

Abstract
Engineered human kidney-on-a-chip platforms show tremendous promise for disease modeling and drug screening. Outstanding challenges exist, however, in reconstructing the complex architecture, cellular make-up, and matrix composition necessary for the proper modeling of kidney function. Here, we report the first fully tunable human kidney-on-a-chip platform that allows the reconstruction of the native architecture of the renal endothelial-epithelial exchange interface using entirely cell-remodelable matrix and patient-derived kidney cells. This platform consists of a double-layer human renal vascular-tubular unit (hRVTU) enabled by a thin collagen membrane that replicates the kidney exchange interface. We show that endothelial and epithelial cells lining their respective lumens remodel the membrane in culture into an approximately 1 µm-thick exchange interface composed of native basement membrane proteins. This interface displays sufficient mechanical integrity for media flow and blood perfusion. As a proof of principle, we demonstrate that our hRVTU performs kidney-specific functions including reabsorption of albumin and glucose from the epithelial channel. By incorporating multiple cell populations from single donors, we demonstrate that our hRVTU may have utility for future precision medicine applications. The success of our system provides new opportunities for the next generation of organ-on-a-chip models.
Organ-on-a-chip engineering has emerged as a promising alternative to animal models for drug safety screening and disease modeling.[1–3] Despite marked technological advances, it remains challenging to recreate complicated and organ-specific structures and functions in vitro. In particular, human kidneys depend upon a complex multicellular architecture for the efficient exchange of solutes between vascular and parenchymal compartments. The exchange process begins with glomerular filtration, followed by tubular secretion and reabsorption. Proper tubular function is critical for human electrolyte regulation, toxin and metabolite excretion, and acid/base homeostasis.[4,5] These processes also result in a high incidence of tubular exposure to concentrated drugs and toxins, leading to kidney injury and substantial subsequent morbidity and mortality.[6] In order to reduce this burden of disease, there is an urgent need for improved preclinical kidney models for pharmaceutical evaluation and disease exploration.[7,8]
The renal tubular system consists of three major structural components: the tubular lumen, the vascular lumen, and a thin layer of basement membrane separating the luminal spaces, surrounded by extracellular matrix (ECM). The system also contains three major cell types: fenestrated endothelial cells, specialized epithelial cells, and interstitial perivascular cells interposed between them. The presence of flow, three-dimensional architecture, close vascular and tubular proximity, and native-like ECM are important for proper cellular and tissue function.[8] Conventional two-dimensional culture fails to replicate the three-dimensional luminal structure and the complexity of the vascular-tubular interface, whereas animal models suffer from significant interspecies variability.[9] The recent development of organ-on-a-chip devices has allowed flow-directed culture and the incorporation of multiple cell types and matrix components. For example, single-channel platforms emulating the renal tubules[5,10–12] or renal vasculature,[4] display physiologic functionality and exhibit confluent cell growth with high viability and nephron segment-specific marker expression. It is technically challenging, however, to achieve the close approximation of vascular and tubular compartments necessary to model solute transfer across the renal tubular exchange interface. A breakthrough came from the development of polydimethylsiloxane (PDMS)-based devices incorporating cell monolayers on either side of ~10 μm thick porous PDMS or polycarbonate membranes.[13–18] Despite their importance for the field, these platforms depend upon the use of artificial materials that are not cell-remodelable and do not support cell incorporation into the bulk matrix, limiting their ability to model the renal tubular functional unit.[8]
To address these challenges, we created a tunable and remodelable human renal vascular-tubular unit (hRVTU) formed entirely in collagen hydrogel. Independently fabricated vascular and tubular lumens are assembled against a solute-permeable and mechanically robust collagen membrane and seeded with human kidney microvascular or epithelial cells, respectively. The collagen membrane within our device is remodeled by cells in culture to resemble the native basement membrane, while maintaining sufficient integrity to support blood perfusion through the vascular lumen. We further demonstrated kidney-specific function in our device, including the selective reabsorption of albumin and glucose. Using cell-remodelable matrix and patient-derived cells, our hRVTU shows promise for the study of human kidney diseases, pharmaceutical screening, and precision medicine applications.
Results:
Engineered hRVTUs were manufactured in collagen and enclosed within an acrylic housing (Figure 1a). First, a thin collagen membrane was compression molded across a circular opening (18 mm in diameter) in a chemically-treated plastic sheet with predefined thickness (25 μm, 50 μm). The membrane’s circular geometry allows for isotropic tethering of collagen at the boundary of the central hole, providing sufficient mechanical support for membrane manipulation. Second, building on previously described methods, the top (vascular) and bottom (tubular) compartments were fabricated with a combination of soft lithography and injection molding.[19] Two separate PDMS molds with lithographically determined geometry were used to imprint microfluidic channels onto each respective collagen layer, with individual inlets and outlets for perfusion positioned in line with wells in the top housing plates. After collagen gelation, PDMS stamps were removed and the plastic sheet was placed on the bottom acrylic platform so that the circular collagen membrane sealed the bottom microfluidic collagen channels, forming the bottom (tubular) layer of our device. The top acrylic housing plate was then added, so that its microfluidic channels were sealed against the superior face of the collagen membrane to form the top (vascular) layer. The assembled device contains separate, orthogonally aligned inlets and outlets for each set of channels, so that cell seeding and media perfusion can be performed independently for each microfluidic layer. Once assembled, our devices were able to consistently withstand flow via syringe pump of up to 10 µl/min, leading to wall shear stress throughout the device ranging from ~1-10 dyne/cm2. For the experiments in this paper, devices were cultured under gravity-driven perfusion with a similar estimated range of shear stress during initial periods of perfusion.[19,20]
Figure 1.

Engineering the human renal vascular-tubular unit (hRVTU). A) Illustration of the components of hRVTU device fabrication. B - C) Device constructed with parallel-channel geometry for the bottom layer and either parallel-channel (B) or grid (C) geometry for the top layer. Channels in each layer were perfused separately with fluorescent beads and 4× (top) and 10× (bottom) magnification widefield fluorescent imaging obtained. D) Orthogonal view of device from C showing complete separation of each channel by a collagen membrane. E) Orthogonal view of 3D confocal images for collagen reflectance for vessels made with 50 µm (top) and 25 µm (bottom) membrane scaffolding sheets. Membranes delineated by yellow arrows. F) Plot of membrane thickness measured by collagen reflectance for 30 hRVTUs shortly after construction on either 25 µm or 50 µm plastic membrane scaffolding. Solid lines indicate the mean and standard deviation for each condition.
Fluorescent microspheres were perfused into the microfluidic channels revealing that lithographic patterns were transferred with high fidelity to the collagen channels within the hRVTU (Figure 1b-c). Orthogonal reconstruction of z-stack images revealed complete separation between the two channels by an intact collagen membrane (Figure 1d). Collagen membrane thickness correlated well with the thickness of plastic sheet used: membranes produced on 25 µm-thick plastic sheets had an average thickness of 30.4 µm (n= 9, SEM = 3.2), significantly lower than an average thickness of 59.3 µm (n= 21, SEM = 4.2) for membranes produced on 50 µm thick sheets (p < .001). To minimize the thickness of the vascular-tubular interface, we chose 25 µm-thick plastic sheets for the following studies of our hRVTU.
Next, we seeded Human Umbilical Vein Endothelial cells (HUVECs) into the top channel, and human kidney epithelial cells into the bottom channel. These epithelial cells were harvested from adult human renal cortical tissue, using methods previously shown to enrich for proximal tubular cells expressing aquaporin 1, aquaporin 2, and sodium-glucose co-transporter-2 (SGLT2).[5,21] Both channel lumens displayed confluent cellular coverage with retention of the intended geometrical features for up to 4 weeks of culture under gravity-driven flow (Figure 2a). Immunofluorescence performed in situ within hRVTUs showed that endothelial channels were populated by cells that elongated in the direction of flow and displayed VE-cadherin staining, signifying intact cellular junctions. Epithelial channels contained a heterogenous population that included tall, E-cadherin-positive cells (Figure 2a-b). No VE-cadherin-positive cells were observed within the epithelial channel, nor were any E-cadherin-positive cells observed within the endothelial channel, as expected. We embedded and cryosectioned the intact hRVTU, revealing the persistence of distinct channels with the intended cellular composition. H&E staining showed that the hRVTU approximated many features of normal human histology, with flat homogenous cells seen in the endothelial channel (Figure 2e) and a population of taller, cuboidal cells containing structures resembling the brush border (Figure 2f) seen in the epithelial channel. Immunohistochemical staining showed matrix remodeling of hRVTUs by the cells, with deposition of basement membrane proteins such as collagen IV and laminin (Figure 2g-h). Collagen membrane thickness decreased over time, from initial thickness of 31.7 µm (n = 9, SEM = 3.9 µm), down to an average of 3.1 µm (n = 7, SEM = 0.8 µm), and a minimum thickness of 0.5 µm after 14 days of culture (p < .0001) (Figure 2i).
Figure 2.
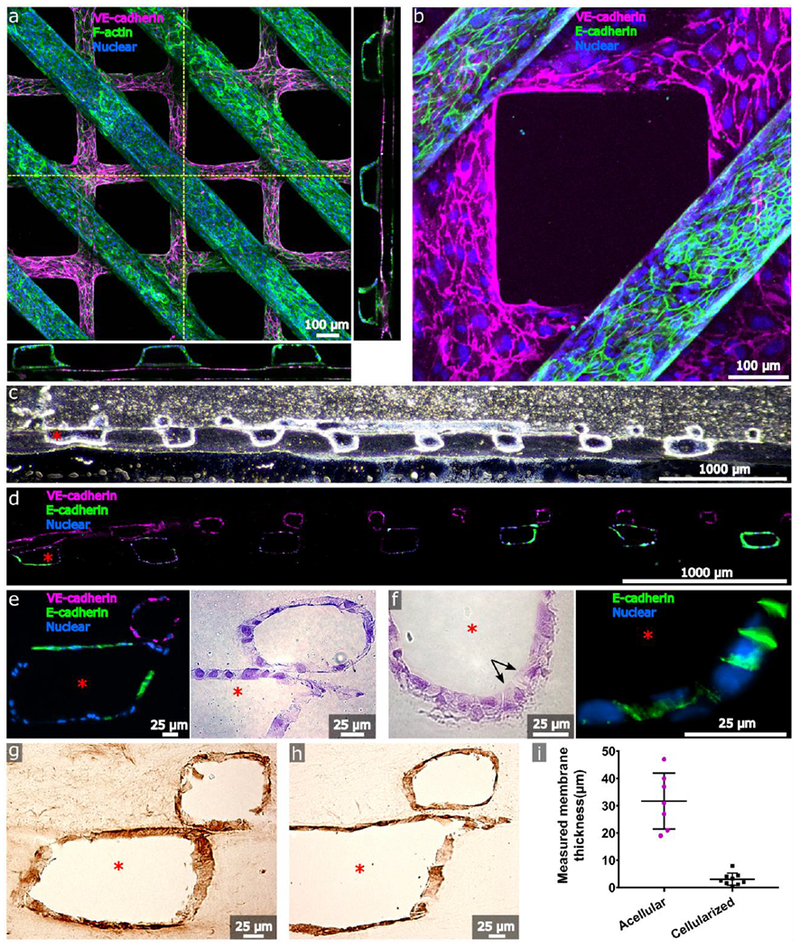
Cellularization of the human renal vascular-tubular unit (hRVTU). Top layers with grid geometry were seeded with P4-P6 human umbilical vein endothelial cells (HUVECs) and bottom layers with parallel-channel geometry were seeded with P1-P3 primary adult human renal cortical epithelial cells. A) Three-dimensional z-stack projection of an hRVTU with epithelial channel stained for F-actin and endothelial channel stained for VE-cadherin. Orthogonal images obtained along indicated planes (dashed yellow lines) are shown flanking the image and highlight the proximity of the two channels. B) 20× close-up of epithelial channel from A, stained for E-cadherin. C and D) Brightfield and fluorescent images of cryosectioned samples from an hRVTU following 14 days of culture and in situ immunostaining, at 10× magnification. E) Zoomed view of hRVTU from D (left), with H&E staining on a similar section shown adjacent (right). Channels show close apposition and heterogeneous junctional staining for E-cadherin. F) H&E staining of cryosectioned hRVTU reveals cuboidal cells with brush border-like structure (arrows) on the luminal face of the epithelium (left). Similar regions expressed basolateral E-cadherin (right). G) IHC staining of cryosectioned hRVTU for collagen IV. H) IHC staining of cryosectioned hRVTU for laminin. I) The membrane thicknesses of acellular devices were compared with devices following 14 days of cell culture. Solid lines indicate the mean and standard deviation for each condition. Red asterisk denotes the epithelial lumen, and representative colors for all staining are indicated within each panel.
In addition to having separate cellular populations in the endothelial and epithelial channels, the collagen matrix used for hRVTU fabrication allows other cell types to be incorporated into the bulk matrix during device manufacturing. Making use of this feature, we next created hRVTUs with HUVECs in the top channel, human adult renal cortical epithelial cells in the bottom channel, and human fetal kidney pericytes incorporated into the collagen matrix (including the membrane). Devices were cultured under gravity-driven perfusion of media through the endothelial- and epithelial-lined channels, as above. After one day of culture, we observed that pericytes interacted with the bulk matrix by extending processes into it. Following one week of culture, pericytes were observed to advance processes along the abluminal surfaces of both the endothelial and epithelial channels. These interactions, however, did not appear to induce any structural changes in the endothelial or epithelial lumens (Figure 3).
Figure 3.
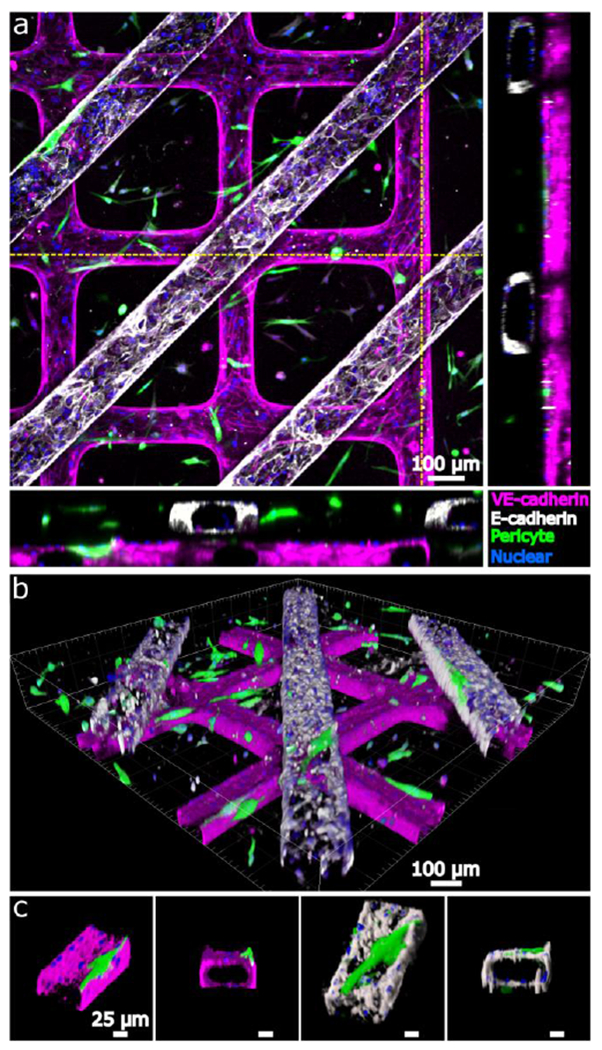
Incorporation of pericytes into the human renal vascular-tubular unit (hRVTU). hRVTUs were formed with P4-P6 Neuron-Glial antigen 2 (NG2)-positive, GFP-transfected primary fetal kidney pericytes incorporated into collagen bulk during formation. Top layers with grid geometry were seeded with P4-P6 HUVECs and bottom layers with straight-channel geometry seeded with P1-P3 primary adult human renal cortical epithelial cells. A) Z-stack projection of an hRVTU showing endothelial junctions (VE-cadherin) and epithelial junctions (E-cadherin) with pericytes visualized with the collagen bulk. Orthogonal images obtained along indicated planes (dashed yellow lines) are shown flanking the image and show pericytes associating with both lumens. B) Three-dimensional blend projection (Imaris, Bitplane) highlighting pericyte association with both channels. C) Close-up of features from B showing pericytes growing along the endothelial (magenta) and epithelial (white) channels.
One promising future possibility of organ-on-a-chip technology is the creation of patient-specific models for precision medicine, incorporating multiple cellular populations from a single donor. Towards this goal, we isolated three cellular populations from a single human fetal kidney donor: epithelial cells via an epithelial cell adhesion molecule (EpCAM)-positive selection, endothelial cells via VE-cadherin-positive / CD45-negative selection, and pericytes via VE-cadherin-negative / Neuron-glial antigen 2 (NG2)-positive selection (Supplemental Figure 1). We have previously shown[4] that human kidney microvascular endothelial cells (HKMECs) isolated in this manner have a unique phenotype and expression pattern, and may therefore better recapitulate organ-specific functions within our device. Isolated epithelial cells in planar culture displayed co-localization of staining for ADP-ribosylation factor-like protein 13B and acetylated tubulin, as expected for primary cilia,[22] and basolateral staining for sodium-potassium-ATPase and E-cadherin (Supplemental Figure 2). hRVTUs were then fabricated using the cells obtained from a single fetal donor. GFP-labeled pericytes were incorporated into the collagen bulk during fabrication, and epithelial cells and HKMECs from the same donor were seeded into their respective channels. All three cellular populations survived over two weeks of co-culture in the same device, via independent perfusion of the appropriate media for each cell type into the vascular and tubular channels. Similar to HUVECs, HKMECs formed robust VE-cadherin junctions and aligned in the direction of flow. As with human adult kidney epithelial cells, the fetal kidney epithelial cells lining the epithelial lumen consisted of a phenotypically heterogeneous population. Pericytes extended their processes, which were observed to associate with the abluminal surface of both the endothelial and epithelial channels (Supplemental Figure 3). We next examined the basic functionality of our hRVTU platform by perfusing either 40kDa Dextran into both lumens, or whole blood into the vascular lumen of devices seeded with fetal HKMECs and renal cortical epithelial cells. Dextran perfusion demonstrated that the epithelial channel has apparent robust barrier function over 5 minutes of perfusion, whereas endothelial channels were shown to be much more permeable, consistent with our prior published results obtained within single-layered devices (Figure 4a, Supplemental Movie 1).[4] Membrane barrier function was maintained during perfusion, and we did not observe any leaking of dextran from one compartment to the other. We perfused whole blood into the endothelial channel for 10 minutes under live fluorescent imaging and confirmed that no red blood cells or platelets leaked from the top endothelial lumen into the bottom epithelial lumen. During perfusion, a small portion of FITC-CD41a-labeled platelets adhered onto the surface of the endothelial channel, but no apparent blood cell aggregation occurred, suggesting that the endothelial channel of the hRVTU maintains a non-thrombogenic surface for blood perfusion (Figure 4b, Supplemental Movie 2).
Figure 4.
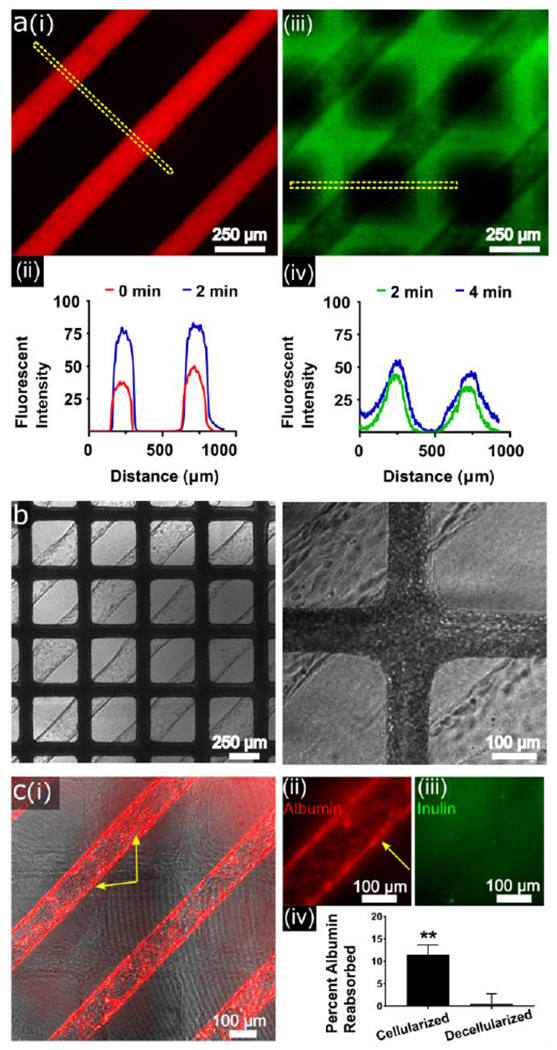
Functional analysis of human fetal and adult single-donor human renal vascular-tubular units (hRVTU). A) hRVTU created from human fetal renal cortical epithelial cells and kidney microvascular endothelial cells from the same donor sets were used for permeability assessment. Texas Red-labeled 70 kDa dextran was perfused into the epithelial channel during real-time imaging with 10 µm FITC-labeled 70 kDa dextran perfused into the endothelial 2 minutes later. (i) Epithelial channel is shown ~1 minute into perfusion and (ii) endothelial channel ~3 minutes into perfusion. (ii and iv) Fluorescent intensity over distance was calculated for the ROI shown in each image (dashed yellow lines) at two time-points using imageJ software (U.S. National Institute of Health, Bethesda, Maryland), demonstrating relatively little increase in width during the measured period. B) Blood from healthy volunteers was perfused into the endothelial channel of a hRVTU constructed as in A, under real-time imaging. No blood components can be seen within epithelial channels throughout ten minutes of blood perfusion. Left panel at 4× magnification, right panel at 10× magnification. C) hRVTU were created from adult renal cortical epithelial cells and human kidney microvascular endothelial cells from the same donor sets. After four days in culture device epithelial inlets were perfused with rhodamine-labeled human serum albumin and FITC-labeled inulin under real-time imaging. Following perfusion, devices were decellularized with trypsin, and perfusion repeated as an internal control. (i) Devices shown 15 minutes into perfusion show albumin (red) accumulating within cells. Yellow arrows highlight intracellular albumin accumulation within cells both on the top and side walls of vessels. (ii) Magnified image of a channel 30 minutes into perfusion shows albumin accumulating within cells, while inulin (iii) is not seen to accumulate intracellularly. Effluent was collected at one hour and run on a spectrophotometer, with albumin reabsorption calculated as noted in the main text. A two-tailed paired T test was done comparing albumin reabsorption in cellularized versus decellularized vessels (n = 3, bars indicate SEM and ** indicates P < 0.01).
To further explore the structure and function of the single donor hRVTU using adult cells, we created devices with matched adult HKMECs and adult human renal cortical cells from single donors (a total of seven devices from four donors were created for the following immunofluorescence and solute-perfusion experiments). These hRVTU showed similar morphology to the above single-donor fetal HRVTU and expressed renal-specific markers including staining for SGLT2 (Supplemental Figure 4). To test the kidney-specific function of our platform, we next examined specific solute reabsorption within these adult hRVTUs. Fluorescent inulin and albumin were perfused through the epithelial channel, and the concentration of both the initial perfusate and the effluent in the epithelial channel were measured spectrophotometrically. As inulin is not reabsorbed by renal cortical epithelial cells,[23] the difference between the ratios of starting to ending concentrations of these substances was used to determine specific albumin reabsorption. At one hour of perfusion, normalized albumin absorption was 11.1% ± 2.0%. (n = 3). To help exclude non-specific differences in filtration, devices were decellularized and perfusion repeated to serve as an internal control. No albumin reabsorption was observed following decellularization (n = 3). The difference in normalized albumin reabsorption between cellularized and decellularized devices was significant (Figure 4c, n = 3, SEM 0.42, p = 0.0015). Fluorescent imaging during perfusion showed albumin, but not inulin, accumulating within cells during perfusion, corroborating these findings (Figure 4c). Barrier function could be observed to be much higher in cellularized devices, as expected (Supplemental Movie 3). Finally, the reabsorption of glucose was tested for through perfusion of the fluorescent glucose analogue, 2-NBDG. While cells were visually observed to uptake 2-NBDG following perfusion (Supplemental Figure 4), attempts to quantify this reabsorption through spectrophotometric analysis of initial perfusate vs effluent did not show a statistically significant difference (not shown).
Discussion:
Approximately 20% of acute kidney injury is due to drugs or toxins,[9] with the most common site of injury in both septic and toxic insult being the renal tubule.[6] The concentration of potential toxins within the tubular lumen, along with the high cardiac output received by the kidney, contributes to the susceptibility of this area to nephrotoxicity.[7] In addition, the renal tubules modulate the pharmacokinetics of administered medications through reabsorption and secretion,[24] and are the site of action for several medications targeting specific transporters.[25] The physiologic functions of the renal tubules depend upon a complex interplay between a polarized renal tubular epithelium lined with numerous specific transporters, the interstitial space and its resident cells, and the specialized endothelium of associated renal blood vessels.[4,5]
While modeling the structure of the renal vascular-parenchymal interface poses significant technical challenges, there is increasing interest in the development of kidney segment-specific platforms for disease modeling and drug screening. In this work, we described the in vitro reconstruction of the renal vascular-tubular exchange unit. We demonstrated the successful fabrication of a thin biological membrane that allowed the close approximation of renal vascular and tubular channels with complex flow and three-dimensional geometry within a single device. We showed that this membrane can be remodeled by luminal cells through long-term culture to create an interface between channels that resembled that of the native matrix in both composition and thickness.[26,27]
Our hRVTU combines several features that have been individually shown to enhance the in vitro modeling of renal tubular function: flow,[8,13,16,28–33] biologic ECM components,[34–36] a three-dimensional tubular structure,[37] and presence of endothelial[38,39] and perivascular cells.[40,41] Previously described renal microphysiological systems include single-channel platforms emulating the renal tubules[5,10–12] or renal vasculature,[4] and dual-channel PDMS-based devices incorporating cell monolayers grown on artificial membranes.[13–18] Our platform is fundamentally different from these prior dual-channel devices in that our hRVTU allows for complete cellularization of a tubular lumen, customizable flow dynamics and three-dimensional geometry, incorporation of stromal cells, and the use of cell-remodelable materials with mechanical properties mimicking those seen in vivo.
Our successful creation of a robust native-like interface with close approximation of epithelial and vascular channels provides a unique opportunity to study interactions between the renal tubules and the associated microcirculation. Perfusion studies within our hRVTU demonstrated kidney-specific function, with specific reabsorption of albumin seen after one hour of perfusion. We observed 11.1% uptake of the perfused albumin, with reabsorption dependent on the presence of renal epithelial cells, but did not observe an increase in albumin within the vascular channel. We believe this finding to be a physiologic result as, in the human body, albumin reabsorption involves both lysosomal degradation within epithelial cells, as well as transcytosis into the extracellular fluid, rather than direct transport from the tubular lumen to the vascular lumen.[42–44] We further showed glucose reabsorption within our devices through perfusion of a fluorescent glucose analogue, 2-NBDG. While we observed microscopic evidence of fluorescent 2-NBDG uptake by epithelial cells (Supplemental Figure 4), we were not able to detect a statistically significant difference between the glucose concentration in the perfusate and the effluent. This may be due to device variability, or to heterogeneity of primary human epithelial cells.
Other limitations of our hRVTU are centered on inter-device variability and difficulties in device manufacturing, leading to a moderate rate of device failure during assembly. Once the device was assembled, however, the membrane and both channels displayed robust mechanical integrity during extended flow-directed culture. While we initially used 7.5 mg/ml collagen throughout, in our later functional experiments of inulin/albumin and glucose reabsorption we were able to achieve greater membrane integrity during assembly using 10 mg/ml collagen for the membrane, while continuing to use 7.5 mg/ml collagen for the top and bottom pieces. We were also able to fabricate membranes in up to 15 mg/ml collagen (data not shown) highlighting the versatility of our platform. Depending on the application, other materials could be used for membrane fabrication to achieve the desired mechanical and biologic characteristics, with decellularized human renal ECM being an intriguing possibility.[27] We observed heterogeneity in our epithelial cell populations, especially with regards to E-cadherin staining, and it is possible that sorting for renal proximal tubule-specific markers such as CD10/CD13[45] would select for a more homogenous population. It is also possible that epithelial exposure to serum from endothelial media may drive cellular differentiation, and experimentation with different media in the future may be helpful. Finally the gravity-driven perfusion protocol we used provides a decaying flow profile with an average shear stress of ~ 0.1 dyne cm−2, different from the more continuous ~ 1 dyne cm−2 experienced by the renal tubules in vivo.[30] While we noted appropriate cellular flow alignment, future work could explore the effect of different flow profiles on hRVTU function.
In summary, our hRVTU system represents the first fully cell-remodelable platform to structurally mimic the close proximity and cellular composition of renal microvascular and tubular compartments. This adaptable platform accommodates different geometries, cell types, hydrogels, and perfusates, while allowing for high-resolution and confocal imaging, histologic analysis, and biochemical analysis of effluent. The fabrication techniques are transferrable, and we anticipate their use in the study of parenchymal-vascular interactions throughout the human body in different states of health and disease. We present promising proof-of-principle functional studies of this device via our quantification of the selective reabsorption of albumin. Future directions include examining the effects of different flow rates on endothelial and epithelial cell function, additional tubular resorption/secretion studies, and detailed characterization of the potential role of pericytes on endothelial cell function and ECM production.[40,41] Improvement in throughput and further characterization of solute transport within our device will enable the use of this platform for preclinical pharmaceutical screening and disease modeling for precision medicine applications.
Methods:
Fabrication of Materials:
Channel geometries (Figure 1b) were created on silicon wafers by photolithographic patterning of SU-8 photoresist, which was transferred onto PDMS stamps via soft lithography.[19,46,47] Top and bottom housings for our microphysiological system were fabricated from poly(methyl methacrylate) by the University of Washington Machine Shop. Top housing pieces were fabricated with a 1 mm deep, 20 mm × 20 mm well on their inferior surface, two injection ports designed to accommodate a 1 ml tuberculin syringe (BD), and two inlets and outlets of 6 mm in diameter. Bottom pieces were designed with a 15 mm × 15 mm square hole in the center, surrounded by a 25 mm × 25 mm ledge recessed 45 µm from the top of the housing. A 50 mm × 50 mm plastic sheet with a central 18 mm diameter hole served as scaffolding for membrane formation, and was custom-ordered from either 25 or 50 µm-thick stock (Practi-shim,™ Accutrex). Aligned holes for 4 × 40mm screws were located in the four corners of the plastic sheet and housing pieces.
Collagen Gel Preparation:
As previously described, type I collagen was extracted from rat tails, lyophilized, and suspended in acetic acid (0.1%) at a concentration of 15 mg/ml to make a collagen stock solution.[19,48] This solution was vigorously mixed, centrifuged at 1000g × 10’ at 4°C, and stored at 4°C. Immediately prior to microvessel fabrication, stock collagen was diluted on ice to 7.5 mg/ml in a buffer made from Medium 199 (10x M199, Lonza) diluted to a concentration of 1x in epithelial media (DMEM/F12 media [Invitrogen], 1x ITS supplement [Invitrogen], 50 nM hydrocortisone [Sigma], and antibiotic/antimycotic supplement [Invitrogen]). For devices utilizing a membrane formed in 10 mg/ml, rather than 7.5 mg/ml collagen, a separate collagen solution was made in this concentration. This working collagen solution was neutralized to a pH of 7.4 using 1 Molar NaOH and mixed thoroughly.
Fabrication and assembly of the human Renal Vascular-Tubular Unit (hRVTU):
All components were either bleach or autoclave sterilized prior to use, and all procedures were performed in sterile fashion within a biosafety hood. hRVTU fabrication can be separated into four steps:
(1) Bottom channel formation - A 22 mm × 22 mm microscope cover glass was seated within the well in the center of the bottom housing. The surface of the cover glass and the surrounding area were treated with oxygen plasma for ~10-30’’ (Corona SB, Elveflow). 1% Polyethyleneimine (PEI) was added to the cover glass and inner well of the bottom housing. After 10 minutes, PEI was aspirated and 0.1% Glutaraldehyde added. After 30 minutes, bottom pieces were rinsed twice with sterile water and allowed to dry. Approximately 200-300 µl of the 7.5 mg/ml collagen working solution was then added uniformly across the cover glass and surrounding well. The patterned surface of a PDMS stamp was treated for 45 seconds with oxygen plasma, 150-200 µl of 7.5 mg/ml collagen solution added uniformly across the patterned area, and the stamp then carefully pressed in place atop the collagen-coated cover glass on the bottom housing piece. Care was taken to ensure that the pattern features were centered along the diagonal line of the coverglass.
(2) Top Channel Formation – Prior to assembly, the well within a top housing piece was treated with oxygen plasma and coated with PEI and glutaraldehyde and then washed, as above. A patterned PDMS stamp was placed within a sterile dish with the patterned side facing up. The patterned surface was treated with oxygen plasma for 30-45’’. A top housing piece was then placed over the top of the patterned surface so that the pattern features on either end lined up with the inlet and outlet of the top housing. The surfaces were pressed together gently to allow them to bond. Sterile dowel pins were placed through the inlets and outlets (to prevent these areas from filling with collagen). Using a 1 ml tuberculin syringe (BD), 7.5 mg/ml collagen was slowly injected through the injection port to fill the space between pattern features.
(3) Membrane fabrication – A plastic sheet served as membrane scaffolding and was placed on top of a 25 mm × 25 mm flat PDMS piece. The area immediately surrounding the central hole in the sheet was treated with PEI and glutaraldehyde, as above. Membranes were then washed as above and allowed to dry completely before use. Approximately 50 µl of collagen was placed in the window within the plastic sheet. A second flat PDMS piece was pressed firmly on top to disperse collagen evenly across the central window of the plastic sheet and allow creation of a collagen membrane via compression molding.
(4) Device Assembly – Above components were transferred to a 37° incubator and collagen allowed to cross-link for 30 min. Following incubation, the bottom housing piece was covered with sterile phosphate-buffered saline (PBS) and removed revealing a patterned collagen piece. Following submersion in PBS, the top PDMS piece was removed from the plastic sheet. The sheet was then carefully worked free from the bottom PDMS piece. This sheet, with an intact collagen membrane now spanning its central hole, was placed on top of the bottom housing piece, with the membrane aligned over the patterned bottom piece. The top housing piece was immersed in PBS and the PDMS stamp removed. Dowel pins were removed, and the inlets/outlets cleared of collagen using a P200 pipette tip. 4 × 40 mm screws were placed through screw holes in its four corners, and the top housing piece slowly lowered onto the top of the plastic sheet/bottom housing, ensuring that pattern features were perpendicular to those of the bottom pattern. Screws were very lightly tightened to mechanically seal all layers together without distorting the pattern features. Holes were poked through the inlet/outlet corresponding to the bottom channel, piercing the membrane in that area and allowing for future perfusion of the bottom channels. Both reservoirs were filled with the desired media, and the device placed in a 37°C incubator.
Cell Isolation and Culture:
Following approval by the Institutional Review Board of the University of Washington (IRB447773EA) and informed consent, human fetal kidney tissue was obtained from voluntary pregnancy interruptions (fetal age 16-20 weeks). Following gross dissection, kidney tissue was minced in endothelial basal medium-2 (Lonza) supplemented with 100 U/ml DNase (Roche) and 0.2 mg/ml Liberase. Tissue homogenate was shaken for 30 min at 37°C and then filtered through a 40 µm cell strainer. EpCAM-positive cells were separated using EpCAM microbeads (Miltenyi Biotec) on a magnetic-activated cell sorting platform, labeled as fetal human renal cortical epithelial cells, and used for experiments between P2-P4. The EpCAM-negative cells were then cultured for 5 days in a 5% oxygen environment in media containing 40 ng/ml VEGF. Fluorescence-activated cell sorting (FACS) was then performed using a BD FACSAria II machine within the University of Washington Cell Analysis Facility and quantitated with FlowJo software (Tree Star). Cells were FACS sorted into a CD144+ population (APC-anti-CD144, eBioscience) which were labeled as fetal Human Kidney Microvascular Endothelial Cells (HKMEC), and a NG2-positive population (FITC-anti-hNG2, R&D Systems) which were labeled as fetal human kidney pericytes (Supplemental Figure 2). HKMEC were cultured in HKMEC media (EBM-2 supplemented with an antibiotic/antimycotic, 10% FBS, 100 μg/ml Endothelial Cell Growth Supplement, 50 μg/ml Heparin, and 20 ng/mL VEGF for fetal HKMEC or 100 ng/ml VEGF for adult HKMEC) on gelatin-coated plates and used for experiments between P2-P4. Fetal kidney pericytes were labeled with GFP via transduction of a GFP-labeled lentiviral vector (PGIPZ non-silencing shRNA, Dharmacon). Pericytes were cultured in pericyte media (DMEM/F12 media [Invitrogen], 1x ITS-A supplement [Invitrogen], 10% FBS, and antibiotic/antimycotic supplement [Invitrogen]) and used for experiments between P4-P6.
For isolation of adult renal tubular epithelial cells and endothelial cells, human kidney cortical biopsy specimens were obtained from a local biorepository (NWBioTrust). Following gross dissection, cortical tissue was finely minced, collagenase (Worthington Biochemical) treated, and the solution incubated in a shaker incubator at 37°C for 30 min. Cell suspension was allowed to settle, and supernatant transferred into a tube containing 5 ml of horse serum. This solution was then mixed and centrifuged for 7 min at 1000 rpm. Following centrifugation, the cell pellet was washed with epithelial media and the suspension then plated and cultured at 37°C. Cells were separated into EPCAM-positive (epithelial) and EPCAM-negative, CD 144-positive (endothelial) populations. Both cell types were used for experiments between P1-P4.
Purchased Human Umbilical Vein Epithelial Cells (HUVEC, Lonza) were cultured in endothelial growth medium (Lonza), with media changed every 48 hours. Cells were used for experiments between P4-P6.
Cell Seeding and hRVTU culture:
To cellularize the collagen channels within our device, cell seeding was performed. Endothelial cells (HUVECs or HKMECs) were detached from their culture flask with trypsin (Invitrogen) and suspended in their respective growth media at a density of 7 × 106 cells/ml. Reservoirs were balanced so that no flow existed through the channels, and 10 µl of cell suspension injected into the designated endothelial channel using a gel-loading pipette tip. hRVTU were placed back in a 37°C incubator for 20 min to allow cells to attach to the channel walls. Additional cells were seeded as needed to obtain complete channel coverage. hRVTU were then placed in the incubator for 6-12 hours, after which time gravity-driven flow was initiated by adding media to the inlet for the seeded channel so that a pressure-drop of 6 mm of water existed across the inlet/outlet reservoirs. 50 ng/ml human fibronectin (Sigma-Aldrich) in epithelial media was added to the inlet for the remaining channel to facilitate adhesion of epithelial cells. After 12 hours either fetal or adult cortical epithelial cells were seeded into the epithelial channel utilizing the same procedure as above, and gravity-driven flow initiated as above. For experiments containing pericytes within the collagen bulk, fetal human kidney pericytes were trypsin-treated, suspended in pericyte media, and mixed with the collagen working solution prior to hRVTU fabrication to give a concentration of 0.5 × 106 cells/ml within a 7.5 mg/ml collagen solution. Following hRVTU seeding, media was changed every 12 hours with a 6 mm height differential between inlet and outlet. These conditions were designed to yield ~2 hours of a decaying, unidirectional Poiseuille flow within the channels, and an average wall shear stress of ~ 0.1 dyne cm−2.[19]
Fluorescent Microsphere Perfusion:
Following hRVTU creation, 1 µm fluorescent polystyrene microspheres (FluoSpheres™, Invitrogen) were perfused at a dilution of 1:100 – 1:200. Red fluorescent beads were perfused into one channel and blue/green spheres into the other, and imaging performed on a Nikon Ti-E inverted microscope and a Nikon A1R confocal microscope.
Immunofluorescence:
For in situ immunofluorescence of hRVTU, devices were cellularized, cultured for the intended duration, and fixed via perfusion of 3.7% paraformaldehyde through the intact device for 20 min followed by washing with PBS three times. For immunofluorescence of isolated fetal kidney cells, P3 cells were cultured on top of glass coverslips for 8 days and then fixed with 3.7% paraformaldehyde. Both hRVTU and coverslips were incubated for one hour in 2% BSA and 0.1% Triton-X in PBS for blocking and membrane permeabilization. Primary antibody staining was performed overnight at 4°C. Primary antibodies and dilutions used were: rabbit anti-VE-cadherin (1:50-1:100 dilutions, Abcam), mouse anti-E-cadherin (1:50, Abcam), Phalloidin-568 primary-conjugated antibody (1:50 dilution, Invitrogen), FITC-conjugated sheep anti-VWF (1:100, Abcam), mouse anti-acetylated tubulin (1:10000 dilution, Sigma-Aldrich), rabbit anti-SGLT2 (1:50 dilution, Abcam), and rabbit anti-ADP-ribosylation factor-like protein 13B (1:10000, Proteintech). Imaging was performed on a Nikon A1R confocal microscope with image analysis conducted in ImageJ software[49] (U.S. National Institute of Health, Bethesda, Maryland) and three-dimensional rendering performed using Imaris software (Bitplane).
Histology:
hRVTU were cellularized, maintained in culture for the intended duration, and fixed via perfusion of 3.7% paraformaldehyde. Following fixation, Tissue-Tek O.C.T. compound (Sakura) was perfused through the devices for 48 hours. Screws were removed, and the intact hRVTU was placed in a −80°C freezer for at least 3 hours. Once frozen, the bottom piece and glass coverslip were then removed, the collagen allowed to soften slightly within the top housing within a −20°C freezer, and the collagen removed en bloc from the top housing with a scalpel. The collagen was then equilibrated in a cassette filled with O.C.T. overnight prior to being frozen in an ethanol slurry. Frozen blocks were then sectioned using a cryostat (Leica CM 1850) into 7 µm sections and sections transferred to glass slides (Superfrost™ Plus Gold, Thermo Fisher). For immunohistochemistry, slides were treated with 3.7% paraformaldehyde, blocked with a mixture of 1.5% natural goat serum (Sigma) in PBS for one hour and then incubated with rabbit anti-Laminin antibody (1:25 dilution, Abcam) or rabbit anti-Collagen IV (1:100 dilution, Abcam) for one hour. A biotinylated goat anti-rabbit antibody (Jackson ImmunoResearch) was then applied for one hour at a 1:400 dilution and developed with DAB solution (Sigma). Hemotoxylin and Eosin staining of fixed slides was also performed. For experiments involving immunofluorescent imaging of cryosections, immunofluorescence was performed in-situ within the hRVTU as described above, prior to embedding and cryosectioning. Histologic specimens were imaged on a Nikon Eclipse 90i microscope within the University of Washington Histology and Imaging Core.
Collagen Membrane Imaging and Quantification:
For high-resolution determination of membrane thickness, the complete light reflectance of the collagen crystal lattice during laser imaging was captured without an emission filter in place. This collagen reflectance imaging was performed on a Nikon A1R confocal microscope using a 488nm laser with intensity of 5-10%. Images were obtained with a step size (z-resolution) of ≤2 µm using Nikon Elements software. Image files were then exported to ImageJ where the membrane was identified in XZ cross-section and measured (Figure 1). Three separate locations were measured and averaged for each hRVTU. For hRVTU with cryosectioning performed, membrane thickness was similarly extracted using analysis of histologic images obtained at 20x or greater resolution on a Nikon Eclipse 90i microscope with the collagen spanning the area between cells lining the endothelial and epithelial channels measured near where channels intersected. The thickness of membranes created on 25 vs 50 µm-thick sheeting was compared using a two-tailed unpaired student’s t-test (significance of p < .05). For devices created using 25 µm-thick plastic sheets, hRVTU were maintained in culture for 7-14 days and their membrane thickness was similarly compared to that of acellular hRVTU. Statistics and graphing were performed using Graphpad software (Prism).
Dextran Perfusion:
hRVTU were fabricated using membranes created on 25 µm-thick plastic sheeting, the top channel seeded with fetal HKMEC, and the bottom channel seeded with fetal cortical epithelial cells. Following six days of culture the device was brought to a Nikon Ti-E inverted microscope where 10 µM FITC-labeled 70 kDa dextran (Thermo Fisher Scientific) was perfused into the endothelial channel during real-time imaging. Perfusion was performed by placing 150 µl of dextran into the inlet and removing all liquid from the outlet. Two minutes into perfusion, Texas Red-labeled 70 kDa dextran (Thermo Fisher Scientific) was similarly perfused through the epithelial channel. In addition to brightfield imaging, images were captured at 1 fps under 532nm laser excitation in the epithelial channel’s focal plane (100 ms exposure) and 488 excitation in the endothelial channel’s focal plane (10 ms exposure). Using ImageJ, fluorescent intensity was calculated across a region of interest at distinct timepoints two minutes apart for each of the channels within their respective focal planes, and this information graphed using Graphpad.
Blood Perfusion:
hRVTU were fabricated using 25 µm-thick plastic sheeting after which the superior channel was seeded with HKMEC and the inferior channel seeded with fetal cortical epithelial cells. Following seven days of culture, blood was drawn from healthy donors into 3.8% sodium citrate (used as an anticoagulant) and centrifuged at 150g for 15 minutes at room temperature. To label platelets, the layer containing platelet-rich plasma was transferred to a separate 14 ml test tube and incubated with 0.86 µg/ml of FITC-labeled mouse anti-human CD41a (BD) for one hour. Platelet-rich plasma was then returned to the original test tube and whole blood reconstituted. 200 µl of whole blood with labeled platelets was then placed into the inlet for the endothelial channel of the hRVTU, liquid removed from the endothelial outlet, and gravity-driven perfusion done for thirty minutes under real-time imaging on a Nikon Ti-E inverted microscope. Images were obtained under brightfield, as well as with excitation by a 488 laser (exposure 125 ms, 1 frame per second).
Albumin and Inulin Perfusion:
hRVTU were fabricated using 25 µm-thick plastic sheeting and 10 mg/ml collagen for membrane formation, after which the superior channel was seeded with primary adult HKMECs and the inferior channel with donor-matched primary adult cortical epithelial cells. A total of two donors were used to create three hRVTUs for this experiment. Following four days of culture, the epithelial channel of each device was perfused with epithelial media containing 1 µM rhodamine-labeled human serum albumin (Abcam) and 1 µM FITC-labeled inulin (Sigma-Aldrich). For perfusion, the epithelial inlet reservoir was filled with 200 µl of the inulin/albumin-containing media, while the endothelial inlet reservoir was filled with regular endothelial media. Immediately after flow was started, real-time imaging was performed using a Nikon Ti2-E inverted fluorescent microscope attached to an ORCA-Flash4.0 V3 sCMOS Camera, with devices kept in a 37° C incubating chamber during imaging. Exposure was set at 100ms for both green and red fluorescent channels for all devices, and images were captured continuously over the first 5 minutes of perfusion for each device, and again at 15 minutes. The inlet-containing sides of devices were elevated 25 mm to maintain gravity-driven perfusion during the times when devices were not being imaged. Following one hour of perfusion, 50 µl of effluent was collected from outlet reservoirs in both the endothelial and epithelial channels and placed into a 96-well plate. A standard curve was prepared for both FITC-inulin and HSA, and all samples were read on a spectrophotometer at excitation/emission wavelengths of 494/520 and 560/590 nm for FITC and Rhodamine, respectively. Following subtraction of blank values (non-fluorescent epithelial or endothelial media), standard curves were used to compute concentration values of inulin and albumin. The percentage of albumin reabsorbed was calculated as follows:
Following the above 1 hour of perfusion, the hRVTU’s were washed with PBS and decellularized by perfusing trypsin through both lumens overnight, followed by washing 6 × 10 minutes with warmed trypsin. This led to near-complete decellularization, with any residual cells no longer appearing to attach to the vessel wall (Supplemental Video 4). Following washing and decellularization, effluent was tested via spectrophotometry to ensure no residual fluorescent material. Perfusion was then repeated in the same manner, and albumin reabsorption calculated as above. These decellularized vessels served as internal controls for the prior perfusion experiment. Using Graphpad Prism, a two-tailed paired T test was performed to compare albumin reabsorption in cellularized vessels to decellularized controls and the results of all analyses graphed using Graphpad Prism software.
Glucose Analogue Perfusion:
Similar hRVTU to the above experiment were fabricated using 25 µm-thick plastic sheeting and 10 mg/ml collagen for membrane formation, with superior channel seeded with primary adult HKMECs and the inferior channel with donor-matched primary adult cortical epithelial cells. An additional two donors were used to create three hRVTUs for these measurements. Following four days of culture, devices were glucose-depleted by perfusing a low glucose formulation of epithelial media (100 mg/dl glucose) into both the endothelial and epithelial channels. Then the devices were perfused with a fluorescent glucose analogue solution, containing 100 µM 2-NBDG (Cayman Chemical) in the same low-glucose epithelial media, for another hour under gravity. 50 µL of effluent from each outlet was collected at the end of perfusion and placed in a 96-well plate, and the concentration of 2-NBDG in the perfusate and collected effluent was calculated using a spectrophotometer set to read excitation/emission of 475/550 nm. Devices were then washed 5 × 5’ with PBS and imaged on a Nikon Ti2-E inverted fluorescent microscope. To test our hypothesis that the epithelial outlet would contain less 2-NBDG than the perfusate or the endothelial outlet, due to active reabsorption of glucose by the epithelial cells, two-tailed paired t-tests were performed comparing the concentration of 2-NBDG in the epithelial outlet to the initial perfusate, and comparing the concentration of 2-NBDG in the epithelial outlet to the endothelial outlet.
Statistical Methods:
Unpaired two-tailed T-tests were performed for graphs found in figures 1 and 2. Paired two-tailed T-tests were performed for the graph in figure 4c and the analyses of albumin and glucose reabsorption, as noted in the text above. A p value < 0.05 was considered significant for all tests. Sample sizes are found in the text above and in accompanying figures. Statistical analysis was performed in Graphpad Prism.
Data Availability:
All relevant data is available from the authors upon request.
Supplementary Material
Acknowledgements:
The authors would like to acknowledge the Lynn and Mike Garvey Imaging Laboratory in the Institute of Stem Cell and Regenerative Medicine, the Washington Nanofabrication Facility, the Histology and Imaging Core, and the Cell Analysis Facility - all located at the University of Washington. The authors would like to thank the laboratory of Professor John D. Scott for their kind gift of the acetylated tubulin and ARL13B antibodies. This work was supported by funding from the National Institutes of Health DP2DK102258 (YZ), UH2/UH3 TR000504, UG3TR002158 (J.H.), UH2/UH3 DK107343 (YZ/SS) and the University of Washington Pulmonary, Critical Care and Sleep Medicine Training Grant (Institutional National Research Service Award, T32675551) (SR).
Footnotes
The authors have declared that no conflict of interest exists
References:
- [1].Bhatia SN, Ingber DE, Nat. Biotechnol 2014, 32, 760. [DOI] [PubMed] [Google Scholar]
- [2].Wikswo JP, Exp. Biol. Med. (Maywood) 2014, 239, 1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rayner SG, Zheng Y, J. Biomech. Eng 2016, 138, 110801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ligresti G, Nagao RJ, Xue J, Choi YJ, Xu J, Ren S, Aburatani T, Anderson SK, MacDonald JW, Bammler TK, Schwartz SM, Muczynski KA, Duffield JS, Himmelfarb J, Zheng Y, J. Am. Soc. Nephrol 2016, 27, 2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Weber EJ, Chapron A, Chapron BD, Voellinger JL, Lidberg KA, Yeung CK, Wang Z, Yamaura Y, Hailey DW, Neumann T, Shen DD, Thummel KE, Muczynski KA, Himmelfarb J, Kelly EJ, Kidney Int. 2016, 90, 627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Liaño F, Pascual J, The Madrid Acute Renal Failure Study Group, Kidney Int. 1996, 50, 811. [DOI] [PubMed] [Google Scholar]
- [7].Nieskens TTGG, Wilmer MJ, Eur. J. Pharmacol 2016, 790, 46. [DOI] [PubMed] [Google Scholar]
- [8].Wilmer MJ, Ng CP, Lanz HL, Vulto P, Suter-Dick L, Masereeuw R, Trends Biotechnol. 2016, 34, 156. [DOI] [PubMed] [Google Scholar]
- [9].Tiong HY, Huang P, Xiong S, Li Y, Vathsala A, Zink D, Mol. Pharm 2014, 11, 1933. [DOI] [PubMed] [Google Scholar]
- [10].Homan KA, Kolesky DB, Skylar-Scott MA, Herrmann J, Obuobi H, Moisan A, Lewis JA, Sci. Rep 2016, 6, 34845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jansen J, Fedecostante M, Wilmer MJ, Peters JG, Kreuser UM, van den Broek PH, Mensink RA, Boltje TJ, Stamatialis D, Wetzels JF, van den Heuvel LP, Hoenderop JG, Masereeuw R, Sci. Rep 2016, 6, 26715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jansen J, De Napoli IE, Fedecostante M, Schophuizen CMSS, Chevtchik NV, Wilmer MJ, van Asbeck AH, Croes HJ, Pertijs JC, Wetzels JFMM, Hilbrands LB, van den Heuvel LP, Hoenderop JG, Stamatialis D, Masereeuw R, Sci. Rep 2015, 5, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Jang K-J, Mehr AP, Hamilton GA, McPartlin LA, Chung S, Suh K-Y, Ingber DE, Integr. Biol 2013, 5, 1119. [DOI] [PubMed] [Google Scholar]
- [14].Vedula EM, Alonso JL, Arnaout MA, Charest JL, PLoS One 2017, 12, e0184330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jang K-J, Suh K-Y, Lab Chip 2010, 10, 36. [DOI] [PubMed] [Google Scholar]
- [16].Ferrell N, Ricci KB, Groszek J, Marmerstein JT, Fissell WH, Biotechnol. Bioeng 2012, 109, 797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ferrell N, Desai RR, Fleischman AJ, Roy S, Humes HD, Fissell WH, Biotechnol. Bioeng 2010, 107, 707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sciancalepore AG, Sallustio F, Girardo S, Passione LG, Camposeo A, Mele E, Di Lorenzo M, Costantino V, Schena FP, Pisignano D, Gioia Passione L, Camposeo A, Mele E, Di Lorenzo M, Costantino V, Schena FP, Pisignano D, PLoS One 2014, 9, e87496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zheng Y, Chen J, Craven M, Choi NW, Totorica S, Diaz-Santana A, Kermani P, Hempstead B, Fischbach-Teschl C, López JA, Stroock AD, Proc. Natl. Acad. Sci. U. S. A 2012, 109, 9342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zheng Y, Chen J, López J. a., Nat. Commun 2015, 6, 7858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ichimura T, V Asseldonk EJPVP, Humphreys BD, Gunaratnam L, Duffield JS, Bonventre JV, J. Clin. Invest 2008, 118, 1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Deane JA, Verghese E, Martelotto LG, Cain JE, Galtseva A, Rosenblum ND, Watkins DN, Ricardo SD, Nephrology 2013, 18, 161. [DOI] [PubMed] [Google Scholar]
- [23].Rennke HG, Denker BM, Renal Pathophysiology: The Essentials, Lippincott Williams & Wilkins, 2006. [Google Scholar]
- [24].Ivanyuk A, Livio F, Biollaz J, Buclin T, Clin. Pharmacokinet 2017, 56, 825. [DOI] [PubMed] [Google Scholar]
- [25].Katzung BG, Trevor AJ, Basic and Clinical Pharmacology, McGraw-Hill Education, 2014. [Google Scholar]
- [26].Chen W-C, Lin H-H, Tang M-J, Am. J. Physiol. Renal Physiol 2014, 307, F695. [DOI] [PubMed] [Google Scholar]
- [27].Nagao RJ, Xu J, Luo P, Xue J, Wang Y, Kotha S, Zeng W, Fu X, Himmelfarb J, Zheng Y, Tissue Eng. Part A 2016, 22, 1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Choucha Snouber L, Jacques S, Monge M, Legallais C, Leclerc E, Genomics 2012, 100, 27. [DOI] [PubMed] [Google Scholar]
- [29].Kaysen JH, Campbell WC, Majewski RR, Goda FO, Navar GL, Lewis FC, Goodwin TJ, Hammond TG, J. Membr. Biol 1999, 168, 77. [DOI] [PubMed] [Google Scholar]
- [30].Maggiorani D, Dissard R, Belloy M, Saulnier-Blache J-S, Casemayou A, Ducasse L, Grès S, Bellière J, Caubet C, Bascands J-L, Schanstra JP, Buffin-Meyer B, PLoS One 2015, 10, e0131416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Essig M, Terzi F, Burtin M, Friedlander G, Am J Physiol Ren. Physiol 2001, 281, F751. [DOI] [PubMed] [Google Scholar]
- [32].Rbaibi Y, Carattino MD, Weisz OA, Proc. Natl. Acad. Sci 2016, 113, E1587.26951650 [Google Scholar]
- [33].Duan Y, Weinstein AM, Weinbaum S, Wang T, Proc. Natl. Acad. Sci 2010, 107, 21860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Maeshima A, Sakurai H, Nigam SK, J. Am. Soc. Nephrol 2005, 17, 188. [DOI] [PubMed] [Google Scholar]
- [35].Discher DE, Science (80-.). 2005, 310, 1139. [DOI] [PubMed] [Google Scholar]
- [36].DesRochers TM, Suter L, Roth A, Kaplan DL, PLoS One 2013, 8, e59219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Shen C, Meng Q, Zhang G, Biotechnol. Bioeng 2013, 110, 2173. [DOI] [PubMed] [Google Scholar]
- [38].Tasnim F, Zink D, Am. J. Physiol. Renal Physiol 2012, 302, F1055. [DOI] [PubMed] [Google Scholar]
- [39].Linas SL, Repine JE, Kidney Int. 1999, 55, 1251. [DOI] [PubMed] [Google Scholar]
- [40].Smith SW, Chand S, Savage COS, Nephrol. Dial. Transplant 2012, 27, 2149. [DOI] [PubMed] [Google Scholar]
- [41].Am S, Péault P, Jj M, Stefańska A, Péault B, Mullins JJ, Pflugers Arch. 2013, 465, 767. [DOI] [PubMed] [Google Scholar]
- [42].Birn H, Christensen EI, Kidney Int. 2006, 69, 440. [DOI] [PubMed] [Google Scholar]
- [43].Tenten V, Menzel S, Kunter U, Sicking E-M, van Roeyen CRC, Sanden SK, Kaldenbach M, Boor P, Fuss A, Uhlig S, Lanzmich R, Willemsen B, Dijkman H, Grepl M, Wild K, Kriz W, Smeets B, Floege J, Moeller MJ, J. Am. Soc. Nephrol 2013, 24, 1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Dickson LE, Wagner MC, Sandoval RM, Molitoris BA, J. Am. Soc. Nephrol 2014, 25, 443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Van der Hauwaert C, Savary G, Gnemmi V, Glowacki F, Pottier N, Bouillez A, Maboudou P, Zini L, Leroy X, Cauffiez C, Perrais M, Aubert S, PLoS One 2013, 8, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Roberts MA, Kotha SS, Phong KT, Zheng Y, J. Vis. Exp 2016, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Morgan JP, Delnero PF, Zheng Y, Verbridge SS, Chen J, Craven M, Choi NW, Diaz-Santana A, Kermani P, Hempstead B, López JA, Corso TN, Fischbach C, Stroock AD, Nat. Protoc 2013, 8, 1820. [DOI] [PubMed] [Google Scholar]
- [48].Rajan N, Habermehl J, Coté M-F, Doillon CJ, Mantovani D, Nat. Protoc 2006, 1, 2753. [DOI] [PubMed] [Google Scholar]
- [49].Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A, Nat. Methods 2012, 9, 676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data is available from the authors upon request.