315485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Aghaeepour N, Kin C, Ganio EA, Jensen KP, Gaudilliere DK, Tingle M, Tsai A, Lancero HL, Choisy B, McNeil LS et al. (2017) Deep immune profiling of an arginine-enriched nutritional intervention in patients undergoing surgery. J Immunol 199, 2171–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fragiadakis GK, Gaudilliere B, Ganio EA, Aghaeepour N, Tingle M, Nolan GP & Angst MS (2015) Patient-specific immune states before surgery are strong correlates of surgical recovery. Anesthesiology 123, 1241–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gaudilliere B, Fragiadakis GK, Bruggner RV, Nicolau M, Finck R, Tingle M, Silva J, Ganio EA, Yeh CG, Maloney WJ et al. (2014) Clinical recovery from surgery correlates with single-cell immune signatures. Sci Transl Med 6, 255ra131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Seshadri A, Brat GA, Yorkgitis BK, Keegan J, Dolan J, Salim A, Askari R & Lederer JA (2017) Phenotyping the immune response to trauma: a multiparametric systems immunology approach. Crit Care Med 45, 1523–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lingblom CMD, Kowli S, Swaminathan N, Maecker HT & Lambert SL (2018) Baseline immune profile by CyTOF can predict response to an investigational adjuvanted vaccine in elderly adults. J Transl Med 16, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Furman D, Chang J, Lartigue L, Bolen CR, Haddad F, Gaudilliere B, Ganio EA, Fragiadakis GK, Spitzer MH, Douchet I et al. (2017) Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nat Med 23, 174–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cheung P, Vallania F, Warsinske HC, Donato M, Schaffert S, Chang SE, Dvorak M, Dekker CL, Davis MM, Utz PJ et al. (2018) Single-cell chromatin modification profiling reveals increased epigenetic variations with aging. Cell 173, 1385–1397.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Stern AD, Rahman AH & Birtwistle MR (2017) Cell size assays for mass cytometry. Cytometry A 91, 14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fienberg HG, Simonds EF, Fantl WJ, Nolan GP & Bodenmiller B (2012) A platinum-based covalent viability reagent for single-cell mass cytometry. Cytometry A 81, 467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Edgar LJ, Vellanki RN, Halupa A, Hedley D, Wouters BG & Nitz M (2014) Identification of hypoxic cells using an organotellurium tag compatible with mass cytometry. Angew Chem 53, 11473–11477. [DOI] [PubMed] [Google Scholar]
- 128.Chang Q, Ornatsky OI, Koch CJ, Chaudary N, Marie-Egyptienne DT, Hill RP, Tanner SD & Hedley DW (2015) Single-cell measurement of the uptake, intratumoral distribution and cell cycle effects of cisplatin using mass cytometry. Int J Cancer 136, 1202–1209. [DOI] [PubMed] [Google Scholar]
- 129.Comsa E, Nguyen KA, Loghin F, Boumendjel A, Peuchmaur M, Andrieu T & Falson P (2018) Ovarian cancer cells cisplatin sensitization agents selected by mass cytometry target ABCC2 inhibition. Future Med Chem 10, 1349–1360. [DOI] [PubMed] [Google Scholar]
- 130.Behbehani GK (2018) Cell cycle analysis by mass cytometry. Methods Mol Biol 1686, 105–124. [DOI] [PubMed] [Google Scholar]
- 131.Ornatsky OI, Baranov VI, Bandura DR, Tanner SD & Dick J (2006) Messenger RNA detection in leukemia cell lines by novel metal-tagged in situ hybridization using inductively coupled plasma mass spectrometry. Transl Oncogenomics 1, 1–9. [PMC free article] [PubMed] [Google Scholar]
- 132.Frei AP, Bava FA, Zunder ER, Hsieh EW, Chen SY, Nolan GP & Gherardini PF (2016) Highly multiplexed simultaneous detection of RNAs and proteins in single cells. Nat Methods 13, 269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mavropoulos A, Allo B, He M, Park E, Majonis D & Ornatsky O (2017) Simultaneous detection of protein and mRNA in jurkat and KG-1a cells by mass cytometry. Cytometry A 91, 1200–1208. [DOI] [PubMed] [Google Scholar]
- 134.Giesen C, Wang HA, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, Schuffler PJ, Grolimund D, Buhmann JM, Brandt S et al. (2014) Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods 11, 417–422. [DOI] [PubMed] [Google Scholar]
- 135.Angelo M, Bendall SC, Finck R, Hale MB, Hitzman C, Borowsky AD, Levenson RM, Lowe JB, Liu SD, Zhao S et al. (2014) Multiplexed ion beam imaging of human breast tumors. Nat Med 20, 436–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chang Q, Ornatsky OI, Siddiqui I, Loboda A, Baranov VI & Hedley DW (2017) Imaging mass cytometry. Cytometry A 91, 160–169. [DOI] [PubMed] [Google Scholar]
- 137.Villarroel-Espindola F, Carvajal-Hausdorf D, Datar I, Esch A, Rashidi N, Nassar A, Ren S, Montgomery RR, Herbst RS, Rimm DL et al. (2017) Abstract 1635: multiplexed analysis of fixed tumor tissues using imaging mass cytometry. Can Res 77, 1635. [Google Scholar]
- 138.Schapiro D, Jackson HW, Raghuraman S, Fischer JR, Zanotelli VRT, Schulz D, Giesen C, Catena R, Varga Z & Bodenmiller B (2017) histoCAT: analysis of cell phenotypes and interactions in multiplex image cytometry data. Nat Methods 14, 873–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Schulz D, Zanotelli VRT, Fischer JR, Schapiro D, Engler S, Lun XK, Jackson HW & Bodenmiller B (2018) Simultaneous multiplexed imaging of mRNA and proteins with subcellular resolution in breast cancer tissue samples by mass cytometry. Cell Syst 6, 25–36.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Keren L, Bosse M, Marquez D, Angoshtari R, Jain S, Varma S, Yang SR, Kurian A, Van Valen D, West R et al. (2018) A structured tumor-immune microenvironment in triple negative breast cancer revealed by multiplexed ion beam imaging. Cell 174, 1373–1387.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chang Q, Ornatsky OI, Siddiqui I, Straus R, Baranov VI & Hedley DW (2016) Biodistribution of cisplatin revealed by imaging mass cytometry identifies extensive collagen binding in tumor and normal tissues. Sci Rep 6, 36641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, Oh EY, Gaber MW, Finklestein D, Allen M et al. (2007) A perivascular niche for brain tumor stem cells. Cancer Cell 11, 69–82. [DOI] [PubMed] [Google Scholar]
- 143.Hambardzumyan D & Bergers G (2015) Glioblastoma: defining tumor niches. Trends Cancer 1, 252–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R & Smibert P (2017) Simultaneous epitope and transcriptome measurement in single cells. Nat Methods 14, 865–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Obier N & Muller AM (2010) Chromatin flow cytometry identifies changes in epigenetic cell states. Cells Tissues Organs 191, 167–174. [DOI] [PubMed] [Google Scholar]
- 146.Van Hoof D, Lomas W, Hanley MB & Park E (2014) Simultaneous flow cytometric analysis of IFN-gamma and CD4 mRNA and protein expression kinetics in human peripheral blood mononuclear cells during activation. Cytometry A 85, 894–900. [DOI] [PubMed] [Google Scholar]
- 147.Bourgne C, Bamdad M, Janel A, Libert F, Gagnieu MC, Rapatel C, Pigeon P, Pereira S, Hermet E, Guerci A et al. (2012) Measurement of imatinib uptake by flow cytometry. Cytometry A 81, 996–1004. [DOI] [PubMed] [Google Scholar]
- 148.Armstrong JS, Steinauer KK, Hornung B, Irish JM, Lecane P, Birrell GW, Peehl DM & Knox SJ (2002) Role of glutathione depletion and reactive oxygen species generation in apoptotic signaling in a human B lymphoma cell line. Cell Death Differ 9, 252–263. [DOI] [PubMed] [Google Scholar]
- 149.Dickinson BC & Chang CJ (2008) A targetable fluorescent probe for imaging hydrogen peroxide in the mitochondria of living cells. J Am Chem Soc 130, 9638–9639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Pozarowski P & Darzynkiewicz Z (2004) Analysis of cell cycle by flow cytometry. Methods Mol Biol 281, 301–311. [DOI] [PubMed] [Google Scholar]
- 151.Belloc F, Belaud-Rotureau MA, Lavignolle V, Bascans E, Braz-Pereira E, Durrieu F & Lacombe F (2000) Flow cytometry detection of caspase 3 activation in preapoptotic leukemic cells. Cytometry 40, 151–160. [DOI] [PubMed] [Google Scholar]
- 152.Perfetto SP, Chattopadhyay PK, Lamoreaux L, Nguyen R, Ambrozak D, Koup RA & Roederer M (2006) Amine reactive dyes: an effective tool to discriminate live and dead cells in polychromatic flow cytometry. J Immunol Methods 313, 199–208. [DOI] [PubMed] [Google Scholar]
- 153.Baran-Gale J, Chandra T & Kirschner K (2018) Experimental design for single-cell RNA sequencing. Brief Funct Genomics 17, 233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.AlJanahi AA, Danielsen M & Dunbar CE (2018) An Introduction to the analysis of single-cell RNA-sequencing data. Mol Ther Methods Clin Dev 10, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Haque A, Engel J, Teichmann SA & Lonnberg T (2017) A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med 9, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM et al. (2015) Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Svensson V, Vento-Tormo R & Teichmann SA (2018) Exponential scaling of single-cell RNA-seq in the past decade. Nat Protoc 13, 599–604. [DOI] [PubMed] [Google Scholar]
- 158.Ziegenhain C, Vieth B, Parekh S, Reinius B, Guillaumet-Adkins A, Smets M, Leonhardt H, Heyn H, Hellmann I & Enard W (2017) Comparative analysis of single-cell RNA sequencing methods. Mol Cell 65, 631–643.e4. [DOI] [PubMed] [Google Scholar]