

Abstract
Inefficient cytosolic delivery has limited the development of many promising biomacromolecular drugs, a long-standing challenge that has prompted extensive development of drug carriers that facilitate endosomal escape. Although many such carriers have shown considerable promise for cytosolic delivery of a diversity of therapeutics, the rupture or destabilization of endo/lysosomal membranes has also been associated with activation of the inflammasome with attendant risk of inflammation and toxicity. In this study, we investigated relationships between pH-dependent membrane destabilization, cytosolic drug delivery, and inflammasome activation using a series of well-defined poly[(ethylene glycol)-block-[(2-(dimethylamino)ethyl methacrylate)-co-(butyl methacrylate)] copolymers of variable second block composition and pH-responsive properties. We found that polymers that demonstrated the most potent membrane-destabilizing activity at early endosomal pH values in an erythrocyte hemolysis assay were most efficient at delivery of siRNA, yet tended to be associated with the least amount of NOD-like related protein 3 (NLRP3) inflammasome activation. By contrast, polymers that displayed minimal hemolysis activity and poor siRNA knockdown, and instead mediated lysosomal rupture likely due to a proton sponge mechanism, strongly induced NLPR3 inflammasome activation in a caspase- and cathepsin-dependent manner. Collectively, these findings reinforce the importance of early endosomal escape in minimizing inflammasome activation and also demonstrate the ability to tune the degree inflammasome activation via control of polymer structure with potential implications for design of vaccine adjuvants and immunotherapeutics.
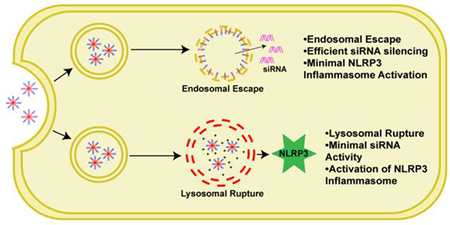
Graphical Abstract
Polymers that are unable to escape the early endosome, and instead cause lysosomal rupture, activate the NLRP3 inflammasome.
Introduction
The cytosol is the site of action for several important classes of therapeutics, including siRNA, miRNA, mRNA, peptides, and enzymes, amongst others; yet, these biomacromolecular drugs have extremely low membrane permeability and minimal access to cytosolic targets.1 This challenge has led to the widespread development of a large diversity of polymeric and nanoparticle drug carriers for cytosolic delivery, the majority of which have been designed to facilitate escape of associated cargo from the endosome and into the cytosol.2–4 These carriers typically mediate destabilization of the endosomal membrane through incorporation of ionizable species that serve as sensors of the lower pH environment within endosomes or lysosomes. Many carriers achieve this through the “proton sponge” mechanism whereby cationic carriers absorb protons, resulting in increased osmotic pressure and eventual rupture or leakage of endo/lysosomes.5–7 Another approach has been to design pH triggerable carriers that unveil or release membrane-destabilizing domains or molecules upon reaching a specific pH within the endosome.1, 4 We, and others, have achieved this active mechanism of endosomal escape through the design of synthetic copolymers comprising both protonatable amino monomers (pKa ~6.2–7) and hydrophobic moieties that mediate disruption of the endosomal membrane.8–13 A key characteristic of these materials is that they are inactive at physiological pH, but rapidly transition to a membrane-destabilizing state at endosomal pH (6.8–5.8). Moreover, by controlling the ratio and/or composition of the cationic and hydrophobic monomers, the magnitude of membrane disruption can be tuned and has been shown to correlate with the efficiency of cytosolic delivery.10, 11, 14 This versatile family of materials has been utilized for delivery of siRNA,10–12, 15 peptides and proteins,8, 16, 17 hydrophilic small molecules,18 and RNA immunotherapeutics,19, 20 with several of these polymers having been demonstrated to be well-tolerated in pre-clinical animal studies.16, 17, 20–22
However, it is also known that many micro- and nanoparticles (e.g., alum,23, 24 and silica25) as well as commonly used cationic drug carriers such as PEI26 and chitosan,27, 28 can induce inflammatory responses via activation of the inflammasome. The inflammasome is a multiprotein complex that is activated in response to cellular stress.29 In the NOD-like related protein 3 (NLRP3) inflammasome, the inflammasome that is best characterized and most associated with disease,30 this complex is made of NOD-like receptors (the sensor), an apoptosis-associated speck-like protein (the adaptor protein), and caspase-1 (the enzymatic component).31 Several mechanisms, all related to cellular stress and infection, have been implicated in the activation of the inflammasome, including K+ efflux, reactive oxygen species (ROS) production, and lysosomal rupture.32–34 In order for the inflammasome to be fully activated, NF-κB needs to be activated first to stimulate the production of pro-IL-1β and pro-IL-18. Next, the second signal (e.g., cellular stress) induces the assembly of the inflammasome, activating caspase 1 which then cleaves the pro-cytokines to IL-1β and IL-18, which are secreted from the cell in their active form.32 Amongst its diverse roles in mediating and shaping innate immune responses to both foreign and endogenous stressors, the inflammasome also serves as an indirect sensor of viral invasion. In one viral infection mechanism, it is endocytosed and the acidic environment allows the virus to fuse to the membrane and form pores to facilitate its replication in the cytosol.35 Several studies have indicated that this viral escape mechanism can activate the inflammasome,36–38 opening the possibility that synthetic carriers with pH-dependent membrane-destabilizing properties may elicit a similar type of response. However, while it has been reported that several commonly used drug carriers can induce inflammasome activation,24, 39–41 the effect of carrier properties on this response has not been widely investigated33, 34, 42 nor is it clear to what extent inflammasome activation is a necessary consequence of endosomal escape and cytosolic drug delivery.
In this study, we aimed to explore how the endosomal escape capacity of pH-responsive polymers correlate to inflammasome activation. To do this, we used a previously described series of diblock copolymers with a poly(ethylene glycol) (PEG) first block and second block comprising a random copolymer dimethylaminoethyl methacrylate (DMAEMA) and butyl methacrylate (BMA).11 We systematically varied the relative amount of BMA in the DMAEMA-co-BMA second block between 0% and 100% to create structurally and chemically similar carriers that exhibit a wide-range of pH-responsive, membrane destabilizing activity, and leveraged these materials to understand relationships between endosomal escape and inflammasome activation. Interestingly, we found little correlation between endosomal escape and inflammasome activation as carriers with the greatest capacity for inflammasome activation (0%−40% BMA) had little endosomolytic activity and were inefficient at delivery of siRNA, and instead mediated lysosomal rupture. By contrast, the most endosomolytic polymers (50%−60% BMA) did not induce appreciable inflammasome activation despite mediating the most efficient siRNA delivery. Collectively, these findings demonstrate that inflammasome activation is not a necessary consequence of effective cytosolic delivery and suggest that early endosomal escape with minimal lysosomal rupture is important for reducing inflammasome-mediated inflammation. Additionally, this work demonstrates that carrier properties can be modulated to tune the degree of inflammasome activation in immune cells, which may be leveraged for the design of more effective carriers for vaccine and immunotherapy applications.
Experimental
Reagents
Ultrapure LPS and Z-VAD-FMK were purchased from Invivogen. The IL-1β ELISA kit, recombinant IL-1β, acridine orange, ethidium homodimer-1, Calcein-AM, and D-luciferin monopotassium salt were purchased from Thermo Fisher Scientific. CA-074 Me, Leu-Leu methyl ester hydrobromide (LLoMe), and phorbol 12-myristate 13-acetate (PMA) were purchased from Sigma-Aldrich. Scrambled siRNA and luciferase siRNA were purchased from Integrated DNA Technologies.
Cell Lines
THP-1 cells were a generous gift from T. Giorgio (Vanderbilt). THP-1 cells were cultured in RPMI 1640 growth media supplemented with 10% heat inactivated FBS, 1% penicillin and streptomycin, 2 mM L-glutamine, 10 mM HEPES, 1X non-essential amino acids, and 125 μM beta-mercaptoethanol. THP1-Def-NLRP3 cells were purchased from Invivogen and cultured in RPMI 1640 growth media supplemented with 10% heat inactivated FBS, 1% penicillin and streptomycin, 2 mM L-glutamine, 25 mM HEPES, 100 μg/mL normacin, and 20 μg/mL hygromycin B gold. Luciferase-expressing MDA-MB-231 cells,22, 43, 44 a generous gift from C. Duvall (Vanderbilt), were cultured in DMEM growth media supplemented with 4.5 g/L D-glucose, 2mM L-glutamine, 10% heat-inactivated FBS, and 1% penicillin and streptomycin.
Polymer Synthesis
Reversible addition-fragmentation chain-transfer (RAFT) polymerization was used to synthesize all polymers as previously described with minor modifications.11 Briefly, dimethylaminoethyl methacrylate (DMAEMA) and butyl methacrylate (BMA) were added to poly(ethylene glycol) 4-cyano-4-(phenylcarbonothioylthio)pentanoate (Sigma), a PEG-modified chain transfer agent (CTA), and dissolved in dioxane with the free radical initiator V70 at a monomer:CTA:initiator ratio of 300:1:0.2. The polymerization was performed under a nitrogen atmosphere at 30°C for 24 hours. The resultant polymer was purified using dialysis (3kDa MWCO) against acetone followed by molecular grade water. The polymers were lyophilized and characterized by 1H NMR (Bruker AV 400) and GPC (Agilent) to determine polymer purity, composition, molecular weight and polydispersity.
Preparation and Characterization of Polymer Solutions
Diblock copolymer solutions were prepared by first dissolving the lyophilized polymers in ethanol at 50 mg/mL followed by rapid dilution in PBS (pH 7.4) to a concentration of 10 mg/mL. Solutions were further diluted as needed for experiments. Particle size in PBS at pH 7.4 and 5.8 was determined using dynamic light scattering analysis (Malvern Zetasizer Nano ZS.)
Erythrocyte Hemolysis Assay
The pH-dependent, membrane-destabilizing activity of polymers was quantified using an erythrocyte hemolysis assay as previously described.9 Briefly, red blood cells (procured from Vanderbilt Hematology Core Lab) were incubated with 5 μg/mL of polymer in 100 mM phosphate buffers at pH values representative of the endo/lysosomal trafficking pathway (7.4, 7.0, 6.6, 6.2, 5.8, 5.2, 4.8). Red blood cell lysis was quantified by amount of hemoglobin released using UV-Vis spectrometry (absorbance of 541 nm) and normalized to 100% lysis (1% Triton X-100).
IL-1β Secretion Assay
Measurement of IL-1β secretion from THP-1 cells were performed as previously described with modifications.45–47 THP-1 and THP1-Def-NLRP3 cells were plated in 96-well plates at a density of 4 × 105 cells/mL with 100 nM PMA for 24 hrs. The media was then replaced with fresh media containing 100 ng/mL LPS for 3 hrs. The media was then replaced with fresh media containing polymers at the indicated concentration for 4 hours. The supernatant was collected and frozen at −80°C until analysis. Secreted IL-1β in cell supernatant was measured using an IL-1β ELISA (Invitrogen) as per manufacturer’s instructions. For studies including the inhibitors Z-VAD-FMK and CA-074 Me, cells were pre-incubated for 30 minutes at a concentration of 20 μM, before addition of polymer solutions.
Microscopy
THP-1 cells were seeded in 4 well chamber slides (Lab-Tek) at a density of 4 × 104 cells/mL with 100 nM PMA. Cells were treated with polymers or LLoMe for 4 hours and then incubated with 2 μg/mL acridine orange for 20 minutes, followed by washing with PBS containing 3% serum twice. Cell were immediately imaged with an Olympus FV-1000 confocal microscope (Vanderbilt Cell Imaging Shared Resource).
Lysosomal Rupture Assay
THP-1 cells were seeded in 12 well plates at a density of 7 × 105 cells/mL and treated with polymer solutions or LLoMe for 4 hours. Cells were then incubated with 2 μg/mL acridine orange for 3 hours, washed 2 times with PBS, and finally re-suspended in PBS with 3% BSA and 0.1% NaN3). Acridine orange release was quantified with flow cytometry (Guava easyCyte HT).24, 39
Cytotoxicity Assay
THP-1 cells were seeded in 12 well plates at a density of 7.5 × 105 cells/mL for 24 hrs and then treated with polymers for 4 hours. Cells were collected, washed and incubated with 8 μM ethidium homodimer-1 and 0.1 μM calcein AM for 20 minutes. Cell viability was analyzed with flow cytometry (Guava easyCyte HT).
Preparation of siRNA Complexed Micelles
For siRNA complexation, polymer dissolved at 50 mg/ml in ethanol was diluted to 3 mg/mL in pH 4 citric acid buffer. This was added to the appropriate amount of siRNA to achieve a N:P complexation ratio of 15:1, and incubated for 30 minutes at room temperature. The pH was then increased to pH 7.4 by adding 5x the volume of pH 8 phosphate buffer.22
Evaluation of Luciferase Knockdown
As a method to evaluate the relative ability of each carrier to mediate cytosolic delivery, luciferase knockdown in luciferase expressing MDA-MB-231 (MDA-MB-231-Luc) cells was used as a model, as previously described.22 Briefly, MDA-MB-231-Luc cells were seeded at a density of 2000 cells per well in a black, clear bottom 96 well plate. The next day, siRNA-polymer complexes were added at a concentration of 10–50 nM. Cells were treated with complexes formulated with either luciferase siRNA or negative control siRNA (DS NC1, Integrated DNA Technologies). After 24 hours, the media was removed and replaced with media containing 150 μg/mL D-luciferin, and bioluminescence was measured with an IVIS Lumina III imaging system.
Statistics
All data is plotted as mean +/− standard deviation unless otherwise noted. Significance was determined using two-way ANOVA unless otherwise noted.
Results and Discussion
Characterization of pH-responsive properties
To examine the effects of polymer composition on inflammasome activation, a series of diblock copolymers of (PEG)-b-(DMAEMA-co-BMA) were synthesized (Table S1). The amount of BMA in the hydrophobic block was varied from 0% BMA to 100% BMA to create a series containing six polymers of variable second block composition, namely 0, 30, 40, 50, 60, and 100% BMA, referred to henceforth as 0B, 30B, 40B, 50B, 60B, and 100B (Fig. 1A). DLS analysis demonstrated that polymers with a sufficiently high amount of hydrophobic BMA in the second block (40B, 50B, 60B, and 100B) formed micellar nanoparticles at pH 7.4, with 40B, 50B, and 60B disassembling at pH 5.8, as indicated by a decrease in particle diameter (Fig. 1B, C). We also evaluated pH-responsive membrane-destabilizing activity in an erythrocyte hemolysis assay, which has been shown previously to correlate well with endosomal escape and cytosolic delivery.11 Consistent with previous reports,11 50B demonstrated the strongest hemolytic activity at pH 6.2–5.8, followed by 60B, 40B, and 30B. As expected 0B, which lacks membrane interactive BMA groups, and 100B, which does not undergo a pH-dependent structural transition, are not hemolytic (Fig. 1D). Therefore, by controlling the amount of BMA in the second block a series of carriers with differing capacities to mediate endosomal escape via active membrane destabilization was generated to allow investigation into how hemolytic activity influences inflammasome activation.
Figure 1. Characterization of diblock copolymer pH-responsive properties.

(A) Structure of and nomenclature for PEG-block-[(DMAEMA)-co-(BMA)x] polymers with differential pH-responsive membrane destabilizing activity achieved through controlling the percentage of BMA (x). (B) Particle size distribution of polymers dissolved in pH 7.4 or 5.8 phosphate buffer measured by dynamic light scattering. (C) Number average diameter of polymers dissolved in pH 7.4 or 5.8 phosphate buffer measured by dynamic light scattering. (D) Relative erythrocyte hemolysis mediated by 5 μg/mL of indicated polymer at pH ranging from 7.4 to 4.8 (n=4).
pH-Dependent Membrane Destabilizing Activity Does Not Correlate with Inflammasome Activation
To examine the relationship between pH-responsive polymer properties and inflammasome activation, the amount of IL-1β secreted by THP-1 cells, a human macrophage line, was measured after treatment with each polymer for 4h at three concentrations (Fig. 2A). Because NF-κB activation is required as a “priming” for inflammasome activation,32 cells were pre-treated with lipopolysaccharide (LPS) prior to introduction of polymer. Interestingly, 30B and 40B stimulated the highest secretion of IL-1β, followed by 0B and 50B, with 60B and 100B resulting in negligible IL-1β production. We validated that polymer-induced IL-1β production was a result of inflammasome activation by measuring IL-1β secretion by THP-1 cells deficient in the NLRP3 inflammasome (THP1-Def-NLRP3) under the same conditions. In all cases, IL-1β secretion was significantly lower in the THP1-Def-NLRP3 cells, indicating that the majority of IL-1β secretion was due to activation of the NLRP3 inflammasome (Fig. 2A). Moreover, to further confirm that IL-1β secretion was inflammasome mediated, cells were pre-treated with a caspase inhibitor, Z-VAD-FMK, to block caspase 1, which is required to cleave pro-IL-1β into IL-1β for secretion. As expected, caspase inhibition returned IL-1β levels to baseline for all polymers (Fig. 2B). Eliminating LPS pre-treatment (Fig. 2C) significantly reduced, but did not entirely abrogate, IL-1β secretion, consistent with the requirement for NF-κB-driven expression of pro-IL-1β. While these results suggest carriers may possess some inherent capacity to induce IL-1β secretion, it is notable that levels of inflammasome activation are generally low for all carriers in the absence of LPS. Such an absence of an exogenous inflammatory stimuli to stimulate NF-κB signaling may, in part, explain why inflammatory responses to this class of pH-responsive carriers has not necessarily been observed.22 Finally, because inflammasome activation can also trigger apoptosis, we evaluated the cytotoxicity of polymers in THP-1 cells. While cytotoxicity did not strictly correlate with the magnitude of inflammasome activation, 30B and 40B, which strongly activated the inflammasome, were also the most toxic whereas 60B and 100B had no effect on cell viability (Fig. 2D). Hence, carriers that activate the inflammasome also tend to be the most cytotoxic, a relationship that may also be reciprocal.
Figure 2. Evaluation of inflammasome activation by PEG-b-(DMAEMA-co-BMA) polymers.

(A) Secretion of IL-1β by THP-1 and THP1-Def-NLRP3 cells treated with LPS (100 ng/mL) for 3 hours and then indicated polymer solution (10–40 μg/mL) for 4 hours as measured with ELISA (n=6; two-way analysis of variance (ANOVA); p-values correspond to comparison between THP1-Def-NLRP3 and THP-1 at same polymer concentration). (B) IL-1β secretion from THP-1 cells treated with LPS (100 ng/mL) for 3 hours and then polymer solutions (10–40 μg/mL) for 4 hours with or without pre-treatment with Z-VAD-FMK (caspase inhibitor), measured with ELISA (n=6; two-way analysis of variance (ANOVA); p-values correspond to Z-VAD-FMK compared to no inhibitor at same concentration for each polymer). (C) Effect of LPS priming on IL-1β secretion in THP-1 cells treated with or without LPS (100 ng/mL) for 3 hours and then 40 μg/mL of polymer solutions for 4 hours, measured with ELISA (n=6; two-way analysis of variance (ANOVA); p-values correspond to no LPS priming compared to LPS priming for each polymer). (D) Cytotoxicity of polymer solutions (40 μg/mL) in THP-1 cells (4 hour treatment), measured by flow cytometry using Calcein (live) AM/Ethidium Homodimer-1 (dead) cell staining (n=2; one-way analysis of variance (ANOVA); p-values correspond to percentage of cell death caused by each polymer compared to no treatment (NT)). *p<0.05, **p<0.01, ***p<0.005 ****p<0.001.
These findings demonstrate, at least within this series of carriers, that potent membrane destabilizing activity at endosomal pH values does not necessarily result in inflammasome activation. It will be important to determine if these trends hold across similar materials comprised of different cationic and hydrophobic monomers.8 These studies further highlight the ability to precisely tune the magnitude of inflammasome induction via control of polymer properties,48 which may have important implications for design of materials where a specified degree of inflammation may be desirable. For example, it has been found that some of the adjuvant activity of alum, the most common vaccine adjuvant, stems from its ability to activate the inflammasome.23, 24 Therefore, polymers that activate the inflammasome while also enhancing intracellular delivery of antigen or adjuvant cargo hold promise for vaccine applications.
Lysosomal Rupture is Correlated with Inflammasome Activation
The introduction of hydrophobic BMA groups into the second block is thought to increase polymer-membrane interactions within early endosomes to facilitate cytosolic delivery prior to trafficking to and/or fusion with highly acidic lysosomes. We therefore hypothesized that cationic carriers with insufficient hydrophobicity to mediate escape from early endosomes (e.g., 0B, 30B, 40B) may instead accumulate within more acidic compartments and cause lysosomal rupture, which has been implicated as a mediator of inflammasome activation for other types of polymers and nanoparticles.24, 33, 34, 39 In order to test this, a lysosomal rupture assay was performed in which a lysosome accumulating dye, acridine orange, was incubated with THP-1 cells following treatment with polymers, and the relevant amount of dye in intact lysosomes was quantified using flow cytometry and fluorescent microscopy. In the low pH environment of the lysosome, acridine orange emits an orange fluorescence, whereas in neutral pH (the cytosol) it emits a green fluorescence, so absence of orange fluorescence indicates loss of intact lysosomes. Flow cytometric analysis indicated that 30B induced the greatest degree of lysosomal rupture, which decreased as the amount of BMA in the second block increased with 60B and 100B mediating negligible rupture even after 24 hours (Fig. 3A, C). These results were confirmed with fluorescence microscopy, where lysosomes that are not ruptured remain orange whereas in cells where lysosomes have ruptured the dye escapes and appears green in the cytosol (Fig. 3B). When the lysosome ruptures, cathepsin is released into the cytosol, a process known to activate the inflammasome.36 Therefore, to further support the role of lysosomal rupture in inflammasome activation, cells were pre-treated with a cathepsin inhibitor, CA-074 Me. Pre-treatment with the inhibitor significantly decreased the amount of IL-1β secretion in all cases, indicating that lysosomal rupture is at least partially responsible for the inflammasome activation (Fig. 3D). Interestingly, 0B (i.e., PEG-b-DMAEMA), which has been used as a nucleic acid carrier and is thought to mediate cytosolic delivery primarily via a proton sponge mechanism,49 induced an intermediate level of lysosomal rupture. This bell-shaped response suggests that increased cationic content or backbone does not necessarily result in more inflammasome activation, and points to the importance of both charge and hydrophobicity in mediating these effects. Interestingly, surface hydrophobicity has been shown to correlate with the magnitude of cytokine secretion induced by gold nanoparticles.50 Nonetheless, we observed a strong correlation between lysosomal rupture and inflammasome activation with this series of carriers, results consistent with recent work by Esser-Kahn and colleagues.33
Figure 3. Evaluation of lysosomal rupture in response to PEG-b-(DMAEMA-co-BMA) polymers.

(A) Representative flow cytometry histograms of THP-1 cells stained with acridine orange (lysosome accumulating dye). THP-1 cells were not treated (NT), incubated with LLoMe (positive control), or treated with indicated polymer solution for 4 hours. (B) Representative fluorescent microscopy images of acridine orange stained THP-1 cells are shown where loss of staining indicates lysosomal rupture. THP-1 cells were not treated (NT), treated with LLoMe (positive control), or treated with polymer solutions for 4 hours and stained with acridine orange for 20 minutes before imaging. Scale bars represent 5 μm. (C) Quantification of lysosomal rupture as measured by flow cytometry in THP-1 cells that were either not treated (NT), treated with LLoMe (positive control), or treated with polymer solutions for 4 or 24 hours and stained with acridine orange for 3 hours. Amount of acridine orange in cells measured using flow cytometry (n=2; one-way analysis of variance (ANOVA); p-value corresponds to percent rupture of each polymer compared to percent rupture of NT, for each time point independently). (D) IL-1β secretion from THP-1 cells treated with LPS (100 ng/mL) for 3 hours and then polymer solutions (10–40 μg/mL) for 4 hours with or without pre-treatment with CA-074 Me (cathepsin inhibitor), measured with ELISA (n=6; two-way analysis of variance (ANOVA); p-value corresponds to CA-074 Me compared to no inhibitor for each polymer). *p<0.05, **p<0.01, ***p<0.005 ****p<0.001.
Efficiency of cytosolic drug delivery does not correlate with carrier-mediated inflammasome activation.
The studies described above suggest that polymers with potent pH-dependent hemolysis activity, and therefore efficient endosomal escape (e.g., 50B, 60B) do not or only weakly activate the inflammasome, whereas carriers that are less hemolytic at early endosomal pH values, but cause lysosomal rupture (30B, 40B), trigger the inflammasome. As the purpose of such carriers is to enhance cytosolic drug delivery, it is important to understand if these trends hold in the context of cargo delivery. To evaluate this, we utilized the cationic DMAEMA groups in the second block to electrostatically complex siRNA, as has been described previously.11, 22 The negatively charged siRNA was complexed to the positively charged DMAEMA at a negative to positive ratio (N:P) of 15:1. First, in comparing inflammasome activation between the different polymers complexed to siRNA, similar trends in IL-1β secretion were seen (Fig. 4A). In this experiment, because a constant siRNA dose and N:P charge ratio of 15:1 was used, for the polymers with less DMAEMA more of the polymer was needed to complex the siRNA. Therefore, the concentration of needed of 50B and 60B was higher than that of 30B and 40B to deliver the same dose of siRNA. Even so, 50B and 60B complexes still induced less IL-1β secretion than 30B. Additionally, siRNA complexation tended to reduce levels of inflammasome activation relative to unloaded carriers (Fig. 4B), potentially reflecting reduction in cellular uptake and/or hindered accessibility of polymers to the endosomal membrane. It is also possible that siRNA complexation reduces the proton sponge effect, since the DMAEMA groups are complexed, and therefore lysosomal rupture and downstream inflammasome activation is decreased.
Figure 4. Evaluation of luciferase knockdown and inflammasome activation with siRNA-loaded carriers.

(A) Secretion of IL-1β by THP-1 cells treated with LPS (100 ng/mL) for 3 hours and then indicated carriers complexed with siRNA (0 – 50nM siRNA) for 4 hours, measured with ELISA (n=6). (B) Secretion of IL-1β by THP-1 cells treated with LPS (100 ng/mL) for 3 hours and then empty polymeric carriers or carriers complexed with siRNA at dose corresponding to 50 nM siRNA for 4 hours, measured with ELISA (n=6). Polymer concentration is matched for both groups ranging from 18 μg/mL (0B) to 38 μg/mL (60B). (C) Percentage of luminescence from MDA-MB-231-Luc cells following 24 hour treatment with indicated carrier complexed to luciferase siRNA, normalized to treatment with the same carrier loaded with scrambled siRNA (n=4; one-way analysis of variance (ANOVA); p-value corresponds to percent luminescence for each polymer compared to percent luminescence of NT, for each concentration independently). *p<0.05, **p<0.01, ***p<0.005 ****p<0.001.
Next, the relative capacity of each carrier to enhance siRNA delivery was tested by evaluating luciferase knockdown in MDA-MB-231 cells expressing luciferase.15 We investigated knockdown in a cancer cell line as a simplified model of siRNA delivery to tumors where the goal is to silence gene expression in cancer cells, yet much of the polymer is endocytosed by myeloid cells, both in the tumor and other sites, which are sensitive to inflammasome activation. DMAEMA-containing polymers were complexed with luciferase siRNA or negative control siRNA, incubated with MDA-MB-231-Luc cells for 24 hours, and luciferase levels were measured using bioluminescent imaging. Consistent with previous reports,11 50B was most efficient at siRNA delivery, resulting in >80% knockdown at 10 nM siRNA, followed by 60B which efficiently silenced luciferase at higher concentrations (50 nM) (Fig. 4C). For the polymers associated with lysosomal rupture (0B, 30B, and 40B), siRNA was not efficiently delivered to the cytosol, likely due to cellular stress and eventual death.
These results further demonstrate that efficient cytosolic delivery via endosomal escape does not necessarily result in significant inflammasome activation. For the majority of applications where maximizing delivery and minimizing inflammatory toxicities is the primary goal, these studies also reinforce the importance of early endosomal escape1, 51 and minimizing lysosomal damage in carrier design. Our results indicate that for cytosolic delivery endosomal delivery via membrane destabilization due to hydrophobic BMA is more effective and less damaging than lysosomal rupture due to the proton sponge effect. It is also notable that 50B, while significantly more efficient at siRNA delivery than 60B, did induce a small, but discernible level of inflammasome activation whereas 60B was entirely inert. As a result of its reduced charge density, it is conceivable that the lower siRNA knockdown observed for 60B is, in part, a consequence of weak and unstable electrostatic complexation of siRNA 44; if so, this carrier may prove useful for other delivery applications owing to its balanced hemolytic activity and lack of inflammasome activation. Moreover, the stark differences in both siRNA delivery and inflammasome activation observed as the BMA content is increased between 40 to 60% BMA suggests that there is a finite, albeit somewhat narrow window, in which efficient cytosolic delivery can be achieved with minimal inflammasome activation. This motivates to the importance of using controlled polymerization techniques (e.g., RAFT) that yield tight control over polymer composition52 since high polydispersity may yield species with high potential for inflammasome activation.
Conclusions
Escape from the endosome is a widely recognized bottleneck for delivery of a large class of promising therapeutics, yet the process of disrupting endo/lysosomal membranes has potential to trigger activation of the inflammasome, resulting in potentially undesirable inflammatory or toxic effects. Using a series of PEG-block-(DMAEMA-co-BMA) polymers with variable second block compositions and differential pH-responsive membrane-destabilizing activity, we have demonstrated that potent endosomal escape activity does not obligate, or directly correlate with, activation of the NLRP3 inflammasome (Fig. 5A). Instead, polymer-mediated lysosomal rupture was more predictive of inflammasome activation (Fig. 5B). Our data suggests that carriers lacking sufficient BMA content to escape early endosomes instead traffic to lysosomes where they mediate lysosomal rupture or membrane damage, potentially via a proton sponge mechanism mediated by DMAEMA groups (Fig. 5C). This results in release of cathepsin, a known activator of the inflammasome. Importantly, the polymers most efficient at cytosolic siRNA delivery (50B and 60B) induced little to no inflammasome activation, consistent with the excellent safety profile of these materials in preclinical mouse models 12, 16, 22. The polymers that do not induce inflammasome activation are likely escaping the endosome early enough that cathepsins, which are activated in the acidic environment of the late endosome53, are not yet activated. This further highlights the importance to design drug delivery vehicles that can escape the endosome early. These studies also reveal the importance of establishing relationships between polymer properties and inflammasome signaling, which is seldom considered when designing drug delivery vehicles. In most cases, inflammasome activation should be minimized as it can lead to undesirable inflammatory responses; however, in some cases this may be desirable, for example, in vaccine delivery. Overall, in addition to more commonly evaluated metrics of drug carrier properties (e.g., toxicity, cellular uptake, transfection efficiency, cytosolic delivery), the intrinsic capacity of materials to trigger inflammasome activation should considered and properties optimized for specific applications.
Figure 5. Lysosomal rupture, but not membrane destabilizing activity at early endosomal pH-value, is correlated to inflammasome activation.

(A) Relationship between percent hemolysis at pH 6.2 and IL-1β secretion after treatment with 40 μg/mL for 4 hours demonstrates there is not a correlation between hemolytic activity and inflammasome activation. (B) Relationship between percentage of lysosomal rupture after 4 hours and IL-1β secretion after treatment with polymers at 40 μg/mL for 4 hours reveals a positive correlation between lysosomal rupture and inflammasome activation. (C) Data support a working hypothesis that PEG-b-(DMAEMA-co-BMA) polymers with 50–60% BMA (50B and 60B) are endocytosed, and disassemble at early endosomal pH values to mediate disruption the endosomal membrane, resulting in release of cargo into the cytosol. By contrast, polymers containing 0–40% BMA do not have enough BMA to actively solvate endosomal membranes, instead trafficking to lysosomal compartments, where they ultimately induce lysosomal rupture and release of cathepsins, which contributes to inflammasome activation and secretion of IL-1β.
Supplementary Material
Acknowledgements
We gratefully acknowledge Dr. Todd Giorgio for providing THP-1 cells, Dr. Craig Duvall for use of gel permeation chromatography and IVIS equipment, and Meredith Jackson for providing MDA-MB-231-Luc cells and for technical advice on siRNA knockdown studies. We would also like to acknowledge the Vanderbilt Hematology Core Lab for providing red blood cell samples, the Vanderbilt Institute of Nanoscale Science and Engineering (VINSE) for use of a Malvern Zetasizer, the Vanderbilt Cell Imaging Shared Resource (CISR) for use of Olympus FV-1000 confocal microscope, and the Small Molecule NMR Facility Core for use of a Bruker NMR spectrometer. This research was supported by grants from the National Science Foundation (CBET-1554623 and DMR-1560414), the National Institutes of Health (5R21AI121626), and the Congressionally-Directed Medical Research Program (W81XWH-161-0063).
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Dowdy SF, Nat Biotechnol, 2017, 35, 222–229. [DOI] [PubMed] [Google Scholar]
- 2.Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR and Anderson DG, Nat Rev Genet, 2014, 15, 541–555. [DOI] [PubMed] [Google Scholar]
- 3.Pack DW, Hoffman AS, Pun S and Stayton PS, Nature Reviews Drug Discovery, 2005, 4, 581–593. [DOI] [PubMed] [Google Scholar]
- 4.Varkouhi AK, Scholte M, Storm G and Haisma HJ, J Control Release, 2011, 151, 220–228. [DOI] [PubMed] [Google Scholar]
- 5.Vermeulen LMP, De Smedt SC, Remaut K and Braeckmans K, Eur J Pharm Biopharm, 2018, 129, 184–190. [DOI] [PubMed] [Google Scholar]
- 6.Boussif O, Lezoualc’h F, Zanta MA, Mergny MD, Scherman D, Demeneix B and Behr JP, Proc Natl Acad Sci U S A, 1995, 92, 7297–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao WW, Chan JM and Farokhzad OC, Mol Pharmaceut, 2010, 7, 1913–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilson JT, Postma A, Keller S, Convertine AJ, Moad G, Rizzardo E, Meagher L, Chiefari J and Stayton PS, Aaps J, 2015, 17, 358–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilson JT, Keller S, Manganiello MJ, Cheng C, Lee CC, Opara C, Convertine A and Stayton PS, Acs Nano, 2013, 7, 3912–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Convertine AJ, Benoit DSW, Duvall CL, Hoffman AS and Stayton PS, Journal of Controlled Release, 2009, 133, 221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nelson CE, Kintzing JR, Hanna A, Shannon JM, Gupta MK and Duvall CL, ACS Nano, 2013, DOI: 10.1021/nn403325f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Werfel TA, Jackson MA, Kavanaugh TE, Kirkbride KC, Miteva M, Giorgio TD and Duvall C, J Control Release, 2017, 255, 12–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peeler DJ, Thai SN, Cheng Y, Horner PJ, Sellers DL and Pun SH, Biomaterials, 2018, 192, 235–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manganiello MJ, Cheng C, Convertine AJ, Bryers JD and Stayton PS, Biomaterials, 2012, 33, 2301–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jackson MA, Werfel TA, Curvino EJ, Yu F, Kavanaugh TE, Sarett SM, Dockery MD, Kilchrist KV, Jackson AN, Giorgio TD and Duvall CL, ACS Nano, 2017, 11, 5680–5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beavers KR, Werfel TA, Shen T, Kavanaugh TE, Kilchrist KV, Mares JW, Fain JS, Wiese CB, Vickers KC, Weiss SM and Duvall CL, Adv Mater, 2016, 28, 7984–7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berguig GY, Convertine AJ, Frayo S, Kern HB, Procko E, Roy D, Srinivasan S, Margineantu DH, Booth G, Palanca-Wessels MC, Baker D, Hockenbery D, Press OW and Stayton PS, Mol Ther, 2015, 23, 907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lane DD, Su FY, Chiu DY, Srinivasan S, Wilson JT, Ratner DM, Stayton PS and Convertine AJ, Polym Chem, 2015, 6, 1255–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elion DL, Jacobson ME, Hicks DJ, Rahman B, Sanchez V, Gonzales-Ericsson PI, Fedorova O, Pyle AM, Wilson JT and Cook RS, Cancer Res, 2018, 78, 6183–6195. [DOI] [PubMed] [Google Scholar]
- 20.Jacobson ME, Wang-Bishop L, Becker KW and Wilson JT, Biomater Sci, 2018, DOI: 10.1039/c8bm01064a. [DOI] [PubMed] [Google Scholar]
- 21.Werfel TA, Wang S, Jackson MA, Kavanaugh TE, Joly MM, Lee LH, Hicks DJ, Sanchez V, Ericsson PG, Kilchrist KV, Dimobi SC, Sarett SM, Brantley-Sieders DM, Cook RS and Duvall CL, Cancer Res, 2018, 78, 1845–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jackson MA, Bedingfield SK, Yu F, Stokan ME, Riles ME, Curvino EJ, Hoogenboezem EN, Bonami RH, Patel SS, Kendall PL, Giorgio TD and Duvall CL, Biomaterials, 2018, 192, 245–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li HF, Willingham SB, Ting JPY and Re F, J Immunol, 2008, 181, 17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA and Latz E, Nat Immunol, 2008, 9, 847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gomez DM, Urcuqui-Inchima S and Hernandez JC, Innate Immun-London, 2017, 23, 697–708. [DOI] [PubMed] [Google Scholar]
- 26.Hu QL, Zhao FH, Guo FL, Wang CC and Fu ZW, Macromol Biosci, 2017, 17. [DOI] [PubMed] [Google Scholar]
- 27.Bueter CL, Lee CK, Wang JP, Ostroff GR, Specht CA and Levitz SM, J Immunol, 2014, 192, 5943–5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bueter CL, Lee CK, Rathinam VAK, Healy GJ, Taron CH, Specht CA and Levitz SM, J Biol Chem, 2011, 286, 35447–35455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schroder K and Tschopp J, Cell, 2010, 140, 821–832. [DOI] [PubMed] [Google Scholar]
- 30.Jo EK, Kim JK, Shin DM and Sasakawa C, Cell Mol Immunol, 2016, 13, 148–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharma D and Kanneganti TD, J Cell Biol, 2016, 213, 617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo HT, Callaway JB and Ting JPY, Nat Med, 2015, 21, 677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manna S, Howitz WJ, Oldenhuis NJ, Eldredge AC, Shen JJ, Nihesh FN, Lodoen MB, Guan ZB and Esser-Kahn AP, Acs Central Sci, 2018, 4, 1593–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li TS, He JY, Horvath G, Prochnicki T, Latz E and Takeoka S, Nanomed-Nanotechnol, 2018, 14, 279–288. [DOI] [PubMed] [Google Scholar]
- 35.Cohen FS, Biophys J, 2016, 110, 1028–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barlan AU, Griffin TM, Mcguire KA and Wiethoff CM, J Virol, 2011, 85, 146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barlan AU, Danthi P and Wiethoff CM, Virology, 2011, 412, 306–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Teigler JE, Kagan JC and Barouch DH, J Virol, 2014, 88, 10354–10363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li AW, Sobral MC, Badrinath S, Choi Y, Graveline A, Stafford AG, Weaver JC, Dellacherie MO, Shih TY, Ali OA, Kim J, Wucherpfennig KW and Mooney DJ, Nat Mater, 2018, DOI: 10.1038/s41563-018-0028-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wegmann F, Gartlan KH, Harandi AM, Brinckmann SA, Coccia M, Hillson WR, Kok WL, Cole S, Ho L-P, Lambe T, Puthia M, Svanborg C, Scherer EM, Krashias G, Williams A, Blattman JN, Greenberg PD, Flavell RA, Moghaddam AE, Sheppard NC and Sattentau QJ, Nature Biotechnology, 2012, 30, 883–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharp FA, Ruane D, Claass B, Creagh E, Harris J, Malyala P, Singh M, O’Hagan DT, Petrilli V, Tschopp J, O’Neill LA and Lavelle EC, Proc Natl Acad Sci U S A, 2009, 106, 870–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fong D, Gregoire-Gelinas P, Cheng AP, Mezheritsky T, Lavertu M, Sato S and Hoemann CD, Biomaterials, 2017, 129, 127–138. [DOI] [PubMed] [Google Scholar]
- 43.Sarett SM, Werfel TA, Lee L, Jackson MA, Kilchrist KV, Brantley-Sieders D and Duvall CL, P Natl Acad Sci USA, 2017, 114, E6490–E6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sarett SM, Werfel TA, Chandra I, Jackson MA, Kavanaugh TE, Hattaway ME, Giorgio TD and Duvall CL, Biomaterials, 2016, 97, 122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.L’homme L, Esser N, Riva L, Scheen A, Paquot N, Piette J and Legrand-Poels S, J Lipid Res, 2013, 54, 2998–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carta S, Tassi S, Pettinati I, Delfino L, Dinarello CA and Rubartelli A, J Biol Chem, 2011, 286, 27069–27080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cullen SP, Kearney CJ, Clancy DM and Martin SJ, Cell Rep, 2015, 11, 1535–1548. [DOI] [PubMed] [Google Scholar]
- 48.Mancini RJ, Stutts L, Ryu KA, Tom JK and Esser-Kahn AP, ACS Chem Biol, 2014, 9, 1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Venkataraman S, Ong WL, Ong ZY, Joachim Loo SC, Ee PL and Yang YY, Biomaterials, 2011, 32, 2369–2378. [DOI] [PubMed] [Google Scholar]
- 50.Moyano DF, Goldsmith M, Solfiell DJ, Landesman-Milo D, Miranda OR, Peer D and Rotello VM, J Am Chem Soc, 2012, 134, 3965–3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gilleron J, Querbes W, Zeigerer A, Borodovsky A, Marsico G, Schubert U, Manygoats K, Seifert S, Andree C, Stoter M, Epstein-Barash H, Zhang L, Koteliansky V, Fitzgerald K, Fava E, Bickle M, Kalaidzidis Y, Akinc A, Maier M and Zerial M, Nat Biotechnol, 2013, 31, 638–646. [DOI] [PubMed] [Google Scholar]
- 52.Chu DS, Schellinger JG, Shi J, Convertine AJ, Stayton PS and Pun SH, Acc Chem Res, 2012, 45, 1089–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chu DS, Johnson RN and Pun SH, J Control Release, 2012, 157, 445–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.