

Abstract
Interleukin-15 (IL-15) and IL-2 drive T-cell malignancies including T-cell large granular lymphocyte leukemia (T-LGLL) and HTLV-1 driven adult T-cell leukemia (ATL). Both cytokines share common γ-chain receptors and downstream signaling pathways. T-LGLL is characterized by clonal expansion of cytotoxic T cells and is associated with abnormal JAK/STAT signaling. ATL is an aggressive CD4+ T cell neoplasm associated with HTLV-1. T-LGLL and ATL share dependence on IL-2 and IL-15 for survival and both diseases lack effective therapies. BNZ-1 is a pegylated peptide designed to specifically bind the γc receptor to selectively block IL-2, IL-15 and IL-9 signaling. We hypothesized that treatment with BNZ-1 would reduce cytokine mediated proliferation and viability. Our results demonstrated that in vitro treatment of a T-LGLL cell line and ex vivo treatment of T-LGLL patient cells with BNZ-1 inhibited cytokine mediated viability. Furthermore, BNZ-1 blocked downstream signaling and increased apoptosis. These results were mirrored in an ATL cell line and in ex vivo ATL patient cells. Lastly, BNZ-1 drastically reduced leukemic burden in an IL-15-driven human ATL mouse xenograft model. Thus, BNZ-1 shows great promise as a novel therapy for T-LGLL, ATL and other IL-2 or IL-15 driven hematopoietic malignancies.
Keywords: Cytokine signaling, JAK/STAT, BNZ-1, LGL, ATL
Introduction
Cytokines that share the common gamma-chain (γc) receptor are essential for development and maturation of lymphocytes as well as immune function and activation. Specifically, IL-2 and IL-15 promote survival and proliferation of T-cells and downstream effects that include induction of the JAK/STAT pathway.1 IL-2 is produced almost exclusively by T cells and signals through the heterotrimeric IL-2 receptor complex, including IL2Rα (CD25), IL2/IL15Rβ (CD122) and the γc receptor (CD132)2. IL-15 is produced mainly by dendritic cells and monocytes, and signals through IL-15Rα together with shared CD122 and γc receptor subunits. The two unique receptor α chains are differentially distributed on cells and are required for high affinity binding.1 Combinations of γc cytokines are involved in the pathogenesis of a diverse array of human diseases. 1,3–5 Implications of IL-2 and IL-15 in several T-cell malignancies have been well established.3,6–14 In this study, we focus on effects of dysregulated IL-2 and IL-15 signaling in large granular lymphocyte leukemia (LGLL) and HTLV-1 driven adult T cell leukemia (ATL).
LGLL is defined by a clonal expansion of large granular lymphocytes in blood. LGL make up 10–15% of peripheral blood mononuclear cells (PBMC), are activated upon antigen recognition, expand quickly, and then undergo apoptosis upon antigen clearance. However, in LGLL, these cells resist apoptosis and clonally expanded leukemic LGL can account for up to 90% of PBMC.15 This LGLL population can be separated into natural killer (NK) cells (typically CD3-CD56+CD16+)15 or T-cells (typically CD3+CD8+CD56-CD57+CD28-TCR-αβ+).16 There is not a cure for LGLL and current treatments include low doses of broad immunosuppressants, including methotrexate, cyclophosphamide and/or cyclosporine, thus indicating a need to develop more specific therapeutic options.17
LGLL is associated with dysregulation of several signaling pathways, including MAPK, PI3K-Akt, sphingolipid metabolism, NF-κB and JAK/STATs.18 Additionally, gain-of-function mutations in STAT319–21 and/or STAT5b22 are found in up to 40% of patients, emphasizing the role of JAK/STATs in the disease. Moreover, independent of STAT3 mutation status, all patients exhibit constitutively activated STAT3 and STAT1.19,23 STAT activating mutations have been shown to provide a growth advantage24 but they are not sufficient for leukemic cell proliferation, rather they enhance upstream signals from cytokine activation of the JAK/STAT pathway and result in a persistence of activated STATs.25 The pathogenesis of a dysregulated JAK/STAT pathway combined with the high prevalence of activating mutations suggests a pathway-directed therapeutic opportunity. IL-15 in particular has been implicated in the pathogenesis of LGLL. Significantly elevated levels of IL-15 mRNA and IL-15Rα are found in patient PBMCs.26,27 Using a systems biology approach, IL-15 was identified as one of two master regulators of the disease28, making IL-15 an attractive target for therapies.
HTLV-1 derived ATL is an aggressive disorder involving Tregs (typically CD2+CD3+CD4+CD25+CD7-)13,29 that also exhibits aberrant cytokine signaling. Many of the immune abnormalities in early ATL have been linked to HTLV-1 Tax that directly and through activation of NF-κB induces the expression of IL-2 and IL-15 and their specific receptors (IL-2Rα and IL-15Rα ) as well as IL-9, leading to excessive signaling through proinflammatory autocrine and paracrine loops.29–31 This leads to activation of JAK1/3 and STAT5 signaling pathways.32 It was previously demonstrated that IL-2/IL-2Rα, IL-9 and IL-15/IL-15Rα are overexpressed in ATL PBMCs and cooperate to yield ex vivo spontaneous proliferation of PBMCs,33 which is an important tool to evaluate potential interventional strategies and specific therapeutic agents. When ex vivo leukemic cells are incubated with antibodies to IL-2R alpha, IL-15 and IL-9, proliferation is profoundly inhibited (over 80%), thereby demonstrating the cytokine dependency of ATL cell proliferation and survival.
Due to the vital role that IL-2 and IL-15 play in LGLL and ATL, there is strong rationale for therapy directed at their signaling pathways. Both diseases have attempted therapies targeting one specific cytokine through monoclonal antibodies. In T-LGLL, a Phase I trial of a humanized monoclonal antibody to the IL2/IL15Rβ (CD122) receptor (Hu-Mikβ1) failed to exhibit clinical efficacy.34 Hu-Mikβ1 blocked trans-presentation of IL-15, the main mechanism of IL-15 presentation.35 However, in the presence of elevated IL-15Rα in LGLL patients, IL-15 can efficiently signal through cis presentation. In ATL, a clinical trial of humanized anti-Tac (daclizumab, anti-IL-2Rα)36 was limited by the fact that the antibody inhibited only IL-2 and had no effect on IL-9 or IL-15 mediated proliferation. An alternative approach using a JAK inhibitor demonstrated unacceptable toxicity when dose and dosing strategies sufficient to block the signaling pathway were utilized.37 To address this challenge, the BNZ-1 PEGylated peptide that targets IL-2, IL-15 and to a lesser extent IL-9 was developed.3 Since the functional redundancy among γc cytokines is largely due to the sharing of the γc subunit, we rationally chose to target this binding interface with the goal of inhibiting multiple γc cytokines.
BNZ-1, formerly known as BNZ 132–1-403, is a helical peptide designed to bind directly to the γc molecule and is PEGylated to increase its half-life. It can selectively block IL-2, IL-15, and IL-9 binding while leaving other γc and non-γc cytokine signaling unaffected.3 Previously, BNZ-1 effectively inhibited ex vivo HTLV-1 associated myelopathy/ tropical spastic paraparesis PBMC proliferation38 and in vivo proliferation of murine CD8+ T cell leukemia in an IL-15 transgenic mouse model.3 In addition, BNZ-1 exhibited no adverse effects on other immune cells and maintained selectivity for Tregs, CD8+ T and NK cells. Furthermore, a recent phase 1 clinical trial proved BNZ-1 to be well tolerated in healthy subjects.39 These positive results prompted us to determine the efficacy and mechanism of BNZ-1 in LGLL and ATL.
In this study, we show the therapeutic potential and mechanism of BNZ-1 in LGLL and HTLV-derived ATL. We hypothesized that attenuation of both IL-2 and IL-15 signaling pathways would result in decreased viability, proliferation, and ultimately death of cancer cells. Here, we not only show the successful treatment using BNZ-1 in vitro and ex vivo in T-LGLL and ATL cell line models and patient PBMCs, but also demonstrate the efficacy of BNZ-1 by reducing leukemic burden in an in vivo HTLV-1 derived ATL mouse model.
Methods
Cell lines
TL-1 is an IL-2-dependent patient-derived T-LGLL cell line.40 NKL is an IL-2-dependent patient-derived NK-LGL cell line.41 32D is an IL-3-dependent murine myeloid precursor cell line that expresses IL-2Rα and γc but not IL-2Rβ.42 32Dβ cell line was established by transfection with an extra-chromosomal DNA expression vector pREP9 (Invitrogen) encoding human IL-2Rβ. ED40515(+) is a human IL-2/IL-15-dependent ATL cell line which was kindly provided by Michiyuki Maeda43 (Kyoto University, Japan). ED40515(+)/luciferase cell line was produced by infection of wild-type ED40515(+) ATL cells with lentivirus expressing luciferase.
Cell viability assay
Cell viability was assessed by the CellTiter-Glo Assay (Promega).
Western blot analysis
After treatment, whole cell lysates were prepared and proteins were electrophoresed on a 4–12% acrylamide gel (ThermoFisher), transferred to PVDF membranes (BioRad) and immunostained with varying antibodies.
Cell proliferation assay
Aliquots (1×104) of the 32Dβ cells were seeded in 96-well plates with medium containing IL-2 (100 U/mL), or IL-15 (5 ng/mL), or murine IL-3 (500 ng/mL) together with serial dilutions of BNZ-1 (Bioniz Therapeutics Inc) and incubated for 48 hours. Aliquots (1×104) of ED40515(+) cells were cultured in medium containing IL-15 (2.5 ng/mL) and serial dilutions of BNZ-1 for 72 hours. The PBMCs isolated by Ficoll-Hypaque density centrifugation from patients with IL-2, IL-9, IL-15 autocrine smoldering ATL at 1 X 106/mL in 96 microtiter plates in RPMI 1640 media supplemented with 10% FBS at 37oC in 5% CO2 were cultured ex vivo for 6 days with and without BNZ-1 or with 10 μg/mL of antibody added at initiation of the cultures. A single antibody directed against the cytokine IL-2, IL-9 or IL-15 was used or with their combination. Cells were pulsed during the last 6 hours with 1 μCi (0.037 MBq) of 3H-thymidine (Perkin Elmer) then harvested and counted in a MicroBeta2 microplate counter.
Apoptosis detection
After treatment, leukemic cells were stained with propidium iodide (PI) and APC-annexin V prior to flow cytometry analysis.
Ex vivo cultures of PBMCs
T-LGLL patient blood samples were obtained and informed consents signed for sample collection according to the Declaration of Helsinki using a protocol approved by the Institutional Review Board of the University of Virginia. Blood was subjected to Ficoll-Hypaque (Sigma Aldrich) gradient centrifugation for PBMC isolation and cells were cultured ex vivo in RPMI 1640 medium with 10% FBS. Patient samples used to assess apoptosis by flow cytometry were previously frozen, and cell viability was determined by trypan blue exclusion assay prior to the start of treatment. Relevant patient data can be found in Supplementary Table 1. ATL patient blood samples were obtained and the PBMCs were cultured ex vivo in RPMI 1640 medium containing 10% FBS with or without therapeutic reagents for 6 days. The human study was approved by the Intramural Review Board of the National Cancer Institute, NIH. Informed consent was obtained from all subjects.
Murine ATL model
The cytokine-dependent ATL model was established by i.v. injection of 1×107 ED40515(+)/luciferase cells into human IL-15 transgenic NSG mice (Taconic). Treatment was initiated seven days later when serum sIL-2Rα levels were >500 pg/mL. Tumor-bearing mice received BNZ-1 (40 mg/kg i.v.) twice a week or ruxolitinib (Selleckchem; 50 mg/kg/day by s.c. osmotic pumps) for 4 weeks. An additional group that received i.v. injections of PBS served as a control. Tumor growth was monitored by measuring serum sIL-2Rα concentrations and by bioluminescence images obtained in vivo and analyzed using Living Imaging software (Xenogen, Alameda, CA).
Statistical analysis
Statistical analyses for all experimental systems utilized unpaired Student’s t test.
Results
BNZ-1 exhibited selective targeting of cytokine-induced proliferation
BNZ-1 mediated effects were exclusively related to IL-2 and IL-15 blockade.3 We have verified the reported results by using 32Dβ cells that express IL-2Rβ and respond to IL-2.42 Both IL-2 and IL-15 stimulated 32Dβ cell proliferation in a dose-dependent manner (Supplementary Figure 1A). The proliferation of the cells was completely inhibited by BNZ-1 while the off-target non-γc-mediated proliferation induced by IL-3 was not inhibited (Supplementary Figure 1B). These studies support the view that BNZ-1 is not generally toxic and has specificity for inhibition of IL-2 and IL-15 mediated proliferation.
BNZ-1 reduced cytokine mediated viability and downstream pathway activation in a T-LGL leukemia cell line
Initial experiments of BNZ-1 efficacy in LGLL were performed on TL-1, an IL-2 dependent T-LGLL patient-derived cell line40. Prior to evaluating effects of BNZ-1, we treated cytokine deprived TL-1 cells with varying concentrations of IL-2 and IL-15 to characterize cytokine mediated survival and expansion. Cell viability decreased in the absence of cytokine supplementation and cell numbers increased upon cytokine addition in a dose-dependent manner, indicating the dependence of survival and proliferation on cytokine treatment (Supplementary Figure 2). To test the effect of BNZ-1 on IL-2 and IL-15 induced cell expansion in vitro, a pretreatment of BNZ-1 (5 μM) was added to TL-1 cells prior to cytokine addition and relative cell numbers were measured at 24, 48 and 72 hours. At all time points, cytokine-only treatment increased viable cells and cytokine stimulation was inhibited by BNZ-1 pretreatment (representative data shown for 48 hours, Figure 1A,B). IL-15 mediated cell expansion was affected to a greater extent than IL-2. Treatment with BNZ-1 alone showed no toxicity in the absence of cytokine, and BNZ-1 was less effective at blocking expansion at higher cytokine doses. The cytokine doses most capable of mediating survival and expansion while maintaining BNZ-1 effectiveness were 12.5 U/mL (190 pM) for IL-2 and 6.25 U/mL (190 pM) for IL-15, which exhibited BNZ-1-mediated reductions of 30% and 92%, respectively. Our next experiment was to determine the BNZ-1 dose response in TL-1 cells treated with varying doses of BNZ-1 prior to IL-2 (12.5 U/mL) or IL-15 (6.25 U/mL) addition. For IL-2 treatment, addition of BNZ-1 at 10 μM resulted in a 50% reduction of cell expansion. For IL-15, BNZ-1 at 1 μM reduced viable cell numbers by more than half and BNZ-1 blocked essentially all IL-15 induced cell survival at 5 and 10 μM (Figure 1C,D). These data show BNZ-1 can significantly inhibit IL-2 and IL-15 induced expansion in a T-LGLL cell line.
Figure 1: BNZ-1 decreased IL-2 and IL-15 mediated viability in a T-LGLL cell line.
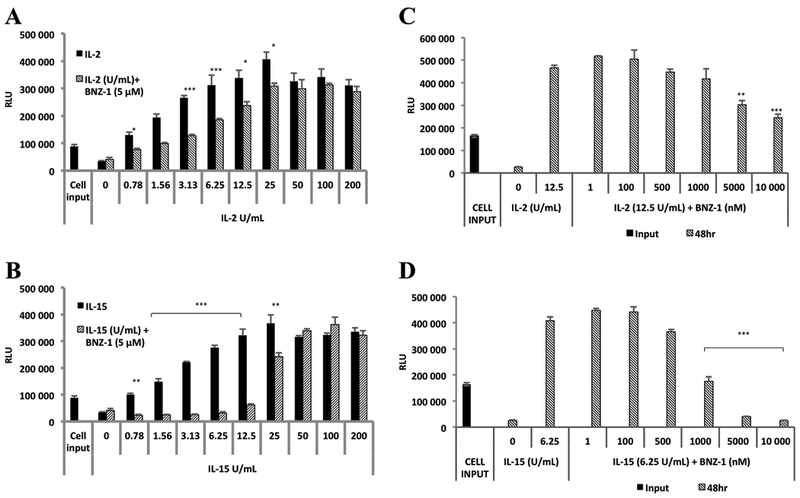
TL-1 cells were cytokine starved for 12 hours then pretreated with BNZ-1 (5 μM) for 20 minutes followed by varying IL-2 [A] and IL-15 [B] to determine effective cytokine doses. IL-2 at 12.5 U/mL and IL-15 at 6.25 U/mL gave a robust increase in TL-1 viability, and BNZ-1 caused significant reduction of cell expansion induced by both cytokines. Cells were then treated with increasing concentrations of BNZ-1 in the presence of these doses of IL-2 [C] and IL-15 [D]. Cell viability induced by IL-2 was halved by 10 μM of BNZ-1 and IL-15-induced cell survival was attenuated by 1 μM of BNZ-1, and fully blocked at 5 and 10 μM. IL-2 at 12.5 U/mL = 0.19 nM; IL-15 at 6.25 U/mL = 0.19nM. Bars represent relative light units (RLU; mean ± standard deviation) of CellTiter Glo assays done at 48h. N = 3; * P < 0.05; ** P < 0.005; *** P < 0.0005.
BNZ-1 blocked signaling pathways downstream of IL-2 and IL-15
BNZ-1 (10 μM) reduced the phosphorylation of downstream IL-2 signaling pathways at 30 minutes post-treatment including STAT3, STAT1, STAT5, Akt and ERK in a cytokine concentration dependent manner in TL-1 cells (Figure 2A). BNZ-1 reduced the IL-2 mediated activation of STAT3, STAT1 and STAT5 close to or below activation levels of untreated, cytokine-deprived cells. BNZ-1 incompletely blocked Akt and ERK activation by IL-2. Conversely, BNZ-1 drastically reduced IL-15 mediated phosphorylation of STAT3, STAT1, STAT5, Akt and ERK (Figure 2C). Additional time points of one, three, and six hours post cytokine addition were tested and results indicated that downstream activation remains blocked at these later time points (Supplementary Figure 3A, B). Therefore, BNZ-1 effectively blocks IL-2 and IL-15 induction of signaling pathways important to leukemic cell survival and proliferation.
Figure 2: BNZ-1 inhibited IL-2 or IL-15 mediated phosphorylation events in a T-LGLL cell line.

TL-1 cells were pretreated with or without BNZ-1 in the presence of varying IL-2 or IL-15 doses. Western blot analyses demonstrated blockage of IL-2 [A] or IL-15 [C] mediated protein phosphorylation with BNZ-1 treatment (10 μM). Graphs of phosphorylated protein over total protein for IL-2 [B] or IL-15 [D] as quantified by ImageLab software. IL-2 at 8 U/mL = 0.12 nM; IL-15 at 8 U/mL = 0.24 nM.
BNZ-1 increased annexin V positive cells in TL-1
To determine whether BNZ-1 treatment would induce apoptosis, cytokine stimulated TL-1 cells with and without BNZ-1 treatment were analyzed for apoptosis by flow cytometry (Figure 3A). BNZ-1 increased the number of cells that were positive for the apoptosis marker annexin V in all cytokine stimulated groups at 72 hours, with similar results at 48 hours (data not shown). Quantification of double negative (live) cells (Figure 3B) demonstrated an expansion of cells upon cytokine addition, especially IL-15, and loss of live cells with BNZ-1 treatment. Conversely, quantification of annexin V positive cells (Figure 3C) showed reduced early and late apoptotic cells with cytokine treatment and increased apoptotic cells upon BNZ-1 addition. Taken together, BNZ-1 reduced viability and enhanced apoptosis in the presence of IL-2, IL-15 or both cytokines.
Figure 3: BNZ-1 increased annexin V positive cells in a T-LGLL cell line.

TL-1 cells were treated with or without BNZ-1 (10 μM) in the presence of IL-2 (2.5 U/mL), IL-15 (2.5 U/mL), or IL-2 and IL-15 (2.5 U/mL each). At 72 hours, cells were incubated with APC-annexin V and PI and analyzed by flow cytometry. [A] In each panel the lower left quadrant shows double negative (live) cells, lower right quadrant shows APC-annexin V positive cells (early apoptotic) and the upper right quadrant shows double positive cells (late apoptotic). [B] Quantification of double negative (live) cells and [C] APC-annexin V positive (apoptotic) cells in the indicated treatment groups. IL-2 at 2.5 U/mL = 0.038 nM; IL-15 at 2.5 U/mL = 0.076 nM; N = 3; * P < 0.05; ** P < 0.005; *** P < 0.0005.
BNZ-1 reduced cytokine-mediated survival in primary T-LGLL PBMCs
With the success of BNZ-1 treatment in the T-LGLL cell line, it was important to translate these findings to patient PBMCs that are known to require cytokine supplementation to survive ex vivo. T-LGLL patient PBMCs of known STAT3 mutational status were pretreated for 20 minutes with BNZ-1 prior to addition of IL-2 (Figure 4A), IL-15 (Figure 4B), or IL-2 and IL-15 (Figure 4C) and percent viability relative to input control was determined at 72 hours. Treatment with cytokine alone stimulated cell survival and expansion in all samples. BNZ-1 (10 μM) impaired cytokine induced survival in all samples. BNZ-1 treatment did not cause any additional loss of viability relative to controls without cytokine supplementation (data not shown). Therefore, BNZ-1 inhibits cytokine-stimulated ex vivo survival and expansion of primary T-LGLL samples, regardless of STAT3 mutational status.
Figure 4: BNZ-1 reduced cytokine mediated viability and blocked IL-15 mediated phosphorylation in T-LGLL patient PBMCs.

PBMC from T-LGL leukemia patients with and without STAT3 mutations were not treated (NTC) or pretreated with BNZ-1 (10 μM) for 20 min followed by IL-2 [A], IL-15 [B] or IL-2 and IL-15 [C] for an additional 24 hrs. Cell viability was measured using CellTiter Glo in triplicate. Results are shown as mean ± SD relative to the input control (100%). BNZ-1 reduced cytokine mediated viability in T-LGLL patient PBMC with and without STAT3 mutations at 72 hours. T1613 was treated at 2.5 U/mL of IL-2 and IL-15. T1954 and T1875 were treated at 6.25 U/mL of IL-2 and 3.13 U/mL of IL-15. T1370 and T2051 were treated with 10 U/mL of IL-2 and IL-15. [D] PBMC from T-LGL leukemia patients with and without STAT3 mutations were pretreated with BNZ-1 (5 or 10 μM) for 20 min followed by IL-15 (2.5 U/mL) for an additional 24 hr. Total protein was harvested and STAT protein phosphorylation was determined by western blot assay. T-LGL leukemia patient PBMCs with and without STAT3 mutations were stimulated with IL-2 [E], IL-15 [F] or IL-2 and IL-15 [G] in the presence or absence of BNZ-1. At 72 hours, cells were stained with APC-annexin V and PI and analyzed by flow cytometry. Percent apoptosis quantified by APC-annexin V positive staining. IL-2 at 2.5 U/mL = 0.038 nM; IL-15 at 2.5 U/mL = 0.076 nM; WT, wild-type; MUT, mutant; N = 3; * P < 0.05; ** P < 0.005; *** P < 0.0005.
BNZ-1 reduced IL-15 mediated STAT phosphorylation in primary T-LGLL PBMCs
To determine whether BNZ-1 blocked downstream phosphorylation in patient samples as observed in TL-1 cells (Figure 2), we tested PBMCs derived from two patients with wild-type STAT3 and two patients with mutant STAT3. Samples were treated with IL-15 after BNZ-1 pretreatment to study the effects on STAT phosphorylation ex vivo. There was a robust and clear decrease in IL-15 induced phosphorylation of STAT1, STAT3 and STAT5 with BNZ-1 pretreatment (5 μM and 10 μM) in all samples, regardless of STAT3 mutational status (Figure 4D).
BNZ-1 induced apoptosis in primary T-LGLL PBMCs.
Lastly, we examined whether BNZ-1 was capable of inducing apoptosis in T-LGLL patient PBMC. Previously frozen T-LGLL patient PBMCs were stimulated with IL-2 (Figure 4E), IL-15 (Figure 4F), or IL-2 and IL-15 (Figure 4G) in the presence or absence of BNZ-1 treatment. After 72 hours, samples were stained with PI and annexin V to assess apoptosis induction. We demonstrated that cytokine treatment reduced the amount of apoptotic cells compared to those not given cytokine. In contrast, co-treatment with BNZ-1 increased apoptotic cells in all five patient samples. In the presence of BNZ-1, the percent of apoptotic cells rose to a level equal to or exceeding the level observed in the no treatment group.
BNZ-1 inhibited cytokine stimulated survival and signaling in an NK-LGL leukemia cell line.
We also tested the effects of BNZ-1 on a NK-LGLL leukemia cell line (NKL). We determined the cytokine concentrations that most efficiently increased cell expansion and maximized potency of BNZ-1 for both IL-2 and IL-15 (Supplementary Figure 4A, B). BNZ-1 (5 μM) inhibited IL-15 induced NKL cell viability by approximately 58% whereas BNZ-1 (10 μM) reduced IL-2 dependent cell viability by roughly 20%. Treatment of NKL cells with BNZ-1 dramatically reduced IL-15 induced phosphorylation of STAT3, STAT1 and ERK. There was a less drastic decrease of STAT5 phosphorylation (Supplementary Figure 4C). Taken together, BNZ-1 showed promise in NK-LGLL model systems.
BNZ-1 inhibited proliferation in a cytokine dependent ATL cell line and ex vivo PBMCs from patients with smoldering/chronic ATL.
Next the efficacy of BNZ-1 was determined in a second T-cell disease model, ATL. Human cytokine-dependent ATL cell lines represent a model of smoldering/chronic ATL, which are clinical disease states that manifest constitutive cytokine production, JAK/STAT activation and spontaneous ex vivo proliferation of ATL cells. The ATL cell line used in the present study requires a supply of extrinsic human IL-2 or IL-15 for their survival in vitro and in vivo. We demonstrated that BNZ-1 inhibited IL-2 or IL-15 or IL-2 plus IL-15 mediated proliferation of the cytokine-dependent ATL cell line ED40515(+) (Figure 5A). We then tested the effects of BNZ-1 on 6-day ex vivo spontaneous proliferation of PBMCs from patients with smoldering/chronic ATL. The ex vivo spontaneous proliferation of PBMCs from 3 patients with smoldering/chronic ATL, as assessed by thymidine uptake, was inhibited by an average of 72 percent by the addition of the anti-IL-2Rα monoclonal antibody, daclizumab (Figure 5B) and by a comparable amount by the combination of daclizumab, anti-IL-15 and anti-IL-9. BNZ-1 (1 μM) inhibited the proliferation of the PBMCs from 3 patients with ATL by an average of 51 percent, whereas BNZ-1 at 10 μM inhibited this ex vivo proliferation by an average of 73 percent (Figure 5B). PBMCs of patients with smoldering ATL were stained with a CD3, a CD4, a CD25 antibody, PI and annexin V (Figure 5C). Increasing doses of BNZ-1 significantly decreased percentages and total numbers of CD3+CD4+CD25+ ATL cells, whereas percentages and numbers of normal CD3+CD4+CD25- T cells were not affected (Figure 5D). However, no CD3+CD4+CD25+ ATL cells were detected positive for annexin V.
Figure 5: BNZ-1 inhibited proliferation of an ATL cell line and of smoldering/chronic ATL PBMCs.

[A] Effect of BNZ-1 on the proliferation of cytokine-dependent ATL cell line ED40515(+). Aliquots of ED40515(+) cells were seeded in 96 well-culture plates in medium with IL-2 (0.1 nM; 12.5 U/mL) or IL-15 (0.2 nM; 2.5 ng/mL) or both. Then the cells were incubated with serial dilutions of BNZ-1 for 72 hours. The cells were pulsed during the last 6 hours with 1μCi 3H-thymidine. BNZ-1 effectively inhibited the proliferation of ED40515(+) cells mediated by IL-2 alone or IL-15 alone or IL-2 plus IL-15. [B] BNZ-1 inhibited the 6-day ex vivo spontaneous proliferation of PBMCs from patients with smoldering ATL. The PBMCs isolated from patients with IL-2, IL-9, IL-15 autocrine smoldering ATL at 1 X 106/mL in 96 microtiter plates in RPMI 1640 media supplemented with 10% FBS at 37oC in 5% CO2 were cultured ex vivo for 6 days with and without BNZ-1 or antibody added at initiation of the cultures. Antibody doses of 10 μg/mL were utilized for a single antibody directed against the cytokine IL-2, IL-9 or IL-15 or 5 μg/mL each when used in combination. 3H-thymidine was added during the last 6 hours of the cultures. Cells were then harvested and analyzed for 3H-thymidine incorporation. The value was calculated as: % inhibition = (cpm of proliferation without BNZ-1 – cpm of proliferation with BNZ-1)/(cpm of proliferation without BNZ-1) X 100. [C] PBMCs of patients with smoldering ATL with IL-2, IL-9, IL-15 autocrine mediated proliferation have been plated at 106 cells/mL, in the presence or absence of BNZ-1 (1 and 10 μM). At day 3, cells have been stained with a CD3, a CD4, a CD25 antibody, PI and annexin V. Percentages and total numbers of CD3+CD4+CD25+ ATL cells (n = 3) significantly decreased in the presence of increasing doses of BNZ, whereas [D] percentages and numbers of normal CD3+CD4+CD25- T cells were not affected.
BNZ-1 showed therapeutic efficacy in an IL-2/IL-15 dependent in vivo model of human ATL.
The majority of the studies of BNZ-1 have evaluated its action on cell lines and ex vivo cells. Nata et al. reported that BNZ-1 effectively inhibited the in vivo proliferation of CD8+ T cells in an IL-15 transgenic mouse model. Here we utilized the cytokine-dependent ED40515(+) xenograft tumor model to provide in vivo validation that BNZ-1 exhibits the acceptable pharmacokinetic and safety profile to be effective in the treatment of an IL-2/IL-15 dependent tumor. The model of IL-2/IL-15 dependent ATL was generated by i.v. injection of 1×107 ED40515(+)/luciferase-expressing cells into female IL-15 transgenic NSG mice. ATL cells express human IL-2Rα and release it into the biological fluids including the serum. One week after injection of the tumor cells, when the average soluble human IL-2Rα was approximately 700 pg/mL (Supplementary Table 2), the animals were randomized to treatment groups and therapy was started. Ruxolitinib (50 mg/kg/day) was administered by pump s.c. for four weeks. BNZ-1 (40 mg/kg) was administered i.v. two times per week for four weeks. Tumor burden over time was assessed by bioluminescence imaging (Figures 6A,B). Leukemic burden was greatly reduced by BNZ-1 when compared to PBS-treated control mice, which was similar to that observed with the JAK1/2 inhibitor, ruxolitinib. To further assess whole animal tumor burden and evaluate antitumor efficacy, serum IL-2Rα levels were quantitated. BNZ-1 therapy was associated with greatly reduced IL-2Rα levels at four and seven weeks of treatment, which is indicative of reduced quantities of prevailing tumor tissue (Figure 6C).
Figure 6: BNZ-1 inhibited IL-15-dependent ATL growth in vivo.

Human IL-15 transgenic NSG mice were injected i.v. with 1×107 ED40515(+)/luciferase cells. One week later, the tumor-bearing mice were divided into 3 groups (n=5–6) with comparable average levels of serum sIL-2Rα and therapy was started. BNZ-1 was given i.v. at a dose of 40 mg/kg twice weekly for 4 weeks and ruxolitinib was continuously administered by a subcutaneous mini-osmotic pump at a dose of 50 mg/kg/day for 4 weeks. (A) Bioluminescence imaging of IL-2/IL-15-dependent ED40515(+)/luciferase ATL-bearing mice confirmed efficacies of BNZ-1 and ruxolitinib. (B) Average total luminescent signals of the tumor-bearing mice. The animals treated with both BNZ-1 and ruxolitinib had significantly lower luminescent signals when compared with those in the PBS control group (*P < 0.05). (C) The mean concentrations of tumor surrogate marker, human sIL-2Rα, levels in the IL-2/IL-15-dependent ED40515(+)/luciferase ATL-bearing mice before and after treatment. The animals treated with BNZ-1 had significantly decreased values of sIL-2Rα when compared with those in the PBS control group (*P < 0.05). Treatment of the tumor-bearing mice with ruxolitinib showed similar therapeutic efficacy as BNZ-1 did. Results of two independent assessments of tumor burden and human sIL-2Rα levels yield similar data, emphasizing the efficacy of BNZ-1 in a human ATL xenograft mouse model.
Discussion
IL-2 and IL-15 are key cytokine drivers in multiple T-cell malignancies including LGLL27, ATL29, cutaneous T-cell lymphoma (CTCL)7,12, gamma-delta T-cell lymphoma (γδTCL)25 and anaplastic large cell lymphoma (ALCL)9. Studies of BNZ-1 in LGLL and ATL allowed us to test the hypothesis that cytokine receptor blockade may exhibit widespread therapeutic benefit in these malignancies. The pathogenesis of both LGLL and ATL depends on the dysregulation of IL-2 and IL-15 signaling, making these cytokines and their signaling pathways attractive therapeutic targets.4,10,18,26,44 In this study, we demonstrated the efficacy of an IL-2, IL-15 and IL-9 inhibitor peptide, BNZ-1, in T-LGLL and HTLV-1 derived ATL. Treatment of T-LGLL and ATL cell lines with BNZ-1 reduced IL-2 and IL-15 mediated cell viability and proliferation. In addition, the loss of STAT 1/3/5, ERK and Akt activation provided a mechanism for the impaired cytokine mediated expansion in T-LGL leukemic cells. Not only did BNZ-1 cause the loss of activation in these critical signaling pathways, we showed that BNZ-1 induced apoptosis in cytokine supplemented T-LGLL cell line and patient PBMCs. We also showed its ability to block the expansion of cytokine dependent and independent ex vivo PBMCs in both T-LGLL and ATL. Lastly, BNZ-1 therapy drastically reduced leukemic burden in an in vivo IL-15 transgenic ATL mouse model. Together, this study proved BNZ-1 to be an effective form of therapy in both T-LGL leukemia and HTLV-1 derived ATL model systems.
To prove the selectivity of BNZ-1 for IL-2 and IL-15, BNZ-1 was evaluated in the 32Dβ cell line. 32Dβ cells are IL-3 dependent and were modified to express all three subunits of the IL-2 receptor complex, including the common γ-chain. Both cytokines utilize the γc receptor, the designated target of BNZ-1. In contrast, IL-3 does not utilize the common γc. BNZ-1 inhibited IL-2 and IL-15 mediated proliferation in 32Dβ but not that of IL-3. The critical importance of this lack of inhibition of IL-3 mediated proliferation is two-fold. First, it shows that BNZ-1 is not universally toxic to cells. Second, these studies provide evidence of specificity of BNZ-1 for IL-2 and IL-15, which are the putative targets of this agent.
Increased γc receptor signaling results in increased activation of downstream targets including STAT1/3/5, ERK and Akt, which are strongly implicated in the pathogenesis of T-LGLL.15,16,23 The loss of STATs, Akt and ERK phosphorylation in leukemic cells upon BNZ-1 treatment indicates the superior ability of BNZ-1 to successfully block multiple signaling pathways activated by cytokine stimulation when compared to conventional JAK inhibitors that only inhibit the JAK/STAT pathway.
To fully examine the efficacy of BNZ-1 in T-LGLL, we utilized parallel studies in both cell lines and patient PBMCs to estimate its putative success in a future clinical trial. We demonstrated that BNZ-1 reduced cytokine mediated viability, blocked downstream pathway activation and induced apoptosis in cell lines and in T-LGLL patient samples. Patient PBMC samples contain varying amount of leukemic LGL, and patient samples used in this study ranged from 38% to 92% LGL in their PBMCs. Regardless of tumor purity, BNZ-1 succeeded at reducing leukemic cell viability, blocking downstream activation and inducing apoptosis, thereby strengthening the evidence of BNZ-1 efficacy.
STAT3 somatic mutations are found in up to 40% of all LGLL patients and constitutive activation of STAT3 is found in all patients, emphasizing the pathogenetic role of this aberrant pathway in T-LGLL.19,20,23 Knowing the importance of STAT3 mutation in T-LGLL, we were curious whether a STAT3 mutation would affect the inhibitory effects of BNZ-1. Regardless of STAT3 mutation status, BNZ-1 reduced the ability of both IL-2 and IL-15 to support ex vivo survival and expansion of leukemic cells, blocked downstream protein activation after cytokine treatment and induced apoptosis. This supports the current understanding that mutant cells still require cytokine signaling at the receptor level and that an activating mutation alone does not induce constitutive phosphorylation of STAT1 or STAT3.20,21 Therefore, BNZ-1 has the capability to be a positive therapy in all T-LGLL patients and would not be limited based on STAT3 mutational status.
In this study, we also demonstrated the efficacy of BNZ-1 on ex vivo primary human ATL leukemic cell spontaneous proliferation, thereby providing the required evidence that BNZ-1 is effective with non-manipulated human cells. We evaluated the BNZ-1 strategy to reduce the excessive immune activation that occurs in HTLV-1 associated ATL by simultaneously inhibiting the effects of multiple pro-inflammatory autocrine and paracrine cytokine loops involved in the pathogenesis of early disease.29 BNZ-1 was demonstrated to be a potent inhibitor of ex vivo spontaneous proliferation of PBMCs from ATL patients and exhibited comparable or superior activity when compared to ruxolitinib. It is of interest that the addition of BNZ-1 to T-cell LGLL PBMCs led to apoptosis of the leukemic cells whereas leukemic cell apoptosis was not observed in ATL PMBCs but rather a reduced persistence of the leukemic cells. This observation may be due to loss of apoptotic cells before evaluation at this 72 hour time point or may reflect the fact that in ATL, HTLV-1 encoded Tax transactivates the anti-apoptotic protein Bcl-X(L).45,46
The PEGylated BNZ-1 peptide was developed to improve the pharmacokinetic properties, increase bioavailability and reduce immunogenicity for clinical studies.3,38 It was critical to determine the efficacy of this agent in in vivo models in addition to the mouse model for CD8+ T cell leukemia based on IL-15 transgenic T cells,3,47 to consider its pharmacokinetics and safety profile in living mice, and to be assured that the BNZ-1 was effective in vivo. In an in vivo mouse model of ED40515(+) ATL leukemia, BNZ-1 exhibited efficacy at least comparable and likely superior to that mediated by the JAK1/2 inhibitor ruxolitinib. It is noteworthy that the superior efficacy of BNZ-1 was achieved with twice weekly dosing as opposed to continuous delivery of ruxolitinib by osmotic pump. This is consistent with our earlier observation that BNZ-1 was superior to tofacitinib, a pan-JAK inhibitor, in protecting host animals from leukemic death in our mouse CD8+ leukemia model.3 Thus, BNZ-1 displayed appropriate pharmacokinetics, safety and efficacy to provide effective treatment in an in vivo model of cytokine-dependent ATL.
Taken together, these preclinical studies indicate that BNZ-1 may be a valuable candidate to treat T-LGLL and the smoldering and chronic forms of ATL, both of which lack safe and curative treatments. In this study BNZ-1 inhibited the proliferation of cytokine dependent ATL and T-LGLL cell lines and patient PBMC’s and induced apoptosis in a T-LGLL cell line and patient PBMC’s. Additionally, it proved effective in reducing leukemic burden in an in vivo model of cytokine-dependent ATL. Early results from a completed single and multiple ascending dose clinical trials in healthy volunteers (NCT03046459 and NCT03239379) demonstrated that BNZ-1 was well tolerated with no major side effects.39 Pharmacodynamic biomarkers showed high specificity to IL-2 and IL-15 (manuscript in preparation). These preclinical and early clinical successes indicate that BNZ-1 represents a promising therapeutic option for multiple disorders with dysregulated γc cytokines and abnormal activation of JAK/STAT pathways.
Supplementary Material
Acknowledgments
LGL leukemia patient samples and clinical information were obtained from the LGL Leukemia Registry at the University of Virginia with the assistance of Holly Davis, Bryna Shemo and Andrea Hines. Alexander Wendling and Matthew Schmachtenberg provided excellent technical support while processing patient samples. We thank Yadeliz Adlin Serrano Matos (University of Puerto Rico, Rio Piedras Campus) for assistance with Supplementary Figure 3. We thank Alexander Wendling in the UVA Flow Cytometry Core for assistance with apoptosis assays. NKL cells, a leukemic LGL NK-cell line, were kindly provided by Dr. Howard Young at the National Cancer Institute. We thank Taconic Inc. (Petersburg, NY) for providing the human IL-15 transgenic mice.
This research was funded by the National Cancer Institute of the National Institutes of Health under award number R01CA098472, R01CA178393 and P30CA044579 (T.P.L.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Additional funding was provided to T.P.L. by the Bess Family Charitable Fund, the LGL Leukemia Foundation and a generous anonymous donor. This research was also supported in part by the Intramural Research Program of the National Cancer Institute, Center for Cancer Research (T.A.W).
Footnotes
Conflict of Interest: Nazli Azimi and Yutaka Tagaya are employed by BIONIZ Therapeutics, the producer of BNZ-1. Thomas Loughran, Jr. and Thomas Waldmann are on the scientific advisory board of BIONIZ Therapeutics. The other authors have no conflicts of interest to disclose.
Supplementary information is available at Leukemia’s website.
References:
- 1.Waldmann TA. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat Rev Immunol. 2006. August;6(8):595–601. [DOI] [PubMed] [Google Scholar]
- 2.Waldmann TA. The interleukin-2 receptor. J Biol Chem. 1991. February 15;266(5):2681–4. [PubMed] [Google Scholar]
- 3.Nata T, Basheer A, Cocchi F, Besien R van, Massoud R, Jacobson S, et al. Targeting the Binding Interface on a Shared Receptor Subunit of a Cytokine Family Enables the Inhibition of Multiple Member Cytokines with Selectable Target Spectrum. J Biol Chem. 2015. September 11;290(37):22338–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yamagishi M, Watanabe T. Molecular Hallmarks of Adult T Cell Leukemia. Front Microbiol [Internet]. 2012 Sep 17 [cited 2017 Sep 12];3. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3444139/ [DOI] [PMC free article] [PubMed]
- 5.Kips JC. Cytokines in asthma. Eur Respir J. 2001. July 2;18(34 suppl):24s–33s. [DOI] [PubMed] [Google Scholar]
- 6.Wasik MA, Sackstein R, Novick D, Butmarc JR, Zhang Q, Vonderheid EC, et al. Cutaneous CD56+ large T-cell lymphoma associated with high serum concentration of IL-2. Hum Pathol. 1996. Jul;27(7):738–44. [DOI] [PubMed] [Google Scholar]
- 7.Marzec M, Halasa K, Kasprzycka M, Wysocka M, Liu X, Tobias JW, et al. Differential effects of interleukin-2 and interleukin-15 versus interleukin-21 on CD4+ cutaneous T-cell lymphoma cells. Cancer Res. 2008. February 15;68(4):1083–91. [DOI] [PubMed] [Google Scholar]
- 8.Ribeiro ST, Ribot JC, Silva-Santos B. Five Layers of Receptor Signaling in γδ T-Cell Differentiation and Activation. Front Immunol [Internet]. 2015 Jan 26 [cited 2018 Mar 23];6. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4306313/ [DOI] [PMC free article] [PubMed]
- 9.Ito M, Zhao N, Zeng Z, Zhou X, Chang C-C, Zu Y. Interleukin-2 Functions in Anaplastic Large Cell Lymphoma Cells through Augmentation of Extracellular Signal-Regulated Kinases 1/2 Activation. Int J Biomed Sci IJBS. 2011. September;7(3):181–90. [PMC free article] [PubMed] [Google Scholar]
- 10.Mishra A, Liu S, Sams GH, Curphey DP, Santhanam R, Rush LJ, et al. Aberrant overexpression of IL-15 initiates large granular lymphocyte leukemia through chromosomal instability and DNA hypermethylation. Cancer Cell. 2012. November 13;22(5):645–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tabbó F, Barreca A, Piva R, Inghirami G. ALK Signaling and Target Therapy in Anaplastic Large Cell Lymphoma. Front Oncol [Internet]. 2012 May 11 [cited 2018 Mar 22];2. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3355932/ [DOI] [PMC free article] [PubMed]
- 12.Krejsgaard T, Ralfkiaer U, Clasen-Linde E, Eriksen KW, Kopp KL, Bonefeld CM, et al. Malignant cutaneous T-cell lymphoma cells express IL-17 utilizing the Jak3/Stat3 signaling pathway. J Invest Dermatol. 2011. June;131(6):1331–8. [DOI] [PubMed] [Google Scholar]
- 13.Dahmoush L, Hijazi Y, Barnes E, Stetler-Stevenson M, Abati A. Adult T-cell leukemia/lymphoma. Cancer Cytopathol. 2002. April 25;96(2):110–6. [DOI] [PubMed] [Google Scholar]
- 14.Malamut G, El Machhour R, Montcuquet N, Martin-Lannerée S, Dusanter-Fourt I, Verkarre V, et al. IL-15 triggers an antiapoptotic pathway in human intraepithelial lymphocytes that is a potential new target in celiac disease-associated inflammation and lymphomagenesis. J Clin Invest. 2010. June;120(6):2131–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shah MV, Zhang R, Loughran TP. Never Say Die: Survival Signaling in Large Granular Lymphocyte Leukemia. Clin Lymphoma Myeloma. 2009;9(0 3): S244–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang D, Loughran TP. Large granular lymphocytic leukemia: molecular pathogenesis, clinical manifestations, and treatment. ASH Educ Program Book. 2012. December 8;2012(1):652–9. [DOI] [PubMed] [Google Scholar]
- 17.Lamy T, Loughran TP. How I treat LGL leukemia. Blood. 2011. March 10;117(10):2764–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steinway SN, Leblanc F, Loughran TP. The pathogenesis and treatment of large granular lymphocyte leukemia. Blood Rev. 2014. May;28(3):87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koskela HLM, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmäki H, Andersson EI, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012. May 17;366(20):1905–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jerez A, Clemente MJ, Makishima H, Koskela H, LeBlanc F, Ng KP, et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood. 2012. October 11;120(15):3048–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andersson E, Kuusanmäki H, Bortoluzzi S, Lagström S, Parsons A, Rajala H, et al. Activating somatic mutations outside the SH2-domain of STAT3 in LGL leukemia. Leukemia. 2016. May;30(5):1204–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rajala HLM, Eldfors S, Kuusanmäki H, Adrichem AJ van, Olson T, Lagström S, et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood. 2013. May 30;121(22):4541–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Epling-Burnette PK, Liu JH, Catlett-Falcone R, Turkson J, Oshiro M, Kothapalli R, et al. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J Clin Invest. 2001. February 1;107(3):351–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rajala HLM, Porkka K, Maciejewski JP, Loughran TP, Mustjoki S. Uncovering the pathogenesis of large granular lymphocytic leukemia-novel STAT3 and STAT5b mutations. Ann Med. 2014. May;46(3):114–22. [DOI] [PubMed] [Google Scholar]
- 25.Küçük C, Jiang B, Hu X, Zhang W, Chan JKC, Xiao W, et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from γδ-T or NK cells. Nat Commun 2015. January 14;6:6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zambello R, Facco M, Trentin L, Sancetta R, Tassinari C, Perin A, et al. Interleukin-15 Triggers the Proliferation and Cytotoxicity of Granular Lymphocytes in Patients With Lymphoproliferative Disease of Granular Lymphocytes. Blood. 1997. January 1;89(1):201–11. [PubMed] [Google Scholar]
- 27.Chen J, Petrus M, Bamford R, Shih JH, Morris JC, Janik JE, et al. Increased serum soluble IL-15Rα levels in T-cell large granular lymphocyte leukemia. Blood. 2012. January 5;119(1):137–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang R, Shah MV, Yang J, Nyland SB, Liu X, Yun JK, et al. Network model of survival signaling in large granular lymphocyte leukemia. Proc Natl Acad Sci U S A. 2008. October 21;105(42):16308–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen J, Petrus M, Bryant BR, Nguyen VP, Goldman CK, Bamford R, et al. Autocrine/paracrine cytokine stimulation of leukemic cell proliferation in smoldering and chronic adult T-cell leukemia. Blood. 2010. December 23;116(26):5948–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tendler CL, Greenberg SJ, Blattner WA, Manns A, Murphy E, Fleisher T, et al. Transactivation of interleukin 2 and its receptor induces immune activation in human T-cell lymphotropic virus type I-associated myelopathy: pathogenic implications and a rationale for immunotherapy. Proc Natl Acad Sci U S A. 1990. July;87(13):5218–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mariner JM, Lantz V, Waldmann TA, Azimi N. Human T cell lymphotropic virus type I Tax activates IL-15R alpha gene expression through an NF-kappa B site. J Immunol Baltim Md 1950. 2001. February 15;166(4):2602–9. [DOI] [PubMed] [Google Scholar]
- 32.Ju W, Zhang M, Jiang J, Thomas CJ, Oh U, Bryant BR, et al. CP-690,550, a therapeutic agent, inhibits cytokine-mediated Jak3 activation and proliferation of T cells from patients with ATL and HAM/TSP. Blood. 2011. February 10;117(6):1938–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Waldmann TA, White JD, Goldman CK, Top L, Grant A, Bamford R, et al. The interleukin-2 receptor: a target for monoclonal antibody treatment of human T-cell lymphotrophic virus I-induced adult T-cell leukemia. Blood. 1993. September 15;82(6):1701–12. [PubMed] [Google Scholar]
- 34.Waldmann TA, Conlon KC, Stewart DM, Worthy TA, Janik JE, Fleisher TA, et al. Phase 1 trial of IL-15 trans presentation blockade using humanized Mik-Beta-1 mAb in patients with T-cell large granular lymphocytic leukemia. Blood. 2013. January 17;121(3):476–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dubois S, Shou W, Haneline LS, Fleischer S, Waldmann TA, Müller JR. Distinct pathways involving the FK506-binding proteins 12 and 12.6 underlie IL-2-versus IL-15-mediated proliferation of T cells. Proc Natl Acad Sci U S A. 2003. November 25;100(24):14169–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berkowitz JL, Janik JE, Stewart DM, Jaffe ES, Stetler-Stevenson M, Shih JH, et al. Safety, efficacy, and pharmacokinetics/pharmacodynamics of daclizumab (anti-CD25) in patients with adult T-cell leukemia/lymphoma. Clin Immunol Orlando Fla. 2014. December;155(2):176–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waldmann TA. JAK/STAT pathway directed therapy of T-cell leukemia/lymphoma: Inspired by functional and structural genomics. Mol Cell Endocrinol. 2017. August 15;451:66–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Massoud R, Enose-Akahata Y, Tagaya Y, Azimi N, Basheer A, Jacobson S. Common γ-chain blocking peptide reduces in vitro immune activation markers in HTLV-1-associated myelopathy/tropical spastic paraparesis. Proc Natl Acad Sci. 2015. September 1;112(35):11030–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frohna P, Tagaya Y, Ratnayake A, Doerr N, Basheer A, Al-Mawsawi L, et al. Results from a First-in-Human Study with Bnz-1: A Novel Peptide Inhibitor of IL-2, IL-9 and IL-15 for the Treatment of T-Cell Malignancies That Safely and Selectively Decreases Regulatory T-Cells, Natural Killer Cells, and CD8+ Central Memory T-Cells. Blood. 2017. December 7;130(Suppl 1):695–695.28798057 [Google Scholar]
- 40.Ren T, Yang J, Broeg K, Liu X, Loughran TP Jr., Cheng H. Developing an in vitro model of T cell type of large granular lymphocyte leukemia. Leuk Res. 2013. December;37(12):1737–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robertson MJ, Cochran KJ, Cameron C, Le JM, Tantravahi R, Ritz J. Characterization of a cell line, NKL, derived from an aggressive human natural killer cell leukemia. Exp Hematol. 1996. February;24(3):406–15. [PubMed] [Google Scholar]
- 42.Otani H, Siegel JP, Erdos M, Gnarra JR, Toledano MB, Sharon M, et al. Interleukin (IL)-2 and IL-3 induce distinct but overlapping responses in murine IL-3-dependent 32D cells transduced with human IL-2 receptor beta chain: involvement of tyrosine kinase(s) other than p56lck. Proc Natl Acad Sci U S A. 1992. April 1;89(7):2789–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maeda M, Shimizu A, Ikuta K, Okamoto H, Kashihara M, Uchiyama T, et al. Origin of human T-lymphotrophic virus I-positive T cell lines in adult T cell leukemia. Analysis of T cell receptor gene rearrangement. J Exp Med. 1985. December 1;162(6):2169–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olson KC, Kulling PM, Olson TL, Tan S-F, Rainbow RJ, Feith DJ, et al. Vitamin D decreases STAT phosphorylation and inflammatory cytokine output in T-LGL leukemia. Cancer Biol Ther. 2017. May 4;18(5):290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nicot C, Mahieux R, Takemoto S, Franchini G. Bcl-XL is up-regulated by HTLV-I and HTLV-II in vitro and in ex vivo ATLL samples. Blood. 2000. July 1;96(1):275–81. [PubMed] [Google Scholar]
- 46.Zhang M, Mathews Griner LA, Ju W, Duveau DY, Guha R, Petrus MN, et al. Selective targeting of JAK/STAT signaling is potentiated by Bcl-xL blockade in IL-2–dependent adult T-cell leukemia. Proc Natl Acad Sci U S A. 2015. October 6;112(40):12480–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sato N, Sabzevari H, Fu S, Ju W, Petrus MN, Bamford RN, et al. Development of an IL-15-autocrine CD8 T-cell leukemia in IL-15-transgenic mice requires the cis expression of IL-15Rα. Blood 2011. April 14;117(15):4032–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.