

Abstract
The Fusarium oxysporum species complex (FOSC) is an economically important group of pathogenic filamentous fungi that are able to infect both animals and plants. Reverse genetic techniques, including gene disruption/deletion methods, to study these fungi are available although limitations exist resulting in decreased efficiency. Herein we describe a gene editing system developed using a F. oxysporum-optimized Cas9 ribonucleoprotein (RNP) and protoplast transformation method. The Cas9 protein and sgRNA were assembled to form a stable RNP in vitro and this complex was transferred into fungal protoplasts for gene editing with PEG-mediated transformation. In order to determine if the Cas9 RNP system is functional in the FOSC protoplasts and assess the efficacy of the system, two genes, URA5 and URA3, were selected for targeted disruption generating uracil auxotroph mutants that are resistant to 5-fluoroorotic acid, 5-FOA. In addition, a gene in a secondary metabolite biosynthetic cluster, the ortholog of BIK1, was mutated using this system and the maximum efficiency of this gene disruption was about 50%. Further analysis of the bik1 mutant confirmed that this polyketide synthase was involved in the synthesis of the red pigment, bikaverin. The mutants generated in this study displayed the strong expected phenotypes, demonstrating this F. oxysporum-optimized CRISPR/Cas9 system is stable and can efficiently disrupt the genes of interest.
Keywords: F. oxysporum, Cas9, gene editing, ribonucleoprotein, bikaverin
1. Introduction
Fungi in the genus Fusarium originated during the Cretaceous period and are currently represented by at least 20 species complexes containing isolates that are able to cause diseases in plants, animals, and humans (Geiser et al., 2013; O’Donnell et al., 2013). They are well known to produce a diverse array of secondary metabolites including mycotoxins that contaminate food and poison livestock leading to reduced production (Geiser et al., 2013; Hansen et al., 2012; O’Donnell et al., 2013). One of these groups, the Fusarium oxysporum species complex (FOSC), are soil-borne filamentous fungi that have an extensive host range from plants to animals. These isolates carry small supernumerary chromosomes that harbor host-specific virulence factors that are capable of being horizontally transferred to other FOSC isolates and increase host range (Ma et al., 2010; van Dam et al., 2017). In agriculture, F. oxysporum is an important plant pathogen that is able to cause significant economic damage on various plant species including tomato, banana, legumes, and cotton (Michielse and Rep, 2009). This fungus infects the root system of a susceptible host and eventually colonizes the xylem system, where it blocks the translocation of water and nutrients, resulting in chlorosis, necrosis, stunting, and wilting. Clinically, members of the FOSC are responsible for ~20% of the disseminated Fusarium infections termed fusariosis (Muhammed et al., 2013; Nucci and Anaissie, 2007) and have been implicated in several disease outbreaks such as nosocomial infections due to contaminated water supplies and contact lens associated keratitis (Chang et al., 2006; Edel-Hermann et al., 2016; O’Donnell et al., 2004).
Reverse genetic approaches utilizing gene deletion/disruption technologies have played an essential role in studies identifying the function of a gene. Recently, the CRISPR (clustered regularly interspaced short palindromic repeats)-Cas9 system, derived from the bacterial and archaeal immune system, has been developed into a powerful gene editing tool (Doudna and Charpentier, 2014; Hsu et al., 2014; Ran et al., 2013). Based on the highly efficient gene targeting editing and low off-target rates, this tool has become popular and has been widely used in model or non-model organisms such as fruit fly, zebrafish, Arabidopsis, yeast, mouse, and human cell cultures (Bassett et al., 2014; Jacobs et al., 2014; Jiang et al., 2013; Li et al., 2016; Platt et al., 2014; Shalem et al., 2014). The simple gene editing system includes two components, a single chimeric guide RNA (sgRNA) and the Cas9 endonuclease enzyme. The sgRNA consists of a protospacer sequence which targets the expected DNA region by base pairing, and is able to interact with Cas9 to become a stable complex. Once the base pairing has occurred, Cas9 cuts the double-stranded DNA in vivo, subsequently activating DNA repair mechanisms. One method of DNA repair involves random insertion or deletion of nucleotides at the DNA cleavage site (Non-Homologous End Joining, NHEJ). The NHEJ mechanism tends to repair the DNA incorrectly, and if this repair occurs in a gene coding region, the associated gene is likely to be inactivated. In contrast, when a template with homologous sequence is present, the cleaved DNA region could be repaired using the homologous sequence as a template. The main benefits of this gene disruption/deletion system are accurate gene modification and highly successful mutation rates (Doudna and Charpentier, 2014; Ran et al., 2013).
The development of CRISPR/Cas9 genome editing tools for fungal research has facilitated functional studies. Several gene deletion systems based on CRISPR/Cas9 have been developed in Saccharomyces cerevisiae, Candida albicans, Trichoderma, Aspergillus, Alternaria and Neurospora (DiCarlo et al., 2013; Liu et al., 2015; Matsu-Ura et al., 2015; Nodvig et al., 2015; Vyas et al., 2015; Wenderoth et al., 2017). While these cases showed successful gene disruptions, some limitations still exist. Trichoderma reesei was the first filamentous fungus used for CRISPR/Cas9 research, where Agrobacterium-mediated fungal transformation was used to insert a codon-optimized Cas9 gene randomly into the genome leading to a potential unintended mutation. Other CRISPR tools have been developed for filamentous fungi that are highly dependent on AMA1-derived plasmids (Nodvig et al., 2015; Pohl et al., 2016; Wenderoth et al., 2017). The AMA1 element is an inverted duplication of a sequence derived from Aspergillus nidulans, and it is able to enhance fungal transformation efficiency (Aleksenko and Clutterbuck, 1997). However, experimental evidence has shown that AMA1 plasmid integration occurred in some filamentous fungi and could lead to long-time retention in fungal transformants (Fierro et al., 1996). Additionally, after successful transformation positive colonies may have to be treated with single spore isolation for several rounds to remove the residual AMA1 plasmids (Nodvig et al., 2015; Wenderoth et al., 2017).
Using the Cas9 protein/sgRNA ribonucleoproteins (RNPs) to perform genome editing has several advantages compared with plasmid transformation and Cas9 mRNA/sgRNA transformation. A major advantage is transformation of Cas9 RNPs alleviates the possibility of integration of genetic material to a non-targeted region of the genome. Additionally, Cas9 and sgRNA are able to form a stable ribonucleoprotein in vitro, so there is less likelihood of RNA degradation compared with Cas9 mRNA/sgRNA transformation. Furthermore, this method is able to evaluate the target cleavage efficiency of different sgRNA in vitro, so a highly efficient sgRNA can be selected for research. This method has been widely used in different organisms such as mouse, human, grape, apple, tobacco, and Arabidopsis (Kim et al., 2014; Malnoy et al., 2016; Woo et al., 2015). In plants, an efficient protoplast transformation method for Cas9 RNPs has been developed and mutant individuals can be generated from the Cas9 RNP-modified protoplasts (Woo et al., 2015); however, few studies involving Cas9 RNP transformation for filamentous fungi have been published.
In this study, we developed and optimized a Cas9 RNP transformation procedure for gene editing in the ascomycete fungus, Fusarium oxysporum. A PEG-mediated transformation method was used to transfer Cas9 RNPs, which efficiently target the locus of interest, into fungal protoplasts. Several genes that have been well characterized in other fungi were disrupted in F. oxysporum with a hygromycin gene cassette using this system, generating mutants displaying strong expected phenotypes based on characterization of ortholgous genes. As a proof of concept, the gene encoding the polyketide synthase PKS4 was disrupted using this system and demonstrated this enzyme is responsible for production of the red pigment bikaverin, and in keeping with the established nomenclature of other Fusarium spp., refer to it as FoBIK1 (Wiemann et al., 2009).
2. Materials and methods
2.1. Fungal isolate and culture media.
All experiments were conducted using F. oxysporum f. sp. vasinfectum obtained from the Fungal Genetics Stock Center (FGSC #10442) (McCluskey et al., 2010). This fungus was cultured in Potato Dextrose Broth (PDB), PDB with agar, and minimal medium (M-100) agar plates (Stevens, 1974). All mutants were strictly determined on M-100 agar plates with a hygromycin concentration of 75 μg/mL or a 5-FOA concentration of 1.5 g/L. The wild type and mutant strains were cultured at room temperature (25 °C).
2.2. Sequence analysis and fluorescence microscopy.
Amino acid multiple sequence alignment analysis was conducted using Clustal Omega (Sievers et al., 2011), and the nuclear localization signal prediction accomplished using NLStradamus (Nguyen Ba et al., 2009). The subcellular localization of NLSH2B-eGFP was carried out using a Zeiss Axiovert 200 fluorescence microscope. The transformant containing NLSH2B-eGFP was cultured in PDB medium on a rotary shaker (160 rpm) at room temperature for three days. Before microscopic examination, the hyphae were stained with 4′,6-diamidino-2-phenylindole (DAPI) at a concentration of 1 μg/mL for 20 min in the dark. The excitation for the DAPI dye was 405 nm while the value for eGFP was 488 nm.
2.3. Plasmid construction.
The donor template DNA loci containing the hygromycin cassette were constructed using a NEBuilder® HiFi DNA Assembly Master Mix kit. All template plasmids were constructed in the background of the pUC19 plasmid with HindIII and BamHI restriction sites. For the construction of the pExFO-NLSH2BeGFP plasmid, the basic plasmid pExFO was constructed in the background of pGEM-T where three fragments, the hygromycin cassette including pgpdA promoter amplified from the plasmid pCWHyg1, the trpC promoter amplified from the plasmid pII99, and the tef1 terminator amplified from the plasmid pFFC332, were simultaneously inserted into the SphI and SacI enzyme sites in pGEM-T. Between the trpC promoter and the tef1 terminator, a multiple cloning site (MCS, HindIII-KpnI-NotI-PacI-SalI-SpeI-XbaI) were inserted. The NLS sequence from histone H2B and eGFP sequence were inserted into this MCS region for the construction of pExFO-NLSH2BeGFP. The assembled plasmids were sequenced to confirm they were correct and plasmids were extracted with the E.Z.N.A® plasmid DNA maxi kit (OMEGA, bio-tek) for PEG-mediated transformation.
The plasmid construction for the E.coli protein expression and purification was performed with the pHis-parallel1 plasmid (Sheffield et al., 1999). First, the nuclear localization sequence from the H2B gene of F. oxysporum was cloned from genomic DNA using primers NLSH2B_BamHIF and NLSH2B_SalIR and inserted into the BamHI and SalI enzyme sites of the pHis-parallel1 plasmid (pHis-paralell1-NLSH2B). The Cas9 gene was cloned from the plasmid pFFC332, and subsequently inserted between the SalI and HindIII sites of the pHis-paralell1-NLSH2B plasmid with the NEBuilder® HiFi DNA Assembly Master Mix kit. The plasmids were sequenced to confirm no mutations for inserted fragments. All the primers used for plasmid constructions and sequencing in this study are listed in Table A.1.
2.4. Cas9 protein purification.
The plasmid pHis-parallel1-NLSH2BCas9 was transformed into Rosetta™(DE3) strain which is able to highly express genes containing rare codons. The correctly transformed E. coli isolate was determined by PCR using with primers DetCas9F and DetCas9R (Table A.1). High expression of this protein in the E. coli containing the pHis-parallel1-NLSH2BCas9 plasmid was obtained by first culturing in 50 mL LB liquid medium for 14 h at 37 °C with 100 μg/mL ampicillin. The cultured E. coli was used to inoculate 1.5 L of fresh LB liquid medium at a dilution of 1:100. When the OD600 value of the E. coli cell culture concentration reached 0.6, IPTG was added to a final concentration of 0.3 mM and the culture incubated for an additional 5 h at room temperature (25 °C) at 160 rpm on a rotary shaker. After determining the proper protein expression using a SDS-PAGE gel, the culture was centrifuged at 13000 rpm for 15 min and the pellet suspended in 30 mL lysis buffer (20 mM NaH2PO4, 300 mM NaCl, 1 mM imidazole, 1% TritonX-100, 1 mM PMSF, pH 7.5) and lysed in an ultrasonic bath (15 s + 15 s, 10 min). The mixture was centrifuged at 13000 rpm, 4 °C for 15 min, and the supernatants were filtered with a 0.22 μm filter, and subsequent protein purification conducted using HisPur™ Ni-NTA Chromatography Cartridges (Thermo Fisher Scientific). The eluted Cas9 protein was further dialyzed against the Cas9 buffer (20 mM NaH2PO4, 300 mM NaCl, 10% glycerol, pH 7.5) for 6–8 h. The dialyzed Cas9 protein was centrifuged and the associated undissolved protein was removed. The Cas9 protein was further concentrated with a 100 K centrifugal filter to a final concentration of 4 mg/mL. The Cas9 nuclease cleavage assay in vitro should be conducted immediately. The final purified Cas9 protein was aliquoted into PCR tubes and stored at −30°C for future use. There was no significant difference in cleavage activity of the purified Cas9 protein after a year at −30°C.
2.5. sgRNA design and in vitro Cas9 nuclease assay.
The sgRNA for gene mutation was designed with the following parameters: 1) the sgRNA cleavage site was located close to the 5′ end of the gene coding region; 2) the protospacer and PAM sequence was (N)20NGG, and if the beginning nucleotide of the target sequence lacked a G, at least one G was added after the T7 promoter; 3) similarity to the protospacer and PAM sequence was searched by BLASTN to the F. oxysporum genome to avoid cleavage at other loci; and 4) the sgRNA forward primer (T7 promoter-(N)20-GTTTTAGAGCTAGAAATAGCAAG) and the sgRNA universal primer (Table A.1) were used to generate the sgRNA DNA templates by PCR, and the sgRNA synthesized using the EnGen® sgRNA Synthesis kit (New England Biolabs).
For the in vitro cleavage assay mediated by Cas9 with different sgRNAs, a PCR fragment ranging from 1000 bp to 3000 bp was first amplified and purified from the agarose gel with E.Z.N.A® Gel Extraction Kit (OMEGA, bio-tek). The purified Cas9 protein was diluted to a concentration of 200 ng/μL. The cleavage assay was conducted in a system with 20 μL total volume (1 μL diluted purified Cas9 protein, 40 ng sgRNA, 1× Cas9 Nuclease Reaction Buffer, 3 μL 20 nM PCR fragment, DEPC-treated water). All items were added together and incubated 37 °C for 1 h. The final products were analyzed on a 0.8% agarose gel and the associated cleavage efficiency was evaluated with the software, ImageJ (https://imagej.nih.gov/ij/). All sgRNAs and the associated targeted genes used in this study are listed in Table A.2.
2.6. PEG-mediated fungal transformation.
PEG-mediated fungal transformation was conducted according as previously described with slight modifications (Coleman et al., 2011b). About 107 conidia were inoculated into 100 mL fresh PDB liquid medium and allowed to germinate at 28 °C at 160 rpm overnight. The fungal germlings were collected and treated with a mixture of 10 mg/mL Driselase (Sigma) and 15 mg/mL β-Glucanase (Sigma) suspended in 0.7M NaCl for 2–3 h. Fungal protoplasts were collected and washed twice with SuTC buffer. The fungal protoplasts were diluted into a concentration of 2 × 107/mL with SuTC buffer, and 200 μL of the protoplast solution was used for each transformation.
The Cas9 RNPs or template donor DNA was prepared during generation of the protoplasts where the Cas9 RNPs were made as follows: a 1:1 mole ratio of Cas9: sgRNA was added into a 50 μL total volume with 5 μL 10× Cas9 Nuclease Reaction Buffer and DEPC-treated water. This mixture was incubated in a 37 °C water bath for 20 min. 200 µL of fungal protoplasts were mixed with 50 μL Cas9 RNPs or donor DNA (template) at room temperature for 20 min. An equal volume of 60 % PEG was added into the above system and incubated at room temperature for 20 min. The total system was transferred into a 15 mL tube containing 5 mL TB3 and placed on a rotary shaker (160 rpm at room temperature) for 18–24 h. The regenerated fungal protoplasts were mixed with the selective medium and poured into sterile plates. For screening ura3 and ura5 mutants with only Cas9 RNPs transformation, the selective plates with uracil, uridine, and 2 g/L 5-FOA were used. For the ura5 mutant generation with Cas9 RNPs and donor DNA transformation, the selective plates contained uracil, uridine, and a final concentration of 150 μg/mL hygromycin. The Fobik1 mutant selection was accomplished using selective plates with 150 μg/mL hygromycin. All transformants were visible in 3 ~ 5 days after plating on selective media. The transformants were then transferred onto the minimum nutrient agar medium (M100) with the associated antibiotics for further determination.
Three pairs of primers were used to detect the hygromycin cassette location for HR-directed mutant determination. The first pair (HinF + HinR) was used to detect the presence of the hygromycin phosphotransferase gene. For HR-directed ura5 mutants, three pairs of primers (HoutF+ FoURA5TR, FoURA5TF + HoutR and FoURA5TF + FoURA5TR) were used to determine the insertion location of the hygromycin phosphotransferase gene. Similarly, Det_bik1g1F + Det_bik1g1R and HoutF+ Det_bik1g2R were used to determine HR-directed Fobik1 gene mutants.
2.7. HPLC and MS analysis.
The wild type and Fobik1 mutants were inoculated into the fresh PDB liquid medium and grown on a rotary shaker (160 rpm) at room temperature for 7 days. The culture was centrifuged at 13000 rpm for 10 min to remove conidia and hyphae. The supernatant was treated with a 0.22 μm filter. The supernatant medium from wild type and the Fobik1 mutant was fractionated on a Sonoma phenyl-hexyl 10-μm 100-Å (25 cm x 4.6 mm) HPLC column (ES Industries, West Berlin, NJ, USA) which was equilibrated in 1% formic acid and acetonitrile (ACN). The HPLC programs were set as follows: a linear gradient from 20% to 50% ACN was applied over 15 minutes, and a linear gradient from 50% to 75% ACN was used over 5 minutes. The column was washed with 100% ACN for 5 minutes and then was re-equilibrated as above. Fractions were collected and submitted to MS analysis which was performed on a Bruker Micro-TOF MS with flow injection.
3. Results and Discussion
3.1. Identification of an efficient endogenous nuclear localization sequence (NLS) from F. oxysporum.
The Cas9 RNPs must enter into the nucleus in order to bind to the targeted DNA regions. The classical nuclear localization sequence (NLS) SV40 has been shown to be functional in many organisms (Liu et al., 2015), however this NLS was not suitable for F. oxysporum and was unable to efficiently translocate GFP into the nucleus (data not shown), similar to observed results with other organisms such as Phytopthora sojae (Fang and Tyler, 2016). In order to facilitate Cas9 entering the F. oxysporum nucleus, the endogenous nuclear localization sequence from histone H2B was fused to the N-terminus of Cas9. Histone H2B is conserved in eukaryotes and involved in the assembly of the nucleosomes (Mosammaparast et al., 2001). The histone H2B NLS amino acid sequence was highly conserved among Fusarium spp., which indicates that this system may be applicable for use in other Fusarium species (Fig. A.1).
Bioinformatic analysis revealed the F. oxysporum histone H2B contained a NLS at its N-terminus (NLSH2B, aa: 6–45). The middle and C-terminus amino acids of histone H2B were highly conserved among a diverse group of organisms including other fungi, plants, and metazoans, but the NLS sequences of these organisms has evolved divergently (Fig. 1A). In order to confirm the predicted NLS of histone H2B was sufficient to localize a protein to the nucleus of F. oxysporum, the first 54 amino acids including the entire NLS sequence were fused to the N-terminus of eGFP under the constitutive promoter of trpC (pExFO-NLSH2BeGFP). Transformants of F. oxysporium expressing the NLSH2B-eGFP construct contained eGFP localized to the nuclei of the cells (Fig. 1B), indicating the NLSH2B was suitable to engineer a protein to be translocated into the nucleus.
Fig. 1.

Determination of an endogenous NLS from the F. oxysporum histone H2B gene. (A) H2B amino acid multiple sequence alignment among F. oxysporum (Fo), yeast, Arabidopsis, human and Drosophila. (B) subcellular localization of F. oxysporum hyphae expressing NLSH2BeGFP showing it localizes to the nucleus. Scale bars, 10 μm.
3.2. Design and protein purification of NLSH2BCas9 and in vitro cleavage assay.
The endogenous NLS from the F. oxysporum histone H2B gene was fused to the N-terminus of the Cas9 gene and cloned into an E. coli protein expression plasmid (pHis parallel1-NLSH2BCas9) which was subsequently transformed into the E. coli Rosetta™ (DE3) strain for protein expression and purification. The resulting recombinant protein with a molecular weight of approximately 165 kDa was soluble in water and reached a yield at least 4 mg per liter of bacterial culture (Fig. 2A). The ability of the purified Cas9 protein to efficiently cleave at a specified target site was evaluated in vitro. A ~2.1 kb PCR fragment containing the coding region of the F. oxysporum URA5 gene was mixed with a single guide RNA targeting this locus and the Cas9 protein. The NLSH2BCas9-FoURA5sgRNA complex was able to cleave almost all of the template in vitro within an hour and the lengths of cleaved fragments were of the expected size, supporting that the NLSH2B sequence fusion had no significant influence on the Cas9 endonuclease cleavage activity (Fig. 2B).
Fig. 2.
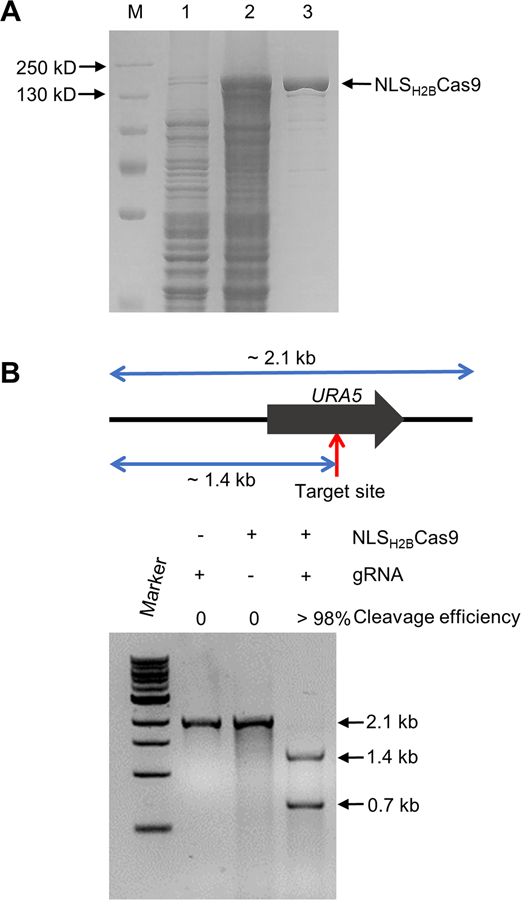
Protein purification and in vitro cleavage activity assay. (A) SDS-PAGE gel depicting protein expression and purification conditions. Lane 1 represents total protein before inducing; Lane 2 total protein after addition of 0.3 mM IPTG for 5 h; Lane 3 represents about 5 μg purified NLSH2BCas9 protein. (B) Top: diagram depicting the template length, the sgRNA cleavage site, and the lengths of expected fragments after cleavage with Cas9; Bottom: assessment of the cleavage efficiency by gel electrophoresis after incubation at 37 °C for one hour.
3.3. A PEG-mediated transformation method can transfer NLSH2BCas9 RNPs into fungal protoplasts.
Recently, Cas9 RNPs have been transferred into plant protoplasts for gene-editing (Malnoy et al., 2016; Woo et al., 2015). This system has at least two advantages in filamentous fungi 1) fungal protoplasts are easier to be regenerated into individual transformants (compared to plants) and 2) Cas9 RNPs will be degraded in the protoplasts, which further minimizes the potential influence from the Cas9 RNPs being maintained in the cell. A RNP mediated transformation method has been developed for two fungi, Aspergillus fumigatus and Penicillium chrysogenum (Al Abdallah et al., 2017; Pohl et al., 2016). A PEG-mediated transformation method was developed for the F. oxysporum optimized NLSH2BCas9. The URA5 gene encoding orotate phosphoribosyltransferase involved in pyrimidine biosynthesis was initially targeted, as mutants of the ortholog in S. cerevisiae and other fungi generates uracil auxotrophs and allows for direct selection on media containing 5-fluoro-orotic acid (5-FOA) (de Montigny et al., 1989). Protoplasts transformed with the NLSH2BCas9-FoURA5sgRNA RNPs should result in cleavage at the target site in the URA5 coding region and subsequent inactivation of the gene due to NHEJ repair. Seven transformants were generated that were able to grow on 5-FOA selective media containing uracil and uridine. As Cas9 RNPs cleave the dsDNA between the last third and fourth nucleotide at the region targeted by the protospacer element, this region was sequenced to characterize the resulting mutations in the transformants. All these ura5 mutants contained short nucleotide deletions ranging in size from a single nucleotide to −123 bp in the target region of the guide RNA (Fig. 3). The ura5 mutants were uracil auxotrophs and are able to grow on media containing 5-FOA (Fig. 4).
Fig. 3.

Disruption of two F. oxysporum genes, URA5 and URA3, using the optimized Cas9 RNP transformation system. (A-C) characterization of individual NHEJ-mediated mutants (A) The multiple sequence alignment of target regions of seven randomly chosen ura5 mutants is shown. (B) DNA sequences of the wild-type isolate and three ura5 mutants. (C) The multiple sequence alignment of target regions of two randomly chosen ura3 mutants is shown. (D) URA5 homologous-directed repair disruption chart depicting insertion site of hygromycin phosphotransferase.
Fig. 4.
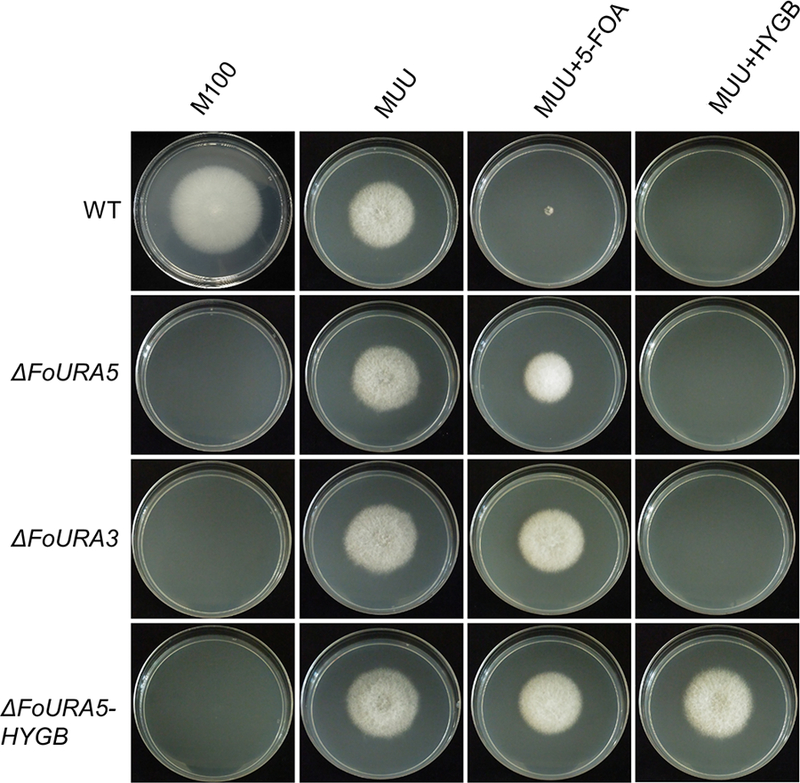
The phenotypes of ura5 and ura3 mutants obtained by NHEJ and HDR methods. About 200 conidia were positioned onto the center of the plates, which were cultured at room temperature. The phenotypes were evaluated after six days. MUU represents M100 medium supplemented with uracil and uridine. The concentrations of 5-FOA and hygromycin are 1.5 g/L and 100 μg/mL, respectively.
In order to further confirm that PEG-mediated transformation of Cas9 RNPs can be used in F. oxysporum protoplasts another sgRNA was generated that targets the homologous gene of URA3 encoding orotidine 5’-phosphate decarboxylase. This gene is involved in de novo UMP biosynthesis and, similar to ura5 mutants, ura3 mutants are uracil auxotrophs and can grow on minimum media containing 5-FOA (Fig. 4) (Boeke et al., 1984). The targeted locus of two randomly selected ura3 transformants were sequenced and contained short deletions at the Cas9 cleavage site (Fig. 3C). Overall, the PEG-mediated transformation of Cas9 RNPs into F. oxysporum protoplasts was successful and the transformed particles can cleave the genomic DNA at the target site generating a mutation via an error-prone DNA repair mechanism. Interestingly, among the sequenced transformants, several of the mutants generated using this gene editing tool contained a single nucleotide deletion instead of double or four nucleotide deletions, which suggests the NHEJ repair system of F. oxysporum prefers the removal of a single nucleotide at the cleavage site causing a frame-shift mutation in the coding sequence.
3.4. Homology directed repair (HDR) can efficiently disrupt targeted genes by insertion of a dominant selectable marker.
To further extend the protocol, experiments were conducted to assess whether donor DNA containing a selective marker and the NLSH2BCas9 RNPs are able to promote CRISPR–Cas9 induced homologous recombination. The NLSH2BCas9-FoURA5sgRNA transformation system used for NHEJ mutation above (Fig. 3) was selected as it was previously confirmed to have activity at the specified target site in URA5. The donor DNA contained the gene encoding hygromycin phosphotransferase (hph), conferring resistance to hygromycin, and had ~ 600 bp of homologous sequence flanking both sides of the cleavage site in the URA5 gene (Fig. 3D). Mutants should grow on the selective medium including uracil, uridine, and hygromycin (Fig. 4). Of the 14 transformants generated, three contained the hph cassette integrated at the gRNA target site (21.4%; Table 1). The ura5 mutants are unable to grow on minimal medium containing hygromycin without uracil and uridine and were resistant to 5-FOA, supporting the integration of the hygromycin resistance cassette at the targeted cleavage site in the F. oxysporum URA5 gene (Fig. 4).
Table 1.
Mutation frequencies of homologous directed repair using Cas9 RNPs
| Gene | Donor DNAa | Cas9/sgRNAa | Pick No. | Positive No. | Frequency (%) |
|---|---|---|---|---|---|
| URA5 | 6 | 0/0 | 32 | 0 | 0 |
| 6 | 10/2 | 14 | 3 | 21.4 | |
| bik1 | 6 | 0/0 | 27 | 0 | 0 |
| 6 | 5/1 | 20 | 4 | 20 | |
| 6 | 10/2 | 13 | 7 | 53.8 | |
| 6 | 15/3 | 25 | 12 | 48 | |
| 6 | 20/4 | 21 | 2 | 9.5 | |
number of μg used of each per reaction.
3.5. Cas9 RNP-mediated disruption of a gene encoding a polyketide synthase in a secondary metabolite biosynthetic gene cluster.
Some filamentous fungi including Aspergillus, Penicillium, and Alternaria, produce a pigment on certain agar medium during the early growth phase (Nodvig et al., 2015; Pohl et al., 2016; Wenderoth et al., 2017), and detection of these pigments have been used as a marker for the rapid development of CRISPR methodologies in these fungi. F. oxysporum is able to produce red pigmented compounds during the later stages of growth. While the gene cluster responsible for the pigment production has not been functionally characterized in F. oxysporum, the biosynthetic genes responsible for a red pigment (bikaverin) biosynthesis in the closely related fungus Fusarium fujikuroi are encoded in a large secondary metabolite gene cluster (Wiemann et al., 2009). Bioinformatic analysis showed that there is a similar secondary metabolite cluster that may be involved in the synthesis of bikaverin in F. oxysporum. This secondary metabolite cluster is composed of six genes, where the ortholog to the BIK1 gene encodes a putative polyketide synthase that could be essential for the biosynthesis of bikaverin. As this method can mediate donor DNA and NLSH2BCas9 RNPs to enter F. oxysporum protoplasts and trigger the CRISPR–Cas9 induced homology directed repair, the bik1 gene was selected for optimizing our CRISPR system.
In order to confirm whether the putative BIK1 gene from F. oxysporum (FOTG_08225) was involved in bikaverin synthesis, a sgRNA site was chosen with a location close to the 5′-terminus of the coding region in the first exon (Fig. 5A). The PCR template cleavage efficiency was close to 100% in vitro, demonstrating the chosen sgRNA had high cleavage activity with NLSH2BCas9 (Fig. 5B). In order to optimize this CRISPR system various concentrations of the assembled NLSH2BCas9-Fobik1sgRNA RNPs were used for transformation. In the absence of the Cas9 RNPs in the transformation system, no bik1 F. oxysporum mutants were generated; however, using concentrations of Cas9/sgRNA between 10/2 (μg/μg) and 15/3 (μg/μg) resulted in generation of Δbik1 transformants of approximately 50% (Table 1). Using a Cas9/sgRNA concentration higher than 15/3 (μg/μg) was unable to further increase the frequency of desired mutants. Six randomly selected Fobik1 mutants were unable to produce the red pigment when cultured in conducive conditions such as PDB medium for 7 days (Fig. 5C), supporting this secondary metabolite gene cluster is responsible for the synthesis of the red pigment. The identity of the red pigment of F. oxysporum was confirmed to be bikaverin by fractionation on HPLC using a phenyl hexyl column and mass spectrometry (MS). The HPLC profile monitoring absorbance at 400 nm indicated the wide-type isolate contained multiple peaks with one major peak at a retention time (RT) of 22 minutes, that appeared different to two large peaks seen under highly similar conditions in F. fujikuroi (Fig. 5D, Fig. A.2). Previous results showed F. fujikuroi synthesized two similarly structured compounds, nor-bikaverin and bikaverin (Wiemann et al., 2009). The peak at RT~22 min from the medium of the wild-type isolate and the associated region from the medium of Fobik1 mutant were collected for MS analysis. A large peak with a molecular weight of 383 was present in the extract of the wild-type F. oxysporum isolate, which was absent in the fraction from the Fobik1 mutant (Fig. A.2). The exact mass determination and isotope ratio suggest a best fit to C20H14O8 corresponded to bikaverin and initial MS/MS fragmentation of the molecule confirmed the identity as bikaverin (ESI-MS m/z 383: MS/MS (40 eV) m/z (%): 270 (100), 340 (70), 256 (60), 297 (25)). This identification combined with the absence of this secondary metabolite in the mutant demonstrates the disrupted gene (FoBIK1) is responsible for the synthesis of bikaverin in F. oxysporum.
Fig. 5.

FoBIK1 gene encoding a polyketide synthase in a secondary metabolite biosynthetic cluster is the core enzyme responsible for the biosynthesis of a red pigment. (A) The BIK1 gene structure, the location of the sgRNA in exon 1, and the construction of the associated HDR template. (B) The cleavage efficiency of the sgRNA for bik1 in vitro after incubation at 37 °C for one hour. (C) The phenotypes of three replicative wild type and six randomly chosen bik1 mutants cultured for seven days. (D) HPLC-DAD analysis of red pigment production between wild type and bik1 mutants.
This CRISPR/Cas9 RNP transformation system is an additional molecular tool to study members of the FOSC. Unlike currently used transformation procedures such as the Agrobacterium tumefaciens-mediated and protoplast transformation methods (Coleman et al., 2011a; Mullins et al., 2001), when utilizing the CRISPR/Cas9 RNP system only short flanking sequences are required for homologous recombination, thereby bypassing the need to construct vectors with >1 kb of homologous sequence flanking the selectable marker. Additionally, this system has the potential to be used to target multiple copies of a single gene if the entire PAM sequence and a significant proportion of the protospacer region between the copies are conserved. While the NLSH2BCas9 protein needs to be expressed and purified in vitro prior to transformation, the advantage of using the purified Cas9 protein is that it does not rely on the integration and expression of the Cas9 gene within the fungal nucleus, and therefore there is less of a chance of unforeseen effects due to the random integration of the Cas9 gene within the fungal genome.
In summary, we have developed an approach to transfer Cas9 RNPs and/or donor DNA into fungal protoplasts for gene editing as demonstrated by generating several mutants having the expected phenotypes (Fig. 6). In addition, we showed experimental evidence that the red-pigmented compound produced by F. oxysporum is bikaverin. This is the first report of a CRISPR system that could be efficiently used to generate mutations in genes of interest in a member of the FOSC and may facilitate functional studies concerning the broad host range, secondary metabolite production, and supernumerary chromosomes of these fungi. Importantly, the high degree of similarity of the NLSH2B sequence between all the fusaria and the NLSH2B sequence that was fused to the Cas9 protein suggests this transformation system may be applicable to the other members within the genus Fusarium.
Fig. 6.
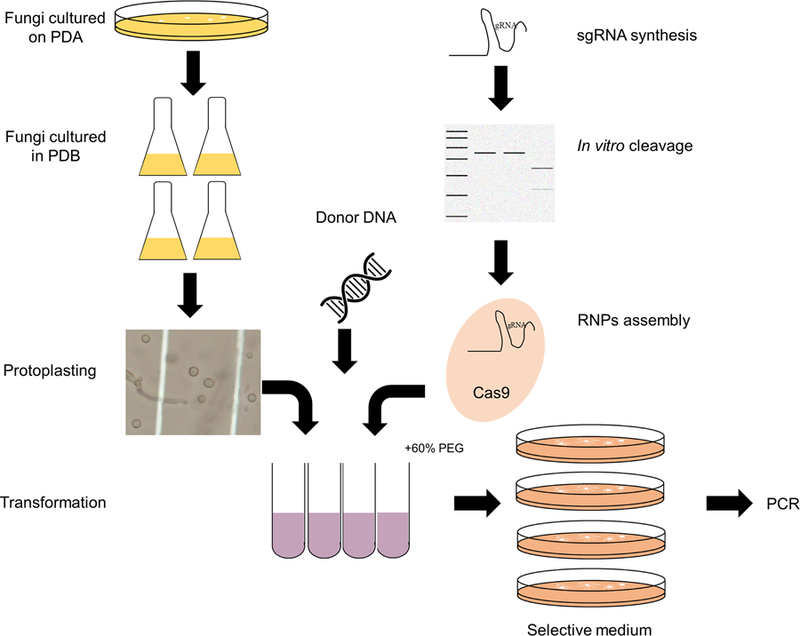
Schematic diagram of Cas9 RNPs-directed delivery method to F. oxysporum protoplasts to efficiently generate gene disruptions/deletions.
Supplementary Material
Fig. A.1. Histone H2B amino acid sequence alignment of different Fusarium species. Abbreviations for specific species are as follows: Fo, F. oxysporum; Fg, F. graminearum; Fv, F. verticillioides; Ff, F. fujikuroi; Fa, F. acuminatum; Fs, F. solani; Fpse, F. pseudograminearum; Flan, F. langsethiae; Fman, F. mangiferae; Fpro, F. proliferatum; and Fpoa, F. poae.
Fig. A.2. Analysis of the compounds in the supernatant of wild type (WT) and a ΔFobik1 transformant by mass spectrometry. The major peak found in the WT at 383.0722 corresponds to bikaverin and is absent in the Fobik1 mutant profile.
Acknowledgements
This work was supported by the National Institute of Allergy and Infectious Diseases (K22AI100983), the Alabama Agricultural Experiment Station, and the Hatch program of the National Institute of Food and Agriculture, USDA. We thank Melissa Boersma at the Auburn University Mass Spectrometry Facility and Uffe Mortensen for providing the plasmid containing Cas9.
References
- Al Abdallah Q, et al. , 2017. A simple and universal system for gene manipulation in Aspergillus fumigatus: in vitro-assembled Cas9-guide RNA ribonucleoproteins coupled with microhomology repair templates. mSphere 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleksenko A, Clutterbuck AJ, 1997. Autonomous plasmid replication in Aspergillus nidulans: AMA1 and MATE elements. Fungal Genet. Biol 21, 373–87. [DOI] [PubMed] [Google Scholar]
- Bassett AR, et al. , 2014. Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell Rep 4, 220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeke JD, et al. , 1984. A positive selection for mutants lacking orotidine-5’-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol. Gen. Genet 197, 345–6. [DOI] [PubMed] [Google Scholar]
- Chang DC, et al. , 2006. Multistate outbreak of fusarium keratitis associated with use of a contact lens solution. JAMA 296, 953–963. [DOI] [PubMed] [Google Scholar]
- Coleman JJ, et al. , 2011a. Characterization of the gene encoding pisatin demethylase (FoPDA1) in Fusarium oxysporum. Mol. Plant-Microbe Interact 24, 1482–1491. [DOI] [PubMed] [Google Scholar]
- Coleman JJ, et al. , 2011b. An ABC transporter and a cytochrome P450 of Nectria haematococca MPVI are virulence factors on pea and are the major tolerance mechanisms to the phytoalexin pisatin. Mol. Plant-Microbe Interact 24, 368–376. [DOI] [PubMed] [Google Scholar]
- de Montigny J, et al. , 1989. Structure and expression of the URA5 gene of Saccharomyces cerevisiae. Mol. Gen. Genet 215, 455–462. [DOI] [PubMed] [Google Scholar]
- DiCarlo JE, et al. , 2013. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res 41, 4336–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA, Charpentier E, 2014. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1258096. [DOI] [PubMed] [Google Scholar]
- Edel-Hermann V, et al. , 2016. A clonal lineage of Fusarium oxysporum circulates in the tap water of different French hospitals. Appl. Environ. Microbiol 82, 6483–6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Tyler BM, 2016. Efficient disruption and replacement of an effector gene in the oomycete Phytophthora sojae using CRISPR/Cas9. Mol. Plant Pathol 17, 127–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierro F, et al. , 1996. Autonomously replicating plasmids carrying the AMA1 region in Penicillium chrysogenum. Curr. Genet 29, 482–489. [DOI] [PubMed] [Google Scholar]
- Geiser DM, et al. , 2013. One fungus, one name: defining the genus Fusarium in a scientifically robust way that preserves longstanding use. Phytopathol 103, 400–408. [DOI] [PubMed] [Google Scholar]
- Hansen FT, et al. , 2012. Quick guide to polyketide synthase and nonribosomal synthetase genes in Fusarium. Int. J. Food Microbiol 155, 128–136. [DOI] [PubMed] [Google Scholar]
- Hsu PD, et al. , 2014. Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs JZ, et al. , 2014. Implementation of the CRISPR-Cas9 system in fission yeast. Nat. Commun 5, 5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang WZ, et al. , 2013. Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res 41, e188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, et al. , 2014. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res 24, 1012–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MY, et al. , 2016. Zebrafish genome engineering using the CRISPR-Cas9 system. Trends Genet 32, 815–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, et al. , 2015. Efficient genome editing in filamentous fungus Trichoderma reesei using the CRISPR/Cas9 system. Cell Discov 1, 15007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L-J, et al. , 2010. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464, 367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malnoy M, et al. , 2016. DNA-free genetically edited grapevine and apple protoplast using CRISPR/Cas9 ribonucleoproteins. Front. Plant Sci 7, 1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsu-Ura T, et al. , 2015. Efficient gene editing in Neurospora crassa with CRISPR technology. Fungal Biol. Biotechnol 2, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCluskey K, et al. , 2010. The Fungal Genetics Stock Center: a repository for 50 years of fungal genetics research. J. Biosci 35, 119–126. [DOI] [PubMed] [Google Scholar]
- Michielse CB, Rep M, 2009. Pathogen profile update: Fusarium oxysporum. Mol. Plant Pathol 10, 311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosammaparast N, et al. , 2001. Nuclear import of histone H2A and H2B is mediated by a network of karyopherins. J. Cell Biol 153, 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhammed M, et al. , 2013. Fusarium infection: report of 26 cases and review of 97 cases from the literature. Medicine (Baltimore) 92, 305–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins ED, et al. , 2001. Agrobacterium-mediated transformation of Fusarium oxysporum: an efficient tool for insertional mutagenesis and gene transfer. Phytopathol 91, 173–180. [DOI] [PubMed] [Google Scholar]
- Nguyen Ba AN, et al. , 2009. NLStradamus: a simple Hidden Markov Model for nuclear localization signal prediction. BMC Bioinformatics 10, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nodvig CS, et al. , 2015. A CRISPR-Cas9 system for genetic engineering of filamentous fungi. PLoS One 10, e0133085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nucci M, Anaissie E, 2007. Fusarium infections in immunocompromised patients. Clin. Microbiol. Rev 20, 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell K, et al. , 2013. Phylogenetic analyses of RPB1 and RPB2 support a middle Cretaceous origin for a clade comprising all agriculturally and medically important fusaria. Fungal Genet. Biol 52, 20–31. [DOI] [PubMed] [Google Scholar]
- O’Donnell K, et al. , 2004. Genetic diversity of human pathogenic members of the Fusarium oxysporum complex inferred from multilocus DNA sequence data and amplified fragment length polymorphism analyses: evidence for the recent dispersion of a geographically widespread clonal lineage and nosocomial origin. J. Clin. Microbiol 42, 5109–5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt RJ, et al. , 2014. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl C, et al. , 2016. CRISPR/Cas9 based genome editing of Penicillium chrysogenum. ACS Synth. Biol 5, 754–764. [DOI] [PubMed] [Google Scholar]
- Ran FA, et al. , 2013. Genome engineering using the CRISPR-Cas9 system. Nature Protocols 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, et al. , 2014. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield P, et al. , 1999. Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein Expr. Purif 15, 34–39. [DOI] [PubMed] [Google Scholar]
- Sievers F, et al. , 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens RB, 1974. Mycology Guidebook University of Washington Press, Seattle. [Google Scholar]
- van Dam P, et al. , 2017. A mobile pathogenicity chromosome in Fusarium oxysporum for infection of multiple cucurbit species. Sci. Rep 7, 9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas VK, et al. , 2015. A Candida albicans CRISPR system permits genetic engineering of essential genes and gene families. Sci. Adv 1, e1500248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenderoth M, et al. , 2017. Establishment of CRISPR/Cas9 in Alternaria alternata. Fungal Genet. Biol 101, 55–60. [DOI] [PubMed] [Google Scholar]
- Wiemann P, et al. , 2009. Biosynthesis of the red pigment bikaverin in Fusarium fujikuroi: genes, their function and regulation. Mol. Microbiol 72, 931–46. [DOI] [PubMed] [Google Scholar]
- Woo JW, et al. , 2015. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nat. Biotechnol 33, 1162–4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. A.1. Histone H2B amino acid sequence alignment of different Fusarium species. Abbreviations for specific species are as follows: Fo, F. oxysporum; Fg, F. graminearum; Fv, F. verticillioides; Ff, F. fujikuroi; Fa, F. acuminatum; Fs, F. solani; Fpse, F. pseudograminearum; Flan, F. langsethiae; Fman, F. mangiferae; Fpro, F. proliferatum; and Fpoa, F. poae.
Fig. A.2. Analysis of the compounds in the supernatant of wild type (WT) and a ΔFobik1 transformant by mass spectrometry. The major peak found in the WT at 383.0722 corresponds to bikaverin and is absent in the Fobik1 mutant profile.