

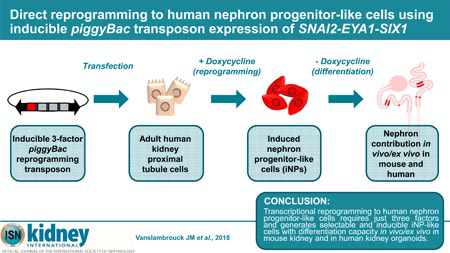
Abstract
All nephrons in the mammalian kidney arise from a transient nephron progenitor population that is lost close to the time of birth. The generation of new nephron progenitors and their maintenance in culture are central to the success of kidney regenerative strategies. Using a lentiviral screening approach, we previously generated a human induced nephron progenitor-like state in vitro using a pool of six transcription factors. Here, we sought to develop a more efficient approach for direct reprogramming of human cells that could be applied in vivo. PiggyBac transposons are a non-viral integrating gene delivery system that is suitable for in vivo use and allows for simultaneous delivery of multiple genes. Using an inducible piggyBac transposon system, we optimized a protocol for the direct reprogramming of HK2 cells to induced nephron progenitor-like cells with expression of only 3 transcription factors (SNAI2, EYA1, and SIX1). Culture in conditions supportive of the nephron progenitor state further increased the expression of nephron progenitor genes. The refined protocol was then applied to primary human renal epithelial cells, which integrated into developing nephron structures in vitro and in vivo. Such inducible reprogramming to nephron progenitor-like cells could facilitate direct cellular reprogramming for kidney regeneration.
Keywords: Reprogramming, direct reprogramming, transposon, nephron progenitor, cap mesenchyme
Graphical Abstract
Introduction
The continued rise in the global incidence of chronic kidney disease (CKD)1 is spurring efforts to develop novel approaches that may benefit future regenerative or cellular therapies. However, progress in this field has been hindered by the complex cellular structure of the mature human kidney. During mammalian development, these multiple kidney cell types are derived from two main progenitor pools: the ureteric bud (UB) and the mesenchymal nephron progenitors (NPs). Through a series of reciprocal inductive signals between these populations, the UB branches to form an intricate collecting duct system for drainage of urine, whilst the NPs undergo an epithelial-to-mesenchymal transition (EMT) to form nephrons (reviewed in Saxen L et al.2). Despite the critical role played by the NP population, these cells exist only transiently during mammalian development, with nephrogenesis ceasing near the time of birth following a final burst of nephron formation.3–6
As such, human NPs are an ideal candidate cell type for kidney regenerative therapies due to their capacity to give rise to all segments of the mammalian nephron. Whilst isolation of putative NPs from human fetal kidneys has been reported,7,8 availability and access to such tissues remains limited. Recently, there has been great interest around the generation of human NPs from pluripotent cell sources,9–14 including our own studies showing the generation of kidney organoids.11,15 While such protocols generate cells possessing phenotypic characteristics of bona fide human NPs, including a capacity to form segmented nephrons, a self-renewing NP population required for continued nephrogenesis is lacking. The long-term maintenance and expansion of these multipotent cells in culture, regardless of source, also continues to be a challenge in the field. We have previously demonstrated the feasibility of direct reprogramming to induced nephron progenitor-like cells (iNP-like cells) using a lentivirus-mediated screen that identified a pool of six transcription factors (SIX1, SIX2, OSR1, HOXA11, EYA1 and SNAI2).16 The resulting iNP-like cells demonstrated appropriate NP gene and protein expression together with a selective capacity to integrate into the endogenous NP population within ex vivo embryonic mouse kidney cultures. However, this screening approach relied on transduction of individual viral constructs, lacking transcription factor inducibility and a selective marker, thus hampering the evaluation of subsequent nephron differentiation capacity.
Here we report the development of a novel piggyBac transposon system for direct transcriptional reprogramming to iNP-like cells. Incorporation of an inducible promoter, selectable marker for cell enrichment and 2A-peptide cleavage signals enabled inducible, simultaneous expression of all six iNP reprogramming factors. Clonal selection and analysis of transposon integration events revealed a requirement for only three of these genes, SNAI2, EYA1 and SIX1, to generate an iNP-like phenotype. The combination of inducible expression of these genes with culture in a previously reported NP-supportive medium, NPSR, proved to be the most effective method to generate human iNP-like cells and also enabled reprogramming of primary proximal tubule cells.17 Such cells were able to contribute to nephron formation in vivo and ex vivo, in neonatal mouse kidney and human iPSC-derived kidney organoids. Not only does this approach represents a significant step towards larger-scale production of NPs, whereby reprogrammable cells may be generated to facilitate bioengineering or drug screening applications, but may also be of significance to in vivo reprogramming approaches in the future.
Results
Development of a piggyBac transposon-mediated reprogramming system
PiggyBac transposons are a non-viral integrating gene delivery system offering several advantages over traditional methods, including a large cargo capacity,18 multiplexed gene delivery,19 flexibility of target cell type20, suitability for in vivo applications21,22, and ability to be excised from the genome.23 To generate a reprogramming system that would provide greater control over reprogramming factor expression compared to lentiviral transduction of individual transcription factors,16 a three-component system was designed that comprised two piggyBac transposons for reprogramming factor delivery (pT-SOH [SIX2, OSR1 and HOXA11] and pT-SES [SNAI2, EYA1 and SIX1]), a separate piggyBac transposon conferring doxycycline inducibility (pT-TetON)21 and a hyperactive piggyBac transposase plasmid (p-EF-1α-HA-m7pB) for efficient mammalian genomic integration (Figure 1 and Table S1).22,24 The multicistronic piggyBac reprogramming transposons were each engineered under the control of a TightTRE doxycycline-inducible promoter, driving the mCherry fluorescent reporter and three reprogramming factors separated by unique 2A peptides (Figure 1 and Table S2). Precise excision of the transposon plasmids from the genome was confirmed (Figure S1A), as was the efficient cleavage of all six intervening 2A-peptides (Figure S1B) and the expression and correct localisation of reprogramming factor proteins for which functional commercial antibodies were available (Figure S1C and Table S3). A transposon with the mCherry reporter alone (pT-mCherry) under the control of the TightTRE promoter was constructed as a control for the transfection and integration process itself (Figure 1). Expression of this reporter confirmed functionality and stringency of the TightTRE promoter (Figure S1D).
Figure 1: piggyBac transposon and transposase plasmid maps.

Transposon plasmids contain the piggyBac 3’ and 5’ terminal repeats (pb3’TR, pb5’TR) flanking the transposon sequence to be integrated permanently into the genome. Abbreviations are as follows: CMV (cytomegalovirus, constitutive viral promoter); rtTA-advanced (advanced reverse tetracycline transactivator protein); SV40 pA (SV40 virus polyadenylation signal); ori (origin of replication); AmpR (ampicillin resistance gene); Tight TRE promoter (tight tetracycline response element promoter); EF-1α promoter (Elongation Factor-1α, constitutive endogenous promoter); HA (N-terminal hemagglutinin tag). White colouring depicts non-transposon portions of the vectors, while black and grey depicts integrating portions of the transposons. Refer to Table S1 for reprogramming factor accession numbers.
In vitro reprogramming of human kidney epithelial cells to a NP-like phenotype
To confirm successful reprogramming to an iNP-like state using the piggyBac transposon system, we utilized the same adult human kidney epithelial cell line (HK2) that was used in our original lentiviral screening approach.16 Following co-transfection of the reprogramming and TetON transposons (pT-SOH, pT-SES and pT-TetON) along with the m7pB hyperactive piggyBac transposase, HK2 cells were exposed to reprogramming conditions with doxycycline and gene expression assessed over a 10 day time course using primers that detect only endogenous gene expression (Figure 2A). A number of NP and EMT markers, including SALL1, SIX2, PAX2, OSR1, MMP2 and MMP9, were found to increase in expression over time. Despite CITED1 RNA levels decreasing dramatically after 4 days of doxycycline exposure, robust CITED1 protein expression was observed between days 6–8 of reprogramming. This data, combined with the spike in stromal marker FOXD1 by day 10, suggested 8 days of doxycycline exposure to be optimal.
Figure 2: HK2 cells show evidence of reprogramming in vitro when transfected with the reprogramming transposons, pT-SOH and pT-SES.

(A) Quantitative RT-PCR (qRT-PCR) time course analysis of EMT, stoma and NP genes in HK2 cells undergoing reprogramming for 4, 6, 8 and 10 days. Normalised gene expression at each time point is presented as a percentage of the maximum expression observed across the experiment duration. Error bars represent ± SD of the mean. (B) CITED1 immunofluorescence time course analysis of HK2 cells from (A), depicting CITED1 and mCherry expression at days 4, 6 and 8 of reprogramming. Scale bars represent 500 μm. (C) Brightfield and fluorescence images of test (transfected with reprogramming transposons) and control (expressing mCherry only) HK2 cells after 8 days of reprogramming. Scale bars represent 500 μm. (D) Immunofluorescence of NP proteins in reprogramming transposon-transfected cells following 8 days of reprogramming. Scale bars represent 30 μm.
By day 8 of reprogramming, the formation of raised structures co-expressing the NP marker CITED1 and mCherry reporter were observed in cultures transfected with the reprogramming transposons (Figure 2 B, C), but were absent from control cells (Figure 2C). Further analysis revealed expression of a range of NP marker proteins in these structures in addition to CITED1, including PAX2, SIX2 and EYA1 (Figure 2D), with co-expression of NP proteins including EYA and SIX2 being confirmed where permitted by antibody species cross-reactivity (Figure S2A). Furthermore, these structures lacked epithelial E-CADHERIN protein expression (Figure S2B) which, along with the increase in MMP2 and MMP9 gene expression, was suggestive of an EMT event required to transform HK2 cells to a mesenchymal state. Taken together, these data suggested the presence of iNP-like cells after reprogramming.
While this represented a polyclonal reprogrammed population with individual cells having different levels and locations of transposon integration, the capacity of these iNP-like cells to form nephrons was assessed using a mouse neonatal kidney injection model which exploits the persistence of the mouse NP population to postnatal day 2 (PND2) (Figure 3A).25 Injections into the cortical renal parenchyma of PND1 mouse kidneys were performed using reprogrammed iNP-like cells (test) or control HK2-mCherry cells (control; HK2 cells transfected with a transposon carrying mCherry only and subjected to the same reprogramming protocol), each combined with fluorescent microspheres for confirmation of injection site. At 3 days post-injection, surviving cells were seen in both test and control kidneys, with a proportion of these cells integrated into developing tubules defined by laminin-bound basement membranes (Figure 3Bi). However, at 7 days post-injection no HK2-mCherry cells persisted in control kidneys. The presence of fluorescent microspheres confirmed that the suspension was delivered successfully and is targeted to the cortical region using this technique (Figure 3C). In contrast, abundant reprogrammed iNP-like cells were integrated into LTL+ proximal and LTL− distal nephron segments (Figure 3Bii). Notable was the formation of extended tubule segments by the iNP-like cells and their binding of LTL on the apical brush border membrane as would be expected for a mature proximal tubule. No reprogrammed cells were observed within the interstitium. Taken together, these data indicated the acquisition of a NP-like phenotype by the reprogrammed HK2 cells and the presence of a subset of cells with nephrogenic differentiation capacity.
Figure 3: iNP-like cells contribute to nephrons in mouse kidney in vivo.

(A) Schematic of the neonatal injection assay used to determine iNP differentiation capacity. (B) Immunofluorescence of sections through mouse kidneys at 3 and 7 days post-injection with iNP-like cells or control HK2-mCherry cells (stained with HuMt, red). Arrows depict examples of integrated HuMt+ cells or HuMt+ tubules. Scale bars represent 30 μm. (C) Immunofluorescence analysis and detection of fluorescence microsphere auto-fluorescence in neonatal kidneys injected with HK2-mCherry control cells and harvested 7 days post-injection. Arrows depict examples of fluorescent microspheres (green). Scale bars represent 30 μm. Dashed lines in A and C indicate the outer edge of the kidney cortex.
Reprogramming is achieved with three factors and is improved with NP-supportive maintenance media
Isolation of individual reprogrammed subclones was performed to examine the relationship between pT-SOH, pT-SES and pT-TetON expression and the presence of a NP-like phenotype. The clone possessing the highest level of endogenous SIX2 and CITED1 expression following reprogramming (Clone 8, Figure S3A) also displayed pronounced mesenchymal morphology from day 5 of reprogramming (Figure S3B). While transposon copy number analysis revealed very low levels of pT-SOH integration in all clones, Clone 8 possessed the highest copy number of pT-SES (Figure S3C). The clone with the highest pT-SOH integration (Clone 12) lacked evidence of NP gene induction. Our previous combinatorial screen for reprogramming factors16 indicated that the three pT-SES genes transduced individually, similar to SIX1 and SNAI2 in combination (Figure S3D), were unable to induce CITED1 expression. This suggests that co-expression of all three genes, SNAI2, EYA1 and SIX1, is sufficient to induce endogenous SIX2 and CITED1 gene expression in HK2 cells and reprogram to an iNP-like state. All additional experiments were performed using this optimally reprogrammable clone.
To examine whether culture of iNP-like cells in recently defined NP maintenance media was feasible, we cultured Clone 8 in three previously described media (CDBLY26, NPEM27 and NPSR25) during reprogramming. Complete replacement of the original basal media for all 8 days of the reprogramming protocol was found to have a negative effect on SIX2 expression (data not shown). Instead, NP-supportive media was added from the time of CITED1 protein expression (approximately day 5 post-doxycycline addition; refer to Figure 2). The addition of NP-supportive media had a significant effect on the expression profile of Clone 8 iNP-like cells (P = 0.008 by two-way ANOVA on normalized [ΔCt] values), improving the expression of a range of NP genes compared to HK2 growth media (HGM) with slightly different outcomes for each media formulation (Figure 4A). Culture in NPEM improved SIX1, PAX2 and MEOX2, while CDBLY culture improved SIX1, PAX2, MEOX2 and CITED1. NPSR resulted in a greater improvement in global NP gene expression, with a larger number of NP markers showing increased average fold change expression above control compared to HGM, CDBLY and NPEM conditions, including SALL1, SIX1, SIX2, EYA1, MEOX2 and OSR. Importantly, control cells lacking the reprogramming transposon but cultured in NPEM or CDBLY exhibited little induction in NP markers, highlighting the importance of having the reprogramming transposons present (Figure S4A). While NPSR alone increased the expression of some NP genes, this induction was lower than in Clone 8 iNPs-like cells for several key NP markers (Figure S4B), suggesting a complementary effect between the media and transposon.
Figure 4: Reprogramming to iNP-like cells is supported by NP-specific maintenance media.

(A) qRT-PCR analysis of NP markers in Clone 8-derived iNP-like cells relative to HK2-mCherry control cells in HGM. Data are presented as mean ± SEM. Statistical significance for each gene across treatment groups was determined using two-way ANOVA and Tukey’s post-test analysis of normalized (ΔCt) values (*P≤0.05, **P≤0.01). (B) RNA-Seq analyses comparing uninduced Clone 8 control cells (blue) with induced Clone 8 iNP-like cells cultured in HGM (red) and NPSR (green), with n = 3 for each condition, showing (i) TREAT analysis heatmap showing relative expression levels of genes identified as significantly differentially expressed between the uninduced and induced conditions (log2 threshold > 1, FDR < 0.05), with expression row scaled to a mean of 0 and standard deviation of 1, (ii) heatmap of gene expression (log2 counts per million) for cap mesenchyme / Crymenriched cap mesenchyme markers, excluding genes present in the reprogramming transposon (SIX1, EYA1 and SNAI2), (iii) heatmap of row scaled gene expression of genes differentially upregulated in human SIX2-enriched NPs and SIX2/MEIS1 interstitial progenitor cells compared to uninduced Clone 8 cells (adjusted p-value < 0.001), and clustered with expression of induced Clone 8 cells, (iv) heatmap of row scaled gene expression of subset of genes that are upregulated in human SIX2-enriched cells compared to uninduced Clone 8 cells that cluster human cells with Clone 8 induced cells, (v) heatmap of row scaled gene expression of genes found in both the subset of genes that cluster human with induced cells, and NP marker genes from single cell analysis of cells from the nephrogenic zone of human fetal kidney.
When developing the NPSR media for NP maintenance, Li et al.17 showed that isolated NPs have the capacity to be cultured as 3D aggregates, a format that was optimal for the maintenance of NP identity in their hands. To investigate the response of reprogrammed cells to this culture system, Clone 8 iNP-like cells were transferred to low attachment 96-well plates and cultured in NPSR. Within 3 days, these cells formed tight spheres while continuing to proliferate, in some cases forming a single floating aggregate by 5 days of culture onwards (Figure S4C). In contrast, Clone-8 iNP-like cells cultured in control media (HGM) and HK2-mCherry control cells cultured in NPSR could not be maintained in this fashion, highlighting the cooperation between the transposon reprogramming and the optimal media conditions.
Induction of a nephron progenitor-like transcriptional profile
In order to further characterize the Clone 8 iNP-like cells at a transcriptional level, RNAseq profiling was performed on parental Clone 8 cells without induction (no doxycycline, HGM media) in comparison to the same lines after 8 days doxycycline induction cultured in either control (HGM) or NP maintenance (NPSR) media. Profiling was performed in triplicate and a PCA analysis showed tight correlation of triplicates for each sample (Figure S4Di). Genes differentially expressed (log2 threshold > 1, FDR < 0.05) between control uninduced Clone 8 and both induced conditions were selected (Table S4). An unbiased clustering revealed strong alignment in gene expression between the induced NPs compared with the uninduced parental clone (Figure 4Bi). GO analysis (https://toppgene.cchmc.org/) using these genes revealed upregulation of a signature of 36 genes from within the top 500 genes enriched in isolated murine NPs (www.gudmap.org.au; P4 KidCapMesRenVes_Crym_top-relative-expression-ranked_500) (Figure 4Bii). This signature also included genes within the reprogramming transposons. While a specific analysis of these 6 transcription factors revealed evidence that all had been induced (Figure S4Dii), expression was significantly higher for SIX1, EYA1 and SNAI2 (Figure S4Diii).
Despite a lack of global RNAseq data from pure populations of embryonic human NPs, to gain insight into the similarities between direct reprogramming-derived iNP-like cells and endogenous human NPs, RNASeq data from the uninduced and induced Clone 8 cells were compared to datasets from NP-enriched cell populations derived from the outer nephrogenic zone of embryonic human kidneys (Figure 4Biii-v).28,29 Using genes from a differential expression comparison between uninduced Clone 8 samples with MARIS/RNA-Seq data from human SIX2-enriched NPs and human SIX2/MEIS1-enriched interstitial progenitor cells (IPCs), induced Clone 8 iNP-like cells were observed to segregate with human endogenous NP populations and showed similar upregulation to human SIX2-enriched cells for multiple gene groups (Figure 4Biii). In subsequent analyses focusing on these gene groups, gene expression was seen to progressively increase from uninduced to induced Clone 8 samples, with overall higher expression in the induced Clone 8 iNP-like cells cultured in NPSR (Figure 4Biv). When comparing to a Single Cell Sequencing dataset obtained from the outer nephrogenic zone of human fetal kidney, iNP-like cells again segregated with the endogenous human progenitor cells rather than the uninduced samples and were enriched for a number of markers that were identified to be differentially expressed in the human NP cluster (Figure 4Bv).
Clone 8 iNP-like cell show nephrogenic potential in mouse and human organoids
While in vitro data supported the role of NP-maintenance media for sustained iNP-like cell culture, it was important to confirm the nephron forming capacity of the resulting cells. To do this, we used two ex vivo differentiation assays; a gold-standard embryonic mouse kidney organoid assay30,31 (Figure 5Ai) and a novel human iPSC-derived kidney organoid assay utilizing our directed differentiation protocol. Of importance, the latter approach enabled the examination of iNP-like cell behavior in a more human context compared to mouse organoids (Figure 5B). Prior to harvesting, organoids were briefly exposed to doxycycline to initiate the re-expression of mCherry by either Clone 8 iNP-like cells or control cells. In mouse kidney organoids, reprogrammed cells were found to integrate into both LTL+ proximal nephron segments as well as LTL− distal nephron segments (co-stained with the epithelial marker E-CADHERIN) when reprogramming was performed in either HGM, CDBLY, NPEM and NPSR (Figure 5Aii). Consistent with the results of gene expression analysis, Clone 8 iNP-like cells cultured in NPSR showed the greatest level of contribution, with extended tubule segments composed of reprogramed cells, predominately in proximal tubules. Within tubules, these integrated cells also showed evidence of mature segment-specific functional proteins, including the distal tubule solute transporter, Slc12a1, and the proximal tubule endocytic receptor, Megalin (Figure S5A). In contrast, HK2-mCherry control cells failed to contribute to nephrons, in some instances clustering together and causing disruption to organoid structure (Figures S5B and S5C). The integration capacity of uninduced Clone 8 control cells (cultured in HGM without doxycycline induction) was similarly evaluated. These cells did not integrate or survive the duration of the experiment (Figure S5C).
Figure 5: iNP-like cells reprogrammed in HGM or NP-supportive media show evidence of nephron-forming capacity in mouse and human organoids.

(Ai) Schematic of the embryonic mouse kidney organoid assay and (Aii) immunofluorescence demonstrating the contribution of Clone 8 iNP-like cells to developing nephrons. Arrows in images depict examples of integrated iNP-like cells (red). (Bi) Schematic of the human iPSC-derived kidney organoid assay and (Bii) immunofluorescence demonstrating the contribution of NPSR-cultured Clone 8 iNP-like cells (red) to developing ECAD+ nephron epithelium (green), NPHS1+ glomeruli (grey) and absence from GATA3+ collecting duct (blue, arrow) . Arrows in left and middle panels depict examples of integrated iNP-like cells (red; magnified in insets). (Biii) Two fields of view from unstained MAFBmTagBFP iPSC-derived organoids demonstrating integration of iNP-like cells (red) into MAFB+ glomeruli (blue) and epithelial structures using endogenous fluorescence of the reporters and phase contrast imaging. Arrows depict examples of iNP-like cells integrated into glomeruli (white arrows) and epithelium (yellow arrows). Scale bars in all panels represent 30 μm.
Our original lentiviral-mediated screen, via quantification of integration into the endogenous nephron progenitor compartment of mouse organoids, estimated reprogramming efficiency to be 0.875%.16 We sought to perform a similar quantification in the current study to determine differentiation efficiency of Clone 8 iNP-like generated using the optimal reprogramming condition in NPSR. Whilst variation existed between organoids, quantification of 6 organoids across 3 independent experiments revealed that of the cells subjected to reprogramming, approximately 39% possessed differentiation efficiency and thus were efficiently reprogrammed (Figure S5D).
In order to further investigate the survival and differentiation capacity of NPSR-cultured Clone 8 iNP-like in a more human setting, these cells were subjected to a human iPSC-derived kidney organoid assay (Figure 5Bi). Similar to mouse kidney organoids, Clone 8 iNP-like cells showed the capacity to differentiate and integrate into developing LTL+ proximal and LTL− distal nephron segments (Figure 5Bii). As would be expected for bona fide NPs, Clone 8 iNP-like cells contributed to NPHS1+ glomeruli but not GATA3+ collecting duct (Figure 5Bii). The use of MAFBmTagBFP iPSC-derived organoids, which report the expression of MAFB-expressing podocytes of the glomeruli (blue),32 enabled further confirmation of the contribution of iNP-like cells to glomeruli and epithelial via endogenous (unstained) expression of mCherry and blue fluorescent protein (BFP), as well as phase contrast imaging showing epithelium (Figure 5Biii). Once again, the ability of uninduced Clone 8 control cells without doxycycline exposure to integrate into developing nephrons was assessed and revealed no integration (Figure S5E). These control cells were seen to cluster together within the interstitium at early time points and either did not survive until the experimental endpoint of 10 days organoid culture or remained clustered within the interstitium.
Refined reprogramming conditions generated iNP-like cells from primary human proximal tubule cells
Primary human renal epithelial cells represent an accessible and reprogrammable cell source that can be isolated from patients through surgery or biopsy or via non-invasive urine collection methods.33,34 We have previously demonstrated that normal human proximal tubule epithelial cells (hRPTECs) transduced with the six iNP reprogramming factors, SIX1, SIX2, OSR1, HOXA11, EYA1 and SNAI2, showed an increase in the expression of NP genes.16 However, these cells did not integrate into endogenous kidney sub-compartments when assessed for nephron progenitor potential in mouse kidney organoid assays (data not shown). Having now developed a refined inducible reprogramming system, hRPTEC cells were subjected to this novel protocol and functionally assessed in the mouse kidney organoid assay (Figure 6).
Figure 6: Refinement of reprogramming factors and conditions enables the generation of iNPlike cells from primary human proximal tubule cells (hRPTECs).

(A) Brightfield images of parental (untransfected) hRPTECs and hRPTEC iNPs that have undergone transfection with pT-SES, pT-TetON and pEF1-HA-m7pB followed by reprogramming. (B) qRT-PCR analysis of NP gene expression in hRPTEC iNP-like cells relative to parental hRPTECs. Data are presented as mean ± SEM. Statistical significance was determined by Student’s t-test (**P≤0.01, n = 3 biological replicates). (C) Immunofluorescence of the mouse kidney organoid assay demonstrating the contribution of hRPTEC iNP-like cells to developing nephrons marked by E-Cadherin (ECad; 692 green). Arrows depict examples of integrated hRPTEC iNP-like cells marked by human nuclear 693 antigen (HuNu; red) and DAPI (nuclei; blue). Scale bar represents 30 μm.
Following electroporation with pT-SES, pT-TetON and hyperactive piggyBac transposase plasmids, hRPTECs were exposed to the optimised reprogramming protocol and media conditions (HGM followed by NPSR) then harvested at day 8 of reprogramming for the mouse kidney organoid assay. These cells showed a marked change in morphology and an increase in the expression of all NP markers except for PAX2 compared to parental hRPTECs (Figure 6A and B). We would note that we have previously noted high endogenous PAX2 expression in parental hRPTECs.16 In functional assays, transfected hRPTECs were observed in three separate kidney recombinations in a variety of locations, including one organoid with abundant hRPTEC iNP-like cells integrated within developing nephron structures (Figure 6C). As anticipated, they were absent from collecting duct (Figure S6A). hRPTEC-mCherry control cells completely failed to integrate into developing nephron structures (Figure S6B). Taken together these data are evidence of the transferability of transposon-mediated reprogramming to hRPTECs, with such cells representing a feasible primary cell source for iNP-like cell derivation.
Discussion
Here we report the use of a piggyBac transposon-mediated system of reprogramming cells to a nephron progenitor-like state with nephrogenic potential. The resulting iNP-like cells showed a capacity to form nephrons both in vivo and in vitro within mouse kidney, as well as integrate into tubules within human kidney organoids. Transcriptional profiling demonstrated the adoption of a NP-like transcriptional profile comparable to the developing human NP population and clonal analysis clarified the requirement for only three transcription factors, SNAI2, EYA1 and SIX1, to recreate an iNP-like phenotype.
The ability to reprogram to iNPs in the absence of SIX2 was surprising given the critical role played by this gene in NP self-renewal and maintenance as shown in mouse kidney,35–37 as well as previous reports associating SIX2 mutation with renal dysplasia and Wilms tumour.38–40 However, it has been recently reported that the NPs of the human fetal kidney express both SIX1 and SIX2, whereas the Six1 protein is not evident in the same region in the mouse.41 Interestingly, SIX1 was found to be one of the most highly regulated targets of SIX2 in human kidney, with both proteins likely to regulate a common set of NP genes through recognition of the same DNA binding motifs. Whilst both proteins complex with EYA1, they are likely to form independent regulatory complexes in vivo. Taken together, successful reprogramming to human iNPs in the absence of SIX2 may be attributed to the functional similarity between these proteins in human kidney and the fact that SIX1 expression may be at least partially activated by SIX2. Genetic loss of Six1 in mouse results in a marked reduction of Six2, Pax2 and Sall1, but not Eya1, leading to apoptosis of the metanephric mesenchyme (MM).42–44 Indeed, data from mouse models suggest that Six1 and Eya1 act upstream of these factors and form a transcriptionally active complex necessary for normal kidney development. This confirms conserved regulatory mechanisms between Six1 and Eya1, also demonstrated by developmental studies of their homologs in drosophila eye45,46 and human fetal kidney,41 as well as co-transfection studies in vitro.47
Expression of Snai2 (Slug) in the endogenous mouse NP population is weak, instead showing strong expression in the cortical stroma.48 SNAI2 was included in the original lentiviral screen due to its activity as an EMT regulatory factor.49 While HK2 cells transduced with SNAI2 alone underwent a marked EMT, they did not adopt a NP-like phenotype,16 suggesting SNAI2’s role the within the reprogramming pool relates to inducing EMT. In support of this concept, studies of murine NP maintenance in vitro highlighted the favorable increase in Snai2 expression when isolated MM cells were treated with low levels of LIF (activating JAK/STAT signaling and maintaining nuclear SIX2) and the Rho kinase inhibitor Y27632 (inhibiting MET through the JNK-mediated differentiation pathway), a combination that is utilized in both CDBLY and NPSR.50 Nevertheless, while inclusion of SNAI2 in the reprogramming cocktail may improve EMT, this factor may not be required for a non-epithelial starting cell population. Similarly, whist having refined the gene set to just three factors for NP induction, it is well-accepted that reprogramming can be imprecise (reviewed in Hendry CE et al. 51), hence there may be several ways to reach a desired endpoint. It will now be possible to use this approach to further characterize and improve reprogramming of primary kidney cells and additional starting cell types.
Inducible transposon approaches are of relevance for transdifferentiation to a range of cellular endpoints, as has been shown with the use of transposons for reprogramming to pluripotency.52 The current study, which is the first demonstration of inducible piggyBac-mediated reprogramming to iNP-like cells with differentiation capacity, represents an incremental yet important advance, potentially inching the field closer to the development of reprogramming strategies as novel disease therapies or for understanding disease pathogenesis. Importantly, this approach may facilitate the large-scale generation of reprogrammable cells that avoids long term culture and expansion of the NPs themselves while retaining their unique phenotype, an issue that continues to be problematic the field. Furthermore, owing to its versatility, this approach not only has the potential to be extended to additional starting cell types, but may also represent a method of precisely generating specific individual cell types of the nephron or achieve direct cellular reprogramming in vivo for tissue regeneration.
Methods
Reprogramming constructs, transfection and clone generation
Reprogramming (pT-SOH and pT-SES) and control (pT-mCherry) transposons were generated using a combination of commercial gene synthesis (GENEWIZ, South Plainfield, New Jersey) and standard cloning procedures. Plasmids pT-TetON21 and pEF-1α-m7pB53 have been described previously. Plasmid DNA transfection was performed with FuGENE (Promega, Madison, Wisconsin) or via electroporation (Neon Transfection system; ThermoFisher Scientific, Waltham, Massachusetts), according to manufacturer’s directions. Additional information can be found in the Supplementary Materials and Methods section. For reprogramming experiments using stably-transfected HK2 cells, transfected cells were cultured for 2 weeks prior to brief exposure to doxycycline and FACS isolation of mCherry-expressing stably transfected cells. Stably transfected clones were generated through serial dilution and expansion of the FACS isolated population.
Cell culture and reprogramming
HK2 cells (ATCC; CRL-2190) were maintained in HK2 growth media (HGM) as described previously. 16,54 hRPTECs (Lonza, Walkersville, Maryland; CC-2553) were maintained in Renal Epithelial Cell Growth Media (REGM; Lonza) according to manufacturer’s instructions. For reprogramming, stably transfected cells were seeded at a density of 4000 cells/cm2. After 48 hours, media was replaced with HGM containing 2 mM valproic acid (VPA; Sigma Aldrich) and 2 μg/mL doxycycline (Sigma Aldrich) for 48 hours. Cells were then exposed to media containing doxycycline without VPA for the duration of reprogramming, refreshing every 24 hours. For NP media experiments, HGM media was removed and replaced with either CDBLY, NPEM or NPSR 17,26,27 containing doxycycline from day 5 onwards.
Immunofluorescence
Cells on coverslips, organoids and tissues were fixed in ice cold 2–4% paraformaldehyde (PFA; Sigma Aldrich). Tissues for sectioning were cryoprotected in 30% sucrose solution (Sigma Aldrich) overnight prior to embedding. Immunofluorescence was performed as previously described, 15 with shortened (1 hour) incubation times for cells and tissue sections. Additional information can be found in the Supplementary Materials and Methods section.
Quantitative RT-PCR
RNA was extracted from cell cultures using RNeasy Mini/Micro Kits (Qiagen) or Bioline Isolate II Mini/Micro Kits (Bioline, New South Wales, Australia) as per manufacturer’s instructions. Synthesis of cDNA and qRT-PCR was performed and analysed as described previously and using the same qRT-PCR primers.16 Each qRT-PCR reaction was performed in triplicate for each biological replicate, with means expressed as +/− SEM. Data was graphed and analysed in GraphPad Prism 7.
RNA-Seq
RNA was sequenced and genes analysed for differential expression (see Supplementary Materials and Methods section). Differential expression was tested between four groups: (1) Clone 8 uninduced and Clone 8 iNP-like cells in HGM, (2) Clone 8 uninduced and Clone 8 iNP-like cells in NPSR, (3) Clone 8 uninduced and both Clone 8 iNP-like cells in HGM and NPSR combined, (4) Clone 8 iNP-like cells in HGM and Clone 8 iNP-like cells in NPSR. The datasets were deposited in the Gene Expression Omnibus(GEO) 55 (accession GSE107410). Additional comparisons between Clone 8 cells and human SIX2-enriched NPs and SIX2/MEIS1 IPCs was calculated by adding samples from GEO accession GSE102596 and using the same methods to perform differential expression tests between the uninduced Clone 8 cells and human SIX-enriched cells as a group.
Neonatal injection assay
Neonatal injections were performed under ultrasound guidance into the renal cortex as described previously 25 in accordance with Institutional animal ethics guidelines (IMB/132/13/NHMRC). Injections were performed in triplicate for control and test cells (approximately 4.5 × 104 cells per injection) and kidneys were harvested at 3 and 7 days post-injection. Reprogramming of the injected test cell population was confirmed via qRT-PCR.
Mouse kidney organoid assay
Assays were performed as previously described,30,31 with 5% exogenous (control or test) cells added to dissociated kidneys prior to aggregation and culture as organoids on 6-well Transwell filter plates (Corning, Corning, New York) for 7 days. A minimum of 3 mouse organoids were generated for each cell type and media condition. Prior to harvesting, organoids were briefly exposed to doxycycline (<6 hours) to re-express mCherry in the exogenous cells.
Human iPSC-derived kidney organoid assay
The CRL1502 clone C32 hiPSC56 and the MAFBmTagBFP hiPSC32 lines were differentiated to renal progenitors as described previously.15 At day 7 differentiation, the iPS-derived progenitor cells were combined with test or control cells to form suspensions (95% iPSC-derived progenitors and 5% exogenous cells). Suspensions were then aggregated and cultured as described previously.15 At least 3 organoids were generated for each cell type and media condition. Prior to harvesting, organoids were briefly exposed to doxycycline (<6 hours) to re-express mCherry in the exogenous cells.
Supplementary Material
Translational Statement:
The detrimental global impact of rising rates of chronic kidney disease and limited patient treatment options is propelling efforts to explore new approaches that may benefit future therapies. In the current study, we identify a combination of three reprogramming factors (SIX1, EYA1 and SNAI2) that, when delivered using a novel inducible piggyBac transposon system, are sufficient to induce reprogramming of adult kidney cells to nephron progenitor-like cells that possess differentiation capacity. It is our hope that advance such as this in the reprogramming field will be of value to the development of novel, regenerative and patient-specific treatment options, as well as studies of disease pathogenesis and drug screening applications, in the future.
Acknowledgements
We acknowledge Associate Professor Jeff Holst for the provision of validated codon-optimised 2A sequences. We thank Han Chiu, Irene Ghobrial and Alex Combes for technical support. This material is the result of work supported with resources and use of facilities at the VA Tennessee Valley Healthcare system and the Molecular Cell Biology Resource Core at Vanderbilt University Medical Center. We acknowledge the Australian Cancer Research Foundation for support of the Institute for Molecular Bioscience Imaging facility. Murdoch Children’s Research Institute is supported by the Victorian Government’s Operational Infrastructure Support Program. This research was funded by the National Health and Medical Research Council of Australia (GNT1041275). L.E.W. was supported by training grants from the National Institutes of Health [2T32DK060445-11], a fellowship from Dr. and Mrs. Harold Seltzman and a Career Development Award from the Department of Veterans Affairs [BX002797]. M.H.W. was supported by the National Institutes of Health [DK093660], Department of Veterans Affairs [BX002190 and BX004258] and Vanderbilt Center for Kidney Disease. M.H.L. was funded by the NHMRC is a NHMRC Senior Principal Research Fellow [APP1042093, APP1136085].
Footnotes
Disclosure
M.H.L. has a research contract with and has consulted for Organovo Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jha V, Garcia-Garcia G, Iseki K et al. Chronic kidney disease: global dimension and perspectives. Lancet 2013; 382: 260–272. [DOI] [PubMed] [Google Scholar]
- 2.Saxen L, Sariola H. Early organogenesis of the kidney. Pediatr Nephrol 1987; 1: 385–392. [DOI] [PubMed] [Google Scholar]
- 3.Hartman HA, Lai HL, Patterson LT. Cessation of renal morphogenesis in mice. Dev Biol 2007; 310: 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hinchliffe SA, Sargent PH, Howard CV et al. Human intrauterine renal growth expressed in absolute number of glomeruli assessed by the disector method and Cavalieri principle. Lab Invest 1991; 64: 777–784. [PubMed] [Google Scholar]
- 5.Rumballe BA, Georgas KM, Combes AN et al. Nephron formation adopts a novel spatial topology at cessation of nephrogenesis. Dev Biol 2011; 360: 110–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ryan D, Sutherland MR, Flores TJ et al. Development of the Human Fetal Kidney from Mid to Late Gestation in Male and Female Infants. EBioMedicine 2018; 27: 275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harari-Steinberg O, Metsuyanim S, Omer D et al. Identification of human nephron progenitors capable of generation of kidney structures and functional repair of chronic renal disease. EMBO Mol Med 2013; 5: 1556–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Da Sacco S, Thornton ME, Petrosyan A et al. Direct Isolation and Characterization of Human Nephron Progenitors. Stem Cells Transl Med 2016; 6: 419–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lam AQ, Freedman BS, Morizane R et al. Rapid and efficient differentiation of human pluripotent stem cells into intermediate mesoderm that forms tubules expressing kidney proximal tubular markers. J Am Soc Nephrol 2014; 25: 1211–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taguchi A, Kaku Y, Ohmori T et al. Redefining the in vivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell Stem Cell 2014; 14: 53–67. [DOI] [PubMed] [Google Scholar]
- 11.Takasato M, Er PX, Becroft M et al. Directing human embryonic stem cell differentiation towards a renal lineage generates a self-organizing kidney. Nat Cell Biol 2014; 16: 118–126. [DOI] [PubMed] [Google Scholar]
- 12.Imberti B, Tomasoni S, Ciampi O et al. Renal progenitors derived from human iPSCs engraft and restore function in a mouse model of acute kidney injury. Sci Rep 2015; 5: 8826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morizane R, Lam AQ, Freedman BS et al. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat Biotechnol 2015; 33: 1193–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morizane R, Bonventre JV. Generation of nephron progenitor cells and kidney organoids from human pluripotent stem cells. Nat Protoc 2017; 12: 195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takasato M, Er PX, Chiu HS et al. Generation of kidney organoids from human pluripotent stem cells. Nat Protoc 2016; 11: 1681–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hendry CE, Vanslambrouck JM, Ineson J et al. Direct transcriptional reprogramming of adult cells to embryonic nephron progenitors. J Am Soc Nephrol 2013; 24: 1424–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Z, Araoka T, Wu J et al. 3D Culture Supports Long-Term Expansion of Mouse and Human Nephrogenic Progenitors. Cell Stem Cell 2016; 19: 516–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li MA, Turner DJ, Ning Z et al. Mobilization of giant piggyBac transposons in the mouse genome. Nucleic Acids Res 2011; 39: e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kahlig KM, Saridey SK, Kaja A et al. Multiplexed transposon-mediated stable gene transfer in human cells. Proc Natl Acad Sci U S A 2010; 107: 1343–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woodard LE, Wilson MH. piggyBac-ing models and new therapeutic strategies. Trends Biotechnol 2015; 33: 525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saridey SK, Liu L, Doherty JE et al. PiggyBac transposon-based inducible gene expression in vivo after somatic cell gene transfer. Mol Ther 2009; 17: 2115–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doherty JE, Huye LE, Yusa K et al. Hyperactive piggyBac gene transfer in human cells and in vivo. Hum Gene Ther 2012; 23: 311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elick TA, Bauser CA, Fraser MJ. Excision of the piggyBac transposable element in vitro is a precise event that is enhanced by the expression of its encoded transposase. Genetica 1996; 98: 33–41. [DOI] [PubMed] [Google Scholar]
- 24.Yusa K, Zhou L, Li MA et al. A hyperactive piggyBac transposase for mammalian applications. Proc Natl Acad Sci U S A 2011; 108: 1531–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li J, Ariunbold U, Suhaimi N et al. Collecting duct-derived cells display mesenchymal stem cell properties and retain selective in vitro and in vivo epithelial capacity. J Am Soc Nephrol 2015; 26: 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanigawa S, Taguchi A, Sharma N et al. Selective In Vitro Propagation of Nephron Progenitors Derived from Embryos and Pluripotent Stem Cells. Cell Rep 2016; 15: 801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown AC, Muthukrishnan SD, Oxburgh L. A synthetic niche for nephron progenitor cells. Dev Cell 2015; 34: 229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lindstrom NO, De Sena Brandine G, Tran T et al. Progressive Recruitment of Mesenchymal Progenitors Reveals a Time-Dependent Process of Cell Fate Acquisition in Mouse and Human Nephrogenesis. Dev Cell 2018; 45: 651–660 e654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lindstrom NO, Guo J, Kim AD et al. Conserved and Divergent Features of Mesenchymal Progenitor Cell Types within the Cortical Nephrogenic Niche of the Human and Mouse Kidney. J Am Soc Nephrol 2018; 29: 806–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davies JA, Unbekandt M, Ineson J et al. Dissociation of embryonic kidney followed by reaggregation as a method for chimeric analysis. Methods Mol Biol 2012; 886: 135–146. [DOI] [PubMed] [Google Scholar]
- 31.Lusis M, Li J, Ineson J et al. Isolation of clonogenic, long-term self renewing embryonic renal stem cells. Stem Cell Res 2010; 5: 23–39. [DOI] [PubMed] [Google Scholar]
- 32.Hale L, Howden SE, Phipson B et al. Human kidney organoid glomeruli provide an improved approach to interrogate podocyte biology and model podocytopathy at scale. Nat Commun 2018; In Press. [Google Scholar]
- 33.Zhou T, Benda C, Dunzinger S et al. Generation of human induced pluripotent stem cells from urine samples. Nat Protoc 2012; 7: 2080–2089. [DOI] [PubMed] [Google Scholar]
- 34.Oliveira Arcolino F, Tort Piella A, Papadimitriou E et al. Human Urine as a Noninvasive Source of Kidney Cells. Stem Cells Int 2015; 2015: 362562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Self M, Lagutin OV, Bowling B et al. Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J 2006; 25: 5214–5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kobayashi A, Valerius MT, Mugford JW et al. Six2 defines and regulates a multipotent selfrenewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell 2008; 3: 169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park JS, Ma W, O’Brien LL et al. Six2 and Wnt regulate self-renewal and commitment of nephron progenitors through shared gene regulatory networks. Dev Cell 2012; 23: 637–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walz AL, Ooms A, Gadd S et al. Recurrent DGCR8, DROSHA, and SIX homeodomain mutations in favorable histology Wilms tumors. Cancer Cell 2015; 27: 286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weber S, Taylor JC, Winyard P et al. SIX2 and BMP4 mutations associate with anomalous kidney development. J Am Soc Nephrol 2008; 19: 891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wegert J, Ishaque N, Vardapour R et al. Mutations in the SIX1/2 pathway and the DROSHA/DGCR8 miRNA microprocessor complex underlie high-risk blastemal type Wilms tumors. Cancer Cell 2015; 27: 298–311. [DOI] [PubMed] [Google Scholar]
- 41.O’Brien LL, Guo Q, Lee Y et al. Differential regulation of mouse and human nephron progenitors by the Six family of transcriptional regulators. Development 2016; 143: 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li X, Oghi KA, Zhang J et al. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature 2003; 426: 247–254. [DOI] [PubMed] [Google Scholar]
- 43.Xu J, Xu PX. Eya-six are necessary for survival of nephrogenic cord progenitors and inducing nephric duct development before ureteric bud formation. Dev Dyn 2015; 244: 866–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu PX, Zheng W, Huang L et al. Six1 is required for the early organogenesis of mammalian kidney. Development 2003; 130: 3085–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen R, Amoui M, Zhang Z et al. Dachshund and eyes absent proteins form a complex and function synergistically to induce ectopic eye development in Drosophila. Cell 1997; 91: 893–903. [DOI] [PubMed] [Google Scholar]
- 46.Pignoni F, Hu B, Zavitz KH et al. The eye-specification proteins So and Eya form a complex and regulate multiple steps in Drosophila eye development. Cell 1997; 91: 881–891. [DOI] [PubMed] [Google Scholar]
- 47.Ohto H, Kamada S, Tago K et al. Cooperation of six and eya in activation of their target genes through nuclear translocation of Eya. Mol Cell Biol 1999; 19: 6815–6824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McMahon AP, Aronow BJ, Davidson DR et al. GUDMAP: the genitourinary developmental molecular anatomy project. J Am Soc Nephrol 2008; 19: 667–671. [DOI] [PubMed] [Google Scholar]
- 49.Savagner P, Yamada KM, Thiery JP. The zinc-finger protein slug causes desmosome dissociation, an initial and necessary step for growth factor-induced epithelial-mesenchymal transition. J Cell Biol 1997; 137: 1403–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tanigawa S, Sharma N, Hall MD et al. Preferential Propagation of Competent SIX2+ Nephronic Progenitors by LIF/ROCKi Treatment of the Metanephric Mesenchyme. Stem Cell Reports 2015; 5: 435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hendry CE, Little MH. Reprogramming the kidney: a novel approach for regeneration. Kidney Int 2012; 82: 138–146. [DOI] [PubMed] [Google Scholar]
- 52.Woltjen K, Michael IP, Mohseni P et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 2009; 458: 766–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Woodard LE, Cheng J, Welch RC et al. Kidney-specific transposon-mediated gene transfer in vivo. Sci Rep 2017; 7: 44904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jones SG, Ito T, Phillips AO. Regulation of proximal tubular epithelial cell CD44-mediated binding and internalisation of hyaluronan. Int J Biochem Cell Biol 2003; 35: 1361–1377. [DOI] [PubMed] [Google Scholar]
- 55.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002; 30: 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Briggs JA, Sun J, Shepherd J et al. Integration-free induced pluripotent stem cells model genetic and neural developmental features of down syndrome etiology. Stem Cells 2013; 31: 467–478. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.