

Gastrointestinal (GI) colonization of 2-day-old (P2) rat pups with Escherichia coli K1 results in translocation of the colonizing bacteria across the small intestine, bacteremia, and invasion of the meninges, with animals frequently succumbing to lethal infection. Infection, but not colonization, is strongly age dependent; pups become progressively less susceptible to infection over the P2-to-P9 period.
KEYWORDS: Escherichia coli, gastrointestinal colonization, knockout rat, meningitis, neonatal pathogens, septicemia, trefoil factor 2
ABSTRACT
Gastrointestinal (GI) colonization of 2-day-old (P2) rat pups with Escherichia coli K1 results in translocation of the colonizing bacteria across the small intestine, bacteremia, and invasion of the meninges, with animals frequently succumbing to lethal infection. Infection, but not colonization, is strongly age dependent; pups become progressively less susceptible to infection over the P2-to-P9 period. Colonization leads to strong downregulation of the gene encoding trefoil factor 2 (Tff2), preventing maturation of the protective mucus barrier in the small intestine. Trefoil factors promote mucosal homeostasis. We investigated the contribution of Tff2 to protection of the neonatal rat from E. coli K1 bacteremia and tissue invasion. Deletion of tff2, using clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9, sensitized P9 pups to E. coli K1 bacteremia. There were no differences between tff2−/− homozygotes and the wild type with regard to the dynamics of GI colonization. Loss of the capacity to elaborate Tff2 did not impact GI tract integrity or the thickness of the small-intestinal mucus layer but, in contrast to P9 wild-type pups, enabled E. coli K1 bacteria to gain access to epithelial surfaces in the distal region of the small intestine and exploit an intracellular route across the epithelial monolayer to enter the blood circulation via the mesenteric lymphatic system. Although primarily associated with the mammalian gastric mucosa, we conclude that loss of Tff2 in the developing neonatal small intestine enables the opportunistic neonatal pathogen E. coli K1 to enter the compromised mucus layer in the distal small intestine prior to systemic invasion and infection.
INTRODUCTION
Although Escherichia coli is a ubiquitous commensal inhabitant of the human gastrointestinal (GI) tract, some strains of this versatile species display a capacity to cause severe GI and extraintestinal infections. Some are well-adapted opportunistic pathogens that cause systemic infection in vulnerable hosts. For example, strains expressing the polysialic acid K1 capsule are harmless constituents of the adult GI tract but may cause life-threatening septicemia, sepsis, and meningitis in the newborn infant following transmission from mother to neonate at or shortly after birth (1, 2). Maternally derived E. coli K1 colonizes the GI tract of the susceptible neonate; bacteria from the GI-colonizing cohort may translocate from the gut lumen to the blood, eliciting symptoms of sepsis and septic shock (2), and then invade the central nervous system to induce inflammation of the meninges (3).
The strong age dependency associated with E. coli K1 human systemic infection can be replicated in the neonatal rat (4, 5). Oral administration of K1 bacteria to 2-day-old (P2) pups initiates stable colonization of the GI tract, and lethal systemic infection develops due to the capacity of a small number of E. coli K1 bacteria to translocate across the epithelium of the middle section of the small intestine (MSI) to the submucosa, avoid capture by the mesenteric lymphatic system, enter the blood circulation, and establish infection in the brain and other organs (6–9). Pups become progressively less susceptible to systemic infection when colonized over the P2-to-P9 period, even though GI colonization can be established as readily in resistant P9 animals as in susceptible P2 pups (5). In P2 animals, the mucus barrier of the small intestine is poorly developed, allowing E. coli K1 bacteria to gain access to the enterocyte surface of the MSI lumen and translocate through the epithelial cell monolayer by an intracellular pathway to the submucosa (8). The protective mucus layer matures to full thickness over P2 to P9, coincident with the development of resistance to infection. At P9, K1 bacteria are physically separated from villi by the mucus layer, and their numbers are controlled by mucus-embedded antimicrobial peptides, preventing invasion of host tissues.
P2 and P9 pups respond differently to the threat posed by colonizing E. coli K1, reflecting substantial changes in developmental gene expression postpartum (10). A large number of genes expressed in the GI tract were upregulated (241 for P2 and 354 for P9) or downregulated (36 for P2 and 240 for P9) following initiation of colonization, but the transcriptomic responses were very different, with virtually no commonality of modulation of gene expression between the two age groups. Notably, the α-defensin genes defa24 and defa-rs1 were upregulated in P9 GI tissues in response to colonization, but no changes in α-defensin gene expression occurred in the P2 GI tract. These mucus-embedded antimicrobial peptides are produced by Paneth cells and contribute to the barrier function of the mucus layer (11), as evidenced by the demonstration that chemical ablation of Paneth cells reduces the GI concentration of α-defensins and sensitizes normally resistant P9 pups to systemic infection (8).
Conversely, developmental expression of the gene encoding trefoil factor 2 (Tff2) was highly downregulated by E. coli K1 colonization in P2, but not in P9, rats; no evidence for modulation of tff1 and tff3, genes for other members of the trefoil factor family, was found (10). Mucin-associated trefoil factor proteins mediate maintenance and restoration of GI mucosal homeostasis, stabilizing the mucus layer, enhancing intestinal epithelial repair, and responding to GI mucosal injury and inflammation (12, 13). As tff2 expression in the GI tract (including the stomach) of noncolonized neonates increases incrementally over P1 to P9, with a substantial decline from P9 to P11 (10), it is likely that tff2 downregulation induced by E. coli K1 colonization further compromises innate GI defenses in susceptible P2 pups. In adult humans (14) and rodents (15), Tff2 appears to be associated primarily with the stomach and duodenum, although intravenously administered Tff2 also distributes rapidly to Paneth cells in the small intestine and to crypt colonic cells before appearing within the associated mucus layer (16). Tff1 is localized predominantly in gastric foveolar cells and surface epithelium throughout the stomach (17), and Tff3 is localized predominantly within goblet cells of the small intestine and colon (18), although species differences are evident (19), and these structurally related proteins may be functionally interchangeable (13). Little is known of the roles or distribution of trefoil factors during GI maturation. Trefoil mRNAs are expressed in early embryonic rat intestine and stomach before overt differentiation of epithelial cells (20); Tff3 appears around gestational day 17 in the rat intestine and increases further postnatally and during the weaning period (21). These observations provide further evidence that the mucosal barrier is not fully formed at birth and may render the neonate vulnerable to infection following colonization of the GI tract.
In this study, we examine the role of the tff2 gene product in protection of the neonatal rat from experimental systemic infection using a tff2−/− knockout rat. We show that P9 pups, normally resistant to infection due to the barrier function of the MSI mucus, become vulnerable to bacteremia following GI colonization with E. coli K1 strain A192PP, and we relate this to changes in GI tract physiology elicited by lack of Tff2.
RESULTS
Tff2 rat knockout.
tff2 knockout Sprague-Dawley rats were generated commercially using the clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 system (22). Nonproprietary details are provided in Materials and Methods. Guide RNA1 (gRNA1) was targeted to exon 1, and gRNA11 was targeted to exon 3 (see Fig. S1A in the supplemental material). Four rats bearing tff2 mutations were identified and confirmed by PCR-mediated amplification and sequencing analysis (see Fig. S1B). We backcrossed founder 2, which carried the most extensive deletion (1,933 bp), with a wild-type (WT) Sprague-Dawley rat (see Fig. S1C). The resulting F1 heterozygotes were identified by PCR-mediated genotyping and were subsequently crossed. F2 tff2−/− homozygotes were identified, and homozygote breeding colonies were maintained. Previous studies have demonstrated higher tff2 expression in the stomach than in intestinal tissues in humans and rats (14, 23). Expression of the rps23 and tff2 genes was therefore measured using quantitative reverse transcriptase PCR (qRT-PCR) analysis of stomach samples from wild-type and tff2−/− animals. There was a 16-fold reduction in stomach tff2 gene expression in tff2−/− homozygotes compared to WT rats (see Fig. S1D), indicative of a knockdown in tff2 gene expression. We were unable to detect sufficient Tff2 protein in tissue extracts by Western blotting and therefore could not directly compare the Tff2 contents of WT and tff2−/− tissues. Homozygote litters comprised 3 to 20 neonatal pups, and all the animals appeared healthy, gained weight identically to WT pups of comparable age, and were maintained in a healthy condition for up to 3 months. As loss of Tff2 could compromise defenses against bacterial invasion of the rodent GI tract (10, 12, 24), we examined changes in expression of genes encoding selected proteins known to impact GI tract protection. Thus, genes encoding Tff1 and Tff3 (tff1 and tff3), the α-defensins DefaRS1 and Defa24 (defaRS1 and defa24), and tumor necrosis factor alpha (tfna) were upregulated 0.5- to 3.5-fold in the proximal (PSI), MSI, and distal (DSI) regions of the P9 small intestine compared to their expression in WT P9 rat pups (Fig. 1). In contrast to tff1, tff3, and tfna, the two α-defensin genes were downregulated in colonic tissue; these antimicrobial peptides are known to play a role in the small intestine rather than the colon (25). Conversely, the interferon gamma gene (ifng) was marginally downregulated in the small intestine and marginally upregulated in the colon. Thus, there appears to be some reprogramming of antimicrobial defenses in the neonatal GI tract following knockout of tff2 against a background of developmental modulation of trefoil factor expression in the stomach, PSI, MSI, DSI, and colon during the immediately postpartum period (Fig. 1; see Fig. S2 in the supplemental material).
FIG 1.
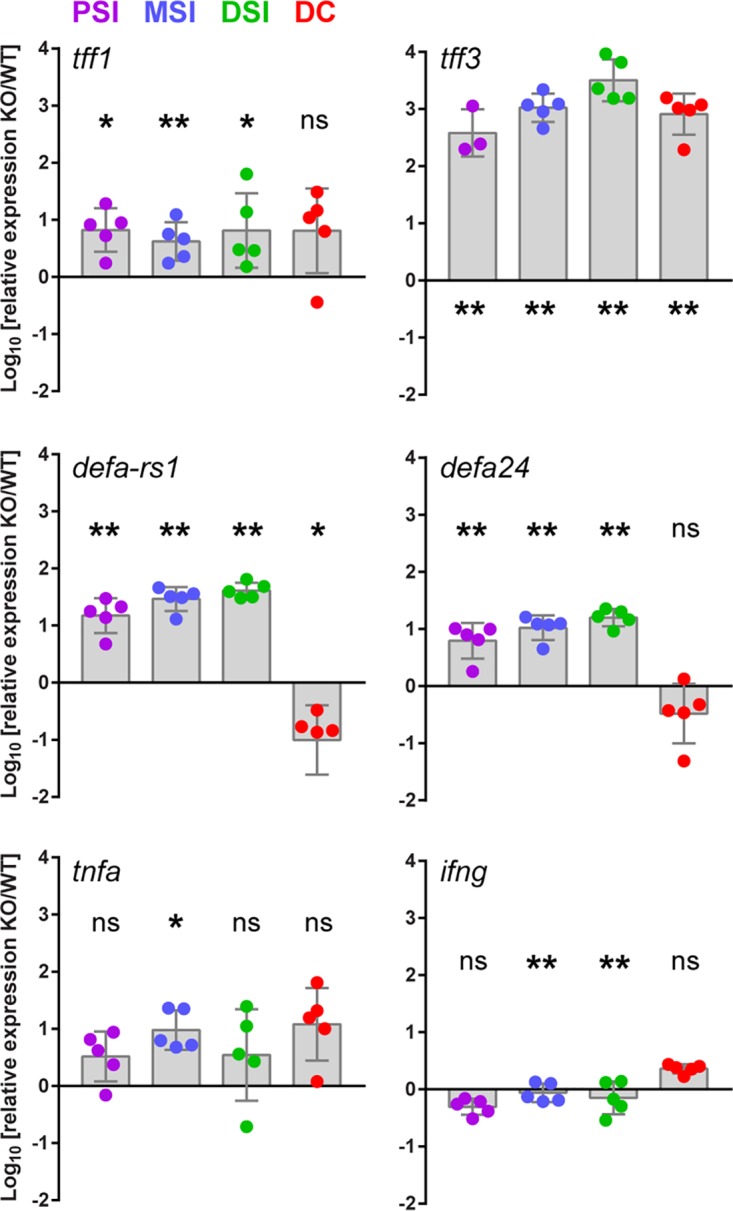
tff2 knockout (KO) rat pups increase expression of intestinal defense proteins. qRT-PCR data for tff1, tff3, defaRS1, defa24, tnfa, and ifng were normalized to the rps23 gene, and data from P9 tff2−/− pups were standardized to expression levels in P9 wild-type animals using the ΔCT method for relative quantification of qPCR data. Shown are means ± standard errors of the mean (SEM); n = 3 to 5. ΔCT values were determined by Student’s t test. ns, nonsignificant; *, P < 0.05; **, P < 0.01. The spots represent data from individual littermates. DC, distal colon.
Susceptibility of tff2−/− rat pups to E. coli K1 infection.
Colonization of each member of a litter of P2 Sprague-Dawley rat pups by oral administration of 2 × 106 to 6 × 106 CFU E. coli K1 strain A192PP bacteria resulted in disseminated systemic infection highly comparably to our previous data obtained using Wistar rats (5, 26). Pups become progressively less susceptible to infection after colonization over the P2-to-P9 period and by P8-P9 are resistant, even though the degrees of GI colonization are comparable to those for P2-susceptible neonates (8, 26). As expected, WT Sprague-Dawley rats that received a single colonizing dose at P9 did not succumb to infection. In contrast, tff2−/− pups colonized at P9 exhibited significantly increased susceptibility to the neonatal pathogen (Fig. 2A). Although a proportion of colonized tff2−/− pups survived for the 25-day observation period without significant symptoms, all the pups examined were bacteremic by day 7 after oral administration of the colonizing dose (Fig. 2B); only a small proportion of WT pups displayed bacteremia during the first 2 days following initiation of colonization. To confirm that systemic invasive disease arose in tff2−/− but not WT neonates, we tracked colonization and invasion in P9 pups using the well-characterized bioluminescent isogenic strain E. coli A192PP-lux (27) and two-dimensional (2D) bioluminescent imaging (Fig. 2C and D). The bioluminescent signals from the torso foci were comparable between WT and tff2−/− mice, suggesting that the total bacterial loads in the intestine and/or colon were comparable. We then measured bioluminescent signals 8 days after colonization, a time point at which we anticipated that tff2−/− pups would be bacteremic and WT pups abacteremic. Wider systemic bioluminescent signals were detected only in tff2−/− pups; notably, the bioluminescent signal from the head region was significantly higher in tff2−/− than in WT pups (Fig. 2C and D), most probably reflecting meningeal colonization and/or invasion (7). These data confirm that tff2−/− P9 pups were more susceptible to systemic invasive infection than wild-type P9 pups. The extent of GI colonization of the PSI, MSI, and DSI, as well as the colon and the mesenteric lymphatic system, of tff2−/− P9 pups was comparable to that of the WT, with only a small but significant difference in the number of CFU in the DSI 24 h after initiation of colonization (Fig. 3).
FIG 2.

Tff2-deficient neonatal rat pups exhibit increased susceptibility to E. coli K1 systemic infection. (A) Survival of wild-type and tff2−/− rats dosed orally with 2 × 106 to 6 × 106 CFU E. coli A192PP at P9; n = 13 (wild type) and 25 (tff2−/−). (B) Presence of E. coli K1 bacteria in blood samples from pups dosed with E. coli A192PP at P9 as determined by culture. E. coli K1 colonies were identified using bacteriophage K1E. Day 1, WT, n = 12; day 2, WT, n = 10; day 1, tff2−/−, n = 11; day 2, tff2−/−, n = 13; day 7, WT, n = 12; day 7, tff2−/−, n = 15. χ2 test; *, P < 0.05. (C and D) 2D bioluminescent imaging of pups dosed orally with 2 × 106 to 6 × 106 CFU E. coli A192PP-lux2 at P9. (C) Images showing photoemissions collected from the entire bodies of live animals at day 8 after initiation of colonization (P17). (D) Distribution of bioluminescent bacteria between torso and head. Bioluminescence values were determined as log10 (flux + 1); flux was measured in photons per second (p/s). Shown are means ± standard deviations (SD) (Student’s t test); *, P < 0.05 (Mann-Whitney U test).
FIG 3.

Comparable degrees of GI colonization in WT and tff2−/− rats administered 2 × 106 to 6 × 106 CFU E. coli A192PP at P9. CFU in PSI, MSI, DSI, mesenteric lymphatics (MLN), and colon are shown 24 h (A) and 48 h (B) after initiation of colonization. *, P < 0.05 (Mann-Whitney U test); all the other values were nonsignificant. Segments of the small intestine were obtained as follows (25). The small intestine was aligned from the midpoint. The last 2 cm of tissue prior to the cecum was collected as the DSI, the tissue from 5 to 7 cm above the midpoint was collected as the PSI, and the tissue from 3 to 5 cm below the midpoint was collected as the MSI.
GI integrity and the mucus layer in tff2−/− neonatal rats.
As colonizing bacteria disseminate from the tff2−/− neonatal gut, the barrier function of the GI tract in noncolonized P9 animals was determined by oral administration of small-molecule (fluorescein isothiocyanate [FITC]; 389-Da) and polymeric (FITC-dextran; 4-kDa) fluorescent probes and quantification of serum fluorescence for 4 h after feeding of the probes (Fig. 4A). There were no significant differences in the uptake of either probe; increased susceptibility to systemic infection of tff2−/− neonates is therefore unlikely to be due to a nonspecific increase in intestinal permeability. The small increases in uptake of both probes over the 4-h incubation period are most likely due to macropinocytosis of luminal content by neonatal enterocytes (8, 28, 29). Similarly, histological analysis demonstrated that there were no differences in the synthesis or secretion of the major mucus structural protein Muc2, as determined by immunostaining using antibodies detecting intracellular Apo-Muc2 (Fig. 4B) or the mature secreted protein (Fig. 4C). Thus, we could detect no differences in the appearance of either the small-intestinal or colonic mucus layer between WT and tff2−/− neonates that would reflect changes in intestinal integrity. In similar fashion, hematoxylin and eosin (H&E) staining failed to reveal any abnormalities in the appearance of tissues from regions of the GI tract (see Fig. S3 in the supplemental material).
FIG 4.

Loss of Tff2 does not affect GI permeability or the appearance of the GI mucus layer. (A) Intestinal permeability in noncolonized P9 wild-type and tff2−/− pups; plasma fluorescence was determined for 4 h following oral administration of FITC or FITC-dextran. Plasma fluorescence was expressed as log10 relative fluorescence units (RFU). (B and C) Representative confocal micrographs of methacarn-fixed PSI, MSI, DSI, and colonic tissue sections from noncolonized P9 WT and tff2−/− rats obtained 48 h after colonization with E. coli A192PP at P9 and probed for Apo-Muc2 (B) and Muc2 (C). In panel C, the red arrowheads in the small-intestine images show secreted Muc2, and the red dashed lines in the colonic images shows the approximate border of the inner mucus layer. Scale bars, 100 μm.
Enterocyte-internalized E. coli A192PP is found only in the distal portion of the small intestine.
E. coli K1 bacteria cross the epithelial barrier and gain access to the blood circulation at low frequency (6, 8, 9). We searched for bacteria expressing the O18 lipopolysaccharide (LPS) antigen in methanol-Carnoy’s fixative (methacarn)-fixed sections of the small intestines and colons of WT and tff2−/− pups 48 h after colonization with E. coli A192PP (O18:K1) at P9. We employed a commercial O18 antibody; bacteria in GI tissues from noncolonized wild-type and tff2−/− P9 animals failed to react with the antibody, indicating that no bacteria expressing the O18 antigen were present in detectable numbers in these tissue samples. No O18 antigen was found in close association with epithelial cells in the small intestines of WT P9 rat pups, and the dense population of O18 antigen-bearing E. coli A192PP in the colon was kept at a distance from the enterocytes lining the lumen (Fig. 5A) by the thick mucus layer in this region of the GI tract (Fig. 4C). In contrast, O18 bacteria were consistently found in close proximity to the GI epithelium in the DSI, but not the PSI, MSI, or colon, in tff2−/− pups, and O18 staining showed bacteria at locations deep within the mucus layer (Fig. 5A and B). Higher-magnification images indicated that some of these bacteria were contained within intracellular compartments and were present in numbers suggestive of replication within vesicles (Fig. 5B). Analysis of DSI tissue sections clearly demonstrated a significantly higher number of tissue-associated O18-positive bacterial cells in tff2−/− than in WT neonates (Fig. 5C). We have previously shown that ablation of Paneth cells sensitizes resistant pups to E. coli A192PP small-intestinal infection (8). We therefore assessed the possibility that Paneth cell function was compromised in the tff2−/− DSI by quantifying Apo-Muc2-positive cells present at the epithelial crypt base (Fig. 5D and E); however, no differences between WT and tff2−/− tissues were detected. Together, these data indicate that colonizing E. coli K1 bacteria gain entry to the systemic circulation in tff2−/− rats by exploiting a transcellular route across the DSI that contrasts with the MSI-restricted transepithelial route adopted by E. coli A192PP in susceptible WT P2 neonatal rat pups (8). Intriguingly, the capacity of the pathogen to infect the DSI of neonates lacking Tff2 appears to be independent of the postnatal mucus barrier formation that is thought to confer resistance in WT neonates.
FIG 5.

E. coli K1 bacteria gain entry to enterocytes only from the DSI. (A) Representative confocal micrographs of methacarn-fixed PSI, MSI, DSI, and colonic tissue sections from WT and tff2−/− neonatal rats administered 2 × 106 to 6 × 106 CFU E. coli A192PP at P9. Tissues were harvested 48 h after initiation of colonization, and sections were stained for O18 LPS and DNA. (B) Enlargements of the boxed areas in panel A. No O18-positive bacteria were visualized in proximity to the enterocyte layer in the DSI or other regions of the WT GI tract. Small numbers of O18-positive bacteria were associated with enterocytes in the tff2−/− DSI, but not with other GI tissues (A), and could be seen in intracellular locations within villi (B). (C) Quantification of O18-positive bacterial cells in contact with DSI tissue of WT and tff2−/− neonates 24 and 48 h after the K1 dose. (D and E) quantification of immature Paneth cells in DSI. Sections were stained for Apo-Muc2 and DNA; the crypt base (green dashed line) is indicated. n = 3 or 4 animals/group, and means ± SEM are shown. *, P < 0.05 as determined by 2-way analysis of variance (ANOVA) and Sidak’s multiple comparison.
DISCUSSION
Initiation of GI colonization of P2 rat pups with E. coli A192PP elicited a substantial (26.4-fold) downregulation of tff2 in the GI tract within 12 hours of bacterial seeding; there was no modulation of expression of other genes encoding trefoil factors or of those involved in GI mucus homeostasis (10). The susceptibility of P9 tff2−/− knockouts to bacteremia and, in the majority of cases, lethality (Fig. 2) supported our earlier microarray data (10) indicating that Tff2 contributes to mucosal barrier function in the small intestine by ensuring that potentially invasive bacteria are unable to make close contact with the epithelial surface. E. coli K1 bacteria were found in close proximity to epithelial surfaces within the DSI region of the P9 tff2−/− GI tract and appeared to gain entry to the blood circulation by an intracellular route (Fig. 5). At least some bacteria accessing the submucosal space were able to avoid capture by the mesenteric lymphatic system to cause systemic infection. Mucosal barrier function appeared to be lost only in the DSI, in contrast to WT P2 pups, in which GI transit occurred in the MSI (8). The lesion enabling E. coli A192PP to interact with the DSI epithelial surface was clearly discrete: we detected no histological changes in this region attributable to loss of tff2 and no increase in overall GI permeability. Although the invading bacteria were able to penetrate the DSI mucus layer, in contrast to the P9 wild type, there were no differences in mucosal thickness, morphological appearance, or Paneth cell numbers, as determined by immunohistochemistry, that could account for increased susceptibility to invasive infection. In addition, there were no major differences in the numbers of colonizing bacteria and their distribution in the GI tract between P9 wild-type and tff2−/− animals (Fig. 3), indicating that Tff2 played no significant role in controlling the population of colonizing E. coli K1 bacteria. It has been reported that basal gastric acid secretion is increased and gastric mucosal thickness is decreased in otherwise healthy tff2−/− mice (30). For technical reasons, tff2−/− homozygotes were generated in Sprague-Dawley rats. Our previous studies of the susceptibility of the neonatal rat to E. coli K1 infection were undertaken in Wistar rats, and although we compared the dynamics of infection following E. coli A192PP colonization in the two breeds and found no qualitative or quantitative differences in susceptibility, this should be borne in mind when comparing our previous work with data from this study.
Tff2, as the naturally occurring dimer, is most frequently found in association with the mammalian gastric mucosa (13, 31) but has been reported in human adult pancreatic, colonic, and skeletal tissues (14). PCR has indicated that tff2 gene expression is less restricted, with relatively low expression in human (14) and rat (15) small-intestinal tissue, and the partial redistribution of intravenously administered Tff2 to the mucus layer of the small intestine (16) suggests a capacity to interact in situ with Muc2, the predominant intestinal mucin. Our data (see Fig. S2) confirm tff2 expression in all regions of the small intestine and suggest that loss of the capacity to elaborate Tff2 in the P9 rat DSI leads to loss of mucosal barrier function, even though the mucus layer appears normal. In the stomach, Tff2 is secreted in concert with Muc6 by deep gastric glands and the gastric glycoform binds with high specificity to O-linked α1,4-GlcNAc-capped hexasaccharides on Muc6-rich stomach mucus (24). These motifs could facilitate Tff2 binding in other regions of the GI tract, particularly during embryonic and early postpartum development. We attempted to localize Tff1, Tff2, and Tff3 in regions of the GI tract from wild-type and tff2−/− homozygotes while preserving the integrity of the mucus layer by methacarn fixation but were unable to do so, as methanol-induced precipitation of the proteins abrogated their capacity to react with polyclonal antibodies (unpublished observations).
Loss of the capacity to elaborate Tff2 led to upregulation in the P9 small intestine of genes associated with mucosal defense (Fig. 1), but these increases in gene expression did not prevent rapid translocation of colonizing E. coli K1 from the DSI to the blood compartment. Interestingly, 60% of P9 pups were bacteremic within 24 h of initiation of colonization (Fig. 2B), a significantly higher rate than for E. coli A192PP-colonized WT P2 pups (5, 8). All the P2 pups succumbed to lethal infection within 7 days of initiation of colonization, but a significant proportion of tff2−/− P9 pups survived for at least 25 days, in all probability reflecting maturation of other components of the GI tract defense repertoire in the older, albeit genetically compromised, animals (8, 10). It is clear that the GI defense in these rapidly maturing animals is a highly cooperative phenomenon and that disruption of one or more key defense determinants provides opportunities to pathogens such as E. coli K1 for invasion of deeper tissues (Fig. 2C), as was also shown by the increased susceptibility of P8-P9 pups to infection induced by chemical ablation of Paneth cells (8).
Tff2 has been used in rodent models to repair gastric injury (32, 33) and inflammation-induced colonic damage (34). The protein is highly resistant to adverse conditions in the GI tract and in these studies was administered intravenously, intrarectally, or orally, in all cases accelerating GI repair. These observations raise the possibility that exogenous administration of recombinant Tff2 could repair the lesions induced by tff2 knockout and eventually accelerate small intestine maturation in neonates deemed at risk from opportunistic infections arising from bacterial colonization of the gut. Such an approach could be particularly relevant if suppression of tff2 expression is a common feature of colonization of the gut of the newborn by neonatal pathogens such as E. coli K1.
MATERIALS AND METHODS
Bacteria.
E. coli A192PP (O18:K1; ST95) was obtained by two rounds of serial passage through P2 rat pups of the septicemia isolate E. coli A192 (5, 35). The passaged derivative was significantly more virulent in the neonatal rat model of infection than the parent isolate (36). The strain was cultured in Luria-Bertani (LB) or Mueller-Hinton (MH) broth or agar at 37°C. Engineering of E. coli A192PP to stably and constitutively express the Photorhabdus luminescens-derived lux operon (luxCDABE) with minimal loss of virulence was described previously (26, 36). E. coli A192PP-lux was cultured in LB medium containing 50 μg/ml kanamycin and used to colonize rat pups as described below.
Generation of tff2 knockout rats.
tff2 knockout Sprague-Dawley rats were generated by Horizon Discovery (St. Louis, MO, USA). In brief, two pairs of single guide RNAs (sgRNAs) were designed to cleave together in order to generate an ∼1.8-kb deletion (4,894 bp) between the target sites, removing the gene encoding Tff2 from the NC 005119 region of the genome. The most active sgRNAs were selected in cultured eukaryotic cells; the design and synthesis of donor DNA containing the desired mutation were facilitated by Horizon in-house bioinformatic programs and were validated by sequencing. Donor DNA, along with Cas9/sgRNA reagent, was delivered by microinjection into fertilized embryos to create the desired mutation; a maximum of two microinjection sessions were conducted. The most potent sgRNA with minimal off-target potential was assembled into a ribonucleoprotein complex with Cas9 endonuclease and, together with the donor DNA, was delivered into zygotes from Sprague-Dawley rats, followed by embryo transfer into pseudopregnant females. For gestation and identification of the founder mutant phase, tissue biopsy specimens were obtained at approximately 2 weeks postpartum. Viable progeny were analyzed for the presence of the desired mutation by genomic PCR and DNA sequencing. Fifteen animals were screened, and four founders contained a 1.8-kb deletion; two rats were bred to the F1 heterozygous stage with wild-type Sprague-Dawley rats, and 19 F1 heterozygotes were identified as carrying the tff2 mutation by PCR-mediated genotyping and DNA sequence analysis. Loss of Tff2 did not impact the wellbeing of tff2−/− animals, at least during the first 14 weeks of life: pups remained healthy, and homozygous adults bred in comparable fashion to the wild type.
PCR.
Genotyping of offspring from tff2 mutant mating pairs was determined by PCR. For upstream sgRNA nonhomologous-end-joining (NHEJ) detection by sequence analysis, the following primers were used: Tff2 Cel1 F1 (5′-GGAGCCATGTCAGCATTTCT) and Tff2 Cel1 R1 (5′-GTCCTTTGCGGGAACATAGA) (expected wild-type band, 349 bp); for downstream sgRNA NHEJ detection by sequence analysis, the following primers were used: Tff2 Cel1 F2 (5′-CCCTAAGAAGGCAGAACTGG) and Tff2 Cel1 R2 (5′-ACAGAGGCACACACAGATGC) (expected wild-type band, 364 bp); for large deletions between sgRNAs by electrophoresis analysis, the following primers were used: Tff2 Cel1 F1 (5′-GGAGCCATGTCAGCATTTCT) and Tff2 Cel1 R2 (5′-ACAGAGGCACACACAGATGC) (expected mutation band, 341 bp). Reaction mixtures (total volume, 25 μl) contained 1 μl DNA template (extracted using Epicentre QuickExtract solution), 2.5 μl of 10 μM forward primer, 2.5 μl of 10 μM reverse primer, 12.5 μl Sigma JumpStart Taq ReadyMix (P2893), and 6.5 μl H2O. Thermal cycling was undertaken at 95°C for 5 min, followed by 35 cycles at 95°C for 30 s, 60°C for 30 s, and 68°C for 40 s, with a final extension step of 5 min at 68°C. PCR products were detected following separation by electrophoresis on 2% agarose gels by staining with ethidium bromide.
Semiquantitative reverse transcriptase PCR (RT-PCR) was used to evaluate expression of key host genes during early postpartum (P2 to P9) development of the neonatal rat. RNA was extracted using an RNeasy Midi kit (Qiagen), and cDNA was amplified from 25 ng RNA by RT-PCR. RNA was mixed with Brilliant II RT-PCR master mix (Agilent), gene-specific forward and reverse primer pairs (see Table S1 in the supplemental material), and AffinityScript RT-RNase block enzyme mixture (Agilent) to a final volume of 25 μl. The thermocycling program comprised 30 min at 50°C and 10 min at 95°C, followed by 35 cycles of 30 s at 95°C, 1 min at 60°C, and 30 s at 72°C. Experiments were performed using Mx3000P v2.0 software (Stratagene) to normalize SYBR1 to ROX (BioRad) fluorescence and to determine cycle threshold values using adaptive baseline and amplification-based threshold algorithm enhancements. Expression was examined by normalizing qRT-PCR data from noncolonized animals to RNA data obtained from noncolonized P2 intestinal tissues. qRT-PCR was undertaken as described previously (10).
Colonization of neonatal rats.
Animal experiments were approved by the Ethical Committee of University College London and were conducted in accordance with national legislation under United Kingdom Home Office Project License PPL 70/7773. All experiments were conducted in accordance with the United Kingdom Animals (Scientific Procedures) Act, 1986, and the Codes of Practice for the Housing and Care of Animals used in Scientific Procedures, 1989. All animal experiments were undertaken with E. coli K1 A192PP or the bioluminescent derivative A192PP-lux (27, 37). Sprague-Dawley rats were used to generate tff2 knockouts and served as WT controls. Susceptibility of genetically unaltered Sprague-Dawley pups to infection following GI colonization was comparable to that obtained with Wistar rats in our previous studies of neonatal rat infection (8). Litters comprised 12 to 14 pups and were retained in a single cage with their natural mothers. The animal model of colonization and infection has been described in detail (26). Each pup was fed 20 μl of MH broth culture of E. coli A192PP (2 × 106 to 6 × 106 CFU; 37°C) from an Eppendorf micropipette. Controls were fed 20 μl of sterile broth. Colonization was determined by MacConkey agar culture of perianal swabs and bacteremia by culture of daily blood samples taken from the footpad. Disease progression was monitored by regular evaluation of symptoms of systemic infection, and pups were culled and recorded as dead once a threshold, described previously (26), had been reached.
Intestinal permeability.
The GI tract permeability of rat pups was assessed using FITC and FITC-dextran (Sigma). The pups were fed 15 μl of probe solution (800 mg/ml in sterile phosphate-buffered saline [PBS]) and culled 1, 2, or 4 h after administration. Plasma fluorescence was determined against plasma from control rat pups, and values were normalized for total blood volume for each pup.
2D bioluminescent imaging.
Quantitative bioluminescent imaging was performed using an IVIS Lumina series III (PerkinElmer) incorporating a prewarmed imaging platform. Anesthesia was induced with 5% isoflurane and maintained with 2.5% isoflurane. Animals were not subjected to repeated anesthesia. Bioluminescence was measured within standardized regions of interest, and the data were expressed as flux (photons per second), adjusted using the following formula to ensure all measurements had positive values: log10 (flux + 1).
Histology, immunohistochemistry, and microscopy.
Neonatal rats were killed by decapitation, and the GI tissues were segmented into stomach, mesentery, colon, and small intestine; 2-cm segments from the proximal, middle, and distal small intestine were excised without washing as described previously (26). Tissue samples were placed in methacarn and maintained at room temperature for at least 3 h. Paraffin-embedded sections were dewaxed and hydrated. Some sections were stained with H&E. Muc2 was stained using polyclonal rabbit antiserum raised against either nonglycosylated Apo-Muc2 protein (PH497) or mature glycosylated Muc2 protein (Muc2C3); Alexa Fluor 555/488-conjugated goat anti-rabbit IgG (ThermoFisher) was used as a secondary antibody. Immunofluorescent sections were counterstained with Hoechst 33258 (Sigma). E. coli A192PP was visualized in tissue sections with rabbit polyclonal antibody raised against the O18 LPS surface antigen (7). Stained sections were mounted in Prolong Anti-Fade (Life Technologies); images were acquired using an LSM 700 confocal microscope equipped with an oil immersion lens and 488-nm and 555-nm lasers (Zeiss). Contact of E. coli A192PP with tissue was quantified by counting the extracellular and intracellular O18-positive cells in contact with the epithelial cell layer in micrograph fields at ×20 magnification.
Supplementary Material
ACKNOWLEDGMENTS
This study was funded by Medical Research Council research grant MR/K018396/1 and by Action Medical Research grant GN2075. The funders had no role in study design, data collection, and interpretation or the decision to submit the work for publication. The National Institute for Health Research University College London Hospitals Biomedical Research Centre provided infrastructural support.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00878-18.
REFERENCES
- 1.Sarff LD, McCracken GH, Schiffer MS, Glode MP, Robbins JB, Ørskov I, Ørskov F. 1975. Epidemiology of Escherichia coli K1 in healthy and diseased newborns. Lancet 1:1099–1104. [DOI] [PubMed] [Google Scholar]
- 2.Simonsen KA, Anderson-Berry AL, Delair SF, Davies HD. 2014. Early-onset neonatal sepsis. Clin Microbiol Rev 27:21–47. doi: 10.1128/CMR.00031-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tunkel AR, Scheld WM. 1993. Pathogenesis and pathophysiology of bacterial meningitis. Clin Microbiol Rev 6:118–136. doi: 10.1128/CMR.6.2.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glode MP, Sutton A, Moxon ER, Robbins JB. 1977. Pathogenesis of neonatal Escherichia coli meningitis: induction of bacteremia and meningitis in infant rats fed E. coli K1. Infect Immun 16:75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mushtaq N, Redpath MB, Luzio JP, Taylor PW. 2004. Prevention and cure of systemic Escherichia coli K1 infection by modification of the bacterial phenotype. Antimicrob Agents Chemother 48:1503–1508. doi: 10.1128/AAC.48.5.1503-1508.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pluschke G, Mercer A, Kusećek B, Pohl A, Achtman M. 1983. Induction of bacteremia in newborn rats by Escherichia coli K1 is correlated with only certain O(lipopolysaccharide) antigen types. Infect Immun 39:599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zelmer A, Bowen M, Jokilammi A, Finne J, Luzio JP, Taylor PW. 2008. Differential expression of the polysialyl capsule during blood-to-brain transit of neuropathogenic Escherichia coli K1. Microbiology 154:2522–2532. doi: 10.1099/mic.0.2008/017988-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Birchenough GMH, Dalgakiran F, Witcomb LA, Johansson MEV, McCarthy AJ, Hansson GC, Taylor PW. 2017. Postnatal development of the small intestinal mucosa drives age-dependent, regio-selective susceptibility to Escherichia coli K1 infection. Sci Rep 7:83. doi: 10.1038/s41598-017-00123-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCarthy AJ, Stabler RA, Taylor PW. 2018. Genome-wide identification of Escherichia coli K1 genes essential for growth in vitro, gastrointestinal colonizing capacity and survival in serum by transposon insertion sequencing. J Bacteriol 200:e00698-17. doi: 10.1128/JB.00698-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Birchenough GMH, Johansson MEV, Stabler RA, Dalgakiran F, Hansson GC, Wren BW, Luzio JP, Taylor PW. 2013. Altered innate defenses in the neonatal gastrointestinal tract in response to colonization by neuropathogenic Escherichia coli. Infect Immun 81:3264–3275. doi: 10.1128/IAI.00268-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lehrer RI, Lu W. 2012. α-defensins in human innate immunity. Immunol Rev 245:84–112. doi: 10.1111/j.1600-065X.2011.01082.x. [DOI] [PubMed] [Google Scholar]
- 12.Kjellev S. 2009. The trefoil factor family—small peptides with multiple functionalities. Cell Mol Life Sci 66:1350–1369. doi: 10.1007/s00018-008-8646-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aihara E, Engevik KA, Montrose MH. 2017. Trefoil factor peptides and gastrointestinal function. Annu Rev Physiol 79:357–380. doi: 10.1146/annurev-physiol-021115-105447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Madsen J, Nielsen O, Tornøe I, Thim L, Holmskov U. 2007. Tissue localization of human trefoil factors 1, 2, and 3. J Histochem Cytochem 55:505–513. doi: 10.1369/jhc.6A7100.2007. [DOI] [PubMed] [Google Scholar]
- 15.Taupin DR, Pang KC, Green SP, Giraud AS. 1995. The trefoil peptides spasmolytic polypeptide and intestinal trefoil factor are major secretory products of the rat gut. Peptides 16:1001–1005. doi: 10.1016/0196-9781(95)00070-Z. [DOI] [PubMed] [Google Scholar]
- 16.Poulsen SS, Thulesen J, Nexø E, Thim L. 1998. Distribution and metabolism of intravenously administered trefoil factor 2/porcine spasmolytic polypeptide in the rat. Gut 43:240–247. doi: 10.1136/gut.43.2.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanby AM, Poulsom R, Singh S, Elia G, Jeffery RE, Wright NA. 1993. Spasmolytic polypeptide is a major antral peptide: distribution of the trefoil peptides human spasmolytic polypeptide and pS2 in the stomach. Gastroenterology 105:1110–1116. doi: 10.1016/0016-5085(93)90956-D. [DOI] [PubMed] [Google Scholar]
- 18.Suemori S, Lynch-Devaney K, Podolsky DK. 1991. Identification and characterization of rat intestinal trefoil factor: tissue- and cell-specific member of the trefoil protein family. Proc Natl Acad Sci U S A 88:11017–11021. doi: 10.1073/pnas.88.24.11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang Z, Lossie AC, Applegate TJ. 2011. Evolution of trefoil factor(s): genetic and spatio-temporal expression of trefoil factor 2 in the chicken (Gallus gallus domesticus). PLoS One 6:e22691. doi: 10.1371/journal.pone.0022691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Familari M, Cook GA, Taupin DR, Marryatt G, Yeomans ND, Giraud AS. 1998. Trefoil peptides are early markers of gastrointestinal maturation in the rat. Int J Dev Biol 42:783–789. [PubMed] [Google Scholar]
- 21.Lin J, Holzman IR, Jiang P, Babyatsky MW. 1999. Expression of intestinal trefoil factor in developing rat intestine. Biol Neonate 76:92–97. doi: 10.1159/000014146. [DOI] [PubMed] [Google Scholar]
- 22.Cong L, Zhang F. 2015. Genome engineering using CRISPR-Cas9 system. Methods Mol Biol 1239:197–217. doi: 10.1007/978-1-4939-1862-1_10. [DOI] [PubMed] [Google Scholar]
- 23.Jeffrey GP, Oates PS, Wang TC, Babyatsky MW, Brand SJ. 1994. Spasmolytic polypeptide: a trefoil peptide secreted by rat gastric mucous cells. Gastroenterology 106:336–345. doi: 10.1016/0016-5085(94)90590-8. [DOI] [PubMed] [Google Scholar]
- 24.Hanisch FG, Bonar D, Schloerer N, Schroten H. 2014. Human trefoil factor 2 is a lectin that binds α-GlcNAc-capped mucin glycans with antibiotic activity against Helicobacter pylori. J Biol Chem 289:27363–27375. doi: 10.1074/jbc.M114.597757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cunliffe RN. 2003. αdefensins in the gastrointestinal tract. Mol Immunol 40:463–467. doi: 10.1016/S0161-5890(03)00157-3. [DOI] [PubMed] [Google Scholar]
- 26.Dalgakiran F, Witcomb LA, McCarthy AJ, Birchenough GMH, Taylor PW. 2014. Non-invasive model of neuropathogenic Escherichia coli infection in the neonatal rat. J Vis Exp 92:e52018. doi: 10.3791/52018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Witcomb LA, Czupryna J, Francis KP, Frankel G, Taylor PW. 2017. Non-invasive three-dimensional imaging of Escherichia coli K1 infection using diffuse light imaging tomography combined with micro-computed tomography. Methods 127:62–68. doi: 10.1016/j.ymeth.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 28.Forbes GB, Reina JC. 1972. Effect of age on gastrointestinal absorption (Fe, Sr, Pb) in the rat. J Nutr 102:647–652. doi: 10.1093/jn/102.5.647. [DOI] [PubMed] [Google Scholar]
- 29.Henning SJ. 1979. Biochemistry of intestinal development. Environ Health Perspect 33:9–16. doi: 10.1289/ehp.79339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Farrell JJ, Taupin D, Koh TJ, Chen D, Zhao CM, Podolsky DK, Wang TC. 2002. TFF2/SP-deficient mice show decreased gastric proliferation, increased acid secretion, and increased susceptibility to NSAID injury. J Clin Invest 109:193–204. doi: 10.1172/JCI0212529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoffmann W. 2015. TFF2, a MUC6-binding lectin stabilizing the gastric mucus barrier and more. Int J Oncol 47:806–816. doi: 10.3892/ijo.2015.3090. [DOI] [PubMed] [Google Scholar]
- 32.Sun Y, Wu W, Zhang Y, Lv S, Wang L, Wang S, Peng X. 2009. Stability analysis of recombinant human TFF2 and its therapeutic effect on burn-induced gastric injury in mice. Burns 35:869–874. doi: 10.1016/j.burns.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 33.Xue L, Aihara E, Podolsky DK, Wang TC, Montrose MH. 2010. In vivo action of trefoil factor 2 (TFF2) to speed gastric repair is independent of cyclooxygenase. Gut 59:1184–1191. doi: 10.1136/gut.2009.205625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tran CP, Cook GA, Yeomans ND, Thim L, Giraud AS. 1999. Trefoil peptide TFF2 (spasmolytic polypeptide) potently accelerates healing and reduces inflammation in a rat model of colitis. Gut 44:636–642. doi: 10.1136/gut.44.5.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Achtman M, Mercer A, Kusećek B, Pohl A, Heuzenroeder M, Aaronson W, Sutton A, Silver RP. 1983. Six widespread bacterial clones among Escherichia coli K1 isolates. Infect Immun 39:315–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCarthy AJ, Negus D, Martin P, Pechincha C, Oswald E, Stabler RA, Taylor PW. 2016. Pathoadaptive mutations of Escherichia coli K1 in experimental neonatal systemic infection. PLoS One 11:e0166793. doi: 10.1371/journal.pone.0166793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Witcomb LA, Collins JW, McCarthy AJ, Frankel G, Taylor PW. 2015. Bioluminescent imaging reveals novel patterns of colonization and invasion in systemic Escherichia coli K1 experimental infection in the neonatal rat. Infect Immun 83:4528–4540. doi: 10.1128/IAI.00953-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.