

Abstract
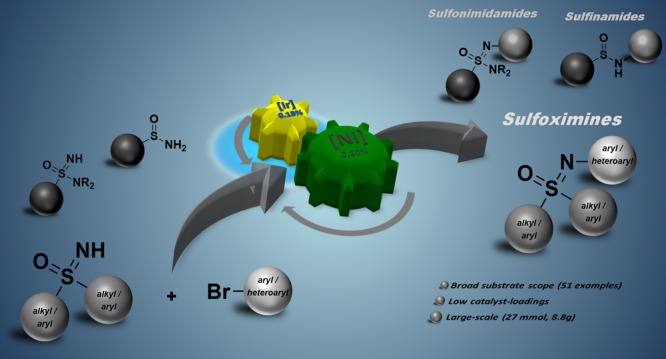
The pharmaceutically underexplored sulfoximine moiety has emerged as a potentially active pharmaceutical ingredient. We developed a scalable synthetic route to N-arylated sulfoximines from the respective “free” NH-sulfoximines and bromoarenes. Our strategy is based on a dual nickel photocatalytic approach, is applicable for a broad scope of substrates, and exhibits a highly functional group tolerance. In addition, we could demonstrate that other sulfoximidoyl derivatives like sulfonimidamides and sulfinamides proceed smoothly under the developed reaction conditions.
Most organic chemists consider sulfoximines mainly as chiral auxiliaries or ligands, being applied in asymmetric reactions or catalysis.1 However, recently, sulfoximines emerged as potentially active pharmaceutical ingredients (APIs) in medicinal and agricultural research.2 Although their bioactivity is already long known, their exploration as APIs was scarce. Recently, it was found that the sulfoximines’ mode of binding to biological receptors can be very different compared to established ligands. For example, the sulfoximine-based insecticide Sulfoxaflor is capable of bypassing many cross-resistances of pest species because of its differing mechanism of binding.3 Discoveries as such call for efficient synthetic routes to sulfoximines. In particular, N-arylated sulfoximines are of interest for medicinal chemists, as they could serve as potent drug analogues.4
Various Pd-, Cu-, or Fe-catalyzed N-arylations of NH-sulfoximines with different types of electrophiles were developed by Bolm, Harmata, and others since the late 1990s (Figure 1A).1i,1n,5 However, demanding reaction conditions such as high catalyst loadings, specialized ligands, elevated reaction temperatures, and long reaction times often limit the practicability or the scope of substrates. This set of limitations already indicates that NH-sulfoximines often behave as a rather special and challenging class of N-nucleophiles for transition-metal-catalyzed N-arylations. In particular, the coupling of pharmaceutically relevant heteroaromatic scaffolds to NH-sulfoximines is rather unexplored. Consequently, there is still a great demand for general, mild, and efficient synthetic solutions toward N-functionalized aliphatic, aromatic, and heteroaromatic sulfoximines. Very recently, we reported the first photocatalytic approach for the N-arylation of NH-sulfoximines.6 At the same time, Meier at al. published a similar method, showing that the mildness of the photocatalytic reaction also allows late-stage sulfoximination of complex molecules in the industrial context.7
Figure 1.

(A) Classic transition-metal-catalyzed N-arylations of NH-sulfoximines. (B) Dual nickel photocatalyzed approach.
Stimulated by the continuous interest in sulfoximines, we wondered whether the N-arylation of NH-sulfoximines could be realized by the combination of classic transition-metal catalysis with visible-light photocatalysis (metallaphotocatalysis) (Figure 1B). Dual nickel photocatalysis has emerged as a powerful strategy and a remarkably efficient tool for organic cross-coupling reactions in the last years.8 In particular, N-arylation was reported for anilines, aliphatic amines, and also sulfonamides.9 We considered that NH-sulfoximines might be suitable substrates for such a strategy, keeping in mind that a practicable synthetic method should work not only on a milligram laboratory scale but also on a preparative multigram scale.
We started our investigations using similar reaction conditions as reported by MacMillan et al.9cNH-sulfoximine 1a (1.5 equiv) and bromoarene 2a (1.0 equiv) as model substrates were reacted with 1.0 mol % of [Ir]-Cat ([Ir(ppy)2(dtbbpy)]PF6) as photocatalyst, 5.0 mol % of [Ni-1]-Cat (NiBr2 and dtbbpy as ligand (1.0:0.2 equiv) added separately), and TMG (1,1,3,3-tetramethylguanidine, 1.5 equiv) as base in dry and degassed DMSO (0.25 M, 1.0 mL) under nitrogen atmosphere. Irradiation with blue light of 455 nm for 3 h at 25 °C yielded the desired N-arylated sulfoximine 3a in an excellent yield of 94% (Table 1, entry 1).10 Further optimization significantly decreased the amount of substrates and catalysts for the transformation. Only 0.15 mol % of [Ir]-Cat was found to be sufficient for the reaction (Table 1, entry 2), and by using already preformed [Ni-2]-Cat, its amount could be decreased to only 0.20 mol % (Table 1, entries 3–5). Finally, the amount of NH-sulfoximine 1a, bromoarene 2a, and TMG could be optimized, reaching an atom-economic ratio of 1.0:1.1:1.2 equiv, respectively (Table 1, entries 5 and 6). In addition, we found that the overall substrate concentration could be increased from 0.25 to 0.75 M. Further studies revealed that also other common organic solvents like MeCN, DMF, DMAc, or THF can be used without any decrease in yield and quinuclidine, DABCO, or KOAc could be used as an alternative base, affording moderate yields. Interestingly, a moderate yield of the product was obtained in the absence of photocatalyst when light of 390 nm was used for irradiation.11 This results indicates that the reaction might proceed via photosensitization processes.9c Control experiments showed that photocatalyst, nickel catalyst, base, and the irradiation with light are all crucial for the reaction.
Table 1. Optimization of the Reaction Conditionsa,b.

| entry | 1a/2a (equiv) | [Ir]-Cat(mol %) | [Ni]-Cat (mol %) | TMG (equiv) | yieldc (%) |
|---|---|---|---|---|---|
| 1 | 1.5:1.0 | 1.0 | [Ni-1]-Cat (5.0) | 1.5 | 94 |
| 2 | 1.5:1.0 | 0.15 | [Ni-1]-Cat (5.0) | 1.5 | 96 |
| 3 | 1.5:1.0 | 0.15 | [Ni-1]-Cat (1.0) | 1.5 | 95 |
| 4 | 1.5:1.0 | 0.15 | [Ni-2]-Cat (0.20) | 1.5 | 76 |
| 5 | 1.0:1.1 | 0.15 | [Ni-2]-Cat (0.20) | 1.5 | 99 |
| 6 | 1.0:1.1 | 0.15 | [Ni-2]-Cat (0.20) | 1.2 | 99 |
| 7 | 1.0:1.1 | 0.15 | [Ni-2]-Cat (0.20) | 1.2 | 99d |
[Ir]-Cat = [Ir(ppy)2(dtbbpy)]PF6, [Ni-1]-Cat = NiBr2 + dtbbpy (1.0:0.20 equiv) added separately, [Ni-2]-Cat = preformed [Ni(dtbbpy]Br2, TMG = 1,1,3,3-tetramethylguanidine,
Reaction conditions: 1a (0.25 mmol, 1.0 equiv), 2a (0.28 mmol, 1.1 equiv), [Ir]-Cat (0.15 mol %), [Ni-2]-Cat (0.20 mol %), TMG (0.30 mmol, 1.2 equiv), dry and degassed DMSO (0.25 M, 1.0 mL), irradiation at 455 nm for 3 h.
Yields were determined by GC analysis with naphthalene as internal standard.
Reaction was up-concentrated to 0.75 M and run for 17 h, and the yield is reported after purification via automated flash-column chromatography.
With the optimized reaction conditions in hand (Table 1, entry 6), we started to explore the scope of the reaction. First, we focused on the scope of brominated arenes and heteroarenes (Figure 2). Both electron-rich and electron-deficient brominated arenes reacted smoothly with NH-sulfoximine 1a, giving the respective N-arylated sulfoximines 3a–r in high to excellent yields (Figure 2A). For this type of brominated substrates, we selected MeCN as solvent as it is easily removed under reduced pressure. Many functional groups, including thioethers (3c), cyanides (3f), ethers (3h and 3j), amides (3k), or carbamates (3r), were tolerated under the reaction conditions. Interestingly, the reaction of 1,3-dibromobenzene stopped after 1-fold substitution, yielding monobrominated 3l as product. This observation could give the opportunity for further functionalizations in other cross-coupling reactions. In particular, the compatibility of pharmaceutically relevant substrate classes like sulfoxides (3m) or sulfones (3n) and bioisosteric scaffolds like −OCF3 (3o), −SCF3 (3p), or −SF5 (3q) was investigated. Gratifyingly, all these moieties were found to be tolerated under the reaction conditions and afforded the respective sulfoximines in moderate to excellent yields. It has to be mentioned that the lower yield of SF5-containing sulfoximine 3q is due to decomposition of the brominated arene during the reaction. In addition, we conducted a large-scale version of the reaction in a custom-made reactor commonly used in our laboratories. The reaction was carried out on a 27 mmol scale, affording 8.8 g (99%) of product, using only 37 mg of [Ir]-Cat (0.15 mol %) and 26 mg of [Ni-2]-Cat (0.20 mol %).12
Figure 2.

Substrate scope of bromoarenes. Reaction conditions: 1a (0.25 mmol, 1.0 equiv), bromo arene (2) (0.275 mmol, 1.1 equiv), [Ir]-Cat (0.15 mol %), [Ni-2]-Cat (0.20 mol %), TMG (1.2 equiv), dry and degassed MeCN (•) or DMA (#) (0.25 M), irradiation at 455 nm for 17 h; (a) 3.5 h; 17 h for the large-scale reaction; (b) 0.5 mol % of [Ir]-Cat, 1.0 mol % of [Ni-2]-Cat; (c) 0.2 mol % of [Ir]-Cat, 1.0 mol % of [Ni-2]-Cat; (d) 1,3-dibromobenzene (0.24 mmol, 1 equiv) as limiting reagent; (e) 0.5 mol % of [Ni-2]-Cat; (f) 0.5 mol % of [Ir]-Cat, 5.0 mol % of [Ni-2]-Cat; (g) 1.0 mol % of [Ir]-Cat, 5.0 mol % of [Ni-2]-Cat; (h) 1.0 mol % of [Ni-2]-Cat; (i) 0.5 mol % of [Ir]-Cat, 3.0 mol % of [Ni-2]-Cat; (j) 0.5 mol % of [Ir]-Cat, 2.0 mol % of [Ni-2]-Cat; (k) 2.0 mol % of [Ni-2]-Cat; (l) 0.15 mol % of [Ir]-Cat, 2.0 mol % of [Ni-2]-Cat, 0.04 M.
The scope of brominated heteroarenes was explored with common heteroaromatic scaffolds, occurring in pharmaceutical agents or natural products (Figure 2B). Introducing the sulfoximine moiety to established bioactive cores like indoles, pyridines, quinolines, pyrimidines, pyrazines, quinoxalines, benzofuranes, oxadiazoles, or benzothiazoles might be of use for pharmaceutical or agricultural research. All of the applied brominated substrates could be coupled with NH-sulfoximine 1a, affording up to yields of 99%.13 However, it has to be noted that these scaffolds showed lower reactivity in the N-arylation reaction, compared to brominated benzene derivatives. Nevertheless, when the conversion to the respective products was incomplete after 17 h and starting substrates were remaining, careful adjustments of the loading of catalysts had beneficial effects on the yield of the reactions.14 Brominated N-Boc-protected indole did react smoothly under the reaction conditions, affording the desired product 3s in 90% yield. The reactions with differently substituted pyridines did generally lead to high product yields, except for acetylated pyridine, where decomposition of the brominated pyridine diminished the outcome of the reaction (3w). Similar to 1,3-dibromobenzene, the reaction with 2,4-dibromopyridine stopped after 1-fold substitution, affording the monobrominated sulfoximine derivative 3x in high yield. Brominated quinolines, pyrimidines, pyrazines, and quinoxalines reacted well under the reaction conditions and afforded the respective N-arylated sulfoximines in moderate to excellent yields. Again, excellent yields were obtained by applying 5-bromobenzofuran (3ae, 97%) and 2-(4-bromophenyl)-1,3,4-oxadiazole (3af, 99%), and the reaction with 2-bromobenzothiazole afforded sulfoximine 3ag in a moderate yield of 44%. Furthermore, methylxanthine alkaloid caffeine was tested as a substrate. The reaction of brominated caffeine afforded the respective N-arylated sulfoximine 3ah in an isolated yield of 29%.
Next, we focused on the scope of different NH-sulfoximines and conducted the reactions using methyl 4-bromobenzoate (2j) as model substrate (Figure 3A). Electron-rich as well as electron-deficient alkyl- and aryl-substituted NH-sulfoximines were suitable for the N-arylation reaction and afforded good to excellent yields of the desired products. Cyclopropyl moieties (3ai), benzylic positions (3ak), and heterocyclic scaffolds (3ap and 3aq) were well tolerated and yielded the respective products in moderate to excellent yields.
Figure 3.

(A) Scope of NH-sulfoximines. (B) Scope of enantiopure substrates. (C) Scope of other sulfoximidoyl derivatives. Reaction conditions: NH-sulfoximine (1) (0.25 mmol, 1.0 equiv), methyl 4-bromobenzoate (2j) (0.275 mmol, 1.1 equiv), [Ir]-Cat (0.15 mol %), [Ni-2]-Cat (0.20 mol %), TMG (1.2 equiv), dry and degassed MeCN (•) or DMA (#) (0.25 M), irradiation at 455 nm for 17 h; (a) 0.5 mol % of [Ir]-Cat, 1.0 mol % of [Ni-2]-Cat; (b) 0.5 mol % of [Ir]-Cat, 5.0 mol % of [Ni-2]-Cat; (c) 1.0 mol % of [Ir]-Cat, 5.0 mol % of [Ni-2]-Cat; (d) 0.5 mol % of [Ir]-Cat, 2.0 mol % of [Ni-2]-Cat.
To further demonstrate the practicability of our method, we investigated whether the chiral information on an enantiopure NH-sulfoximine is conserved throughout the reaction to yield the respective enantiopure N-arylated sulfoximine. This allows the rapid generation of enantiopure substrate libraries. We investigated the reaction of an enantiopure NH-sulfoximine with various brominated arenes and heteroarenes (Figure 3B) and verified the optical purity of the products by chiral HPLC analysis. To our delight, the reaction of enantiopure NH-sulfoximine yielded the respective chiral cross-coupling products ((S)-3at–(S)-3ax), and no racemization was observed.15
Finally, we decided to also test other sulfoximidoyl derivatives under the N-arylation conditions, optimized for NH-sulfoximines (Figure 3C). NH2-Sulfinamide 4 was reacted with methyl 4-bromobenzoate (2j) and afforded the respective product 5 in an excellent yield of 93%. Furthermore, applying NH-sulfonimidamide 6 yielded the respective N-arylated sulfonimidamide 7 in a yield of 96%.
In conclusion, we demonstrated that NH-sulfoximines can be N-arylated with brominated arenes and heteroarenes as coupling partners, by using a dual nickel photocatalyzed strategy. For the conversion of most of the benzene-based NH-sulfoximines and brominated arenes, catalyst loadings of only 0.15 mol % of [Ir]-Cat and 0.20 mol % of [Ni-2]-Cat were sufficient and afforded up to 99% yield of the desired products. In addition, by careful adjustment of the catalyst loadings a diverse range of heteroaromatic substrates could be applied, including a series of relevant scaffolds occurring in natural products and bioactive compounds. Additionally, the reaction was carried out on a preparative scale of 27 mmol (8.8 g product) without any decrease in yield. Furthermore, it was shown that enantiopure products can be obtained by using enantiopure NH-sulfoximines as starting materials. Finally, we demonstrated that the same reaction conditions are suitable for structurally related sulfoximidoyl derivatives, like NH2-sulfinamides and NH-sulfonimidamides. The method extends the synthetic toolbox for the synthesis of sulfoximidoyl derivatives, and applications in the development of molecules for use in pharmaceutical industry or crop protection can be readily envisaged.
Acknowledgments
This project has received funding from the European Research council (ERC) under the European Union’s Horizon 2020 research and innovation programme (Grant Agreement No. 741623). We thank Dr. Rudolf Vasold (University of Regensburg) for his assistance with the GC–MS measurements and Roxane Harteis (University of Regensburg) for her assistance with the chiral HPLC measurements.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.9b00698.
Experimental details, characterization data, and NMR spectra (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Otocka S.; Kwiatkowska M.; Madalinska L.; Kielbasinski P. Chem. Rev. 2017, 117 (5), 4147–4181. 10.1021/acs.chemrev.6b00517. [DOI] [PubMed] [Google Scholar]; b Shen X.; Liu Q.; Zhang W.; Hu J. Eur. J. Org. Chem. 2016, 2016, 906–909. 10.1002/ejoc.201501611. [DOI] [Google Scholar]; c Shen X.; Miao W.; Ni C.; Hu J. Angew. Chem., Int. Ed. 2014, 53 (3), 775–779. 10.1002/anie.201308484. [DOI] [PubMed] [Google Scholar]; d Craig D.; Grellepois F.; White A. J. J. Org. Chem. 2005, 70 (17), 6827–6832. 10.1021/jo050747d. [DOI] [PubMed] [Google Scholar]; e Langner M.; Remy P.; Bolm C. Chem. - Eur. J. 2005, 11 (21), 6254–6265. 10.1002/chem.200500497. [DOI] [PubMed] [Google Scholar]; f Reetz M. T.; Bondarev O. G.; Gais H. J.; Bolm C. Tetrahedron Lett. 2005, 46 (34), 5643–5646. 10.1016/j.tetlet.2005.06.107. [DOI] [Google Scholar]; g Gais H. J.; Babu G. S.; Gunter M.; Das P. Eur. J. Org. Chem. 2004, 2004 (7), 1464–1473. 10.1002/ejoc.200300726. [DOI] [Google Scholar]; h Langner M.; Bolm C. Angew. Chem., Int. Ed. 2004, 43 (44), 5984–5987. 10.1002/anie.200460953. [DOI] [PubMed] [Google Scholar]; i Harmata M.; Hong X. J. Am. Chem. Soc. 2003, 125 (19), 5754–5756. 10.1021/ja034744z. [DOI] [PubMed] [Google Scholar]; j Koep S.; Gais H. J.; Raabe G. J. Am. Chem. Soc. 2003, 125 (43), 13243–13251. 10.1021/ja030324y. [DOI] [PubMed] [Google Scholar]; k Bolm C.; Martin M.; Simic O.; Verrucci M. Org. Lett. 2003, 5 (4), 427–429. 10.1021/ol027273e. [DOI] [PubMed] [Google Scholar]; l Bolm C.; Verrucci M.; Simic O.; Cozzi P. G.; Raabe G.; Okamura H. Chem. Commun. 2003, (22), 2826–2827. 10.1039/B309556H. [DOI] [PubMed] [Google Scholar]; m Bolm C.; Simić O. J. Am. Chem. Soc. 2001, 123 (16), 3830–3831. 10.1021/ja004261k. [DOI] [PubMed] [Google Scholar]; n Harmata M.; Ghosh S. K. Org. Lett. 2001, 3 (21), 3321–3323. 10.1021/ol016546n. [DOI] [PubMed] [Google Scholar]; o Reggelin M.; Zur C. Synthesis 2000, 2000 (1), 1–64. 10.1055/s-2000-6217. [DOI] [Google Scholar]; p Bolm C.; Felder M.; Müller J. Synlett 1992, 1992 (05), 439–441. 10.1055/s-1992-21373. [DOI] [Google Scholar]; q Johnson C. R. Acc. Chem. Res. 1973, 6 (10), 341–347. 10.1021/ar50070a003. [DOI] [Google Scholar]
- Lucking U. Angew. Chem., Int. Ed. 2013, 52 (36), 9399–9408. 10.1002/anie.201302209. [DOI] [PubMed] [Google Scholar]
- a Babcock J. M.; Gerwick C. B.; Huang J. X.; Loso M. R.; Nakamura G.; Nolting S. P.; Rogers R. B.; Sparks T. C.; Thomas J.; Watson G. B.; Zhu Y. Pest Manage. Sci. 2011, 67 (3), 328–334. 10.1002/ps.2069. [DOI] [PubMed] [Google Scholar]; b Zhu Y.; Loso M. R.; Watson G. B.; Sparks T. C.; Rogers R. B.; Huang J. X.; Gerwick B. C.; Babcock J. M.; Kelley D.; Hegde V. B.; Nugent B. M.; Renga J. M.; Denholm I.; Gorman K.; DeBoer G. J.; Hasler J.; Meade T.; Thomas J. D. J. Agric. Food Chem. 2011, 59 (7), 2950–2957. 10.1021/jf102765x. [DOI] [PubMed] [Google Scholar]; c Bacci L.; Convertini S.; Rossaro B. J. Entomol. Acarol. Res. 2018, 50 (3), 51–71. 10.4081/jear.2018.7836. [DOI] [Google Scholar]
- a Frings M.; Bolm C.; Blum A.; Gnamm C. Eur. J. Med. Chem. 2017, 126, 225–245. 10.1016/j.ejmech.2016.09.091. [DOI] [PubMed] [Google Scholar]; b Sirvent J. A.; Lucking U. ChemMedChem 2017, 12 (7), 487–501. 10.1002/cmdc.201700044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bolm C.; Hildebrand J. P. Tetrahedron Lett. 1998, 39 (32), 5731–5734. 10.1016/S0040-4039(98)01199-X. [DOI] [Google Scholar]; b Harmata M.; Pavri N. Angew. Chem., Int. Ed. 1999, 38 (16), 2419–2421. 10.1002/(SICI)1521-3773(19990816)38:16<2419::AID-ANIE2419>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]; c Bolm C.; Hildebrand J. P. J. Org. Chem. 2000, 65 (1), 169–175. 10.1021/jo991342u. [DOI] [PubMed] [Google Scholar]; d Bolm C.; Hildebrand J. P.; Rudolph J. Synthesis 2000, 2000 (07), 911–913. 10.1055/s-2000-6287. [DOI] [Google Scholar]; e Bolm C.; Martin M.; Gibson L. Synlett 2002, 2002 (5), 832–834. 10.1055/s-2002-25362. [DOI] [Google Scholar]; f Harmata M.; Hong X.; Ghosh S. K. Tetrahedron Lett. 2004, 45 (27), 5233–5236. 10.1016/j.tetlet.2004.05.027. [DOI] [Google Scholar]; g Yongpruksa N.; Calkins N. L.; Harmata M. Chem. Commun. 2011, 47 (27), 7665–7667. 10.1039/c1cc12444g. [DOI] [PubMed] [Google Scholar]; h Zhou H.; Chen W.; Chen Z. Org. Lett. 2018, 20 (9), 2590–2594. 10.1021/acs.orglett.8b00776. [DOI] [PubMed] [Google Scholar]; i Yang Q.; Choy P. Y.; Zhao Q.; Leung M. P.; Chan H. S.; So C. M.; Wong W.-T.; Kwong F. Y. J. Org. Chem. 2018, 83 (18), 11369–11376. 10.1021/acs.joc.8b01599. [DOI] [PubMed] [Google Scholar]; j Cho G. Y.; Remy P.; Jansson J.; Moessner C.; Bolm C. Org. Lett. 2004, 6 (19), 3293–3296. 10.1021/ol048806h. [DOI] [PubMed] [Google Scholar]; k Sedelmeier J.; Bolm C. J. Org. Chem. 2005, 70 (17), 6904–6906. 10.1021/jo051066l. [DOI] [PubMed] [Google Scholar]; l Correa A.; Bolm C. Adv. Synth. Catal. 2007, 349 (17–18), 2673–2676. 10.1002/adsc.200700408. [DOI] [Google Scholar]; m Macé Y.; Pégot B.; Guillot R.; Bournaud C.; Toffano M.; Vo-Thanh G.; Magnier E. Tetrahedron 2011, 67 (39), 7575–7580. 10.1016/j.tet.2011.07.060. [DOI] [Google Scholar]; n Liu Z. J.; Vors J. P.; Gesing E. R. F.; Bolm C. Green Chem. 2011, 13 (1), 42–45. 10.1039/C0GC00296H. [DOI] [Google Scholar]; o Moessner C.; Bolm C. Org. Lett. 2005, 7 (13), 2667–2669. 10.1021/ol050816a. [DOI] [PubMed] [Google Scholar]; p Vaddula B.; Leazer J.; Varma R. S. Adv. Synth. Catal. 2012, 354 (6), 986–990. 10.1002/adsc.201100808. [DOI] [Google Scholar]; q Kim J.; Ok J.; Kim S.; Choi W.; Lee P. H. Org. Lett. 2014, 16 (17), 4602–4605. 10.1021/ol502174n. [DOI] [PubMed] [Google Scholar]; r Zhu H.; Teng F.; Pan C.; Cheng J.; Yu J.-T. Tetrahedron Lett. 2016, 57 (22), 2372–2374. 10.1016/j.tetlet.2016.04.042. [DOI] [Google Scholar]; s Jiang Y.; You Y.; Dong W.; Peng Z.; Zhang Y.; An D. J. Org. Chem. 2017, 82 (11), 5810–5818. 10.1021/acs.joc.7b00633. [DOI] [PubMed] [Google Scholar]; t Correa A.; Bolm C. Adv. Synth. Catal. 2008, 350 (3), 391–394. 10.1002/adsc.200700508. [DOI] [Google Scholar]
- Wimmer A.; König B. Adv. Synth. Catal. 2018, 360 (17), 3277–3285. 10.1002/adsc.201800607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lämmermann H.; Sudau A.; Rackl D.; Weinmann H.; Collins K.; Wortmann L.; Candish L.; Hog D. T.; Meier R. Synlett 2018, 29 (20), 2679–2684. 10.1055/s-0037-1609656. [DOI] [Google Scholar]
- a Twilton J.; Le C.; Zhang P.; Shaw M. H.; Evans R. W.; MacMillan D. W. C. Nature Reviews Chemistry 2017, 1, 0052. 10.1038/s41570-017-0052. [DOI] [Google Scholar]; b Terrett J. A.; Cuthbertson J. D.; Shurtleff V. W.; MacMillan D. W. C. Nature 2015, 524 (7565), 330–334. 10.1038/nature14875. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Tellis J. C.; Primer D. N.; Molander G. A. Science 2014, 345 (6195), 433–436. 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Zuo Z.; Ahneman D. T.; Chu L.; Terrett J. A.; Doyle A. G.; MacMillan D. W. C. Science 2014, 345 (6195), 437–440. 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Oderinde M. S.; Jones N. H.; Juneau A.; Frenette M.; Aquila B.; Tentarelli S.; Robbins D. W.; Johannes J. W. Angew. Chem., Int. Ed. 2016, 55 (42), 13219–13223. 10.1002/anie.201604429. [DOI] [PubMed] [Google Scholar]; b Corcoran E. B.; Pirnot M. T.; Lin S.; Dreher S. D.; DiRocco D. A.; Davies I. W.; Buchwald S. L.; MacMillan D. W. C. Science 2016, 353 (6296), 279–283. 10.1126/science.aag0209. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kim T.; McCarver S. J.; Lee C.; MacMillan D. W. C. Angew. Chem., Int. Ed. 2018, 57 (13), 3488–3492. 10.1002/anie.201800699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A comprehensive version of the optimization of the reaction conditions and all of the control reactions is displayed in the SI.
- For a mechanistic proposal and further experimental details, see the SI. Electron-transfer processes between the applied photocatalyst and nickel catalyst species cannot be ruled out.
- For further experimental details, see the SI.
- Due to the higher solubility of the brominated heteroarenes, mainly DMAc was used as solvent in this part of the substrate scope.
- The amounts of [Ir]-Cat and [Ni-2]-Cat used in these cases vary between 0.15–1.0 mol % and 0.20–5.0 mol %, respectively, and are displayed for every substrate in Figure 2.
- For further experimental details, see SI.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.