

Plague is a rapidly lethal human disease caused by the bacterium Yersinia pestis. This study demonstrated that the Y. pestis plasminogen activator Pla, a protease that promotes fibrin degradation, thwarts T cell-mediated defense against fully virulent Y. pestis.
KEYWORDS: YopE, Pla, plasminogen activator, T cell-mediated protection, Yersinia pestis, fibrin, fibrin degradation, fibrinolysis, immunization, plague
ABSTRACT
Plague is a rapidly lethal human disease caused by the bacterium Yersinia pestis. This study demonstrated that the Y. pestis plasminogen activator Pla, a protease that promotes fibrin degradation, thwarts T cell-mediated defense against fully virulent Y. pestis. Introducing a single point mutation into the active site of Pla suffices to render fully virulent Y. pestis susceptible to primed T cells. Mechanistic studies revealed essential roles for fibrin during T cell-mediated defense against Pla-mutant Y. pestis. Moreover, the efficacy of T cell-mediated protection against various Y. pestis strains displayed an inverse relationship with their levels of Pla activity. Together, these data indicate that Pla functions to thwart fibrin-dependent T cell-mediated defense against plague. Other important human bacterial pathogens, including staphylococci, streptococci, and borrelia, likewise produce virulence factors that promote fibrin degradation. The discovery that Y. pestis thwarts T cell defense by promoting fibrinolysis suggests novel therapeutic approaches to amplifying T cell responses against human pathogens.
INTRODUCTION
Plague is a rapidly lethal human disease. Outbreaks of plague have caused several major pandemics, including Europe’s devastating Black Death (1). The etiologic agent of plague is Yersinia pestis, a Gram-negative bacterium transmitted by fleas to rodents and humans. Experimental infection of mice with Y. pestis bacilli provides an opportunity to study immune defense against one of the world’s most lethal human pathogens in a natural host. The bacteria grow logarithmically to high titer after their inoculation into mice, resulting in death within a few days (2–5). Mechanistic studies have established that Y. pestis dampens inflammation, incapacitates phagocytes, and fully thwarts innate immune defense (6–9).
Infection by fleabite typically leads to the bubonic form of human plague, characterized by painful swelling of lymph nodes draining the site of infection (1). When bubonic plague is left untreated, bacilli can disseminate to the blood, causing septicemic plague. If bacilli colonize the lungs, the disease may progress to pneumonic plague, which is particularly fulminant and can be spread from person to person via infectious respiratory droplets (1, 2). Purposeful dissemination of weaponized Y. pestis is a prominent bioterrorism concern given that antibiotic-resistant Y. pestis strains exist and that there is no licensed vaccine against plague in the United States (10–12). A live attenuated vaccine is available for human use in Russia and China but not elsewhere (13, 14). It induces robust antibody and T cell responses (15), but safety concerns have prevented its approval in Western countries (16).
Y. pestis-specific antibodies can protect against pneumonic plague in rodent models (9, 13). However, robust antibody-mediated protection is more difficult to demonstrate in nonhuman primate models (11, 17, 18). It is not known whether Y. pestis-specific antibodies would suffice to protect humans (9, 11, 12).
Cellular immunity can be sufficient to protect against pneumonic plague in rodent models that challenge the rodents with D27, a modestly attenuated laboratory strain of Y. pestis (19–21). For example, immunizing B cell-deficient μMT mice with D27-pLpxL, a highly attenuated vaccine strain, confers robust T cell-mediated protection against lethal intranasal challenge with D27 (20–22). Much of this protection is mediated by CD8 T cells recognizing a dominant antigen encoded by the Y. pestis YopE gene and restricted by the murine Kb major histocompatibility complex (MHC) molecule (19). Indeed, immunizing C57BL/6 wild-type (WT) mice with a 9-amino-acid YopE69−77 peptide primes YopE-specific CD8 T cells that protect against lethal intranasal challenge with D27. The protection mediated by YopE69−77 immunization involves production of tumor necrosis factor alpha (TNF-α) and gamma interferon (IFN-γ) by CD8 T cells but not perforin-dependent cytotoxicity (23). Immunization with YopE69−77 also protects mice against lethal intragastric or intravenous challenge with fully virulent Yersinia pseudotuberculosis (23–25), a cause of severe human gastroenteritis (26). Despite providing robust protection against fully virulent Y. pseudotuberculosis strains and attenuated Y. pestis strain D27, immunization with YopE69−77 delays time to mortality only minimally after challenge of mice with fully virulent Y. pestis strains, such as CO92 (23).
Y. pestis and Y. pseudotuberculosis are closely related at the genetic level (26, 27). However, Y. pestis has acquired an additional plasmid, pPCP1, which contributes to its exceptional virulence (27). pPCP1 encodes Pla, a protease expressed on the outer membrane of Y. pestis bacilli that activates mammalian plasminogen (1, 28, 29). Pla is critical for the lethality of subcutaneous Y. pestis infections in naive mice but is dispensable for lethality when bacilli are administered intravenously or intranasally (29–33).
The activation of plasminogen by Pla generates plasmin, an enzyme that degrades fibrin. Fibrin is a provisional extracellular matrix laid down at sites of trauma and infection (34). Interestingly, fibrin plays critical roles during T cell-mediated defense against Y. pestis strain D27 in mice; genetic or pharmacologic depletion of fibrin abrogates YopE69−77 immunization-induced protection (35).
Given that fibrin facilitates T cell-mediated defense against Y. pestis and that Y. pestis produces Pla, a factor that promotes fibrin degradation (i.e., fibrinolysis), studies were designed to investigate whether Pla functions to antagonize T cell-mediated defense against plague. The data presented here establish that the presence of primed CD8 T cells can suffice to protect against a lethal dose (LD) of a virulent Y. pestis strain rendered deficient in Pla activity. Further, this defense is fibrin dependent and the efficacy of CD8 T cell-mediated defense against Y. pestis strains shows an inverse relationship with the level of Pla activity elaborated by the strains. These data demonstrate that Pla thwarts fibrin-dependent CD8 T cell-mediated defense against virulent Y. pestis and suggest that suppressing Pla-induced fibrinolysis may constitute a novel therapeutic means to amplifying T cell defense against plague, a strategy which may have broad applicability as a countermeasure against other human pathogens that antagonize fibrin.
RESULTS
T cells suffice to protect against Y. pestis when Pla is inactivated.
T cells effectively protect against intranasal challenge with modestly attenuated, pigmentation locus-negative (pgm−) Y. pestis strain D27 (19–21). To evaluate the capacity of T cells to defend against fully virulent Y. pestis, vaccination experiments were performed using intranasal challenge with fully virulent pgm-positive (pgm+) Y. pestis strain CO92. B cell-deficient μMT mice were immunized with D27-pLpxL, or WT C57BL/6 mice were immunized with YopE69−77 peptide. Both of these immunization regimens prime robust CD8 T cell responses that suffice to dramatically improve the overall survival rates of mice challenged intranasally with D27 (19, 20). Consistent with a prior study (23), these regimens marginally extended the median survival time of mice challenged intranasally with CO92 but did not substantially improve their overall survival to day 20 (Fig. 1A and B).
FIG 1.

T cells protect against Pla-deficient Y. pestis. (A, C, and E) B cell-deficient μMT (μMT) mice were left unvaccinated (unvac) or were primed and boosted intranasally with D27-pLpxL. (B, D, and F) WT mice were immunized intranasally with CT adjuvant alone (CT) or CT mixed with YopE69−77 peptide (CT+YopE) or control OVA257–264 peptide (CT+OVA). Mice were then challenged intranasally with approximately 10 MLD strain CO92 (1 × 104 CFU; A and B), strain CO92 ΔPla (1 × 105 CFU; C and D), or strain CO92 Pla-D206A (1.5 × 104 CFU; E and F). In comparison with unimmunized mice, immunized mice exhibited significantly increased survival after Pla-deficient CO92 challenge. Data for all panels are representative of results from 2 independent experiments (ns, not significant).
Prior studies established that fibrin is essential for T cell-mediated defense against Y. pestis D27 (35). Y. pestis produces Pla, a factor that promotes fibrin degradation. To assess whether Pla contributes to the relative inability of T cells to combat fully virulent Y. pestis, mice were immunized to prime Y. pestis-specific T cells and were then challenged intranasally with isogenic CO92 strains that either lack expression of the Pla protein (strain CO92 ΔPla) or carry a single point mutation in the active site of Pla that specifically abolishes its plasminogen-activating protease activity (strain CO92 Pla-D206A) (33). These Pla-deficient strains are known to be attenuated when administered subcutaneously but to retain exceptional virulence when administered intranasally or intravenously (29–33). Indeed, the median lethal dose (MLD) of intranasally administered strain CO92 Pla-D206A was indistinguishable from that of CO92 (102.7 ± 100.8 and 103.0 ± 100.3 CFU, respectively), and that of strain CO92 ΔPla was only slightly higher (103.8 ± 100.4 CFU). While the D27-pLpxL-immunized μMT mice (Fig. 1A) as well as YopE-immunized WT mice (Fig. 1B) were poorly protected against challenge with CO92, they readily withstood challenge with either Pla-mutant CO92 strain (Fig. 1C to F). These data demonstrate that T cells can suffice to protect against Pla-deficient Y. pestis and suggest that Pla functions to thwart T cell defense against Y. pestis.
T cell protection against Pla-deficient Y. pestis is fibrin dependent.
The Pla protein expressed by Y. pestis activates mammalian plasminogen to generate plasmin, a protease that degrades fibrin (36). However, Pla also has other substrates, including metalloproteinases (28, 37), and plasmin can degrade other extracellular matrix proteins (38, 39). To assess whether CD8 T cell-mediated defense against CO92 Pla-D206A is fibrin dependent, mice with deficiencies in fibrin production were evaluated. First, gene-targeted fibrinogen-deficient (fibrinogen knockout [FibKO]) mice and littermate control fibrinogen-heterozygous (FibHet) mice were immunized with YopE69−77 and then challenged intranasally with CO92 Pla-D206A. Prior studies established that immunization with YopE69−77 primes similar levels of YopE-specific CD8 T cells in FibHet and FibKO mice (35). While YopE-immunized FibHet mice were well protected against CO92 Pla-D206A, almost all YopE-immunized FibKO mice succumbed (Fig. 2A), indicating that CD8 T cell-mediated defense against Pla-mutant Y. pestis requires fibrinogen.
FIG 2.

T cell-mediated protection against Pla-deficient Y. pestis is fibrin dependent. Fibrinogen-deficient mice (FibKO) and littermate control heterozygous mice (FibHet) (A), mice expressing very low levels of tissue factor activity (mTFKO) and littermate control heterozygous mice (mTFHet) (B), PAI-1/TAFI-deficient mice (P/T DKO) (C), and WT mice treated with Coumadin (D) were immunized intranasally with CT+YopE and then challenged intranasally with 10 MLD CO92 Pla-D206A. In comparison with fibrin-sufficient mice, fibrin-deficient mice exhibited significantly decreased survival after Pla-D206A CO92 challenge. Survival rates for control mice that received CT adjuvant only or CT+OVA in each genotype or treatment were 0% to 20% (not shown). Data for all panels are pooled from 2 to 3 independent experiments.
The fibrinogen dependency of T cell defense against CO92 Pla-D206A could potentially reflect activities that are mediated by fibrinogen, the precursor to fibrin, but are independent of fibrin per se. To further establish a role of fibrin, mice possessing normal levels of fibrinogen but reduced capacities to generate fibrin were examined. Fibrin is generated when thrombin cleaves fibrinogen (34), and thrombin is generated by coagulation pathways activated by tissue factor (TF) (40). Gene-targeted mice lacking expression of the mouse TF gene and instead expressing a human TF transgene produce very low levels of TF activity and thus display a decreased capacity for thrombin-initiated conversion of fibrinogen to fibrin (41–43). Reduced fibrin production is also observed in mice with increased fibrinolysis, such as those rendered deficient with respect to both plasminogen activator inhibitor 1 (PAI-1) and thrombin-activatable fibrinolysis inhibitor (TAFI) (43, 44). As shown in Fig. 2B and C, the protective capacity of YopE-specific CD8 T cells against CO92 Pla-D206A was abrogated in mice with low levels of TF activity (mTFKO mice; Fig. 2B) as well as in PAI-1/TAFI double knockout (P/T DKO) mice (Fig. 2C). In addition, YopE-specific CD8 T cells poorly protected WT mice treated with Coumadin (Fig. 2D), a pharmacologic anticoagulant that reduces production of fibrin (45). Taken together, these results indicate that fibrin plays critical roles during CD8 T cell-mediated defense against CO92 Pla-D206A and suggest that Pla functions to thwart fibrin-dependent T cell-mediated defense against plague.
Although the MLDs of intranasally administered mutants CO92 ΔPla and CO92 Pla-D206A were similar to those seen with strain CO92, the diseases induced by these strains differ somewhat, with slightly delayed mortality in the setting of Pla-deficient Y. pestis (33) (Fig. 1). Indeed, the mortality caused by Pla-deficient Y. pestis may be due to systemic dissemination of bacteria rather than to pneumonia (33). Accordingly, differences in pathogenesis, rather than differences in fibrin-dependent immune defense, could potentially contribute to the different efficacies of YopE-specific CD8 T cells after intranasal challenge with CO92 and Pla-mutant CO92. To address this possibility, follow-up studies employed an intravenous model of septicemic plague, which eliminates any potential differences in dissemination and where the virulence of CO92 and Pla-deficient CO92 are indistinguishable (29–31).
Consistent with a prior report (31), the MLDs for intravenously administered strains CO92, CO92 ΔPla, and CO92 Pla-D206A were found to be nearly identical in naive WT mice (approximately 10 CFU). Moreover, control mice immunized with adjuvant alone or adjuvant and an irrelevant OVA257–264 peptide succumbed with indistinguishable kinetics after intravenous challenge with each of these strains (Fig. 3A to C). YopE immunization failed to protect mice against intravenous challenge with strain CO92 (Fig. 3A); however, it readily protected against intravenous challenge with strain CO92 ΔPla (Fig. 3B) or strain CO92 Pla-D206A (Fig. 3C). These findings are consistent with those observed after intranasal challenge (Fig. 1) and indicate that T cells suffice to protect against systemic challenge with Pla-deficient Y. pestis. Moreover, the T cell-mediated protection against systemic challenge is also dependent on fibrin, as it was abrogated in FibKO mice (Fig. 3D), mTFKO mice, and P/T DKO mice (Fig. 3E).
FIG 3.

T cells protect against systemic challenge with Pla-deficient Y. pestis in a fibrin-dependent manner. WT (A to C), FibKO and FibHet (D), or mTFKO, mTFHet, and P/T DKO (E) mice were immunized intranasally with CT, CT+YopE, or CT+OVA and then challenged intravenously with 10 MLD strain CO92 (A), strain CO92 ΔPla (B), or strain CO92 Pla-D206A (C to E). YopE immunization provides fibrin-sufficient mice with significant protection against systemic infection with Pla-deficient Y. pestis. Data for all panels are pooled from 2 to 3 independent experiments (ns, not significant).
T cell-mediated protection against plague shows an inverse relationship with Y. pestis Pla activity.
Prior studies and the data presented thus far establish that T cells can effectively combat Y. pestis strains D27 (19–21), CO92 ΔPla (Fig. 1C and D; see also Fig. 3B), and CO92 Pla-D206A (Fig. 1E and F; see also Fig. 3C) but not strain CO92 (Fig. 1A and B; see also Fig. 3A) (23). Since the Pla activity of D27 had not been evaluated previously, it was of interest to assess whether there might be correlations between Pla activity and T cell-mediated defense against Y. pestis. In vitro comparison of plasminogen-activating capacity showed CO92 to have far greater activity than D27 (Fig. 4A). As expected, the Pla-mutant CO92 ΔPla and CO92 Pla-D206A strains exhibited minimal activity (Fig. 4A). Notably, a pgm-negative strain of CO92 (CO92 pgm−) also exhibited reduced plasminogen-activating capacity compared with CO92 (Fig. 4A), and YopE-specific CD8 T cells sufficed to protect against lethal intranasal challenge with CO92 pgm− (Fig. 4B). The greater Pla activity of pgm-positive CO92 could not be ascribed to formation of bacterial aggregates (data not shown). Analyzing the data together, the rate at which YopE-immunized mice survived Y. pestis challenge revealed an evident inverse relationship with the Pla activity of the various challenge strains, with those exhibiting higher Pla activity in vitro thwarting T cell defense more effectively in vivo (Fig. 4C). These observations further implicate Y. pestis Pla as an antagonist of T cell-mediated defense against plague.
FIG 4.
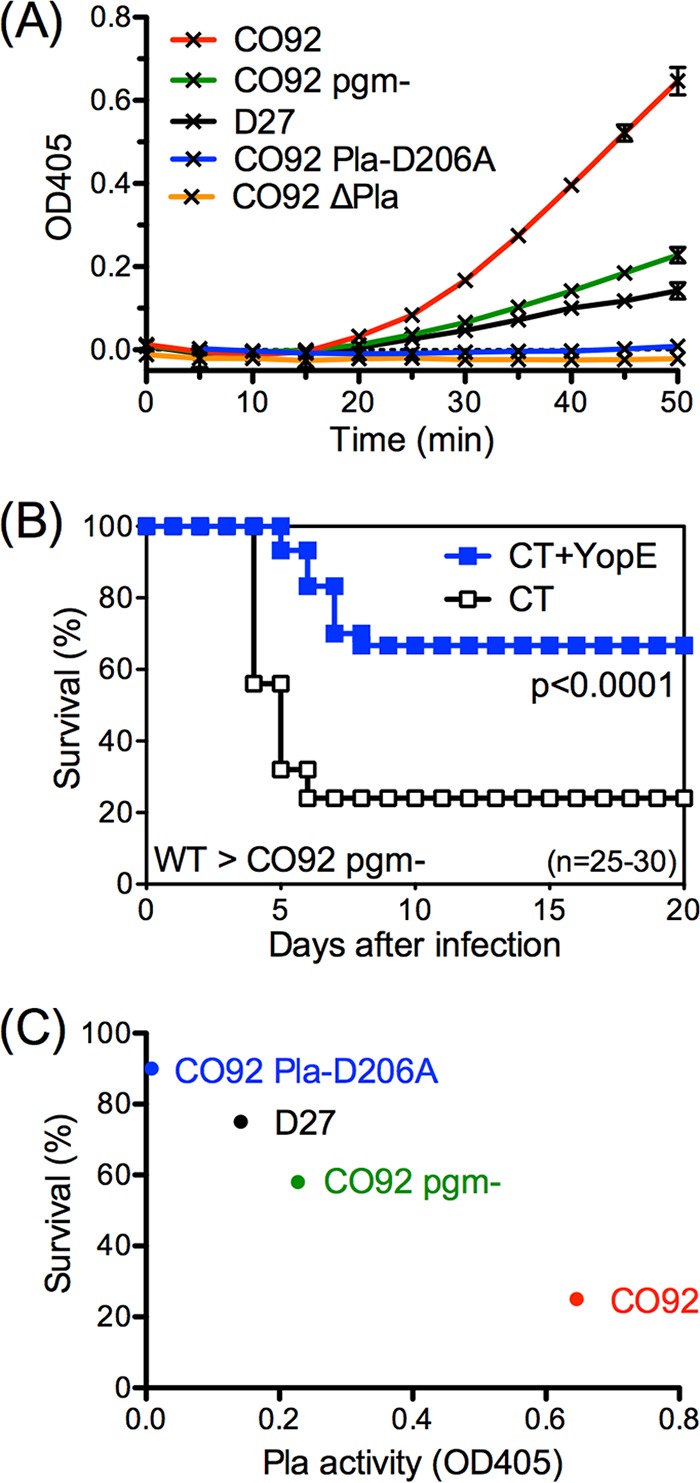
T cell-mediated protection against plague displays an inverse relationship with Y. pestis Pla activity. (A) Plasminogen-activating capacity of indicated Y. pestis strains measured in vitro using a chromogenic assay for plasmin and presented as the absorbance at 405 nm at the indicated time points. Data are representative of results from 2 independent experiments. (B) WT mice were immunized intranasally with CT or CT+YopE and challenged intranasally with 20 MLD strain CO92 pgm−. In comparison with control mice, YopE-immunized mice exhibited significantly increased survival after CO92 pgm− challenge. Data shown are pooled from 3 independent experiments. (C) Inverse relationship between Pla activity and the survival rate of YopE-immunized WT mice challenged intranasally with 10 to 20 MLD of the indicated Y. pestis strains. Survival data were pooled from 2 to 3 independent experiments. Pla activity data are representative of results from 2 independent experiments and are presented as the OD value measured at 50 min.
DISCUSSION
Despite steady advances in our understanding of immune defense, we have yet to harness that knowledge in the pursuit of vaccines for many virulent human pathogens. Highly effective antibody-based vaccines have been developed for some bacterial pathogens, but such approaches have failed for many others. When primed by vaccination or infection, antigen-specific T cells can be key orchestrators of effective immune defense. Not surprisingly, many successful pathogens have evolved mechanisms to overcome T cell-mediated defense. For example, viruses frequently encode proteins that downregulate host expression of class I MHC molecules, thereby preventing CD8 T cells from recognizing infected cells. Less is known about the mechanisms by which bacteria evade T cell-mediated defense.
Infection rapidly activates a robust acute-phase reaction in all animals. In vertebrates, this innately encoded response elevates circulating levels of fibrinogen, the precursor to fibrin, and PAI-1, a suppressor of fibrinolysis (46). This response facilitates the deposition of fibrin at sites of infection, where coagulation pathways are activated locally by pathogen-associated molecular pattern- and cytokine-mediated induction of TF (40). Although best known for its roles in blood clotting and wound healing, fibrin is also a ligand for a number of cell surface receptors that facilitate leukocyte adhesion and activation, including CD11b, CD11c, TLR4, and ICAM-1, suggesting roles during immune defense (34, 47–52).
Mouse studies have indicated that fibrin critically supports immune defense against a variety of human pathogens (35, 43, 45, 47, 53, 54). The underlying details remain to be fully elucidated, but there appear to be several distinct mechanisms. In the case of infection by the protozoan parasite Toxoplasma gondii, fibrin limits immunopathology that manifests as hemorrhagic bleeding and anemia in fibrin-deficient mice (53). Fibrin similarly suppresses hemorrhagic pathology during infection by the intracellular bacterium Listeria monocytogenes but also dramatically reduces bacterial burden in that setting (45). Fibrin-mediated control of bacterial burden is likewise observed in mice infected with extracellular bacteria such as Yersinia enterocolitica (43), group A streptococci (54), and Staphylococcus aureus (45, 47), as well as Y. pestis (35), which can replicate extracellularly or intracellularly.
The precise mechanisms by which fibrin limits bacterial burden have yet to be established. It is often hypothesized that fibrin physically traps bacteria at subcutaneous sites of infection, thereby limiting their dissemination (55, 56). In mouse models of bubonic plague, where Y. pestis bacteria are delivered subcutaneously or intradermally, deletion of Pla dramatically attenuates Y. pestis virulence (29–31). The Pla-mutant Y. pestis grows to high titer at the injection sites but fails to disseminate efficiently to draining lymph nodes or distal tissues (29–31). These findings are consistent with the hypothesis that bacteria such as Y. pestis produce fibrinolysis-promoting factors such as Pla to facilitate their escape from fibrin matrices at peripheral sites of infection. The observation that Pla-deficient Y. pestis regains high levels of virulence when inoculated subcutaneously into fibrinogen-deficient mice (57) further supports the hypothesis that Pla thwarts innate defense during bubonic plague by degrading fibrin matrices that act as physical barriers to dissemination.
Pla appears to play multiple roles that manifest differentially across the various forms of plague. While Pla is critical for pathogenesis during bubonic plague, it is dispensable for lethality in models of pneumonic plague, where Y. pestis is delivered intranasally or by aerosol. In those models, Pla-mutant Y. pestis levels are not attenuated by MLD analysis (32, 58). However, the mice inoculated intranasally with Pla-mutant Y. pestis succumbed with slightly delayed kinetics compared to CO92 inoculation (Fig. 1) and apparently succumbed due to bacterial dissemination and sepsis rather than pneumonia (33). Thus, unlike its dissemination from subcutaneous sites, dissemination of Y. pestis from the lung is not dependent on Pla.
Little is known about the function of bacterial fibrinolysis-promoting proteins in the context of adaptive immune defense. This report demonstrates key roles for Pla in thwarting fibrin-dependent T cell-mediated defense against Y. pestis using the intranasal model of pneumonic plague (Fig. 1 and 2), where lethality is associated with dissemination and sepsis. It also demonstrates critical roles for Pla in thwarting fibrin-dependent T cell-mediated defense against intravenously delivered Y. pestis (Fig. 3), where the obstacles to dissemination are experimentally bypassed and where Pla-sufficient and Pla-deficient Y. pestis strains are equally virulent (29–31) and kill mice with similar kinetics (Fig. 3). Together, these observations suggest that Pla functions to thwart T cell defense against disseminated Y. pestis bacteria.
Prior studies using other challenge models suggested that fibrin can be essential for innate immune defense against plague (29–31, 35). Indeed, fibrin, in conjunction with cytokines produced by CD8 T cells, helps phagocytes to withstand encounters with Y. pestis in hepatic tissue (23, 35). Immunohistologic observations suggested that phagocytes are recruited to sites of hepatic infection in both fibrin-deficient and fibrin-sufficient mice but that the phagocytes fail to clear bacteria in fibrin-deficient mice (35). Instead, the phagocytes succumb to the bacteria in fibrin-deficient mice. One hypothesis is that the deposition of fibrin matrices at sites of hepatic infection functions to ligate signaling receptors on phagocytes, such as the integrins CD11b and CD11c, thus transmitting signals that enhance cell survival and/or increase expression of bactericidal activities. In this model, fibrin may specifically flag sites of infection and license cytokine-activated phagocytes to deploy their destructive arsenals at these sites. In response, bacteria apparently evolved to promote fibrinolysis as a means to thwart fibrin-dependent immunity.
This report does not establish or exclude specific roles for phagocytes or other elements of innate immunity in fibrin-dependent T cell-mediated defense against Pla-deficient Y. pestis. However, fibrin-dependent innate defense is not sufficient to protect against Pla-deficient Y. pestis since naive fibrin-sufficient and fibrin-deficient mice both rapidly succumbed with indistinguishable kinetics to intravenous or intranasal challenge with D206A (data not shown). Notably, prior studies indicated that the failure of T cell-mediated defense against plague in fibrin-deficient mice did not result from the differences in the number or function of YopE-specific CD8 T cells primed in the FibHet and FibKO mice (35).
In further support of the hypothesis that Pla thwarts T cell defense against Y. pestis, this report reveals an inverse relationship between the efficacy of T cell-mediated protection and the levels of Pla activity. Y. pestis strains exhibiting higher Pla activity in vitro overcome T cell defense more effectively in vivo (Fig. 4). It is notable and interesting that both pgm-negative strains (i.e., D27 and CO92 pgm−) expressed reduced Pla levels of activity in comparison to pgm-positive strain CO92 (Fig. 4A). The deletion of the pgm locus is commonly observed in passaged Y. pestis strains (59) and, like Pla deficiency, has been associated with the reduced virulence seen in mice when Y. pestis was inoculated subcutaneously in comparison to the results seen when the bacteria were inoculated intravenously. The pgm locus has not previously been associated with Pla activity. Further studies will be required to determine whether one or more proteins encoded by the pgm locus might interact with Pla directly or indirectly to enhance its activity.
This study demonstrated that Pla suppresses the capacity of T cells to protect mice against plague. The precise mechanisms by which T cells combat Y. pestis, and by which Pla thwarts T cell defense, have yet to be established. Nevertheless, prior studies, along with the observations presented here, suggest that antigen-specific T cells produce cytokines that activate phagocytes and that fibrin then licenses the activated phagocytes to clear bacteria at sites of infection (23, 35). Y. pestis and other human pathogens, including streptococci, staphylococci, and borrelia, apparently evolved to evade this host response by robustly activating and exploiting fibrinolysis (38, 46, 56, 57). The fibrinolytic potency of these bacterial enzymes is evidenced by their use in human thrombolytic therapies, for example, streptokinase from Streptococcus pneumoniae and staphylokinase from Staphylococcus aureus (60, 61). Future studies should assess whether antagonists of infection-associated fibrinolysis can bolster T cell defense against Y. pestis and the many other important human pathogens that promote fibrinolysis.
MATERIALS AND METHODS
Mice.
C57BL/6 wild-type (WT) and B cell-deficient (μMT; B6.129S2-Igh-6tm1cgn) mice were originally obtained from the Jackson Laboratory (Bar Harbor, ME). Breeder stocks of fibrinogen-deficient mice were generously supplied by Jay L. Degen (Children’s Hospital Medical Center, Cincinnati, OH) (62). Breeder stocks of mice with very low levels of tissue factor (TF) activity were generously supplied by Nigel Mackman (University of North Carolina, Chapel Hill, NC) (41). These mice lack expression of mouse TF due to its inactivation by gene targeting and instead express a human TF transgene, which imparts low levels of TF activity (mTFKO; mTF−/− hTF+). Littermates expressing human TF that were heterozygous for mouse TF (mTFHet; mTF+/− hTF+) were used as controls. Plasminogen activator inhibitor 1 (PAI-1) and thrombin-activatable fibrinolysis inhibitor (TAFI) doubly deficient (P/T DKO) mice were generated at Trudeau Institute (43). All strains are on the C57BL/6 background and were bred in the specific-pathogen-free Trudeau Institute Animal Breeding Facility. Experimental mice were matched for age and sex, and they received their first immunization at between 6 and 10 weeks of age. The fibrinogen-deficient or TF transgene mice used for comparisons were cohoused with littermate controls, and investigators were blind with regard to the animals’ genotypes. Where indicated, WT mice were subjected to pharmacological anticoagulation by supplementation of drinking water with 2 mg/liter Coumadin [3-(α-acetonylbenzyl)-4-hydroxycoumarin; Sigma-Aldrich] beginning 3 days prior to infection, with replenishment every 48 h. This anticoagulant regimen reduces fibrin deposition in mice during infection (45).
All animal studies were conducted in accordance with the Guide for Care and Use of Laboratory Animals of the National Institutes of Health and were approved by Trudeau Institute Animal Care and Use Committee (IACUC protocols 02-022 and 02-161).
Bacteria.
Pigmentation locus (pgm)-deficient strain D27 was generously provided by Robert Brubaker (Michigan State University, East Lansing, MI). Highly attenuated vaccine strain D27-pLpxL was generated by transforming strain D27 with plasmid pLpxL (20), which was generously provided by Egil Lien (University of Massachusetts Medical School, Worcester, MA). The pgm-deficient variant of strain CO92 (CO92 pgm−) was generously provided by James B. Bliska (State University of New York, Stony Brook, NY). Fully virulent pgm-positive strain CO92 NR-641 was obtained through the NIH Biodefense and Emerging Infections Research Resources Repository, NIAID, NIH. Pla-deficient strains CO92 ΔPla and CO92 Pla-D206A were generously provided by Wyndham W. Lathem (33). Y. pestis bacilli from frozen stocks were grown overnight at 26°C in Bacto heart infusion broth (Difco Laboratories, Detroit, MI) with (strains D27, D27-pLpxL, and CO92 pgm−) or without (strains CO92, CO92 ΔPla, and CO92 Pla-D206A) 2.5 mM CaCl2. Cultures were diluted to an optical density (OD) of approximately 0.1 at 620 nm, regrown for 2.5 to 4 h at 26°C, quantified by OD measurement, washed with saline solution, and adjusted to the desired concentration. The levels of bacterial CFU in the inocula were confirmed by plating.
Measurement of median lethal dose (MLD).
For the MLD studies, 10-fold dilutions of bacteria ranging between 101 to 106 CFU were administered intranasally to WT naive C57BL/6 mice (5 mice per group in 2 independent experiments). The MLD was calculated by plotting the percentages of survival of mice at each dose for each bacterial strain against the log10 dose to obtain the data, which were then fitted to dose-response curves. The log10 MLD, log10 MLD standard error, log10 95% confidence interval for MLD, and n levels of doses for intranasally administered bacteria were as follows for each strain: for strain CO92, 2.95, 0.28, 2.2 to 3.7, and 9; for strain CO92 ΔPla, 3.79, 0.36, 2.96 to 4.62, and 12; for strain CO92 Pla-D206A, 2.71, 0.75, 0.64 to 4.78, and 8; and for strain CO92 pgm−, 4.13, 0.15, 3.7 to 4.55, and 8. Pairwise comparisons of the strains by one-way analysis of variance (ANOVA) followed by Tukey’s multiple-comparison test revealed no statistically significant differences between any pairs of strains.
Immunization and challenge.
Mice were lightly anesthetized by the use of isoflurane for intranasal administration. For immunization with D27-pLpxL, mice were primed with intranasal inoculation of 2 × 106 CFU D27-pLpxL in 30 μl saline solution, boosted with the same dose of D27-pLpxL 30 days later, and rested for 60 days before challenge. For peptide immunizations, mice were inoculated intranasally with a 15-μl solution containing 1 μg cholera toxin (CT; List Biological Laboratory, Campbell, CA) as the adjuvant and 1 μg YopE69−77 peptide (H2N-SVIGFIQRM-OH; New England Peptide, Gardner, MA) or control OVA257–264 peptide (H2N-SIINFEKL-OH). Mice were immunized on days 0, 7, and 21 and were challenged on day 37. For challenge, mice were infected intranasally with approximately 10 MLD strain CO92 (1 × 104 CFU), CO92 ΔPla (1 × 105 CFU), or CO92 Pla-D206A (1.5 × 104 CFU) or 20 MLD strain CO92 pgm− (2 × 105 CFU) mixed with 30 μl saline solution. In some experiments, mice were infected intravenously via the retro-orbital route with approximately 10 MLD (100 CFU) strain CO92, CO92 ΔPla, or CO92 Pla-D206A mixed with 100 μl saline solution. Mice were monitored at least once daily after infection. Unresponsive or recumbent mice were considered moribund and were euthanized.
Plasminogen activation assay.
To measure bacterial plasminogen-activating activity, Y. pestis bacilli were grown overnight at 26°C in Bacto heart infusion broth. Cultures were diluted to an OD of 0.05 at 620 nm and regrown at 37°C to the logarithmic-growth phase (range of OD at 620 nm, 0.55 to 0.65). Culture concentrations were estimated by interpolating from previously established strain-specific standard curves and were then diluted to 2.5 × 108 CFU/ml in a mixture containing 50 mM Tris and 100 mM NaCl (pH 7.4) and assay buffer containing 0.1% NaN3 to prevent further bacterial growth. Each reaction mixture was set up in triplicate on a 96-well microplate and contained 100 μl of bacterial suspension, 10 μl of 400 μg/ml human glu-plasminogen (Enzyme Research Labs, South Bend, IN), and 10 μl of 5 mM Spectrozyme PL plasmin substrate (Sekisui Diagnostics, Lexington, MA). Reaction mixtures were incubated for 1 h at 37°C with absorbance at 405 nm measured every 5 min in a Spectromax 190 microplate reader (Molecular Devices, San Jose, CA). Plate setup also included a parallel set of control samples containing no plasminogen, which were used to subtract out both bacterial turbidity and any direct impacts that the bacteria might have on the chromogenic substrate. Culture concentrations were confirmed by plating serially diluted culture aliquots that were taken from each strain prior to addition of NaN3.
Statistics.
Statistical analyses were performed using Prism 5 (GraphPad Software). Survival data were analyzed by log rank tests. Intranasal log10 MLD data were calculated by nonlinear regression and compared by one-way ANOVA followed by Tukey’s multiple-comparison test.
ACKNOWLEDGMENTS
We thank Lawrence Johnson for assistance with statistical analyses and critical reading of the manuscript and James Bliska, Robert Brubaker, Jay Degen, Egil Lien, Nigel Mackman, and Wyndham Lathem for providing bacterial strains, reagents, and/or mice. We thank the employees of the Trudeau Institute animal facilities for dedicated breeding and care of the mice used in this study. We acknowledge the Trudeau Institute Select Agent Program and personnel for enabling studies of the fully virulent Y. pestis.
This work was supported by funding from the Trudeau Institute (IHP-886 to S.T.S. and J.-S.L.) and by Public Health Service grants R01-AI061577 (to S.T.S. and J.-S.L.) and R01-AI071295 (to S.T.S.).
REFERENCES
- 1.Perry RD, Fetherston JD. 1997. Yersinia pestis–etiologic agent of plague. Clin Microbiol Rev 10:35–66. doi: 10.1128/CMR.10.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pechous RD, Sivaraman V, Stasulli NM, Goldman WE. 2016. Pneumonic plague: the darker side of Yersinia pestis. Trends Microbiol 24:190–197. doi: 10.1016/j.tim.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 3.Sebbane F, Gardner D, Long D, Gowen BB, Hinnebusch BJ. 2005. Kinetics of disease progression and host response in a rat model of bubonic plague. Am J Pathol 166:1427–1439. doi: 10.1016/S0002-9440(10)62360-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lathem WW, Crosby SD, Miller VL, Goldman WE. 2005. Progression of primary pneumonic plague: a mouse model of infection, pathology, and bacterial transcriptional activity. Proc Natl Acad Sci U S A 102:17786–17791. doi: 10.1073/pnas.0506840102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bubeck SS, Cantwell AM, Dube PH. 2007. Delayed inflammatory response to primary pneumonic plague occurs in both outbred and inbred mice. Infect Immun 75:697–705. doi: 10.1128/IAI.00403-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cornelis GR. 2002. Yersinia type III secretion: send in the effectors. J Cell Biol 158:401–408. doi: 10.1083/jcb.200205077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Viboud GI, Bliska JB. 2005. Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu Rev Microbiol 59:69–89. doi: 10.1146/annurev.micro.59.030804.121320. [DOI] [PubMed] [Google Scholar]
- 8.Heesemann J, Sing A, Trulzsch K. 2006. Yersinia's stratagem: targeting innate and adaptive immune defense. Curr Opin Microbiol 9:55–61. doi: 10.1016/j.mib.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 9.Smiley ST. 2008. Immune defense against pneumonic plague. Immunol Rev 225:256–271. doi: 10.1111/j.1600-065X.2008.00674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inglesby TV, Dennis DT, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Friedlander AM, Hauer J, Koerner JF, Layton M, McDade J, Osterholm MT, O'Toole T, Parker G, Perl TM, Russell PK, Schoch-Spana M, Tonat K. 2000. Plague as a biological weapon: medical and public health management. JAMA 283:2281–2290. doi: 10.1001/jama.283.17.2281. [DOI] [PubMed] [Google Scholar]
- 11.Smiley ST. 2008. Current challenges in the development of vaccines for pneumonic plague. Expert Rev Vaccines 7:209–221. doi: 10.1586/14760584.7.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verma SK, Tuteja U. 2016. Plague vaccine development: current research and future trends. Front Immunol 7:602. doi: 10.3389/fimmu.2016.00602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feodorova VA, Sayapina LV, Corbel MJ, Motin VL. 2014. Russian vaccines against especially dangerous bacterial pathogens. Emerg Microbes Infect 3:e86. doi: 10.1038/emi.2014.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feodorova VA, Motin VL. 2012. Plague vaccines: current developments and future perspectives. Emerg Microbes Infect 1:e36. doi: 10.1038/emi.2012.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feodorova VA, Lyapina AM, Khizhnyakova MA, Zaitsev SS, Sayapina LV, Arseneva TE, Trukhachev AL, Lebedeva SA, Telepnev MV, Ulianova OV, Lyapina EP, Ulyanov SS, Motin VL. 2018. Humoral and cellular immune responses to Yersinia pestis Pla antigen in humans immunized with live plague vaccine. PLoS Negl Trop Dis 12:e0006511. doi: 10.1371/journal.pntd.0006511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meyer KF. 1970. Effectiveness of live or killed plague vaccines in man. Bull World Health Organ 42:653–666. [PMC free article] [PubMed] [Google Scholar]
- 17.Pitt ML. (ed). 2004. Non-human primates as a model for pneumonic plague. Anim Model Correlates of Prot Plague Vaccin Workshop 2004, Gaithersburg, MD, 13–14 October 2004. [Google Scholar]
- 18.Quenee LE, Ciletti NA, Elli D, Hermanas TM, Schneewind O. 2011. Prevention of pneumonic plague in mice, rats, guinea pigs and non-human primates with clinical grade rV10, rV10-2 or F1-V vaccines. Vaccine 29:6572–6583. doi: 10.1016/j.vaccine.2011.06.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin JS, Szaba FM, Kummer LW, Chromy BA, Smiley ST. 2011. Yersinia pestis YopE contains a dominant CD8 T cell epitope that confers protection in a mouse model of pneumonic plague. J Immunol 187:897–904. doi: 10.4049/jimmunol.1100174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szaba FM, Kummer LW, Wilhelm LB, Lin JS, Parent MA, Montminy-Paquette SW, Lien E, Johnson LL, Smiley ST. 2009. D27-pLpxL, an avirulent strain of Yersinia pestis, primes T cells that protect against pneumonic plague. Infect Immun 77:4295–4304. doi: 10.1128/IAI.00273-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parent MA, Berggren KN, Kummer LW, Wilhelm LB, Szaba FM, Mullarky IK, Smiley ST. 2005. Cell-mediated protection against pulmonary Yersinia pestis infection. Infect Immun 73:7304–7310. doi: 10.1128/IAI.73.11.7304-7310.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin JS, Kummer LW, Szaba FM, Smiley ST. 2011. IL-17 contributes to cell-mediated defense against pulmonary Yersinia pestis infection. J Immunol 186:1675–1684. doi: 10.4049/jimmunol.1003303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szaba FM, Kummer LW, Duso DK, Koroleva EP, Tumanov AV, Cooper AM, Bliska JB, Smiley ST, Lin JS. 2014. TNFalpha and IFNgamma but not perforin are critical for CD8 T cell-mediated protection against pulmonary Yersinia pestis infection. PLoS Pathog 10:e1004142. doi: 10.1371/journal.ppat.1004142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Mena P, Romanov G, Lin JS, Smiley ST, Bliska JB. 2012. A protective epitope in type III effector YopE is a major CD8 T cell antigen during primary infection with Yersinia pseudotuberculosis. Infect Immun 80:206–214. doi: 10.1128/IAI.05971-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fonseca DM, Hand TW, Han SJ, Gerner MY, Glatman Zaretsky A, Byrd AL, Harrison OJ, Ortiz AM, Quinones M, Trinchieri G, Brenchley JM, Brodsky IE, Germain RN, Randolph GJ, Belkaid Y. 2015. Microbiota-dependent sequelae of acute infection compromise tissue-specific immunity. Cell 163:354–366. doi: 10.1016/j.cell.2015.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wren BW. 2003. The yersiniae–a model genus to study the rapid evolution of bacterial pathogens. Nat Rev Microbiol 1:55–64. doi: 10.1038/nrmicro730. [DOI] [PubMed] [Google Scholar]
- 27.Achtman M, Zurth K, Morelli G, Torrea G, Guiyoule A, Carniel E. 1999. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proc Natl Acad Sci U S A 96:14043–14048. doi: 10.1073/pnas.96.24.14043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korhonen TK, Haiko J, Laakkonen L, Jarvinen HM, Westerlund WB. 2013. Fibrinolytic and coagulative activities of Yersinia pestis. Front Cell Infect Microbiol 3:35. doi: 10.3389/fcimb.2013.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sodeinde OA, Subrahmanyam YV, Stark K, Quan T, Bao Y, Goguen JD. 1992. A surface protease and the invasive character of plague. Science 258:1004–1007. doi: 10.1126/science.1439793. [DOI] [PubMed] [Google Scholar]
- 30.Welkos SL, Friedlander AM, Davis KJ. 1997. Studies on the role of plasminogen activator in systemic infection by virulent Yersinia pestis strain C092. Microb Pathog 23:211–223. doi: 10.1006/mpat.1997.0154. [DOI] [PubMed] [Google Scholar]
- 31.Sebbane F, Jarrett CO, Gardner D, Long D, Hinnebusch BJ. 2006. Role of the Yersinia pestis plasminogen activator in the incidence of distinct septicemic and bubonic forms of flea-borne plague. Proc Natl Acad Sci U S A 103:5526–5530. doi: 10.1073/pnas.0509544103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Welkos S, Pitt ML, Martinez M, Friedlander A, Vogel P, Tammariello R. 2002. Determination of the virulence of the pigmentation-deficient and pigmentation-/plasminogen activator-deficient strains of Yersinia pestis in non-human primate and mouse models of pneumonic plague. Vaccine 20:2206–2214. doi: 10.1016/S0264-410X(02)00119-6. [DOI] [PubMed] [Google Scholar]
- 33.Lathem WW, Price PA, Miller VL, Goldman WE. 2007. A plasminogen-activating protease specifically controls the development of primary pneumonic plague. Science 315:509–513. doi: 10.1126/science.1137195. [DOI] [PubMed] [Google Scholar]
- 34.Mosesson MW. 2005. Fibrinogen and fibrin structure and functions. J Thromb Haemost 3:1894–1904. doi: 10.1111/j.1538-7836.2005.01365.x. [DOI] [PubMed] [Google Scholar]
- 35.Luo D, Lin JS, Parent MA, Mullarky-Kanevsky I, Szaba FM, Kummer LW, Duso DK, Tighe M, Hill J, Gruber A, Mackman N, Gailani D, Smiley ST. 2013. Fibrin facilitates both innate and T cell-mediated defense against Yersinia pestis. J Immunol 190:4149–4161. doi: 10.4049/jimmunol.1203253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rijken DC, Lijnen HR. 2009. New insights into the molecular mechanisms of the fibrinolytic system. J Thromb Haemost 7:4–13. doi: 10.1111/j.1538-7836.2008.03220.x. [DOI] [PubMed] [Google Scholar]
- 37.Caulfield AJ, Lathem WW. 2012. Substrates of the plasminogen activator protease of Yersinia pestis. Adv Exp Med Biol 954:253–260. doi: 10.1007/978-1-4614-3561-7_32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bergmann S, Hammerschmidt S. 2007. Fibrinolysis and host response in bacterial infections. Thromb Haemost 98:512–520. doi: 10.1160/TH07-02-0117. [DOI] [PubMed] [Google Scholar]
- 39.Boyle MD, Lottenberg R. 1997. Plasminogen activation by invasive human pathogens. Thromb Haemost 77:1–10. [PubMed] [Google Scholar]
- 40.Tilley R, Mackman N. 2006. Tissue factor in hemostasis and thrombosis. Semin Thromb Hemost 32:5–10. doi: 10.1055/s-2006-933335. [DOI] [PubMed] [Google Scholar]
- 41.Parry GC, Erlich JH, Carmeliet P, Luther T, Mackman N. 1998. Low levels of tissue factor are compatible with development and hemostasis in mice. J Clin Invest 101:560–569. doi: 10.1172/JCI814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mullarky IK, Szaba FM, Winchel CG, Parent MA, Kummer LW, Mackman N, Johnson LL, Smiley ST. 2006. In situ assays demonstrate that interferon-gamma suppresses infection-stimulated hepatic fibrin deposition by promoting fibrinolysis. J Thromb Haemost 4:1580–1587. doi: 10.1111/j.1538-7836.2006.02010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luo D, Szaba FM, Kummer LW, Plow EF, Mackman N, Gailani D, Smiley ST. 2011. Protective roles for fibrin, tissue factor, plasminogen activator inhibitor-1, and thrombin activatable fibrinolysis inhibitor, but not factor XI, during defense against the gram-negative bacterium Yersinia enterocolitica. J Immunol 187:1866–1876. doi: 10.4049/jimmunol.1101094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vercauteren E, Peeters M, Hoylaerts MF, Lijnen HR, Meijers JC, Declerck PJ, Gils A. 2012. The hyperfibrinolytic state of mice with combined thrombin-activatable fibrinolysis inhibitor (TAFI) and plasminogen activator inhibitor-1 gene deficiency is critically dependent on TAFI deficiency. J Thromb Haemost 10:2555–2562. doi: 10.1111/jth.12036. [DOI] [PubMed] [Google Scholar]
- 45.Mullarky IK, Szaba FM, Berggren KN, Parent MA, Kummer LW, Chen W, Johnson LL, Smiley ST. 2005. Infection-stimulated fibrin deposition controls hemorrhage and limits hepatic bacterial growth during listeriosis. Infect Immun 73:3888–3895. doi: 10.1128/IAI.73.7.3888-3895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tapper H, Herwald H. 2000. Modulation of hemostatic mechanisms in bacterial infectious diseases. Blood 96:2329–2337. [PubMed] [Google Scholar]
- 47.Flick MJ, Du X, Witte DP, Jirouskova M, Soloviev DA, Busuttil SJ, Plow EF, Degen JL. 2004. Leukocyte engagement of fibrin(ogen) via the integrin receptor alphaMbeta2/Mac-1 is critical for host inflammatory response in vivo. J Clin Invest 113:1596–1606. doi: 10.1172/JCI20741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Loike JD, Sodeik B, Cao L, Leucona S, Weitz JI, Detmers PA, Wright SD, Silverstein SC. 1991. CD11c/CD18 on neutrophils recognizes a domain at the N terminus of the A alpha chain of fibrinogen. Proc Natl Acad Sci U S A 88:1044–1048. doi: 10.1073/pnas.88.3.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smiley ST, King JA, Hancock WW. 2001. Fibrinogen stimulates macrophage chemokine secretion through Toll-like receptor 4. J Immunol 167:2887–2894. doi: 10.4049/jimmunol.167.5.2887. [DOI] [PubMed] [Google Scholar]
- 50.Tsakadze NL, Zhao Z, D'Souza SE. 2002. Interactions of intercellular adhesion molecule-1 with fibrinogen. Trends Cardiovasc Med 12:101–108. doi: 10.1016/S1050-1738(01)00157-8. [DOI] [PubMed] [Google Scholar]
- 51.Holmback K, Danton MJ, Suh TT, Daugherty CC, Degen JL. 1996. Impaired platelet aggregation and sustained bleeding in mice lacking the fibrinogen motif bound by integrin alpha IIb beta 3. EMBO J 15:5760–5771. doi: 10.1002/j.1460-2075.1996.tb00962.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Flick MJ, Du X, Degen JL. 2004. Fibrin(ogen)-alpha M beta 2 interactions regulate leukocyte function and innate immunity in vivo. Exp Biol Med (Maywood) 229:1105–1110. doi: 10.1177/153537020422901104. [DOI] [PubMed] [Google Scholar]
- 53.Johnson LL, Berggren KN, Szaba FM, Chen W, Smiley ST. 2003. Fibrin-mediated protection against infection-stimulated immunopathology. J Exp Med 197:801–806. doi: 10.1084/jem.20021493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun H, Wang X, Degen JL, Ginsburg D. 2009. Reduced thrombin generation increases host susceptibility to group A streptococcal infection. Blood 113:1358–1364. doi: 10.1182/blood-2008-07-170506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Esmon CT, Xu J, Lupu F. 2011. Innate immunity and coagulation. J Thromb Haemost 9:182–188. doi: 10.1111/j.1538-7836.2011.04323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Peetermans M, Vanassche T, Liesenborghs L, Lijnen RH, Verhamme P. 2016. Bacterial pathogens activate plasminogen to breach tissue barriers and escape from innate immunity. Crit Rev Microbiol 42:866–882. doi: 10.3109/1040841X.2015.1080214. [DOI] [PubMed] [Google Scholar]
- 57.Degen JL, Bugge TH, Goguen JD. 2007. Fibrin and fibrinolysis in infection and host defense. J Thromb Haemost 5:24–31. doi: 10.1111/j.1538-7836.2007.02519.x. [DOI] [PubMed] [Google Scholar]
- 58.Samoilova SV, Samoilova LV, Yezhov IN, Drozdov IG, Anisimov AP. 1996. Virulence of pPst+ and pPst- strains of Yersinia pestis for guinea-pigs. J Med Microbiol 45:440–444. doi: 10.1099/00222615-45-6-440. [DOI] [PubMed] [Google Scholar]
- 59.Lahteenmaki K, Virkola R, Saren A, Emody L, Korhonen TK. 1998. Expression of plasminogen activator pla of Yersinia pestis enhances bacterial attachment to the mammalian extracellular matrix. Infect Immun 66:5755–5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Collen D, Lijnen HR. 2005. Thrombolytic agents. Thromb Haemost 93:627–630. doi: 10.1160/TH04-11-0724. [DOI] [PubMed] [Google Scholar]
- 61.Tapson VF. 2013. Thrombolytic therapy for acute pulmonary embolism. Semin Thromb Hemost 39:452–458. doi: 10.1055/s-0033-1334145. [DOI] [PubMed] [Google Scholar]
- 62.Suh TT, Holmback K, Jensen NJ, Daugherty CC, Small K, Simon DI, Potter S, Degen JL. 1995. Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen-deficient mice. Genes Dev 9:2020–2033. doi: 10.1101/gad.9.16.2020. [DOI] [PubMed] [Google Scholar]