

Abstract
Chagas disease (CD), or American trypanosomiasis, causes more than 10,000 deaths per year in the Americas. Current medical therapy for CD has low efficacy in the chronic phase of the disease and serious adverse effects; therefore, it is necessary to search for new pharmacological treatments. In this work, the ZINC15 database was filtered using the N-acylhydrazone moiety and a subsequent structure-based virtual screening was performed using the cruzain enzyme of Trypanosoma cruzi to predict new potential cruzain inhibitors. After a rational selection process, four compounds, Z2 (ZINC9873043), Z3 (ZINC9870651), Z5 (ZINC9715287), and Z6 (ZINC9861447), were chosen to evaluate their in vitro trypanocidal activity and enzyme inhibition. Compound Z5 showed the best trypanocidal activity against epimatigote (IC50 = 36.26 ± 9.9 μM) and trypomastigote (IC50 = 166.21 ± 14.5 μM and 185.1 ± 8.5 μM on NINOA and INC-5 strains, respectively) forms of Trypanosoma cruzi. In addition, Z5 showed a better inhibitory effect on Trypanosoma cruzi proteases than S1 (STK552090, 8-chloro-N-(3-morpholinopropyl)-5H-pyrimido[5,4-b]-indol-4-amine), a known cruzain inhibitor. This study encourages the use of computational tools for the rational search for trypanocidal drugs.
Keywords: Trypanosoma cruzi, molecular docking, cruzain, Zinc15, inhibitors
1. Introduction
According to the World Health Organization (WHO), the Americas are one of the primary regions with a high prevalence of Chagas disease (CD). CD, also known as American trypanosomiasis, affects approximately 8 million people worldwide with more than 10,000 deaths annually, mainly in endemic areas of Latin America [1,2,3]. The causal agent for CD is the parasite Trypanosoma cruzi (T. cruzi), which is transmitted mainly by triatomine insects to mammals, including human beings [4,5,6].
Trypanocidal therapy currently involves the use of two drugs: nifurtimox (Nfx) and benznidazole (Bzn). However, these drugs have several limitations that include uncertainty about their efficacy, poor tolerance, and severe adverse effects [7,8,9]. The low effectiveness of both drugs in the chronic phase of the disease, as well as the lack of interest of pharmaceutical companies in developing new drugs, has generated the need for new chemical compounds against T. cruzi.
A pharmacological target widely studied in T. cruzi is cruzain (Cz), which belongs to the family of proteases or peptide hydrolases. Proteases play an important and indispensable role in parasitic organisms, allowing them to participate in key catabolic functions such as parasite immunoevasion, encystment, exanthema, and tissue cell invasion [10]. Moreover, cysteine proteases of parasites have immunogenic properties that make them suitable targets for vaccine developments or as biomarker candidates [10].
Cz is the main cysteine protease of T. cruzi, which is expressed through all stages of the parasite life cycle. It allows parasite survival within the host. It also participates in penetration of the parasite into the host cell, in the digestion of immunoglobulins, and other varied biological functions [11,12,13]. The search for new Cz enzyme inhibitors continues to be very important because of their success in blocking the parasite life cycle in animal models [11,12,13].
A variety of chemical structures identified as Cz inhibitors have been studied, such as those derived from ureas, carbazones, chalcones, carboxyamides, carboxylic acids, triazoles, vinylsulfones, hydrazones, and others (Figure 1) [14,15,16,17,18]. Among these compounds, thiazolylhydrazones have been proven powerful agents with a capacity to inhibit replication of the epimastigote (IC50 = 0.3 μM) with a higher trypanocidal activity than benznidazole (IC50 = 1.8 μM) [4]. Other authors state that hybrids of N-acylhydrazone and furoxane are active in amastigotes of T. cruzi. Additionally, this class of compounds, by permeability screening in Caco-2 cells and cytotoxicity assays in human cells, showed less cytotoxicity, good permeability, and a higher selectivity index than the reference drug benznidazole [19]. Additionally, the N-acylhydrazone moiety is considered a privileged structure in medicinal chemistry, since it has the potential to interact with different biological targets, including the T. cruzi cysteine protease [20,21].
Figure 1.

Chemical structure of urea, thiocarbazone, chalcone, amide, nitrile, and hydrazine derivatives identified as Cz inhibitors.
In this work, the N-acylhydrazone moiety was used as the main filter in the ZINC15 database. This filtering step allowed obtention of a subset of chemicals in order to predict new potential Cz inhibitors by a structure-based virtual screening. Finally, four chemical compounds were chosen to test their trypanocidal activity in an in vitro model against epimastigotes and trypomastigotes of T. cruzi and their enzymatic inhibition effects in an extract of cysteine proteases.
2. Results and Discussion
2.1. Virtual Screening
After screening using the moiety (C=NNC(C)=O), a total of 2221 compounds that met our inclusion criteria were retrieved from the ZINC15 database. These compounds were cyclic N-acylhydrazone derivatives (Table S1, Table 1). The reason for this high number was because the small size of the moiety resulted in a high probability of it appearing during screening. However, we decided to continue the work with these compounds because they belong to the primary category known to be used as initial starting points in medicinal chemistry efforts [22]. This category is also one of the broadest libraries used in virtual screening analyses since these structures have simple chemical characteristics that are susceptible to chemical optimization [23]. Hence, these compounds were used in the next analysis.
Table 1.
Structures of the top ten compounds with the best binding energies obtained from a clean lead database of ZINC15.
| Compound | Structure | Binding Energy (kcal/mol) | Activity Reported |
|---|---|---|---|
| Z1 ZINC129684040 |
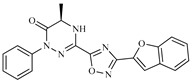
|
−8.6 | None |
| Z2 ZINC9873043 |
|
−8.3 | Inhibitor of cannabinoid receptor type 2 |
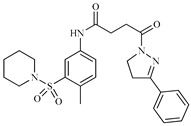
| Z3 ZINC9870651 |
|
−8.2 | None |
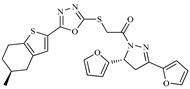
| Z4 ZINC9835465 |
|
−8.2 | Amine oxidase inhibitor |
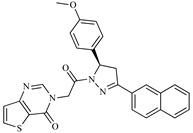
| Z5 ZINC9715287 |
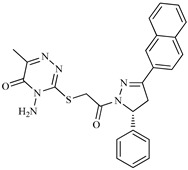
|
−8.1 | Amine oxidase inhibitor |
| Z6 ZINC9861447 |
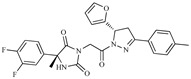
|
−8.1 | Amine oxidase inhibitor |
| Z7 ZINC9873693 |
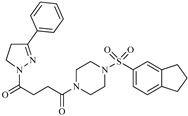
|
−8.1 | Inhibitor of aldo keto reductases |
| Z8 ZINC14741665 |
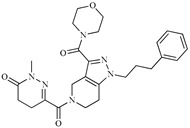
|
−8.0 | Inhibitor of cathepsin S |
| Z9 ZINC60293658 |
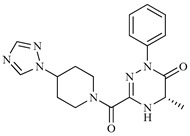
|
−8.0 | None |
| Z10 ZINC9867137 |
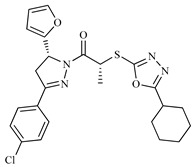
|
−8.0 | Amine oxidase inhibitor |
| 186 |
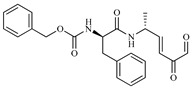
|
−6.6 | Cz inhibitor |
Cz is a cysteine protease responsible for protein degradation. Its enzyme activity is related to the presence of cysteine (Cys25) and histidine (His172) residues in the active site. The kind of covalent bond formed between the ligand and active site at Cys25 is very important to determine if the inhibition is reversible or irreversible. For example, vinylsulfones and chalcones are covalently bound to the active thiol from Cys25 and act as irreversible inhibitors. In molecular docking analyses, the Cz structure 1U9Q with the ligand 186 (Table 1), a benzyl ester, was used. This was because in other studies, relevant interactions have been shown with amino acids of the catalytic triad (Cys25, His159, and Asn175) and with the amino acids Gln19, Asp158, Trp177, and Gly66, which have been reported in interactions showing a large series of hydrazone derivatives and competitive inhibitors of Cz [4,19,24]. With the ProteinsPlus server and the DoGSiteScorer tool, the binding site of 1U9Q was identified (Figure 2a). This site has a surface of 820.85 Aº, where 15 hydrogen donors, 37 hydrogen acceptors, 35 hydrophobic interactions, 47% non-polar amino acids, 33% polar amino acids, and 7% positively charged and 13% negatively charged amino acids are located. Residues contained in the pocket of the binding site are shown in Figure 2b.
Figure 2.

Binding site detection of the binding site on the crystallographic structure 1U9Q. This pocket, yellow region (a) was detected by DoGSiteScorer tool using ProteinsPlus server. (b) Interactions recorded in cruzain-ligand 186 complex.
The coordinates of this binding site in 1U9Q were used to dock the retrieved 2221 ligands. A total of 682 ligands showed a negative binding energy (<−6.6 Kcal/mol) lower than that obtained for ligand 186 (Table S1). Table 1 shows the best compounds (top ten with binding energies <−8.0 Kcal/mol) containing the moiety (C=NNC(C)=O).
The grouping analysis of 2221 ligands made with AuPosSOM showed 20 groups (Figure S1). No grouping pattern correlated with the binding energy, a finding similar to that reported by Fereidoonnezhad et al. [25]. In the present work, we selected compounds with the highest binding energy within each clade and reviewed their commercial availability, as previously done in [26]. Of the 20 clades (Figure S1), 9 contained compounds with the best scores distributed in groups 4, 6, 10, 11, 14, 16, 17, 18, and 19, and four groups contained compounds with lower scores (2, 12, 13, and 20). Within those 20 clades, 6 compounds Z1, Z2, Z3, Z4, Z5, and Z6 showed the highest negative scores (Table 1); however, the best compound, Z1, was not commercially available, therefore, it was not considered in further selection analyses. Compound Z4 was not considered for in vitro analyses either because it had a binding energy similar to Z3, and the interactions presented were very similar. A detailed visual inspection for amino acid interactions (Figure 3) was performed for compounds Z2 and Z3. We observed that both compounds interacted in the catalytic site in a manner similar to the reference inhibitor 186. The compounds Z5 and Z6 interacted in an opposite binding site, which indicated that they could be non-competitive inhibitors and warrant further Cz binding site explorations.
Figure 3.

Analysis of coupling postures between the pose of ligand 186 interacting on the catalytic site (green) and compound Z2 in pink, Z3 in light blue, Z5 in green lemon and Z6 in blue, alignment of structures performed with AutoDock Tools programm.
The amino acid interactions for Z2 involved hydrophilic interactions with the amino acids His159 and Cys25; both amino acids are essential for the catalytic process. Other hydrophilic interactions observed for the Z2 compound near the catalytic site were Gly66, Gln19, and Asp158. In contrast, the compound Z3 only presented a hydrophobic interaction with the catalytic residue Cys25 as well as hydrophobic interactions with Gly66, Gln19, and Asp158. Both compounds Z2 and Z3 presented interactions through hydrophobic bonds with amino acids (Ala133, Leu157, Leu67, Gly23, Trp177, Gly66, and Glu205). On the other hand, the compound Z5 showed hydrophilic interactions with the residues Asn69, Ser208, Ser207, and Val111 as well as hydrophobic interactions with the residues Phe72, Asn70, Gly109, Glu73, Gln77, Trp108, and Val210. The compound Z6 showed hydrophilic interactions with Asn69, Ser208, and Val210 as well as hydrophobic interactions with Asn70, Phe72, Gly109, Glu112, Leu 113, Trp123, Ala209, and Ser207. Most of the hydrophilic interactions with the amino acids Cys25, His159, Gln19, Ser207, and Val210 are involved in the binding of oxygen atoms with the carbonyl groups present in the four compounds; this class of interactions has been reported for irreversible Cz inhibitors that contain ketones. Such chemical reactions provide the electrophilia necessary to interact with the enzyme conferring the mechanism of enzyme inhibition to elapse the reaction [27]. Moreover, it was also observed that the residues Asn69, Ser208, and Asp158 interacted with hydrogen atoms present in the methyl group and NH groups because they were polar acceptor residues or proton donors. Compounds Z5 and Z6 presented interactions with residues Asn69, Ser208, Ser207, Val111, and Val210, which are represented as covalent bonds (Figure 4). Based on the above findings, the compounds Z2, Z3, Z5, and Z6 were selected for subsequent experimental studies.
Figure 4.

Amino acid interactions of the four (Z2, Z3, Z5 and Z6) selected compounds and the reference ligand 186 on Cz. In the five figures, arcs with red lines represent hydrophobic amino acid contacts, green dashed lines represent hydrogen bonds and purple lines represent covalent bonds between protein and ligand or “elastic” bonds within the ligand. The image was produced with LigPlot software.
2.2. In Vitro Evaluation
The compounds S1 (STK552090, 8-chloro-N-(3-morpholinopropyl)-5H-pyrimido[5,4-b]-indol-4-amine, a known Cz inhibitor), Z2, Z3, Z5 and Z6 were evaluated in vitro as potential trypanocidal agents against epimastigotes (INC-5 strain) and trypomastigotes (NINOA and INC-5 strains) of T. cruzi, using Bzn and Nfx as reference drugs. The results are shown in Table 2. Bzn had a better trypanocidal effect (LC50 = 9.64 µM) than Nfx (LC50 = 38.36 µM) in the epimastigote. In the trypomastigote, however, the best biological activity was produced by Nfx (LC50 < 117.16 µM) in both strains. This was an expected variance for the reference drugs studied in vitro and in vivo [28,29,30,31]. The variation between different parasitic stages could be because the epimastigote is not the infectious stage. Its susceptibility is not reflected in the other stages (trypomastigotes and amastigotes) or to a behavior clearly dependent on the dose, in both parasitic stages, to cell viability [29]. In other words, therapeutic success seems to depend on the susceptibility of the parasites to the trypanocidal drug, the access and accumulation of the drug in different environments (plasma levels and specific tissues), and the host immune response [30].
Table 2.
Half maximal lytic concentration of the compounds Z2, Z3, Z5, Z6 and S1 and the reference drugs on epimastigotes and trypomastigotes of T. cruzi.
| Compound | Epimastigotes | Bloodstream Trypomastigotes | |
|---|---|---|---|
| LC50 (µM) T. cruzi INC-5 |
LC50 (µM) | ||
| T. cruzi NINOA | T. cruzi INC-5 | ||
| Z2 | 239.40 ± 9.3 | >250 | >250 |
| Z3 | >250 | >250 | >250 |
| Z5 | 36.26 ± 9.9 | 166.21 ± 14.5 | 185.1 ± 8.5 |
| Z6 | >250 | >250 | >250 |
| S1 | >250 | >250 | >250 |
| Nfx | 38.36 ± 5.2 | 99.41± 11.1 | 117.16 ± 16.36 |
| Bnz | 9.64 ± 4.2 | 183.1 ± 16.2 | 225.40 ± 26.5 |
The evaluated compounds Z2, Z3, Z5, and Z6 showed different trypanocidal effects. In the epimastigote form, the compounds Z3 and Z6 did not show trypanocidal effects (LC50 > 250 µM). The compound S1, a Cz inhibitor, also did not show an effect. Compounds Z2 and Z5 only showed trypanocidal activity. Z2 had a low activity, but Z5 showed a trypanocidal effect similar to Nfx, though it was four times less potent than Bzn.
Biological behavior did not vary between the compounds described above and the form of T. cruzi evaluated. Against bloodstream trypomastigotes, the compounds Z2, Z3, Z6 and the inhibitor S1 showed LC50 values greater than 250 μM, resulting in a concentration twice as high as that obtained with the reference drugs for both strains (INC-5 and NINOA). The trypanocidal results obtained from the S1 inhibitor differ from that reported by Ferreira et al. (2010) and Pinto et al. (2017), because these authors indicated that the compound S1 showed IC50 values of 2.5 μM in infected mouse fibroblasts (L929) with trypomastigotes of T. cruzi of the Tulahúen strain.
The most active compound in both stages and both strains was compound Z5, which in epimastigotes of the strain INC-5, showed the same trypanocidal activity as that of Nfx. On the other hand, in trypomastigotes, Z5 presented LC50 values lower than Bnz in both strains with concentrations 1.6 times lower than those present with the drug Nfx; therefore, compound Z5 is a promising structure in the search for new agents to treat Chagas disease.
2.3. Enzyme Inhibition
To confirm the predictive study of new potential Cz inhibitors and confirm the mechanism of action, enzymatic inhibition with cysteine proteases of T. cruzi was done. The results of the mean inhibitory concentration of the enzyme activity are shown in Table 3. A behavior similar to that of the in vitro evaluation on epimastigotes and trypomastigotes of T. cruzi can be seen. The compounds Z2 and S1 showed weak inhibitory activities (IC50 > 200 µM). Z3 showed an IC50 value of 84.37 µM in protease inhibition, but this compound did not have a trypanocidal effect in epimastigotes and trypomastigotes. In this evaluation, we observed that the compounds Z5 and Z6 were characterized by a better inhibitory activity with IC50 values of 56.23 μM and 50.35 μM, respectively. However, Z6 also did not have a trypanocidal effect. In contrast, Z5 was the best compound with trypanocidal activity against epimastigotes and trypomastigotes. Although these results confirm an inhibition of cysteine proteases as mechanism of action, a specific study on the Cz enzyme is necessary to determine the kind of inhibition that these compounds could have.
Table 3.
IC50 values for cysteine proteases from epimastigotes of Trypanosoma cruzi strain INC-5.
| Compound | IC50 (µM) |
|---|---|
| Z2 | 1410.05 |
| Z3 | 84.37 |
| Z5 | 56.23 |
| Z6 | 50.35 |
| SI | >200 |
According to the results obtained in this work, Z5 presents interesting biological activity that provides a guide to analyze analogous compounds as well as to continue analyzing the interactions and the place where these occur since, as can be seen in Figure 5, it is a different catalytic site. This could possibly favor the activity of compound Z5.
Figure 5.

Analysis of the interaction site between ligand 186 and compound Z5 on Cz.
For the above, the binding mode and enzyme activity were not correlated, as we expected. The lack of biological relation could be due to the physicochemical properties of the molecules. This could interfere in the penetration of compounds into cell membranes or limit evaluation of Cz enzyme inhibition on T. cruzi only, not in an extract of proteases as we did [11].
3. Materials and Methods
3.1. Structure-Based Virtual Screening
Compounds were selected from the clean lead folder (n = 4,591,276 ligands) available in the ZINC15 database (http://zinc.docking.org, accessed on: 5 August 2018). Filtration of the clean lead compounds was done using the general structure of an N-acylhydrazone, with the format formula (C=NNC(C)=O), including chemical structures that had up to 50% similarity and complied with Lipinski’s rules. Ligands that met the criteria were downloaded and prepared as ligands by adding hydrogens and a subsequent energy minimization process with the obminimize command from Babel software [32]. Then, Gasteiger charges were added during the transformation to pdbqt files with the “prepare_ligand4.py” script of AutoDock Tools [33]. The ligands in pdbqt format were used for the further structure-based virtual screening through a molecular docking analysis with the software Autodock vina. For molecular docking, the three-dimensional structure of Cz (PDBID: 1U9Q) obtained from the Protein Data Bank database (http://www.pdb.org, accessed on: 15 July 2018) was used. This structure was used as a receptor to remove the ligand 186 (1-(1-methyl-4,5-dioxo-pent-2-enylcarbamoyl)-2-phenyl-ethyl]-carbamic acid benzyl ester) and the water molecules. Then, the receptor molecule was prepared using the AutoDock Tools software, also by the addition of polar hydrogens, Gasteiger charges, and the generation of the receptor in pdbqt format.
To determine the size of the search spaces in the active site of Cz, the online program Proteins Plus and the tool DoGSiteScorer https://proteins.plus/ (accessed on 11 December 2018) were employed to confirm the binding site in order to design the most optimal grid-box [34]. With the obtained information, a grid-box was determined with the following sizes: X = 18 Å, Y = 15 Å, and Z = 20 Å and with the center coordinates X = 7.605, Y = 9.788, and Z = 8.779. This search space was used for docking the retrieved ZINC15 compounds. The binding energy score obtained from the first reference docking with ligand 186 was used as a cutoff value for the selection of potential inhibitors on the basis of the most negative energy binding higher than that of ligand 186. A total of 682 ligands were chosen because these showed the best binding energies (<−6.6 Kcal/mol). The compounds were further analyzed according to their amino acid contact with Cz, using the software AuPosSOM (Automatic analysis of poses using SOM) [35], with the aim of generating clusters that could be related across the binding energy and the structure of the ligand. The AuPosSOM results contained in the Newick tree file were plotted with TreeGraph2 [36] and analyzed visually. In addition, a more detailed analysis of the interactions between the best selected compounds that docked onto the protein was performed using the LigPlot program [37].
3.2. In Vitro Evaluation
For biological evaluation, the compounds Z2 (ZINC9873043), Z3 (ZINC9870651), Z5 (ZINC9715287), and Z6 (ZINC9861447) were selected and purchased from Molport https://www.molport.com (accessed on: 03 March 2018). For evaluation of trypanocidal activity of the selected compounds, two strains (NINOA and INC-5) in the epimastigote or trypomastigote stage of T. cruzi were used. Each strain was used to infect CD-1 mice (18–20 g) intraperitoneally with a concentration of 1 × 106 trypomastigotes/mL of blood. In the maximum peak of parasitemia, blood was obtained by cardiac puncture using heparin as an anticoagulant. The parasite concentration was then adjusted with isotonic saline (0.85% NaCl), and 90 μL of blood was used to achieve a concentration of 1 × 106 trypomastigotes/mL of blood. A 10 μL sample of each compound was evaluated in 96-well plates. The treatments performed consisted of a negative control, which contained dimethylsulfoxide (DMSO 2.5%) and the four compounds selected from virtual screening. As positive controls, the reference drugs Bnz (Rochagan, Roche) and Nfx (Lampit, Bayer) were used as well as the compound STK552090 (8-chloro-N-(3-morpholinopropyl)-5H-pyrimido[5,4-b]indol-4-amine), which was named internally as S1 and reported as a Cz inhibitor in T. cruzi in the ZINC15 library. Once the selected compounds were added after being homogenized with blood, the plates were incubated at 4 °C for 24 h [38]. Once the incubation time had elapsed, the plates were kept at room temperature for 30 min, and then an aliquot of 5 μL was taken from each well, a fresh preparation was made, and the viable trypomastigotes were counted using the Pizzi method [39,40]. A calculation of the number of viable trypomastigotes per mL of blood was carried out, and the survival percentages were obtained taking 100% of the negative control. Each test was done in triplicate. To obtain 50% lysis concentration (LC50) of the population, the following concentrations were used: 200, 100, 50, 25, and 12.5 μg/mL. The LC50 values were determined using Probit statistical analysis of the dose-response, and the results were expressed as the mean of the standard deviation. The results were later converted to micromolar units.
3.3. Enzyme Inhibition
T. cruzi epimastigotes of the INC-5 strain were used to obtain a protein extract quantified by the Bradford method. Evaluation of the activity was carried out using the synthetic fluorogenic peptide Z-Phe-Arg-MCA as a substrate at 5 µM, with 1 μg of the enzyme extract and the corresponding compounds (Z2, Z3, Z5, and Z6) at different concentrations (6.25–200 µM) using a reaction buffer (50 mM Na2HPO4, 100 mM NaCl, 5 mM EDTA, 2.5 mM dithiothreitol; pH 6.5) at room temperature. As a negative control of enzyme inhibition, the substrate without compounds was evaluated with 2% DMSO (concentration corresponding to the highest dilution of the compounds). Hydrolysis of the substrate was monitored in a continuous assay for 1 h in the Spectramax M5 spectrofluorometer (Molecular Devices) at 380 nm excitation and 440 nm emission. The mean of fluorescence changes was normalized taking the control (without inhibitor) as 100%. The IC50 values were determined from semilog concentration-response plots using the nonlinear curve-fitting method [41].
The relationship of the biological evaluation with the binding mode of the selected molecules was inferred based on in vitro results.
4. Conclusions
Filtering the ZINC15 database using the N-acyl hydrazone moiety and further structure-based virtual screening allowed us to select four compounds as potential Cz enzyme inhibitors with trypanocidal activity. Compounds Z5 showed the best trypanocidal activity, with LC50 values in epimastigotes of 36.26 ± 9.9 μM and in trypomastigotes of 166.21 ± 14.5 μM for the strain NINOA, and 185.1 ± 8.5 μM for the strain INC-5. In addition, this compound showed inhibition of T. cruzi proteases with an IC50 value of 56.23 μM. According to the in silico study of its interaction in the active site of the enzyme, Z5 could be a non-competitive inhibitor. Therefore, the results obtained encourage us to continue using computational tools to search for anti-Trypanosoma cruzi drugs and to continue exploring the behavior of these derivatives on the Cz enzyme.
Acknowledgments
We wish to express our gratitude to the CONACyT and the Secretaría de Investigación y Posgrado del Instituto Politécnico Nacional for the support granted.
Abbreviations
| Nfx | Nifurtimox |
| Bzn | Benznidazole |
| Cz | Cruzain |
| CD | Chagas disease |
| IC50 | Inhibitory concentration |
| LC50 | Lytic concentration |
Supplementary Materials
Supplementary materials can be found at https://www.mdpi.com/1422-0067/20/7/1742/s1. Figure S1: Clustering pattern obtained with AuPosSOM based on the amino acid-ligand contact footprint. Table S1: Compounds obtained from virtual screening, binding energies and chemical properties.
Author Contributions
Conceptualization, supervision, funding acquisition, G.R. and B.N.-T.; Project management, V.B.-G.; Methodology and Investigation, K.F.C.-V., and C.A.-A.; Software, Data curation and Supervision E.E.L-R. and F.R.-E.; Funding acquisition and Formal Analysis, L.R.-P. and V.A.-F.; Methodology and writing original draft, V.H.-M and J.C.-M.
Funding
This research was funded by Consejo Nacional de Ciencia y Tecnologia (CONACyT), grant number CB-2014-01, 241615.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Bern C., Kjos S., Yabsley M.J., Montgomery S.P. Trypanosoma cruzi and Chagas’ Disease in the United States. Clin. Microbiol Rev. 2011;24:655–681. doi: 10.1128/CMR.00005-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization (WHO) The Global Burden of Disease: 2004 Update. World Health Organization; TDR; Geneva, Switzerland: 2008. p. 160. [Google Scholar]
- 3.World Health Organization (WHO) Chagas Disease (American Trypanosomiasis) [(accessed on 11 August 2018)];2016 Available online: https://www.who.int/chagas/en/
- 4.Zaldini H.M., Montenegro R.M., Lima L.A.C., Oliveira C.M.V., Magalhaes M.D.R., Brondani D.J., Simone C.A., Campos R.L., Assis S.M., Alves P.V.R., et al. Studies toward the structural optimization of novel thiazolylhydrazone-based potent antitrypanosomal agents. Bioorg. Med. Chem. 2010;18:7826–7835. doi: 10.1016/j.bmc.2010.09.056. [DOI] [PubMed] [Google Scholar]
- 5.Nichi S.A.R., Kimoto K.Y., Steinde M., Grisard E.C., Gomes M.L. Limit of detection of PCR/RFLP analysis of cytochrome oxidase II for the identification of genetic groups of Trypanosoma cruzi and Trypanosoma rangeli in biological material from vertebrate hosts. Parasitol. Res. 2018;117:2403–2410. doi: 10.1007/s00436-018-5928-1. [DOI] [PubMed] [Google Scholar]
- 6.Maguire J.H. Trypanosoma. In: Gorbach S., Bartlett J., Blacklow N., editors. Infectious Diseases. 2nd ed. Lippincott Williams & Wilkins; Philadelphia, PA, USA: 2004. pp. 2327–2334. [Google Scholar]
- 7.Villar J.C., Marin N.J.A., Ebrahim S., Yusuf S. Trypanocidal drugs for chronic asymptomatic Trypanosoma cruzi infection. Cochrane Database Syst. Rev. 2002;1:1–22. doi: 10.1002/14651858.CD003463. [DOI] [PubMed] [Google Scholar]
- 8.Siles R., Chen E.C., Zhou M., Pinney K.G., Trawick M.L. Design, synthesis, and biochemical evaluation of novel cruzain inhibitors with potential application in the treatment of Chagas’ disease. Bioorg Med. Chem. Lett. 2006;16:4405–4409. doi: 10.1016/j.bmcl.2006.05.041. [DOI] [PubMed] [Google Scholar]
- 9.Muñoz C., Zulantay I., Apt W., Ortiz S., Schijman A.G., Bisio M., Ferrada V., Herrera C., Martínez G., Solarib A. Evaluation of nifurtimox treatment of chronic Chagas disease by means of several parasitological methods. Antimicrob. Agents Chemother. 2013;57:4518–4523. doi: 10.1128/AAC.00227-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sajid M., McKerrow J.H. Cysteine proteases of parasitic organisms. Mol. Biochem. Parasitol. 2002;120:1–21. doi: 10.1016/S0166-6851(01)00438-8. [DOI] [PubMed] [Google Scholar]
- 11.Choe Y., Brinen L.S., Price M.S., Engel J.C., Lange M., Grisostomi C., Weston S.G., Pallai P.V., Cheng H., Hardy L.W., et al. Development of a-keto-based inhibitors of cruzain, a cysteine protease implicated in Chagas disease. Bioorg Med. Chem. 2005;13:2141–2156. doi: 10.1016/j.bmc.2004.12.053. [DOI] [PubMed] [Google Scholar]
- 12.Delcroix M., Sajid M., Caffrey C.R., Lim K.C., Dvorak J., Hsieh I., Bahgat M., Dissous C., McKerrow J.H. A multienzyme network functions in intestinal protein digestion by a platyhelminth parasite. J. Biol. Chem. 2006;281:39316–39329. doi: 10.1074/jbc.M607128200. [DOI] [PubMed] [Google Scholar]
- 13.McKerrow J.H., Rosenthal P.J., Swenerton R., Doyle P. Development of protease inhibitors for protozoan infections. Curr. Opin. Infect. Dis. 2008;21:668–672. doi: 10.1097/QCO.0b013e328315cca9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Du X., Hansell E., Engel J.C., Caffrey C.R., Cohen F.E., McKerrow J.H. Aryl ureas represent a new class of anti-trypanosomal agents. Chem. Biol. 2000;7:733–742. doi: 10.1016/S1074-5521(00)00018-1. [DOI] [PubMed] [Google Scholar]
- 15.Roush W.R., Alvarez H.A., McKerrow J.H., Selzer P.M., Hansell E., Engel J.C. Design, Synthesis and Evaluation of d-Homophenylalanyl Epoxysuccinate Inhibitors of the Trypanosomal Cysteine Protease Cruzain. Tetrahedron. 2000;56:9747–9762. doi: 10.1016/S0040-4020(00)00882-6. [DOI] [Google Scholar]
- 16.Zanatta N., Madruga C.C., Marisco P.C., Da Rosa L.S., Fernandes L.D.S., Flores D.C., Martins M.A.P. Synthesis and structural study of a new series of 2-methylsulfanyl-tetrahydropyrimidines from β-alkoxyvinyl trihalomethyl ketones. J. Heterocyclic Chem. 2008;45:221–227. doi: 10.1002/jhet.5570450127. [DOI] [Google Scholar]
- 17.Borchhardt D.M., Mascarello A., Chiaradia L.D., Nunes R.J., Oliva G., Yunes R.A., Andricopulo A.D. Biochemical evaluation of a series of synthetic chalcone and hydrazide derivatives as novel inhibitors of cruzain from Trypanosoma cruzi. J. Braz. Chem. Soc. 2010;21:142–150. doi: 10.1590/S0103-50532010000100021. [DOI] [Google Scholar]
- 18.Beaulieu C., Isabel E., Fortier A., Massé F., Mellon C., Méthot N., Black W.C. Identification of potent and reversible cruzipain inhibitors for the treatment of Chagas disease. Bioorg Med. Chem. Lett. 2010;20:7444–7449. doi: 10.1016/j.bmcl.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 19.Massarico S.R.A., Gonçalves J.E., Pereira S.F., de Melo L.A.P., Storpirtis S., Krogh R., Defini A.A., Dias L.C., Igne F.E. Design, synthesis and biological evaluation of hybrid bioisoster derivatives of N-acylhydrazone and furoxan groups with potential and selective anti-Trypanosoma cruzi activity. Eur. J. Med. Chem. 2014;82:418–425. doi: 10.1016/j.ejmech.2014.05.077. [DOI] [PubMed] [Google Scholar]
- 20.Romeiro N.C., Aguirre G., Hernández P., González M., Cerecetto H., Aldana I., Pérez S.S., Monge A., Barreiro E.J., Lima L.M. Synthesis, trypanocidal activity and docking studies of novel quinoxaline-N-acylhydrazones, designed as cruzain inhibitors candidates. Bioorg Med. Chem. 2009;17:641–652. doi: 10.1016/j.bmc.2008.11.065. [DOI] [PubMed] [Google Scholar]
- 21.Moreira M.C., Wang Y., Heringer W.S., Wessel N., Walther T. Prognostic value of natriuretic peptides in Chagas’ disease: A head-to-head comparison of the 3 natriuretic peptides. Congest Heart Fail. 2009;15:75–81. doi: 10.1111/j.1751-7133.2009.00051.x. [DOI] [PubMed] [Google Scholar]
- 22.Oprea T.I., Davis A.M., Teague S.J., Leeson P.D. Is there a difference between leads and drugs? A historical perspective. J. Chem. Inf. Comput. Sci. 2001;41:1308–1315. doi: 10.1021/ci010366a. [DOI] [PubMed] [Google Scholar]
- 23.Sterling T., Irwin J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015;55:2324–2337. doi: 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferreira R.S., Simeonov A., Jadhav A., Eidam O., Mott B.T., Keiser M.J., McKerrow J.H., Maloney D.J., Irwin J.J., Shoichet B.K. Complementarity between a docking and a high-throughput screen in discovering new cruzain inhibitors. J. Med. Chem. 2010;53:4891–4905. doi: 10.1021/jm100488w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fereidoonnezhad M., Mostoufi A., Zali S., Eskandari M., Afshar D., Aliyan F. Molecular Docking and PLIF Studies of Novel Tacrine-Naphtoquinone Hybrids Based on Multi-Target-Directed Ligand Approach for Alzheimer’s Disease. Jundishapur. J. Nat. Pharm. Prod. 2018;13:1–10. doi: 10.5812/jjnpp.65048. [DOI] [Google Scholar]
- 26.Lara-Ramírez E.E., López-Cedillo J.C., Nogueda-Torres B., Kashif M., Garcia-Perez C., Bocanegra-García V., Agusti R., Uhrig M.L., Rivera G. An in vitro and in vivo evaluation of new potential trans-sialidase inhibitors of Trypanosoma cruzi predicted by a computational drug repositioning method. Eur. J. Med. Chem. 2017;26:249–261. doi: 10.1016/j.ejmech.2017.03.063. [DOI] [PubMed] [Google Scholar]
- 27.Mahler G., Moreira L.L. Cisteína endopeptidasa de T. cruzi: Cruzipaína. In: Cerecetto M.H., González M., editors. Enfermedad de Chagas: Estrategias En La Búsqueda de Nuevos Medicamentos, Una Visión Iberoamericana. 1st ed. Laboratorio Silanes; Mexico City, México: 2012. pp. 45–63. [Google Scholar]
- 28.Villarreal D., Barnabé C., Sereno D., Tibayrenc M. Lack of correlation between in vitro susceptibility to Benznidazole and phylogenetic diversity of Trypanosoma cruzi, the agent of Chagas disease. Exp. Parasitol. 2004;108:24–31. doi: 10.1016/j.exppara.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 29.Toledo M.J.O., Bahia M.T., Veloso V.M., Carneiro C.M., Machado-Coelho G.L.L., Alves M.H.R., Cruz R.E., Tafuri W.L., Lana M. Effects of specific treatment on parasitological and histopathological parameters in mice infected with different Trypanosoma cruzi clonal genotypes. J. Antimicrob. Chemother. 2004;53:1045–1053. doi: 10.1093/jac/dkh224. [DOI] [PubMed] [Google Scholar]
- 30.Moreno M., D’ávila D.A., Silva M.N., Galvão L.M., Macedo A.M., Chiari E., Gontijo E.D., Zingales B. Trypanosoma cruzi benznidazole susceptibility in vitro does not predict the therapeutic outcome of human Chagas disease. Mem. Inst. Oswaldo. Cruz. 2010;105:918–924. doi: 10.1590/S0074-02762010000700014. [DOI] [PubMed] [Google Scholar]
- 31.Muñoz-Calderón A., Santaniello A., Pereira A., Yannuzzi J., Díaz-Bello Z., Alarcón de Noya B. Susceptibilidad in vitro a Nifurtimox y Benznidazol de aislados de Trypanosoma cruzi obtenidos de pacientes venezolanos con enfermedad de Chagas infectados por mecanismos de transmisión oral y vectorial. Rev. Ibero-Latinoam. Parasitol. 2012;71:14–22. [Google Scholar]
- 32.Boyle N.O.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchison G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011;3:1–14. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trott O., Olson A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Volkamer A., Kuhn D., Grombacher T., Rippmann F., Rarey M. Combining global and local measures for structure-based druggability predictions. J. Chem. Inf. Model. 2012;52:360–372. doi: 10.1021/ci200454v. [DOI] [PubMed] [Google Scholar]
- 35.Mantsyzov A.B., Bouvier G., Evrard-Todeschi N., Bertho G. Contact-based ligand-clustering approach for the identification of active compounds in virtual screening. Adv. Appl. Bioinform. Chem. 2012;5:61–79. doi: 10.2147/AABC.S30881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stöver B.C., Müller K.F. TreeGraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC Bioinform. 2010;11:1–9. doi: 10.1186/1471-2105-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laskowski R.A., Swindells M.B. LigPlot+: Multiple Ligand-Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 38.Paveto C., Güida M.C., Esteva M.I., Martino V., Coussio J., Flawiá M.M., Torres H.N. Anti-Trypanosoma cruzi Activity of Green Tea (Camellia sinensis) Catechins. Antimicrob. Agents Chemother. 2004;48:69–74. doi: 10.1128/AAC.48.1.69-74.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brener Z. Biology of Trypanosoma cruzi. Annu. Rev. Microbiol. 1973;27:347–382. doi: 10.1146/annurev.mi.27.100173.002023. [DOI] [PubMed] [Google Scholar]
- 40.Pizzi T. Inmunología de la enfermedad de Chagas. Estado actual del problema. Boletín de la Oficina Sanitaria Panamericana. 1961;51:450–464. [PubMed] [Google Scholar]
- 41.Santos C.C., Sant’Anna C., Terres A., Cunha-e-Silva N.L., Scharfstein J., Lima A.P.C.D.A. Chagasin, the endogenous cysteine-protease inhibitor of Trypanosoma cruzi, modulates parasite differentiation and invasion of mammalian cells. J. Cell Sci. 2005;118:901–915. doi: 10.1242/jcs.01677. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.