

Human Q fever is caused by the intracellular bacterial pathogen Coxiella burnetii. Q fever presents with acute flu-like and pulmonary symptoms or can progress to chronic, severe endocarditis.
KEYWORDS: Coxiella burnetii, intracellular pathogen, macrophage
ABSTRACT
Human Q fever is caused by the intracellular bacterial pathogen Coxiella burnetii. Q fever presents with acute flu-like and pulmonary symptoms or can progress to chronic, severe endocarditis. After human inhalation, C. burnetii is engulfed by alveolar macrophages and transits through the phagolysosomal maturation pathway, resisting the acidic pH of lysosomes to form a parasitophorous vacuole (PV) in which to replicate. Previous studies showed that C. burnetii replicates efficiently in primary human alveolar macrophages (hAMs) in ex vivo human lung tissue. Although C. burnetii replicates in most cell types in vitro, the pathogen does not grow in non-hAM cells of human lung tissue. In this study, we investigated the interaction between C. burnetii and other pulmonary cell types apart from the lung environment. C. burnetii formed a prototypical PV and replicated efficiently in human pulmonary fibroblasts and in airway, but not alveolar, epithelial cells. Atypical PV expansion in alveolar epithelial cells was attributed in part to defective recruitment of autophagy-related proteins. Further assessment of the C. burnetii growth niche showed that macrophages mounted a robust interleukin 8 (IL-8), neutrophil-attracting response to C. burnetii and ultimately shifted to an M2-polarized phenotype characteristic of anti-inflammatory macrophages. Considering our findings together, this study provides further clarity on the unique C. burnetii-lung dynamic during early stages of human acute Q fever.
INTRODUCTION
The human lung is a complex environment, containing numerous cell types with distinct functions. Epithelial cells are the main structural component of the airway, allowing the passage of oxygen to the alveoli for respiration (1). Type I epithelial cells form the alveolar wall, and type II epithelial cells along the wall produce components, such as surfactants, that interact with pathogens and trigger immune cell activation (2). Alveolar epithelial and endothelial cells, while serving structurally as the blood-air barrier, are necessary for gas exchange via ion transport and tissue remodeling, particularly when exposed to pathogens (1). The extracellular matrix provides structural support and is composed mainly of fibroblasts, which also aid in wound repair (3). Macrophages present within the lung allow rapid innate responses to foreign material. Two major types of pulmonary macrophages are present within interstitial and alveolar spaces, and they represent the first line of defense against inhaled microorganisms (4). Alveolar macrophages adhere loosely to epithelial cells, where they engage and remove foreign particles, including microbes (5). Finally, a limited number of neutrophils are present in the alveolar region, and additional neutrophils can be recruited to amplify the innate response to pathogens. Thus, the lung is well situated to respond rapidly to inhaled material but has been exploited by multiple bacterial pathogens as a targeted replication niche.
Coxiella burnetii is a highly infectious intracellular pulmonary bacterial pathogen that inhabits livestock in nature and causes human Q fever. C. burnetii has a global distribution and causes significant disease in outbreaks associated with infected livestock (6–8). Natural human transmission occurs by spread of contaminated aerosols, resulting in an initial pulmonary infection and additional flu-like symptoms during acute Q fever. By an unknown mechanism, C. burnetii escapes the lung environment and causes chronic disease, typically presenting as endocarditis. C. burnetii is a category B select agent with potential use as a bioweapon, and no worldwide vaccine is currently approved for public use (9). Due to these characteristics and the existence of an environmentally stable form outside host cells, it is critical to fully understand human pulmonary infection by C. burnetii in order to design appropriate therapeutic approaches to combat Q fever.
Following inhalation of C. burnetii, human alveolar macrophages (hAMs) are targeted for intracellular survival and replication within a host membrane-derived parasitophorous vacuole (PV). Trafficking through the phagolysosomal maturation pathway, the PV fuses with endosomes, autophagosomes, and acidic lysosomes (pH ∼5.0) to create a large, lysosome-like PV. C. burnetii uses a Dot/Icm type IV secretion system (T4SS) to secrete bacterial proteins into the host cytoplasm and generate the PV (10, 11). The pathogen also uses T4SS effectors to prevent apoptosis and alter host signaling events to promote infection (12, 13). Two major variants of C. burnetii exist: phase I and phase II (14). Phase I organisms are virulent, producing a full-length lipopolysaccharide (LPS) to evade innate immune recognition. Phase II organisms have a truncated O antigen, resulting in avirulent bacteria that are cleared by an immunocompetent host yet are widely used to model interactions with the host cell in vitro (15). The T4SS and LPS represent the two best-characterized C. burnetii virulence determinants, and animal models of infection have confirmed the importance of both factors in C. burnetii pathogenesis (14, 16). However, the pathogen does not replicate efficiently or cause severe disease in immunocompetent mouse models, suggesting that C. burnetii is a human-adapted pathogen. Therefore, human-derived models of infection are critical to defining the host response to C. burnetii.
The early stages of Q fever occur in the lung, specifically within the alveoli. We recently developed a novel human tissue infection system to assess C. burnetii interactions with lung tissue and cells, and we found that avirulent organisms trigger a robust hAM-mediated interleukin 1β (IL-1β) inflammatory response to infection (11, 17). This system consists of postmortem human lungs that are used to harvest primary pulmonary cells and are processed into tissue slices in order to study the innate response to bacteria. Interestingly, C. burnetii replicates efficiently in hAMs but does not replicate in other cell types in lung tissue, such as epithelial cells and fibroblasts. In contrast, C. burnetii replicates in most cell types tested in vitro, including macrophages, epithelial cells, and fibroblasts (10, 18), suggesting that some pulmonary cells restrict infection within the lung environment. These reports and our previous results suggest that C. burnetii-infected hAMs or other lung components affect the ability of other pulmonary cells to support bacterial growth in human lung tissue.
To investigate the ability of nonmacrophage lung cells to suppress C. burnetii replication, we used established human pulmonary cell lines and primary cells to assess infection. The results indicate that C. burnetii replicates poorly in alveolar epithelial cells, while displaying robust replication in macrophages and primary lung fibroblasts. The autophagy-related proteins LC3 and p62 are recruited to PVs in all cell types examined except alveolar epithelial cells, suggesting defective heterotypic PV fusion in these cells. Finally, C. burnetii-infected hAMs produce significant levels of the neutrophil attractant IL-8 while shifting from an M1 to M2 phenotype. Together, these results define novel interactions between C. burnetii and distinct pulmonary cell types within the human alveolar region, and they allow enhanced modeling of initial stages of Q fever.
RESULTS
C. burnetii preferentially replicates within hAMs in human lung tissue.
We reported previously that C. burnetii replicates to high numbers in primary hAMs (11, 17). Although the pathogen enters and resides within other pulmonary cell types, it does not display robust replication when these cells are contained within human precision-cut lung slices (hPCLS). However, the nature of other cell types infected by C. burnetii within human lung tissue has not been defined. To define pulmonary cells that harbor nonreplicating C. burnetii within hPCLS, we used antibodies directed against defined cell type-specific proteins (see Fig. S1 in the supplemental material). As shown in Fig. 1, C. burnetii formed a large vacuole and replicated to high numbers in hAMs, as observed previously (17), while only individual bacteria were present in other cell types. Antibody-based labeling indicated that C. burnetii was engulfed by, but did not replicate within, epithelial cells, fibroblasts, and interstitial macrophages. These results suggest that nonalveolar macrophages have the capacity to suppress C. burnetii replication within the lung environment.
FIG 1.

C. burnetii preferentially replicates within alveolar macrophages in human lung tissue. Human precision-cut lung slices (hPCLS) were infected with mCherry-expressing C. burnetii (red) for 72 h. Samples were processed for confocal microscopy using DAPI (blue) to stain nuclei and antibodies directed against CD10 (fibroblasts), cytokeratin (epithelial cells), CD206 (alveolar macrophages), or CD169 (interstitial macrophages). All cell marker proteins are shown in green. AS, alveolar space. Images are representative of experiments from three different donors. Bars, 20 μm. C. burnetii is located in alveolar macrophages and areas containing fibroblasts, epithelial cells, and interstitial macrophages, but only alveolar macrophages contain expanded PVs filled with bacteria.
Alveolar epithelial cells support inefficient C. burnetii replication in vitro.
Numerous cell types have been used in vitro that support C. burnetii replication. Thus, C. burnetii-infected hAMs may produce molecules that influence bacterial replication in other cell types when they are present together in the lung. To assess this possibility, we first assessed C. burnetii infection of individual cultures of the THP-1 (human macrophage-like), A549 (human alveolar epithelial), and H292 (human airway epithelial) cell lines and of primary human lung fibroblasts (HLFs). Attempts to investigate primary epithelial cells isolated from human lung tissue were unsuccessful due to the technical difficulty of obtaining sufficient numbers of cells for analysis. As seen in Fig. 2A, THP-1 cells, a commonly used model of C. burnetii-macrophage interactions (19–21), supported the formation of large CD63-positive PVs that expanded from 2 to 96 h postinfection (hpi). In contrast, A549 and H292 cells supported the generation of significantly smaller, atypical PVs across the same time course despite the uptake of similar numbers of bacteria at 2 hpi (data not shown). Interestingly, primary HLFs supported C. burnetii growth in large PVs similar to those in THP-1 cells. To quantify cell type-specific differences in PV size, the vacuole area was determined in each cell type. As shown in Fig. 2B, PVs in THP-1 cells and HLFs were similar in size, whereas A549 and H292 cells contained significantly smaller PVs.
FIG 2.

PV formation by C. burnetii differs by cell type. (A) THP-1, H292, and A549 cells and HLFs were infected with C. burnetii for 96 h. Cells were prepared for fluorescence microscopy using DAPI (blue) to stain nuclei, a CD63 antibody (green) to visualize PVs, and a C. burnetii antibody (red) to observe bacteria. Images are representative of experiments performed in triplicate. Bars, 20 μm. Large, C. burnetii-filled PVs are present in THP-1 cells and HLFs; smaller PVs are present in H292 cells, and small, atypical PVs containing few bacteria are seen in A549 cells. (B) The areas of individual PVs in each cell type were quantified and averaged in triplicate. ****, P < 0.0001; n.s., not significant. PV areas in H292 cells, A549 cells, and HLFs were compared to those in THP-1 cells for statistical analysis. PVs in A549 and H292 cells are significantly smaller than those in THP-1 cells, while the size of PVs in HLFs is not significantly different from that in THP-1 cells.
In order to confirm that cell type-dependent differences in PV expansion correlate with bacterial numbers, focus-forming unit (FFU) assays were performed to quantify infectious bacteria in each cell type (22). As shown in Fig. 3, the numbers of FFUs in THP-1 cells and HLFs were comparable. In contrast, the numbers of FFUs in A549 and H292 cells were significantly lower than those in THP-1 cells and HLFs, confirming that C. burnetii does not replicate to the same extent in the former cells. Furthermore, the number of FFUs was significantly lower in A549 cells than in H292 cells. These results indicate that fibroblasts apart from the lung environment support C. burnetii replication, while alveolar epithelial cells do not support C. burnetii replication to the same extent as other pulmonary cells.
FIG 3.
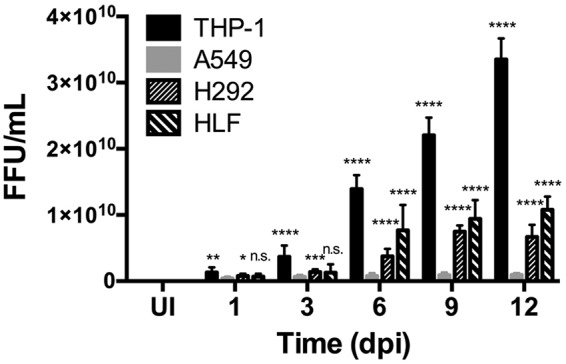
C. burnetii replication differs by cell type. THP-1, A549, and H292 cells and HLFs were infected with C. burnetii for 1, 3, 6, 9, or 12 days. At each time point, samples were harvested, sonicated, and incubated with Vero cells for 4 days; then they were processed for fluorescence microscopy using an antibody against C. burnetii in triplicate in order to quantify the bacterial focus-forming units (FFUs). UI, uninfected cells. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; n.s., not significant. C. burnetii foci are present in the highest numbers in THP-1 cells, followed by HLFs, H292 cells, and A549 cells, respectively. From day 6 to day 12, THP-1 cells, H292 cells, and HLFs contained significantly higher numbers of C. burnetii than A549 cells, indicating more-efficient replication in nonalveolar epithelial cells.
C. burnetii generates a lysosome-like vacuole in each pulmonary cell type.
A factor contributing to reduced C. burnetii replication within epithelial cells could be failure to generate a prototypical PV that supports bacterial growth. The PV is a degradative compartment that matures via interaction with lysosomes, which have an acidic pH and contain proteases (23), including the aspartyl protease cathepsin D. This environment is required for the activation of C. burnetii metabolism and resultant growth (24). To determine if C. burnetii generates a prototypical PV in distinct pulmonary cell types, THP-1, A549, and H292 cells and HLFs were infected for 72 h and then processed for microscopy to visualize cathepsin D and C. burnetii within the PV (Fig. 4). Regardless of cell type or PV size, C. burnetii was harbored within a vacuole that was decorated with cathepsin D, indicating that PV maturation is not altered in these cell types.
FIG 4.

C. burnetii forms a lysosome-like PV in all pulmonary cell types. THP-1, A549, and H292 cells and HLFs were infected with C. burnetii for 72 h. Cells were processed for fluorescence microscopy using DAPI (blue) to stain nuclei, a cathepsin D antibody (red) as a lysosomal protein marker, and a C. burnetii antibody (violet) to observe bacteria. Images are representative of three separate experiments. Bars, 20 μm. PVs in all cells are decorated with the lysosomal marker cathepsin D, indicating that PV maturation occurs regardless of cell type.
The autophagy-related proteins LC3 and p62 do not localize to PVs in alveolar epithelial cells.
Decreased C. burnetii replication in alveolar epithelial cells could be due to active suppression of the pathogen or, alternatively, to reductions in fusogenic events that promote the expansion of the PV required for bacterial growth. In addition to lysosomal fusion, addressed above, C. burnetii actively engages host cell autophagy, recruiting LC3-positive autophagosomes and the cargo adaptor p62 to the PV (19, 20). Autophagosome fusion with the PV is actively directed by the C. burnetii T4SS and is predicted to allow the acquisition of nutrients and a membrane for the expanding PV (25–27). Thus, a reduced autophagic response could prevent optimal PV expansion and bacterial growth. In order to test this possibility, we assessed the localization of LC3 and p62 by fluorescence microscopy to determine if autophagy-related proteins are recruited to the PV in each pulmonary cell type. PVs in THP-1 and H292 cells and HLFs associated with LC3 and p62, while PVs in A549 cells were devoid of both proteins (Fig. 5). The absence of LC3 and p62 at the PV in A549 cells is not due to an absence of total protein, since a signal is observed in the cytoplasm of infected cells distinct from the PV. Additionally, Western blot analysis confirmed that both proteins, including lipidated LC3-II, indicative of active autophagy, are present in all uninfected and C. burnetii-infected cell types (data not shown). These results suggest that C. burnetii is unable to recruit autophagosomes to the PV in alveolar epithelial cells and provide one reason why the PV remains atypically small in these cells.
FIG 5.

The autophagy-related proteins LC3 and p62 do not localize to PVs in alveolar epithelial cells. (A) THP-1 and A549 cells were infected with C. burnetii for 72 h. Cells were processed for fluorescence microscopy using antibodies against LC3 (green), p62 (green), CD63 (red), and C. burnetii (violet), with DAPI to stain nuclei (blue). Images are representative of three separate biological experiments. Bars, 20 μm. THP-1 and H292 cells and HLFs contain PVs that are decorated with LC3 and p62, while PVs in A549 cells are devoid of both proteins. (B) The colocalization of LC3 or p62 with PVs in each cell type at 72 hpi was quantified and averaged. ***, P < 0.001; ****, P < 0.0001; n.s., not significant. A549, H292, and HLF data were compared to THP-1 data for statistical analysis. PVs in A549 cells are devoid of LC3 and p62, whereas PVs in HLFs are not significantly different from those in THP-1 cells.
C. burnetii replicates in HLFs exposed to infected hAM supernatants.
While alveolar epithelial cells do not support efficient C. burnetii growth within or apart from the lung environment, primary HLFs supported robust C. burnetii replication in isolated cultures but not when present within hPCLS. To determine if infected hAMs produce signals that suppress replication in HLFs via cell-cell communication, supernatants from C. burnetii-infected hAMs containing any secreted macrophage components were incubated with HLFs; then cells were infected and PV formation assessed at 72 hpi. As shown in Fig. S2 in the supplemental material, the addition of hAM supernatants to HLFs had no significant impact on PV formation, suggesting that another mechanism prevents C. burnetii replication in fibroblasts within hPCLS.
SP-D antagonizes PV expansion and C. burnetii replication in HLFs.
To investigate components in hPCLS that prevent C. burnetii replication in HLFs, we assessed the impact of surfactant protein D (SP-D) on this interaction. SP-D is constitutively produced by alveolar epithelial cells (2, 28) and has been shown to decrease bacterial replication and increase bacterial degradation (2). Therefore, we assessed the effect of SP-D on PV expansion and C. burnetii replication in HLFs as a potential mechanism for suppressing pathogen growth in hPCLS fibroblasts. HLFs were incubated with increasing concentrations of recombinant SP-D during infection and were then analyzed using fluorescence microscopy and FFU analysis. As shown in Fig. 6A, SP-D significantly decreased PV expansion in a dose-dependent manner. Previous SP-D studies had assessed the impact of the protein on C. burnetii growth in macrophages (29). To determine if SP-D also serves an antibacterial role in human macrophages, hAMs were incubated with SP-D at the same concentrations used with HLFs, and PV formation was assessed. As shown in Fig. 6B, PV formation was unaltered by SP-D at the concentrations tested, indicating that the protein does not prevent C. burnetii infection or replication in hAMs and suggesting that SP-D affects other cells, such as fibroblasts, in hPCLS. Correlating with the PV expansion results, higher concentrations of SP-D significantly decreased C. burnetii replication in HLFs but not in hAMs (Fig. 6C), suggesting a role for SP-D in suppressing growth in pulmonary fibroblasts. Finally, we assessed whether SP-D prevention of typical PV expansion was due to a lack of recruitment of autophagy proteins, as in the A549 results presented above. As shown in Fig. 6D, SP-D did not alter the localization of p62 or LC3 to the PV, suggesting that a different mechanism suppresses PV expansion during SP-D treatment.
FIG 6.

Surfactant protein D antagonizes PV expansion and C. burnetii replication. (A and B) C. burnetii was incubated with surfactant protein D (SP-D) at the indicated concentrations for 1 h at 37°C. HLFs (A) or hAMs (B) were infected with either WT or SP-D-treated C. burnetii for 72 h. Cells were processed for fluorescence microscopy using antibodies against CD63 and C. burnetii, with DAPI to stain nuclei. PV areas under each condition were quantified and averaged from triplicate biological experiments. SP-D antagonizes PV expansion in HLFs in a dose-dependent manner but does not impact PV expansion in hAMs. ***, P < 0.001; n.s., not significant. (C) HLFs or hAMs were infected with WT or SP-D-treated C. burnetii for 6 days, harvested, sonicated, and plated onto Vero cells for 4 days. Cells were processed for fluorescence microscopy and FFUs quantified using a C. burnetii antibody, with DAPI to stain nuclei. C. burnetii replication decreases significantly in the presence of increasing concentrations of SP-D. (D) HLFs or hAMs were infected with either WT or SP-D-treated C. burnetii for 72 h and were then processed for fluorescence microscopy using antibodies against p62, LC3, and C. burnetii, with DAPI to stain nuclei. p62 and LC3 localization to individual PVs was quantified and averaged from triplicate experiments. SP-D treatment does not alter the recruitment of p62 or LC3 to the PV in either cell type.
hAMs secrete the neutrophil attractant IL-8 in response to C. burnetii.
The results presented above further suggest that hAMs are the preferred replication niche for C. burnetii during pulmonary infection. During natural infection, neutrophil influx plays a key role in C. burnetii clearance (30), but the role of neutrophils has not been assessed within the context of human lungs. In humans, the chemokine IL-8 is produced by macrophages in response to infection and serves as a neutrophil-attracting signal (31). Thus, to generate a clearer picture of the C. burnetii-lung dynamic and cellular response to infection, hAMs were assessed by an enzyme-linked immunosorbent assay (ELISA) using cells from three separate donors to analyze the IL-8 response to C. burnetii. As shown in Fig. 7, all donor hAMs displayed significant IL-8 production from 2 to 72 hpi, indicating that hAMs respond appropriately to C. burnetii by generating signals that recruit other immune cells to the infected region.
FIG 7.
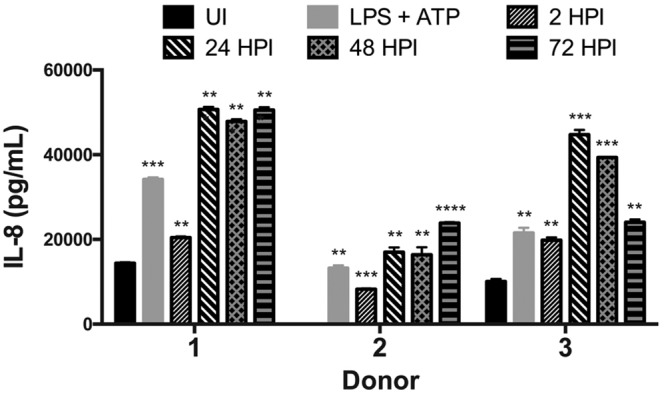
hAMs secrete substantial levels of the chemokine IL-8 in response to C. burnetii. hAMs from three separate donors were infected with C. burnetii for 2, 24, 48, or 72 h. At each time point, supernatants were harvested and assessed for the presence of IL-8 via ELISA in triplicate. UI, uninfected cell supernatants. Supernatants from cells treated with LPS and ATP served as a positive control. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. All donor hAMs display significant IL-8 secretion during C. burnetii infection.
C. burnetii-infected hAMs shift to an M2 polarization phenotype.
Our previous work (11), combined with the IL-8 data presented above, suggests that hAMs are initially able to mount an effective inflammatory response to C. burnetii. Other researchers have suggested that C. burnetii must combat this response to establish primary infection and promote chronic infection (14, 16). Proinflammatory hAMs typically display markers of M1 polarization, while anti-inflammatory macrophages produce M2 polarization markers and are often more hospitable for intracellular pathogen proliferation. To determine if C. burnetii-infected macrophages are primarily M1- or M2-polarized, hAMs were infected with C. burnetii for 72 h and were then prepared for fluorescence microscopy using antibodies directed against the M1 marker CXCL10 or the M2 marker TGM2. As shown in Fig. 8, C. burnetii infection triggers a significant phenotypic switch from primarily M1 to M2 hAMs, as evidenced by decreased CXCL10 production (Fig. 8A) and increased TGM2 production (Fig. 8B). These data agree with our previous results showing that the M2 cytokine IL-10 is produced as early as 24 hpi by hAMs (11). Furthermore, hAMs respond to C. burnetii by secreting significant amounts of the M1 cytokine tumor necrosis factor alpha (TNF-α) at 24 to 48 hpi; however, TNF-α drops to near-undetectable levels by 72 hpi (not shown), the time at which TGM2 expression increases. Taken together, these results suggest that M2-polarized macrophages are the preferred macrophage niche for C. burnetii replication in the human lung.
FIG 8.
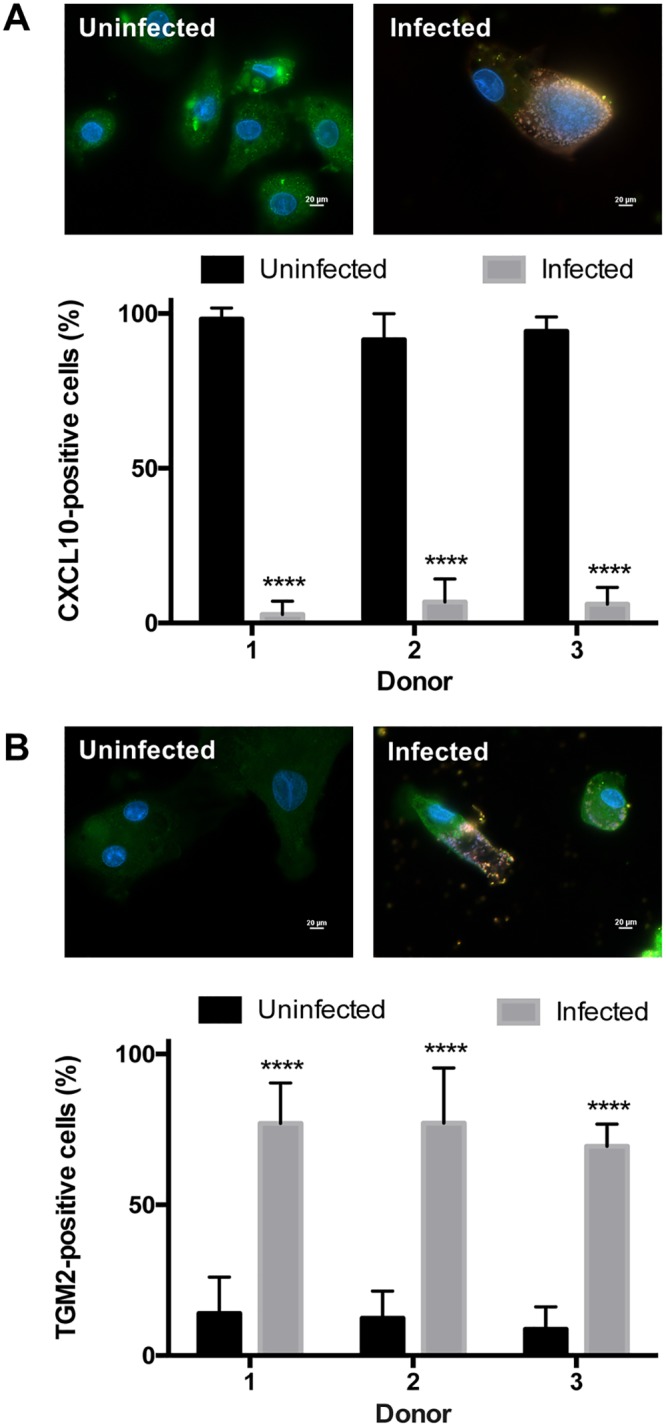
hAMs convert from an M1 to an M2 phenotype during C. burnetii infection. hAMs from three separate donors were infected with C. burnetii for 72 h. Samples were processed for fluorescence microscopy using antibodies against the M1 macrophage marker CXCL10 (green) (A), the TGM2 macrophage marker (green) (B), and C. burnetii (red), with DAPI to stain nuclei (blue), in three separate experiments. ****, P < 0.0001. Representative images are shown for each marker. Bars, 20 μm. During C. burnetii infection, hAMs contain significantly decreased CXCL10 expression and significantly increased TGM2 expression.
DISCUSSION
In the current study, we refine our understanding of the interaction between C. burnetii and the human lung by using our hPCLS infection system. Previous studies showed that C. burnetii replicates to high numbers specifically within hAMs (11, 17), but other infected cell types were not defined. Using cell type-specific antibodies, we observe C. burnetii within hAMs, alveolar epithelial cells, interstitial macrophages, and interstitial fibroblasts. Interestingly, as seen previously, C. burnetii does not replicate within any cell type other than hAMs in the lung environment, suggesting that non-hAM cells suppress replication. Alternatively, infected hAMs, or other cells, may produce antireplication signals that are transmitted to other alveolar cells, preventing bacterial growth. These findings are intriguing, since C. burnetii replicates in all other cell types reported (10).
Although C. burnetii does not grow efficiently in nonalveolar macrophages in human lung tissue, the pathogen forms a large PV and replicates within primary human pulmonary fibroblasts when removed from the lung environment. Due to the difficulty of obtaining sufficient numbers of primary alveolar epithelial cells, the A549 alveolar epithelial cell line was used as an alternative infection model. As with alveolar epithelial cells in hPCLS, C. burnetii does not replicate efficiently in A549 cells, forming small, atypical PV. In contrast, replication occurs in H292 airway epithelial cells, albeit in atypical PVs, suggesting that C. burnetii growth is specifically diminished in alveolar epithelial cells. Airway epithelial cells and fibroblasts represent potential niches for C. burnetii growth in the lung, further suggesting the presence of antireplication components in intact lung tissue, where bacterial replication occurs only in hAMs. These findings agree with a recent report investigating C. burnetii infection of bovine epithelial cells isolated from diverse organ sites. That study showed that, of five different bovine cell origins, C. burnetii replicates most poorly in lung epithelial cells (32).
The inability of C. burnetii to replicate efficiently in A549 cells is surprising, since many cell types, including epithelial cell lines, are used to model the pathogen’s cellular infectious process (33, 34). C. burnetii forms small, atypical PVs in A549 cells that are decorated with lysosomal cathepsin D, indicating that the pathogen resides within a lysosomal compartment in these cells. Similar PV maturation also occurs in airway epithelial cells, primary fibroblasts, and THP-1 macrophage-like cells, as seen previously (19, 23, 35). Thus, C. burnetii is able to form a PV with lysosomal characteristics but is unable to replicate efficiently in alveolar epithelial cells. PV fusion with autophagosomes is directed by the C. burnetii T4SS and is required for optimal PV expansion (19, 20, 36). Interestingly, the PV is decorated with LC3 in each cell type other than A549 cells, suggesting that autophagosome recruitment is defective in alveolar epithelial cells. Moreover, the autophagy adaptor p62 is present on PVs in all cell types except A549 cells. T4SS-mediated recruitment of p62 is predicted to benefit the pathogen by delivering ubiquitinated proteins to the vacuole (19, 20). The absence of p62 at the PV in A549 cells further suggests that this protein is required for optimal vacuole expansion, and proteins delivered by p62 are currently being investigated. Together, these results suggest that defective autophagosome recruitment contributes to suboptimal PV expansion in human alveolar epithelial cells.
One major aspect of the innate immune response that is difficult to mimic in hPCLS is the neutrophil influx that occurs during acute Q fever (30). Animal model studies indicate that C. burnetii can infect, and remain viable within, neutrophils but does not use these cells as a replication niche (30, 37). However, neutrophils are recruited to the lungs of C. burnetii-infected mice, supporting the importance of this innate immune response (30). To initially address neutrophil importance during human lung infection, we determined if C. burnetii-infected primary hAMs produce neutrophil-recruiting signals. Indeed, IL-8 (CXCL8) levels in infected hAM supernatants increase significantly from 2 to 24 hpi and remain elevated through 72 hpi, indicating that hAMs respond appropriately to the pathogen by producing neutrophil attractants. Neutrophils are not detected within the alveoli of C. burnetii-infected mice until 7 days postinfection, suggesting a delayed response to C. burnetii that can now be assessed using our hPCLS platform, long-term IL-8 analysis, and isolated human neutrophils. This approach establishes a readout that can be used to further understand the hAM response to differing C. burnetii pathotypes that cause distinct forms of Q fever (11).
Intracellular macrophage-targeting pathogens typically attempt to disarm the initial cell in which they reside so as to avoid detection by the immune system. Thus, we also assessed whether C. burnetii alters the polarization status of hAMs. M1 macrophages are highly inflammatory and produce several signals, including IL-1β, IL-6, and TNF-α, that activate downstream immune responses (38). Interestingly, C. burnetii triggers significant conversion of infected hAMs to an M2 phenotype, which is anti-inflammatory and is typically accompanied by IL-10 production (39). C. burnetii-infected hAMs produce substantial levels of IL-10 at 24 hpi (11), correlating with the shift to M2 polarization observed here. Together, these results indicate that hAMs initially respond to C. burnetii by secreting neutrophil attractants, while the pathogen combats macrophage activation by stimulating M2 polarization. This activity is predicted to promote the progression of infection to chronic Q fever, since IL-10-deficient mice are unable to clear C. burnetii (39), further validating the use of our human-derived infection system to inform us about downstream infection events.
Finally, surfactant proteins play a major role in the pulmonary response to microbial pathogens (28). Surfactants are produced by type II alveolar epithelial cells and affect phagocytosis and innate immune signaling events that respond to microbial infection (2). Specifically, surfactant protein D coats bacteria and enhances phagocytosis, leading to efficient destruction of engulfed organisms (2). Although C. burnetii is not degraded in nonmacrophage pulmonary cells, as evidenced by sustained expression of mCherry in hPCLS, surfactants may suppress bacterial replication in non-hAM cells by altering signaling and/or vesicular fusion events. Indeed, PVs in SP-D-treated fibroblasts are significantly smaller than PVs in untreated cells and contain fewer bacteria, suggesting that this protein antagonizes optimal vacuole expansion. These results correlate with a recent study showing that SP-D prevents normal PV formation in mouse macrophages (29). In contrast, SP-D does not impact PV formation in primary hAMs, indicating an important difference between these cells and macrophages of murine origin. This finding provides another plausible reason for C. burnetii to target hAMs, which appear to be refractory to common antibacterial activities of SP-D.
Taking our findings together, the current study defines novel aspects of C. burnetii interactions with human lung cells and tissue and supports the prediction that the pathogen alters hAM activation to trigger an anti-inflammatory environment supportive of chronic infection. Using our disease-relevant hPCLS and hAM infection systems, we can assess numerous innate immune response events that allow enhanced modeling of pulmonary pathogenesis. Combining previous results and the current data regarding hAM production of neutrophil attractants and the role of surfactant proteins in bacterial infection, we are continually uncovering new aspects of the hPCLS and hAM infection platforms that can be used to investigate diverse pulmonary pathogens in a human-disease-relevant ex vivo setting.
MATERIALS AND METHODS
Eukaryotic cell culture.
THP-1 human monocytes (TIB-202) were acquired from the American Type Culture Collection (ATCC) and were cultured in RPMI 1640 medium with 10% fetal bovine serum (FBS) at 37°C under 5% CO2. By use of phorbol 12-myristate 13-acetate (PMA; 200 nM), cells were differentiated into macrophage-like cells overnight. The medium containing PMA was exchanged for a medium lacking PMA 4 h prior to infection. A549 human alveolar epithelial cells (CCL-185; ATCC), NCI-H292 human airway epithelial cells (CRL-1848; ATCC), primary human lung fibroblasts (FC-0049; LifeLine Cell Tech), and Vero green monkey epithelial cells (CCL-81; ATCC) were cultured in RPMI 1640 medium with 10% FBS at 37°C under 5% CO2.
Primary human alveolar macrophages (hAMs) were obtained via bronchoalveolar lavage (BAL) from postmortem human lung donors (National Disease Research Interchange). Lungs and BAL fluid were processed as described previously (11). hAMs were cultured at 37°C under 5% CO2 in Dulbecco’s modified Eagle/F-12 (DMEM/F12) medium containing 10% FBS, penicillin (50 U/ml), streptomycin sulfate (50 μg/ml), gentamicin sulfate (50 μg/ml), and amphotericin B (0.25 μg/ml). Before infection with C. burnetii, the antibiotic/antimycotic-containing medium was removed and replaced with DMEM/F12 medium containing 10% FBS for 24 h.
hPCLS preparation.
Low-melting-temperature agarose was instilled into postmortem human lungs, which were then incubated at 4°C for solidification. Human precision-cut lung slices (hPCLS) (diameter, 8 mm; thickness, 750 μm) were obtained using a microtome (Compresstome; Precisionary Instruments) to section cores from 1-in-thick lung slices. hPCLS were cultured at 37°C under 5% CO2 in DMEM/F12 medium containing 10% FBS, penicillin (50 U/ml), streptomycin sulfate (50 μg/ml), gentamicin sulfate (50 μg/ml), and amphotericin B (0.25 μg/ml). The antibiotic/antimycotic-containing medium was replaced with DMEM/F12 medium containing 10% FBS 24 h prior to infection with mCherry-expressing C. burnetii.
C. burnetii cultivation.
Wild-type C. burnetii and mCherry-expressing C. burnetii (Nine Mile II; RSA439, clone 4) were cultured in acidified citrate cysteine medium (ACCM) for 7 days at 37°C under 5% CO2 and 2.5% O2. mCherry-expressing bacterial cultures contained chloramphenicol (3 μg/ml) for plasmid maintenance. After 7 days of growth, bacteria were pelleted by centrifugation, washed in sucrose phosphate (SP) buffer, and stored at –80°C in SP buffer. All cell and tissue infections were performed using a multiplicity of infection of 10.
Immunofluorescence microscopy.
THP-1, A549, or H292 cells or HLFs were cultured in 24-well plates on 12-mm glass coverslips. After infection with C. burnetii, cells were washed three times with cold phosphate-buffered saline (PBS) and were fixed either with cold methanol for 3 min or with formaldehyde for 15 min. Cells were washed three times with cold PBS after fixing and were blocked in a buffer containing either 0.5% bovine serum albumin (BSA) alone or 0.5% BSA containing 0.3% Triton X-100 for 1 h at room temperature. Samples were then incubated in a blocking buffer containing primary antibodies directed against CD63 (BD Biosciences), CD10 (BioLegend), pan-cytokeratin (Abcam), CD206 (BioLegend), CD169 (Abcam), cathepsin D (Sigma-Aldrich), CXCL10 (Abcam), LC3 (Cell Signaling), p62 (Sigma-Aldrich), immunofluorescence-specific p62 (Cell Signaling), TGM2 (Abcam), or C. burnetii for 1 h at room temperature. Samples were then washed three times with cold PBS and were incubated with secondary antibodies conjugated to Alexa Fluor 488, Alexa Fluor 594, or Alexa Fluor 647 (Invitrogen) for 1 h at room temperature. Finally, samples were washed three times with cold PBS for 5 min and were stained with 4',6-diamidino-2-phenylindole (DAPI; Invitrogen) for 5 min at room temperature. Coverslips were mounted onto slides using Mowiol and were incubated at room temperature overnight. Cells were visualized using a Ti-U microscope (Nikon), a 60× objective, and a D5-Qi1Mc digital camera (Nikon), or a Ti Eclipse confocal microscope (Nikon). Images were analyzed using NIS Elements software (Nikon). PV areas were quantified and averaged from at least 15 infected cells by use of NIS Elements software.
Bacterial growth analysis.
For focus-forming unit (FFU) assays (22), THP-1, A549, or H292 cells or HLFs were cultured in a 24-well plate and were infected with C. burnetii for 1 to 12 days. Cells were then harvested by scraping and were sonicated twice for 15 s on ice. Sonicated samples containing released bacteria were applied to Vero cells in a 24-well plate on coverslips. At 4 days postinfection, cells were fixed using cold methanol for 3 min and were processed for microscopy as described above using an anti-C. burnetii primary antibody and an Alexa Fluor 488-conjugated secondary antibody. After coverslips were mounted onto slides, foci of infection were quantified from three independent coverslips (10 fields/coverslip) using a 40× objective (Nikon).
Recombinant SP-D treatment.
C. burnetii was incubated with recombinant surfactant protein D (SP-D; 1 or 10 μg/ml; R&D Systems) for 1 h at 37°C. Cells were then infected with SP-D-treated C. burnetii or untreated C. burnetii for 72 h or 6 days. The medium was changed daily to replenish SP-D. After C. burnetii infection, cells were prepared for immunofluorescence microscopy or FFU analysis, and the PV area was quantified as described above.
IL-8 secretion analysis.
A standard enzyme-linked immunosorbent assay (ELISA) for IL-8 (BD Biosciences) was performed using supernatants from C. burnetii-infected hAMs. Supernatants from cells treated with LPS (6 h) and ATP (30 min) were used as a positive control, and uninfected cells served as the negative control. ELISAs were performed according to the manufacturer’s instructions.
Statistical analysis.
Results were analyzed and graphs generated using Prism, version 6.0b. Statistical analysis of results from triplicate experiments was performed using a Student t test.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by funding to D.E.V. from the NIH/NIAID (grant R21AI127931), the Arkansas Biosciences Institute, and the Center for Microbial Pathogenesis and Host Inflammatory Responses (NIH/NIGMS grant P20GM103625). R.C.K. was supported by the Arkansas Biosciences Institute and the NIH/NCRR/NCATS (grant UL1TR000039).
We have no conflicts of interest to declare.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00028-19.
REFERENCES
- 1.Rackley CR, Stripp BR. 2012. Building and maintaining the epithelium of the lung. J Clin Invest 122:2724–2730. doi: 10.1172/JCI60519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lawson PR, Reid KB. 2000. The roles of surfactant proteins A and D in innate immunity. Immunol Rev 173:66–78. doi: 10.1034/j.1600-065X.2000.917308.x. [DOI] [PubMed] [Google Scholar]
- 3.Wuyts WA, Agostini C, Antoniou KM, Bouros D, Chambers RC, Cottin V, Egan JJ, Lambrecht BN, Lories R, Parfrey H, Prasse A, Robalo-Cordeiro C, Verbeken E, Verschakelen JA, Wells AU, Verleden GM. 2013. The pathogenesis of pulmonary fibrosis: a moving target. Eur Respir J 41:1207–1218. doi: 10.1183/09031936.00073012. [DOI] [PubMed] [Google Scholar]
- 4.Schyns J, Bureau F, Marichal T. 2018. Lung interstitial macrophages: past, present, and future. J Immunol Res 2018:5160794. doi: 10.1155/2018/5160794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guth AM, Janssen WJ, Bosio CM, Crouch EC, Henson PM, Dow SW. 2009. Lung environment determines unique phenotype of alveolar macrophages. Am J Physiol Lung Cell Mol Physiol 296:L936–L946. doi: 10.1152/ajplung.90625.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Enserink M. 2010. Infectious diseases. Questions abound in Q-fever explosion in the Netherlands. Science 327:266–267. doi: 10.1126/science.327.5963.266-a. [DOI] [PubMed] [Google Scholar]
- 7.Van den Brom R, Moll L, van Schaik G, Vellema P. 2013. Demography of Q fever seroprevalence in sheep and goats in The Netherlands in 2008. Prev Vet Med 109:76–82. doi: 10.1016/j.prevetmed.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 8.Schimmer B, Schotten N, van Engelen E, Hautvast JL, Schneeberger PM, van Duijnhoven YT. 2014. Coxiella burnetii seroprevalence and risk for humans on dairy cattle farms, the Netherlands, 2010–2011. Emerg Infect Dis 20:417–425. doi: 10.3201/eid2003.131111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oyston PC, Davies C. 2011. Q fever: the neglected biothreat agent. J Med Microbiol 60:9–21. doi: 10.1099/jmm.0.024778-0. [DOI] [PubMed] [Google Scholar]
- 10.Voth DE, Heinzen RA. 2007. Lounging in a lysosome: the intracellular lifestyle of Coxiella burnetii. Cell Microbiol 9:829–840. doi: 10.1111/j.1462-5822.2007.00901.x. [DOI] [PubMed] [Google Scholar]
- 11.Graham JG, MacDonald LJ, Hussain SK, Sharma UM, Kurten RC, Voth DE. 2013. Virulent Coxiella burnetii pathotypes productively infect primary human alveolar macrophages. Cell Microbiol 15:1012–1025. doi: 10.1111/cmi.12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crabill E, Schofield WB, Newton HJ, Goodman AL, Roy CR. 2018. Dot/Icm-translocated proteins important for biogenesis of the Coxiella burnetii-containing vacuole identified by screening of an effector mutant sublibrary. Infect Immun 86:e00758-17. doi: 10.1128/IAI.00758-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luhrmann A, Nogueira CV, Carey KL, Roy CR. 2010. Inhibition of pathogen-induced apoptosis by a Coxiella burnetii type IV effector protein. Proc Natl Acad Sci U S A 107:18997–19001. doi: 10.1073/pnas.1004380107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moos A, Hackstadt T. 1987. Comparative virulence of intra- and interstrain lipopolysaccharide variants of Coxiella burnetii in the guinea pig model. Infect Immun 55:1144–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Narasaki CT, Toman R. 2012. Lipopolysaccharide of Coxiella burnetii. Adv Exp Med Biol 984:65–90. doi: 10.1007/978-94-007-4315-1_4. [DOI] [PubMed] [Google Scholar]
- 16.van Schaik EJ, Case ED, Martinez E, Bonazzi M, Samuel JE. 2017. The SCID mouse model for identifying virulence determinants in Coxiella burnetii. Front Cell Infect Microbiol 7:25. doi: 10.3389/fcimb.2017.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graham JG, Winchell CG, Kurten RC, Voth DE. 2016. Development of an ex vivo tissue platform to study the human lung response to Coxiella burnetii. Infect Immun 84:1438–1445. doi: 10.1128/IAI.00012-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller JD, Curns AT, Thompson HA. 2004. A growth study of Coxiella burnetii Nine Mile Phase I and Phase II in fibroblasts. FEMS Immunol Med Microbiol 42:291–297. doi: 10.1016/j.femsim.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 19.Winchell CG, Dragan AL, Brann KR, Onyilagha FI, Kurten RC, Voth DE. 2018. Coxiella burnetii subverts p62/sequestosome 1 and activates Nrf2 signaling in human macrophages. Infect Immun 86:e00608-17. doi: 10.1128/IAI.00608-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Winchell CG, Graham JG, Kurten RC, Voth DE. 2014. Coxiella burnetii type IV secretion-dependent recruitment of macrophage autophagosomes. Infect Immun 82:2229–2238. doi: 10.1128/IAI.01236-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.MacDonald LJ, Kurten RC, Voth DE. 2012. Coxiella burnetii alters cyclic AMP-dependent protein kinase signaling during growth in macrophages. Infect Immun 80:1980–1986. doi: 10.1128/IAI.00101-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cockrell DC, Beare PA, Fischer ER, Howe D, Heinzen RA. 2008. A method for purifying obligate intracellular Coxiella burnetii that employs digitonin lysis of host cells. J Microbiol Methods 72:321–325. doi: 10.1016/j.mimet.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Howe D, Shannon JG, Winfree S, Dorward DW, Heinzen RA. 2010. Coxiella burnetii phase I and II variants replicate with similar kinetics in degradative phagolysosome-like compartments of human macrophages. Infect Immun 78:3465–3474. doi: 10.1128/IAI.00406-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hackstadt T, Williams JC. 1981. Biochemical stratagem for obligate parasitism of eukaryotic cells by Coxiella burnetii. Proc Natl Acad Sci U S A 78:3240–3244. doi: 10.1073/pnas.78.5.3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Colombo MI. 2007. Autophagy: a pathogen driven process. IUBMB Life 59:238–242. doi: 10.1080/15216540701230503. [DOI] [PubMed] [Google Scholar]
- 26.Colombo MI, Gutierrez MG, Romano PS. 2006. The two faces of autophagy: Coxiella and Mycobacterium. Autophagy 2:162–164. doi: 10.4161/auto.2827. [DOI] [PubMed] [Google Scholar]
- 27.Colombo MI. 2005. Pathogens and autophagy: subverting to survive. Cell Death Differ 12(Suppl 2):1481–1483. doi: 10.1038/sj.cdd.4401767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hartl D, Griese M. 2006. Surfactant protein D in human lung diseases. Eur J Clin Invest 36:423–435. doi: 10.1111/j.1365-2362.2006.01648.x. [DOI] [PubMed] [Google Scholar]
- 29.Soltysiak KA, van Schaik EJ, Samuel JE. 2015. Surfactant protein D binds to Coxiella burnetii and results in a decrease in interactions with murine alveolar macrophages. PLoS One 10:e0136699. doi: 10.1371/journal.pone.0136699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elliott A, Peng Y, Zhang G. 2013. Coxiella burnetii interaction with neutrophils and macrophages in vitro and in SCID mice following aerosol infection. Infect Immun 81:4604–4614. doi: 10.1128/IAI.00973-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Struyf S, Gouwy M, Dillen C, Proost P, Opdenakker G, Van Damme J. 2005. Chemokines synergize in the recruitment of circulating neutrophils into inflamed tissue. Eur J Immunol 35:1583–1591. doi: 10.1002/eji.200425753. [DOI] [PubMed] [Google Scholar]
- 32.Sobotta K, Hillarius K, Jimenez PH, Kerner K, Heydel C, Menge C. 2017. Interaction of Coxiella burnetii strains of different sources and genotypes with bovine and human monocyte-derived macrophages. Front Cell Infect Microbiol 7:543. doi: 10.3389/fcimb.2017.00543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McDonough JA, Newton HJ, Klum S, Swiss R, Agaisse H, Roy CR. 2013. Host pathways important for Coxiella burnetii infection revealed by genome-wide RNA interference screening. mBio 4:e00606-12. doi: 10.1128/mBio.00606-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weber MM, Chen C, Rowin K, Mertens K, Galvan G, Zhi H, Dealing CM, Roman VA, Banga S, Tan Y, Luo ZQ, Samuel JE. 2013. Identification of Coxiella burnetii type IV secretion substrates required for intracellular replication and Coxiella-containing vacuole formation. J Bacteriol 195:3914–3924. doi: 10.1128/JB.00071-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Winchell CG, Steele S, Kawula T, Voth DE. 2016. Dining in: intracellular bacterial pathogen interplay with autophagy. Curr Opin Microbiol 29:9–14. doi: 10.1016/j.mib.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Newton HJ, Kohler LJ, McDonough JA, Temoche-Diaz M, Crabill E, Hartland EL, Roy CR. 2014. A screen of Coxiella burnetii mutants reveals important roles for Dot/Icm effectors and host autophagy in vacuole biogenesis. PLoS Pathog 10:e1004286. doi: 10.1371/journal.ppat.1004286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cherla R, Zhang Y, Ledbetter L, Zhang G. 2018. Coxiella burnetii inhibits neutrophil apoptosis by exploiting survival pathways and antiapoptotic protein Mcl-1. Infect Immun 86:e00504-17. doi: 10.1128/IAI.00504-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murray PJ. 2017. Macrophage polarization. Annu Rev Physiol 79:541–566. doi: 10.1146/annurev-physiol-022516-034339. [DOI] [PubMed] [Google Scholar]
- 39.Meghari S, Bechah Y, Capo C, Lepidi H, Raoult D, Murray PJ, Mege JL. 2008. Persistent Coxiella burnetii infection in mice overexpressing IL-10: an efficient model for chronic Q fever pathogenesis. PLoS Pathog 4:e23. doi: 10.1371/journal.ppat.0040023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.