

Abstract
Genetic and epigenetic changes drive carcinogenesis, and their integrated analysis provides insights into mechanisms of cancer development. Computational methods have been developed to measure copy number variation (CNV) from methylation array data, including ChAMP-CNV, CN450K, and, introduced here, Epicopy. Using paired single nucleotide polymorphism (SNP) and methylation array data from the public The Cancer Genome Atlas repository, we optimized CNV calling and benchmarked the performance of these methods. We optimized the thresholds of all three methods and showed comparable performance across methods. Using Epicopy as a representative analysis of Illumina450K array, we show that Illumina450K-derived CNV methods achieve a sensitivity of 0.7 and a positive predictive value of 0.75 in identifying CNVs, which is similar to results achieved when comparing competing SNP microarray platforms with each other.
Keywords: CNV, copy number variation, methylation microarray, microarray, TCGA
1. Introduction
Both genetic and epigenetic alterations are implicated in the development of cancer (You and Jones, 2012). Genetic lesions, such as copy number variation (CNV) and point mutations, can lead to the development of cancer through the alteration of expression or structure of tumor suppressors and oncogenes (Stratton et al., 2009). Epigenetic modifications, through DNA methylation or histone modifications, lead to the silencing or reactivation of genes. Further, genetic and epigenetic modifications can complement each other as a second hit to critical loci (Esteller, 2008). Integration of genetic and epigenetic data can provide a more complete view of underlying pathogenic mechanisms (Verhaak et al., 2010). Performing concurrent genetic and epigenetic characterization of tumors may, however, be limited by cost and availability of sufficient material. The ability to read out multiple data types from a single platform both minimizes cost and ensures a single source of test material and a common biology.
The Illumina Infinium Human Methylation microarray is the leading methylation microarray platform and uses fluorescent probes to identify genetic and epigenetic variants of a given genome locus, technically very similar to the approach used to detect single nucleotide polymorphisms (SNPs) in microarrays (Supplementary Fig. S1). The bisulfite treatment of DNA that precedes hybridization on methylation arrays chemically creates induced SNPs at unmethylated CpG dinucleotide sites. This allows a similar analytical approach to the two array platforms, which has driven efforts to obtain CNV information from DNA methylation microarrays (Sturm et al., 2012; Aryee et al., 2014; Feber et al., 2014).
The cumulative intensity values from the unmethylated probe, U, and the methylated probe, M, are a proxy of total DNA copy number at that locus, and they were used by Sturm et al. (2012) to characterize the CNV in a series of glioblastomas. The “getCN” function from the minfi package on Bioconductor sums the raw intensities of U and M to obtain total intensities (Aryee et al., 2014), and Feber et al. (2014) most recently proposed a statistical pipeline that provides copy number information from the Illumina arrays. However, there has been no study to date investigating in detail the circumstances in which it is possible to reliably obtain CNV information from methylation arrays.
Herein, we study the experimental parameters allowing the reliable assessment of CNV using DNA methylation arrays, presenting the first comprehensive comparison of copy number estimates from methylation arrays with gold standard results obtained from high-density SNP arrays. We consider several previously published algorithms for estimating copy number from DNA methylation arrays as well as an optimized method, Epicopy, which is introduced here. Performance is evaluated on various The Cancer Genome Atlas (TCGA) datasets where both methylation and CNV data are available for several tumor types. A better understanding of when one can efficiently use and how to interpret CNV information from methylation arrays will help in deciding when reliable CNV and methylation calls can be made by using a single platform.
1.1. The challenges
Although in theory, the Illumina DNA methylation array can be used just like an SNP array to estimate DNA abundance across the genome, the platform presents unique challenges that may be difficult to overcome in practice. Coverage is perhaps the primary concern—CpG dinucleotides are not uniformly distributed throughout the genome, and methylation arrays necessarily reflect this fact. Another factor likely to affect performance is that the Illumina methylation array is designed with two different probe chemistries, with unique distributions of the probe intensities (Dedeurwaerder et al., 2011). The probe type is closely correlated to the guanine-cytosine (CG) content of the probe sequence, so that different regions of the genome are enriched for each probe type (Supplementary Fig. S2).
2. Methods
2.1. Sample selection
Three TCGA datasets, thyroid carcinoma (THCA) (Cancer Genome Atlas Research, 2014), breast carcinoma (BRCA) (Cancer Genome Atlas, 2012), and lung squamous cell carcinoma (LUSC) (Cancer Genome Atlas Research, 2012), were chosen for model development and validation because a large number of samples with paired Affymetrix SNP6 Array and Illumina HM450K methylation array data were available. THCA, which has few, but frequently recurrent CNVs (Cancer Genome Atlas Research, 2014) was used for model development; whereas BRCA and LUSC, representing cancers with many CNVs per sample, were used for testing. Combined, these three datasets are representative of the spectrum of CNV seen in human cancer (Ciriello et al., 2013).
2.2. Data download and analysis
Although Illumina has recently released the high-density Infinium MethylationEPIC kit, the vast majority of public data, including the National Cancer Institute's Cancer Genome Atlas Project (TCGA), is currently derived from the older 450K platform, which includes 485,577 CpG probes that are widely distributed across both intra- and intergenic regions of the genome.
The Illumina Human Methylation HM450K .idat files (TCGA level 1 data) were downloaded from Broad Institute's Firehose Genome Data Analysis Center (GDAC) server (data freeze December, 2013). Processed Affymetrix SNP6 array data (TCGA Level 3 data) and the accompanying CNV results from the Genomic Identification of Significant Targets in Cancer (GISTIC) for the same tumors were downloaded from the same server, for comparison. Data were analyzed by using the R statistical environment (Version 3.1.1), packages from Bioconductor, and custom functions.
ChAMP-CNV was performed on Level-1 TCGA data by using recommended parameters. The same post-segmentation processing of identifying optimal thresholds was performed to obtain comparable datasets between Epicopy, ChAMP-CNV, and CN450K.
2.3. Estimating copy number using Epicopy
Raw methylation data were processed by using the functional normalization algorithm (funnorm) adapted from the developer's version of the minfi package (Aryee et al., 2014) to return red-green channel data. Log2 signal intensities for both the methylated, Mi, and unmethylated, Ui, channels, as calculated in minfi, were summed together to obtain total signal intensity, τi, of genomic position i. Normal samples (n = 55) from the THCA dataset were used to represent the diploid genome. Specifically, at each genomic position i represented on the array, we calculated γi = mode (normal log2 intensities at i) estimated by using the naive estimator from the modeest package (Poncet, 2012). These values were then used to calculate the log ratio of the intensities, Δτij, for genomic position i in sample j.
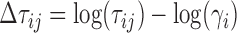 |
Finally, the mean Δτij was centered at zero and subjected to circular binary segmentation, implemented by the DNAcopy package (Seshan and Olshen, 2012), using the sdundo option with a value of 2, to obtain copy number (CN) estimates λ. These steps are shown in a flowchart in Supplementary Figure S3.
2.4. Calling copy number events
GISTIC2.0 (Mermel et al., 2011) was performed to (1) generate gene-level copy number estimates, λgj, for gene g in each sample j and (2) identify focal and arm-level events that are recurrently amplified or deleted in each tumor type. Default parameters were used when applying GISTIC to SNP arrays, including the requirements that segments include at least five tags and that log R ratios (LRRs), |θ| ≥ 0.3, when calling amplifications and deletions. For Illumina450k-based methods, optimal parameters were derived by maximizing area under the curve (AUC) in the THCA dataset as training data (described in Supplementary Methods and Results) for each method. On the basis of those analyses, we required that segments contain at least 200 tags for all three methods and LRRs, |θ| ≥ 0.15 for Epicopy, ≥0.11 for ChAMP-CNV, and ≥0.13 for CN450K, specifically for GISTIC2.0 analysis.
Recurrent copy number events were estimated for each tumor type for the SNP microarray and Epicopy. Reciprocal overlaps between recurrently amplified and deleted peaks between SNP and Illumina450K for each tumor type were calculated by using GenomicRanges.
2.5. Performance metrics
Several measures were used to compare results across algorithms or platforms, including: concordance, sensitivity, specificity, positive predictive value, negative predictive value, and area under the receiver-operating characteristic (ROC) calculated by using the pROC package (Robin et al., 2011). We also used a reproducibility index calculated, at the gene level, as the Jaccard similarity coefficient (Shi, 1993). This measure is similar to sensitivity but treats methods symmetrically rather than assuming that one method represents a benchmark. The reproducibility index for sample j is calculated as;
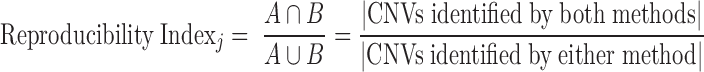 |
Local regression to highlight trend in figures was performed by using the locfit function from the locfit R package (Loader, 2013).
To simplify the analysis, all comparisons were performed on thresholded gene-level estimates from GISTIC2.0.
3. Results and Discussion
3.1. Probe coverage
HM450K has a sparser genomic coverage than the current generation of SNP arrays, consisting of 485,577 probes compared with 1.8 million probes (906,600 SNP probes and 946,000 non-polymorphic copy number probes) in the SNP6 platform from Affymetrix (Supplementary Table ST1). Further, although the probes in the HM450K array are distributed across both inter- and intragenic regions (Fig. 1), they are concentrated in intragenic regions and particularly in gene promoters. This can result in varying sensitivities for making CNV calls between array platforms in different regions across the genome.
FIG. 1.

Probe coverage of HM450K. Probe coverage of Illumina Infinium Human Methylation 450K microarray (450K) across the human genome. To better highlight the distribution of probes across the genome, chromosomes are not to scale compared with each other. Despite having only 485,577 probes across the genome, there is good coverage of all but 1 autosome (Ch 21) and Chromosome X. This implies that CNV can be estimated well for most of the genome. Epicopy is written to profile the CNV on autosomes, excluding Chromosome X and Y. CNV, copy number variation; HM450K, Illumina Human Methylation 450K microarray.
3.2. Comparison of algorithms
Feber et al. (2014) recently published a method to identify CNV from HM450K array data by using the ChAMP pipeline (Morris et al., 2014), and another method, CopyNumber450k (CN450K), is available as an R package as well (http://bioconductor.org/packages/CopyNumber450k/) (Papillon-Cavanagh et al., 2013). We compared the performance of ChAMP-CNV and CN450K, as well as our own Epicopy approach, to understand how the various features of the algorithms influence results.
We plotted the ROC curves for correct classification of gene-level CNV and calculated the AUC for gene-level amplification and deletions in each of the TCGA tumor types (Supplementary Fig. S4 and Table 1) to assess the overall performance of these methods against CN value thresholds. The performance of these methods was equal in the THCA training set, whereas Epicopy showed a marginally increased performance compared with ChAMP-CNV and CN450K in the BRCA and LUSC validation datasets. This can be attributed to slight differences in data normalization and the use of reference samples. Epicopy uses functional normalization followed by probe type-specific normalization, whereas ChAMP-CNV uses quantile normalization and CN450K uses functional normalization. Epicopy uses a series of normal samples as reference intensities, whereas ChAMP-CNV and CN450K use the median intensities across all tumor samples. This leads to a difference in reference intensities, which manifests itself when the LRRs are calculated, thereby allowing for differences in called CNVs.
Table 1.
Epicopy and CHAMP-CNV Area Under the Curves in Three The Cancer Genome Atlas Datasets
| Dataset | Method | AUC |
|---|---|---|
| THCA | Epicopy | 0.97 |
| THCA | CHAMP-CNV | 0.04 |
| BRCA | Epicopy | 0.90 |
| BRCA | CHAMP-CNV | 0.85 |
| LUSC | Epicopy | 0.88 |
| LUSC | CHAMP-CNV | 0.76 |
AUC, area under the curve.
3.3. Detailed comparison with single nucleotide polymorphism arrays
Next, we performed a detailed comparison between results obtained on SNP arrays and those from DNA methylation by using our own Epicopy approach.
As shown in Figure 2a, the LRR, or magnitude change in copy number compared with reference, of the Illumina450 segment means are lower than their SNP6 counterparts, which are approximately twice as large, although the direction of copy number change remains the same. Further, Illumina450-derived segments are more fragmented than SNP6 segments due to the concentration of HM450K probes around CpG islands. For example, a single CNV event identified as a single segment in SNP6 may be represented by multiple adjacent segments in Illumina450. In spite of these differences, Illumina450 results closely approximate results obtained by SNP6 CNV analysis. This is illustrated in a representative comparison of SNP and Illumina450 CN profiles from a breast tumor sample showing that Illumina450 is able to detect chromosomal, arm, and focal copy number changes (Fig. 3a).
FIG. 2.

Epicopy results and metrics. (a) Representative example of copy number profiles of the same breast cancer TCGA sample from SNP array (top) and Epicopy generated segmentation values (bottom). Note the lower copy number values on the Epicopy-derived CNV information. The y-axis represents copy number or LRR. The x-axis represents genomic location with each dotted red line signaling a transition across chromosomes. The horizontal blue and black bars represent the segments of that sample with alternating colors signifying chromosome transition. The dotted horizontal line represents the threshold of making a CNV call. (b) Performance of gene-level Epicopy calls against SNP analysis in three TCGA datasets; THCA, BRCA, and LUSC using a copy number (CN) threshold of 0.15 and 200 probes per segment. In the top panels, the tan line represents specificity whereas the black line represents sensitivity. The bottom panel shows the concordance, or accuracy, of gene-level data. BRCA, breast carcinoma; CN; LRR, log R ratio; LUSC, lung squamous cell carcinoma; SNP, single nucleotide polymorphism; TCGA, The Cancer Genome Atlas; THCA, thyroid carcinoma. (c) Reproducibility Index as a function of CN-altered genes for a given sample across 3 TCGA datasets. Each point is a sample and the blue line represents the local regression line generated using the Locfit function in R. The vertical dotted grey line represents the average number of CN-altered genes of samples for a tumor type.
FIG. 3.

Percent CNV from the SNP6 array detected by Epicopy as a function of LRR from SNP6 microarray. (a) Each point represents the average of all segments identified by SNP array in the THCA dataset, disregarding the length or probe number within the segment. The x-axis represents the LRR of the segment in the SNP array, and the y-axis represents the percent of those segments identified by Epicopy. The blue line is the local regression line fitted using the locfit function in R. (b) Comparison of the GISTIC results obtained by SNP analysis and Epicopy-derived values. Left panel: Frequent (recurrent) amplifications identified by SNP (left)- and Epicopy (right)-derived results. Right panel: Frequent deletions identified by SNP (left)- and Epicopy (right)-derived CNV results. There was 72% overlap between the recurrently altered peaks identified across both platforms. GISTIC, Genomic Identification of Significant Targets in Cancer.
3.4. Performance on gene-wise correlations
We tested the performance of Illumina450 at the gene level, on the BRCA and LUSC datasets, by using the CNV results from the SNP arrays as the standard of comparison. Measures of performance were evaluated at the gene level, and they included overall accuracy, sensitivity, and specificity. Of note, we used thyroid normal tissue from TCGA as the reference diploid samples for both BRCA and LUSC, reflecting the situation that well-matched reference samples are often not available.
With the thresholds of LRR, or magnitude of change, at 0.15 and number of probes per segment at 200 (see Supplementary Methods and Results for the derivation of these values), the accuracy of the method in the THCA, BRCA, and LUSC datasets was 99%, 86%, and 83%, respectively. The sensitivity of Illumina450 was 84%, 72%, and 69%, respectively (Table 2 and Fig. 2b), whereas the specificity was 99%, 92%, and 90%, respectively.
Table 2.
Epicopy Performance in Three The Cancer Genome Atlas Datasets
| Dataset | THCA | BRCA | LUSC |
|---|---|---|---|
| Concordance | 0.99 | 0.86 | 0.83 |
| Sensitivity | 0.84 | 0.72 | 0.69 |
| Specificity | 0.99 | 0.92 | 0.90 |
| PPV | 0.73 | 0.78 | 0.79 |
| NPV | 1.00 | 0.89 | 0.85 |
BRCA, breast carcinoma; LUSC, lung squamous cell carcinoma; NPV, negative predictive value; PPV, positive predictive value, THCA, thyroid carcinoma.
We further calculated a reproducibility index between SNP and Illumina450 gene CNVs (Fig. 2c). This measure, which is based on the Jaccard distance (Shi, 1993), describes the probability that a copy number alteration identified on either platform is found on both and has the advantage of treating the SNP and Illumina450 results symmetrically. We observed an average reproducibility between CN calls from the SNP6 platform and Epicopy of 27%, 57%, and 51% for THCA, BRCA, and LUSC, respectively.
To put these reproducibility results in perspective, studies evaluating the CN detection reproducibility across SNP array platforms and even across different CNV-calling algorithms on the same platform have shown that reproducibility in replicate experiments ranges between 39% and 79%, even within the same platform, whereas reproducibility across platforms ranges between 25% and 50% (Baumbusch et al., 2008; Curtis et al., 2009; Pinto et al., 2011). Specifically, the maximum reproducible copy number alterations detected by the SNP6 platform using the same algorithm assessed by Pinto et al. (2011) was 79%.
Thyroid cancer is distinctive among these three tumor types due to its very low copy number changes overall, and the low level of agreement may be attributable to this (Ciriello et al., 2013; Cancer Genome Atlas Research, 2014). The reproducibility index weighs CNV events being called by both Illumina450 and SNP analysis. In samples with no events identified by SNP analysis, as often occurred in the case in the THCA dataset, even a single CNV event identified by Epicopy causes the reproducibility index to be zero.
Based on this, we hypothesize that there is an upper limit to the reproducible CNV detection rate, given the present array technologies, and that the performance of Illumina450, as assessed by the agreement between Illumina450 and SNP6 measurements of CNV from high-density microarrays, is comparable to that seen between different SNP array platforms.
3.5. Performance on recurrent amplifications and deletions
We used GISTIC2.0 (Mermel et al., 2011), which employs a probabilistic method to identify peaks of recurrent CNV events that occur in a set of samples, to focus on the most frequently recurring events that are likely to be driving the development and progression of disease.
TCGA SNP6 datasets were processed by using the Broad Institute's Copy Number Pipeline-analyzed SNP6 data (Saksena et al., 2012), which uses circular binary segmentation (CBS) to obtain CN segments (Olshen et al., 2004), using the GISTIC 2.0 output, which contains both the gene-level CNVs and recurrent CNVs. Since characterization of recurrent CNVs can be used to identify driver events, we compared Illumina450-based calls and SNP6 arm-level events identified by GISTIC to assess Illumina450's ability to detect recurrent CNVs.
In BRCA and LUSC, two tumor types that are characterized by a high number of CNVs, Epicopy is able to identify 70% (Fig. 3 and Supplementary Fig. S5) of peaks identified in the SNP6 platform. Interestingly, there are peaks identified in the Illumina450 data that are not seen by SNP6 in the THCA dataset. These may be false positives, which would reflect limitations in using HM450K for CNV profiling, or true positive calls that are detected by Illumina450 and missed by SNP6-based analysis. As discussed, there are regions of the genome where HM450K probe coverage is denser than in the SNP6 array and these peaks may fall into such regions. Indeed, when we investigate the probe density in these peaks for SNP6 and HM450K arrays, HM450K had more probes in 9 out of 12 peaks, suggesting that these are regions where HM450K is more sensitive at detecting CNV than SNP6.
Further, when we investigated the probe density of Illumina HM450K methylation array compared with the Affymetrix SNP6 array, we were able to show that Illumina CpG probes were enriched around transcriptional start sites and exons (Supplementary Fig. S6a, b). The enrichment of CpG probes in and around gene bodies suggests that in regions of the genome where the functional consequences of CNV is well understood, Illumina450-derived CNV profiles may be as sensitive or more sensitive than SNP arrays, especially for focal CNVs. In support of that, Feber et al. (2014) have shown that the HM450K CpG array is able to identify a PTCH1 focal deletion undetected by the Illumina CytoSNP array. Of note, some biologically relevant cancer driver genes are present in these peaks; for example, TERT and AKT1 are amplified whereas BRCA2 is deleted. TERT amplification has been shown to be significant in familial papillary thyroid cancer patients (Capezzone et al., 2008), and genetic alterations in all three genes have been described by TCGA (Cancer Genome Atlas Research, 2014).
In addition, there is a distinct peak at chromosome 6p22 detected by Illumina450 but not the SNP array in the THCA dataset. The Illumina probes in this peak are situated in human leukocyte antigen (HLA) genes, a known hypervariable region (Supplementary Fig. S6c, red boxes). Further, nearby probes outside of this hypervariable region show no CNV, suggesting that the deletion in this peak is indeed unique to probes within HLA genes. As such, this implies a lack of probe binding due to probe mismatch rather than the actual loss of copy number in this region. Therefore, we recommend that these probes be removed from analysis when using the HM450K platform to profile CNV, and we have accordingly implemented this option in the Epicopy package.
4. Conclusion
We have conducted the first comprehensive comparison of HM450K and SNP6 on the same samples, and we have presented a series of new tools in the Epicopy pipeline to identify CNV by using the Illumina HM450K methylation array with high probe density across the genome. With ample probe coverage across the genome, especially within promoter and exonic regions of genes, HM450K can be used to obtain CNV information in the human genome. Using publicly available paired SNP and methylation array data from TCGA, we show that agreement between HM450K and SNP6 is nearly as good as previously published results comparing SNP array results across labs, on common samples and platforms (Baumbusch et al., 2008; Curtis et al., 2009; Pinto et al., 2011). Our new Epicopy pipeline promises to be a useful addition to the analyst's toolbox, performing as well as or better than similar methods.
Some of the pressing questions in cancer biology can be answered by using multiplatform analyses of clinical samples with long-term follow-up information. Such studies are often limited to archival samples where available tissues are frequently scarce. Being able to analyze both genomic and epigenomic data from a single DNA input will allow for more samples to be analyzed and also allow for better correlation of genomic and epigenomic data, since both analyses are performed on the same sample. As such, we believe that methods such as Epicopy, ChAMP-CNV, and CN450K will allow a more complete characterization of molecular changes across multiple platforms.
Supplementary Material
Acknowledgments
The authors would like to thank Rob Scharpf for advice in the early development of the Epicopy method and insight into DNA copy number analysis. They would also like to thank Jean-Philippe Fortin and Kasper D. Hansen for sharing the developer's version of the funnorm code and advice on normalization methods. They are grateful to Elana J. Fertig for testing early versions of the Epicopy software. Data used for the study were obtained from the TCGA effort. Funding: This work was supported in part by grants from the Susan G. Komen Foundation (KG 110094) and the NIH (R01CA140331) awarded to C.B.U.
Authors' Contributions
S.C., H.S.K., C.B.U., and L.M.C. developed the method. S.C. and L.M.C. devised comparison metrics. H.S.K. downloaded relevant data files from TCGA. S.C. and H.S.K. analyzed the data. S.C., K.S.H., M.A.Z., C.B.U., and L.M.C. interpreted results. S.C. wrote the software. S.C., H.S.K., M.A.Z., C.B.U., and L.M.C. wrote the article. All authors read and approved the final article.
Author Disclosure Statement
The authors declare that no competing financial interests exist.
Supplementary Material
References
- Aryee M.J., Jaffe A.E., Corrada-Bravo H., et al. 2014. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumbusch L.O., Aaroe J., Johansen F.E., et al. 2008. Comparison of the Agilent, ROMA/NimbleGen and Illumina platforms for classification of copy number alterations in human breast tumors. BMC Genomics 9, 379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas, N. 2012. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. 2012. Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. 2014. Integrated genomic characterization of papillary thyroid carcinoma. Cell 159, 676–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capezzone M., Cantara S., Marchisotta S., et al. 2008. Short telomeres, telomerase reverse transcriptase gene amplification, and increased telomerase activity in the blood of familial papillary thyroid cancer patients. J. Clin. Endocrinol. Metab. 93, 3950–3957 [DOI] [PubMed] [Google Scholar]
- Ciriello G., Miller M.L., Aksoy B.A., et al. 2013. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 45, 1127–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis C., Lynch A.G., Dunning M.J., et al. 2009. The pitfalls of platform comparison: DNA copy number array technologies assessed. BMC Genomics 10, 588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedeurwaerder S., Defrance M., Calonne E., et al. 2011. Evaluation of the infinium methylation 450K technology. Epigenomics 3, 771–784 [DOI] [PubMed] [Google Scholar]
- Esteller M. 2008. Epigenetics in cancer. N. Engl J. Med. 358, 1148–1159 [DOI] [PubMed] [Google Scholar]
- Feber A., Guilhamon P., Lechner M., et al. 2014. Using high-density DNA methylation arrays to profile copy number alterations. Genome Biol. 15, R30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loader C: locfit: Local Regression, Likelihood and Density Estimation. CRAN: CRAN; 2013. [Software] https://cran.r-project.org/web/packages/locfit/index.html [Google Scholar]
- Mermel C.H., Schumacher S.E., Hill B., et al. 2011. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris T.J., Butcher L.M., Feber A., et al. 2014. ChAMP: 450k chip analysis methylation pipeline. Bioinformatics 30, 428–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshen AB, Venkatraman ES, Lucito R, Wigler M: Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 2004, 5:557–572 [DOI] [PubMed] [Google Scholar]
- Papillon-Cavanagh S, Fortin J, Jay ND J.M: CopyNumber 450k: an R package for CNV inference using Illumina 450k DNA methylation assay., vol. 3.1, 1.4 edition: Bioconductor; 2013. [Software] http://bioconductor.org/packages/3.2/bioc/html/CopyNumber450k.html [Google Scholar]
- Pinto D., Darvishi K., Shi X., et al. 2011. Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat. Biotechnol. 29, 512–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poncet P: modeest: Mode Estimation. R package version 2.1.; 2012. [Software] https://cran.r-project.org/web/packages/modeest/index.html
- Robin X., Turck N., Hainard A., et al. 2011. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinf. 12, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saksena G, Tabak B, Gentry J, Broad Institute: Copy Number Inference Pipeline Documentation. Broad Institute; Cambridge, MA; 2012. [Software] http://genepattern.broadinstitute.org/gp/pages/index.jsf?Isid=CopyNumberInferencePipeline [Google Scholar]
- Seshan VE, Olshen A: DNAcopy: DNA copy number data analysis. R package version 1.40.0. Bioconductor: Bioconductor; 2012. [Software] https://www.bioconductor.org/packages/release/bioc/html/DNAcopy.html [Google Scholar]
- Shi G. 1993. Multivariate data analysis in palaeoecology and palaeobiology—A review. Palaeogeogr. Palaeoclimatol. Palaeoecol. 105, 199–234 [Google Scholar]
- Stratton M.R., Campbell P.J., and Futreal P.A. 2009. The cancer genome. Nature 458, 719–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm D., Witt H., Hovestadt V., et al. 2012. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22, 425–437 [DOI] [PubMed] [Google Scholar]
- Verhaak R.G., Hoadley K.A., Purdom E., et al. 2010. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You J.S., and Jones P.A. 2012. Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell 22, 9–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.