

The first line of defense in cells during viral infection is the innate immune system, which is activated by different viral products. PKR is a part of this innate immune system and is induced by interferon and activated by dsRNA produced by DNA and RNA viruses. PKR is such an important part of the antiviral response that many viral families have gene products to counteract its activation or the resulting effects of its activity. Although a few RNA viruses degrade PKR, this method of counteracting PKR has not been reported for any DNA viruses. MAV-1 does not encode virus-associated RNAs, a human adenoviral defense against PKR activation. Instead, MAV-1 degrades PKR, and it is the first DNA virus reported to do so. The innate immune evasion by PKR degradation is a previously unidentified way for a DNA virus to circumvent the host antiviral response.
KEYWORDS: PKR, adenoviruses, eIF2alpha kinase, proteasome, protein degradation, protein stability
ABSTRACT
Protein kinase R (PKR) plays a major role in activating host immunity during infection by sensing double-stranded RNA (dsRNA) produced by viruses. Once activated by dsRNA, PKR phosphorylates the translation factor eukaryotic initiation factor 2α (eIF2α), halting cellular translation. Many viruses have methods of inhibiting PKR activation or its downstream effects, circumventing protein synthesis shutdown. These include sequestering dsRNA or producing proteins that bind to and inhibit PKR activation. Here we describe our finding that in multiple cell types, PKR was depleted during mouse adenovirus type 1 (MAV-1) infection. MAV-1 did not appear to be targeting PKR at the transcriptional or translational level, because total PKR mRNA levels and levels of PKR mRNA bound to polysomes were unchanged or increased during MAV-1 infection. However, inhibiting the proteasome reduced the PKR depletion seen in MAV-1-infected cells, whereas inhibiting the lysosome had no effect. This suggests that proteasomal degradation alone is responsible for PKR degradation during MAV-1 infection. Time course experiments indicated that the degradation occurs early after infection. Infecting cells with UV-inactivated virus prevented PKR degradation, whereas inhibiting viral DNA replication did not. Together, these results suggest that an early viral gene is responsible. Degradation of PKR is a rare mechanism to oppose PKR activity, and it has been described in only six RNA viruses. To our knowledge, this is the first example of a DNA virus counteracting PKR by degrading it.
INTRODUCTION
Activation of protein kinase R (PKR) is a major innate immune response to viral infection. PKR is an interferon (IFN)-induced protein that is comprised of two major domains, namely, an N-terminal double-stranded RNA binding domain and a C-terminal serine/threonine kinase domain (1, 2). PKR binds to double-stranded RNA (dsRNA) (3–5), and, once bound, it becomes activated by dimerizing and autophosphorylating (6–9). When activated, PKR phosphorylates eukaryotic translation initiation factor 2α (eIF2α), causing inhibition of protein synthesis and reduced viral replication (10–13). Many viruses encode gene products that block PKR activation or inhibit its ability to phosphorylate eIF2α (14). A common mechanism is that of producing a viral protein that binds and sequesters dsRNA, blocking its interaction with PKR. Examples of this are vaccinia virus E3L (15–17), influenza virus NS1 (18, 19), and Ebola virus protein VP35 (20). Other viruses produce proteins or RNAs that bind directly to PKR to inhibit its activation, such as herpes simplex virus US11 (21, 22), HIV-1 Tat protein (23, 24) or trans-activation response element (TAR) RNA (25), and human adenovirus (hAd) virus-associated (VA) RNAs (10, 26–28).
Degradation of PKR by viruses is a less extensively documented method of regulating PKR. To date, PKR degradation has been reported in six RNA viruses: Toscana virus (TOSV) (29), Rift Valley fever virus (RVFV) (30–32), poliovirus (33, 34), foot-and-mouth disease virus (FMDV) (35, 36), encephalomyocarditis virus (EMCV [strain mengovirus]) (37, 38), and enterovirus 71 (39). RVFV and TOSV both degrade PKR via proteasomal mechanisms involving a viral nonstructural protein (NSs) (29, 32, 40). RVFV NSs recruits a SCF (SKP1-CUL1-F-box)FBXW11 E3 ubiquitin ligase to ubiquitinate PKR and target it to the proteasome, though PKR ubiquitination could not be demonstrated (32, 40). The mechanism for PKR proteasomal degradation by NSs has not been described for TOSV (29). FMDV uses the other major cellular protein degradation pathway, the lysosome, to degrade PKR during infection (36). Though the mechanism is unclear, expression of major FMDV protease 3Cpro is required for PKR degradation by the lysosome. However, 3Cpro does not interact with PKR, and its protease activity is not required for PKR degradation. Enterovirus A71 3Cpro causes PKR degradation by direct interaction (39). The mechanism of PKR depletion by poliovirus is unclear, though gene expression is required and the major poliovirus proteases (2A and 3C) are not directly involved (33). The mechanism by which mengovirus depletes PKR during infection is unknown (37, 38).
Adenoviruses are species specific, making the study of hAd pathogenesis difficult in an animal model. MAV-1 represents a useful alternative to study adenovirus pathogenesis (41–45). MAV-1 has molecular, genetic, and pathogenic similarities to and differences from hAd. Their genomic structures are similar at a gross level, and both contain early genes involved in pathogenesis and immune evasion. Pathogenically, their tropisms differ, with hAd infecting epithelial cells, leading to upper respiratory and GI tract infections and to conjunctivitis, while MAV-1 infects endothelial cells and monocytes, causing encephalitis and myocarditis. We and others have been investigating the adaptive and innate immune responses to MAV-1.
Human adenovirus VA RNAs bind PKR as a monomer, preventing its transautophosphorylation (46). However, MAV-1 does not produce VA RNAs (47), and whether MAV-1 induces PKR activation is not known. In our studies of MAV-1 pathogenesis and the innate response, we discovered that during MAV-1 infection, PKR was depleted from cells as early as 12 h postinfection (hpi). Total PKR mRNA levels and levels of PKR mRNA bound to polysomes were unchanged or increased during MAV-1 infection, suggesting that MAV-1 did not appear to be targeting PKR at a transcriptional or translational level. However, inhibiting the proteasome blocked the PKR depletion seen in MAV-1-infected cells, indicating that proteasomal degradation is responsible for PKR depletion during MAV-1 infection. We report results indicating that an early viral gene is likely responsible for mediating PKR degradation. To our knowledge, this is the first example of a DNA virus counteracting PKR by degrading it.
RESULTS
Viral DNA yield is increased in PKR−/− mouse embryonic fibroblasts.
While PKR is an important part of the innate immune response, PKR−/− cells in culture are not always more susceptible to viral infection than wild-type (WT) cells (48–50). PKR−/− mouse embryonic fibroblasts (MEFs) show increased viral yields compared to wild-type MEFs when infected with vesicular stomatitis virus and influenza A virus (48, 49), but there is no change in viral yield during vaccinia virus infection compared to the results seen with wild-type cells (50). However, it was later discovered that the PKR−/− MEF lines used were not complete PKR knockouts (51). There are two categories of PKR−/− MEFs derived from knockout mice: N-PKR−/− MEFs and C-PKR−/− MEFs (51). The PKR−/− MEFs derived from mice created in the Weissmann laboratory (52) are designated N-PKR−/− MEFs, because the C-terminal fragment of PKR is still expressed and can be detected by immunoblotting when there is IFN induction (51). The fragment has the kinase catalytic activity of PKR, but it does not bind dsRNA (51). The PKR−/− MEFs derived from mice created in the Bell laboratory (53) are designated C-PKR−/− MEFs, because the N-terminal fragment of PKR is still expressed and can be detected by immunoblotting with specific PKR antibodies (51). The fragment is catalytically inactive, but it can still bind dsRNA (51). Susceptibility of these PKR−/− MEFs to specific viruses may be dependent on the PKR mutation and the mechanism used by each virus to circumvent PKR.
To determine whether PKR plays an important role during MAV-1 infection, we tested the susceptibility of both PKR−/− MEF lines to MAV-1 infection. We infected wild-type MEFs, N-PKR−/− MEFs, and C-PKR−/− MEFs with MAV-1 at a multiplicity of infection (MOI) of 1 PFU/cell and collected cell pellets at 48 and 72 hpi. DNA was purified from the cell pellets and analyzed for MAV-1 genome copies by quantitative PCR (qPCR). N-PKR−/− MEFs produced a significantly higher viral DNA yield than wild-type MEFs at 48 hpi, and both PKR mutant MEF lines had a significantly higher viral DNA yield than wild-type MEFs at 72 hpi (Fig. 1). Although we have not confirmed the production of truncated PKR proteins in the cells in our laboratory, the results shown in Fig. 1 indicate that PKR activation is an important antiviral response during MAV-1 infection in vitro.
FIG 1.
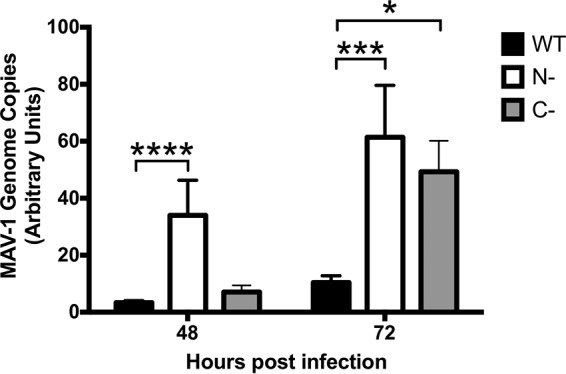
Viral DNA yield is increased in PKR−/− mouse embryonic fibroblasts. PKR WT MEFs (WT), N-PKR−/− MEFs (N-), and C-PKR−/− MEFs (C-) were infected with MAV-1 at an MOI of 1 and collected at 48 and 72 hpi. DNA was purified from cell pellets and analyzed for MAV-1 genome copies by qPCR. The graph is representative of results from three experiments (14 biological replicates per cell line per time point). Error bars represent standard errors of the means (SEM). *, P ≤ 0.05; ***, P ≤ 0.0002; ****, P ≤ 0.0001.
Mouse PKR is depleted during MAV-1 infection.
To determine whether MAV-1 affects PKR during infection, we infected several cell types and analyzed PKR protein expression. We infected immortalized C57BL/6 MEFs, C57BL/6 primary peritoneal macrophages, and CMT93 cells (mouse rectal carcinoma cells) with MAV-1 at an MOI of 10 and collected cell lysates 24, 48, and 72 hpi. We analyzed cell lysates for the presence of PKR by immunoblotting using a polyclonal antibody that detects mouse PKR. We probed blots with antibodies to actin as a loading control. To our surprise, in C57BL/6 MEFs, PKR was almost completely depleted from lysates at 24 hpi and remained depleted through 72 hpi (Fig. 2A and B). PKR was also depleted compared to the levels seen with mock infection at 24 and 48 hpi in C57BL/6 MEFs infected at MOIs of 2 and 5 (see Fig. S1 in the supplemental material). We also observed depletion of PKR in other cell types. In CMT93 cells, PKR was nearly undetectable at 24 hpi (Fig. 2A). PKR levels were decreased in C57BL/6 primary peritoneal macrophages at 48 hpi compared to mock-infected lysates, and PKR was absent in infected lysates at 72 hpi (Fig. 2A). This indicates that MAV-1 causes PKR depletion during infection.
FIG 2.

Mouse PKR is depleted during MAV-1 infection. (A) Cells (indicated at left) were infected with MAV-1 (MAV) at an MOI of 10 or were mock infected (mock). Cell lysates were collected at the indicated times and analyzed by immunoblotting with antibodies for PKR (B-10 for C57BL/6 MEFs and D-20 for C57BL/6 primary peritoneal macrophages and CMT93 cells) and actin. Blots are representative of results from a minimum of three independent experiments per cell line. (B) Densitometry quantitation of five of the C57BL/6 MEF immunoblots represented in panel A. Error bars represent SEM. n = 5 for 24 hpi, n = 4 for 48 hpi, and n = 2 for 72 hpi. **, P ≤ 0.01. (C) Untreated SV40 MEFs or kinase-dead (K271R) SV40 MEFs were infected with MAV-1 at an MOI of 10. Cell lysates were immunoblotted as described for panel A with PKR D-20. The numbers below the blots represent the proportion of PKR protein for each time point, normalized to actin, and the mock PKR protein levels from the corresponding time point.
PKR is depleted at MOIs of 2, 5, and 10. C57BL/6 MEFs were infected with MAV-1 at an MOI of 2, 5, or 10 or were subjected to mock infection (mock). Cell lysates were analyzed by immunoblotting with antibodies for PKR (B-10) and actin. Data from densitometry quantitations performed using ImageJ are listed below each lane; for each time point, the infected samples were normalized to the mock-infected samples. The PKR blot image was uniformly adjusted to a brightness of 150 in Adobe Photoshop. Download FIG S1, PDF file, 2.2 MB (2.3MB, pdf) .
Copyright © 2019 Goodman et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To determine whether the kinase activity of PKR is important for the depletion, we assayed infection of MEFs expressing a mutant form of mouse PKR with a point mutation in the kinase domain (K271R) (54). These cells, designated K271R simian virus 40 (SV40) MEFs, showed an even higher rate of depletion of PKR than the WT SV40 MEFs (Fig. 2C). At 24 hpi, 28% of PKR remained in K271R SV40 MEFs compared to 60% in the WT SV40 MEFs. The fraction remaining at 72 hpi in K271R SV40 MEFs was 10% compared to 30% in WT SV40 MEFs (Fig. 2C). This indicates that the PKR kinase does not have to be functional to be depleted during MAV-1 infection. Also, comparing the PKR immunoblot bands in the mock-infected WT SV40 MEFs and mutant K271R SV40 MEFs suggests that the upper band of the PKR doublet usually seen in wild-type cells is a phospho-PKR band, because only the lower band of the PKR doublet is seen in kinase-dead mutant K271R SV40 MEFs. The data in Fig. 2A and C thus indicate that both PKR and phospho-PKR are depleted during MAV-1 infection.
MAV-1 does not cause PKR depletion by reducing steady-state levels of PKR mRNA.
To determine the mechanism of PKR depletion, we first assayed whether the reduction in the level of PKR protein during MAV-1 infection was due to reduced PKR mRNA steady-state levels. We mock-infected or infected C57BL/6 MEFs and primary peritoneal macrophages at an MOI of 10 and collected cell lysates at 24, 48, and 72 hpi. We synthesized cDNA from RNA purified from these cell lysates and assayed for PKR mRNA by qPCR. The PKR mRNA levels in C57BL/6 MEFs were similar between mock-infected and infected lysates at 24 hpi (Fig. 3A), a time point at which the PKR protein levels were already greatly reduced in the infected lysates compared to mock-infected lysates (Fig. 2). Although the PKR mRNA levels were depleted 33% at 48 hpi and 40% at 72 hpi in MAV-1-infected lysates compared to mock-infected lysates, this does not correlate to the 84% and 94% reductions, respectively, in PKR protein levels seen at those time points (Fig. 2B). PKR mRNA levels in C57BL/6 primary peritoneal macrophages in the infected lysates were 2 to 3 times higher than the levels in the mock-infected lysates at all three time points assayed (Fig. 3B), even though the PKR protein was almost completely depleted in infected lysates at 72 hpi (Fig. 2). This represents evidence that MAV-1 was not causing PKR protein depletion by reducing PKR steady-state mRNA levels during infection.
FIG 3.
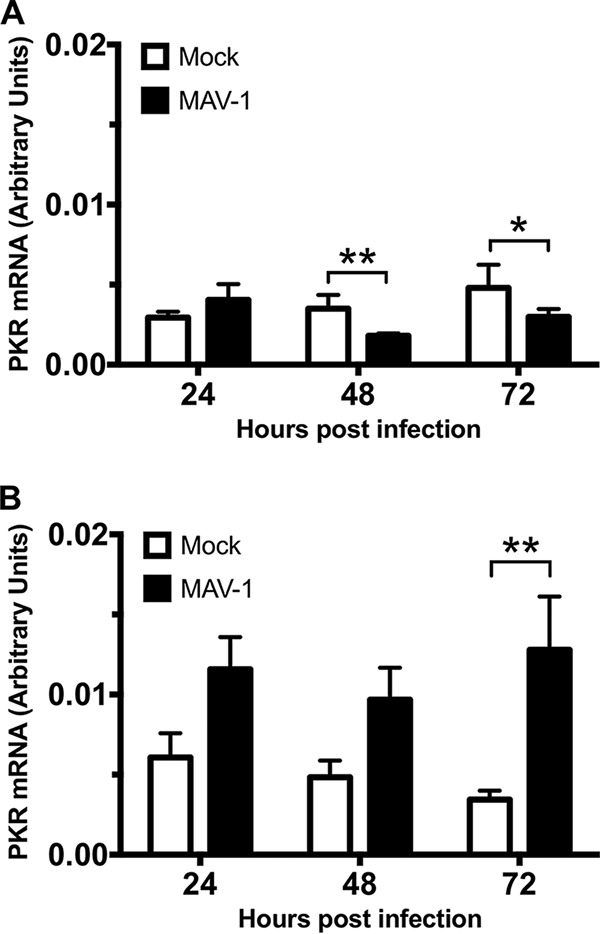
MAV-1 does not cause PKR depletion by reducing steady-state levels of PKR mRNA at times when the protein levels are already reduced. MEFs (A) or isolated primary peritoneal macrophages (B) were harvested and infected with MAV-1 at an MOI of 10 or mock infected. The cell pellets were collected, and RNA was isolated. cDNA was generated from the RNA, and qPCR was used to quantitate PKR mRNA levels. Each graph represents 5 to 7 replicates for each time point from three pooled experiments. Error bars show the SEM. *, P ≤ 0.05; **, P ≤ 0.01.
MAV-1 infection effects on PKR translation.
Because MAV-1 did not cause reductions in the PKR mRNA steady-state levels, we determined whether MAV-1 causes PKR depletion by reducing translation of its mRNA. We first assayed total PKR mRNA bound to ribosomes during infection. C57BL/6 MEFs were mock infected or infected at an MOI of 5, and lysates were collected at 48 hpi in the presence of cycloheximide to keep the mRNA bound to the ribosomes (55). Lysates were centrifuged through 25% sucrose to pellet ribosomes, and RNA was purified from the pellets. The purified RNAs were used to generate cDNA, which we assayed for PKR mRNA by qPCR. As a control for pelleting of ribosomes, we assayed the pellets and sucrose cushion supernatants by immunoblotting with antibodies to ribosomal protein RPL7. We confirmed that RPL7 was present only in the pellets and not in the supernatants (Fig. S2). There was no significant difference between the amount of PKR mRNA in the ribosome pellet of mock-infected lysates and the amount in that of MAV-1-infected lysates (Fig. 4A).
FIG 4.

MAV-1 infection does not affect PKR translation. (A) C57BL/6 MEFs were infected with MAV-1 (MAV) at an MOI of 5 or were mock infected (mock) and collected at 48 hpi. Cells were lysed, and cleared lysates from three 10-cm-diameter plates were layered onto 25% sucrose and centrifuged to pellet ribosomes. RNA was purified from the pellets, cDNA was generated from the RNA, and qPCR was used to quantitate the PKR mRNA levels. The graph represents 9 replicates for each time point, pooled from 3 independent experiments. Error bars show the SEM. (B) C57BL/6 MEFs were infected with MAV-1 (MAV) at an MOI of 2 or were mock infected (mock). Cells were collected at 25 hpi and lysed; cleared lysates were layered onto 10%-to-50% sucrose gradients and centrifuged. Gradients were collected from the top and pumped through a UV spectrophotometer, and 34 fractions were collected. The gradients are displayed with the bottom fractions indicated to the right. The UV trace of the first 10 fractions (including 40S and 60S ribosomal subunits) is not shown. (C) RNA was purified from each fraction of the gradients represented in panel B. cDNA was generated from the RNA, qPCR was used to quantitate PKR mRNA in each fraction, and the results are displayed as the percentages of total PKR mRNA associated with ribosomes. Panels B and C present results from one experiment representative of 3 independent experiments. (D) Percentage of total PKR mRNA associated with monosomes (fractions 1 to 6) and polysomes (fractions 7 to 19) from the trial represented in panels B and C (pooled for mock-infected and infected samples). The percentages represented by each bar are displayed below each bar. (E) Pooled monosome and polysome data (as described for panel D) from three independent experiments. Error bars show the SEM. The percentages represented by each bar are displayed below each bar. There were no significant differences between the mock-infected samples and the infected samples (A and E).
Ribosome pelleting. To confirm that most ribosomes ended up in the pellet after centrifugation through sucrose (Fig. 4A), a sample of the pellet and a sample of the corresponding supernatant were analyzed by immunoblotting with antibodies for RPL7 (ribosomal protein L7; Abcam catalog no. ab72550) (1:2,000). Download FIG S2, PDF file, 3.2 MB (3.2MB, pdf) .
Copyright © 2019 Goodman et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To confirm the results seen in total mRNA bound to ribosomes, we also centrifuged cell extracts on sucrose gradients to generate polysome profiles. This enabled us to analyze levels of PKR mRNA associated with actively translating ribosomes during infection. C57BL/6 MEFs were mock infected or infected at an MOI of 2, and lysates were collected at 24 hpi in the presence of cycloheximide, as described above. The levels of RNA content for mock-infected and infected lysates were estimated by NanoDrop spectrophotometry, and equivalent optical density (OD) amounts of RNA were centrifuged on 10% to 50% sucrose gradients to sediment 40S and 60S ribosomal subunits, 80S ribosomes (monosomes), and polyribosomes (polysomes). A typical polysome profile was obtained (Fig. 4B). RNA was purified from fractions containing monosomes and polysomes and then used to generate cDNA, which we assayed for PKR mRNA by qPCR (Fig. 4C). As a control, GAPDH mRNA was measured by qPCR, and PKR mRNA levels in each fraction were normalized to the GAPDH mRNA content. When the data representing the percentages of PKR mRNA bound to ribosomes were pooled into monosome and polysome fractions and analyzed (Fig. 4D), 90.1% and 91.8% were bound to polysomes (fractions 7 to 19) for mock-infected and infected samples, respectively, compared to 9.9% and 8.2% bound to monosomes (fractions 1 to 6). We performed two additional polysome gradient analyses. The pooled data from all three analyses (Fig. 4E) were similar to the data shown in Fig. 4D; i.e., 82.7% and 92.7% of PKR mRNA were bound to mock-infected and infected polysomes, respectively, compared to 17.3% and 7.3% bound to monosomes. Thus, PKR protein depletion during MAV-1 infection does not appear to stem from a decrease in PKR mRNA translation.
We also assayed whether PKR mRNA might have a signal that would reduce its translation during MAV-1 infection. We constructed a plasmid that positioned sequence corresponding to the 5′ untranslated region (UTR) of PKR mRNA upstream of a reporter nanoluciferase gene (56), transfected it into C57BL/6 MEFs, and then infected with MAV-1. Compared to cells transfected with a control plasmid with the human β-globin 5′ UTR upstream of the reporter nanoluciferase, there was no significant difference in luciferase activity between mock-infected and infected samples (Fig. S3). These data suggest that MAV-1 was not affecting PKR translation through interaction with the 5′ UTR of PKR. The data are consistent with the ribosome pellet and polysome data indicating that MAV-1 infection does not reduce PKR mRNA translation.
PKR mRNA 5′ UTR presence does not result in altered reporter protein levels upon MAV-1 infection. (A) C57BL/6 MEFs or (B) CMT93 cells were cotransfected with pmPKR5UTRfullNL or AUG-NL-3xFLAG and pGL4.13 using jetPRIME reagents (Polyplus catalog no. 114-15) and the standard Polyplus protocol, with 200 ng total of plasmid and 300 µl of jetPRIME reagent per 35-mm-diameter well. At 24 h after transfection, the cells were infected with MAV-1 at an MOI of 10. At 24 hpi, cells were lysed in Glo lysis buffer (Promega Corp.) (70 µl/well). After lysing, 25 µl of each lysed sample and 25 µl of OneGlo or NanoGlo (Promega Corp.) were added to two wells in a black 96-well plate (Fisher Scientific catalog no. 07-000-634). After 5 min, the plate was read on a Promega GloMax luminometer. Levels of relative light units from the pmPKR5UTRfullNL plasmid were normalized to the firefly luciferase and positive-control plasmids. Graphs are representative of 7 to 9 biological replicates per treatment group. Download FIG S3, PDF file, 0.1 MB (71.5KB, pdf) .
Copyright © 2019 Goodman et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
PKR is depleted by proteasomal degradation during MAV-1 infection.
There are two main proteolysis pathways in cells: proteasomal degradation and lysosomal degradation (57). To determine whether MAV-1 depletes PKR by either protein degradation pathway, we first assayed whether PKR is lysosomally degraded as follows. CMT93 cells were mock infected or infected with MAV-1 and treated at the time of infection with a lysosome inhibitor (ammonium chloride or chloroquine) or water (as a control). At 24 hpi, we collected lysates and analyzed them by immunoblotting with antibodies to PKR. In the presence of the lysosomal degradation inhibitors, PKR was depleted by 24 hpi (Fig. 5A), indicating that lysosomal degradation was not the cause of PKR depletion during MAV-1 infection. We confirmed that the inhibitor treatment did block lysosomal degradation by incubating cells with dye-quenched bovine serum albumin (DQ BSA) in addition to the lysosomal inhibitors. DQ BSA is self-quenched until it is digested in the lysosome (58, 59), and imaging confirmed that the cells treated with lysosome inhibitors did not fluoresce but that the cells treated with the vehicle control (H2O) did, as expected (Fig. 5B).
FIG 5.
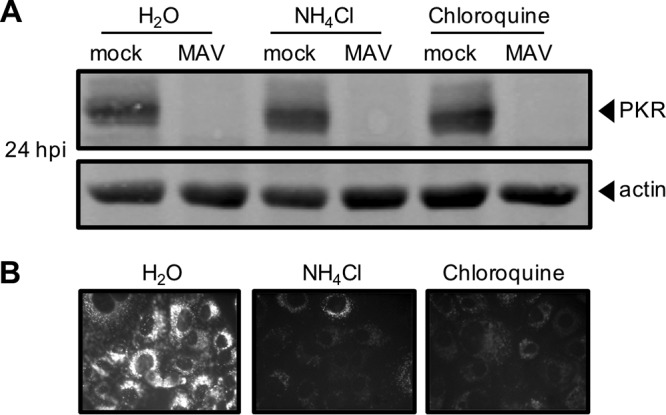
PKR is not depleted by lysosomal degradation during MAV-1 infection. (A) CMT93 cells were infected with MAV-1 (MAV) at an MOI of 10 or were mock infected (mock) and treated with 10 mM ammonium chloride or 60 µM chloroquine to inhibit lysosomal degradation (or with water as a control). Cell lysates were analyzed by immunoblotting with antibodies for PKR (D-20) and actin. Blots are representative of results from three independent experiments. (B) Inhibitors were tested for activity using a DQ BSA assay; the DQ BSA molecule fluoresces only if lysosomal degradation is functional. Uninfected cells were treated as indicated and imaged by fluorescence microscopy.
Next, we examined whether proteasomal degradation is responsible for the degradation of PKR by using proteasome inhibitors MG132 and bortezomib. These inhibit proteasome activity by binding to the active sites in the 20S subunit and blocking the proteolytic activity (60–62). We mock infected or infected C57BL/6 MEFs with MAV-1 and treated with MG132 or bortezomib and dimethyl sulfoxide (DMSO) at the time of infection. At 24 hpi, we collected lysates and analyzed them by immunoblotting for PKR protein levels. While PKR was depleted in the control DMSO-treated MAV-1-infected cells as expected, PKR protein was present in the MG132- and bortezomib-treated cells at levels comparable to those in the mock-infected cells (Fig. 6). To rule out the possibility that PKR was present (not depleted) because the virus infection itself was inhibited by MG132 or bortezomib, we assayed viral replication of MAV-1 with MG132 and bortezomib treatment by qPCR of viral DNA. Viral replication levels were equivalent in all three treatment groups (Fig. S4), indicating that the treatments did not affect the ability of the virus to productively infect the cells. Taken together, these data indicate that MAV-1 infection results in PKR depletion by causing PKR to be degraded by the proteasome during infection.
FIG 6.
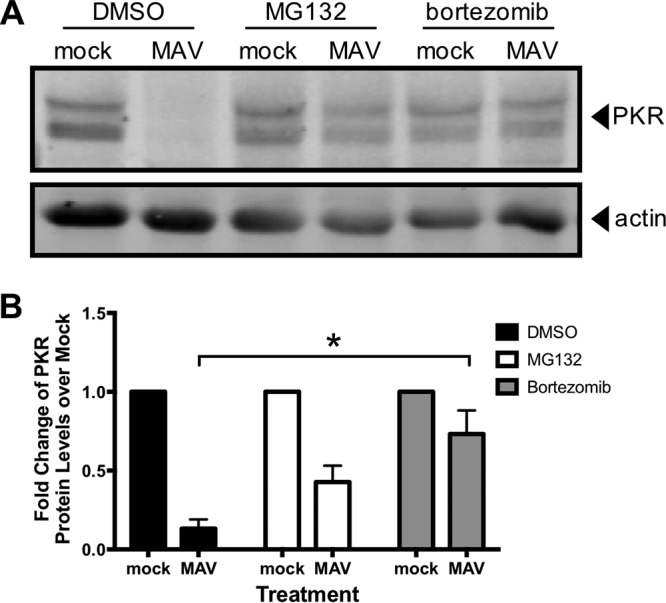
PKR is depleted by proteasomal degradation during MAV-1 infection. (A) C57BL/6 MEFs were infected with MAV-1 (MAV) at an MOI of 10 or were mock infected (mock) and treated with DMSO (vehicle for inhibitors), 1 µM MG132, or 1 µM bortezomib. Cell lysates were then analyzed by immunoblotting with antibodies for PKR (D-20) and actin. Blots are representative of results from four independent experiments. (B) Densitometry quantitation of four independent experiments. Treatment with bortezomib significantly inhibited PKR depletion in MAV-1-infected cells. *, P ≤ 0.05.
Treatment with MG132 and bortezomib does not affect MAV-1 replication at 24 hpi. C57BL/6 MEFs were infected with MAV-1 at an MOI of 10 and treated with DMSO (vehicle for inhibitors) or with 1 µM MG132 or bortezomib and were collected at 24 hpi. DNA was purified from cell pellets and analyzed for MAV-1 genome copies by qPCR. The graph is representative of results from five biological replicates per treatment group. Error bars represent standard errors of the means (SEM). *, P ≤ 0.05. Download FIG S4, PDF file, 0.1 MB (63.7KB, pdf) .
Copyright © 2019 Goodman et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
A signal for proteasomal degradation is the conjugation of ubiquitin to a protein (63, 64). We examined by immunoblotting whether PKR is ubiquitinated. We detected ubiquitination of a positive control, mouse p53, which is degraded in the presence of MAV-1 proteins (65). However, even with the use of epitope-tagged ubiquitin (66) and MG132 treatment, we were unable to detect PKR ubiquitination during infection (Fig. S5). This is consistent with an inability to detect PKR ubiquitination when it is degraded during RVFV infection (32). Although RVFV NSs is known to recruit an E3 ligase to PKR, the authors of that study reported that ubiquitinated PKR is undetectable. Therefore, the cellular degradation signal for PKR remains unclear.
PKR is not detectably ubiquitinated during MAV-1 infection. CMT93 cells were transfected with a GFP-ubiquitin plasmid using standard Polyplus transfection protocols. At 24 h posttransfection (hpt), the cells were infected with MAV-1 (MAV) at an MOI of 5 or were mock infected (mock) and treated with 10 µM MG132 at 6 hpi. Cell lysates were collected at 12 hpi and immunoprecipitated with (A) PKR (D-20) or an isotype control antibody or (B) p53 (DO-1) or an isotype control antibody. Immunoprecipitated samples were analyzed by immunoblotting with GFP, ubiquitin, isotype, PKR (B-10), or p53 (1801) antibodies. The input lane (mock input) contained a 0.008 volume of mock-infected lysate (relative to the volume in the immunoprecipitations). For panel B, GFP and p53 were probed on separate duplicate blots. Download FIG S5, PDF file, 6.8 MB (7MB, pdf) .
Copyright © 2019 Goodman et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
PKR is actively depleted early in infection.
We investigated the time point at which proteasomal degradation of PKR occurs. Early viral proteins are expressed prior to viral DNA replication, which is then followed by late viral protein expression. First, we examined the kinetics of PKR degradation to determine whether an early or late viral protein was likely responsible. We mock infected and infected CMT93 cells with MAV-1 at an MOI of 10, collected lysates every 6 h for 24 h, and analyzed them by immunoblotting with antibodies to PKR or MAV-1 early region 1A (E1A) protein, the first viral protein made during infection (67). PKR degradation in the infected cells was first detected at 12 hpi (Fig. 7A), and quantitation of the results of five independent experiments showed that ∼20% of the starting levels of PKR protein remained at 24 hpi (Fig. 7B).
FIG 7.

PKR is actively depleted early in infection. (A) CMT93 cells were infected with MAV-1 (MAV) at an MOI of 10 or were mock infected (mock) (top), or uninfected cells were treated with 50 µg/ml cycloheximide (CHX, bottom) to inhibit elongation of protein synthesis. Cell lysates were analyzed by immunoblotting with antibodies for PKR (D-20) and actin. Blots are representative of results from five independent experiments. (B) Densitometry quantitation of five independent experiments; **, P ≤ 0.01. (C) CMT93 cell lysates from the experiment performed as described in the panel A legend were analyzed with a second immunoblotting step with antibodies for E1A and actin. Blots are representative of results from four replicates from two independent experiments. The E1A blot image was uniformly adjusted to a brightness value of 30 and a contrast value of 5 in Adobe Photoshop.
In parallel, to determine the half-life of PKR in uninfected CMT93 cells, we treated CMT93 cells with cycloheximide to halt protein translation and thus production of new PKR. We collected lysates every 6 h for 24 h and analyzed by immunoblotting with antibodies to PKR. After 24 h of cycloheximide treatment, approximately 90% of the starting levels of PKR protein remained (Fig. 7A, bottom, and 7B). Comparing the results from MAV-1 infection (Fig. 7A, top, and 7B) and cycloheximide treatment of uninfected cells (Fig. 7A, bottom, and 7B), we conclude that MAV-1 was actively depleting PKR protein early in infection. E1A was detected by immunoblotting at 18 hpi (Fig. 7C), whereas viral DNA replication was first detected at 24 hpi in CMT93 cells (Fig. S6). Thus, the 18-hpi time point is considered an early time point during MAV-1 infection of CMT93 cells, prior to DNA replication, suggesting the involvement of an early viral protein in PKR depletion.
Viral DNA replication can be detected at 24 hpi by qPCR. CMT93 cells were infected with MAV-1 (MAV) at an MOI of 10 and collected every 6 h for 24 h. DNA was purified from cell pellets and analyzed for MAV-1 genome copies by qPCR. The graph is representative of results from four to five biological replicates per treatment group. Error bars represent standard errors of the means (SEM). Download FIG S6, PDF file, 0.1 MB (60.2KB, pdf) .
Copyright © 2019 Goodman et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
An early viral function is required for PKR depletion by MAV-1.
To determine whether viral gene expression or DNA replication is required for PKR degradation during infection, we infected C57BL/6 MEFs and CMT93 cells with UV-inactivated MAV-1 (which does not replicate viral DNA; Fig. S7). We infected cells at an MOI of 10 with WT MAV-1 or UV-inactivated MAV-1 and analyzed lysates from 24 and 48 hpi by immunoblotting for PKR protein levels. In both cell types, while PKR was degraded by 24 hpi in the cells infected with WT MAV-1, PKR protein levels were unaffected at both time points in cells infected with UV-inactivated MAV-1 (Fig. 8A). This suggested that either gene expression or DNA replication was required for PKR degradation during MAV-1 infection.
FIG 8.

Early gene expression is required for PKR depletion by MAV-1. (A) Cells (as indicated at left) were infected with WT MAV-1 (WT MAV) or UV-inactivated MAV-1 (UV MAV) at an MOI of 10 or were mock infected (mock). Cell lysates were analyzed by immunoblotting with antibodies for PKR (D-20) and actin. Two independent wells were infected for each condition at both time points. (B) CMT93 cells were infected with WT MAV-1 (MAV) at an MOI of 10 or were mock infected (mock). Infected cells were also treated (+) or not (-) with 20 µg/ml cytosine arabinoside (AraC), an inhibitor of DNA synthesis. Cell lysates were analyzed with antibodies for PKR and actin.
UV-inactivated virus does not replicate viral DNA. (A) C57BL/6 MEFs or (B) CMT93 cells were infected with WT MAV-1 (WT) or UV-inactivated MAV-1 (UV) at an MOI of 10 and collected at the indicated times. DNA was purified from cell pellets and analyzed for MAV-1 genome copies by qPCR. Graphs are representative of results from three to four biological replicates per treatment group. Error bars represent standard errors of the means (SEM). Download FIG S7, PDF file, 0.1 MB (87.5KB, pdf) .
Copyright © 2019 Goodman et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We addressed whether viral DNA replication is needed for PKR degradation. We mock infected or infected CMT93 cells with MAV-1 at an MOI of 10 and treated them with cytosine arabinoside (araC) at the time of infection to inhibit DNA synthesis (68, 69). This would allow the virus to infect the cell and produce early viral proteins, but would inhibit viral DNA replication and prevent late protein synthesis. We collected lysates at 20 and 40 hpi and analyzed them by immunoblotting. We confirmed that araC treatment resulted in no late protein synthesis by immunoblotting for late virion proteins (Fig. S8). In samples treated with araC, PKR degradation was seen at 20 and 40 hpi (Fig. 8B), indicating that DNA replication was not required for PKR degradation. Taken together, the results shown in Fig. 8 are consistent with early viral gene expression prior to DNA replication being involved in induction of PKR degradation by MAV-1.
AraC treatment inhibited late protein expression. CMT93 cells were infected with WT MAV-1 (MAV) at an MOI of 10 or were mock infected (mock). Infected cells were also treated (+) or not (-) with 20 µg/ml cytosine arabinoside (araC), an inhibitor of DNA synthesis. Cell lysates were analyzed with antibodies for late virion proteins (AKO1-103; 1:1,000). Arrowheads indicate late viral proteins. Download FIG S8, PDF file, 2.3 MB (2.3MB, pdf) .
Copyright © 2019 Goodman et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DISCUSSION
We have demonstrated here that PKR is antiviral in MAV-1 infections of cultured cells. Surprisingly, MAV-1 infection of primary and established cultured cells depleted PKR. The depletion was not due to reduced steady-state levels or reduced translation of PKR mRNA. Instead, we showed that PKR depletion is inhibited by proteasome inhibitors, implicating proteasomal degradation of PKR. Several lines of evidence suggest that the degradation is due to a viral early function.
PKR is an IFN-inducible gene product that is an important component of the innate immune response (1, 48). However, not all viruses, including EMCV and vaccinia virus, have increased virulence in PKR−/− MEFs (50, 70). While hAds produce VA RNAs that inhibit PKR antiviral activity during infection (10, 71), MAV-1 does not produce such VA RNAs, and how MAV-1 infection is affected by PKR is first described in this report. When we infected PKR−/− MEFs with MAV-1, viral DNA yields were 5 to 6 times higher than the viral DNA yields from wild-type MEFs (Fig. 1), indicating that PKR plays an antiviral role during MAV-1 infection. The viral DNA yield from the N-PKR−/− MEFs was nearly 4 times higher than the yield from the C-PKR−/− MEFs at 48 hpi, but by 72 hpi the viral DNA yields from the two types of PKR−/− MEFs were similar to each other and were significantly increased compared to the yield seen with wild-type MEFs (Fig. 1). This difference in viral replication kinetics between the two types of PKR−/− MEFs may be due to differences in the expression and activity levels of the PKR fragments reportedly produced by them; we have not assayed PKR fragment production in our cells.
We examined PKR protein levels during MAV-1 infection and found that PKR was depleted from the cells as early as 12 hpi (Fig. 7). Depletion was seen in a wide variety of cell types, including immortalized C57BL/6 MEFs, primary C57BL/6 peritoneal macrophages, and CMT93 mouse colon carcinoma cells. Once depleted, the PKR protein levels never returned to the levels measured for the mock-infected cells during infection. Activation (transautophosphorylation) of PKR (6–9) was not required for this depletion, because kinase-dead mouse PKR was also depleted from K271R SV40 MEFs during infection (Fig. 2C). Both PKR and phospho-PKR were depleted in all cell types examined.
We examined several possible explanations for the depletion of PKR protein, including PKR mRNA levels and alterations in translation. PKR mRNA levels remained unchanged during MAV-1 infection in C57BL/6 MEFs at 24 hpi and were increased in primary C57BL/6 peritoneal macrophages during MAV-1 infection (Fig. 3), corresponding to the times when the PKR protein levels were depleted (Fig. 2A). While the PKR mRNAs in C57BL/6 MEFs were depleted 33% at 48 hpi and 40% at 72 hpi compared to mock-infected lysates, this was not sufficient to explain the 84% and 94% reductions, respectively, in PKR protein levels at those time points (Fig. 2B). More likely, the reduction in PKR steady-state mRNA levels at the late infection time points can be attributed to other effects from viral infection, including the degradation or inhibition of proteins that induce PKR expression. For example, p53 is capable of binding to the PKR promoter and inducing its expression (72), but MAV-1 proteins cause p53 proteolysis (65). Together, our results from C57BL/6 MEFs and macrophages suggest that the virus does not cause PKR protein depletion by reducing PKR steady-state mRNA levels.
The differences in total PKR mRNA levels between C57BL/6 MEFs and primary peritoneal macrophages during infection (Fig. 3) were possibly due to the fact that macrophages are immune cells whereas MEFs are not. PKR protein took almost 3 times as long to be completely degraded during infection in macrophages than in MEFs (72 h versus 24 h) (Fig. 2A). Total PKR mRNA levels were 2 to 3 times higher in MAV-1-infected macrophages than in mock-infected macrophages, unlike the MEFs, where total PKR mRNA levels were unchanged or were reduced 33% to 40% during MAV-1-infection compared to the mock-infected MEFs. Since PKR is an IFN-stimulated gene (1, 2), the higher levels of total PKR mRNA seen during infection in the macrophages suggest IFN induction. This suggests that the immune response mounted by the macrophages was greater than the immune response in the MEFs and could help explain why PKR took longer to degrade in macrophages than in MEFs.
We considered whether the reduced PKR levels were due to reduced PKR protein translation. There was no change in the total amount of PKR mRNA bound to ribosomes during infection compared to uninfected cells, and there was also no significant change in the amount of actively translating PKR mRNA during infection (Fig. 4). We also found that the 5′ UTR of mouse PKR placed upstream of a reporter gene produced the same amounts of reporter with and without MAV-1 infection. These data indicate that there are not translational effects of MAV-1 infection on PKR protein levels that could explain the depleted PKR levels that we observed.
Inhibiting lysosomal degradation resulted in no change in PKR depletion in infected cells (Fig. 5A), but adding proteasome inhibitors preserved PKR protein within cells (Fig. 6A and B). This indicates that PKR is degraded not by lysosomal degradation during viral infection but by proteasomal degradation. Though PKR degradation was due to proteasome activity during MAV-1 infection, we were unable to demonstrate PKR ubiquitination, although we did detect ubiquitination of mouse p53 (see Fig. S5 in the supplemental material). This inability to demonstrate PKR ubiquitination could be explained if at any given moment there were only low levels of ubiquitinated PKR present in the cell. Perhaps increasing the time spent under conditions of MG132 treatment could increase the amounts of ubiquitinated proteins enough that PKR ubiquitination could be seen. However, our inability to detect ubiquitinated PKR is consistent with a similar inability to identify PKR ubiquitination by RVFV NSs, even though NSs is known to recruit an E3 ligase to PKR (32). Alternatively, it is possible that in MAV-1 infection, PKR is degraded in a ubiquitin-independent manner, possibly because of the presence of intrinsic disordered regions of PKR or binding of regulating proteins to PKR that target proteins to the proteasome (73, 74).
Our experiments indicated that MAV-1 actively depletes PKR early in infection. Ongoing experiments are focused on determining the MAV-1 early protein(s) responsible for PKR degradation. Two possibilities are represented by E4 proteins, the homologs of hAd E4orf6 and E4orf3, which we originally termed E4orfa/b and E4orfa/c, respectively (75). In human adenovirus, E4orf6 interacts with another early hAd protein, E1B 55K, to participate in an E3 ligase complex that ubiquitinates and degrades p53 via proteasomal degradation (76, 77). When MAV-1 E4orf6, E1B 55K, and mouse p53 are introduced by transfection into human cells, all three proteins interact and mouse p53 is degraded (65). If MAV-1 E4orf6 and E1B 55K form a similar complex in mouse cells, it may also degrade PKR. We have preliminary evidence indicating that mouse p53 is ubiquitinated in C57BL/6 MEFs during MAV-1 infection, which suggests that the mouse p53 degradation seen in human cells could be paralleled by degradation of endogenous mouse p53 and mouse PKR in mouse cells, mediated by MAV-1 E4orf6 and E1B 55K during infection. Another hAd E4 protein, E4orf3, causes proteasomal degradation of transcriptional intermediary factor 1γ (78) and general transcription factor II-I (79) in a manner independent of hAd E4orf6 and E1B 55K. E4orf3 has SUMO E3 ligase and E4 elongase activity and induces sumoylation of general transcription factor II-I, leading to its proteasome-dependent degradation (79). MAV-1 E4orf3 may similarly have sumoylation activity that results ultimately in proteasome-dependent PKR degradation. Another possibility of a viral protein involved in PKR degradation is the protease encoded by MAV-1. The hAd protease is encapsidated in virions and proteolytically processes viral proteins IIIa, VI, VII, VIII, mu, and TP (80–83). However, we think it is unlikely that the MAV-1 protease degrades PKR, because we showed that UV-inactivated virus was unable to degrade PKR. We assume that UV treatment would not destroy the MAV-1 protease activity, just as HSV-1 VP16 activity is not altered by UV inactivation of HSV-1 (84), but we have not tested this directly.
In summary, we demonstrated that PKR has an antiviral role during MAV-1 infection in vitro, because when PKR is mutated, viral replication in MEFs is significantly higher than that seen in wild-type MEFs. Analysis of global PKR steady-state protein levels during infection showed complete PKR depletion by 72 hpi in multiple cell types, including immortalized and primary cells, with even faster kinetics in some. PKR transcription and translation were not decreased by MAV-1 infection, whereas proteasomal inhibition prevented PKR degradation. Taken together, these data suggest that MAV-1 causes PKR to be proteasomally degraded at a posttranslational level. This work provides new insight into possible mechanisms of adenovirus inhibition of PKR by DNA viruses. PKR degradation may be induced by other adenoviruses that do not produce VA RNA, which includes all animal adenoviruses except primate adenoviruses and one type of fowl adenovirus (85).
MATERIALS AND METHODS
Cells, virus, and infections.
CMT93 cells (CCL-223) and C57BL/6 MEFs (SCRC-1008) were obtained from the American Type Culture Collection and passaged in Dulbecco’s modified Eagle medium (DMEM) containing 5% and 10% heat-inactivated fetal bovine serum (FBS), respectively, before use. Primary peritoneal macrophages were obtained from 6-to-10-week-old C57BL/6 mice purchased from Jackson Laboratory (catalog no. 000664) as described previously (86). Briefly, 6-to-10-week-old C57BL/6 mice were injected intraperitoneally with 1.2 ml 3% thioglycolate and euthanized 3 to 5 days later. The abdominal skin was carefully removed, exposing the peritoneum, which was then injected with 5 ml of sterile phosphate-buffered saline (PBS). The abdomen was massaged gently, and then the PBS containing the peritoneal macrophages was carefully withdrawn. The macrophages were centrifuged at 100 × g for 4 min, and red blood cells were lysed in lysis buffer (0.15 M ammonium chloride, 1 mM potassium bicarbonate, 0.1 mM EDTA disodium salt) for 2 min at room temperature, centrifuged at 100 × g for 4 min, washed twice in PBS, resuspended in DMEM–5% heat-inactivated FBS, and plated in 6-well plates. WT and PKR−/− MEFs (termed PKR WT MEFs and N-PKR−/− MEFs, respectively, throughout this paper) were obtained from Robert Silverman, Cleveland Clinic (87), and were passaged in DMEM containing 10% heat-inactivated FBS before use. PKR−/− MEFs stably transfected with empty vector (termed C-PKR−/− MEFs throughout this paper) were obtained from Gokhan Hotamisligil, Harvard University (88), and were passaged in DMEM containing 10% heat-inactivated FBS before use. WT (SV40 MEFs) and K271R PKR mutant (K271R SV40 MEFs) MEFs were obtained from Anthony Sadler, Hudson Institute of Medical Research (54), and were passaged in DMEM containing 10% heat-inactivated FBS before use.
Wild-type mouse adenovirus type 1 (MAV-1) stock was prepared, and titers were determined on mouse NIH 3T6 fibroblasts as described previously (89). WT MAV-1 was subjected to UV inactivation by UV treatment of 200 µl of virus for 10 min at 800 mJ/cm2. UV inactivation was confirmed by qPCR and plaque assay.
For infection assays, medium was removed from cells and adsorption procedures were performed with 0.4 ml of inocula in 6-well plates with 35-mm-diameter wells (unless otherwise noted) for 1 h at 37°C at the indicated MOIs (PFU/cell). After 60 min, 2 ml of DMEM–5% FBS was added without removal of inocula; that time point was designated 0 hpi. For araC experiments, 20 µg/ml araC (Sigma C1768) was added at 0 hpi and replenished every 12 to 16 h.
Immunoblotting.
At room temperature, cells were washed once with PBS, and Pierce radioimmunoprecipitation assay (RIPA) lysis buffer (Thermo Scientific catalog no. 89900) with 1× protease inhibitors (protease inhibitor cocktail kit; Thermo Scientific catalog no. 78410) was added to the plate. The cells were allowed to lyse at room temperature for 10 min before being harvested and centrifuged at 4°C at 14,000 × g for 10 min to remove debris. Equivalent amounts of protein, determined by a bicinchoninic acid (BCA) assay (Pierce BCA protein assay kit; Thermo Scientific catalog no. 23227), were subjected to acetone precipitatation by incubation with a 4× volume of ice-cold acetone overnight at −20°C. Precipitated proteins were pelleted at 4°C at 13,000 × g for 10 min, and the pellets were dried for 30 min at room temperature. Pellets were resuspended in a mixture of 10 µl Pierce RIPA lysis buffer (Thermo Scientific catalog no. 89900), 3.25 µl NuPAGE 4× lithium dodecyl sulfate (LDS) sample buffer (Invitrogen catalog no. NP0007), and 1.25 µl 1 M dithiothreitol (DTT). Samples were incubated at 37°C for 10 min and then loaded into a well of an 8% acrylamide gel (8.3 cm wide by 7.3 cm high by 0.1 cm thick) with a 2.5% stacking gel, electrophoresed for 30 min at 50 V and 85 min at 150 V, and then transferred to a polyvinylidene difluoride (PVDF) membrane (Bio-Rad catalog no. 1620177) for 1 h at 100 V at 4°C. Blots were blocked in 5% bovine serum albumin (BSA; Sigma catalog no. A7906)–Tris-buffered saline (Bio-Rad catalog no. 1706435)–0.1% Tween 20 (Sigma catalog no. P1379). Blots were probed with primary antibodies to detect mouse PKR (Santa Cruz D-20 sc-708 [1:2,000] or B-10 sc-6282 [1:200]), mouse actin (Santa Cruz sc-1616-R [1:1,000]), MAV-1 E1A (AKO-7-147 [1:1,000]; described previously [67]), or MAV-1 late viral proteins (AKO 1-103 [1:1,000]; described previously [90, 91]). The secondary antibodies used were IRDye 800CW anti-rabbit (Li-Cor 925-32213 [1:15,000]) or IgG peroxidase-conjugated anti-mouse (Jackson Immuno 515-035-062 [1:20,000]) antibody. Blots were visualized by the use of Li-Cor Odyssey imaging (Li-Cor Biosciences) or enhanced chemiluminescent substrates (Pierce ECL Western blotting substrate; catalog no. 32106) and X-ray film (Dot Scientific catalog no. BDB810). Densitometric quantification was performed using .tif files and ImageJ software from NIH (92).
To attempt to demonstrate PKR ubiquitination status during MAV-1 infection, C57BL/6 MEFs were transfected with green fluorescent protein (GFP) (Addgene catalog no. 11928)-tagged or hemagglutinin (HA) epitope (Addgene catalog no. 18712)-tagged ubiquitin plasmids 24 h before infection. We used Polyplus jetPRIME transfection reagent (Polyplus catalog no. 114-15) with 10 µg plasmid DNA and 30 µl jetPRIME reagent per 10-cm-diameter plate. At 12 hpi (36 h posttransfection), we treated mock-infected and infected C57BL/6 MEFs with 10 µM MG132 (Sigma M7449) for 6 h before collecting lysates at 18 hpi in HCN buffer (50 mM HEPES, 150 mM NaCl, 2 mM CaCl2, 1% Triton X-100 [Sigma T9284]), 1× protease inhibitors [protease inhibitor cocktail kit; Thermo Scientific catalog no. 78410], 5 mM N-ethylmaleimide). The lysates were split into two aliquots, and 3 µg PKR (Santa Cruz D-20 sc-708; discontinued) or 3 µg isotype rabbit polyclonal antibody (Jackson Immuno catalog no. 011-000-002) was added to lysates. After rocking samples overnight at 4°C, 20 µl of a protein A agarose suspension (Calbiochem/Millipore catalog no. IP02-1.5ML) was added to each sample and the samples were rocked at 4°C for 2 h. After incubation, the agarose was washed 3 times with 1 ml HCN buffer, resuspended in 40 µl 2× Laemmli buffer (Bio-Rad catalog no. 161-0737)–5% 2-mercaptoethanol (Sigma M6250), and boiled for 10 min. The lysate supernatants remaining after the initial PKR immunoprecipitation were then immunoprecipitated again using the same procedure but with 4 µg anti-p53 mouse monoclonal antibody (DO-1; Santa Cruz sc-126) or 4 µg isotype mouse monoclonal antibody (ThermoFisher Scientific catalog no. 02-6200). Immunoprecipitated proteins were subjected to immunoblotting for GFP or HA epitope-tagged ubiquitin with antibodies for GFP (Roche catalog no. 11814460001) (1:3,000) or HA (Abcam catalog no. ab9110) (1:4,000). No ubiquitin signal was detected from the PKR immunoprecipitations, though the positive control, p53, showed ubiquitin signal with both epitope-tagged ubiquitins (Fig. S5 in the supplemental material). Blots were also probed for PKR (PKR B-10 sc-6282) (1:200), p53 (anti-p53 sc-98) (1:200), and IRDye 800CW anti-mouse (Li-Cor 925-32212) (1:15,000) to confirm that the immunoprecipitations had been successful, and signals for both proteins were detected (Fig. S5).
Viral DNA yield analysis by qPCR.
Cells were washed twice with room temperature PBS and harvested by scraping into PBS, centrifuging at 100 × g for 4 min at 4°C, and resuspending in PBS. Total cellular DNA was purified using an Invitrogen PureLink DNA purification kit (Thermo Scientific catalog no. K1820-02) and quantitated using a NanoDrop spectrophotometer. A 10-ng volume of total cellular DNA was analyzed by qPCR using custom primers specific to MAV-1 E1A (mE1Agenomic Fwd [5′ GCA CTC CAT GGC AGG ATT CT 3′] and mE1Agenomic Rev [5′ GGT CGA AGC AGA CGG TTC TTC 3′]), and the results were normalized to GAPDH (glyceraldehyde-3-phosphate dehydrogenase), which was analyzed using a GAPDH-specific primer/probe set (Thermo Fisher Scientific Mm99999915_g1; catalog no. 4331182).
mRNA analysis by qPCR.
Cells were harvested by scraping into media, centrifuging at 100 × g for 4 min at 4°C, and washing the cell pellet three times with ice-cold PBS. RNA was purified using a Qiagen RNeasy minikit (Qiagen catalog no. 74134) and stored at −80˚C. A 125-ng volume of RNA per sample was used to make cDNA using a High-Capacity cDNA reverse transcription (RT) kit (Applied Biosystems catalog no. 4368814), and 2 µl of the cDNA was analyzed by qPCR using a primer/probe set specific to mouse PKR sequence (Thermo Fisher Mm01235643_m1; catalog no. 4331182). The results were normalized to GAPDH, which was analyzed using a GAPDH-specific primer/probe set (Thermo Fisher Scientific Mm99999915_g1; catalog no. 4331182). Arbitrary units were calculated as follows: Mean threshold cycle (CT) PKR − mean CT GAPDH = ΔCT for sample (arbitrary unit = 2−ΔCT).
Proteasome inhibition.
C57BL/6 MEFs were infected at an MOI of 10, and DMSO, 1 µM MG132 (Sigma M7449), or 1 µM bortezomib (Selleckchem catalog no. S1013) was added to the media after a 1 h adsorption. At 24 hpi, cells were washed once with room temperature PBS, and Pierce RIPA lysis buffer (Thermo Scientific catalog no. 89900) with 1× protease inhibitors (protease inhibitor cocktail kit; Thermo Scientific catalog no. 78410) was added to the plate. The cells were allowed to lyse at room temperature for 10 min before being harvested and centrifuged at 4°C at 14,000 × g for 10 min to remove debris.
Lysosome inhibition and DQ BSA assay.
CMT93 cells were infected at an MOI of 10. After 1 h of adsorption, 10 µl water, 10 mM (final concentration) NH4Cl (Baker Chemical Company catalog no. 0660-1), or 60 µM (final concentration) chloroquine (Sigma C6628) was added to the media. At 24 hpi, at room temperature, cells were washed once with PBS, and Pierce RIPA lysis buffer (Thermo Scientific catalog no. 89900) with 1× protease inhibitors (protease inhibitor cocktail kit; Thermo Scientific catalog no. 78410) was added to the plate. The cells were allowed to lyse at room temperature for 10 min before being harvested and centrifuged at 4°C at 14,000 × g for 10 min to remove debris.
The DQ BSA assay was performed as described previously (59). Briefly, C57BL/6 MEFs and CMT93 cells were plated at 1.5 × 105 cells/plate and 3 × 105 cells/plate, respectively, in MatTek glass-bottom microwell dishes (part no. P35G-1.5-14C) with 2 ml of DMEM–10% and DMEM–5% FBS, respectively. The next day, the cell medium was treated with 10 µl water, 10 mM (final concentration) NH4Cl, or 60 µM (final concentration) chloroquine (Sigma C6628). Four hours after addition of inhibitors, DQ red BSA (Invitrogen catalog no. D12051) was added to the media to reach a final concentration of 5 µg/ml in 2 ml DMEM plus 10% and 5% FBS, respectively. At 24 h posttreatment, the cells were imaged on a Nikon TE300 inverted microscope equipped with a mercury arc lamp; a Plan-Apochromat 60×, 1.4-numerical-aperture (NA) objective; a cooled digital charge-coupled-device (CCD) camera (Quantix Photometrics, Tucson, AZ); and a temperature-controlled stage (set at 37°C). To image the DQ-BSA, we used an excitation filter centered at 572 nm and an emission filter centered at 635 nm. The exposure times were the same for all images.
Ribosome pelleting.
Ribosomes were pelleted as described previously (93). Briefly, C57BL/6 MEFs were plated on 10-cm-diameter plates at 3 × 105 cells per plate. The next day, the cells (∼90% confluent) were infected with MAV-1 at an MOI of 5. C57BL/6 MEF lysates were collected at 48 hpi by scraping the cells in ice-cold PBS containing 100 µg/ml cycloheximide (Sigma C7698), pelleting, and resuspending in lysis buffer, which contained 10 mM HEPES (pH 7.5), 100 mM KCl, 5 mM MgCl2, 4 mM DTT, 0.5% NP-40, 100 µg/ml cycloheximide, 20 U/ml RNasin (Promega catalog no. N2511), 10% sucrose, and 1× protease inhibitors (protease inhibitor cocktail kit; Thermo Scientific catalog no. 78410). Cells were lysed by passage through a chilled 26-gauge needle five times and were cleared by centrifugation for 10 min at 21,000 × g at 4°C. A 400-µl volume of cleared lysate (optical density at 260 nm [OD260] of 10) was layered onto 25% sucrose and centrifuged at 29,500 rpm in an SW41 rotor (average relative centrifugal force [rcf], 107,458) for 4 h at 4°C. After pelleting, the supernatant was removed by the use of a micropipette, and 350 µl of Buffer RLT Plus (from Qiagen RNeasy minikit) (4°C) was added to the pellet for collection of the RNA. The RNA was purified immediately using an Qiagen RNeasy minikit (Qiagen catalog no. 74134) and stored at −80˚C until analysis.
Polyribosome gradients.
C57BL/6 MEFs were plated on 10-cm-diameter plates (2 × 106 cells per plate). The next day, the cells were infected with MAV-1 at an MOI of 2. Following a standard protocol (94), 5 min prior to collection, cycloheximide was added at a final concentration of 100 µg/ml and the reaction mixture was incubated at 37°C. Cells were collected at 24 hpi by scraping in ice-cold PBS containing 100 µg/ml cycloheximide, pelleting, and resuspending in 500 µl lysis buffer (20 mM Tris-Cl, 150 mM NaCl, 15 mM MgCl2, 8% glycerol, 20 IU/ml SUPERase•In [Thermo Fisher Scientific catalog no. AM2696], 80 IU/ml murine RNase inhibitor [New England BioLabs catalog no. M0314S], 0.1 mg/ml heparin [Sigma H3393-50], 0.1 mg/ml cycloheximide, 1 mM DTT, 1× protease inhibitor [protease inhibitor cocktail kit; Thermo Scientific catalog no. 78410], 20 IU/ml Turbo DNase [Thermo Fisher Scientific catalog no. AM2238], 1% Triton X-100 [Sigma T9284]). Cells were lysed by passage through a chilled 26-gauge needle 10 times, vortex mixing for 30 s, and then incubating on ice for 5 min. Lysates were cleared by centrifugation for 5 min at 14,000 × g at 4°C. A 500-µl volume of cleared lysate (OD260 of 10) was layered onto a 10%-to-50% sucrose gradient and centrifuged at 35,000 rpm in an SW41 rotor (151,000 rcf) for 3 h at 4°C. After centrifugation, gradients were pumped out of the top with a Brandel BR-188 density gradient fractionation system with a continuous reading of the OD254. From 24 to 34 fractions (350 to 500 µl) were collected. RNA was purified from selected fractions immediately using a Qiagen RNeasy minikit (Qiagen catalog no. 74134) and stored at −80˚C until analysis by RT-qPCR.
Statistical analyses.
Data were analyzed with GraphPad Prism 7.0 software. For qPCR and densitometry analyses, the data were analyzed by individual Mann-Whitney tests. A P value of <0.5 was considered significant.
ACKNOWLEDGMENTS
We thank Robert Silverman, Gokhan Hotamisligil, and Anthony Sadler for PKR mutant and wild-type cells. We thank Katelyn Green, Becky Billmire, and Peter Todd for the use of and assistance with the Brandel BR-188 density gradient fractionation system and for providing the nanoluciferase and firefly luciferase plasmids. We thank Zachary Mendel and Joel Swanson for assistance with imaging the DQ BSA assay. We thank Jason Weinberg, Michael Imperiale, Christiane Wobus, Billy Tsai, Andrew Mehle, Mitch Ledwith, Mike Mathews, Chuck Samuel, and Britt Glaunsinger for helpful discussions. We thank Michael Imperiale and Christiane Wobus for comments on the manuscript.
This work was supported by HHS/NIH/National Institute of Allergy and Infectious Diseases (NIAID) (5 R01AI091721 and 1 R01AI133935 [K.R.S.]), by NIH Cellular and Molecular Biology training grant T32 GM007315, and by Rackham Pre-Candidate and Candidate Student Research grants (D.E.G.).
Footnotes
Citation Goodman DE, Pretto CD, Krepostman TA, Carnahan KE, Spindler KR. 2019. Enhanced replication of mouse adenovirus type 1 following virus-induced degradation of protein kinase R (PKR). mBio 10:e00668-19. https://doi.org/10.1128/mBio.00668-19.
Contributor Information
Thomas Shenk, Princeton University.
Patrick Hearing, Stony Brook University.
Andrew Turnell, University of Birmingham.
REFERENCES
- 1.Meurs E, Chong K, Galabru J, Thomas NS, Kerr IM, Williams BR, Hovanessian AG. 1990. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell 62:379–390. doi: 10.1016/0092-8674(90)90374-N. [DOI] [PubMed] [Google Scholar]
- 2.McCormack SJ, Thomis DC, Samuel CE. 1992. Mechanism of interferon action: identification of a RNA binding domain within the N-terminal region of the human RNA-dependent P1/eIF-2 alpha protein kinase. Virology 188:47–56. doi: 10.1016/0042-6822(92)90733-6. [DOI] [PubMed] [Google Scholar]
- 3.Feng GS, Chong K, Kumar A, Williams BR. 1992. Identification of double-stranded RNA-binding domains in the interferon-induced double-stranded RNA-activated p68 kinase. Proc Natl Acad Sci U S A 89:5447–5451. doi: 10.1073/pnas.89.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuhen KL, Samuel CE. 1997. Isolation of the interferon-inducible RNA-dependent protein kinase Pkr promoter and identification of a novel DNA element within the 5'-flanking region of human and mouse Pkr genes. Virology 227:119–130. doi: 10.1006/viro.1996.8306. [DOI] [PubMed] [Google Scholar]
- 5.Patel RC, Sen GC. 1992. Identification of the double-stranded RNA-binding domain of the human interferon-inducible protein kinase. J Biol Chem 267:7671–7676. [PubMed] [Google Scholar]
- 6.Dabo S, Meurs EF. 2012. dsRNA-dependent protein kinase PKR and its role in stress, signaling and HCV infection. Viruses 4:2598–2635. doi: 10.3390/v4112598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mundschau LJ, Faller DV. 1994. Endogenous inhibitors of the dsRNA-dependent eIF-2 alpha protein kinase PKR in normal and ras-transformed cells. Biochimie 76:792–800. doi: 10.1016/0300-9084(94)90083-3. [DOI] [PubMed] [Google Scholar]
- 8.Hovanessian AG, Galabru J. 1987. The double-stranded RNA-dependent protein kinase is also activated by heparin. Eur J Biochem 167:467–473. doi: 10.1111/j.1432-1033.1987.tb13360.x. [DOI] [PubMed] [Google Scholar]
- 9.Galabru J, Hovanessian A. 1987. Autophosphorylation of the protein kinase dependent on double-stranded RNA. J Biol Chem 262:15538–15544. [PubMed] [Google Scholar]
- 10.Berk AJ. 2013. Chapter 55, Adenoviridae: the viruses and their replication, p 1704–1731. In Knipe DM, Howley P (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 11.Wek RC. 2 July 2018, posting date. Role of eIF2alpha kinases in translational control and adaptation to cellular stress. Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a032870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farrell PJ, Balkow K, Hunt T, Jackson RJ, Trachsel H. 1977. Phosphorylation of initiation factor elF-2 and the control of reticulocyte protein synthesis. Cell 11:187–200. doi: 10.1016/0092-8674(77)90330-0. [DOI] [PubMed] [Google Scholar]
- 13.Tahara SM, Traugh JA, Sharp SB, Lundak TS, Safer B, Merrick WC. 1978. Effect of hemin on site-specific phosphorylation of eukaryotic initiation factor 2. Proc Natl Acad Sci U S A 75:789–793. doi: 10.1073/pnas.75.2.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dzananovic E, McKenna SA, Patel TR. 2018. Viral proteins targeting host protein kinase R to evade an innate immune response: a mini review. Biotechnol Genet Eng Rev 34:33–59. doi: 10.1080/02648725.2018.1467151. [DOI] [PubMed] [Google Scholar]
- 15.Chang HW, Jacobs BL. 1993. Identification of a conserved motif that is necessary for binding of the vaccinia virus E3L gene products to double-stranded RNA. Virology 194:537–547. doi: 10.1006/viro.1993.1292. [DOI] [PubMed] [Google Scholar]
- 16.Rice AD, Turner PC, Embury JE, Moldawer LL, Baker HV, Moyer RW. 2011. Roles of vaccinia virus genes E3L and K3L and host genes PKR and RNase L during intratracheal infection of C57BL/6 mice. J Virol 85:550–567. doi: 10.1128/JVI.00254-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carroll K, Elroy-Stein O, Moss B, Jagus R. 1993. Recombinant vaccinia virus K3L gene product prevents activation of double-stranded RNA-dependent, initiation factor 2 alpha-specific protein kinase. J Biol Chem 268:12837–12842. [PubMed] [Google Scholar]
- 18.Lu Y, Wambach M, Katze MG, Krug RM. 1995. Binding of the influenza virus NS1 protein to double-stranded RNA inhibits the activation of the protein kinase that phosphorylates the elF-2 translation initiation factor. Virology 214:222–228. doi: 10.1006/viro.1995.9937. [DOI] [PubMed] [Google Scholar]
- 19.Tan SL, Katze MG. 1998. Biochemical and genetic evidence for complex formation between the influenza A virus NS1 protein and the interferon-induced PKR protein kinase. J Interferon Cytokine Res 18:757–766. doi: 10.1089/jir.1998.18.757. [DOI] [PubMed] [Google Scholar]
- 20.Cardenas WB, Loo YM, Gale M Jr, Hartman AL, Kimberlin CR, Martinez-Sobrido L, Saphire EO, Basler CF. 2006. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J Virol 80:5168–5178. doi: 10.1128/JVI.02199-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poppers J, Mulvey M, Khoo D, Mohr I. 2000. Inhibition of PKR activation by the proline-rich RNA binding domain of the herpes simplex virus type 1 Us11 protein. J Virol 74:11215–11221. doi: 10.1128/JVI.74.23.11215-11221.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cassady KA, Gross M, Roizman B. 1998. The herpes simplex virus US11 protein effectively compensates for the gamma1 (34.5) gene if present before activation of protein kinase R by precluding its phosphorylation and that of the alpha subunit of eukaryotic translation initiation factor 2. J Virol 72:8620–8626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cai R, Carpick B, Chun RF, Jeang KT, Williams BR. 2000. HIV-I TAT inhibits PKR activity by both RNA-dependent and RNA-independent mechanisms. Arch Biochem Biophys 373:361–367. doi: 10.1006/abbi.1999.1583. [DOI] [PubMed] [Google Scholar]
- 24.McMillan NA, Chun RF, Siderovski DP, Galabru J, Toone WM, Samuel CE, Mak TW, Hovanessian AG, Jeang KT, Williams BR. 1995. HIV-1 Tat directly interacts with the interferon-induced, double-stranded RNA-dependent kinase, PKR. Virology 213:413–424. doi: 10.1006/viro.1995.0014. [DOI] [PubMed] [Google Scholar]
- 25.Park H, Davies MV, Langland JO, Chang HW, Nam YS, Tartaglia J, Paoletti E, Jacobs BL, Kaufman RJ, Venkatesan S. 1994. TAR RNA-binding protein is an inhibitor of the interferon-induced protein kinase PKR. Proc Natl Acad Sci U S A 91:4713–4717. doi: 10.1073/pnas.91.11.4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thimmappaya B, Weinberger C, Schneider RJ, Shenk T. 1982. Adenovirus VAI RNA is required for efficient translation of viral mRNAs at late times after infection. Cell 31:543–551. doi: 10.1016/0092-8674(82)90310-5. [DOI] [PubMed] [Google Scholar]
- 27.Mathews MB, Grodzicker T. 1981. Virus-associated RNAs of naturally occurring strains and variants of group C adenoviruses. J Virol 38:849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reich PR, Forget BG, Weissman SM. 1966. RNA of low molecular weight in KB cells infected with adenovirus type 2. J Mol Biol 17:428–439. doi: 10.1016/S0022-2836(66)80153-5. [DOI] [PubMed] [Google Scholar]
- 29.Kalveram B, Ikegami T. 2013. Toscana virus NSs protein promotes degradation of double-stranded RNA-dependent protein kinase. J Virol 87:3710–3718. doi: 10.1128/JVI.02506-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Habjan M, Pichlmair A, Elliott RM, Overby AK, Glatter T, Gstaiger M, Superti-Furga G, Unger H, Weber F. 2009. NSs protein of Rift Valley fever virus induces the specific degradation of the double-stranded RNA-dependent protein kinase. J Virol 83:4365–4375. doi: 10.1128/JVI.02148-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ikegami T, Narayanan K, Won S, Kamitani W, Peters CJ, Makino S. 2009. Rift Valley fever virus NSs protein promotes post-transcriptional downregulation of protein kinase PKR and inhibits eIF2alpha phosphorylation. PLoS Pathog 5:e1000287. doi: 10.1371/journal.ppat.1000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mudhasani R, Tran JP, Retterer C, Kota KP, Whitehouse CA, Bavari S. 2016. Protein kinase R degradation is essential for Rift Valley fever virus infection and is regulated by SKP1-CUL1-F-box (SCF)FBXW11-NSs E3 ligase. PLoS Pathog 12:e1005437. doi: 10.1371/journal.ppat.1005437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Black TL, Barber GN, Katze MG. 1993. Degradation of the interferon-induced 68,000-M(r) protein kinase by poliovirus requires RNA. J Virol 67:791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Black TL, Safer B, Hovanessian A, Katze MG. 1989. The cellular 68,000-Mr protein kinase is highly autophosphorylated and activated yet significantly degraded during poliovirus infection: implications for translational regulation. J Virol 63:2244–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li W, Zhu Z, Cao W, Yang F, Zhang X, Li D, Zhang K, Li P, Mao R, Liu X, Zheng H. 2016. Esterase D enhances type I interferon signal transduction to suppress foot-and-mouth disease virus replication. Mol Immunol 75:112–121. doi: 10.1016/j.molimm.2016.05.016. [DOI] [PubMed] [Google Scholar]
- 36.Li C, Zhu Z, Du X, Cao W, Yang F, Zhang X, Feng H, Li D, Zhang K, Liu X, Zheng H. 2017. Foot-and-mouth disease virus induces lysosomal degradation of host protein kinase PKR by 3C proteinase to facilitate virus replication. Virology 509:222–231. doi: 10.1016/j.virol.2017.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rabouw HH, Langereis MA, Knaap RC, Dalebout TJ, Canton J, Sola I, Enjuanes L, Bredenbeek PJ, Kikkert M, de Groot RJ, van Kuppeveld FJ. 2016. Middle East respiratory coronavirus accessory protein 4a inhibits PKR-mediated antiviral stress responses. PLoS Pathog 12:e1005982. doi: 10.1371/journal.ppat.1005982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hovanessian AG, Galabru J, Meurs E, Buffet-Janvresse C, Svab J, Robert N. 1987. Rapid decrease in the levels of the double-stranded RNA-dependent protein kinase during virus infections. Virology 159:126–136. doi: 10.1016/0042-6822(87)90355-2. [DOI] [PubMed] [Google Scholar]
- 39.Chang YH, Lau KS, Kuo RL, Horng JT. 2017. dsRNA binding domain of PKR is proteolytically released by enterovirus A71 to facilitate viral replication. Front Cell Infect Microbiol 7:284. doi: 10.3389/fcimb.2017.00284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kainulainen M, Lau S, Samuel CE, Hornung V, Weber F. 2016. NSs virulence factor of Rift Valley fever virus engages the F-box proteins FBXW11 and beta-TRCP1 to degrade the antiviral protein kinase PKR. J Virol 90:6140–6147. doi: 10.1128/JVI.00016-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kring SC, King CS, Spindler KR. 1995. Susceptibility and signs associated with mouse adenovirus type 1 infection of adult outbred Swiss mice. J Virol 69:8084–8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spindler KR, Moore ML, Cauthen AN. 2007. Mouse adenoviruses, p 49–65. In The mouse in biomedical research, 2nd ed, vol 2 Academic Press, New York, NY. [Google Scholar]
- 43.Blailock ZR, Rabin ER, Melnick JL. 1968. Adenovirus myocarditis in mice. An electron microscopic study. Exp Mol Pathol 9:84–96. doi: 10.1016/0014-4800(68)90053-1. [DOI] [PubMed] [Google Scholar]
- 44.McCarthy MK, Procario MC, Twisselmann N, Wilkinson JE, Archambeau AJ, Michele DE, Day SM, Weinberg JB. 2015. Proinflammatory effects of interferon gamma in mouse adenovirus 1 myocarditis. J Virol 89:468–479. doi: 10.1128/JVI.02077-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guida JD, Fejer G, Pirofski LA, Brosnan CF, Horwitz MS. 1995. Mouse adenovirus type 1 causes a fatal hemorrhagic encephalomyelitis in adult C57BL/6 but not BALB/c mice. J Virol 69:7674–7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mathews MB, Shenk T. 1991. Adenovirus virus-associated RNA and translation control. J Virol 65:5657–5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meissner JD, Hirsch GN, LaRue EA, Fulcher RA, Spindler KR. 1997. Completion of the DNA sequence of mouse adenovirus type 1: sequence of E2B, L1, and L2 (18–51 map units). Virus Res 51:53–64. doi: 10.1016/S0168-1702(97)00079-8. [DOI] [PubMed] [Google Scholar]
- 48.Balachandran S, Roberts PC, Brown LE, Truong H, Pattnaik AK, Archer DR, Barber GN. 2000. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 13:129–141. doi: 10.1016/S1074-7613(00)00014-5. [DOI] [PubMed] [Google Scholar]
- 49.Stojdl DF, Abraham N, Knowles S, Marius R, Brasey A, Lichty BD, Brown EG, Sonenberg N, Bell JC. 2000. The murine double-stranded RNA-dependent protein kinase PKR is required for resistance to vesicular stomatitis virus. J Virol 74:9580–9585. doi: 10.1128/JVI.74.20.9580-9585.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiang Y, Condit RC, Vijaysri S, Jacobs B, Williams BR, Silverman RH. 2002. Blockade of interferon induction and action by the E3L double-stranded RNA binding proteins of vaccinia virus. J Virol 76:5251–5259. doi: 10.1128/JVI.76.10.5251-5259.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baltzis D, Li S, Koromilas AE. 2002. Functional characterization of pkr gene products expressed in cells from mice with a targeted deletion of the N terminus or C terminus domain of PKR. J Biol Chem 277:38364–38372. doi: 10.1074/jbc.M203564200. [DOI] [PubMed] [Google Scholar]
- 52.Yang YL, Reis LF, Pavlovic J, Aguzzi A, Schafer R, Kumar A, Williams BR, Aguet M, Weissmann C. 1995. Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO J 14:6095–6106. doi: 10.1002/j.1460-2075.1995.tb00300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abraham N, Stojdl DF, Duncan PI, Methot N, Ishii T, Dube M, Vanderhyden BC, Atkins HL, Gray DA, McBurney MW, Koromilas AE, Brown EG, Sonenberg N, Bell JC. 1999. Characterization of transgenic mice with targeted disruption of the catalytic domain of the double-stranded RNA-dependent protein kinase, PKR. J Biol Chem 274:5953–5962. doi: 10.1074/jbc.274.9.5953. [DOI] [PubMed] [Google Scholar]
- 54.Yim HC, Wang D, Yu L, White CL, Faber PW, Williams BR, Sadler AJ. 2016. The kinase activity of PKR represses inflammasome activity. Cell Res 26:367–379. doi: 10.1038/cr.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Felicetti L, Colombo B, Baglioni C. 1966. Inhibition of protein synthesis in reticulocytes by antibiotics. II. The site of action of cycloheximide, streptovitacin A and pactamycin. Biochim Biophys Acta 119:120–129. doi: 10.1016/0005-2787(66)90044-X. [DOI] [PubMed] [Google Scholar]
- 56.Kearse MG, Green KM, Krans A, Rodriguez CM, Linsalata AE, Goldstrohm AC, Todd PK. 2016. CGG repeat-associated non-AUG translation utilizes a cap-dependent scanning mechanism of initiation to produce toxic proteins. Mol Cell 62:314–322. doi: 10.1016/j.molcel.2016.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cooper GM. 2000. Protein degradation, the cell: a molecular approach, 2nd ed Sinauer Associates, Sunderland, MA. [Google Scholar]
- 58.Marwaha R, Sharma M. 5 October 2017, posting date. DQ-Red BSA trafficking assay in cultured cells to assess cargo delivery to lysosomes. Bio Protoc doi: 10.21769/BioProtoc.2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Palm W, Park Y, Wright K, Pavlova NN, Tuveson DA, Thompson CB. 2015. The utilization of extracellular proteins as nutrients is suppressed by mTORC1. Cell 162:259–270. doi: 10.1016/j.cell.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guo N, Peng Z. 2013. MG132, a proteasome inhibitor, induces apoptosis in tumor cells. Asia Pac J Clin Oncol 9:6–11. doi: 10.1111/j.1743-7563.2012.01535.x. [DOI] [PubMed] [Google Scholar]
- 61.Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP. 2011. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets 11:239–253. doi: 10.2174/156800911794519752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Berkers CR, Verdoes M, Lichtman E, Fiebiger E, Kessler BM, Anderson KC, Ploegh HL, Ovaa H, Galardy PJ. 2005. Activity probe for in vivo profiling of the specificity of proteasome inhibitor bortezomib. Nat Methods 2:357–362. doi: 10.1038/nmeth759. [DOI] [PubMed] [Google Scholar]
- 63.Ciechanover A, Heller H, Elias S, Haas AL, Hershko A. 1980. ATP-dependent conjugation of reticulocyte proteins with the polypeptide required for protein degradation. Proc Natl Acad Sci U S A 77:1365–1368. doi: 10.1073/pnas.77.3.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hershko A, Ciechanover A, Heller H, Haas AL, Rose IA. 1980. Proposed role of ATP in protein breakdown: conjugation of protein with multiple chains of the polypeptide of ATP-dependent proteolysis. Proc Natl Acad Sci U S A 77:1783–1786. doi: 10.1073/pnas.77.4.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gilson T, Blanchette P, Ballmann MZ, Papp T, Pénzes JJ, Benkő M, Harrach B, Branton PE. 2016. Using the E4orf6-Based E3 ubiquitin ligase as a tool to analyze the evolution of adenoviruses. J Virol 90:7350–7367. doi: 10.1128/JVI.00420-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bernardi KM, Williams JM, Inoue T, Schultz A, Tsai B. 2013. A deubiquitinase negatively regulates retro-translocation of nonubiquitinated substrates. Mol Biol Cell 24:3545–3556. doi: 10.1091/mbc.E13-06-0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Smith K, Ying B, Ball AO, Beard CW, Spindler KR. 1996. Interaction of mouse adenovirus type 1 early region 1A protein with cellular proteins pRb and p107. Virology 224:184–197. doi: 10.1006/viro.1996.0520. [DOI] [PubMed] [Google Scholar]
- 68.Mayne LV. 1984. Inhibitors of DNA synthesis (aphidicolin and araC/HU) prevent the recovery of RNA synthesis after UV-irradiation. Mutat Res 131:187–191. [DOI] [PubMed] [Google Scholar]
- 69.Zittoun J, Marquet J, David JC, Maniey D, Zittoun R. 1989. A study of the mechanisms of cytotoxicity of Ara-C on three human leukemic cell lines. Cancer Chemother Pharmacol 24:251–255. [DOI] [PubMed] [Google Scholar]
- 70.Zhou A, Paranjape JM, Der SD, Williams BR, Silverman RH. 1999. Interferon action in triply deficient mice reveals the existence of alternative antiviral pathways. Virology 258:435–440. doi: 10.1006/viro.1999.9738. [DOI] [PubMed] [Google Scholar]
- 71.Ma Y, Mathews MB. 1996. Structure, function, and evolution of adenovirus-associated RNA: a phylogenetic approach. J Virol 70:5083–5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yoon CH, Lee ES, Lim DS, Bae YS. 2009. PKR, a p53 target gene, plays a crucial role in the tumor-suppressor function of p53. Proc Natl Acad Sci U S A 106:7852–7857. doi: 10.1073/pnas.0812148106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baugh JM, Viktorova EG, Pilipenko EV. 2009. Proteasomes can degrade a significant proportion of cellular proteins independent of ubiquitination. J Mol Biol 386:814–827. doi: 10.1016/j.jmb.2008.12.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Maupin-Furlow J. 2011. Proteasomes and protein conjugation across domains of life. Nat Rev Microbiol 10:100–111. doi: 10.1038/nrmicro2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kring SC, Ball AO, Spindler KR. 1992. Transcription mapping of mouse adenovirus type 1 early region 4. Virology 190:248–255. doi: 10.1016/0042-6822(92)91211-C. [DOI] [PubMed] [Google Scholar]
- 76.Querido E, Blanchette P, Yan Q, Kamura T, Morrison M, Boivin D, Kaelin WG, Conaway RC, Conaway JW, Branton PE. 2001. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev 15:3104–3117. doi: 10.1101/gad.926401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Harada JN, Shevchenko A, Shevchenko A, Pallas DC, Berk AJ. 2002. Analysis of the adenovirus E1B-55K-anchored proteome reveals its link to ubiquitination machinery. J Virol 76:9194–9206. doi: 10.1128/JVI.76.18.9194-9206.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Forrester NA, Patel RN, Speiseder T, Groitl P, Sedgwick GG, Shimwell NJ, Seed RI, Catnaigh PO, McCabe CJ, Stewart GS, Dobner T, Grand RJ, Martin A, Turnell AS. 2012. Adenovirus E4orf3 targets transcriptional intermediary factor 1gamma for proteasome-dependent degradation during infection. J Virol 86:3167–3179. doi: 10.1128/JVI.06583-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sohn SY, Hearing P. 2016. The adenovirus E4-ORF3 protein functions as a SUMO E3 ligase for TIF-1gamma sumoylation and poly-SUMO chain elongation. Proc Natl Acad Sci U S A 113:6725–6730. doi: 10.1073/pnas.1603872113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Weber J. 1976. Genetic analysis of adenovirus type 2 III. Temperature sensitivity of processing viral proteins. J Virol 17:462–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Anderson CW, Baum PR, Gesteland RF. 1973. Processing of adenovirus 2-induced proteins. J Virol 12:241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Weber JM, Anderson CW. 1988. Identification of the gene coding for the precursor of adenovirus core protein X. J Virol 62:1741–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Anderson CW. 1990. The proteinase polypeptide of adenovirus serotype 2 virions. Virology 177:259–272. doi: 10.1016/0042-6822(90)90479-B. [DOI] [PubMed] [Google Scholar]
- 84.Sanfilippo CM, Chirimuuta FN, Blaho JA. 2004. Herpes simplex virus type 1 immediate-early gene expression is required for the induction of apoptosis in human epithelial HEP-2 cells. J Virol 78:224–239. doi: 10.1128/JVI.78.1.224-239.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (ed). 2011. Virus Taxonomy. Classification and of nomenclature viruses. Ninth report of the International Committee on Taxonomy of Viruses, p 128 Elsevier Academic Press, San Diego, CA. [Google Scholar]
- 86.Ashley SL, Welton AR, Harwood KM, Van Rooijen N, Spindler KR. 2009. Mouse adenovirus type 1 infection of macrophages. Virology 390:307–314. doi: 10.1016/j.virol.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sawicki DL, Silverman RH, Williams BR, Sawicki SG. 2003. Alphavirus minus-strand synthesis and persistence in mouse embryo fibroblasts derived from mice lacking RNase L and protein kinase R. J Virol 77:1801–1811. doi: 10.1128/JVI.77.3.1801-1811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nakamura T, Kunz RC, Zhang C, Kimura T, Yuan CL, Baccaro B, Namiki Y, Gygi SP, Hotamisligil GS. 2015. A critical role for PKR complexes with TRBP in immunometabolic regulation and eIF2alpha phosphorylation in obesity. Cell Rep 11:295–307. doi: 10.1016/j.celrep.2015.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cauthen AN, Welton AR, Spindler KR. 2007. Construction of mouse adenovirus type 1 mutants. Methods Mol Med 130:41–59. [DOI] [PubMed] [Google Scholar]
- 90.Kajon AE, Brown CC, Spindler KR. 1998. Distribution of mouse adenovirus type 1 in intraperitoneally and intranasally infected adult outbred mice. J Virol 72:1219–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cauthen AN, Spindler KR. 1999. Novel expression of mouse adenovirus type 1 early region 3 gp11K at late times after infection. Virology 259:119–128. doi: 10.1006/viro.1999.9713. [DOI] [PubMed] [Google Scholar]
- 92.Schindelin J, Rueden CT, Hiner MC, Eliceiri KW. 2015. The ImageJ ecosystem: an open platform for biomedical image analysis. Mol Reprod Dev 82:518–529. doi: 10.1002/mrd.22489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sheets MD, Fritz B, Hartley RS, Zhang Y. 2010. Polyribosome analysis for investigating mRNA translation in Xenopus oocytes, eggs and embryos. Methods 51:152–156. doi: 10.1016/j.ymeth.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Morita M, Alain T, Topisirovic I, Sonenberg N. 2013. Polysome profiling analysis. Bio Protoc 3:e833. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PKR is depleted at MOIs of 2, 5, and 10. C57BL/6 MEFs were infected with MAV-1 at an MOI of 2, 5, or 10 or were subjected to mock infection (mock). Cell lysates were analyzed by immunoblotting with antibodies for PKR (B-10) and actin. Data from densitometry quantitations performed using ImageJ are listed below each lane; for each time point, the infected samples were normalized to the mock-infected samples. The PKR blot image was uniformly adjusted to a brightness of 150 in Adobe Photoshop. Download FIG S1, PDF file, 2.2 MB (2.3MB, pdf) .
Copyright © 2019 Goodman et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Ribosome pelleting. To confirm that most ribosomes ended up in the pellet after centrifugation through sucrose (Fig. 4A), a sample of the pellet and a sample of the corresponding supernatant were analyzed by immunoblotting with antibodies for RPL7 (ribosomal protein L7; Abcam catalog no. ab72550) (1:2,000). Download FIG S2, PDF file, 3.2 MB (3.2MB, pdf) .
Copyright © 2019 Goodman et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
PKR mRNA 5′ UTR presence does not result in altered reporter protein levels upon MAV-1 infection. (A) C57BL/6 MEFs or (B) CMT93 cells were cotransfected with pmPKR5UTRfullNL or AUG-NL-3xFLAG and pGL4.13 using jetPRIME reagents (Polyplus catalog no. 114-15) and the standard Polyplus protocol, with 200 ng total of plasmid and 300 µl of jetPRIME reagent per 35-mm-diameter well. At 24 h after transfection, the cells were infected with MAV-1 at an MOI of 10. At 24 hpi, cells were lysed in Glo lysis buffer (Promega Corp.) (70 µl/well). After lysing, 25 µl of each lysed sample and 25 µl of OneGlo or NanoGlo (Promega Corp.) were added to two wells in a black 96-well plate (Fisher Scientific catalog no. 07-000-634). After 5 min, the plate was read on a Promega GloMax luminometer. Levels of relative light units from the pmPKR5UTRfullNL plasmid were normalized to the firefly luciferase and positive-control plasmids. Graphs are representative of 7 to 9 biological replicates per treatment group. Download FIG S3, PDF file, 0.1 MB (71.5KB, pdf) .
Copyright © 2019 Goodman et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Treatment with MG132 and bortezomib does not affect MAV-1 replication at 24 hpi. C57BL/6 MEFs were infected with MAV-1 at an MOI of 10 and treated with DMSO (vehicle for inhibitors) or with 1 µM MG132 or bortezomib and were collected at 24 hpi. DNA was purified from cell pellets and analyzed for MAV-1 genome copies by qPCR. The graph is representative of results from five biological replicates per treatment group. Error bars represent standard errors of the means (SEM). *, P ≤ 0.05. Download FIG S4, PDF file, 0.1 MB (63.7KB, pdf) .
Copyright © 2019 Goodman et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
PKR is not detectably ubiquitinated during MAV-1 infection. CMT93 cells were transfected with a GFP-ubiquitin plasmid using standard Polyplus transfection protocols. At 24 h posttransfection (hpt), the cells were infected with MAV-1 (MAV) at an MOI of 5 or were mock infected (mock) and treated with 10 µM MG132 at 6 hpi. Cell lysates were collected at 12 hpi and immunoprecipitated with (A) PKR (D-20) or an isotype control antibody or (B) p53 (DO-1) or an isotype control antibody. Immunoprecipitated samples were analyzed by immunoblotting with GFP, ubiquitin, isotype, PKR (B-10), or p53 (1801) antibodies. The input lane (mock input) contained a 0.008 volume of mock-infected lysate (relative to the volume in the immunoprecipitations). For panel B, GFP and p53 were probed on separate duplicate blots. Download FIG S5, PDF file, 6.8 MB (7MB, pdf) .
Copyright © 2019 Goodman et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Viral DNA replication can be detected at 24 hpi by qPCR. CMT93 cells were infected with MAV-1 (MAV) at an MOI of 10 and collected every 6 h for 24 h. DNA was purified from cell pellets and analyzed for MAV-1 genome copies by qPCR. The graph is representative of results from four to five biological replicates per treatment group. Error bars represent standard errors of the means (SEM). Download FIG S6, PDF file, 0.1 MB (60.2KB, pdf) .
Copyright © 2019 Goodman et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
UV-inactivated virus does not replicate viral DNA. (A) C57BL/6 MEFs or (B) CMT93 cells were infected with WT MAV-1 (WT) or UV-inactivated MAV-1 (UV) at an MOI of 10 and collected at the indicated times. DNA was purified from cell pellets and analyzed for MAV-1 genome copies by qPCR. Graphs are representative of results from three to four biological replicates per treatment group. Error bars represent standard errors of the means (SEM). Download FIG S7, PDF file, 0.1 MB (87.5KB, pdf) .
Copyright © 2019 Goodman et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
AraC treatment inhibited late protein expression. CMT93 cells were infected with WT MAV-1 (MAV) at an MOI of 10 or were mock infected (mock). Infected cells were also treated (+) or not (-) with 20 µg/ml cytosine arabinoside (araC), an inhibitor of DNA synthesis. Cell lysates were analyzed with antibodies for late virion proteins (AKO1-103; 1:1,000). Arrowheads indicate late viral proteins. Download FIG S8, PDF file, 2.3 MB (2.3MB, pdf) .
Copyright © 2019 Goodman et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.