

Abstract
Background:
Virtual screening is vital for contemporary drug discovery but striking performance fluctuations are commonly encountered, thus hampering error-free use.
Results and Methodology:
A conceptual framework is suggested for combining screening algorithms characterized by orthogonality (docking-scoring calculations, 3D shape similarity, 2D fingerprint similarity) into a simple, efficient and expansible python-based consensus ranking scheme. An original experimental dataset is created for comparing individual screening methods versus the novel approach. Its utilization leads to identification and phosphoproteomic evaluation of a cell-active DYRK1α inhibitor.
Conclusion:
Consensus ranking considerably stabilizes screening performance at reasonable computational cost, whereas individual screens are heavily dependent on calculation settings. Results indicate that the novel approach, currently available as a free online tool, is highly suitable for prospective screening by nonexperts.
Keywords: : screening enrichment, CREB1, docking-scoring calculations, fingerprint similarity, NCI diversity set-II, NSC379099, p53, phosphoproteomics, analysis of residuals, shape-based similarity
The contribution of computational methodologies toward rationalization and acceleration of contemporary drug discovery is nowadays indisputable. Among the wide range of available in silico methods, a key role in the early discovery stages is held by virtual screening (VS) techniques [1]. VS tools span a wide array of methodological principles, but they all share the common objective of timely and cost–effective evaluation of compound collections and identification of the most promising initial hits. However, as each VS approach comes with their own theoretical background and practical implementation, they also have their own limitations and weaknesses. Consequently, performance of VS is strongly dependent on calculation settings and, to this respect, preoptimization is in most cases a fundamental requirement for carrying out successful screening endeavors [2–4]. Indeed, an increasing number of benchmarks describe elaborate procedures for unbiased evaluation or optimization of VS algorithms [2,5–11]. Nonetheless, the process of preoptimizing VS calculations is not effortless and usually it depends on retrospective analyses based on already published datasets which, notably, cannot always be successfully integrated even when global measures such as Ki values are provided [12]. The intense need for optimal VS procedures is illustrated in community-wide initiatives like the Grand Challenge competitions held by Drug Design Data Resources (D3R) [13,14].
In contrast to such sophisticated approaches, a simple literature search shows that in most instances where VS is applied, a rather elementary approach is ultimately followed. This is especially true for the vast majority of projects undertaken by groups that focus their research efforts on medicinal rather than computational chemistry and on prospective discovery rather than retrospective methodology-oriented studies. The approach usually involves a single VS method either as standalone or by introducing a limited amount of methodological variation. Consensus processing of results may follow but is limited to either sequential implementation of different methods or minor modifications over the originally used scoring method [4,15–17]. To this respect, one of the most common pitfalls of prospective VS endeavors based on a single method is robustness [18]. We understand robustness as the capacity of a VS method to operate at a relatively stable performance level independent from small differentiations in calculation settings and preparation. Our group and others have shown that VS performance is heavily dependent on factors that cannot always be fully rationalized, such as screening template selection, model construction, setup and fine-tuning of calculations [3,19–26]. Small variations in those factors can introduce large deviations in resulting enrichment and overall success of VS, underlying the need for careful assessment of any VS protocol prior to its implementation.
It can, thus, be reasonably suggested that there exists a conceptual gap between research efforts that implement VS for pursuing methodological advancement and others having as their top priority derivation of primary experimental results, in other words, bioactive hits for follow-up optimization studies. We hereby present a VS protocol that is characterized by enhanced robustness and combines computational efficiency, scalability and capacity for effortless integration within typical medicinal chemistry projects with no requirements for expertise on computational method optimization. The protocol represents an advancement of our consensus methodology used with success in previous studies [19,27–29]. It is stepwise and consists of performing different screens based on orthogonal VS methodologies, linearly integrating results and, finally, implementing an exponential ranking scheme for deriving a single, consensus-sorted dataset. Three VS methods are utilized in the present study, namely docking-scoring calculations implementing a rigid protein representation and an empirical scoring function (structure-based method, Glide software, Schrodinger, Inc.), 2D similarity searches based on hashed fingerprints that encode chemical patterns (ligand-based method, Canvas software, Schrodinger, Inc.) and 3D similarity searches based on molecular shape and distinct pharmacophoric points comparison (ligand-based method, ROCS software, Openeye, Inc.) [8,30–34]. For validation reasons, we create a self-consistent dataset by screening in vitro a collection of 1364 compounds against disease-relevant protein kinases. The protocol application enhances overall VS performance compared with individual methods, especially with respect to screening stability and enrichment. Its implementation assists in identifying a number of compounds with kinase inhibitory capacity and a promising DYRK1α inhibitor is further subjected to a phosphoproteomic analysis for evaluating its potential as a cell-active modulator of key signaling pathways.
Materials & methods
Virtual screening
Preparation of the compound collection, proteins and ligand queries were performed as previously described [29]. The crystal structures used are: CDK5: 4au8, 3o0g, 1unl, 1ung; DYRK1α: 4ylk, 5a4t, 4mq1, 3anr; GSK3β: 1q5k, 4afj, 4pte, 1q41; CK1: 4twc, 4hgt, 3uzp, 4kb8. Cocrystallized inhibitors were extracted from crystal structures and used as query templates for 2D- and 3D-similarity searches. Docking grid preparations, docking calculations and similarity screening were performed as previously described [19,29]. All grids were prepared using no scaling for protein VdW radii and an extended ligand centering box of 12 × 12 × 12Å. Docking calculations were performed by using VdW scaling of 0.8 for ligand atoms. All 3D-based ROCS screens were performed as previously described [19]. With respect to 2D-similarity Canvas screening, the NCI Diversity set-II and the 16 query inhibitors were introduced in the spreadsheet and the complete set of physicochemical, topological, Ligfilter and Qikprop descriptors were determined for all entries. Then, six fingerprints, (linear, radial, dendritic, MOLPRINT2D, topological, MACCS) were calculated with default atom typing and scaling parameters and Tanimoto similarity was used for ranking.
Consensus ranking methodology & web application
The programming environment used was Python v. 3.5 with additional packages. The input dataset was ported to Sqlite3 for ease of querying by use of the xls2db package. Query results from the database were stored in pandas data frames. For numerical calculations, the numpy package was used for better performance. For the receiver operating characteristic (ROC) calculation, the scikit-learn package with the corresponding functions was deployed. The results were visualized using matplotlib and seaborn. The web application is written in pure R. It accepts .xlsx files as input and outputs, the sorted molecules based on the consensus score assigned to each molecule. The result can be downloaded as .xlsx file. The web application can be found at ‘www.consensus-calculator.online.’
Kinase inhibition & phosphoproteomics assays
The kinase assays for DYRK1α, CDK5 and GSK3β were performed as described previously [19,64,65]. All compounds were screened at a concentration of 10 μM, a commonly used concentration threshold in high-throughput screening settings utilizing isolated targets. For phosphoproteomics, cell lines were provided by ProtAtOnce Ltd. and cell viability assays were performed as described previously [19]. Bead-based sandwich ELISA xMAP assays were performed on a Luminex FlexMAP 3D platform (Luminex, USA). An array of 25 phosphorylated protein targets, (bovine serum albumine (BSA), negative control) and PE (phycoerythrin (PE), positive control), was measured in this multiplex antibody-based ELISA. The custom phosphoprotein antibody-coupled beads were developed by ProtAtOnce Ltd. (Athens, Greece) and phosphoprotein levels were measured as previously described [19]. The UniProt accession numbers of the targeted phosphorylated proteins are provided as Supplementary Information.
Results & discussion
Concept & design of experiments
A wide range of VS methods is currently available. However, their theoretical foundations can be conceptually reduced to the key issue of searching for structural requisites on a potentially bioactive small molecule that imply optimal intermolecular interactions with its macromolecular target. By taking into account the extensive limitations and systematic errors of underlying algorithms (e.g., in terms of empirical parameterization and inherent approximations for structure-based methods), it is fair to hypothesize that the information content of each method is incomplete and as such, it entails an aspect of orthogonality over other methods. To this end, our primary intention was to evaluate whether this orthogonality could be exploited through a consensus approach for enhancing VS robustness and performance. The aforementioned primary VS methods that are incorporated in the present protocol (docking-scoring calculations, 3D-shape similarity, 2D-fingerprint similarity) are widely used and, to our view, are orthogonal to a good degree with each other. Furthermore, they represent both categories of available VS techniques, the structure-based (docking calculations, Glide) as well as the ligand-based approach (3D-shape similarity, ROCS; 2D-fingerprint similarity, Canvas).
We then sought to devise a scalable VS workflow that would provide the basis of our consensus protocol. The various steps of the procedure are summarized in Figure 1. In short, the workflow consisted by preprocessing the compound collection (see the Materials & Methods section) and then by implementing the three aforementioned VS methods for performing a series of 44 different screens for each kinase target. More specifically, four crystal structure templates were selected for each of the kinases by applying three criteria, namely high resolution, presence in the active site of an ATP-competitive inhibitor and structural diversity between these cocrystallized ligands. Each crystal structure was used to perform four distinct docking runs by independently modifying two basic parameters, the presence or not of crystallographic water molecules and the scaling of the Van der Waals radii of atoms, a functionality provided by Glide software for simulating induced-fit effects (16 structure-based screens in total for each kinase target). In parallel, each of the four cocrystallized ligands present in the aforementioned crystal structures was subjected to a 3D-shape similarity search using ROCS and six distinct 2D-fingerprint similarity searches using equally numbered fingerprint derivation methods as implemented in Canvas (four 3D-based and 24 2D-based screens making a total of 27 ligand-based screens for each kinase). This sums to 44 individual screens performed for each protein target by applying small variations on the three primary VS methods presented before. This task would need approximately 120 CPU hours for a collection at the order of magnitude of the NCI Diversity set-II, including all necessary preparation steps. As discussed later, this protocol entails a minimal increase in computational requirements when compared with a simple, stand-alone VS experiment, yet this cost is fully compensated by the resulting enrichment and efficiency of the consensus method.
Figure 1. . The workflow for the proposed consensus ranking approach.

Three orthogonal primary VS methods are involved. Four protein–ligand complexes are utilized, giving rise to equal numbers of protein templates for docking calculations and ligand queries for similarity screening. Templates and settings are varied so as to increase sampling and information content. Results are averaged, normalized, whereas the log transformation ensures that compounds attaining top ranks in individual screens will be favored upon incorporation in consensus ranking. The tool is freely available at www.consensus-calculator.online.
VS: Virtual screening.
With respect to quantification of VS performance, the analysis of each individual screen as well as the consensus approach was undertaken in terms of widely used and easily comprehensible metrics such as the ROC and the related area under the ROC curve (ROC-AUC) analysis, the enrichment factors for specific percentages of the screened collection (EFx%), while we intentionally avoided more complex or less intuitive metrics [35–38]. Details regarding the conceptual framework and related formalism of the utilized metrics can be found in the Supplementary Information section.
In vitro kinase assays
Use of a complete and self-consistent set with experimentally determined biological activities is an absolute necessity for properly validating VS protocols [12,39]. To this end, we created an original dataset by evaluating in vitro the structurally diverse NCI Diversity set-II collection of 1364 compounds against three disease relevant protein kinases, namely GSK3β, DYRK1α and CDK5. The dataset was further enhanced by incorporating a recently published study targeting the closely related kinase CK1 performed in an identical experimental context [17]. The final set comprised a total of 1364 × 4 evaluations unbiasedly ranking all compounds according to their inhibitory potential against the four kinase targets. Compounds were screened at a single concentration of 10 μM and percentage residual enzyme activities were determined. Subsequently, molecules were partitioned between ‘actives’ and ‘inactives’ by applying a threshold of 50% as the minimum enzyme inhibition for annotating each compound as an inhibitor, thus giving rise to our VS validation dataset. In total, 13 molecules were found within the 50% threshold of residual activity in the case of CDK5, 65 in DYRK1α, 14 in GSK3β and 31 in CK1. The most potent inhibitors are presented in Table 1, whereas the complete dataset is freely provided as an effort to facilitate similar independent unbiased benchmarks. In addition, the experimental ranking of all NCI Diversity-II entries also enabled a straightforward analysis of residuals, in other words, difference between experimental and predicted ranks. To our knowledge, such an analysis is never applied in VS benchmarks due to lack of knowledge concerning the actual ranks of inactive compounds. Availability of such a feature in the dataset presented herein facilitated a more thorough estimation concerning the error limits of each method as to their capacity to not only discriminate between actives and inactives (usually partitioned by an arbitrary activity threshold) but to rank order all molecules of the screened collection as accurately as possible.
Table 1. . Structures and NSC numbers of the most potent NCI Diversity set-II compounds along with corresponding residual enzyme activities at 10 μM final concentration and ranking of compounds based on the Norm. Log Cons. version of the novel consensus approach.
| NCI/DTP compound code | Structure | Residual kinase activity – (Ranking determined by VS) | |||
|---|---|---|---|---|---|
| DYRK1α | GSK3α/β | CDK5/p25 | CK1δ/ε | ||
| NSC10428 (1) |  |
8% (555/1364) | 74% (578/1364) | 100% (1108/1364) | 82% (973/1364) |
| NSC13653 (2) |  |
5% (352/1364) | 46% (731/1364) | 79% (501/1364) | 93% (591/1364) |
| NSC16873 (3) |  |
12% (380/1364) | 68% (383/1364) | 69% (278/1364) | 95% (590/1364) |
| NSC21678 (4) |  |
17% (1090/1364) | 51% (304/1364) | 47% (1025/1364) | 95% (1157/1364) |
| NSC71795 (5)† |  |
9% (7/1364) | 46% (12/1364) | 22% (74/1364) | 65% (710/1364) |
| NSC72292 (6)† |  |
2% (3/1364) | 95% (16/1364) | 50% (45/1364) | 45% (380/1364) |
| NSC99034 (7) |  |
9% (655/1364) | 51% (625/1364) | 100% (641/1364) | 95% (882/1364) |
| NSC134199 (8)† |  |
96% (1146/1364) | 25% (940/1364) | 60% (929/1364) | 100% (1272/1364) |
| NSC150821 (9) |  |
19% (123/1364) | 94% (517/1364) | 49% (627/1364) | 83% (508/1364) |
| NSC155698 (10) |  |
11% (40/1364) | 77% (152/1364) | 30% (277/1364) | 97% (658/1364) |
| NSC156565 (11) | 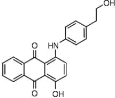 |
17% (256/1364) | 99% (243/1364) | 100% (110/1364) | 27% (84/1364) |
| NSC163920 (12) | 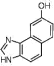 |
12% (22/1364) | 33% (143/1364) | 7% (88/1364) | 20% (61/1364) |
| NSC283845 (13)† | 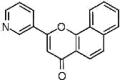 |
14% (80/1364) | 56% (133/1364) | 52% (328/1364) | 79% (145/1364) |
| NSC378719 (14) | 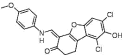 |
12% (302/1364) | 53% (93/1364) | 100% (608/1364) | 78% (301/1364) |
| NSC379099 (15)† | 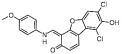 |
3% (319/1364) | 32% (889/1364) | 74% (1210/1364) | 60% (962/1364) |
| NSC400938 (16) |  |
30% (10091/364) | 17% (1024/1364) | 24% (1123/1364) | 47% (1211/1364) |
| NSC657149 (17) |  |
17% (15/1364) | 77% (6/1364) | 40% (244/1364) | 80% (592/1364) |
Five compounds including ellipticine (5) and harmol (6) have been reported in the past as kinase inhibitors (marked by an †), while the rest are reported here for the first time (bold characters indicate the most potent inhibitors, i.e. of residual enzyme activity below 15%).
Performance assessment of individual screening methods
As a first step, we tried to determine the level of accuracy that a stand-alone VS method and its corresponding single screen could reach, in other words, the average procedure followed by a typical prospective VS campaign. The mean ROC-AUC and EF values of individual screens are summarized in Table 2. A rather anticipated initial observation was that the structure-based screens outperform those derived by the ligand-based methods in an almost systematic fashion, with the latter demonstrating rather comparable metrics and a marginally better performance for the 2D-similarity method. Indeed, the mean ROC-AUC value of all structure-based screens performed against the four kinases (Glide, n = 64) was 0.702 in contrast to 0.520 achieved by 3D-similarity ligand-based (ROCS, n = 16) and 0.561 by 2D-similarity ligand-based screens (Canvas, n = 96), respectively. This trend in ROC-AUC values favoring the structure-based approach was consistently propagated in results, indicating that the moderately higher overall success rate of the docking-scoring method is likely not case dependent (Table 2).
Table 2. . Mean receiver operating characteristic-area under the curve, enrichment factor and related statistics determined for individual screens based on the three primary virtual screening methodologies utilized herein.
| ROC-AUC | EF1% | EF2% | EF5% | EF10% | |
|---|---|---|---|---|---|
| GLIDE | |||||
| Cumulative | 0.702 ± 0.570 | 4.41 ± 4.33 | 4.04 ± 2.45 | 3.60 ± 1.31 | 2.98 ± 0.86 |
| (min–max) | (0.563–0.806) | (0.00–14.98) | (0.00–14.23) | (1.43–7.16) | (1.43–5.01) |
| DYRK1α | 0.750 ± 0.029 | 3.32 ± 1.33 | 3.49 ± 1.35 | 3.75 ± 1.07 | 3.29 ± 0.55 |
| (min–max) | (0.669–0.806) | (0.00–4.84) | (0.77–5.43) | (1.85–5.86) | (2.47–4.47) |
| CDK5 | 0.689 ± 0.064 | 6.08 ± 6.60 | 4.96 ± 3.57 | 3.76 ± 1.51 | 2.98 ± 1.02 |
| (min–max) | (0.563–0.786) | (0.00–14.98) | (0.00–14.23) | (1.43–7.16) | (1.43–5.01) |
| GSK3β | 0.691 ± 0.051 | 4.21 ± 3.71 | 4.06 ± 2.16 | 3.67 ± 1.43 | 2.86 ± 0.72 |
| (min–max) | (0.592–0.770) | (0.00–7.49) | (0.00–7.21) | (1.43–7.16) | (1.43–4.29) |
| CK1 | 0.676 ± 0.044 | 4.01 ± 4.33 | 3.66 ± 1.86 | 3.24 ± 1.09 | 2.77 ± 0.98 |
| (min–max) | (0.607–0.769) | (0.00–13.53) | (1.62–8.14) | (1.94–5.82) | (1.61–4.85) |
| ROCS | |||||
| Cumulative | 0.520 ± 0.103 | 2.26 ± 3.10 | 2.40 ± 2.55 | 1.72 ± 1.69 | 1.46 ± 0.92 |
| (min–max) | (0.385–0.707) | (0.00–8.07) | (0.00–7.21) | (0.00–5.73) | (0.00–3.39) |
| DYRK1α | 0.614 ± 0.106 | 4.44 ± 2.88 | 4.27 ± 2.60 | 3.08 ± 1.57 | 2.12 ± 1.10 |
| (min–max) | (0.437–0.707) | (0.00–8.07) | (0.00–6.99) | (0.62–4.93) | (0.46–3.39) |
| CDK5 | 0.498 ± 0.095 | 0.00 | 2.70 ± 2.99 | 2.15 ± 2.15 | 1.43 ± 0.88 |
| (min–max) | (0.405–0.648) | (0.00) | (0.00–7.21) | (0.00–5.73) | (0.72–2.86) |
| GSK3β | 0.430 ± 0.053 | 3.74 ± 3.74 | 1.80 ± 1.80 | 0.72 ± 0.72 | 1.07 ± 0.36 |
| (min–max) | (0.385–0.519) | (0.00–7.49) | (0.00–3.61) | (0.00–1.43) | (0.72–1.43) |
| CK1 | 0.537 ± 0.031 | 0.85 ± 1.46 | 0.81 ± 0.81 | 1.13 ± 0.70 | 1.21 ± 0.80 |
| (min–max) | (0.502–0.586) | (0.00–3.38) | (0.00–1.63) | (0.00–1.94) | (0.65–2.59) |
| Canvas | |||||
| Cumulative | 0.561 ± 0.074 | 0.85 ± 1.46 | 1.02 ± 1.31 | 0.89 ± 0.93 | 0.98 ± 0.81 |
| (min–max) | (0.234–0.749) | (0.00–8.07) | (0.00–7.21) | (0.00–5.24) | (0.00–4.30) |
| DYRK1α | 0.625 ± 0.102 | 2.62 ± 2.41 | 2.39 ± 2.00 | 2.21 ± 1.57 | 1.86 ± 1.14 |
| (min–max) | (0.356–0.749) | (0.00–8.07) | (0.00–6.21) | (0.00–5.24) | (0.15–3.70) |
| CDK5 | 0.563 ± 0.086 | 0.00 | 1.20 ± 1.99 | 1.61 ± 1.19 | 1.43 ± 0.85 |
| (min–max) | (0.377–0.731) | (0.00) | (0.00–7.21) | (0.00–4.30) | (0.00–4.30) |
| GSK3β | 0.451 ± 0.081 | 0.94 ± 2.48 | 1.05 ± 1.64 | 0.84 ± 0.82 | 0.72 ± 0.62 |
| (min–max) | (0.234–0.669) | (0.00–7.49) | (0.00–3.61) | (0.00–2.86) | (0.00–2.15) |
| CK1 | 0.561 ± 0.073 | 0.85 ± 1.46 | 1.02 ± 1.31 | 0.89 ± 0.93 | 0.98 ± 0.81 |
| (min–max) | (0.426–0.707) | (0.00–3.38) | (0.00–3.26) | (0.00–3.32) | (0.00–2.59) |
Suboptimal robustness in screening performance is clearly illustrated on extensive fluctuations of means and related standard deviations. Scores are partitioned by primary VS method, while cumulative scores describe enrichment achieved by each VS method over all four kinases (cumulative scores: Glide, n = 64; ROCS, n = 16; Canvas, n = 96; kinase-specific scores: Glide, n = 16; ROCS, n = 4; Canvas, n = 24)
EF: Enrichment factor; ROC-AUC: Area under the ROC curve; ROC: Receiver operating characteristic; VS: Virtual screening.
However, a key outcome of this first step was the strikingly high degree of variability in performance metrics among the primary VS methods. While the mean ROC-AUC of structure-based screens was 0.702 (Glide), there were individual screens where the same value was as low as 0.563 (CDK5, docking template: 4AU8), indicating performance marginally above average. On the other extreme, the best performing structure-based screen afforded a notably high ROC-AUC of 0.806 (DYRK1α, docking template: 3ANR). In the same direction, the moderately lower overall success of 2D-similarity ligand-based screens (Canvas) was accompanied by strikingly high fluctuations. Indeed, the poorest performing individual 2D-screen afforded a fairly unacceptable ROC-AUC value of 0.234 (GSK3β, query: 1Q41 ligand), while the best result achieved by the same method was 0.749 (DYRK1α, query: 4YLK ligand), a value in direct comparison with the top-performing structure-based screen. On the other hand, although 3D-similarity ligand-based screens (ROCS) typically afforded the lowest overall scores, their performance was much more stable than the respective 2D-similarity screens (Canvas) but not as stable as the structure-based screens, with minimum and maximum values in this case equal to 0.385 (GSK3β, query: 1Q5K) and 0.707 (DYRK1α, query: 3ANR), respectively. Concerning instances of poor performance, none of 64 total structure-based screens performed below average (ROC-AUC < 0.500), while three of 64 screens were borderline with ROC-AUC values between 0.500 and 0.600. The number of below-average screens was larger in 3D-similarity ligand-based screens (6 out of 16) and notably larger in their 2D-similarity counterparts (32 out of 96), while borderline cases were 6 of 16 and 30 of 96, respectively.
The same variations were outlined in a more dramatic way when examining EF values, particularly those of the early enrichment domain at levels of 1 and 2% (Table 2). Regarding EF1%, the mean value for structure-based screens was 4.41, with corresponding values for 3D-similarity ligand-based and 2D-similarity ligand-based screens significantly lower, at 2.26 and 0.85, respectively. A similar trend was determined for EF2% where Glide afforded a moderate value of 4.04, while respective scores of ROCS and Canvas were 2.40 and 1.02. With respect to Canvas, early enrichment performance actually tended to zero. In agreement with observations made for the ROC-AUC metric in the individual screens, large dispersions were observed on EF values as well. At EF1%, the maximum value among structure-based screens was 14.98 representing a fairly satisfactory degree of enrichment but at the same time, the minimum value was 0. Concerning both ligand-based screens (ROCS and Canvas), the EF1% fluctuated between 0 and 8.07. These differences tended to smoothen along higher EF thresholds. For example, the mean EF10% was 2.98, 1.46 and 0.98 for Glide-, ROCS- and Canvas-based screens, respectively. Even at this level, deviations between individual screens based on different primary VS methods were considerable (from 1.43 to 5.01 in Glide, from 0 to 3.39 in ROCS, from 0 to 4.30 in Canvas, respectively).
All different values of ROC-AUC and EF derived by the above-presented systematic analysis collectively converge to the following notion; performance of each individual VS method may indeed follow a general trend distinguishing methods between more (structure-based screening) or less (ligand-based screening) efficient, however, this distinction cannot be deducted with high certainty a priori without any knowledge on the specific system or, in other words, a retrospective, training set-based analysis that would fine-tune the protocol. This consistently suboptimal performance recorded for individual VS methods clearly shows that in cases where a single nonoptimized VS method is applied, then one is liable of getting results of extremely poor quality and questionable reliability.
Performance assessment of the consensus ranking protocol & residual analysis
In light of this outcome, we devised a consensus ranking scheme as outlined conceptually in the previous section. The first stage was derivation of the mean rank of each compound over all 44 utilized screens performed for each kinase target. The key hypothesis here was that the dispersion of inactive compounds over the possible ranks is wider than the corresponding effect observed for actives. This notion was an initial guess based on previous experience, but it was fully justified by the array of recorded mean ROC-AUC values presented above (Table 2). Hence, it was hypothesized that by summing up results obtained from individual screens, an amplification of the signal -or a higher enrichment rate- could be possibly achieved. This effect would be further enhanced due to the presence of orthogonal structural or energetic information derived by each primary VS method. Mean ranks were determined by averaging all individual ranks followed by normalization over the three primary methodologies (referred to as Norm. Cons. ranking hereafter). Normalization was applied due to the different number of alternative screens originating from each of the three primary methods, with structure-based (Glide), 3D-similarity ligand-based (ROCS) and 2D-similarity ligand-based (Canvas) calculations entailing 16, 4 and 24 distinct screens for each kinase, respectively. For exploring the effect of normalization, mean ranks were also calculated by simple averaging without normalizing individual ranks (referred to as Cons. ranking).
Another aspect of key importance was promotion of high ranks against lower ones. It was obvious that compounds that showed a completely distinct ranking profile across the various individual screens could end up in having an identical consensus rank. This would conceal important information derived by the different methods and possibly cancel out the orthogonality aspect of the approach. A method for guaranteeing that compounds achieving top positions in any of the individual screens might retain an advantage upon incorporation of these rankings into the consensus process was devised. A simple exponential weighting scheme was applied through the transformation of all ranks into their logarithms, thus conferring a smoothing decay to the boosting effect. The transformation was applied to both normalized and raw rankings as derived from the previous step and resulting consensus ranks are referred to as Norm. Log. Cons. and Log. Cons., respectively.
Data from all individual screens were processed by the consensus ranking scheme and enrichment metrics were calculated for the four consensus variations (Cons., Norm. Cons., Log. Cons. and Norm. Log. Cons.). Results are summarized in Table 3. Overall, the consensus approach succeeded in introducing a considerable degree of stability on screening performance. Among the four variations, those incorporating the log transformation (Log. Cons. and Norm. Log. Cons.) afforded the best cumulative metrics. Their ROC-AUC values were 0.730 and 0.723, respectively, slightly higher than those achieved by simple averaging (Cons.: 0.695, Norm. Cons.: 0.686). This was an indication that orthogonality is indeed present among the three primary VS methods and, moreover, that exponential factorization can indeed capture and exploit a measurable fraction of this information. The cumulative ROC-AUC score of 0.723 as derived by Norm. Log. Cons. was directly comparable with the corresponding cumulative scores obtained for structure-based (0.702, Glide), 3D ligand-based (0.520, ROCS) and 2D ligand-based screens (0.561, Canvas). Yet, the importance of this comparison lies at the fact that in the consensus approach no fluctuations are applicable. Thus, the ROC-AUC Norm. Log. Cons. describes the performance of a singular screening experiment in contrast to the cumulative scores of individual screens that are means characterized by wide deviations. The worth in this aspect of consensus ranking can be more clearly illustrated in the screening success as determined for each kinase separately. Concerning DYRK1α, for example, the Norm. Log. Cons. consensus ROC-AUC is 0.821, whereas the most successful primary VS method affords values deviating between 0.669 and 0.806 (structure-based screen, Table 2, Glide min–max). For the same enzyme, the remaining ligand-based screening methods fluctuate even wider, with values ranging from 0.437 and 0.356 (Table 2, ROCS and Canvas min, respectively) to 0.749 or 0.707 (Table 2, ROCS and Canvas max, respectively). The stability trend propagates in an almost consistent manner across the other targets and it is quite similar in CDK5. In this instance, Log. Cons. reaches 0.756, whereas individual methods may perform somehow better at exceptional instances (max Glide ROC-AUC: 0.786), but may also result to ROC-AUC values as poor as 0.563, giving ultimately rise to a mediocre mean AUC of 0.689, a value markedly lower than the corresponding consensus score. An equally interesting aspect is that consensus ranking outperforms ROCS- and Canvas-based screens in a systematic and striking manner. With respect to GSK3β, the target affording the lowest success rate, consensus scores and more specifically Norm. Log. Cons. results to a ROC-AUC of 0.630, far beyond the below-average values obtained by ROCS (min: 0.385, mean: 0.430) or Canvas (min: 0.234, mean: 0.451). Even though there are single instances of structure-based or ligand-based screens in GSK3β that perform better that consensus (Glide-based max ROC-AUC: 0.770, docking template: 1Q5K; Canvas-based max: 0.669, query: 1Q41 ligand), the corresponding mean values show that such successful individual screens are rather rare and tend to diminish in light of the already discussed restraint that no error-free selection of screening templates might be applied in advance of a VS campaign. Comparison of the mean performance of primary VS methodologies versus the best-performing consensus schemes is shown in Figures 2 (ROC curves) and 3A (box plots). Enhancement of screening robustness is evident in all cases, while graphs clearly show that consensus is not a simplistic process of merging different ranking schemes and obtaining an average result, but it enables a constructive interference that ultimately introduces a multiplier effect in individual VS rankings.
Table 3. . Mean receiver operating characteristic-area under the curve, enrichment factor and related statistics as determined for the four versions of the consensus ranking approach. .
| ROC-AUC | EF1% | EF2% | EF5% | EF10% | |
|---|---|---|---|---|---|
| CONS. | |||||
| Cumulative | 0.707 ± 0.090 | 3.92 ± 2.70 | 2.28 ± 1.58 | 1.88 ± 1.27 | 2.56 ± 1.32 |
| DYRK1α | 0.808 | 4.84 | 3.88 | 4.01 | 4.01 |
| CDK5 | 0.729 | 7.49 | 3.61 | 1.43 | 3.58 |
| GSK3β | 0.561 | 0.00 | 0.00 | 1.43 | 0.72 |
| CK1 | 0.730 | 3.38 | 1.63 | 0.65 | 1.94 |
| NORM. CONS. | |||||
| Cumulative | 0.691 ± 0.093 | 1.61 ± 2.79 | 2.37 ± 2.59 | 2.48 ± 1.89 | 2.36 ± 1.42 |
| DYRK1α | 0.807 | 6.45 | 6.21 | 4.32 | 4.63 |
| CDK5 | 0.672 | 0.00 | 0.00 | 4.30 | 2.15 |
| GSK3β | 0.553 | 0.00 | 0.00 | 0.00 | 0.72 |
| CK1 | 0.731 | 0.00 | 3.26 | 1.29 | 1.94 |
| LOG. CONS. | |||||
| Cumulative | 0.734 ± 0.069 | 4.73 ± 3.28 | 2.67 ± 2.05 | 3.79 ± 1.08 | 2.89 ± 0.98 |
| DYRK1α | 0.820 | 8.07 | 5.44 | 4.63 | 3.55 |
| CDK5 | 0.756 | 7.49 | 3.61 | 4.30 | 4.30 |
| GSK3β | 0.629 | 0.00 | 0.00 | 4.30 | 2.15 |
| CK1 | 0.733 | 3.38 | 1.63 | 1.94 | 1.94 |
| NORM. LOG. CONS. | |||||
| Cumulative | 0.723 ± 0.069 | 6.61 ± 1.88 | 4.86 ± 2.35 | 3.99 ± 1.05 | 3.08 ± 0.775 |
| DYRK1α | 0.821 | 8.07 | 6.99 | 5.55 | 4.01 |
| CDK5 | 0.702 | 7.49 | 3.61 | 4.30 | 3.58 |
| GSK3β | 0.630 | 7.49 | 7.21 | 2.86 | 2.15 |
| CK1 | 0.740 | 3.38 | 1.63 | 3.23 | 2.59 |
The stability in screening performance and overall enrichment quality is evident by comparing the values especially of log-based consensus schemes with the respective means, minimum and maximum values of individual screens as these are presented in Table 2.
EF: Enrichment factor; ROC-AUC: Area under the ROC curve; ROC: Receiver operating characteristic.
Figure 2. . Screening results and consensus ranking evaluation.

(A) A comparison of the ROC curves and metrics determined for the mean performance of each primary VS method (Glide, blue; ROCS, orange; Canvas, green) versus the two optimal consensus variations (Norm. Cons., red; Norm. Log. Cons., purple marked). In all cases except GSK3β consensus outperforms individual screening, whereas in GSK3β consensus retains a good early recovery of true actives and a considerably improved stability and performance over ligand-based methods. (B) The cumulative performance comparison of consensus ranking against individual methods over all kinases.
ROC: Receiver operating characteristic; VS: Virtual screening.
Figure 3. . Screening results and consensus ranking evaluation.

(A) Box plots showing performance of individual screens versus consensus ranking as illustrated by the ROC-AUC metric. (B) A graph showing the enrichment factors calculated at several levels of the screened library. The gain in early enrichment achieved by the consensus approach is evident particularly with respect to the ligand-based VS methods.
ROC-AUC: Area under the ROC curve; ROC: Receiver operating characteristic; VS: Virtual screening.
Moreover, consensus retains an advantage at the critical aspect of early recovery of actives. Indeed, increased stability of consensus ranking was evident on early enrichment as captured by EF1% and EF2% (Table 3 & Figure 3B). The top-performing approach in this case also was Norm. Log. Cons., which reached in EF1%, a value of 6.61 compared with 4.41 determined for the best-performing individual VS method. It should be stressed that the overall enrichment enhancement is substantial, given that EF values of Table 2 are means entailing striking deviations and including, as already mentioned, several totally unsuccessful individual screens with EFs of 0. The positive impact of log-based consensus ranking on early enrichment was even more clearly reflected at the gap between EF values derived by either Log. Cons. or Norm. Log. Cons. and corresponding EF scores of individual ROCS- and Canvas-based screens (Figure 3B). Consensus ranks emphatically outperformed early enrichment rates obtained by the ligand-based methods. For ROCS, the mean EF1% was 2.26 and the minimum 0 while for Canvas corresponding metrics were 0.85 and 0. Similar differences were calculated at EF2%, with means of ROCS-based and Canvas-based screens at 2.40 and 1.02, respectively, and minima at 0. On the other hand, consensus variations where no log transformation was applied (Cons. and Norm. Cons.) failed to improve early enrichment, thus providing additional evidence that the exponential transformation is of key importance for optimally extracting information included in individual screens, in line with the orthogonality hypothesis. The constructive effect of log-based consensus gradually decayed beyond the 5% level, where in any case the recovery of active molecules is not expected to be exceedingly rapid (Figure 3B).
Finally, an analysis of residuals was performed for all screens undertaken in this study by determining the differences between experimental and predicted ranks and corresponding root-mean-square deviation (RMSD) values for each of the screened compounds. Graphs of residuals were created and their inspection revealed random distribution patterns consistently throughout the different screens (Figure 4). The absence of any nonrandom motifs was considered as a good indication that no concealed parameters were involved in calculations and, most importantly, that the error of prediction in each screen truly represent the methodological limit of the corresponding VS method. The RMSD values once more underlined the gain in performance achieved by consensus ranking over most of the ligand-based and a good fraction of the structure-based screens.
Figure 4. . Screening results and consensus ranking evaluation.


(A) Typical graphs of residuals (experimental–predicted ranks) as determined for the four kinases. The plots depicted herein were derived by implementing the Norm. Log. Cons. consensus approach but similar random distribution of residuals was identified for all screens performed in this study. (B) A heat map of the RMSD values calculated by the residual analysis for all screens reported in the study.
Phosphoproteomic evaluation of NSC379099
The in vitro screening of the NCI Diversity set-II led to identification of several new chemotypes with kinase-inhibitory activity along with known inhibitors (Table 1). Among the most interesting molecules, the dibenzofuranone 15 (NSC379099) was selected to be subjected in a phosphoproteomic analysis due to its potent inhibitory activity (3% residual enzyme activity against DYRK1α at 10 μM) and its interesting structural resemblance with sunitinib, a kinase inhibitor currently in clinical use. Although 15 has been previously studied to an extent as a CK2 inhibitor [40], its cellular effects with respect to DYRK1α inhibition have not been evaluated so far. Hence, a panel of 25 potential DYRK1α-related downstream targets and signaling modules was assembled and the effect of 15 on the phosphoproteome of one normal (HEK293 human embryonic kidney cells) and three pathological (WM115 primary melanoma, HepG2 hepatoblastoma and PC3 prostate cancer) cell lines was analyzed (Figure 5). Phosphoprotein levels were measured using bead-based sandwich ELISA (Luminex platform, ProtATonce Ltd.), while prior to analysis, it was verified that 15 did not induce significant alterations on cell viability.
Figure 5. . Screening results and consensus ranking evaluation.

(A) Results from phosphoproteomic analysis on the effect of 15 in different cell lines. Administration of the compound induces significant upregulation of specific gene products such as the tumor-suppressor p53 and the transcription factor CREB1 in a consistent fashion over three of four studied systems. An increase of kinases MAPK3, MAPK12, MEK1, transcription factor c-JUN, NF-κβ inhibitor-α and metalloproteinase ADAM-TS1 was also observed in melanoma and hepatoblastoma cell lines. The only statistically significant downregulated gene product was tyrosine kinase LCK on PC3 prostate cells (n = 6; *, p < 0.5; **, p < 0.01; ***, p < 0.001).
Apart from its well-established relation with the pathology of Down syndrome, development of CNS and neurogenesis, DYRK1α is involved in a number of signaling pathways related with survival, quiescence or apoptosis [41]. Among such DYRK1α substrates, of special interest are the NFAT, components of DREAM and possibly STAT [42,43]. A particularly interesting aspect of DYRK1α biology lies at its ambiguous role in cancer, where data supporting its contribution both as an oncogene and a tumor suppressor are available [44]. Indeed, the DYRK1α gene is suggested to be strongly dosage-dependent and this feature may in part explain such contrasting conclusions regarding its functionality [44]. Cases have been reported where both downregulation and overexpression of DYRK1α demonstrate similar results [45]. The data presented herein provide evidence that compound 15 can alter in a cell-type-dependent manner phosphorylation levels of specific signaling proteins participating to the AKT, RAS/B-RAF/MEK/ERK and JNK signaling pathways, the tumor-suppressor p53, the transcription factor CREB1 and proteins of the NF-κB family. More specifically, treatment with 15 induced an increasing trend in levels of p53 in a significant and consistent manner over all studied cell lines with the exception of PC3 cells (HEK293, 1.5-fold; WM115, 1.7-fold; HepG2, 1.9-fold). Concerning DYRK1α-mediated phosphorylation of p53, it has been reported that it leads to induction of the endogenous CDK inhibitor p21 [46], while regulation of p53 by DYRK1α can be furthermore mediated through epigenetic mechanisms involving SIRT1 [47]. Interestingly, p53 can also regulate DYRK1α through a negative feedback loop [48]. A similar pattern was observed for CREB, a protein downstream of DYRK1α with crucial role in cellular responses including immunity and differentiation [49]. Levels of phosphorylated CREB1 were significantly increased in HEK293, WM115 and HepG2 cells (1.5-, 1.7- and 1.9-folds, respectively). In addition to the pronounced effects of 15 on p53 and CREB1, increased levels were recorder in the melanoma and hepatoblastoma cells for proteins MAPK3 (WM115, 1.5-fold; HepG2, 1.7-fold), MAPK12 (HepG2, 1.6-fold), MEK1 (WM115, 1.5-fold; HepG2, 2-fold), JUN (HepG2, 2.1-fold) and NF-κBIA (both WM115 and HepG2, 1.5-fold), key players of the above-mentioned signaling pathways.
An interesting result from the analysis in melanoma cells was that DYRK1α inhibition leads to 1.5-fold upregulation of the disintegrin and metalloproteinase with thrombospondin motif protein ADAM-TS1, a relatively underexplored factor involved in angiogenesis [50,51]. Deregulation of this protease holds a role in angiogenesis processes in several cancer cell types [52]. Notably, the mRNA levels of DYRK1α are downregulated in melanoma cell lines [53]. An additional feature linking DYRK1α activity to angiogenesis is its possible involvement in the newly discovered NDY1/EZH2-miR-101-EZH2 pathway. In this pathway, downstream of DYRK1α is CREB that subsequently activates FGF-2 leading to NDY1 induction [54]. In a study involving the DYRK1α inhibitor harmine, it was shown that angiogenesis in tumors was strongly inhibited as a result of DYRK1α inhibition-related activation of p53 [55]. Several studies additionally correlate DYRK1α with angiogenesis through either the calcineurin inhibitor DSCR1 [56] or EGFR [57]. The only significant downregulation upon treatment with 15 was observed in prostate cells, where LCK demonstrated a decrease of approximately 37% from control levels. The tyrosine kinase LCK is a member of the Src family and, like many other kinases, its activation requires phosphorylation at a priming tyrosine site [58]. Not much data are available concerning involvement of DYRK1α in prostate cancer or its relation with LCK, but the closely related DYRK1β has been recently identified as a negative regulator of the tumor-suppressor NKX3.1 and is considered as an attractive target [59]. Overall, the discussed results point to a notable implication of 15 in many cellular processes. The possibility that part of changes determined on the phosphoproteome are caused by concurrent inhibition of CK2 cannot be ruled out, provided that CK2 is a highly promiscuous and multifaceted kinase [60]. Yet, on the basis of existing literature and the panel of proteins analyzed, we believe that DYRK1α represents a principal target of 15, especially with respect to upregulation effects determined for CREB, p53 and ADAM-TS1. However, more experiments and larger scale proteomic studies need to further clarify the exact role of 15 in regulating key signaling molecules including kinases upstream of the particular proteins. Finally, to gain structural insight to the potential interaction between 15 and the kinase active site, MD simulations of 40ns were performed (see Supplementary Information), indicating that the proposed binding pose of 15 (Supplementary Figure 1) represents a physically relevant geometry, which can be used as a basis for rational optimization of the dibenzofuranone hit into potent and selective DYRK1α inhibitors.
Conclusion
Robustness is a factor of pivotal importance for trouble-free incorporation of VS protocols in medicinal chemistry-oriented research groups that have no special expertise in computational chemistry or cheminformatics. As systematically shown in this study, serious fluctuations characterize the performance of VS experiments based on singe methods, especially when those that are utilized without any prior fine-tuning by use of a training set. In several cases, dramatic deviations from a better-than-average performance were noted, giving rise to screens of awkwardly low success. Such fluctuations on performance represent the risk associated with a prospective hit discovery endeavor that is exclusively based on a single, not preoptimized VS approach. The consensus ranking protocol presented herein introduces a context for substantially improving stability of VS performance. Consensus is based on the utilization of a simple process for amplifying the signal of true positives as produced by a convenient array of orthogonal VS methods and their variations. Performance of the consensus ranking scheme was in every case directly comparable to the best-performing individual screening method while, given the contribution of each individual method in the consensus result, the novel approach should be more appropriately considered as a member of the family of structure-based VS methods. In most instances, consensus ranking outperformed individual structure-based or ligand-based screens and even in those cases where higher enrichment rates were not achieved by it, consensus afforded the highest possible enrichment, further enhanced by a substantial decrease in screening uncertainty. Stated differently, the analysis reported here clearly shows that selecting the single most appropriate template for either docking calculations, 3D- or 2D-similarity screens are a rather low-probability move and, to this direction, the consensus approach offers a framework for circumventing such a stochastic behavior by embedding a rational dimension leading to robust screening endeavors. As such, consensus ranking comprises a powerful and flexible tool for optimizing any available VS setting, especially with respect to prospective screening.
With respect to its possible drawbacks, although the number of individual screens needed for implementing the proposed consensus approach can be regarded as fairly high, all preparation efforts can be minimized due to the interoperability aspect of involved algorithms. Upon selection of the most suitable crystal structure templates, preparation of protein structures for docking calculations and of ligand structures for similarity searches is straightforward and involves steps that proceed rapidly. The time-limiting step of consensus ranking is neither preparation nor postprocessing but calculations. However, the effort for performing a number of different screens instead of a single screen is completely offset by the accuracy of consensus results. On the long run, undertaking a consensus screen rather saves time by ensuring the maximal outcome from many individual screens that are, however, performed only once in contrast to the typical procedure of using a single screening method that nevertheless in most instances needs to be performed repeatedly for several times to ensure that reasonable results finally emerge. Additional drawbacks of the novel approach may be identified with respect to bias that are introduced by the individual VS methods integrated to consensus ranking. For example, such biasing can arise by using the bioactive conformation as a query structure for 3D-similarity screens or by using a compound collection with narrow coverage of the chemical space for benchmarking. Although the impact on the performance of 3D-similarity screening by querying against the biologically relevant conformation instead of a different rationally selected low-energy geometry have been shown to be limited, such factors should be systematically addressed in the future as to their impact on the performance gain achieved by the consensus scheme [8,61–63]. Finally, by implementing the consensus ranking protocol, a number of chemotypes with kinase-inhibitory properties were discovered and are reported herein. Most interestingly, it was shown that compound 15 (NSC379099), a dibenzofuranone structurally resembling the clinically used kinase inhibitor sunitinib, can affect the endogenous levels of a series of gene products related with key signaling pathways such as p53 and CREB1. As these effects seem to be strongly correlated with the capacity of 15 to inhibit DYRK1α kinase, they suggest its utilization as a promising lead for further development of analogs with potential value either as chemical biology tools or drug candidates.
Future perspective
Since their establishment as sine qua non components of most contemporary drug discovery campaigns, VS tools have gained respect by offering a collective increase in screening success between 2 and 3 orders of magnitude. Despite their collective achievements, these computational tools are still characterized by low performance stability and dependence from poorly understood factors. This is in contrast to the impressive progress in computational capacities recorded the last years. Such a notion may indicate that the collective bottleneck of VS methods is not to be found on computational efficiency but rather on suboptimal parameterization, with critical features in a drug–target interaction event likely not taken into account at the appropriate way. One such key factor is solvent thermodynamics and the underlying entropic contribution to binding. Quantitatively considering solvation effects when modeling binding of small molecules to macromolecules can have a dramatic impact on modeling accuracy and currently it comprises a highly promising field of research in computational chemistry related to drug discovery. Several cutting-edge algorithms for solvent thermodynamics mapping have opened new routes toward high-accuracy free energy estimations and their contribution to improving precision of molecular modeling is catalytic. However, no such method is currently a functional part of any in silico screening tool apart from investigational cases. We consider that incorporation of solvation-mapping modules may offer great enhancement in VS accuracy by enabling modeling of factors currently not properly addressable. We anticipate that integrating such components in VS algorithms will be accomplished in due time and with good success, thus defining a landmark in the respective field. Intense research efforts will be needed to adjust the demanding computational machinery of solvation mapping and entropy estimation to the high-throughput needs and overall orientation of VS. Moreover, large-scale optimization studies may be required for facilitating fine-tuning of the newly developed algorithms and to this end, collective training sets based on databases like ChEMBL could be used for deriving global models. Given the undeniably constructive impact of in silico screening in life sciences and in light of the emerging technological advances on computing, we believe that in the near future VS tools dealing with entropic contributions of binding as accurately as they are capable of performing today for corresponding enthalpy counterparts will be an everyday reality.
Summary points.
Virtual screening (VS) is invaluable in accelerating the early stages of drug discovery but its performance is generally characterized by broad fluctuations depending on calculation settings.
Although all available VS methods are based on empirical parameterization, they may demonstrate orthogonality with each other by capturing different aspects of the drug–target interaction requisites.
A framework is proposed for exploiting the orthogonality aspect by introducing a consensus ranking scheme that enables improved stability of screening performance over individual VS approaches.
For the purpose of benchmarking the novel protocol, an original dataset is created by evaluating in vitro a compound collection against a panel of protein kinases.
Among the identified actives molecules, compound NSC379099 is a potent inhibitor of DYRK1α kinase with the capacity to affect levels of critical modules in signaling pathways related to the target kinase such as p53, CREB1 and ADAM-TS1 in cells.
Consensus ranking is a user-friendly, efficient and expansible protocol with markedly enhanced robustness and improved enrichment with respect to individual VS approaches. It is freely available on the web at www.consensus-calculator.online.
Supplementary Material
Footnotes
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.future-science.com/doi/full/10.4155/fmc-2018-0198
Financial & competing interests disclosure
The screened compounds were provided free of charge from NCI/DTP repository (https://dtp.cancer.gov). V Myrianthopoulos and E Mikros acknowledge financial support by the H2020-INFRADEV-01-2017 project EPTRI (777554). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest
- 1.Shoichet BK. Virtual screening of chemical libraries. Nature. 2004;432:862. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]; • A seminal work on the capabilities and limitations on virtual screening (VS) algorithms.
- 2.Lagarde N, Zagury J-F, Montes M. Benchmarking data sets for the evaluation of virtual ligand screening methods: review and perspectives. J. Chem. Inf. Model. 2015;55(7):1297–1307. doi: 10.1021/acs.jcim.5b00090. [DOI] [PubMed] [Google Scholar]
- 3.Madhavi Sastry G, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013;27(3):221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]; • A very interesting and useful report on the impact of calculations setup on structure-based VS performance.
- 4.Sheridan RP, Mcgaughey GB, Cornell WD. Multiple protein structures and multiple ligands: effects on the apparent goodness of virtual screening results. J. Comput. Aided Mol. Des. 2008;22(3):257–265. doi: 10.1007/s10822-008-9168-9. [DOI] [PubMed] [Google Scholar]
- 5.Chaput L, Martinez-Sanz J, Saettel N, Mouawad L. Benchmark of four popular virtual screening programs: construction of the active/decoy dataset remains a major determinant of measured performance. J. Cheminform. 2016;8(1):56. doi: 10.1186/s13321-016-0167-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cross JB, Thompson DC, Rai BK, et al. Comparison of several molecular docking programs: pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2009;49(6):1455–1474. doi: 10.1021/ci900056c. [DOI] [PubMed] [Google Scholar]
- 7.Kirchmair J, Markt P, Distinto S, Wolber G, Langer T. Evaluation of the performance of 3D virtual screening protocols: RMSD comparisons, enrichment assessments, and decoy selection – what can we learn from earlier mistakes? J. Comput. Aided Mol. Des. 2008;22(3):213–228. doi: 10.1007/s10822-007-9163-6. [DOI] [PubMed] [Google Scholar]; • An excellent review dealing with issues arising from performance comparison between different VS algorithms.
- 8.Hawkins PCD, Skillman AG, Nicholls A. Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 2007;50(1):74–82. doi: 10.1021/jm0603365. [DOI] [PubMed] [Google Scholar]
- 9.Huang N, Shoichet B, Irwin J. Benchmarking sets for molecular docking. J. Med. Chem. 2006;49(23):6789–6801. doi: 10.1021/jm0608356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rueda M, Bottegoni G, Abagyan R. Recipes for the selection of experimental protein conformations for virtual screening. J. Chem. Inf. Model. 2010;50(1):186–193. doi: 10.1021/ci9003943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang Z, Wong CF. Inexpensive method for selecting receptor structures for virtual screening. J. Chem. Inf. Model. 2016;56(1):21–34. doi: 10.1021/acs.jcim.5b00299. [DOI] [PubMed] [Google Scholar]
- 12.Kramer C, Kalliokoski T, Gedeck P, Vulpetti A. The experimental uncertainty of heterogeneous public Ki data. J. Med. Chem. 2012;55(11):5165–5173. doi: 10.1021/jm300131x. [DOI] [PubMed] [Google Scholar]
- 13.Wingert BM, Oerlemans R, Camacho CJ. Optimal affinity ranking for automated virtual screening validated in prospective D3R grand challenges. J. Comput. Aided Mol. Des. 2018;32(1):287–297. doi: 10.1007/s10822-017-0065-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ye Z, Baumgartner MP, Wingert BM, Camacho CJ. Optimal strategies for virtual screening of induced-fit and flexible target in the 2015 D3R Grand Challenge. J. Comput. Aided Mol. Des. 2016;30(9):695–706. doi: 10.1007/s10822-016-9941-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee HS, Choi J, Kufareva I, et al. Optimization of high throughput virtual screening by combining shape-matching and docking methods. J. Chem. Inf. Model. 2008;48(3):489–497. doi: 10.1021/ci700376c. [DOI] [PubMed] [Google Scholar]
- 16.Feher M. Consensus scoring for protein–ligand interactions. Drug Discov. Today. 2006;11(9):421–428. doi: 10.1016/j.drudis.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 17.Yunierkis P-C, Aliuska Morales H, Cordeiro MNDS, et al. Fusing docking scoring functions improves the virtual screening performance for discovering Parkinson's disease dual target ligands. Curr. Neuropharmacol. 2017;15(8):1107–1116. doi: 10.2174/1570159X15666170109143757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sheridan RP. Alternative global goodness metrics and sensitivity analysis: heuristics to check the robustness of conclusions from studies comparing virtual screening methods. J. Chem. Inf. Model. 2008;48(2):426–433. doi: 10.1021/ci700380x. [DOI] [PubMed] [Google Scholar]
- 19.Myrianthopoulos V, Lozach O, Zareifi D, et al. Combined virtual and experimental screening for CK1 inhibitors identifies a modulator of p53 and reveals important aspects of in silico screening performance. Int. J. Mol. Sci. 2017;18(10):2102. doi: 10.3390/ijms18102102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mcgaughey GB, Sheridan RP, Bayly CI, et al. Comparison of topological, shape, and docking methods in virtual screening. J. Chem. Inf. Model. 2007;47(4):1504–1519. doi: 10.1021/ci700052x. [DOI] [PubMed] [Google Scholar]
- 21.Scior T, Bender A, Tresadern G, et al. Recognizing pitfalls in virtual screening: a critical review. J. Chem. Inf. Model. 2012;52(4):867–881. doi: 10.1021/ci200528d. [DOI] [PubMed] [Google Scholar]
- 22.Kumar A, Zhang KYJ. A cross docking pipeline for improving pose prediction and virtual screening performance. J. Comput. Aided Mol. Des. 2018;32(1):163–173. doi: 10.1007/s10822-017-0048-z. [DOI] [PubMed] [Google Scholar]
- 23.Cuzzolin A, Sturlese M, Malvacio I, Ciancetta A, Moro S. DockBench: an integrated informatic platform bridging the gap between the robust validation of docking protocols and virtual screening simulations. Molecules. 2015;20(6):9977. doi: 10.3390/molecules20069977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug Discov. 2004;3:935. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]; • An interesting review on the challenges and applications of various structure-based screening methods in drug design.
- 25.Perola E, Walters WP, Charifson PS. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins. 2004;56(2):235–249. doi: 10.1002/prot.20088. [DOI] [PubMed] [Google Scholar]
- 26.Warren GL, Andrews CW, Capelli A-M, et al. A critical assessment of docking programs and scoring functions. J. Med. Chem. 2006;49(20):5912–5931. doi: 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- 27.Myrianthopoulos V, Gaboriaud-Kolar N, Tallant C, et al. Discovery and optimization of a selective ligand for the switch/sucrose nonfermenting-related bromodomains of polybromo protein-1 by the use of virtual screening and hydration analysis. J. Med. Chem. 2016;59(19):8787–8803. doi: 10.1021/acs.jmedchem.6b00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Myrianthopoulos V, Cartron PF, Liutkevičiūtė Z, et al. Tandem virtual screening targeting the SRA domain of UHRF1 identifies a novel chemical tool modulating DNA methylation. Eur. J. Med. Chem. 2016;114:390–396. doi: 10.1016/j.ejmech.2016.02.043. [DOI] [PubMed] [Google Scholar]
- 29.Myrianthopoulos V, Lamprinidis G, Mikros E. In silico screening of compound libraries using a consensus of orthogonal methodologies. In: Mavromoustakos T, Kellici T, editors. Rational Drug Design: Methods and Protocols. Humana Press; New York, USA: 2018. pp. 261–277. [DOI] [PubMed] [Google Scholar]
- 30.Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004;47(7):1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 31.Halgren TA, Murphy RB, Friesner RA, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004;47(7):1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 32.Friesner RA, Murphy RB, Repasky MP, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein—ligand complexes. J. Med. Chem. 2006;49(21):6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 33.Duan J, Dixon SL, Lowrie JF, Sherman W. Analysis and comparison of 2D fingerprints: insights into database screening performance using eight fingerprint methods. J. Mol. Graph. Model. 2010;29(2):157–170. doi: 10.1016/j.jmgm.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 34.Sastry M, Lowrie JF, Dixon SL, Sherman W. Large-scale systematic analysis of 2D fingerprint methods and parameters to improve virtual screening enrichments. J. Chem. Inf. Model. 2010;50(5):771–784. doi: 10.1021/ci100062n. [DOI] [PubMed] [Google Scholar]
- 35.Zhao W, Hevener KE, White SW, Lee RE, Boyett JM. A statistical framework to evaluate virtual screening. BMC Bioinformatics. 2009;10(1):225. doi: 10.1186/1471-2105-10-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Truchon J-F, Bayly CI. Evaluating virtual screening methods: good and bad metrics for the “early recognition” problem. J. Chem. Inf. Model. 2007;47(2):488–508. doi: 10.1021/ci600426e. [DOI] [PubMed] [Google Scholar]; • A research article including a comprehensive overview of metrics applied for measuring screening performance with special focus on the early enrichment.
- 37.Empereur-Mot C, Guillemain H, Latouche A, Zagury J-F, Viallon V, Montes M. Predictiveness curves in virtual screening. J. Cheminform. 2015;7(1):52. doi: 10.1186/s13321-015-0100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nicholls A. What do we know and when do we know it? J. Comput. Aided Mol. Des. 2008;22(3–4):239–255. doi: 10.1007/s10822-008-9170-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; • A critical insight to a number of pitfalls and sensitivities of VS methods.
- 39.Mysinger MM, Carchia M, Irwin JJ, Shoichet BK. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J. Med. Chem. 2012;55(14):6582–6594. doi: 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guerra B, Hochscherf J, Jensen NB, Issinger O-G. Identification of a novel potent, selective and cell permeable inhibitor of protein kinase CK2 from the NIH/NCI Diversity Set Library. Mol. Cell. Biochem. 2015;406(1):151–161. doi: 10.1007/s11010-015-2433-z. [DOI] [PubMed] [Google Scholar]
- 41.Abbassi R, Johns TG, Kassiou M, Munoz L. DYRK1A in neurodegeneration and cancer: molecular basis and clinical implications. Pharmacol. Ther. 2015;151:87–98. doi: 10.1016/j.pharmthera.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 42.Arron JR, Winslow MM, Polleri A, et al. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature. 2006;441(7093):595–600. doi: 10.1038/nature04678. [DOI] [PubMed] [Google Scholar]
- 43.Sadasivam S, Decaprio JA. The DREAM complex: master coordinator of cell cycle-dependent gene expression. Nat. Rev. Cancer. 2013;13(8):585–595. doi: 10.1038/nrc3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fernández-Martínez P, Zahonero C, Sánchez-Gómez P. DYRK1A: the double-edged kinase as a protagonist in cell growth and tumorigenesis. Mol. Cell. Oncol. 2015;2(1):e970048. doi: 10.4161/23723548.2014.970048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu M, Zheng L, Han B, et al. REST regulates DYRK1A transcription in a negative feedback loop. J. Biol. Chem. 2011;286(12):10755–10763. doi: 10.1074/jbc.M110.174540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park J, Oh Y, Yoo L, et al. Dyrk1A phosphorylates p53 and inhibits proliferation of embryonic neuronal cells. J. Biol. Chem. 2010;285(41):31895–31906. doi: 10.1074/jbc.M110.147520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo X, Williams JG, Schug TT, Li X. DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. J. Biol. Chem. 2010;285(17):13223–13232. doi: 10.1074/jbc.M110.102574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Liao JM, Zeng SX, Lu H. p53 downregulates Down syndrome-associated DYRK1A through miR-1246. EMBO Rep. 2011;12(8):811–817. doi: 10.1038/embor.2011.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wen AY, Sakamoto KM, Miller LS. The role of the transcription factor CREB in immune function. J. Immunol. 2010;185(11):6413–6419. doi: 10.4049/jimmunol.1001829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vázquez F, Hastings G, Ortega M-A, et al. METH-1, a human ortholog of ADAMTS-1, and METH-2 are members of a new family of proteins with angio-inhibitory activity. J. Biol. Chem. 1999;274(33):23349–23357. doi: 10.1074/jbc.274.33.23349. [DOI] [PubMed] [Google Scholar]
- 51.Luque A, Carpizo DR, Iruela-Arispe ML. ADAMTS1/METH1 inhibits endothelial cell proliferation by direct binding and sequestration of VEGF165. J. Biol. Chem. 2003;278(26):23656–23665. doi: 10.1074/jbc.M212964200. [DOI] [PubMed] [Google Scholar]
- 52.Tan IA, Ricciardelli C, Russell DL. The metalloproteinase ADAMTS1: a comprehensive review of its role in tumorigenic and metastatic pathways. Int. J. Cancer. 2013;133(10):2263–2276. doi: 10.1002/ijc.28127. [DOI] [PubMed] [Google Scholar]
- 53.De Wit NJW, Burtscher HJ, Weidle UH, Ruiter DJ, Van Muijen GNP. Differentially expressed genes identified in human melanoma cell lines with different metastatic behaviour using high density oligonucleotide arrays. Melanoma Res. 2002;12(1):57–69. doi: 10.1097/00008390-200202000-00009. [DOI] [PubMed] [Google Scholar]
- 54.Kottakis F, Polytarchou C, Foltopoulou P, Sanidas I, Kampranis SC, Tsichlis PN. FGF-2 regulates cell proliferation, migration and angiogenesis through an NDY1/KDM2B-miR-101-EZH2 pathway. Mol. Cell. 2011;43(2):285–298. doi: 10.1016/j.molcel.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dai F, Chen Y, Song Y, et al. A natural small molecule harmine inhibits angiogenesis and suppresses tumour growth through activation of p53 in endothelial cells. PLoS ONE. 2012;7(12):e52162. doi: 10.1371/journal.pone.0052162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baek K-H, Zaslavsky A, Lynch RC, et al. Down syndrome suppression of tumor growth and the role of the calcineurin inhibitor DSCR1. Nature. 2009;459(7250):1126–1130. doi: 10.1038/nature08062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pozo N, Zahonero C, Fernández P, et al. Inhibition of DYRK1A destabilizes EGFR and reduces EGFR-dependent glioblastoma growth. J. Clin. Invest. 2013;123(6):2475–2487. doi: 10.1172/JCI63623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamaguchi H, Hendrickson WA. Structural basis for activation of human lymphocyte kinase Lck upon tyrosine phosphorylation. Nature. 1996;384:484. doi: 10.1038/384484a0. [DOI] [PubMed] [Google Scholar]
- 59.Song L-N, Silva J, Koller A, Rosenthal A, Chen EI, Gelmann EP. The tumor suppressor NKX3.1 is targeted for degradation by DYRK1B kinase. Mol. Cancer Res. 2015;13(5):913–922. doi: 10.1158/1541-7786.MCR-14-0680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pinna LA. Protein kinase CK2: a challenge to canons. J. Cell Sci. 2002;115(20):3873–3878. doi: 10.1242/jcs.00074. [DOI] [PubMed] [Google Scholar]
- 61.Koeppen H, Kriegl J, Lessel U, Tautermann C-S, Wellenzohn B. Ligand-based virtual screening. In: Sotriffer C, editor. Virtual Screening. Wiley-VCH Verlag GmbH & Co.; Weinheim, Germany, KGaA: 2011. pp. 61–85. [Google Scholar]; • A concise overview of ligand-based VS methods with case studies and examples.
- 62.Renner S, Schwab C-H, Gasteiger J, Schneider G. Impact of conformationa flexibility on three-dimensional similarity searching using correlation vectors. J. Chem. Inf. Model. 2006;46(6):2324–2332. doi: 10.1021/ci050075s. [DOI] [PubMed] [Google Scholar]
- 63.Zhang Q, Muegge I. Scaffold hopping through virtual screening using 2D and 3D similarity descriptors: ranking, voting and consensus scoring. J. Med. Chem. 2006;49(5):1536–1548. doi: 10.1021/jm050468i. [DOI] [PubMed] [Google Scholar]
- 64.Bettayeb K, Oumata N, Echalier A, et al. CR8, a potent and selective, roscovitine-derived inhibitor of cyclin-dependent kinases. Oncogene. 2008;27:5797. doi: 10.1038/onc.2008.191. [DOI] [PubMed] [Google Scholar]
- 65.Bach S, Knockaert M, Reinhardt J, et al. Roscovitine targets, protein kinases and pyridoxal kinase. J. Biol. Chem. 2005;280(35):31208–31219. doi: 10.1074/jbc.M500806200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.