

Abstract
Objective
The mixed lineage kinase domain like (MLKL) protein, receptor interacting protein (RIPK) 1, and RIPK3 are key regulators of necroptosis, a highly pro-inflammatory mode of cell death that has been implicated in various pathological processes and human diseases. However, the role of these necroptotic regulators in diabetes remains unknown. Here we sought to delineate the role of MLKL in insulin resistance and type 2 diabetes (T2D).
Methods
We first analyzed the expression of key necroptotic regulators in obese/diabetic mouse models. We then utilized MLKL knockout (MLKL−/−) mice to evaluate the effects of MLKL on obesity-induced metabolic complications. We further determined the consequences of MLKL inhibition on hepatic insulin signaling and explored the underlying mechanism. Finally, we assessed the potential therapeutic effects of necroptotic inhibitor, necrostatin-1 (Nec-1), in ob/ob mice.
Results
In wild-type or obese mice (ob/ob, db/db, or diet-induced obesity), MLKL was increased in certain obesity-associated tissues, particularly in the liver. Whole-body deficiency of MLKL prevented obesity-induced insulin resistance and glucose intolerance. Inhibition of MLKL or other key necroptotic regulators enhanced hepatic insulin sensitivity. MLKL modulated insulin-stimulated PI(3,4,5)P3 production in liver cells but did not affect the expression of inflammatory genes in vitro and in vivo. Nec-1 administration ameliorated insulin resistance and glucose intolerance in ob/ob mice.
Conclusions
These findings reveal MLKL as a regulator of insulin sensitivity and suggest necroptotic regulators might be potential therapeutic targets for insulin resistance and T2D.
Keywords: MLKL, Necroptosis, Diabetes, Insulin sensitivity, Inflammation
Highlights
-
•
Hepatic MLKL is increased in diabetic mouse models.
-
•
MLKL deficiency prevents obesity-induced metabolic complications.
-
•
Inhibition of MLKL enhances insulin sensitivity independent of inflammation.
-
•
Pharmacological inhibition of RIPK1 in ob/ob mice ameliorates insulin resistance.
Abbreviations
- T2D
type 2 diabetes
- Nec-1
necrostatin-1
- NSA
necrosulfonamide
- GSK’872
GSK2399872A
- PI(3,4,5)P3
phosphatidylinositol (3,4,5)-trisphosphate
- PI(4,5)P2
phosphatidylinositol (4,5)-bisphosphate
- SPF
specific pathogen-free
- DIO
diet-induced obesity
- CD
chow diet
- HFD
high-fat diet
- GTT
glucose tolerance test
- ITT
insulin tolerance test
- VAT
visceral adipose tissue
- SAT
subcutaneous adipose tissue
- BAT
brown adipose tissue
- HGP
hepatic glucose production
- AUC
area under the curve
1. Introduction
Type 2 diabetes (T2D), an alarmingly fast-growing epidemic, is hallmarked by insulin resistance in peripheral tissues, including liver, adipose, and skeletal muscle. Although many genetic and epigenetic factors integrate to initiate and promote insulin resistance, the molecular mechanisms underlying insulin resistance are only partially understood [1], [2], [3], [4], [5]. The PI3K/AKT signaling mediates multiple physiological functions of insulin and is central to proper glucose homeostasis. Briefly, insulin receptor (IR) on the cell membrane is activated upon the binding of insulin, which subsequently phosphorylates insulin-receptor substrate (IRS) proteins. IRS phosphorylation leads to the activation of phosphatidylinositol-3-kinase (PI3K), resulting in PI(3,4,5)P3 production at the plasma membrane. This promotes the recruitment and interaction between PDK1 and protein kinase B/AKT, which in turn results in phosphorylation of AKT that exerts most of the metabolic actions of insulin. Therefore, improved understanding of the wiring of the AKT signaling is critical for elucidating the pathophysiology of insulin resistance and T2D.
Inflammation and cell death contribute to the development of T2D [6], [7]. Necroptosis, a recently characterized necrosis integrated with the extrinsic apoptosis pathway, is widely viewed as a highly pro-inflammatory mode of cell death, due to rapid release of immune-stimulatory intracellular components after cell-membrane rupture [8], [9]. Classically, necroptosis relies on three key proteins: RIPK1, RIPK3, and MLKL. The best characterized mechanism of necroptosis involves RIPK1-induced activation of RIPK3 through the formation of RIPK1/RIPK3 necrosomes, which in turn phosphorylate the pseudokinase MLKL, a substrate of RIPK3 [8], [10]. This phosphorylation facilitates MLKL oligomerization, which then translocates and damages the plasma membrane by interacting with phosphatidylinositol phosphate (PIP) phospholipids, leading to mitochondrial uncoupling, lipid peroxidation, and eventually cell death [11], [12]. Apart from promoting necroptosis, RIPK1 can alternatively participate in caspase-8-mediated apoptosis [13], and mediate cell survival and inflammation by kinase-independent scaffolding functions [14], [15]. In addition, several biological stimuli and some viruses have shown to trigger necroptosis independent of RIPK1 [9]. Similarly, RIPK3 also modulates other processes through apoptosis or cytokine production independent of MLKL [15], [16], [17]. By contrast, MLKL is currently believed to be the major and perhaps the only substrate of RIPK3 that executes necroptosis [9]. Necroptosis has been demonstrated to be an important regulator of immunity and implicated in various inflammatory diseases, such as acute pancreatitis, cancers, dermatitis, psoriasis, septic shock, and ischemia reperfusion [18], [19], [20], [21]. However, the involvement of necroptotic markers in insulin resistance and T2D remains unclear.
Here we show that the loss of MLKL in mice prevents obesity-induced insulin resistance and glucose intolerance. Blockage of MLKL, as well as RIPK1 and RIPK3, leads to increased hepatic insulin sensitivity in vivo and in vitro. Furthermore, chemical inhibition of RIPK1 in mice relieves obesity-associated metabolic disturbances. Taken together, these findings reveal a role of MLKL in insulin sensitivity and suggest the potential involvement of necroptotic regulators in the physiopathology of T2D.
2. Material and methods
2.1. Animals
Generation of MLKL knockout hemizygous (MLKL+/−) mice has been described previously [22]; hemizygotes were backcrossed to C57BL/6 background for nine generations. MLKL+/− mice were crossed to generate MLKL−/− mice and wild type (WT) littermates (MLKL+/+) for experiments. Db/db (C57BLKS/J) and ob/ob (C57BL/6J) mice were purchased from Model Animal Research Center of Nanjing University. All mice were maintained in specific pathogen-free (SPF) environment with constant temperature and humidity and 12-hour light/12-hour dark cycle with free access to standard diet and water. Age- and body weight-matched male MLKL−/− and their WT littermates were kept on standard chow diet (CD) (Beijing Hua Fu Kang Bioscience, # 1025) or high-fat diet (HFD) consisting of 60% kcal from fat (Research Diets, # D12492) for at least 16 weeks to establish diet-induced obesity (DIO) mouse models. All mouse-related experiments were repeated independently and conducted according to the protocols approved by the Institutional Animal Care and Use Committee of Sichuan University.
2.2. Nec-1 administration
Ob/ob mice at the age of 6 weeks were intraperitoneally injected with Nec-1 (2.5 mg/kg body weight, Selleck, # S8037) in 0.1% DMSO saline solution or vehicle once a day persisting for one month. GTT, ITT, and blood glucose measuring were performed during this duration.
2.3. Plasmids
Human MLKL cDNA sequence was amplified and subsequently cloned into pcDNA3.1 vector. Primer sequences for human MLKL overexpressing vector are as follows: Forward, 5’-CTAGCTAGCATGGAAAATTTGAAGCATAT-3’; Reverse, 5’-TATGCTCGAGCTACTTAGAAAAGGTGGAGAG-3’.
2.4. Cell culture
HepG2 cells were purchased from ATCC and cultured in complete DMEM medium supplemented with 10% FBS and 1% Penicillin-Streptomycin. For gene knockdown, cells were transfected with indicated siRNAs (50 nM) for at least 24 h, according to manufacturer's guidelines (QIAGEN, HiPerFect Transfection Reagent, # 301705). For insulin signaling examination, cells were serum-starved for 6 h before treating with 100 nM insulin (Sigma, # I5500) for the indicated times. The sequences of siRNAs were shown as follows: human MLKL, 5’-CAAACUUCCUGGUAACUCA-3’; human RIPK1, 5’-CCACUAGUCUGACGGAUAA-3’; murine MLKL, 5’-GAGAUCCAGUUCAACGAUA-3’; murine RIPK1, 5’-CCACUAGUCUGACU GAUGA-3’; and murine RIPK3, 5’-CCCGACGAUGUCUUCUGUCAA-3’.
2.5. Murine hepatocyte isolation and culture
Primary hepatocytes were isolated by two-step collagenase perfusion technique and cultured as described previously [23]. Briefly, mice were anesthetized and perfused with 50 ml 1 × EBSS solution (Gibco, # 14155063) containing 0.5 mM EGTA per mouse under portal vein rupturing condition, followed by 60 ml 1 × HBSS solution (Gibco, # 14175103) containing 0.3 mg/ml collagenase II (Thermo Fisher, # 17101015) and 40 μg/ml trypsin inhibitor (Sigma, # T6522). Digested liver was crushed into single cells and centrifuged with Percoll (Sigma, # P4937) at 600 rpm for 10 min. Collected cell pellet was resuspended with complete DMEM medium with 10% FBS after twice of washing and seeded into cell culture plates. For insulin signaling examination, hepatocytes were treated similarly as HepG2 except that 10 nM insulin was used for simulation. To test the effect of necroptotic inhibitors on insulin signaling, indicated inhibitors were added to medium for 2 h with Nec-1 (50 μM), or for 3 h with GSK’872 (6 μM. Biovision, # 2673-5) and NSA (1 μM. Calbiochem, # 480073) prior to insulin stimulation.
2.6. Glucose production assay
Glucose production assay was performed as described previously [2]. Briefly, isolated primary hepatocytes were cultured in glucose- and FBS-free DMEM medium for 30 min, and the medium was replaced with glucose production buffer (glucose-free DMEM, 20 mM L-sodium lactate, 2 mM sodium pyruvate, pH 7.4) and incubated for 6 h. Cultured medium was collected and centrifuged, then the supernatants were collected and glucose concentration determined by Glucose Colorimetric/Fluorometric Assay Kit (Biovision, # K606-100). At the same time, cells were harvested with RIPA (Pierce, # 89901) for protein quantification.
2.7. Blood glucose and insulin measurement
Unless otherwise specified, random blood glucose was measured in free-feeding condition, while fasting blood glucose was measured after starvation for 16 h. Blood glucose was determined through tail vein bleeding with use of portable glucometer (Abbot Laboratories). For insulin measurement, blood samples were collected from the tail vein and centrifuged at 3000 rpm, 4 °C for 10 min. Serum insulin levels were determined using Ultra-Sensitive Mouse Insulin ELISA Kit (Crystal Chem, # 90080) according to manufacturer's instructions. Homeostasis model assessment for insulin resistance index (HOMA-IR) was calculated as the following formula: fasting blood glucose (mmol/L) × fasting blood insulin (μU/mL)/22.5.
2.8. Glucose tolerance test (GTT) and insulin tolerance test (ITT)
GTT and ITT were performed as described previously [2]. For GTT, mice were fasted for 16 h and intraperitoneally injected with D-glucose (Sigma, # G7021, 2 g/kg BW) in sterile water; then, blood glucose was monitored at pre-set time points after injection (0, 15, 30, 60, 90, and 120 min). For ITT, mice were fasted for 6 h and injected with human insulin (Lilly, # HI0219, 1 U/kg BW) in saline; then, blood glucose was determined at pre-set time points after injection (0, 15, 30, 60, and 90 min).
2.9. In vivo insulin stimulation and analysis of AKT activation
In vivo insulin stimulation and AKT analysis were performed as described previously, with minor modifications [2]. Briefly, mice were anesthetized, and insulin (0.25 U/kg BW) was injected through portal vein. Five-, eight-, and ten-minutes post infusion, liver tissues, visceral fat, and muscle were excised orderly and used for total protein extraction. Western blot analyses were performed to test AKT activation.
2.10. Western blot
For western blot analysis, frozen tissues or collected cells were homogenized on ice in RIPA buffer supplemented with protease and phosphatase inhibitors (Pierce, # 88668). Protein concentration was determined by Bradford assay and equal quantity of total protein of each sample was used for denaturalized samples. The prepared samples were resolved by SDS-PAGE, and then were transferred to PVDF membrane. Membranes were blocked for 1 h at room temperature, and incubated in the primary antibodies for 16 h at 4 °C. Then membranes were washed and incubated for 2 h at room temperature with HRP-conjugated secondary antibodies. Membranes were washed and developed using the ECL kit (ThermoFisher, # 34075 and # 34580). Antibodies used in western blot were listed in Table S1.
2.11. Gene expression
Gene expression was determined by real-time quantitative polymerase chain reaction (QRT-PCR) as previously described [2]. Total RNA was isolated using Trizol-Reagent (MRC, # TR118). Complementary DNA was synthesized using M-MLV reverse transcriptase (Invitrogen, # 28025) and QRT-PCR was performed according the Power SYBR Green PCR Master Mix protocol (Applied Biosystems, # 4473369). Sequences for the QRT-PCR primers were provided in Table S2.
2.12. Immunofluorescence (IF) staining
HepG2 cells were transfected with MLKL-overexpression or empty vectors for 48 h, and treated with insulin (100 nM) for 3 min. Primary hepatocytes were isolated from 6 to 8 weeks old MLKL−/− mice and WT littermates, respectively, cultured overnight, and then treated with insulin (10 nM) for 3 min. After insulin stimulation, cells were fixed with 4% formaldehyde for 15 min at room temperature, rinsed three times in 1 × PBS, and blocked in 5% (w/v) BSA/TBST buffer for 1 h at room temperature. The blocked specimens were incubated with the anti-human pMLKL (phosphor S358) (Abcam, # ab187091), anti-mouse pMLKL (phosphor S345) (Abcam, # ab196436), or anti-PI(3,4,5)P3 (Echelon, # Z-P345) antibodies overnight at 4 °C. Then the specimens were washed with TBST for three times, incubated in fluorochrome-conjugated secondary antibody solution for 2 h at room temperature and protected from light, and then stained with DAPI for 5–10 min. After washing three times, the slides were mounted using VECTASHIELD mounting medium (Vector Laboratories, # H-1000) then collected the images by laser confocal scanning microscopy.
2.13. Histological analysis
Tissues were collected immediately from sacrificed mice and fixed with 4% formaldehyde for 48 h at room temperature. The fixed samples were embedded in paraffin and cut into 4–6 μm sections. The sections were used for Hematoxylin and Eosin (H&E) staining, and immunohistochemical (IHC) staining for MLKL (Abcam, # ab194699), phosphorylated MLKL (Abcam, # ab196436), CD45 (Proteintech, # 20103-1-AP), or F4/80 (Proteintech, # 27044-1-AP).
2.14. Statistical analysis
All data represent at least three independent experiments unless otherwise indicated. Statistical analyses were performed using Graphpad Prism 6. All data were shown as means ± SEM and p < 0.05 was considered statically significant. Analyses performed included 2-way ANOVA, Student's t-test, and paired t-test where appropriate.
3. Results
3.1. MLKL expression is increased in diabetic mouse models
Initially, the levels of RIPK1, RIPK3, and MLKL in peripheral tissues associated with T2D were determined by QRT-PCR analysis. It revealed that they were enriched in liver and adipose (Figure 1A). We then measured the levels of RIPK1, RIPK3, and MLKL in 3 diabetic mouse models (Figure S1A, B). In diet-induced obesity (DIO) mouse model, for which wild-type (WT) mice were fed a high-fat diet (HFD) for 16 weeks as compared with mice fed a standard chow diet (CD), RIPK1 and MLKL were upregulated in obesity-associated organs including liver, adipose, and muscle, whereas RIPK3 was specifically increased in adipose (Figure 1B and Figure S1C, D). Interestingly, RIPK1, RIPK3, and MLKL were upregulated in the livers of leptin-deficient (ob/ob) obese mice (Figure 1C), but only the increase of MLKL was recapitulated in the livers of leptin receptor-deficient (db/db) diabetic mice (Figure 1D).
Figure 1.

MLKL is increased in diabetic mouse models. (A) Expression of RIPK1, RIPK3, and MLKL in obesity-associated organs of WT mice (n = 4–5). (B–D) Expression of RIPK1, RIPK3, and MLKL in the livers of WT DIO mice fed a CD (n = 5–6) or a HFD (n = 5–6) (B), WT lean (n = 5) and ob/ob mice (n = 4) (C), and WT control (n = 3) and db/db mice (n = 4) (D). (E and F) Levels of phosphorylated and total MLKL proteins in the livers of WT DIO mice fed a CD or a HFD (n = 3) (E), and WT control and db/db mice (n = 3) (F). (G–J) Representative IHC staining for total (G and H) and phosphorylated MLKL (I and J) in the livers of WT DIO mice fed a CD (n = 5) or a HFD (n = 6), and WT control (n = 3) and db/db mice (n = 4). Scale bars, 100 μm. Data are shown as mean ± SEM. *P < 0.05, **P < 0.01. Student's t-test was used.
Independent of the triggering mechanisms, MLKL is the executive molecule of necroptosis and its activation is the most proximal step to membrane disruption and cell death [18]. Moreover, the upregulation of hepatic MLKL was most consistent and notable in 3 diabetic mouse models (Figure 1B–D). Therefore, we chose MLKL for further analysis. Western blot (WB) (Figure 1E,F) and immunohistochemistry (IHC) (Figure 1G,H and Figure S1E) showed that the protein levels of MLKL in the livers of diabetic mice were higher than that of control mice. The levels of MLKL phosphorylation were also increased in diabetic mice, as assessed by western blot (Figure 1E,F) and IHC (Figure 1I,J). Taken together, these results suggest that MLKL might have a potential role in the development of obesity and T2D.
3.2. MLKL deficiency ameliorates obesity-induced metabolic disturbances
We investigated the function of MLKL in vivo using the recently established MLKL knockout (MLKL−/−) mouse line [22]. MLKL−/− mice and MLKL+/+ (wild type, WT) littermates of 8–12 weeks of age fed a CD had no significant differences in body weight (BW), glucose disposal, glucose tolerance, or insulin sensitivity (Figure S2A–D). However, when MLKL−/− mice and their WT littermates were fed a HFD, MLKL−/− mice had significantly lower BWs than WT littermates (Figure 2A), although both genotypes had comparable food intake (Figure 2B). The analyses of tissue weight showed that MLKL−/− mice had less visceral adipose tissue (VAT) accumulation than WT littermates (Figure 2C). When mice of both genotypes fed a HFD for either 8 or 16 weeks, MLKL−/− mice presented significantly better glucose and insulin tolerance than WT littermates did, as analyzed by the glucose tolerance test (GTT) and insulin tolerance test (ITT), respectively (Figure 2D–G). Accordingly, MLKL−/− mice fed a HFD had significantly lower random and fasting blood-glucose levels (Figure 2H,I). However, insulin levels of both genotypes were comparable during GTT analysis (Figure 2J), indicating that improved glucose metabolism in MLKL−/− mice results from enhanced insulin sensitivity but not elevated insulin secretion. Consistent with this notion, the homeostatic model assessment index of insulin resistance (HOMA-IR) was significantly lower in MLKL−/− mice (Figure 2K). Taken together, these results suggest that MLKL deficiency enhances insulin sensitivity and prevents obesity-induced metabolic complications.
Figure 2.

Deficiency of MLKL ameliorates obesity-induced insulin resistance and glucose intolerance. Mice were fed a HFD beginning at 6–8 weeks of age. Measurements were performed during the course of the HFD, as shown below. (A) Total body weight (n = 10 in each group). AUC, area under the curve. (B) Food intake (n = 6 for WT, n = 5 for MLKL−/−). (C) Ratio of organ weight (n = 6 for WT, n = 5 for MLKL−/−). Panc: pancreas. (D and E) GTT (n = 7 for WT, n = 8 for MLKL−/−) (D) and ITT (n = 8 for WT, n = 9 for MLKL−/−) (E), performed after 8 weeks of HFD. (F and G) GTT (n = 7 for WT, n = 8 for MLKL−/−) (F) and ITT (n = 12 for WT, n = 10 for MLKL−/−) (G), performed after 16 weeks of HFD. (H and I) Blood glucose levels of mice fed a HFD for 8 weeks (n = 13 for WT, n = 8 for MLKL−/−) (H), or for 16 weeks (n = 15 for WT, n = 8 for MLKL−/−) (I). (J) Blood insulin levels of mice fed a HFD for 16 weeks during GTT (n = 5 for WT, n = 7 for MLKL−/−). (K) HOMA-IR (n = 5 for WT, n = 7 for MLKL−/−). Data are shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. 2-way ANOVA or Student's t-test were used.
3.3. Inhibition of MLKL improves insulin-stimulated AKT activation
As shown above, MLKL−/− mice were resistant to obesity-induced insulin resistance (Figure 2). To further verify this, we directly evaluated the effect of MLKL on insulin signaling in mouse peripheral tissues stimulated with insulin. Under obese conditions, MLKL−/− mice had greater responsiveness to insulin stimulation than their WT littermates in peripheral tissues, including liver, adipose, and muscle (Figure 3A and Figure S3). A cell-intrinsic improvement in the ability of insulin to stimulate AKT activity was confirmed in primary hepatocyte from MLKL−/− mice (Figure 3B). Furthermore, knockdown of RIPK1, RIPK3, or MLKL by small interfering RNAs (siRNAs) led to an increase in insulin-stimulated AKT activation in murine primary hepatocytes (Figure 3C–E). This improvement was recapitulated by treatment with Nec-1 and GSK′872 (Figure 3F), which are chemical inhibitors for RIPK1 and RIPK3, respectively [10], [16], [24]. Necrosulfonamide (NSA), which specifically inhibits human MLKL but not mouse MLKL [25], had no effect on AKT activation in murine hepatocytes (Figure 3F). This effect of necroptotic markers on insulin-stimulated AKT activity was recapitulated in HepG2 cells (Figure 3G–I).
Figure 3.

MLKL regulates insulin signaling. (A) AKT phosphorylation in livers of MLKL−/− mice and WT littermates fed a HFD for 16 weeks and infused with insulin (0.25 U/kg) through the portal vein. (B) AKT phosphorylation in primary hepatocytes isolated from MLKL−/− mice and WT littermate controls fed a HFD for 18 weeks and stimulated with insulin (10 nM) for the indicated times. (C and D) mRNA (C) or protein (D) levels of RIPK1, RIPK3, and MLKL in primary hepatocytes transfected with indicated siRNAs were determined by QRT-PCR or western blot. (E and F) Insulin signaling in primary hepatocytes isolated from WT mice transfected with siRNAs (E) or treated with chemical inhibitors (F) prior to insulin stimulation. (G and H) mRNA (G) or protein (H) levels of RIPK1, RIPK3, and MLKL in HepG2 cells transfected with indicated siRNAs were determined by QRT-PCR or western blot. (I) AKT phosphorylation in HepG2 cells transfected with siRNAs prior to insulin stimulation. (J) Expression of FBP1, G6PC, and PCK1 in livers from MLKL−/− mice and WT littermates (n = 7 per group). (K) Glucose production in primary hepatocytes isolated from MLKL−/− mice (n = 3) and WT littermates (n = 3) fed a HFD for 16 weeks. Western blots are representative of three independent experiments. Data are shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. Student's t-test was used.
Insulin inhibits gluconeogenesis and hepatic glucose production. In line with increased insulin signaling, expression of gluconeogenetic genes was significantly downregulated in the livers of MLKL−/− mice fed a HFD (Figure 3J). Accordingly, primary hepatocytes from MLKL−/− mice fed a HFD produced less glucose than that of WT littermates (Figure 3K). Together, these results suggest that MLKL, probably RIPK1 and RIPK3, regulates the sensitivity and action of insulin.
3.4. MLKL regulates insulin signaling by modulating PIP3 production
MLKL as well as RIPK1 and RIPK3 affected insulin-stimulated AKT phosphorylation but not IR phosphorylation (Figure 3E), suggesting that they may function downstream of IR but upstream of AKT. Upon the activation of IR, PI(3,4,5)P3 catalyzed from PI(4,5)P2 is critical for insulin-stimulated AKT phosphorylation. Interestingly, PI(4,5)P2 is the most preferred binding-partner of oligomerized MLKL [12]. Therefore, we wondered if MLKL could affect PI(3,4,5)P3 production in insulin signaling. To this end, we transiently overexpressed MLKL in HepG2 cells (Figure 4A), which were subsequently stimulated with insulin. Immunofluorescence analysis showed that HepG2 cells overexpressing MLKL had lower PI(3,4,5)P3 levels than control cells (Figure 4B). In contrast, insulin-stimulated PI(3,4,5)P3 levels were higher in isolated primary hepatocytes from MLKL−/− mice than in that of WT littermates (Figure 4C,D). These results suggest that MLKL may modulate PI(3,4,5)P3 production, thereby regulating insulin sensitivity.
Figure 4.

MLKL regulates insulin-stimulated PI(3,4,5)P3 production. (A and B) Protein levels of MLKL in HepG2 cells transfected with empty or MLKL overexpressing vectors were determined by western blot (A). PI(3,4,5)P3 levels were determined by immunofluorescence (B). Scale bars, 50 μm. (C and D) Protein levels of MLKL in primary hepatocytes isolated from MLKL−/− mice and WT littermates (C). PI(3,4,5)P3 levels were determined by immunofluorescence (D). Scale bars, 20 μm. Images are representative of three independent experiments.
3.5. The effect of MLKL on insulin signaling is not associated with inflammation and cell death
Next, we determined if the effect of MLKL on insulin sensitivity is associated with inflammation. MLKL−/− and WT littermates fed a HFD had no obvious differences in the expression of inflammatory genes, including IL-6, IL-1β, MCP-1 and TNFα (Figure 5A). Moreover, CD45 and F4/80 immunostaining analyses showed comparable levels of granulocyte infiltrate and macrophagocyte residence in the livers of MLKL−/− and their WT littermates (Figure 5B,C). Importantly, mRNA levels of inflammatory genes in isolated primary hepatocytes stimulated with insulin were comparable in both genotypes (Figure 5D). TUNEL assay showed that MLKL−/− mice and their WT littermates had similar amounts of cell death (Figure 5E and Figure S4A), indicating that MLKL deficiency might have minor effect on eventual cell death in vivo. Taken together, these data suggest that MLKL may modulate insulin signaling independent of inflammation and cell death.
Figure 5.

Inhibition of MLKL has minor effects on hepatic inflammation. (A) Expression of chemokines and pro-inflammatory cytokines in livers from MLKL−/− mice (n = 4–6) and WT littermates (n = 5–9) fed a HFD for 18 weeks. (B and C) Representative IHC staining for CD45 (B) and F4/80 (C) in livers of MLKL−/− mice (n = 6) and WT littermates (n = 5) fed a HFD for 18 weeks. Scale bars, 50 μm. (D) Expression of MCP-1, TNFα and IL-1β in primary hepatocytes isolated from MLKL−/− mice (n = 3) and WT littermates (n = 3) stimulated with insulin (10 nM) for the indicated time. (E) Quantification of TUNEL positive area in livers from MLKL−/− mice and WT littermates (n = 4). Data are shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. Student's t-test was used.
3.6. Nec-1 ameliorates insulin resistance and glucose intolerance in ob/ob mice
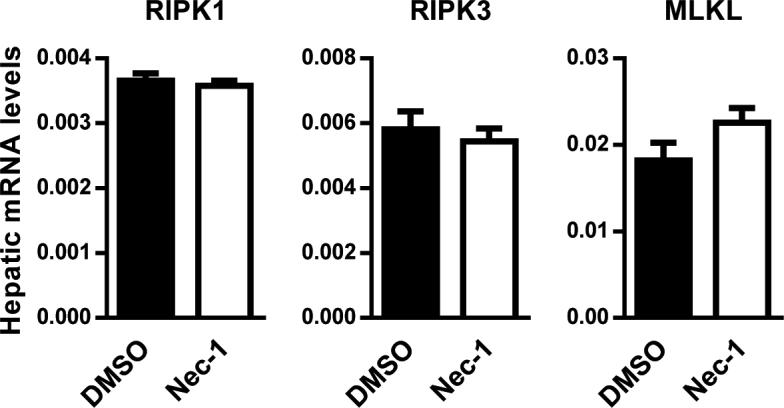
Given that MLKL impairs insulin sensitivity, we hypothesized that its inhibition may be beneficial for T2D treatment. Unfortunately, NSA cannot target murine MLKL, and there is no effective MLKL inhibitor for in vivo mouse studies thus far [25]. Meanwhile, we have showed a critical role of MLKL in DIO mice, but the conservation of this regulation in genetic obese mouse model remains elusive. In addition, we noticed that inhibition of RIPK1 and RIPK3, the upstream regulators of MLKL, can improve insulin sensitivity in vitro. Therefore, we administrated ob/ob mice with the RIPK1 inhibitor Nec-1 (Figure 6A), which has been widely used in in vivo studies [26], [27]. Nec-1 administration had no effect on body weight and food intake (Figure 6B,C) but significantly decreased the fasting blood glucose (Figure 6F) and increased glucose tolerance (Figure 6D). ITT analysis revealed that insulin resistance was ameliorated in ob/ob mice administrated with Nec-1 (Figure 6E). Insulin levels for mice of both groups were similar during GTT analysis (Figure 6G), supporting that Nec-1 administration enhanced insulin sensitivity, but not increased insulin secretion. Consistent with this, the HOMA-IR was significantly lower in ob/ob mice administrated with Nec-1 (Figure 6H). Moreover, Nec-1-treated ob/ob mice had increased AKT activation compared with control mice (Figure 6I). In addition, Nec-1-treated ob/ob mice had slightly less fat deposition (Figure 6J) and significantly reduced hepatic triglyceride (Figure 6K). However, mRNA levels of inflammatory genes were comparable in the livers of both groups (Figure 6L), indicating that metabolic improvements by Nec-1 treatment might be independent of inflammation. In line with these observations, Nec-1-treated ob/ob mice exhibited lower MLKL phosphorylation than control mice (Figure 6M), although both groups had comparable mRNA levels of necroptotic regulators (Figure S5A). Taken together, these results suggest that Nec-1 administration could ameliorate obesity-induced metabolic complications, partially recapitulating the consequences of MLKL deficiency in mice fed a HFD.
Figure 6.

Nec-1 attenuates the metabolic abnormalities of ob/ob mice. (A) Protocol for Nec-1 administration. (B) Body weight (n = 6 for DMSO, n = 8 for Nec-1). (C) Food intake (n = 6 for DMSO, n = 8 for Nec-1). (D) GTT (n = 5 per group). (E) ITT (n = 7 for DMSO, n = 6 for Nec-1). (F) Blood glucose levels (n = 6 per group). (G) Blood insulin levels (n = 4–5 per group). (H) HOMA-IR (n = 4 for DMSO, n = 5 for Nec-1). (I) AKT phosphorylation in mouse livers (n = 3 per group). Quantification of pAKT were shown (right panel). (J) Representative H&E staining of mouse livers (n = 4 per group). Scale bar, 100 μm. (K) Hepatic triglyceride levels (n = 6 for DMSO, n = 8 for Nec-1). (L) Expression of inflammatory genes in mouse livers (n = 6 per group). (M) Representative IHC staining for pMLKL in mouse livers (n = 3 for DMSO, n = 4 for Nec-1). Scale bar, 50 μm. Data are shown as mean ± SEM. *P < 0.05, **P < 0.01. Student's t-test was used.
4. Discussion
We have shown that MLKL regulates insulin sensitivity and participates in the development of T2D. We provided several lines of evidence supporting the involvement of necroptotic regulators in T2D. First, MLKL, to some extent RIPK1 and RIPK3, was upregulated in 3 diabetic mouse models. Second, inhibition of RIPK1, RIPK3, or MLKL by RNAi or pharmacological inhibitors enhanced insulin signaling in vitro and in vivo. Third, genetic deficiency of MLKL in mice significantly prevented obesity-induced insulin resistance and glucose intolerance. Finally, Nec-1 (a RIPK1 inhibitor) improved insulin sensitivity and glucose tolerance in ob/ob mice. These results suggest that these three necroptotic regulators may participate in the pathophysiology of insulin resistance and T2D.
It has been shown that oligomerized MLKL can translocate to plasma membrane and bind phosphatidylinositol phosphates. PI(4,5)P2, the most preferred binding-ligand of MLKL [12], is indispensable to insulin-stimulated AKT phosphorylation through controlling PI(3,4,5)P3 production. Indeed, we observed that MLKL impaired insulin-stimulated PI(3,4,5)P3 production in both primary hepatocytes and HepG2 cells. On the basis of these results and recent literature [12], we proposed that elevated hepatic MLKL may interact with PI(4,5)P2, resulting in impaired insulin signaling and the pathophysiology of T2D. Given a conventional way for MLKL activation, it is possible that RIPK1 and RIPK3 may regulate insulin sensitivity through MLKL. Of note, MLKL−/− mice exhibited significantly lower BWs than WT littermates under HFD conditions, indicating a potential contribution of energy metabolism to the phenotype of MLKL deficiency. It is interesting for future studies to explore the effect of MLKL on energy metabolism independently of hepatic insulin sensitivity.
Necroptosis is widely considered to be a highly immunogenic activity and the majority of its functions are known to be associated with inflammation [28], [29]. Intriguingly, genetic deficiency of MLKL or pharmacological inhibition of RIPK1 in mice significantly relieved obesity-induced hepatic insulin resistance but had no effect on hepatic inflammation. Moreover, inhibition of MLKL, RIPK1, or RIPK3 in liver cells in vitro did not affect the expression of inflammatory genes. These results suggest that MLKL may regulate insulin signaling independent of inflammation.
In the traditional view, obesity-induce MLKL activation could lead to cell death. However, we showed that MLKL deficiency did not change HFD-induced liver cell death in vivo. These results suggest that MLKL activation in hepatocytes may not result in cell death under obese conditions. This notion not only provides a reasonable explanation for the disassociation of MLKL activation from hepatic inflammation but also is consistent with recent findings. It has been shown that the endosomal sorting complexes required for transport (ESCRT)-III machinery can interact with MLKL, which results in exosome formation and ectosome budding, and subsequently reduces MLKL phosphorylation-induced cytotoxicity and maintains cell survival [30], [31], [32]. It will be interesting for future studies to investigate whether ESCRT-III interacts with MLKL and contributes to the development of insulin resistance and T2D.
In summary, our findings expand knowledge of the physiological functions of MLKL in human diseases and suggest inhibition of necroptotic regulators as an attractive therapeutic strategy for treatment of insulin resistance and T2D. Our study also provides a paradigm for exploring the potential involvement of necroptotic makers in biological processes that are not associated with inflammation and cell death.
Author contributions
X.F. conceived the project, oversaw research, and wrote the manuscript. X.F. H.X., and X.D. designed the experiments, analyzed and interpreted the data and made the figures. H.X., X.D., G.L., S.H., W.D., S.Z., D.T., C.F., and Y.T. performed the experiments, analyzed and interpreted the data. Y.W., Y.T., and Y.X. helped conceive the project and interpret the data.
Acknowledgments
We thank Dr. Jiahuai Han (Xiamen University) for providing the MLKL−/− mice. This work was supported by the Ministry of Science and Technology of China (2018X092018-005 to X.F.), the National Natural Science Foundation of China (91540113 and 81570527 to X.F., 81502631 to Y.T), and 1.3.5 Project for Disciplines of Excellence, West China Hospital, Sichuan University.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molmet.2019.02.003.
Conflicts of interest
The authors declare no competing interests.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
figs1.

figs2.

figs3.

figs4.

figs5.
References
- 1.Tian Y., Xu J., Du X., Fu X. The interplay between noncoding RNAs and insulin in diabetes. Cancer Letters. 2018;419:53–63. doi: 10.1016/j.canlet.2018.01.038. [DOI] [PubMed] [Google Scholar]
- 2.Fu X., Dong B., Tian Y., Lefebvre P., Meng Z., Wang X. MicroRNA-26a regulates insulin sensitivity and metabolism of glucose and lipids. Journal of Clinical Investigation. 2015;125:2497–2509. doi: 10.1172/JCI75438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haeusler R.A., McGraw T.E., Accili D. Biochemical and cellular properties of insulin receptor signalling. Nature Reviews Molecular Cell Biology. 2018;19:31–44. doi: 10.1038/nrm.2017.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koh E.H., Chernis N., Saha P.K., Xiao L., Bader D.A., Zhu B. miR-30a remodels subcutaneous adipose tissue inflammation to improve insulin sensitivity in obesity. Diabetes. 2018;67:2541–2553. doi: 10.2337/db17-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tian Y., Peng B., Fu X. New ADCY3 variants dance in obesity etiology. Trends in Endocrinology and Metabolism TEM. 2018;29:361–363. doi: 10.1016/j.tem.2018.02.004. [DOI] [PubMed] [Google Scholar]
- 6.Reilly S.M., Saltiel A.R. Adapting to obesity with adipose tissue inflammation. Nature Reviews Endocrinology. 2017;13:633–643. doi: 10.1038/nrendo.2017.90. [DOI] [PubMed] [Google Scholar]
- 7.Lackey D.E., Olefsky J.M. Regulation of metabolism by the innate immune system. Nature Reviews Endocrinology. 2016;12:15–28. doi: 10.1038/nrendo.2015.189. [DOI] [PubMed] [Google Scholar]
- 8.Weinlich R., Oberst A., Beere H.M., Green D.R. Necroptosis in development, inflammation and disease. Nature Reviews Molecular Cell Biology. 2017;18:127–136. doi: 10.1038/nrm.2016.149. [DOI] [PubMed] [Google Scholar]
- 9.Wallach D., Kang T.B., Dillon C.P., Green D.R. Programmed necrosis in inflammation: toward identification of the effector molecules. Science (New York, N.Y.) 2016;352:aaf2154. doi: 10.1126/science.aaf2154. [DOI] [PubMed] [Google Scholar]
- 10.Sun L., Wang H., Wang Z., He S., Chen S., Liao D. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 11.Dondelinger Y., Declercq W., Montessuit S., Roelandt R., Goncalves A., Bruggeman I. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Reports. 2014;7:971–981. doi: 10.1016/j.celrep.2014.04.026. [DOI] [PubMed] [Google Scholar]
- 12.Quarato G., Guy C.S., Grace C.R., Llambi F., Nourse A., Rodriguez D.A. Sequential engagement of distinct MLKL phosphatidylinositol-binding sites executes necroptosis. Molecular Cell. 2016;61:589–601. doi: 10.1016/j.molcel.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takahashi N., Vereecke L., Bertrand M.J., Duprez L., Berger S.B., Divert T. RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature. 2014;513:95–99. doi: 10.1038/nature13706. [DOI] [PubMed] [Google Scholar]
- 14.Dannappel M., Vlantis K., Kumari S., Polykratis A., Kim C., Wachsmuth L. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature. 2014;513:90–94. doi: 10.1038/nature13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Newton K., Dugger D.L., Wickliffe K.E., Kapoor N., de Almagro M.C., Vucic D. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science (New York, N.Y.) 2014;343:1357–1360. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- 16.Mandal P., Berger S.B., Pillay S., Moriwaki K., Huang C., Guo H. RIP3 induces apoptosis independent of pronecrotic kinase activity. Molecular Cell. 2014;56:481–495. doi: 10.1016/j.molcel.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newton K. RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends in Cell Biology. 2015;25:347–353. doi: 10.1016/j.tcb.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 18.Galluzzi L., Kepp O., Chan F.K., Kroemer G. Necroptosis: mechanisms and relevance to disease. Annual Review of Pathology. 2017;12:103–130. doi: 10.1146/annurev-pathol-052016-100247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krysko O., Aaes T.L., Kagan V.E., D'Herde K., Bachert C., Leybaert L. Necroptotic cell death in anti-cancer therapy. Immunological Reviews. 2017;280:207–219. doi: 10.1111/imr.12583. [DOI] [PubMed] [Google Scholar]
- 20.Yuan J., Amin P., Ofengeim D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nature Reviews Neuroscience. 2019;20:19–33. doi: 10.1038/s41583-018-0093-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kondylis V., Pasparakis M. RIP kinases in liver cell death, inflammation and cancer. Trends in Molecular Medicine. 2019;25:47–63. doi: 10.1016/j.molmed.2018.10.007. [DOI] [PubMed] [Google Scholar]
- 22.Wu J., Huang Z., Ren J., Zhang Z., He P., Li Y. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Research. 2013;23:994–1006. doi: 10.1038/cr.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen W.D., Fu X., Dong B., Wang Y.D., Shiah S., Moore D.D. Neonatal activation of the nuclear receptor CAR results in epigenetic memory and permanent change of drug metabolism in mouse liver. Hepatology. 2012;56:1499–1509. doi: 10.1002/hep.25766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Degterev A., Hitomi J., Germscheid M., Ch'en I.L., Korkina O., Teng X. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nature Chemical Biology. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Conrad M., Angeli J.P., Vandenabeele P., Stockwell B.R. Regulated necrosis: disease relevance and therapeutic opportunities. Nature Reviews Drug Discovery. 2016;15:348–366. doi: 10.1038/nrd.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang S.H., Lee D.K., Shin J., Lee S., Baek S., Kim J. Nec-1 alleviates cognitive impairment with reduction of Abeta and tau abnormalities in APP/PS1 mice. EMBO Molecular Medicine. 2017;9:61–77. doi: 10.15252/emmm.201606566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin-Sanchez D., Fontecha-Barriuso M., Carrasco S., Sanchez-Nino M.D., Massenhausen A.V., Linkermann A. TWEAK and RIPK1 mediate a second wave of cell death during AKI. Proceedings of the National Academy of Sciences of the United States of America. 2018;115:4182–4187. doi: 10.1073/pnas.1716578115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pasparakis M., Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–320. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- 29.Cuchet-Lourenco D., Eletto D., Wu C., Plagnol V., Papapietro O., Curtis J. Biallelic RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science (New York, N.Y.) 2018;361:810–813. doi: 10.1126/science.aar2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gong Y.N., Guy C., Olauson H., Becker J.U., Yang M., Fitzgerald P. ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell. 2017;169:286–300. doi: 10.1016/j.cell.2017.03.020. e216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoon S., Kovalenko A., Bogdanov K., Wallach D. MLKL, the protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity. 2017;47:51–65. doi: 10.1016/j.immuni.2017.06.001. e57. [DOI] [PubMed] [Google Scholar]
- 32.Guo H., Kaiser W.J. ESCRTing necroptosis. Cell. 2017;169:186–187. doi: 10.1016/j.cell.2017.03.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.