

Abstract
Patients with idiopathic pulmonary fibrosis (IPF) often experience precipitous deteriorations, termed “acute exacerbations” (AE), marked by diffuse alveolitis and altered gas exchange resulting in significant loss of lung function or mortality. The missense isoleucine to threonine substitution at position 73 [I73T] in the alveolar type-2 cell-restricted Surfactant Protein-C [SP-C] gene [SFTPC], has been linked to clinical IPF. To better understand the sequence of events that impact AE-IPF, we leveraged a murine model of inducible SP-CI73T (SP-CI73T/I73TFlp+/−) expression. Following administration of tamoxifen to 8–12 week old mice, an upregulation of SftpcI73T initiated a diffuse lung injury marked by increases bronchoalveolar lavage fluid (BALF) protein and histochemical evidence of CD45+ and CD11b+ cells infiltrates. Flow cytometry of collagenase digested lung cells revealed a transient, early reduction in SiglecFhiCD11blowCD64hiCD11chi macrophages, countered by sequential accumulation of SiglecFloCD11b+CD64−CD11c−CCR2+Ly6C+ immature macrophages (3 days), Ly6G+ neutrophils (7 days) and SiglecFhiCD11bhiCD11clo eosinophils (2 weeks). By mRNA analysis, BALF cells demonstrated a time dependent phenotypic shift from a pro-inflammatory (3 days) to an anti-inflammatory / profibrotic activation state, along with serial elaboration of monocyte and eosinophil recruitment factors. Intravenous administration of clodronate effectively reduced total BALF cell numbers, CCR2+ immature macrophages, and eosinophil influx while improving survival. In contrast, resident macrophage depletion from intratracheal delivery of clodronate liposomes enhanced SftpcI73T induced mortality. These results using SftpcI73T mice provide a detailed ontogeny for AE-IPF driven by alveolar epithelial dysfunction that induces a polycellular inflammation initiated by the early influx of pro-inflammatory CCR2+Ly6Chi immature macrophages.
INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) is a devastating interstitial lung disease (ILD) characterized by disruption of distal lung architecture that ultimately leads to scar formation, abnormal gas exchange, and respiratory failure. The IPF lung is marked by the pathognomonic histology of Usual Interstitial Pneumonitis (UIP) composed of temporally and spatially heterogeneous areas of fibroblast/myofibroblast accumulation coupled with extracellular matrix deposition, disrupted alveolar architecture, and subpleural honeycombing (1–3). In part, due to incomplete understanding of its pathogenesis, the current “gold-standard” therapy is based on “antifibrotic” (i.e. Pirfenidone; Nintedanib) strategies) which at best only slow the deterioration in pulmonary function (4)
While initial preclinical and translational modeling focused on aberrant mesenchymal expansion in IPF pathogenesis, alveolar type-2 (AT2) epithelial cell dysfunction has re-emerged as a key mechanism driving aberrant lung remodeling through refined crosstalk with all parenchymal populations (5). This notion is bolstered by epidemiological studies and case reports that identified both inherited and sporadic monogenetic mutations in key genes related to telomere length and pulmonary surfactant system proteins (3, 6). Over 60 mutations in the surfactant protein C (SP-C) gene (SFTPC), an AT2 specific product, have been described in both adult and pediatric ILD patients. Functionally, SP-C is known to intimately interact with surfactant lipids due to its hydrophobic structure resulting in surface tension modulation in the distal lung. The missense substitution (g.1286T>C), resulting in a change of isoleucine to threonine at position 73 in the SFTPC proprotein (“SP-CI73T”), represents the most common human ILD mutation (7). In vitro studies of epithelial cell lines expressing mutant SP-CI73T offered mechanistic insights into the cellular changes seen in SFTPCI73T patients, which were each marked by organellar disruption, defective proteostasis, and disruption of cell survival pathways (macroautophagy and mitophagy) (8). To link epithelial dysfunction with fibrotic remodeling in vivo, we developed a novel preclinical allelic knock-in murine model designed to express mutant SftpcI73T in an inducible manner (9), thus allowing temporal characterization of key pathways and cellular constituents that drive the aberrant injury/repair response.
Acute exacerbations represent severe clinical deteriorations in patients with chronic ILD (AE-ILD or AE-IPF) that are associated with a high mortality and characterized by extensive inflammatory cell infiltrates, hypoxic respiratory failure, and histological evidence of diffuse alveolar damage superimposed upon the existing PF pathology. Loss of pulmonary function and increased fibrogenic burden represent key downstream effects associated with survivors, corroborating the notion that components of innate and/or adaptive immunity can contribute to aberrant lung remodeling. Although longitudinal data from IPF cohorts are challenging to acquire, analysis of bronchoalveolar lavage fluid (BALF) has shown that elevated levels of chemokines involved in myeloid cell recruitment (MIP-3, CCL22, and CCL17) correlate with poor disease outcomes (10–12). These biomarkers have also been observed in several preclinical models of lung fibrosis (13, 14), therefore supporting the notion that peripheral myeloid cells mobilized during inflammation have the potential to persist in the tissue and participate in lung remodeling (15–18). Given that traditional approaches to AE-PF such as corticosteroids have unproven efficacy (19), a deeper understanding of the immune cell dynamics driving tissue remodeling are needed to improve efficacy and specificity of current anti-inflammatory strategies.
To address the unmet need for temporal characterization of the inflammation contributing to lung remodeling, the current studies aim to define the sequence of early events resulting in polycellular inflammation during AE-PF. Leveraging the SftpcI73T mouse model, we assessed the ontogeny of the recruitment of these immune cell subsets in lung tissue and BALF following induction of mutant SP-CI73T expression. Furthermore, using depletion protocols, we provide evidence for a key role of peripheral Ly6Chi monocytes in the initiation of the injury. Taken together these studies contribute to a refined understanding of epithelial-immune cell crosstalk in acute episodes of sterile inflammation occurring in PF exacerbations.
MATERIALS AND METHODS
Reagents
Tamoxifen (non-pharmaceutical grade) was purchased from Sigma-Aldrich (St Louis, MO). Clodronate Liposomes were purchased from Encapsula Nanoscience (Encapsula Nanoscience LLC, Brentwood, TN). Giemsa cytological stain was purchased from Sigma-Aldrich. Antibodies used for immunohistochemical and flow cytometric analysis were: CD45 (1:250, Santa Cruz); Eisoniphil Peroxidase (1:150, Santa Cruz); Myeloperoxidase (1:150, Novus Biologics); iNOS (1:200, Abcam); CD11b (1:1500, Abcam); FIZZ-1 (1:800, Stem Cell Technologies); CD64 (1:250, Bioss Antibodies); CCR2 (1:400, Abcam); CCR4 (1:400, Abcam); CX3CR1 (Bioss Antibodies); CD16/32 (eBiosciences), CD11b (eFluo450, eBiosciences); Fixable Viability dye (eFluo780, eBiosciences); SiglecF (PE-CF594, BD Biosciences); CD45 (PerCP5.5, Biolegend); CD11c (BV705, Biolegend); Ly6G (AF700, biolegend); Ly6C (BV510, Biolegend); CD64 (PE/Cy7, Biolegend); CD3 (BUV395, Biolegend); CD43 (PE, Biolegend); CCR2 (AF647, Biolegend). All other reagents were purchased from Thermo Fisher Scientific, Inc. (Waltham, MA), or Sigma-Aldrich.
Murine Model of Tamoxifen Mediated SftpcI73T Expression
Tamoxifen inducible SP-CI73TFlp mice were generated as previously reported (9). Briefly the HA-SP-CI73T-Neo hypomorphic founder line was crossed with a mouse line expressing a FLPe recombinase variant (Flp-O) under control of the mutated ER2 receptor knocked into the Rosa26 locus (Jackson Laboratory, Bar Harbor, ME) to generate the inducible-SP-CI73TFlp line. Tamoxifen treatment of adult SP-CI73T/I73TFlp+/− mice was initiated at 8–12 weeks of age. Both male and female animals were used for the studies. Control groups mice are represented as pooled data from tamoxifen treated SP-CI73T/I73TFlp−/− genotypes or vehicle (oil) treated SP-CI73T/I73TFlp+/−mice. All mice were housed under pathogen free conditions in an AALAC approved barrier facility at the Perelman School of Medicine, University of Pennsylvania. All experiments were approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania.
Lung Histology and Immunohistochemistry
Whole lungs were fixed by tracheal instillation of 10% neutral buffer formalin at a constant pressure (25 cm H2O). Following paraffin embedding, 6 uM sections were cut and stained with Hematoxylin & Eosin (H&E) by the Pathology Core Laboratory of Children’s Hospital of Philadelphia. Immunostaining of deparaffinized tissue sections was performed as previously described (20). `Briefly, after antigen retrieval using citrate buffer (10.2 mM sodium citrate, pH 6.0, for 20 minutes) and quenching of endogenous peroxidase with 3% hydrogen peroxide in methanol (30 minutes), non-specific binding was blocked with 10% goat or rabbit serum according to primary antibody origin. Appropriate serum/IgG controls each diluted in blocking buffer were applied for overnight incubation at 4°C in a humidified chamber. Following incubation with biotinylated secondary antisera (Vectastain Elite ABC kit, Vector Labs, Burlingame, CA) for 30 minutes (room temperature), staining was visualized using a Peroxidase Substrate Kit DAB (Vector Labs) and counterstained with Harris Modified Hematoxylin (Thermo Fisher Scientific, Inc.).
Measurement of Pulmonary Function
Invasive measurement of inspiratory capacity and static lung compliance was performed as part of a terminal harvest. Mice were anesthetized with intraperitoneal pentobarbital, tracheas cannulated with a 18-gauge metal stub adapter, and connected to a small-rodent Flexi Vent ventilator (SCIREQ, Inc. Toronto, Ontario CA). Following establishment of standardized basal ventilatory support at a respiratory rate of 150 breaths per min and a tidal volume of 10 mL/kg of body weight, static lung compliance was measured (using PVs-P [27 cmH2O] maneuver) utilizing the manufacturer’s software.
Bronchoalveolar Lavage Fluid (BALF) Analysis
BALF was collected from mice using five sequential lavages of 1 ml sterile saline ad processed for analysis as previously described (9). Briefly, cell pellets obtained by centrifuging BALF samples at 400 × g for 6 minutes were re-suspended in 1 ml of PBS, and total cell counts determined using a NucleoCounter (New Brunswick Scientific, Edison, NJ). Differential cell counts were determined manually from BALF cytospins stained with modified Giemsa for 20 minutes to identify macrophages, lymphocytes, eosinophils and neutrophils. Total protein content of cell free BALF was determined by the Bradford method with bovine IgG as a standard as described (9).
Cytokine Analysis
First-return aliquots of cell-free BALF were analyzed for CX3CL1 levels using a Luminex platform (Millipore Sigma, Burlington, MA) by the Human Immunology Core at Perelman School of Medicine.
Flow Cytometry and Cell Sorting For Identification of Immune Populations
Following BALF collection, lungs were cleared of blood by cardiac perfusion with saline solution, removed from the chest cavity, minced, and transferred into a 50 ml conical tube and incubated (37°C, 30’) in DMEM + 5% FBS + 2 mg/ml Collagenase D (Cat #11088866001, Roche, Indianapolis, IN). Digested lungs were passed through 70-μm nylon mesh to obtain a single-cell suspension, counted and mixed with ACK Lysis Buffer (Thermo Fisher Scientific) to remove any remaining red blood cells. BALF and tissue cell pellet (5X105 cells) were resuspended in 100μl staining buffer (PBS+0.1% sodium azide) and incubated with anti-mouse CD16/32 antibody (Fc block, eBiosciences, San Diego, CA) for 10 min at 4°C to block nonspecific binding. This was followed by 30-min incubation with fluorescently-tagged antibodies or appropriate isotype controls (0.25–1.5 μg/106 cells) for 30 minutes (4ºC). Cells were then spun and resuspended in staining buffer for viability staining (30 minutes at 4ºC). Cells were fixed in 2% paraformaldehyde and analyzed with an LSR Fortessa (BD Biosciences, San Jose, CA) or FACS ARIA (BD Biosciences) for cell sorting experiments. Immune cells were identified by forward and side scatter followed by doublet discrimination of CD45+ viable cells. Specific subsets were identified using a gating strategy modified from Misharin, et al (21) as we have published (9), using FlowJo software (FlowJo, LLC, Ashland, Oregon).
Quantitative Real-Time Polymerase Chain Reaction
RNA extracted from BALF cell pellets was purified using RLT buffer and used as template for cDNA preparation using the RetroScript Kit (Qiagen, Valencia, CA) following the manufacturer’s protocol. Quantitative single-plex polymerase chain reactions (qPCR) were performed using Taq polymerase and TaqMan qPCR kits (Applied Biosystems/ Thermo Fisher Scientific) on a QuantStudio 7 Flex Real-Time PCR System. The commercial primer sets (all from Applied Biosystems / Thermo Fisher Scientific) were: Ccl2 (MCP-1) (catalog #Mm00441242_m1), Ccl17 (TARC) (catalog #Mm01244826_g1), Cxcl1 (GroA/KC) (catalog #Mm04207460_m1), Il6 (catalog #Mm00446190_m1), Il5 (catalog #Mm00439646_m1), Ccl11 (eotaxin) (catalog #Mm0041238_m1), and 18s (catalog #Mm03928990_g1). Results are expressed as relative quantities of RNA (versus untreated or wild type controls) after normalization of all values to the corresponding 18s RNA content.
Macrophage Depletion
Clodronate, encapsulated in liposomes, was used to deplete monocytes/macrophages in blood and lung (22). Control liposomes contained phosphate-buffered saline (PBS) only. For intratracheal treatment each animal received 50 μL (5 mg of clodronate per mL of total suspension volume) of clodronate or control liposomes. For intravenous administration, mice were restrained so that the tail vein was accessible and an insulin syringe was used to administer liposomes (150 mg/kg), 2 h after i.p. tamoxifen injections.
Statistics
All data are presented with dot-plots and group mean ± SEM unless otherwise indicated. Statistical analyses were performed with GraphPad Instat (GraphPad Software, San Diego, CA). Student’s t-test were used for paired data; for analyses involving multiple groups, one-way or two way analysis of variance (ANOVA) was performed with post hoc testing as indicated, and survival analyses performed using Log Rank (Mantel-Cox) test. In all cases statistical significance was considered at p ≤ 0.05.
RESULTS
SP-CI73T expression results in an influx of inflammatory cells into the lung.
Induction of mutant SP-CI73T expression by intraperitoneal tamoxifen administration resulted in significant changes in lung histology, barrier function, and physiology. Whereas minimal histological changes were observed at 3 days post-induction, marked increases in alveolar septal thickening accompanied by epithelial hyperplasia were notable beginning at 7 days (Fig. 1A). In addition, time-dependent increases in alveolar septal disruption, tissue edema, and hemorrhage occurred in association with increased perivascular cellularity. Consistent with prior observations (9), we also found increased BALF total protein at 7 days post induction, along with reduced total inspiratory capacity and increased tissue stiffness, as measured by quasistatic lung compliance (Table 1).
Figure 1. Parenchymal lung injury and alteration in immune cell composition induced by SP-CI73T expression.
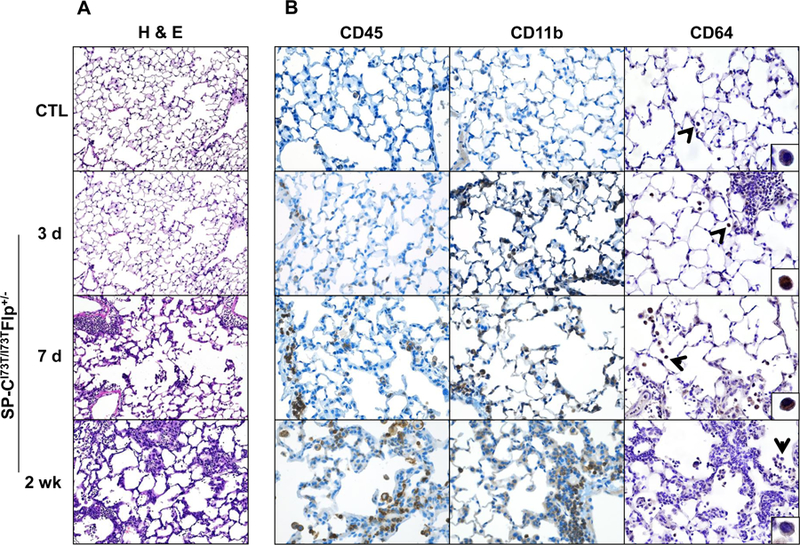
(A) High power images of Hematoxylin & Eosin stained sections (100x) of control (tamoxifen treated SP-CWT/WTFlp+/+ or oil treated SP-CI73T/I73TFlp+/− mice) and SP-CI73T/I73TFlp+/− mice 3 days, 7 days and 2 weeks following intraperitoneal tamoxifen administration (250 mg/kg). (B) Immunohistochemical staining of lungs stained for CD45/immune cells (left panels), CD11b/migrating cells (center panels) and CD64/mature macrophages (right panels). Magnification: 200x; inset magnification 600x. Images shown are representative of 3–5 animals per group.
Table 1. Tissue inflammation, injury and lung mechanics.
Summary analysis for BALF protein content, BALF cell counts and in vivo pulmonary function testing (inspiratory capacity, static compliance and tissue damping) from control (tamoxifen treated SP-CWT/WTFlp+/+ or oil treated SP-CI73T/I73TFlp+/− mice) and tamoxifen treated (250 mg/kg;i.p.) SP-CI73T/I73TFlp+/− mice at 3 d, 7 d and 2 wk. Note that control data represents pooled data from 3 d (N=3), 7 d (N=4), and 2 wk (N=4). Data is represented as mean ± SEM.
| Table 1 | Protein Content (µg/ml) | Cell Count (106) | Inspiratory Capacity (ml) | Static Compliance (ml/cmH2O) | Tissue Damping (G, cmH2O/ml) | |
|---|---|---|---|---|---|---|
| CTL (N=11) | 438.2 ± 38.3 | 0.26 ± 0.02 | 0.74 ± 0.02 | 0.067 ± 0.002 | 4.59 ± 0.15 | |
| SP-CI73T/I73TFlp+/− | 3 d (N=8) | 455.9 ± 61.6 | 0.19 ± 0.03 | 0.66 ± 0.03 | 0.057 ± 0.003 | 5.11 ± 0.28 |
| 7 d (N=7) | 2266.6 ± 422.5* | 0.99 ± 0.18* | 0.51 ± 0.05* | 0.040 ± 0.005* | 7.62 ± 0.74* | |
| 2 wk (N=14) | 2245.2 ± 245.0* | 2.66 ± 0.32* | 0.46 ± 0.03* | 0.031 ± 0.004* | 8.84 ± 1.35* | |
p < 0.05 compared to control SP-CWT/WTFlp+/+ or oil treated SP-CI73T/I73TFlp+/− mice SP-CI73T/I73TER−/− by ANOVA, using Tukey post-hoc test.
These alterations were accompanied by increases in BALF cell counts, highlighting a mounting inflammatory response that peaked at 2 weeks post tamoxifen (Table 1). This finding was mirrored by immunohistochemistry showing accumulation of CD45+ cells in the lung tissue (Fig. 1B, left panels) and perivascular clusters of CD11b+ immune cells, suggesting their peripheral origin (Fig. 1B, center panels). Using the Fc receptor CD64 as a marker of macrophage maturity, early time dependent decreases in fully differentiated macrophages were observed for up to 2 weeks (Fig. 1B, right panels).
Induction of SP-CI73T expression results in monocyte recruitment and a shift in macrophage polarization towards an anti-inflammatory phenotype.
Examination of BALF cytospins obtained from SP-CI73T mice post tamoxifen treatment revealed dynamic changes in a mixed population of effector cells occurring over the course of 2 weeks (Supp. Fig. 1). To further characterize these cells, BALF and digested lung tissue were each analyzed by flow cytometry utilizing a previously described antibody panel and sorting strategy (9, 21) to identify resident SigF+CD11b− alveolar macrophages, Ly6G+ neutrophils, SigF+CD11c− eosinophils, CD3+ lymphocytes, and Ly6Chi immature macrophages (Fig. 2). Correct population gating was validated using Giemsa staining of FACS derived cytospins (Fig. 2, insets).
Figure 2. Flow cytometric analysis of CD45+ populations in BALF and lung tissue.

Gating strategy used to identify myeloid subsets in control (tamoxifen treated SP-CWT/WTFlp+/+ or oil treated SP-CI73T/I73TFlp+/− mice) and SP-CI73T/I73TFlp+/− mice 3 d, 7 d and 2 weeks following intraperitoneal tamoxifen administration (250 mg/kg). BALF and enzymatically digested tissue immune cells were gated on CD45+ following exclusion of doublets. Populations were identified based on a modified protocol from Misharin et al., as previously described (9, 20). Alveolar macrophages were gated based SigF+CD11b− expression and confirmed using CD11c, CD64; neutrophils (CD11b+Ly6G+); eosinophils (SigF+CD11b+CD11c− ); lymphocytes (CD3+CD11b−); and immature macrophages (CD11b+CD64−Ly6Chi). Insets represent Giemsa stained cytospins of FACS sorted subsets.
Using this strategy, we first observed a significant diminution of SigF+CD11c+ CD64+CD11b− alveolar macrophages in BALF and lung tissue digests within 3 days of SP-CI73T induction (Fig. 3A and Supp. Fig. 2B). Whereas the number of these cells returned to control levels by 7 days in the BALF, their relative abundance continued to remain low for up to 2 weeks in the lung tissue (Supp. Fig. 2B). Conversely, BALF and tissue Ly6C+ immature macrophages were increased during this same early inflammatory phase (Fig. 3B and Supp. Fig. 2B). Previously we had shown that SP-CI73T expression increased BALF levels of cytokines involved in the recruitment of monocytes and activated macrophages (CCL2; CCL17) which was mirrored by similar changes in cytokine mRNA expression in AT2 cells (9). qPCR analysis of BALF immune cells indicate that these populations also contribute to Ccl2/Mcp1 production (Fig. 3C) whereas Ccl17 mRNA expression remained unchanged 3 days and 2 weeks following mutant SP-CI73T induction (Fig. 3D). Immunohistochemical analysis of lung sections also showed increased expression of the CCL17 receptor, CCR4, on macrophages at 3 days and returned to control levels at 2 weeks (Fig. 3E). By comparison, progressive increases in numbers of CCR2+ mononuclear cells in the lung were found beginning 3 days post induction, while CCR2 expression was absent on neutrophils and eosinophils (Fig. 3F). Similarly, numbers of monocytes/macrophages expressing the chemokine receptor CX3CR1 were incrementally increased in the lung during the evolution of injury (Fig. 3G), mirrored by commensurate increases in BALF levels of its ligand CX3CL1 (Fig. 3H).
Figure. 3. Characterization of lung macrophage and peripheral monocyte dynamics in SP-CI73T mutant mice.

(A–B) Flow cytometric analysis and quantification of BALF for (A) absolute numbers of SigF+CD11c+CD11b− macrophages and (B) Ly6C+ immature macrophages (absolute numbers left y-axis and relative percentage right y-axis) collected from control (tamoxifen treated SP-CWT/WTFlp+/+ or oil treated SP-CI73T/I73TFlp+/− mice) and SP-CI73T/I73TFlp+/− mice 3 d, 7 d, 2 wk and 4 wk following intraperitoneal tamoxifen administration (250 mg/kg). (C-D) qPCR analysis of BALF cells collected from control (tamoxifen treated SP-CWT/WTFlp+/+ or oil treated SP-CI73T/I73TFlp+/− mice) and SP-CI73T/I73TFlp+/− mice 3 d, and 2 weeks following intraperitoneal tamoxifen administration (250 mg/kg) for markers associated with recruitment of (C) monocytes (Mcp1) and (D) monocyte/macrophages (Ccl17). Data are represented as mean ± SEM (N=3–8). * p < 0.05 compared to control SP-CWT/WTFlp+/+ or oil treated SP-CI73T/I73TFlp+/− mice SP-CI73T/I73TER−/− by One-Way ANOVA, using Tukey post-hoc test. (E-G) Immunohistochemical staining of lungs stained for (E) CCR4, (F) CCR2 and (G) CX3CR1 at the indicated time points showing prominent monocyte/macrophage staining (arrowheads). Note peak expression for CCR4 3 d post mutant induction, and time dependent increases in numbers of CCR2+ and CX3CR1+ macrophages. Magnification: 200x; inset magnification: 600x. Representative images are shown (N=4). (H) CX3CL1 ELISA of BALF from control (tamoxifen treated SP-CWT/WTFlp+/+ or oil treated SP-CI73T/I73TFlp+/− mice) and SP-CI73T/I73TFlp+/− mice 3 d, and 2 weeks following intraperitoneal tamoxifen administration (250 mg/kg). Data are represented as mean ± SEM (N=6–11). Controls were pooled from controls at all 3 time points. *p< 0.05 versus control group using 1-way ANOVA followed by Tukey’s post hoc test.
Macrophages are known to participate in both acute lung injury and resolution/fibrosis (23). Therefore, we sought to temporally define phenotypic changes that occurred after SP-CI73T induced injury. To this end, we measured mRNA expression of markers canonically associated with pro-inflammatory (M1; inducible nitric oxide synthase [Nos2]; Il6) and anti-inflammatory/profibrotic (M2; Arg1, Fizz1) polarization. Due to low baseline expression of inflammatory genes, we expressed quantitated gene expression based on their delta-cycle threshold (dCt) relative to the reference gene 18s (lower dCT ~ higher expression). Although M1 gene expression was increased starting at 3 days after tamoxifen administration, the most dynamic changes observed were on M2 macrophage markers including Arg1 and Fizz1 at later time points (2 weeks) (Fig. 4A). Using 2-ΔΔCt method, we also measured Arg1:Nos2 expression ratio, a surrogate for macrophage activation (24, 25), and found time dependent shift to an anti-inflammatory state (3 days ratio = 10.0; 7 days ratio = 30.4; 2 weeks ratio = 72.7). We further validated these findings at the protein level by staining lung sections and cytospins for iNOS, as well as FIZZ1. Consistent with mRNA analysis, there was upregulation of iNOS in BALF macrophages at 3 days, which persisted up to 2 weeks (Fig. 4B). Conversely, staining for M2 markers increased steadily through the first 2 weeks (Fig. 4C).
Figure 4. Phenotypic shift of BALF cells following SP-CI73T–induced injury.

(A) qPCR analysis of BALF cells collected from control (tamoxifen treated SP-CWT/WTFlp+/+ or oil treated SP-CI73T/I73TFlp+/− mice) and SP-CI73T/I73TFlp+/− mice 3 d, and 2 wk following intraperitoneal tamoxifen administration (250 mg/kg) for markers associated with M1 (Nos2 and Il6) and M2 (Arg1, Fizz1) activation. Data are represented as dCt relative to 18s and shown as mean ± SEM (N=3–6). *p< 0.05 compared to control SP-CWT/WTFlp+/+ or oil treated SP-CI73T/I73TFlp+/− mice SP-CI73T/I73TER−/− by One-Way ANOVA, followed by Tukey post-hoc test. (B-C) Immunohistochemical and cytospin staining of lungs for (B) iNOS and (C) FIZZ-1. Representative images are shown (200x; N=3–5) from control (tamoxifen treated SP-CWT/WTFlp+/+ or oil treated SP-CI73T/I73TFlp+/− mice) and SP-CI73T/I73TFlp+/− mice 3 d and 2 wk following intraperitoneal tamoxifen administration (250 mg/kg).
SP-CI73T expression promotes a multiphasic and multicellular alveolitis.
Using the same gating strategy (Figure 2), analysis of BALF and tissue-derived cells both demonstrated a steady accumulation of Ly6G+ neutrophils peaking at 7 days (Fig. 5A and Supp. Fig. 2B) and SigF+CD11c− eosinophils, peaking at 2 weeks (Fig. 5D and Supp. Fig. 2B). Consistent with these findings, immunohistochemical staining of tissue sections showed increased numbers of myeloperoxidase (MPO) positive neutrophils at 7 days (Fig. 5B), as well as eosinophil peroxidase (EPX) positive eosinophils 2 weeks post induction (Fig. 5E). Furthermore, consistent with our BALF differential analysis, the number of BALF CD3+ cells also steadily increased, though the relative percentage of these cells remained unchanged in the tissue (Suppl. Fig. 2A).
Figure 5. Characterization of BALF neutrophil and eosinophil recruitment in SP-CI73T mutant mice.

Flow cytometric analysis and quantification of BALF (A) Ly6G+ neutrophil and (D) SigF+CD11c− eosinophil subsets collected from BALF in control (tamoxifen treated SP-CWT/WTFlp+/+ or oil treated SP-CI73T/I73TFlp+/− mice) and SP-CI73T/I73TFlp+/− mice 3 d, 7 d, 2 wk and 4 wk following intraperitoneal tamoxifen administration (250 mg/kg). Absolute quantitation was performed by multiplying the relative abundance of each myeloid subset by the BALF cell counts. Data are represented as mean ± SEM (N=25–51). Immunohistochemical staining of lungs stained for (B) myeloperoxidase and (E) eosinophil peroxidase (EPX). Magnification: 200x; inset magnification 600x. Representative images are shown (N=3–5). qPCR analysis of BALF cells collected from control (tamoxifen treated SP-CWT/WTFlp+/+ or oil treated SP-CI73T/I73TFlp+/− mice) and SP-CI73T/I73TFlp+/− mice 3 d, and 2 weeks following intraperitoneal tamoxifen administration (250 mg/kg) for markers associated with (C) neutrophil (Cxcl1) and (F) eosinophil (Eotaxin/Ccl11 and Il5) recruitment. *p<0.05 compared to control SP-CWT/WTFlp+/+ or oil treated SP-CI73T/I73TFlp+/− mice SP-CI73T/I73TFlp−/− by One-Way ANOVA, followed by Tukey post-hoc test.
As previous work from our group identified a central role for epithelial cells in the early recruitment of peripheral subsets of these granulocytes through elaboration of key cytokines (9), we sought to determine whether BALF immune cells could also participate in this process. Interestingly, while BALF cells were not a source of Cxcl1 (Fig. 5C), we did observe increased and persistent expression of Ccl11/Eotaxin and Il5 up to 2 weeks post-tamoxifen (Fig. 5F).
Peripheral monocyte depletion alters the injury course by improving survival and reducing cell influx in SP-CI73T mice.
As a result of this detailed temporal characterization, we identified peripheral Ly6Chi monocytes as the first peripheral subset infiltrating into the tissue through activation of the MCP-1/CCR2 axis and hypothesized that their participation was crucial in driving lung injury and alveolitis caused by SftpcI73T expression. We next tested whether pharmacological depletion of resident alveolar macrophages or peripheral monocytes with control (CL) and clodronate loaded liposomes (CLOD) would alter the course of early and late inflammatory processes. Consistent with a role of resident macrophages in maintenance of homeostasis, depletion of alveolar macrophages using intratracheal CLOD resulted in a significant increase in mortality within 7 days post SftpcI73T induction (Fig. 6A). At this time point, while histological analysis of lungs from tamoxifen induced mice showed relatively mild cellular infiltration, resident macrophage depletion revealed extensive parenchymal remodeling and appearance of conspicuous pockets of inflammatory cells, resembling bronchoalveolar lymphoid tissue (BALT) (Fig. 6B). Of note, neither control nor clodronate loaded liposomes affected lung architecture. Conversely, intravenous delivery of clodronate liposomes, previously shown to alter peripheral monocyte homeostasis, resulted in enhanced survival post SftpcI73T induction (90% TAM+CLOD vs. 54% TAM, at 2 weeks) (Fig. 6C). This protective effect on mortality was reflected in the histology with animals receiving i.v. CLOD demonstrating reduction in inflammatory cells particularly in the alveolar and perivascular regions and overall attenuation of parenchymal lung damage (Fig. 6D). The blunted inflammatory response was also reflected in diminished numbers of total BALF cells in mice treated at both 7 days and 2 weeks post tamoxifen induction (Fig. 6E).
Figure 6. The effect of depletion of alveolar macrophages or peripheral monocytes with clodronate liposomes on lung injury and survival of SP-CI73T mutant mice is dependent upon route of administration.

Kaplan–Meier survival analysis from control (tamoxifen treated SP-CWT/WTFlp+/+ or oil treated SP-CI73T/I73TFlp+/− mice) and tamoxifen induced (250 mg/kg; i.p.) SP-CI73T/I73TFlp+/− mice following (A) intratracheal (50ul/mouse given 48h prior to induction) or (C) intravenous (150 µg/kg given 2 h post induction) clodronate (CLOD) or control (CL) liposome administration (N=3 for control liposome group collected at terminal day; n =10–32 for all other groups). Mean survival using death or body weight < 75% on 2 consecutive days as endpoints is shown. *p<0.05 compared to control mice; #p<0.05 compared to tamoxifen treated mice by Log-Sum (Mantel-Cox) Rank test.
Hematoxylin & Eosin stained sections (40x) from SP-CI73T cohorts treated with i.t. or i.v. clodronate as indicated. (B) i.t. CL treated control mice, i.t. CLOD treated control mice or SP-CI73T/I73TFlp+/− mice 6 days following TAM induction alone or TAM plus i.t. CLOD as labeled. Representative images are shown (N=1–5). (D) i.v. CL treated control mice, CLOD treated control mice or SP-CI73T/I73TFlp+/− mice 6 days following TAM induction alone or TAM plus i.v. CLOD. Representative images are shown (N=4–5).
(E) BALF cell counts from SP-CI73T/I73TFlp+/− mice (N =10–32) harvested 3 days, 7 days and 2 weeks after tamoxifen (“TAM”) induction. A “TAM + CLOD” group was co-treated with intravenous CLOD liposomes (150 µg/kg administered at 2 hours post tamoxifen). Control mice (tamoxifen treated SP-CWT/WTFlp+/+ or oil treated SP-CI73T/I73TFlp+/− mice) received intravenous CLOD or CL liposomes (150 µg/kg administered at 2 hours post tamoxifen or oil injection) as indicated. Data are represented as mean ± SEM (N=3 for each control liposome group harvested at terminal day; N=8–18 for all other groups). *p<0.05 compared to control SP-CWT/WTFlp+/+ or oil treated SP-CI73T/I73TFlp+/− mice; **p<0.05 compared to tamoxifen treated SP-CI73T/I73TFlp+/− mice by One-Way ANOVA, using Tukey post-hoc test.
Intravenous clodronate effectively reduces accumulation of Ly6ChiCCR2+ immature macrophages and eosinophils in the lungs of SftpcI73T expressing mice.
The protective effects of systemic clodronate (i.v. CLOD) administration on the early injury were reflected in alterations in the dynamics of the immune cell composition of BALF and lung tissue. As noted previously, induction of SftpcI73T mutant was associated with early reduction in SigF+CD11b− alveolar macrophages paired with increased Ly6Chi immature macrophages in the BALF (Fig. 3). Systemic clodronate had no effects on alveolar macrophage numbers in BALF (Supp. Fig. 3A) and tissue digests (data not shown). Conversely, i.v. CLOD resulted in reduced immature macrophage numbers in the BALF during the early (3 days) inflammatory phase, but not in total Ly6Chi cells in the tissue (Figs. 7A, 7C). Further analysis on Ly6Chi immature macrophages subpopulations revealed that CLOD had significant effects on BALF and tissue burden of pro-inflammatory Ly6C+CCR2+ immature macrophages at 3 days (Figs. 7B, 7D). Histochemical analysis of lung sections further confirmed reduction of CCR2+ cells in SP-CI73T induced mice following CLOD treatment (Fig. 7E). Additional qPCR analysis of recovered BALF immune cells showed that CLOD treated SftpcI73T mice exhibited no alteration in the gene expression of factors involved in monocyte recruitment (Mcp1), nor macrophage activation (Fizz1, Arg1, Nos2, and Il6) (Supp. Fig. 4). CLOD depletion also had no effect on neutrophil infiltration (Sup. Fig. 3B), while there was significant reduction in eosinophils in both BALF and tissue at 2 weeks assessed by flow cytometry analysis of SigF+CD11c− cells (Figs. 8A, 8B), as well as immunohistochemistry for eosinophil peroxidase (EPX) (Fig. 8C).
Figure 7. Intravenous monocyte depletion with clodronate liposomes results in altered monocyte/macrophage composition in SP-CI73T mice.

(A; C) Changes in relative percentage Ly6Chi immature macrophages in BALF (A) and lung tissue (C) from control (tamoxifen treated SP-CWT/WTFlp+/+ or oil treated SP-CI73T/I73TFlp+/− mice) and SP-CI73T/I73TFlp+/− mice 3 d, 7 d and 2 weeks following intraperitoneal tamoxifen (250 mg/kg) and intravenous clodronate (150 µg/kg, 2 hours post tamoxifen injection) administration. Data are represented as mean ± SEM (N=5–12).
(B; D) Changes in relative percentage of Ly6ChiCCR2+ immature macrophages in BALF (B) and tissue (D) from control (tamoxifen treated SP-CWT/WTFlp+/+ or oil treated SP-CI73T/I73TFlp+/− mice) and SP-CI73T/I73TFlp+/− mice 3 d, 7 d and 2 weeks following intraperitoneal tamoxifen (250 mg/kg) and intravenous clodronate (150 µg/kg, 2 h post tamoxifen injection) administration. Data are represented as mean ± SEM (N=3–8). All analysis was considered significant *p< 0.05 compared to control SP-CWT/WTFlp+/+ or oil treated SP-CI73T/I73TFlp+/− mice SP-CI73T/I73TFlp−/− by One-Way ANOVA, using Tukey post-hoc test.
(E) Representative immunohistochemical staining for CCR2 expression of lung sections from SP-CI73T mice (4 lungs/condition) harvested 14 days after tamoxifen induction with (“TAM + CLOD”) or without (“TAM”) clodronate co-treatment as indicated. “CLOD” represents SP-CWT/WTFlp+/+ control mice co-treated with clodronate alone.
Figure 8. Peripheral monocyte depletion with intravenous clodronate liposomes results in altered eosinophil numbers in SP-CI73T mice.
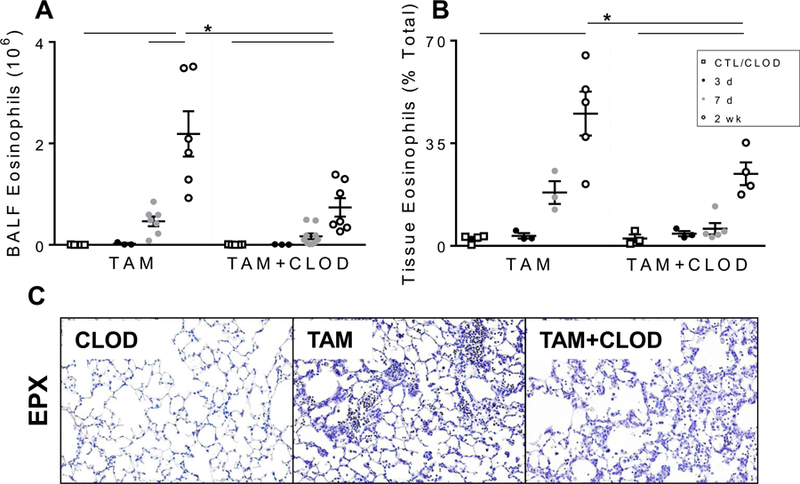
(A-B) Changes in BAL and tissue SigFhiCD11b+CD11c− eosinophils from control (tamoxifen treated SP-CWT/WTFlp+/+ or oil treated SP-CI73T/I73TFlp+/− mice) and SP-CI73T/I73TFlp+/− mice 3 d, 7 d and 2 weeks following intraperitoneal tamoxifen (TAM, 250 mg/kg) and intravenous clodronate (CLOD, 150 µg/kg, 2 h post tamoxifen injection) administration. Data are represented as mean ± SEM (N=3–8). All analysis was considered significant *p< 0.05 compared to control SP-CWT/WTFlp+/+ or oil treated SP-CI73T/I73TFlp+/− mice SP-CI73T/I73TER−/− by One-Way ANOVA, using Tukey post-hoc test. C) Immunohistochemical analysis of vehicle and clodronate (150 µg/kg) treated control (tamoxifen treated SP-CWT/WTFlp+/+ or oil treated SP-CI73T/I73TFlp+/− mice) and SP-CI73T/I73TFlp+/− mice 2 weeks following intraperitoneal tamoxifen administration (250 mg/kg) lungs stained for eosinophil peroxidase (EPX). Representative 40x images from 3–5 separate animals at each condition are shown.
DISCUSSION
Mutations of the SFTPC gene have been linked to various forms of diffuse parenchymal lung disease (DPLD) in humans, namely idiopathic pulmonary fibrosis (IPF) and chILD in adults and pediatric patients, respectively. The clinical course of these DPLDs is often punctuated by acute exacerbations (AE) which are marked by overt spikes in the inflammatory burden to the lung associated with high mortality from superimposed acute lung injury which often results in a significant reduction of pulmonary function among survivors (26). There is an unmet need for new therapies for IPF/ILD due to incomplete understanding of the pathogenesis of both the underlying fibrosis and acute exacerbations, as prior pharmacological interventions (corticosteroids, cytokine modulation, anti-fibrotics) have focused on attenuating downstream responses that at best result in delay, rather than prevention, of pulmonary function decline (27–29).
Part of this challenge to development of effective therapies has been the absence of preclinical experimental models that translate to IPF/chILD patients. Our laboratory has recently developed two murine models of spontaneous pulmonary fibrosis each driven by a distinct functional class of SFTPC mutation that localize within either the BRICHOS (C121G mutant) or ‘linker’ (I73T) domains of the SP-C proprotein (9, 30). Mutant SFTPC BRICHOS isoforms are misfolded and produce extensive AT2 ER stress and apoptosis (30). In contrast, Non-BRICHOS mutants such as SFTPCI73T mistraffic to the AT2 plasma membrane, accumulate in late endosomes, and generate a different toxic gain of function AT2 cell phenotype marked by an acquired block in macroautophagy and hyperproliferation (8–9). Importantly, although expression of these two mutants result in divergent molecular, biochemical, and cellular phenotypes, each is characterized by an overlapping lung phenotype which includes inflammation and extensive tissue remodeling that established a central role for AT2 cells in the initiation of the inflammatory response through release of a diverse array of factors involved in recruitment and activation of immune cell subsets. Herein, the present work extends these prior observations as we now utilize the SP-CI73T Non-BRICHOS model to temporally define and characterize the role of resident (alveolar macrophages) and peripheral (monocytes, neutrophils, and eosinophils) immune cell subsets accumulating in the lung prior to and during the alveolitis, parenchymal injury, lung injury and early remodeling following the induction of mutant SftpcI73T expression. Furthermore, utilizing clodronate depletion we establish a detrimental role for pro-inflammatory Ly6C+ monocytes in injury progression.
Understanding the AT2-immune cell crosstalk coordinating both recruitment of CD45+ effector cells and systemic inflammation is pivotal to expanding and validating new therapeutic paradigms for IPF and chILD. A large part of our understanding of the mechanisms of acute and chronic inflammatory lung disease is the product of experimental models that rely on exogenous exposures (ozone, LPS, or bleomycin) which can non-specifically affect a myriad of parenchymal subsets. Conversely, the SP-CI73T model represents a new and clinically relevant approach to characterize endogenous sterile inflammation and fibrogenesis initiated by epithelial cell stress (9, 31). Leveraging this approach, the induction of mutant SftpcI73T expression was associated with an initial tissue inflammatory response (Fig. 1) in association with a transient decrease in SigF+CD11b− resident alveolar macrophages (Fig. 2). Alveolar macrophages represent the first line of defense against pathogen exposure and may be important for maintaining homeostasis of the alveolar niche, a response achieved in coordination with the lung epithelium and mesenchyme (32, 33). Due to their extensive phenotypic plasticity through adaption to the surrounding chemical milieu (34, 35), macrophages have been classified into two major activation states: classically activated (M1) macrophages, essential in the early response to pro-inflammatory signals (TNF-α, IFN-γ); and alternatively activated (M2) macrophages, important in termination of inflammation as well as in tissue remodeling following Th2 stimulation (IL-10, TGF-β, IL-4 and IL-13) (23). Consistent with this notion we found sequential M1 (3 d; Nos2, Il6) and M2 (2 wk; Arg1, Fizz1) activation following induction of mutant SftpcI73T expression (Fig. 4). It remains to be determined whether the change in activation status observed over time is the result of phenotypic shift of resident macrophages, influx of activated peripheral subsets or rather a combination of the two which will be elucidated in future studies.
Under homeostatic conditions, the alveolar macrophage pool self-maintains through proliferation (36), while a combination of recruitment and maturation of peripheral monocytes is indispensable to overcome loss of resident subsets following injury (37, 38). Bone marrow chimeras and monocyte reporter mice have been necessary to pinpoint and study the phenotypic changes taking place during the shift from monocyte, to immature macrophages, and ultimately mature alveolar macrophage (15, 39). In accord with this notion, the initial reduction in BALF and tissue CD11c+CD64+SigF+CD11b− alveolar macrophages upon SP-CI73T mutant protein induction was countered by accumulation of CD11b+Ly6Chi immature macrophages. This immature myeloid subset was previously shown to be recruited through a CCL2/MCP-1 axis (40) and is congruent with our previous findings of increased MCP-1 levels in the BALF within 3 days post-induction of the SP-CI73T mutant (9). Although the BALF levels of CCL2/MCP-1 returned to control values by 7 days, we found sustained Ccl2 transcript levels in epithelial and immune cells, accompanied by the steady accumulation of CD11b+Ly6ChiCCR2+ immature macrophages in the lung for up to 2 weeks (Fig. 3B and 3F). While this could likely generate a more localized gradient for CCL2/MCP-1 not detectable in whole BALF, we cannot exclude the existence of parallel monocyte recruitment signals. Indeed, our results support this notion by showing increased levels of CCL17 protein and CCR4 expression, thus complementing previous cytokine analysis proposing CCL17/CCR4 axis participation in the development of a non-canonical M2/Th2 inflammatory phenotype (9) in clinical IPF (12, 41). Our data also show that while both epithelial and immune cells can contribute to persistent CCL2/MCP-1 production (9), AT2 cells were the sole source of Ccl17 expression, thus confirming a central role for the epithelium in the immune response in this model and likely in clinical IPF (10, 12, 13).
As monocytes were the first peripheral population recruited in the lung following SP-CI73T induction (Fig. 3), we asked whether disruption of this process could alter injury progression. Intratracheal (i.t.) and intravenous (i.v.) clodronate liposomes represent widely used experimental methodologies to reduce monocyte/macrophage numbers in the compartment of interest (42). Although its mechanism of action is not targeted to specific macrophage subsets, neither route of administration has been shown to affect interstitial cells (43, 44). Perhaps one of the most interesting findings to emerge from this work is represented by the dichotomous effects of clodronate depending upon route of administration. Notable was observation that resident alveolar macrophage depletion, resulted in significantly decreased survival (Fig. 6A), that was accompanied by histological evidence of severe inflammation within 6 days of SP-CI73T induction (Fig. 6B). At higher power, we noted the presence of clusters of neutrophils and lymphocytes. The morphology of these lesions is consistent with a possible acquisition of induced Bronchus-associated Lymphoid Tissue (iBALT), however its definitive identification will require microdissection and flow cytometric characterization of these regions to properly define such structures as published by Harmsen’s group (45). Regardless, the current data intriguingly suggest a pivotal role for resident alveolar macrophages in lung immune homeostasis and in containing the inflammatory burst that develops in response to mutant SP-CI73T induction in AT2 cells (Fig 6B).
Conversely, we found that when systemically administered, clodronate effectively blunted the accumulation of CCR2+Ly6C+ immature macrophages during acute injury, while also significantly improving overall survival and modulating inflammatory endpoints such as total BALF cell numbers, CCR2+ cell influx, and expansion of eosinophils (Figs. 6 and 7). These findings suggest that recruitment of this blood borne subset plays a deleterious role early in SP-CI73T lung injury, a notion consistent with other experimental models of acute and chronic injury in the lung and elsewhere (15, 46, 47). In addition, our data are consistent with findings indicating a pro-inflammatory and pro-injurious function of peripheral monocytes due, at least in part, to their distinct ontogeny from that of resident macrophages (48). This notion is further supported by experimental models of bleomycin-induced injury showing significant contributions of monocyte-derived alveolar macrophages to the aberrant injury and remodeling (15).
In this model, we observed a consistent and well defined dynamic accumulation of eosinophils in tissue and BALF after tamoxifen induced SftpcI73T induction (Fig. 5D; Supp. Fig. 2B). An exhaustive literature describes the role of eosinophils in parasitic infection, allergic disease, and asthma (49, 50). Complementary, clinical evidence has clearly established eosinophil presence in several subtypes of DPLD including IPF (18, 51, 52) and in acute exacerbations (53), however their exact functions are currently unknown. Eosinophils have been shown to promote parenchymal destruction via release of highly toxic granules containing hypobromite, superoxide, and peroxide; yet, they are also source of hematopoietic and growth factors involved in tissue remodeling/fibrosis (e.g. TGF-β) (54, 55). Using this model we previously showed that AT2 epithelial cells represent a source of cytokines associated with eosinophil recruitment (CCL11) and maintenance (IL-5) upon induction of SP-CI73T (9). In the current studies we show by qPCR that BALF immune cells also participate in eosinophil mobilization albeit at a later time point (2 weeks), suggesting they may act to amplify ongoing inflammatory cell recruitment. Notably, as Ly6C+ monocytes have been shown to recruit eosinophils in a CCL11-dependent manner (56), our observations that peripheral monocyte depletion results in reduced eosinophil numbers in the lung after SP-CI73T mutant induction further support the notion that these cells could be important in injury progression.
Through analysis of both tissue and BALF, we demonstrated that these compartments are comparable when characterizing mobile effector cell subsets and for which BALF analysis in patients with a variety of ILDs has often been used to assess disease activity. However, due to the complexity of acute exacerbations, analysis of tissue digests offers additional information of interstitial populations that may contribute to injury initiation and progression. While our data has focused on the role of myeloid immune cell populations, it is likely that effector cells of lymphoid origin such as regulatory T cells, or the recently characterized lineage negative innate lymphoid cells (ILCs), could also play a role. In support to this notion, Tregs have been previously shown to participate to both acute exacerbations of lung disease and aberrant lung remodeling (57, 58), while ILC2s, despite their elusive nature, have been heavily linked with pulmonary disease states enriched in non-canonical Th2 factors (i.e., IL-5, IL-6 and IL-13) (59, 60). Based on our findings of conspicuous levels of these cytokines after SP-CI73T mutant induction, it is intriguing to propose that this regulatory subset could play a role in the dynamic inflammatory events associated with this preclinical model.
In summary, using a novel model of endogenous, sterile lung injury, the present study demonstrates that expression of a mutant Sftpc protein in the pulmonary epithelium of mice recapitulates many features of acute exacerbations of DPLD in human patients. In addition to characterizing the ontogeny of the immune cell response to AT2 cell stress, these results offer important insight into the role of peripheral pro-inflammatory monocytes in the initiation and amplification/progression of inflammatory events in ILD pathogenesis. The complexity of the effector cell cascades observed during SP-CI73T induced acute exacerbations supports the notion that a multi-target therapeutic approach will be required to make significant inroads into the treatment of IPF and chILD.
Supplementary Material
KEY POINTS.
In a murine model of lung fibrosis, an ontogeny of inflammatory events was defined
Mutant SftpcI73T expression caused early lung injury marked by a polycelluar alveolitis
Depletion of peripheral monocytes was protective from SftpcI73T injury
ACKNOWLEDGEMENTS
The authors thank Drs. Debra Laskin, and Andrew Gow for valuable input. MF Beers is the Albert M. Rose Established Investigator of the Pulmonary Fibrosis Foundation.
Grant Support: VA Merit Review 1I01BX001176 (MFB), NIH RO1 HL119436 (MFB), NIH P30 ES013508 (MFB) and HL129150 (SM); JK was supported by NIH 2T32 HL007586
Non-Standard Abbreviations:
- AT2
alveolar type-2 cell
- BRICHOS
Bri family of proteins
- SftpcI73T
surfactant protein-C I73T mutant
- HA
hemaglutinin antigen protein
- ILD
interstitial lung disease
- IPF
idiopathic pulmonary fibrosis
- BALF
bronchoalveolar lavage fluid
- FACS
fluorescence-activated cell sorting
- CLOD
clodronate liposomes
Footnotes
Conflict of Interest: The authors declare that no conflict of interest exists
REFERENCES
- 1.Cameron HS, Somaschini M, Carrera P, Hamvas A, Whitsett JA, Wert SE, Deutsch G, and Nogee LM. 2005. A common mutation in the surfactant protein C gene associated with lung disease. J Pediatr 146: 370–375. [DOI] [PubMed] [Google Scholar]
- 2.Abou Taam R, Jaubert F, Emond S, Le Bourgeois M, Epaud R, Karila C, Feldmann D, Scheinmann P, and de Blic J. 2009. Familial interstitial disease with I73T mutation: A mid- and long-term study. Pediatr Pulmonol 44: 167–175. [DOI] [PubMed] [Google Scholar]
- 3.Crossno PF, Polosukhin VV, Blackwell TS, Johnson JE, Markin C, Moore PE, Worrell JA, Stahlman MT, Phillips JA 3rd, Loyd JE, Cogan JD, and Lawson WE. 2010. Identification of early interstitial lung disease in an individual with genetic variations in ABCA3 and SFTPC. Chest 137: 969–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noble PW, Albera C, Bradford WZ, Costabel U, du Bois RM, Fagan EA, Fishman RS, Glaspole I, Glassberg MK, Lancaster L, Lederer DJ, Leff JA, Nathan SD, Pereira CA, Swigris JJ, Valeyre D, and King TE. 2016. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J 47: 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Selman M, and Pardo A. 2014. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. an integral model. Am J Respir Crit Care Med 189: 1161–1172. [DOI] [PubMed] [Google Scholar]
- 6.Povedano JM, Martinez P, Flores JM, Mulero F, and Blasco MA. 2015. Mice with Pulmonary Fibrosis Driven by Telomere Dysfunction. Cell Rep 12: 286–299. [DOI] [PubMed] [Google Scholar]
- 7.Brasch F, Griese M, Tredano M, Johnen G, Ochs M, Rieger C, Mulugeta S, Muller KM, Bahuau M, and Beers MF. 2004. Interstitial lung disease in a baby with a de novo mutation in the SFTPC gene. Eur Respir J 24: 30–39. [DOI] [PubMed] [Google Scholar]
- 8.Hawkins A, Guttentag SH, Deterding R, Funkhouser WK, Goralski JL, Chatterjee S, Mulugeta S, and Beers MF. 2015. A non-BRICHOS SFTPC mutant (SP-C(I73T)) linked to interstitial lung disease promotes a late block in macroautophagy disrupting cellular proteostasis and mitophagy. Am J Physiol Lung Cell Mol Physiol 308: L33–L47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nureki SI, Tomer Y, Venosa A, Katzen J, Russo SJ, Jamil S, Barrett M, Nguyen V, Kopp M, Mulugeta S, and Beers MF. 2018. Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J Clin Invest 128: 4008–4024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shinoda H, Tasaka S, Fujishima S, Yamasawa W, Miyamoto K, Nakano Y, Kamata H, Hasegawa N, and Ishizaka A. 2009. Elevated CC chemokine level in bronchoalveolar lavage fluid is predictive of a poor outcome of idiopathic pulmonary fibrosis. Respiration 78: 285–292. [DOI] [PubMed] [Google Scholar]
- 11.Capelli A, Di Stefano A, Gnemmi I, and Donner CF. 2005. CCR5 expression and CC chemokine levels in idiopathic pulmonary fibrosis. Eur Respir J 25: 701–707. [DOI] [PubMed] [Google Scholar]
- 12.Yogo Y, Fujishima S, Inoue T, Saito F, Shiomi T, Yamaguchi K, and Ishizaka A. 2009. Macrophage derived chemokine (CCL22), thymus and activation-regulated chemokine (CCL17), and CCR4 in idiopathic pulmonary fibrosis. Respir Res 10: 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Belperio JA, Dy M, Murray L, Burdick MD, Xue YY, Strieter RM, and Keane MP. 2004. The Role of the Th2 CC Chemokine Ligand CCL17 in Pulmonary Fibrosis. J Immunol 173: 4692. [DOI] [PubMed] [Google Scholar]
- 14.Inoue T, Fujishima S, Ikeda E, Yoshie O, Tsukamoto N, Aiso S, Aikawa N, Kubo A, Matsushima K, and Yamaguchi K. 2004. CCL22 and CCL17 in rat radiation pneumonitis and in human idiopathic pulmonary fibrosis. Eur Respir J 24: 49. [DOI] [PubMed] [Google Scholar]
- 15.Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, Chen CI, Anekalla KR, Joshi N, Williams KJN, Abdala-Valencia H, Yacoub TJ, Chi M, Chiu S, Gonzalez-Gonzalez FJ, Gates K, Lam AP, Nicholson TT, Homan PJ, Soberanes S, Dominguez S, Morgan VK, Saber R, Shaffer A, Hinchcliff M, Marshall SA, Bharat A, Berdnikovs S, Bhorade SM, Bartom ET, Morimoto RI, Balch WE, Sznajder JI, Chandel NS, Mutlu GM, Jain M, Gottardi CJ, Singer BD, Ridge KM, Bagheri N, Shilatifard A, Budinger GRS, and Perlman H. 2017. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med 214: 2387–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Obayashi Y, Yamadori I, Fujita J, Yoshinouchi T, Ueda N, and Takahara J. 1997. The role of neutrophils in the pathogenesis of idiopathic pulmonary fibrosis. Chest 112: 1338–1343. [DOI] [PubMed] [Google Scholar]
- 17.Gregory AD, Kliment CR, Metz HE, Kim KH, Kargl J, Agostini BA, Crum LT, Oczypok EA, Oury TA, and Houghton AM. 2015. Neutrophil elastase promotes myofibroblast differentiation in lung fibrosis. J Leukoc Biol 98: 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brix N, Rasmussen F, Poletti V, and Bendstrup E. 2016. Eosinophil alveolitis in two patients with idiopathic pulmonary fibrosis. Respiratory Medicine Case Reports 19: 61–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raghu G, Anstrom KJ, King TE Jr., Lasky JA, and Martinez FJ. 2012. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 366: 1968–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Venosa A, Malaviya R, Choi H, Gow AJ, Laskin JD, and Laskin DL. 2015. Characterization of Distinct Macrophage Subpopulations During Nitrogen Mustard-Induced Injury and Fibrosis. Am J Respir Cell Mol Biol [DOI] [PMC free article] [PubMed]
- 21.Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, and Perlman H. 2013. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol 49: 503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rooijen NV, and Sanders A. 1994. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods 174: 83–93. [DOI] [PubMed] [Google Scholar]
- 23.Mantovani A, and Sica A. 2010. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol 22: 231–237. [DOI] [PubMed] [Google Scholar]
- 24.Munder M, Eichmann K, Moran JM, Centeno F, Soler G, and Modolell M. 1999. Th1/Th2-regulated expression of arginase isoforms in murine macrophages and dendritic cells. J Immunol 163: 3771–3777. [PubMed] [Google Scholar]
- 25.Shi JD, Golden T, Guo CJ, Tu SP, Scott P, Lee MJ, Yang CS, and Gow AJ. 2013. Tocopherol supplementation reduces NO production and pulmonary inflammatory response to bleomycin. Nitric Oxide 34: 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hambly N, Cox G, and Kolb M. 2017. Acute exacerbations of idiopathic pulmonary fibrosis: tough to define; tougher to manage. Eur Respir J 49. [DOI] [PubMed] [Google Scholar]
- 27.Behr J, and Richeldi L. 2013. Recommendations on treatment for IPF. Respir Res 14: S6–S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richeldi L, Davies H, Spagnolo P, and Luppi F. 2003. Corticosteroids for idiopathic pulmonary fibrosis. Cochrane Database Syst Rev [DOI] [PMC free article] [PubMed]
- 29.Papiris SA, Manali ED, Kolilekas L, Triantafillidou C, Tsangaris I, and Kagouridis K. 2012. Steroids in idiopathic pulmonary fibrosis acute exacerbation: defenders or killers? Am J Respir Crit Care Med 185: 587–588. [DOI] [PubMed] [Google Scholar]
- 30.Katzen J, Wagner BD, Venosa A, Kopp M, Tomer Y, Russo SJ, Headen AC, Basil MC, Stark JM, Mulugeta S, Deterding RR, and Beers MF. 2019. A SFTPC BRICHOS mutant links epithelial ER stress and spontaneous lung fibrosis. JCI Insight doi: 10.1172/jci.insight.126125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blackwell TS 2018. Lung injury and fibrosis induced by a mutant form of surfactant protein C. J Clinl Invest 128: 3745–3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herold S, Mayer K, and Lohmeyer J. 2011. Acute Lung Injury: How Macrophages Orchestrate Resolution of Inflammation and Tissue Repair. Front Immunol 2: 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Westphalen K, Gusarova GA, Islam MN, Subramanian M, Cohen TS, Prince AS, and Bhattacharya J. 2014. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature 506: 503–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu H, Wu X, Gang N, Wang S, Deng W, Zan L, and Yu S. 2015. Macrophage functional phenotype can be consecutively and reversibly shifted to adapt to microenvironmental changes. Int J Clin Exp Med 8: 3044–3053. [PMC free article] [PubMed] [Google Scholar]
- 35.Stout RD, and Suttles J. 2004. Functional plasticity of macrophages: reversible adaptation to changing microenvironments. J Leukoc Biol 76: 509–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, Greter M, Mortha A, Boyer SW, Forsberg EC, Tanaka M, van Rooijen N, Garcia-Sastre A, Stanley ER, Ginhoux F, Frenette PS, and Merad M. 2013. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 38: 792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, Brija T, Gautier EL, Ivanov S, Satpathy AT, Schilling JD, Schwendener R, Sergin I, Razani B, Forsberg EC, Yokoyama WM, Unanue ER, Colonna M, Randolph GJ, and Mann DL. 2014. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40: 91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maus UA, Janzen S, Wall G, Srivastava M, Blackwell TS, Christman JW, Seeger W, Welte T, and Lohmeyer J. 2006. Resident alveolar macrophages are replaced by recruited monocytes in response to endotoxin-induced lung inflammation. Am J Respir Cell Mol Biol 35: 227–235. [DOI] [PubMed] [Google Scholar]
- 39.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, Butte AJ, and Bhattacharya M. 2019. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 20: 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gelman AE, Okazaki M, Sugimoto S, Li W, Kornfeld CG, Lai J, Richardson SB, Kreisel FH, Huang HJ, Tietjens JR, Zinselmeyer BH, Patterson GA, Miller MJ, Krupnick AS, and Kreisel D. 2010. CCR2 regulates monocyte recruitment as well as CD4 T1 allorecognition after lung transplantation. Am J Transplant 10: 1189–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pignatti P, Brunetti G, Moretto D, Yacoub MR, Fiori M, Balbi B, Balestrino A, Cervio G, Nava S, and Moscato G. 2006. Role of the chemokine receptors CXCR3 and CCR4 in human pulmonary fibrosis. Am J Respir Crit Care Med 173: 310–317. [DOI] [PubMed] [Google Scholar]
- 42.van Rooijen N, and Hendrikx E. 2010. Liposomes for specific depletion of macrophages from organs and tissues. Methods Mol Biol 605: 189–203. [DOI] [PubMed] [Google Scholar]
- 43.Mathie SA, Dixon KL, Walker SA, Tyrrell V, Mondhe M, O’Donnell VB, Gregory LG, and Lloyd CM. 2015. Alveolar macrophages are sentinels of murine pulmonary homeostasis following inhaled antigen challenge. Allergy 70: 80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sabatel C, Radermecker C, Fievez L, Paulissen G, Chakarov S, Fernandes C, Olivier S, Toussaint M, Pirottin D, Xiao X, Quatresooz P, Sirard JC, Cataldo D, Gillet L, Bouabe H, Desmet CJ, Ginhoux F, Marichal T, and Bureau F. 2017. Exposure to Bacterial CpG DNA Protects from Airway Allergic Inflammation by Expanding Regulatory Lung Interstitial Macrophages. Immunity 46: 457–473. [DOI] [PubMed] [Google Scholar]
- 45.Richert LE, Harmsen AL, Rynda-Apple A, Wiley JA, Servid AE, Douglas T, and Harmsen AG. 2013. Inducible bronchus-associated lymphoid tissue (iBALT) synergizes with local lymph nodes during antiviral CD4+ T cell responses. Lymphat Res Biol 11: 196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sung SA, Jo SK, Cho WY, Won NH, and Kim HK. 2007. Reduction of Renal Fibrosis as a Result of Liposome Encapsulated Clodronate Induced Macrophage Depletion after Unilateral Ureteral Obstruction in Rats. Nephron Exp Nephrol 105: e1–e9. [DOI] [PubMed] [Google Scholar]
- 47.Misharin AV, Cuda CM, Saber R, Turner JD, Gierut AK, Haines GK 3rd, Berdnikovs S, Filer A, Clark AR, Buckley CD, Mutlu GM, Budinger GR, and Perlman H. 2014. Nonclassical Ly6C(−) monocytes drive the development of inflammatory arthritis in mice. Cell Rep 9: 591–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu X, Buttgereit A, Lelios I, Utz SG, Cansever D, Becher B, and Greter M. 2017. The Cytokine TGF-beta Promotes the Development and Homeostasis of Alveolar Macrophages. Immunity 47: 903–912.e904. [DOI] [PubMed] [Google Scholar]
- 49.O’Connell EM, and Nutman TB. 2015. Eosinophilia in Infectious Diseases. Immunol Allergy Clin North Am 35: 493–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Durham SR, and Kay AB. 1985. Eosinophils, bronchial hyperreactivity and late-phase asthmatic reactions. Clin Allergy 15: 411–418. [DOI] [PubMed] [Google Scholar]
- 51.Birring SS, Parker D, McKenna S, Hargadon B, Brightling CE, Pavord ID, and Bradding P. 2005. Sputum eosinophilia in idiopathic pulmonary fibrosis. Inflamm Res 54: 51–56. [DOI] [PubMed] [Google Scholar]
- 52.Peterson MW, Monick M, and Hunninghake GW. 1987. Prognostic role of eosinophils in pulmonary fibrosis. Chest 92: 51–56. [DOI] [PubMed] [Google Scholar]
- 53.Kakugawa T, Sakamoto N, Sato S, Yura H, Harada T, Nakashima S, Hara A, Oda K, Ishimoto H, Yatera K, Ishimatsu Y, Obase Y, Kohno S, and Mukae H. 2016. Risk factors for an acute exacerbation of idiopathic pulmonary fibrosis. Respir Res 17: 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ohkawara Y, Tamura G, Iwasaki T, Tanaka A, Kikuchi T, and Shirato K. 2000. Activation and transforming growth factor-beta production in eosinophils by hyaluronan. Am J Respir Cell Mol Biol 23: 444–451. [DOI] [PubMed] [Google Scholar]
- 55.Tanaka H, Komai M, Nagao K, Ishizaki M, Kajiwara D, Takatsu K, Delespesse G, and Nagai H. 2004. Role of interleukin-5 and eosinophils in allergen-induced airway remodeling in mice. Am J Respir Cell Mol Biol 31: 62–68. [DOI] [PubMed] [Google Scholar]
- 56.Waddell A, Ahrens R, Steinbrecher K, Donovan B, Rothenberg ME, Munitz A, and Hogan SP. 2011. Colonic eosinophilic inflammation in experimental colitis is mediated by Ly6C(high) CCR2(+) inflammatory monocyte/macrophage-derived CCL11. J Immunol 186: 5993–6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Birjandi SZ, Palchevskiy V, Xue YY, Nunez S, Kern R, Weigt SS, Lynch JP 3rd, Chatila TA, and Belperio JA. 2016. CD4(+)CD25(hi)Foxp3(+) Cells Exacerbate Bleomycin-Induced Pulmonary Fibrosis. Am J Pathol 186: 2008–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hams E, Armstrong ME, Barlow JL, Saunders SP, Schwartz C, Cooke G, Fahy RJ, Crotty TB, Hirani N, Flynn RJ, Voehringer D, McKenzie ANJ, Donnelly SC, and Fallon PG. 2014. IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proc Natl Acad Sci U S A 111: 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, Furusawa J, Ohtani M, Fujii H, and Koyasu S. 2010. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 463: 540–544. [DOI] [PubMed] [Google Scholar]
- 60.Barlow JL, Peel S, Fox J, Panova V, Hardman CS, Camelo A, Bucks C, Wu X, Kane CM, Neill DR, Flynn RJ, Sayers I, Hall IP, and McKenzie AN. 2013. IL-33 is more potent than IL-25 in provoking IL-13-producing nuocytes (type 2 innate lymphoid cells) and airway contraction. J Allergy Clin Immunol 132: 933–941. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.