

Abstract
Although chronic obstructive pulmonary disease (COPD) is regarded as a chronic inflammatory lung disease, the disease mechanism is still not known. Intriguingly, aging lungs are quite similar to COPD-affected lungs in many ways, and COPD has been viewed as a disease of accelerated premature aging of the lungs. In this paper, based on a literature review, we would like to propose immunosenescence, age-associated decline in immunity, as a critical mechanism for the development of COPD. Immunosenescence can cause a low-grade, systemic inflammation described as inflammaging. This inflammaging may be directly involved in the COPD pathogenesis. The potential contributors to the development of inflammaging in the lungs possibly leading to COPD are discussed in the review paper. A notable fact about COPD is that only 15% to 20% of smokers develop clinically significant COPD. Given that there is a substantial inter-individual variation in inflammaging susceptibility, which is genetically determined and significantly affected by the history of the individual's exposure to pathogens, immunosenescence and inflammaging may also provide the answer for this unexpectedly low susceptibility of smokers to clinically significant COPD.
Keywords: COPD, immunosenescence, inflammaging, inflammation, aging
INTRODUCTION
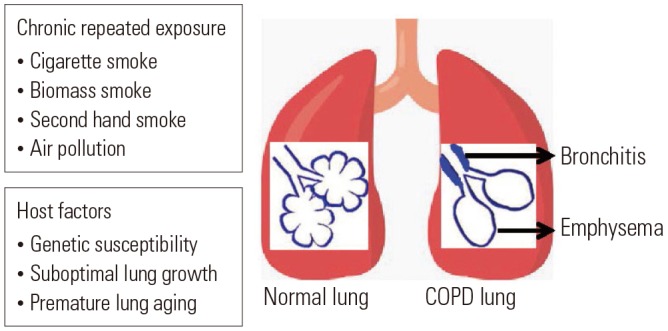
Chronic obstructive pulmonary disease (COPD) is characterized by persistent respiratory symptoms and airflow limitation due to airway and/or alveolar abnormalities usually caused by prolonged exposure to noxious particles or gases.1 Cigarette smoking is the most common cause of COPD; however, environmental factors, such as polluted air, second-hand smoke, or biomass smoke, could also cause COPD. In addition, genetic factors or suboptimal lung growth in childhood may be associated with the development of COPD (Fig. 1).2 Currently, COPD is the fourth leading cause of deaths worldwide, and by 2020, it is expected to be the third leading cause of deaths worldwide.1 Approximately three million people died of COPD in 2015, accounting for 5% of all deaths worldwide in that year.3 Due to the high prevalence and chronicity of COPD, it has been a global burden for many years, and the mechanisms of its development remain unidentified. Intriguingly, the incidence of COPD becomes higher with aging, and peak incidence is observed in patients aged 65–74 years.4,5 Therefore, aging is considered a critical factor in the development of COPD.6 It has been proposed that accelerated premature aging of the lungs may be the mechanism of COPD development.7 Once a patient is suspected as having COPD, the diagnosis is confirmed based on spirometry results. For diagnosis of COPD, patients should have irreversible airflow limitation, which is defined by post-bronchodilator forced expired volume in 1 second (FEV1)/forced vital capacity (FVC) ratio of <0.70 according to the Global Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines.8 It is well documented that lung function typically declines with aging. Specifically, FEV1 decreases, and aging lungs, just like COPD-affected lungs, often have FEV1/FVC ratio <0.70 and increased residual volume.9 This is because the physiology of aging lungs and COPD-affected lungs are similar, showing decreased elastic recoil of the lungs, stiffened chest wall, and altered gas exchange.6 In addition, similarly to COPD-affected lungs, aging lungs have decreased mucociliary clearance, compromised vascular regulation, and alveolar enlargement.10 However, alveolar enlargement in aging lungs, so-called “senile emphysema,” differs from that in COPD in terms of the lack of alveolar wall destruction and inflammation.9 It has been suggested that the main driving force for COPD development could differ among individuals. For instance, suboptimal lung growth in childhood has also been shown to play an important role in causing COPD in some individuals.11 Taken together, although premature lung aging may not be a pathogenic mechanism in all COPD patients, many aspects of aging lungs and COPD-affected lungs are similar, implying that the mechanisms of aging may contribute to the development of COPD. In this study, based on our literature review, we propose that immunosenescence, which is the gradual deterioration of the immune system brought on by natural age advancement, is the key pathogenetic mechanism of COPD.
Fig. 1. Causes and risk factors for COPD. Besides exposure to noxious particles or gases, host factors determine the susceptibility of individuals to develop COPD. COPD, chronic obstructive pulmonary disease.
COPD AS A CHRONIC INFLAMMATORY DISORDER
COPD is a chronic inflammatory disease induced by repeated exposure to respiratory irritants, mostly cigarette smoke, and inflammation in COPD-affected lungs continues to be observed even after stopping smoking.12 Normal immune responses during inflammatory processes are expected to prevent lungs from further injury or infection; however, chronic inflammation in COPD-affected lungs is associated with abnormal immune responses leading to increased tissue damage, resulting in remodeling of the lung.12 Inflammation in COPD is predominantly driven by type 1 immunity and type 3 immunity.13 Both innate and adaptive immune systems are involved in the development of chronic inflammation leading to COPD. The role of innate immunity in COPD pathogenesis has been extensively studied recently in terms of the induction and acute exacerbation of the disease.11,14 Interleukin (IL) 1-like cytokines are reportedly elevated in the lungs of COPD patients, implying the role of an inflammasome in the pathogenesis of COPD.11,14 The inflammasome is a multiprotein intracellular complex that activates proinflammatory caspases, such as caspase 1, leading to the cleavage of proinflammatory cytokines and production of active IL-1β and IL-18 in response to pathogenic microorganisms and sterile stressors.11,14 To activate the inflammasome, innate immune cells should recognize pathogen- associated molecular patterns (PAMPs) of microbial pathogens or damage-associated molecular patterns (DAMPs) of sterile stressors that are categorized based on their subcellular origin (e.g., cytoplasm, endoplasmic reticulum, nucleus, and mitochondria). This recognition of PAMP or DAMP is achieved through several families of pattern recognition receptors (PRRs) expressed in innate immune cells.11,14 The PRRs include Toll-like receptors (TLRs), nucleotide-binding domain leucine-rich repeat-containing receptors (NLRs), C-type lectin receptors, and RIG-I-like receptors.11,14 In the lungs, TLRs are expressed on various cells, including alveolar macrophages, lymphocytes, dendritic cells, and bronchial epithelial cells.11,14 Among different TLRs, TLR4 reportedly plays an important role in murine models of COPD.15,16,17 TLRs recognize molecular patterns on the cell surface of a microbial pathogen, whereas NLRs identify molecular patterns in the cytosol of the host cell.18 It has been shown that the DAMPs generated by cigarette smoke-induced death of airway epithelial cells or the PAMPs induced by microbial antigens activate the NLR family pyrin domain containing 3 (NLRP3) inflammasome, leading to the release of IL-1β with subsequent neutrophilic inflammation in the lungs of COPD patients.19,20 However, the role of the inflammasome in COPD development has been challenged recently. One study reported that, although innate immunity reportedly correlates with disease progression in COPD patients, the NLRP3 inflammasome is not activated in patients with stable COPD,21 thus, the exact roles of innate immunity, including that of the inflammasome, in COPD pathogenesis remain unclear. Several studies have reported on adaptive immune responses in the lungs of COPD patients. Active participation of cytotoxic CD8+ T cells, T helper 1, and 17 CD4+ cells and B-cell response leading to the production of antibodies have been reported in COPD-affected lungs.22 Upon activation, CD8+ T cells release proteolytic enzymes, such as perforin or granzymes, which cause cell death of structural cells by apoptosis or necrosis.22 Th1 cells secrete interferon gamma and Th17 releases IL-17, thus leading to increased inflammation at the site of injury in response to cigarette exposure.23,24 In addition, smokers with COPD have significantly fewer T-regulatory cells in the lungs; this may contribute to the perpetuation of inflammation and the induction of autoimmunity. The development of autoimmunity has been mostly reported in patients with severe COPD.25 Further, B cells are also increased in large airways, and peribronchial lymphoid follicles are often observed, which are due to lymphoid neogenesis in COPD-affected lungs.26 Taken together, it is very likely that the interaction between innate and adaptive immune cells may orchestrate the onset, progression, and severity of lower airway inflammation in COPD.21
IMMUNOSENESCENCE
Given that COPD, a chronic inflammatory lung disease, is probably caused by accelerated lung aging, the changes in immune systems with aging may be associated with COPD pathogenesis. The immune system also undergoes an aging process termed immunosenescence. Both innate immunity and adaptive immunity are affected by aging.27 Aging innate immune cells, such as neutrophils, macrophages, dendritic cells, and natural killer cells, undergo functional decline in phagocytosis, chemotaxis, their ability to secrete inflammatory cytokine, antigen- presenting capacity, and bactericidal ability.28,29,30,31 Declining function of innate immune cells with aging also contributes to the dysregulation of the adaptive immune system via molecular cross-talk.32 The decline in cell-mediated immunity forms a part of the aging immune profile, which is primarily characterized by reduced thymic output leading to reduced T-cell repertoire and increased oligoclonal expansion of memory cells.33 In other words, reduced responsiveness to new antigen load, owing to the reduced naïve to memory cell ratio and expansion of mature cell clones, characterizes immunosenescence of cell-mediated immunity.34 Further, aging is also associated with shifts from Th1 cytokine profile to Th2 cytokine profile in response to immune stimulation. The overproduction of Th2 cytokines in this setting may augment B-cell-mediated autoimmune disorders in older adults.35 Humoral immune function also changes significantly with aging; these changes include decreased antibody responses and diminished production of high-affinity antibodies related to defective surface immunoglobulin/B-cell receptor affinity, decreased signaling, and reduced B-cell proliferation.36 There is also a loss of naïve B-cells and an increase in memory cells with age,37 resulting in a reduced ability to respond to new antigens.38 Overall, the immunosenescence of adaptive immunity results from the depletion of the reservoir of naïve cells over time as a result of repeated exposure to pathogens and their conversion to memory cells.33
INFLAMMAGING (CHRONIC INFLAMMATION)
Immunosenescence leads not only to impaired immune responses but also to low-grade, chronic, systemic inflammation, which is described as inflammaging.39 Inflammaging is characterized by elevated proinflammatory cytokines, such as IL-1, IL-6, and tumor necrosis factor (TNF)-α.40 Inflammaging is believed to be a cumulative result of lifetime exposure to antigenic load from infections, as well as exposure to noninfectious antigens.41 Key genes that are known to regulate inflammaging include the insulin-like growth factor (IGF-1) signaling pathway, target of rapamycin (TOR), and AMP-activated protein kinase (AMPK).42 Not surprisingly, inflammaging has been proposed as one of the contributors to the development of most age-related disorders and as a common biological factor responsible for the decline and onset of diseases in older adults.43,44 Many factors are likely involved in inflammaging. First, intrinsic dysregulation of immune cells simply may be a cause of inflammaging. Regarding the innate immune system, monocytes and macrophages are suggested to contribute to inflammaging significantly by the active generation of inflammatory cytokines.29 In addition, T cell-mediated immunity can induce inflammaging by changing their phenotype into a more proinflammatory phenotype in a manner dependent on previous exposure, reactivation of antigenic challenges, and exhaustion of T-cell repertoire.29 The CD8+ T cell population is thought to be a bigger contributor in this regard than the CD4+ population.45 Second, senescent cells can produce proinflammatory mediators and proteases, which are collectively termed as senescence-associated secretory phenotypes (SASPs).46 SASPs may activate inflammatory responses by recruiting immune cells possibly to cause inflammaging.47 Third, age-related changes in adiposity and hormones (e.g., estrogen) reportedly cause inflammation. Proinflammatory cytokines and free fatty acids secreted from inflamed adipose tissue can activate inflammation, which may be a part of inflammaging.48,49 Fourth, age-associated changes in gut microbe concentrations may foster an imbalance in vivo that affects inflammaging.50 Fifth, increased cell death or damage to cellular components induced by cellular senescence and age-related stress, such as metabolic stress, may exceed the capacity limit of the phagocytic system of aged myeloid cells, resulting in the accumulation of DAPMs and the activation of inflammasome.51,52 This process can be further intensified as aging is associated with decreases in autophagy.53 Autophagy is a cellular housekeeping mechanism responsible for the removal of dysfunctional and damaged intracellular proteins via lysosomal degradation. Thus, one of the roles of autophagy is to prevent the activation of inflammasomes. The consequence of a decline in autophagy with aging is therefore increased activation of the inflammasome and greater proinflammatory responses.54 All these abovementioned age-related changes can contribute to inflammaging among older adults.
COPD AND INFLAMMAGING
Given that inflammaging in the context of immunosenescence has been implicated in the pathogenesis of most age-related chronic diseases, COPD could be one of the target diseases of inflammaging.43,44 Previous study demonstrated that aged mice developed more inflammation leading to emphysema when exposed to cigarette smoke, compared to younger mice.55 If inflammaging plays a significant role in the generation of COPD, the specific mechanisms for inflammaging-induced COPD need to be explored. Airway epithelial cells seem to be a key player in this regard. Airway epithelial cells are pivotal innate immune cells and serve as a physical and molecular barrier for particulate matter against microbes, as these cells entrap all dangerous molecules via mucociliary clearance, and then, these molecules ultimately get degraded in the gastrointestinal tract. By this process, airway epithelial cells can prevent the access of molecules that can activate TLR deployed on their basolateral surface. Once TLR agonists breach this epithelial barrier, they become critical danger signals for activating the strategically arrayed cellular components of the innate immune defense.56 The airway epithelial cells can serve as a physical and molecular barrier as they are polarized and maintain a tight junction that prevents paracellular transport of microbes and harmful substances into underlying lung tissues in normal conditions. Aging is associated with decreased epithelial barrier function,57 abnormalities in both cilia structure and function,58 and reduced production of antimicrobial and anti-inflammatory peptides produced by epithelial cells, including secretory leukocyte protease inhibitor (SLPI).59 Not surprisingly, both alveolar and airway epithelial cells of smokers with COPD reportedly have increased numbers of senescent cells relative to healthy controls.60 Another key cell in terms of the generation of inflammaging in the lungs might be the airway and alveolar macrophages. Communication between airway epithelial cells and airway macrophages seems to be critical for immune homeostasis within the lungs. For instance, airway epithelial cells and airway macrophage cooperate to phagocytose particulates or microorganisms.61 Aging macrophages undergo the functional decline in phagocytosis, chemotaxis, ability to secrete inflammatory cytokine, antigen-presenting capacity, and bactericidal ability.28,29,30,31 Therefore, the capability to protect the lungs against inhaled particles or infectious agents will be decreased with aging, resulting in more inflammation in the lungs. Recently, changes in the airway microbiome have been implicated in various lung diseases.62,63,64,65 Due to difficulties in sampling the airways while preventing contamination by concurrent oropharyngeal microbes, understanding of the airway microbiome was initially not easy; however, a series of studies utilizing DNA sequencing approaches have demonstrated the microbiota of the lower airways. Further, it is now clearly accepted that variations in bacterial abundance, content, and structure occur in chronic inflammatory airway diseases, such as cystic fibrosis,62 COPD,63 and asthma.64,65 This means that a particular microbiome may be characteristic of certain airway pathology. Taken together, the interaction between the microbiome and the host immune system within the airway, as well as the consequent loss of immune function with age, may have implications for the onset and progression of various age-related chronic airway diseases.66 Additionally, increased oxidative stress has been strongly implicated in both COPD and aging lungs as determined by reactive oxygen species levels, oxidized DNA, lipid peroxidation and nitric oxide levels.67,68 Certainly, this increased level of oxidative stress can contribute to the development of inflammaging. Other factors associated with inflammaging, which have been described earlier, including intrinsically abnormal senescent immune cells, accumulation of DAMPs from the dying senescent cells in the setting of decreased autophagy in aging cells, or SASP produced by senescent cells, can also contribute to the development of inflammaging in the lungs of smokers, thus, possibly, leading to COPD. As described above, aging lungs seem to be prone to developing inflammation in response to certain stimuli irrespective of whether they are related to inflammaging or not. From the COPD standpoint, chronic inflammation can develop when inflammation-prone aging lungs are repeatedly exposed to respiratory irritants, such as cigarette smoke, ultimately leading to tissue destruction and the development of COPD. Notably, COPD patients often undergo acute exacerbation. In the cases of acute exacerbation of COPD, which is typically caused by a viral or bacterial infection, chronic inflammation can be further intensified.69 Intriguingly, only 15% to 20% of smokers develop clinically significant COPD. Thus, the majority of smokers do not develop COPD.70 This is different from the mouse model of emphysema, which gives a predictable dose-response relationship between smoking amount and emphysema.70 Currently, we do not know the reason behind only a small percentage of smokers developing COPD. If immunosenescence and/or inflammaging play a critical role in the development of COPD, the reasoning for COPD occurring in a small percentage of smokers can perhaps be explained from immunosenescence or inflammaging or both perspectives. There is reportedly a huge inter-individual variation in inflammaging susceptibility, which seems to be, in part, genetically determined through polymorphisms of promoter regions of cytokine genes or the length of telomeres.29 A negative correlation between telomere length and the levels of C-reactive protein71 and IL-672 has also been observed, and shorter telomere length in leukocytes from COPD patients has been demonstrated.73 In addition, enhanced telomere erosion has been observed in the small airway epithelium from COPD patients.74 Therefore, it is quite possible that susceptibility to inflammaging, which is genetically determined, may explain the inter-individual vulnerability to the development of COPD among smokers. In addition, thymic involution, a factor associated with immunosenescence, begins early in life, and the size of the naïve cell pool is mostly determined before puberty.33 Thus, it is possible that immunosenescence is significantly affected by the history of an individual's exposure to pathogens. In other words, the cumulative effects of antigenic stressors may determine immunosenescence or inflammaging or both and thus explain the differences among smokers in their susceptibility to the development of COPD (Fig. 2).
Fig. 2. Inflammaging: a new mechanism of age-related disease. Low-grade inflammation called inflammaging during the ageing process may contribute to the pathogenesis of most age-related diseases. SLPI, secretory leukocyte protease inhibitor; PAMP, pathogen-associated molecular pattern; DAMP, damage-associated molecular pattern; TLR, Toll-like receptor; NLR, NOD-like receptor; SASP, senescence-associated secretory phenotype.

CONCLUSION
Based on our literature review, we have proposed immunosenescence and inflammaging as the driving mechanisms of the development of COPD and have outlined why only a minority of smokers develop COPD (Fig. 3). If this is true, we could slow the progression of COPD by regulating immunosenescence and inflammaging in the lungs. Future studies will be needed to support our hypotheses.
Fig. 3. Overview of immune hypotheses on COPD. Different levels of inflammaging caused by immunosenescence determine the susceptibility of individuals to COPD. COPD, chronic obstructive pulmonary disease.

ACKNOWLEDGEMENTS
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), which is funded by the Ministry of Education (NRF-2016R1D1A1B01015292 to L.K.K.).
Footnotes
The authors have no potential conflicts of interest to disclose.
AUTHOR CONTRIBUTIONS: Won-Kyung Cho performed an initial conceptualization and wrote the manuscript. Chun Geun Lee revised the manuscript. Lark Kyun Kim performed the initial conceptualization and revised the manuscript.
References
- 1.Weiner A, Chen HV, Liu CL, Rahat A, Klien A, Soares L, et al. Systematic dissection of roles for chromatin regulators in a yeast stress response. PLoS Biol. 2012;10:e1001369. doi: 10.1371/journal.pbio.1001369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Salvi SS, Barnes PJ. Chronic obstructive pulmonary disease in nonsmokers. Lancet. 2009;374:733–743. doi: 10.1016/S0140-6736(09)61303-9. [DOI] [PubMed] [Google Scholar]
- 3.Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3:e442. doi: 10.1371/journal.pmed.0030442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mannino DM, Homa DM, Akinbami LJ, Ford ES, Redd SC. Chronic obstructive pulmonary disease surveillance--United States, 1971-2000. MMWR Surveill Summ. 2002;51:1–16. [PubMed] [Google Scholar]
- 5.Faner R, Rojas M, Macnee W, Agustí A. Abnormal lung aging in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;186:306–313. doi: 10.1164/rccm.201202-0282PP. [DOI] [PubMed] [Google Scholar]
- 6.Ito K, Barnes PJ. COPD as a disease of accelerated lung aging. Chest. 2009;135:173–180. doi: 10.1378/chest.08-1419. [DOI] [PubMed] [Google Scholar]
- 7.Fletcher C, Peto R. The natural history of chronic airflow obstruction. Br Med J. 1977;1:1645–1648. doi: 10.1136/bmj.1.6077.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vestbo J, Hurd SS, Agustí AG, Jones PW, Vogelmeier C, Anzueto A, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2013;187:347–365. doi: 10.1164/rccm.201204-0596PP. [DOI] [PubMed] [Google Scholar]
- 9.Janssens JP. Aging of the respiratory system: impact on pulmonary function tests and adaptation to exertion. Clin Chest Med. 2005;26:469–484. doi: 10.1016/j.ccm.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 10.Brandenberger C, Mühlfeld C. Mechanisms of lung aging. Cell Tissue Res. 2017;367:469–480. doi: 10.1007/s00441-016-2511-x. [DOI] [PubMed] [Google Scholar]
- 11.Rycroft CE, Heyes A, Lanza L, Becker K. Epidemiology of chronic obstructive pulmonary disease: a literature review. Int J Chron Obstruct Pulmon Dis. 2012;7:457–494. doi: 10.2147/COPD.S32330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhat TA, Panzica L, Kalathil SG, Thanavala Y. Immune dysfunction in patients with chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2015;12(Suppl 2):S169–S175. doi: 10.1513/AnnalsATS.201503-126AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barnes PJ. Targeting cytokines to treat asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. 2018;18:454–466. doi: 10.1038/s41577-018-0006-6. [DOI] [PubMed] [Google Scholar]
- 14.Rovina N, Koutsoukou A, Koulouris NG. Inflammation and immune response in COPD: where do we stand? Mediators Inflamm. 2013;2013:413735. doi: 10.1155/2013/413735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maes T, Bracke KR, Vermaelen KY, Demedts IK, Joos GF, Pauwels RA, et al. Murine TLR4 is implicated in cigarette smoke-induced pulmonary inflammation. Int Arch Allergy Immunol. 2006;141:354–368. doi: 10.1159/000095462. [DOI] [PubMed] [Google Scholar]
- 16.Doz E, Noulin N, Boichot E, Guénon I, Fick L, Le Bert M, et al. Cigarette smoke-induced pulmonary inflammation is TLR4/MyD88 and IL-1R1/MyD88 signaling dependent. J Immunol. 2008;180:1169–1178. doi: 10.4049/jimmunol.180.2.1169. [DOI] [PubMed] [Google Scholar]
- 17.Freeman CM, Martinez FJ, Han MK, Washko GR, Jr, McCubbrey AL, Chensue SW, et al. Lung CD8+ T cells in COPD have increased expression of bacterial TLRs. Respir Res. 2013;14:13. doi: 10.1186/1465-9921-14-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franchi L, McDonald C, Kanneganti TD, Amer A, Núñez G. Nucleotide-binding oligomerization domain-like receptors: intracellular pattern recognition molecules for pathogen detection and host defense. J Immunol. 2006;177:3507–3513. doi: 10.4049/jimmunol.177.6.3507. [DOI] [PubMed] [Google Scholar]
- 19.Birrell MA, Eltom S. The role of the NLRP3 inflammasome in the pathogenesis of airway disease. Pharmacol Ther. 2011;130:364–370. doi: 10.1016/j.pharmthera.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 20.Wanderer AA. Interleukin-1beta targeted therapy in severe persistent asthma (SPA) and chronic obstructive pulmonary disease (COPD): proposed similarities between biphasic pathobiology of SPA/COPD and ischemia-reperfusion injury. Isr Med Assoc J. 2008;10:837–842. [PubMed] [Google Scholar]
- 21.Di Stefano A, Caramori G, Barczyk A, Vicari C, Brun P, Zanini A, et al. Innate immunity but not NLRP3 inflammasome activation correlates with severity of stable COPD. Thorax. 2014;69:516–524. doi: 10.1136/thoraxjnl-2012-203062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hogg JC. Pathophysiology of airflow limitation in chronic obstructive pulmonary disease. Lancet. 2004;364:709–721. doi: 10.1016/S0140-6736(04)16900-6. [DOI] [PubMed] [Google Scholar]
- 23.Di Stefano A, Caramori G, Gnemmi I, Contoli M, Vicari C, Capelli A, et al. T helper type 17-related cytokine expression is increased in the bronchial mucosa of stable chronic obstructive pulmonary disease patients. Clin Exp Immunol. 2009;157:316–324. doi: 10.1111/j.1365-2249.2009.03965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grumelli S, Corry DB, Song LZ, Song L, Green L, Huh J, et al. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS Med. 2004;1:e8. doi: 10.1371/journal.pmed.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caramori G, Ruggeri P, Di Stefano A, Mumby S, Girbino G, Adcock IM, et al. Autoimmunity and COPD: clinical implications. Chest. 2018;153:1424–1431. doi: 10.1016/j.chest.2017.10.033. [DOI] [PubMed] [Google Scholar]
- 26.Brusselle GG, Demoor T, Bracke KR, Brandsma CA, Timens W. Lymphoid follicles in (very) severe COPD: beneficial or harmful? Eur Respir J. 2009;34:219–230. doi: 10.1183/09031936.00150208. [DOI] [PubMed] [Google Scholar]
- 27.Castelo-Branco C, Soveral I. The immune system and aging: a review. Gynecol Endocrinol. 2014;30:16–22. doi: 10.3109/09513590.2013.852531. [DOI] [PubMed] [Google Scholar]
- 28.Fulop T, Larbi A, Kotb R, de Angelis F, Pawelec G. Aging, immunity, and cancer. Discov Med. 2011;11:537–550. [PubMed] [Google Scholar]
- 29.Baylis D, Bartlett DB, Patel HP, Roberts HC. Understanding how we age: insights into inflammaging. Longev Healthspan. 2013;2:8. doi: 10.1186/2046-2395-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang CQ, Udupa KB, Xiao H, Lipschitz DA. Effect of age on marrow macrophage number and function. Aging (Milano) 1995;7:379–384. doi: 10.1007/BF03324349. [DOI] [PubMed] [Google Scholar]
- 31.Solana R, Tarazona R, Gayoso I, Lesur O, Dupuis G, Fulop T. Innate immunosenescence: effect of aging on cells and receptors of the innate immune system in humans. Semin Immunol. 2012;24:331–341. doi: 10.1016/j.smim.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 32.Aspinall R, Del Giudice G, Effros RB, Grubeck-Loebenstein B, Sambhara S. Challenges for vaccination in the elderly. Immun Ageing. 2007;4:9. doi: 10.1186/1742-4933-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pawelec G. Hallmarks of human “immunosenescence”: adaptation or dysregulation? Immun Ageing. 2012;9:15. doi: 10.1186/1742-4933-9-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weiskopf D, Weinberger B, Grubeck-Loebenstein B. The aging of the immune system. Transpl Int. 2009;22:1041–1050. doi: 10.1111/j.1432-2277.2009.00927.x. [DOI] [PubMed] [Google Scholar]
- 35.Ongrádi J, Kövesdi V. Factors that may impact on immunosenescence: an appraisal. Immun Ageing. 2010;7:7. doi: 10.1186/1742-4933-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Whisler RL, Grants IS. Age-related alterations in the activation and expression of phosphotyrosine kinases and protein kinase C (PKC) among human B cells. Mech Ageing Dev. 1993;71:31–46. doi: 10.1016/0047-6374(93)90033-n. [DOI] [PubMed] [Google Scholar]
- 37.Macallan DC, Wallace DL, Zhang Y, Ghattas H, Asquith B, de Lara C, et al. B-cell kinetics in humans: rapid turnover of peripheral blood memory cells. Blood. 2005;105:3633–3640. doi: 10.1182/blood-2004-09-3740. [DOI] [PubMed] [Google Scholar]
- 38.Haynes L, Eaton SM, Burns EM, Randall TD, Swain SL. Newly generated CD4 T cells in aged animals do not exhibit age-related defects in response to antigen. J Exp Med. 2005;201:845–851. doi: 10.1084/jem.20041933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- 40.Vasto S, Candore G, Balistreri CR, Caruso M, Colonna-Romano G, Grimaldi MP, et al. Inflammatory networks in ageing, age-related diseases and longevity. Mech Ageing Dev. 2007;128:83–91. doi: 10.1016/j.mad.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 41.De Martinis M, Franceschi C, Monti D, Ginaldi L. Inflamm-ageing and lifelong antigenic load as major determinants of ageing rate and longevity. FEBS Lett. 2005;579:2035–2039. doi: 10.1016/j.febslet.2005.02.055. [DOI] [PubMed] [Google Scholar]
- 42.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cevenini E, Monti D, Franceschi C. Inflamm-ageing. Curr Opin Clin Nutr Metab Care. 2013;16:14–20. doi: 10.1097/MCO.0b013e32835ada13. [DOI] [PubMed] [Google Scholar]
- 44.Caramori G, Casolari P, Barczyk A, Durham AL, Di Stefano A, Adcock I. COPD immunopathology. Semin Immunopathol. 2016;38:497–515. doi: 10.1007/s00281-016-0561-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Effros RB, Dagarag M, Spaulding C, Man J. The role of CD8+ T-cell replicative senescence in human aging. Immunol Rev. 2005;205:147–157. doi: 10.1111/j.0105-2896.2005.00259.x. [DOI] [PubMed] [Google Scholar]
- 46.Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123:966–972. doi: 10.1172/JCI64098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15:482–496. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- 48.Eguchi K, Manabe I, Oishi-Tanaka Y, Ohsugi M, Kono N, Ogata F, et al. Saturated fatty acid and TLR signaling link β cell dysfunction and islet inflammation. Cell Metab. 2012;15:518–533. doi: 10.1016/j.cmet.2012.01.023. [DOI] [PubMed] [Google Scholar]
- 49.Shen H, Eguchi K, Kono N, Fujiu K, Matsumoto S, Shibata M, et al. Saturated fatty acid palmitate aggravates neointima formation by promoting smooth muscle phenotypic modulation. Arterioscler Thromb Vasc Biol. 2013;33:2596–2607. doi: 10.1161/ATVBAHA.113.302099. [DOI] [PubMed] [Google Scholar]
- 50.Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–236. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- 51.Fontana L, Zhao E, Amir M, Dong H, Tanaka K, Czaja MJ. Aging promotes the development of diet-induced murine steatohepatitis but not steatosis. Hepatology. 2013;57:995–1004. doi: 10.1002/hep.26099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shaw AC, Goldstein DR, Montgomery RR. Age-dependent dysregulation of innate immunity. Nat Rev Immunol. 2013;13:875–887. doi: 10.1038/nri3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pallauf K, Rimbach G. Autophagy, polyphenols and healthy ageing. Ageing Res Rev. 2013;12:237–252. doi: 10.1016/j.arr.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 54.Salminen A, Kaarniranta K, Kauppinen A. Inflammaging: disturbed interplay between autophagy and inflammasomes. Aging (Albany NY) 2012;4:166–175. doi: 10.18632/aging.100444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.John-Schuster G, Günter S, Hager K, Conlon TM, Eickelberg O, Yildirim AÖ. Inflammaging increases susceptibility to cigarette smoke-induced COPD. Oncotarget. 2016;7:30068–30083. doi: 10.18632/oncotarget.4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Whitsett JA, Alenghat T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol. 2015;16:27–35. doi: 10.1038/ni.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ghadially R, Brown BE, Sequeira-Martin SM, Feingold KR, Elias PM. The aged epidermal permeability barrier. Structural, functional, and lipid biochemical abnormalities in humans and a senescent murine model. J Clin Invest. 1995;95:2281–2290. doi: 10.1172/JCI117919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ho JC, Chan KN, Hu WH, Lam WK, Zheng L, Tipoe GL, et al. The effect of aging on nasal mucociliary clearance, beat frequency, and ultrastructure of respiratory cilia. Am J Respir Crit Care Med. 2001;163:983–988. doi: 10.1164/ajrccm.163.4.9909121. [DOI] [PubMed] [Google Scholar]
- 59.Shugars DC, Watkins CA, Cowen HJ. Salivary concentration of secretory leukocyte protease inhibitor, an antimicrobial protein, is decreased with advanced age. Gerontology. 2001;47:246–253. doi: 10.1159/000052808. [DOI] [PubMed] [Google Scholar]
- 60.Tsuji T, Aoshiba K, Nagai A. Alveolar cell senescence in patients with pulmonary emphysema. Am J Respir Crit Care Med. 2006;174:886–893. doi: 10.1164/rccm.200509-1374OC. [DOI] [PubMed] [Google Scholar]
- 61.Lloyd CM, Marsland BJ. Lung homeostasis: influence of age, microbes, and the immune system. Immunity. 2017;46:549–561. doi: 10.1016/j.immuni.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 62.Chmiel JF, Aksamit TR, Chotirmall SH, Dasenbrook EC, Elborn JS, LiPuma JJ, et al. Antibiotic management of lung infections in cystic fibrosis. I. The microbiome, methicillin-resistant Staphylococcus aureus, gram-negative bacteria, and multiple infections. Ann Am Thorac Soc. 2014;11:1120–1129. doi: 10.1513/AnnalsATS.201402-050AS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang YJ, Sethi S, Murphy T, Nariya S, Boushey HA, Lynch SV. Airway microbiome dynamics in exacerbations of chronic obstructive pulmonary disease. J Clin Microbiol. 2014;52:2813–2823. doi: 10.1128/JCM.00035-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chotirmall SH, Burke CM. Aging and the microbiome: implications for asthma in the elderly? Expert Rev Respir Med. 2015;9:125–128. doi: 10.1586/17476348.2015.1002473. [DOI] [PubMed] [Google Scholar]
- 65.Han MK, Huang YJ, Lipuma JJ, Boushey HA, Boucher RC, Cookson WO, et al. Significance of the microbiome in obstructive lung disease. Thorax. 2012;67:456–463. doi: 10.1136/thoraxjnl-2011-201183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Segal LN, Rom WN, Weiden MD. Lung microbiome for clinicians. New discoveries about bugs in healthy and diseased lungs. Ann Am Thorac Soc. 2014;11:108–116. doi: 10.1513/AnnalsATS.201310-339FR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Caramori G, Adcock IM, Casolari P, Ito K, Jazrawi E, Tsaprouni L, et al. Unbalanced oxidant-induced DNA damage and repair in COPD: a link towards lung cancer. Thorax. 2011;66:521–527. doi: 10.1136/thx.2010.156448. [DOI] [PubMed] [Google Scholar]
- 68.Rahman I, van Schadewijk AA, Crowther AJ, Hiemstra PS, Stolk J, MacNee W, et al. 4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2002;166:490–495. doi: 10.1164/rccm.2110101. [DOI] [PubMed] [Google Scholar]
- 69.Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med. 2008;359:2355–2365. doi: 10.1056/NEJMra0800353. [DOI] [PubMed] [Google Scholar]
- 70.Rennard SI, Vestbo J. COPD: the dangerous underestimate of 15% Lancet. 2006;367:1216–1219. doi: 10.1016/S0140-6736(06)68516-4. [DOI] [PubMed] [Google Scholar]
- 71.Aviv A, Valdes A, Gardner JP, Swaminathan R, Kimura M, Spector TD. Menopause modifies the association of leukocyte telomere length with insulin resistance and inflammation. J Clin Endocrinol Metab. 2006;91:635–640. doi: 10.1210/jc.2005-1814. [DOI] [PubMed] [Google Scholar]
- 72.Shiels PG, McGlynn LM, MacIntyre A, Johnson PC, Batty GD, Burns H, et al. Accelerated telomere attrition is associated with relative household income, diet and inflammation in the pSoBid cohort. PLoS One. 2011;6:e22521. doi: 10.1371/journal.pone.0022521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morlá M, Busquets X, Pons J, Sauleda J, MacNee W, Agustí AG. Telomere shortening in smokers with and without COPD. Eur Respir J. 2006;27:525–528. doi: 10.1183/09031936.06.00087005. [DOI] [PubMed] [Google Scholar]
- 74.Walters MS, De BP, Salit J, Buro-Auriemma LJ, Wilson T, Rogalski AM, et al. Smoking accelerates aging of the small airway epithelium. Respir Res. 2014;15:94. doi: 10.1186/s12931-014-0094-1. [DOI] [PMC free article] [PubMed] [Google Scholar]