

Abstract
The last three decades have seen a dwindling number of novel antibiotic classes approved for clinical use and a concurrent increase in levels of antibiotic resistance, necessitating alternative methods to combat the rise of multi-drug resistant bacteria. A promising strategy employs antibiotic adjuvants, non-toxic molecules that disarm antibiotic resistance. When co-dosed with antibiotics, these compounds restore antibiotic efficacy in drug-resistant strains. Herein we identify derivatives of tryptamine, a ubiquitous biochemical scaffold containing an indole ring system, capable of disarming colistin resistance in the Gram-negative bacterial pathogens Acinetobacter baumannii, Klebsiella pneumoniae, and Escherichia coli while having no inherent bacterial toxicity. Resistance was overcome in strains carrying endogenous chromosomally-encoded colistin resistance machinery, as well as resistance conferred by the mobile colistin resistance – 1 (mcr-1) plasmid-borne gene. These compounds restore a colistin minimum inhibitory concentration (MIC) below the Clinical Laboratory Sciences Institute (CLSI) breakpoint in all resistant strains.
Graphical Abstract
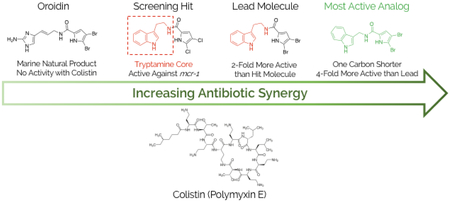
1. Introduction
The proliferation of antibiotic resistant bacteria threatens a collapse of modern medicine. As multi-drug resistant (MDR) bacterial strains continue to grow in both strength and number while the antibiotic clinical arsenal remains stagnate, a “Post-Antibiotic World” has become increasingly tangible. Antibiotic resistant infections currently claim more than 700,000 lives world-wide each year.1 These infections are most often caused by one or more of the ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species). Efforts to combat these six agents have been met with limited success, as only two novel antibiotic classes, lipopeptides and oxazolidinones, have been deployed clinically in the last two decades. Both are active only against Gram-positive bacteria, and offer a transient solution as bacterial resistance to each has been documented.2 Given the lack of treatment options to combat MDR Gram-negative bacteria, colistin (polymyxin E), a decades-old cationic polypeptide, has reemerged as salvage therapy for MDR Gram-negative bacterial infections despite causing severe nephrotoxicity.3 This “last resort” antibiotic is not exempt from resistance development, as numerous primary clinical isolates have shown resistance that far exceeds the Clinical Laboratory Standards Institute (CLSI) breakpoint for safe and effective treatment.4
Colistin elicits bacterial cell lysis by displacing divalent cations chelated by phosphate moieties on lipid A, a component of lipopolysaccharide (LPS), which destabilizes the outer membrane leading to envelope rupture and cell death.5 Bacteria become resistant to colistin by modifying lipid A in a number of ways, including modification of the phosphate group with positively charged residues. Addition of aminoarabinose, galactosamine, and phosphoethanolamine residues to the terminal phosphate moieties of lipid A, the membrane anchor of lipopolysaccharide (LPS) has been documented.6 These modifications all impart decreased electrostatic attraction toward the polycationic polymyxin, leading to drug resistance. This resistance mechanism is typically evolved under intense selection pressure, comes at a fitness cost, and is controlled and encoded by endogenous genomic material.7 The latter of the three modifications, however, can also be acquired from the mobile colistin resistance – 1 (mcr-1) or related genes. These compact, plasmid-borne transcripts can be rapidly disseminated amongst pathogenic bacteria.8 While mcr-1 was first identified in China, several additional mcr gene variants (mcr-1–8) have since been found in bacteria across the globe.9
Antibiotic adjuvant therapy offers an alternative to novel antibiotic discovery by targeting non-essential bacterial systems and restoring susceptibility to a given antibiotic in a concurrent treatment strategy. Our research group has demonstrated diverse modulation of bacterial behavior using compounds derived from and inspired by marine natural products. Among these active compounds, a 2-aminoimidazole (2-AI) or 2-aminobenzimidazole (2-ABI) moiety has been a key component of the adjuvant scaffold in previous reports.10, 11 Herein, we expand the breadth of active scaffolds and report that tryptamine, a well-established biogenic amine and component of the amino acid tryptophan, serves as a synthetic platform for novel, non-toxic small molecules that successfully disarm colistin resistance, and report a lead molecule active against multiple colistin-resistant (colR) strains.
2. Results and Discussion
In a previous report from our group, a comprehensive screening of our in-house library identified that compound 1 (Figure 1) disarms colistin resistance in Gram-negative strains carrying the mcr-1 gene.12 When dosed at 60 μM, 1 returned up to a 32-fold reduction in the colistin minimum inhibitory concentration (MIC) against five engineered mcr-1 colR Gram-negative strains.13 This compound was one of six tryptamine derivatives (1–6, Figure 1) of the natural product oroidin first prepared in a previous report from our group aimed toward discovery of anti-biofilm agents against P. aeruginosa.14 Interestingly, biofilm dispersion or inhibition activity was not noted for any of these six molecules. When we screened the remaining five compounds from this original study (2-6) for synergy with colistin in four colR Gram-negative strains, two that harbor chromosomally encoded resistance (A. baumannii 4106, K. pneumoniae B9), and two transfected with the mcr-1 gene contained in a plasmid vector (A. baumannii ATCC 17978+mcr−1, and E. coli ATCC 25922+mcr−1) (Table 1), we found consensus activity in the molecule subset. Of these, compound 2 was consistently twice as active as 1 and returned MICs below the CLSI breakpoint (4 μg/mL) and thus became our lead molecule and the subject of a structure activity relationship (SAR) study.
Figure 1.

Structures of compounds 1-6 and structural segmentation.
Table 1.
MIC (μg/mL (fold reduction)) of colistin with in-house library adjuvants dosed at 60 μM.
| A. baumannii 4106 | K. pneumoniae B9 | A. baumannii ATCC 17978+mcr−1 | E. coli ATCC 25922 mcr−1 | |
|---|---|---|---|---|
| No compound | 1024 μg/mL | 1024 μg/mL | 16 μg/mL | 8 μg/mL |
| 1 (19.3 μg/mL) | 4 (256) | 1 (1024) | 0.5 (32) | 1 (8) |
| 2 (24.7 μg/mL) | 2 (512) | 0.5 (2048) | 0.5 (32) | 1 (8) |
| 3 (19.9 μg/mL) | 16 (64) | 8 (128) | 1 (16) | 4 (2) |
| 4 (20.8 μg/mL) | 32 (32) | 256 (4) | 4 (4) | 4 (2) |
| 5 (16.1 μg/mL) | 512 (2) | 1024 (0) | 16 (0) | 8 (0) |
| 6 (15.2 μg/mL) | 512 (2) | 1024 (0) | 8 (2) | 8 (0) |
With lead compound 2 in hand, we envisioned an SAR strategy that focused on dividing the molecule along the amide bond and launching two separate diversification campaigns, one focused on the indole head group and carbon linker, and the other on the brominated carboxypyrrole tail (Figure 1). As each of the thirty-three analogs were synthesized, their activity with colistin was determined in the same four Gram-negative colR strains used previously (Table 2). Interestingly, all analogs synthesized in this study showed no antimicrobial activity in monotherapy (MIC = >200 μM) and were initially tested at 60 μM when co-dosed with colistin. We synthesized a panel of analogs with a variety of changes to the head group scaffold via condensing 7 with commercially available amines under basic conditions (Scheme 1).14 The two-carbon linker of 2 was first both shortened and lengthened using 1H-indole-3-methanamine and homotryptamine as reactive amines to afford compounds 8 and 9 respectively, both of which showed increased activity over 2 in several strains. Reactions with histamine, benzylamine, 2-pyridineethanamine, and 2-aminoindane yielded compounds 10, 11, 12, and 13, respectively, all of which were inactive. We moved the tail group from the 3- position to the 2- position of the indole ring (14) via reaction with 1H-indole-2-methanamine, and moved it to the 6- position (15) using 1H-indole-6-methanamine as the amine source. Both 14 and 15 were roughly equipotent with 2. Using this same methodology, we then employed both 5-hydroxytryptamine and 5-methoxytryptamine to afford the corresponding indole ring substitution in final products 16 and 17, which were both upwards of eight-fold less active than 2.
Table 2.
Initial screen of head-group substituted analogs dosed at 60 μM with colistin given as MIC (μg/mL (fold reduction)).
| A. baumannii 4106 | K. pneumoniae B9 | A. baumannii ATCC 17978+mcr−1 | E. coli ATCC 25922+mcr−1 | |
|---|---|---|---|---|
| No compound | 1024 μg/mL | 1024 μg/mL | 16 μg/mL | 8 μg/mL |
| 8 (23.8 μg/mL) | 0.5 (2048) | 0.5 (2048) | 0.5 (32) | 0.5 (16) |
| 9 (25.5 μg/mL) | 2 (512) | 0.25 (4096) | 0.5 (32) | 0.5 (16) |
| 10 (21.7 μg/mL) | 128 (8) | 1024 (0) | 16 (0) | 8 (0) |
| 11 (21.5 μg/mL) | 512 (2) | 512 (2) | 16 (0) | 8 (0) |
| 12 (22.4 μg/mL) | 1024 (0) | 512 (2) | 16 (0) | 8 (0) |
| 13 (23.0 μg/mL) | 128 (8) | 1024 (0) | 16 (0) | 8 (0) |
| 14 (23.8 μg/mL) | 0.5 (2048) | 0.25 (4096) | 0.5 (32) | 0.5 (16) |
| 15 (23.8 μg/mL) | 4 (256) | 0.5 (2048) | 0.5 (32) | 1 (8) |
| 16 (25.6 μg/mL) | 256 (4) | 128 (8) | 4 (4) | 2 (4) |
| 17 (26.5 μg/mL) | 8 (128) | 8 (128) | 2 (8) | 4 (2) |
| 19 (25.7 μg/mL) | 32 (32) | 64 (16) | 2 (8) | 2 (4) |
| 24 (25.7 μg/mL) | 2 (512) | 16 (64) | 0.5 (32) | 1 (8) |
| 25 (25.5 μg/mL) | 4 (256) | 2 (512) | 0.5 (32) | 1 (8) |
| 29a (25.5 μg/mL) | 256 (4) | 1024 (0) | 16 (0) | 8 (0) |
| 29b (28.0 μg/mL) | 1024 (0) | 1024 (0) | 16 (0) | 8 (0) |
| 29c (30.1 μg/mL) | 1024 (0) | 1024 (0) | 16 (0) | 8 (0) |
Scheme 1.

Reaction scheme and structures of compounds 8 through 17. (a): Br2, CHCl3, 2h, RT. (b): RCH2NH2, K2CO3, DMF, 18h, RT.
Further modifications to the head group required preparation of derivatized amines (Scheme 2). We first sought to substitute benzothiophene for indole. Starting from commercially available benzo[b]thiophene-3-acetonitrile, reduction with SmI2 gave the corresponding ethylamine 18,15 which was then condensed with 7 to give 19, a product with greatly reduced (32 fold) activity compared to 2. To access additional substitutions of the indole ring, synthesis of substituted tryptamine precursors was necessary due to either lack of commercial availability or cost. Briefly, a nitrovinyl group was appended to the 3-position of the appropriately substituted indole via an addition-elimination reaction with 1-(dimethylamino)-2-nitroethylene (DMANE) in trifluoroacetic acid and dichloromethane. The nitroalkenes were then reduced with lithium aluminum hydride at room temperature to give the corresponding tryptamine derivatives.16 Starting from 6-fluoroindole and 7-methylindole, this route gave both 6-fluorotryptamine (22) and 7-methyltryptamine (23), which were each condensed with 7 to afford 24 and 25 in three steps overall. This same route also proved useful to access N-alkylated analogs of 2. N-methyl, N-butyl, and N-benzyl indole (26a-c) were each accessed by N-alkylating with the appropriate alkyl halide, and were then converted to the nitroalkene intermediate (27a-c) and finally reduced to the tryptamine derivative (28a-c). Each was then condensed with 7 to afford 29a-c.
Scheme 2.

Reaction scheme and structures of compounds 18 through 29c; (a): SmI2, TEA, H2O, THF; (b): K2CO3, DMF, 18h, RT (c): NaH, DMF, 0°C, 30 min, then R-X, 18h, RT; (d): DMANE, TFA, DCM; (e): LAH, THF.
From the biological data, we see that in general, an indole ring is required for activity though its orientation with respect to the aliphatic linker is flexible, as is the case for the overall length of the aliphatic linker itself. Substitutions about the indole ring are well tolerated in some cases but do not return increases in activity over the original lead 2, while indole N-alkylation renders a complete loss of activity.
Turning our attention to the pyrrole tail moiety, tryptamine was either directly reacted with commercially available acid chlorides, or coupled with commercially available carboxylic acids through activation with PyBOP, to access compounds 30–39 (Scheme 3, biological data given in Table 3). These compounds offer diversification of the 4,5-dibromopyrrole with multiple five and six membered heterocycles with varying halide substituents, and revealed that alteration of the 4,5-dibromopyrrole moiety is deleterious for activity with few exceptions. Isolated activity was seen with 37 in K. pneumoniae B9, returning a colistin MIC of 1 μg/mL (1024 fold reduction) when dosed at 60 μM, while remaining inactive in other Gram-negative strains.
Scheme 3.

Reaction scheme and structures of compounds 30 through 39; (a): RCOCl, TEA, DMF; (b): RCOOH, TEA, PyBOP, DMF.
Table 3.
Colistin MIC (μg/mL (fold reduction)) of colR strains co-challenged with tail-group substituted and shortened analogs of 2 dosed at 60 μM.
| A. baumannii 4106 | K. pneumoniae B9 | A. baumannii ATCC 17978+mcr−1 | E. coli ATCC 25922+mcr−1 | |
|---|---|---|---|---|
| 1024 μg/mL | 1024 μg/mL | 16 μg/mL | 8 μg/mL | |
| No compound | ||||
| 30 (16.2 μg/mL) | 128 (8) | 1024 (0) | 16 (0) | 8 (0) |
| 31 (17.3 μg/mL) | 64 (16) | 128 (8) | 8 (2) | 4 (2) |
| 32 (24.7 μg/mL) | 16 (64) | 2 (512) | 1 (16) | 4 (2) |
| 33 (25.7 μg/mL) | 32 (32) | 2 (512) | 1 (16) | 4 (2) |
| 34 (20.0 μg/mL) | 64 (16) | 16 (64) | 8 (2) | 8 (0) |
| 35 (21.0 μg/mL) | 16 (64) | 8 (128) | 4 (4) | 4 (2) |
| 36 (25.4 μg/mL) | 128 (8) | 8 (128) | 16 (0) | 8 (0) |
| 37 (20.7 μg/mL) | 64 (16) | 1 (1024) | 8 (2) | 4 (2) |
| 38 (20.7 μg/mL) | 256 (4) | 128 (8) | 16 (0) | 8 (0) |
| 39 (15.9 μg/mL) | 128 (8) | 1024 (0) | 16 (0) | 8 (0) |
| 41a (25.5 μg/mL) | 32 (32) | 4 (256) | 1 (16) | 4 (2) |
| 41b (28.0 μg/mL) | 64 (16) | 64 (16) | 8 (2) | 8 (0) |
| 41c (30.1 μg/mL) | 128 (8) | 64 (16) | 8 (2) | 8 (0) |
| 44 (30.3 μg/mL) | 8 (128) | 0.5 (2048) | 1 (16) | 2 (4) |
| 45 (22.8 μg/mL) | 16 (64) | 0.5 (2048) | 1 (16) | 4 (2) |
| 46 (20.7 μg/mL) | 32 (32) | 32 (32) | 16 (0) | 8 (0) |
| 47 (29.5 μg/mL) | 1024 (0) | 1024 (0) | 16 (0) | 8 (0) |
Mirroring efforts towards the indole N-alkylated analogs 29a-c, we accessed N-alkylated derivatives of the 4,5-dibromopyrrole moiety, namely N-methyl, N-butyl, and N-benzyl analogs of 7. Each of these derivatives was condensed with tryptamine to afford compounds 41a-c (Scheme 4). We observed diminished biological activity with increased steric bulk. To further explore the impact that halogenation patterns of the pyrrole tail had upon activity, the 4,5-diiodopyrrole analog 44 was synthesized from 7 (Scheme 4). Iodination of the 2-trichloroacetyl pyrrole using molecular iodine and silver trifluoroacetate generated the 4,5-diiodinated pyrrole along with the 4-iodopyrrole as a side product as an inseparable mixture.17 Condensation of this mixture with tryptamine gave both the 4,5-diiodoinated product 44, as well as the 4-iodinated 45, which were readily isolated on silica gel. Both 44 and 45 were most effective in K. pneumoniae B9 and returned a 2048 fold reduction in colistin MIC when dosed at 60 μM.
Scheme 4.

Reaction scheme and structures of compounds 41a through 47; (a): NaH, DMF, 30 mins, 0°C, then R-X, RT, 18h; (b): tryptamine, K2CO3, DMF, 18h; (c): I2, AgCOCF3, CHCl3; (d): 6-bromopicolinic acid, PyBOP, TEA, DMF; (e): 42, K2CO3, DMF.
Preliminary screening showed promising results for compounds 8, 37, and 44 (MIC fold-reduction of >512 in any tested strain, Tables 2 and 3) and thus we elected to combine successful alterations on both sides of the amide bond (Scheme 4). Coupling 1H-indole-3-methanamine with 6-bromopicolinic acid delivered 46, a shortened analog of 37. We also generated 47, the analogous shortened derivative of 44 via condensation of 1H-indole-3-methanamine with the corresponding 4,5-diiodopyrrole to afford 47. Compound 46 had modest activity (32-fold MIC colistin reduction) against two strains (A. baumannii 4106, K. pneumoniae B9), while compound 47 was completely inactive (Table 3). All analogs having with equal or greater activity compared to 2 in any of the four test strains were then taken forward for further testing. These criteria eliminated twenty-three analogs and identified a cohort of nine molecules of interest (8, 9, 14, 15, 24, 25, 37, 44, and 45.)
These nine compounds were then subjected to further screening with nine additional bacterial strains: five primary clinical isolates that harbor chromosomally-encoded colistin resistance (A. baumannii 3941, A. baumannii 3942, A. baumannii 4112, K. pneumoniae A5, and K. pneumoniae C3) as well as the mcr-1 parent strains without plasmid (A. baumannii ATCC 17978, and E. coli ATCC 25922) and two other colistin sensitive strains (A. baumannii 5075 and K. pneumoniae ATCC BAA 2146). These results are tabulated in Table 4.
Table 4.
MIC (μg/mL (fold reduction)) of colistin with adjuvants dosed at 60 μM against nine Gram-negative strains.
| Colistin MIC | 2 (24.7 μg/mL) |
8 (23.8 g/mL) |
9 (25.5 μg/mL) |
14 (23.8 μg/mL) |
15 (23.8 μg/mL) |
24 (25.7 μg/mL) |
25 (25.5 μg/mL) |
37 (20.7 μg/mL) |
44 (30.3 μg/mL) |
45 (22.8 μg/mL) |
|
|---|---|---|---|---|---|---|---|---|---|---|---|
| A. baumannii 3941 | 512 μg/mL | 2 (256) | 0.5 (1024) | 2 (256) | 2 (256) | 4 (128) | 1 (512) | 2 (256) | 64 (8) | 4 (128) | 8 (64) |
| A. baumannii 3942 | 512 μg/mL | 1 (512) | 0.25 (2048) | 2 (256) | 2 (256) | 4 (128) | 1 (512) | 2 (256) | 64 (8) | 4 (128) | 16 (32) |
| A. baumannii 4112 | 1024 μg/mL | 2(512) | 0.5 (2048) | 4 (256) | 2 (512) | 4 (256) | 2 (512) | 4 (256) | 128 (8) | 8 (128) | 32 (32) |
| K. pneumoniae A5 | 1024 μg/mL | 4 (256) | 2 (512) | 4 (256) | 2 (512) | 4 (256) | 4 (256) | 4 (256) | 1024 (0) | 4 (256) | 32 (32) |
| K. pneumoniae C3 | 512 μg/mL | 0.5 (1024) | 0.25 (2048) | 0.5 (1024) | 0.25 (2048) | 1 (512) | 1 (512) | 1 (512) | 1 (512) | 1 (512) | 4 (128) |
| A. baumannii 5075 | 1 μg/mL | 0.5 (2) | 0.25 (4) | 0.25 (4) | 0.5 (2) | 0.5 (2) | 0.5 (2) | 0.5 (2) | 1 (0) | 0.5 (2) | 1 (0) |
| A. baumannii ATCC 17978parent | 1 μg/mL | 0.25 (4) | 0.25 (4) | 0.25 (4) | 0.5 (2) | 0.5 (2) | 0.5 (2) | 0.5 (2) | 0.5 (2) | 0.5 (2) | 0.5 (2) |
| E. coli ATCC 25922parent | 1 μg/mL | 0.25 (4) | 0.25 (4) | 0.5 (2) | 0.5 (2) | 0.5 (2) | 0.5 (2) | 0.5 (2) | 0.5 (2) | 0.5 (2) | 0.5 (2) |
| K. pneumoniae ATCC 2146NDM−1 | 1 μg/mL | 0.5 (2) | 1 (0) | 0.5 (2) | 1 (0) | 1 (0) | 0.5 (2) | 0.5 (2) | 0.5 (2) | 0.5 (2) | 0.5 (2) |
This second round of screening established that all molecules were efficacious in colR primary clinical isolates of A. baumannii and K. pneumoniae, and identified 8 as the most active colistin modulator, in some cases returning a 2048-fold reduction in colistin MIC and a four-fold increase in activity compared to the original lead molecule 2. A subsequent dose-response study with 8 (Table 5) showed concentration-dependent activity in two Gram-negative clinical isolates (A. baumannii 3942, K. pneumoniae C3). To explore potential aggregate effects, this same experiment was repeated supplementing Cation-Adjusted Mueller Hinton Broth (CAMHB) with 0.001% Triton X100. No significant changes in colistin MIC were observed in the presence of detergent (Table S1, Supporting Information). Additionally, growth curves were constructed for A. baumannii 3942 treated with 60 μM 8, (Figure S1) which showed that 8 did not perturb bacterial proliferation. To identify changes in membrane permeability, the BacLight cell viability assay was used. This duplex colorimetric fluorescence screen offers quantized detection of potential increases in cell permeability caused by xenobiotics. The ratio of nucleic-acid bound STYO-9 (green-fluorescent), which readily crosses healthy cellular membranes, to the redfluorescence of propidium iodide, which only enters cells with compromised cellular envelopes, is determined by scanning the distinct emission wavelengths of the two dyes. In endotoxin-free water dosed with 60 μM 8, membrane permeability was increased by a factor of 1.9 in A. baumannii 3942 and of 1.5 in K. pneumoniae C3 compared to untreated controls. Conversely, no significant change in cell permeability was detected in either strain treated with the inactive compound 29b at the same concentration. In a separate assay, neither 8 nor 29b lysed red blood cells (<1%) when dosed in phosphate-buffered saline (PBS) at 100 μM.
Table 5.
Colistin MIC (μg/mL (fold-reduction) of A. baumannii 3942 and K. pneumoniae C3 treated with various concentrations of 8.
| A. baumannii 3942 | K. pneumoniae C3 | |
|---|---|---|
| 60 μM 8 (23.8 μg/mL) | 0.25 (2048) | 0.25 (2048) |
| 50 μM 8 (19.8 μg/mL) | 1 (512) | 1 (512) |
| 40 μM 8 (15.9 μg/mL) | 2 (256) | 2 (256) |
| 30 μM 8 (11.9 μg/mL) | 4 (128) | 4 (128) |
| 20 μM 8 (7.9 μg/mL) | 32 (16) | 4 (128) |
| 15 μM 8 (6.0 μg/mL) | 64 (8) | 8 (64) |
| 10 μM 8 (4.0 μg/mL) | 256 (2) | 16 (32) |
| 5 μM 8 (2.0 μg/mL) | 512 (0) | 128 (4) |
In conclusion, we have presented the synthesis of thirty-three analogs of a tryptamine-based lead-molecule previously observed to suppress colistin resistance. This diverse panel allowed for preliminary relationships between structure and activity to be uncovered indicating that there are rigid constraints on the scaffold to retain suppression of colistin resistance. The indole head group is necessary for activity, and neither substitution about the ring nor N-alkylation enhance efficacy. We further saw that deviation from the 4,5-dibromopyrrole to other five and six membered heterocycles greatly reduced activity. These molecules are non-toxic to bacteria, each having an MIC >200 μM in all strains that were studied. Based on activity, this panel of thirty-three analogs was narrowed to nine compounds for further screening against an additional nine bacterial strains. This second round of screening revealed that while they had unremarkable activity in colistin-sensitive strains, they were effective in both colR primary clinical isolates of A. baumannii and K. pneumoniae, as well as strains transfected with the mcr-1 gene, and returned sub-breakpoint colistin MICs, in some cases as low as 0.25 μg/mL.
In the end, shortening the aliphatic linker by one carbon accessed compound 8 with fourfold greater activity than the parent molecule. In subsequent studies we observed dose-dependent activity of 8, and when given as monotherapy, it caused no abnormalities in bacterial growth and did not lyse red blood cells. Currently, the mechanism-of-action of this family of compounds is unknown. Previous reports from our research group have disclosed colistin adjuvants containing a 2-AI heterocycle reversed lipid A modification leading to re-sensitization to colisin. These tryptamine-based adjuvants, however, do not reverse lipid A modification, thus appear to act via a novel mechanism. In all, this study identified several scaffolds that can be tasked in future probes of colistin resistance pathways and potentially new leads for further SAR studies while concurrent in vivo investigations are pursued.
3. Experimental Methods
General Biological Assay Methods:
All antibiotics, media, and other biological reagents were purchased from commercial sources and used without further purification. Compounds synthesized in this study were dosed from DMSO stocks.
Single Compound MIC Determination via Broth Microdilution:
This procedure follows the guidelines set by the Clinical Laboratory Sciences Institute.4 Bacteria were cultured for 4 to 6 hours in Cation-Adjusted Mueller-Hinton Broth (CAMHB) and subcultured to 5 ×105 CFU/mL in fresh CAMHB. For each compound to be tested, a 1 mL aliquot of subculture was taken and dosed with compound to a final concentration of 200 μM. Samples were then dispensed (200 μL) into the first row of a 96-well microtiter plate in which subsequent wells were prefilled with 100 μL of subculture. 100 μL of dosed subculture was then serially diluted a total of 6 times in each subsequent row of the plate save for the last row which served as a control. Plates were then sealed and incubated stationary at 37°C. After 18 hours, the plates were removed and MIC values were recorded. All compounds tested had a purity of >95%.
Antibiotic Resensitization MIC Determination via Broth Microdilution:
This procedure was adapted from the guidelines set by the Clinical Laboratory Sciences Institute.4 Bacteria were cultured for 4 to 6 hours in Cation-Adjusted Mueller-Hinton Broth (CAMHB) and subcultured to 5 ×105 CFU/mL in fresh CAMHB. For each compound to be tested, a 5 mL aliquot of subculture was taken and dosed with adjuvant to a set concentration. A 1 mL aliquot of each dosed subculture was collected and dosed with antibiotic to a set concentration. Co-dosed aliquots were then dispensed (200 μL) into the first row of a 96-well microtiter plate in which subsequent wells were prefilled with 100 μL of the corresponding dosed subculture. 100 μL of co-dosed subculture was then serially diluted a total of 6 times in each subsequent row of the plate save the last row as a control to afford serial dilution of the antibiotic while holding a constant concentration of adjuvant. Plates were then sealed and incubated stationary at 37°C. After 18 hours, the plates were removed and MIC values were recorded. All compounds tested had a purity of >95%.
Bacterial Growth Curve:
Bacteria were cultured overnight in CAMHB and subcultured to 5 × 105 CFU/mL in fresh CAMHB. The subculture was then transferred to culture tubes in 5 mL aliquots, which were dosed with adjuvant to a set concentration, save one aliquot as a control. All subcultures were then incubated at 37°C with shaking. At 2, 4, 6, 8, and 24 hours, 100 μL samples of each culture was serially diluted in 900 μL aliquots of CAMHB for a total of 5 to 7 times. 100 μL of each dilution point were plated on nutrient agar and incubated stationary overnight. The total number of bacterial colonies on each plate was recorded.
BacLight Cell Permeability Assay:
Bacteria were cultured overnight in CAMHB at 37°C and diluted 1:10 in fresh CAMHB and grown for an additional 4 hours to an OD600 of ~0.3. Cultures were centrifuged at 10,000 g for 15 mins, supernatants were discarded, and cell pellets were washed once with sterile water and resuspended in 1/10th the original volume in sterile water or water dosed with compounds. Suspensions were incubated for 1 hour at 37°C with shaking and centrifuged at 10,000 g for 15 mins, washed with sterile water and resuspended in sterile water supplemented with 1:1 SYTO-9 and propidium iodide (3 μL/mL, from Invitrogen BacLight Kit). 100 μL of each test condition were added to a 96 well plate which was covered and allowed to stand at room temperature. After 15 mins, the plate was read at 530 nm and 645 nm (excitation 485 nm). The ratio of green to red fluorescence was calculated as a percentage of the control.
Ovine Erythrocyte Lysis Assay:
Hemolysis assays were performed on mechanically difibrinated sheep blood (Hemostat Labs: DSB100). Difibrinated blood (1.5 mL) was placed into a microcentrifuge tube and centrifuged for 10 min at 10,000 rpm. The supernatant was then removed and then the cells were resuspended in 1 mL of phosphate-buffered saline (PBS). The suspension was centrifuged, the supernatant was removed and cells were resuspended two additional times in PBS. The final cell suspension was then diluted 10-fold in PBS dosed with test compounds from DMSO stock solutions. DMSO was used as a negative control and a zero hemolysis marker. Triton X100 (1%) was used as a positive control serving as the 100% lysis marker. Samples were incubated at 37°C with shaking. After one hour, the samples were centrifuged for 10 min at 10,000 rpm. The supernatant was diluted 1:40 in distilled water. The absorbance of the supernatant at 540 nm was then measured with a UV spectrometer.
General Synthetic Methods:
All reagents used for chemical synthesis were purchased from commercial sources and used without further purification. Flash chromatography was performed using 60Å mesh silica gel (Sorbtech, Norcross, GA). Both 1H (300 and 400 MHz) and 13C NMR (75 MHz and 100 Mhz) spectra were collected on Varian Mercury spectrometers at 25°C. Ultraviolet spectra were obtained from a Genesys 10 Scanning UV/Vis spectrophotometer (λmax in nm). Infrared spectra were obtained on a solid-phase FT-IR-4100 spectrophotometer (νmax in cm−1). High-resolution mass spectra were obtained by the NCSU Molecular Education, Technology, and Research Innovation Center (METRIC). All NMR spectra are available in this article’s supplemental information document. The purity of all reported compounds was determined to be greater than or equal to 95% both by NMR spectra and subsequent liquid chromatography UV/Vis trace analysis.
General Procedure for Condensing Primary Amines with 2,2,2-trichloroacetylpyrroles:
This method was adapted from methods described by Richards et al.14 One equivalent of the appropriate 2,2,2-trichloroacetylpyrrole was combined with 2 equivalents of the appropriate primary amine along with 3 equivalents of anhydrous potassium carbonate and dissolved in 4 mLs of anhydrous dimethylformamide (DMF) under argon and stirred for 18 hours. The reaction was poured into 75 mLs EtOAc and 25 mLs of deionized water, the aqueous layer was discarded and the organic was further washed thrice with water, twice with 25 mLs 1N HCl, and once with 25 mLs brine. The organic layer was then dried with anhydrous magnesium sulfate and evaporated under reduced pressure. The crude solid was purified by flash chromatography (5–20% EtOAc/Hexanes) to yield the final products.
General Procedure for Synthesis of 3-(2-nitrovinyl) indoles:
This procedure was adapted from the methods described by Holloway.16 The corresponding indole (0.9 equiv) was dissolved in 4 mLs anhydrous dichloromethane (DCM) under argon and stirred for 15 mins at RT. In a separate flask, N,N-dimethyl-2-nitroethen-1-amine (DMANE) (1 equiv) was dissolved in 3 mLs trifluoroacetic acid (TFA) and stirred for 15 mins at RT. The solution of indole in DCM was added dropwise to the stirring solution of TFA and stirred for an additional 30 mins at RT. The reaction was quenched with the addition of water (10 mLs) and was poured into 25 mLs water and extracted three times with 25 mLs DCM. The organic layers were combined and dried with anhydrous magnesium sulfate and evaporated under reduced pressure. The resulted crude solid was recrystallized from chloroform to give the products with no further purification necessary.
General Procedure for N-alkylation of Indole:
Indole (1 g, 8.54 mmols) was dissolved in 15mLs DMF and cooled to 0°C under argon. Sodium hydride (410 mg, 10.2 mmols) was added in portions to the solution which was then stirred at 0°C for 30 mins. Two equivalents of the appropriate alkyl halide were then added to the solution which was allowed to warm to RT for 18 hours. The reaction was poured into 75 mLs EtOAc and 25 mLs of deionized water, the aqueous layer was discarded and the organic was further washed thrice with water, twice with 25mLs 1N HCl, and once with 25 mLs brine. The organic layer was then dried with anhydrous magnesium sulfate and evaporated under reduced pressure. The crude solid was purified by flash chromatography (5–20% EtOAc/Hexanes) to yield the final products.
General Procedure for the Reduction of N-alkylated 3-nitrovinyl Indoles:
This procedure was adapted from the methods described by Muratore.18 The corresponding N-alkylated 3-nitrovinyl indole was dissolved in 15 mLs anhydrous tetrahydrofuran (THF) and cooled to −78°C for 20 mins. A 2.4M solution (5 mLs) of lithium aluminum hydride (LAH) in THF) was added dropwise to the stirring solution which was then allowed to come to room temperature for 18 hours. The stirring solution was then cooled to 0°C and the following solutions were added dropwise in sequence: 0.86 mL of water, 0.86 mL of 15% aq. NaOH, and finally, 2.2 mL of deionized water. After 30 mins, magnesium sulfate was added to the slurry which was subsequently filtered through a pad of celite and washed twice with 20 mLs DCM. The organics were evaporated under reduced pressure and the crude amine was taken to further transformations with no further purification.
General Procedure for the Coupling of Carboxylic Acids to Primary Amines using PyBOP:
The corresponding carboxylic acid (1 equiv), benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) (1 equiv), and triethylamine (TEA) (3 equiv) were dissolved in anhydrous DMF under argon and stirred for 15 mins at RT. The appropriate primary amine (2 equiv) was then added in one portion and the reaction was allowed to stir for 18 hours at RT and then poured into 75 mLs EtOAc and 25 mLs deionized water. The aqueous layer was discarded and the organic layer was washed twice with water (25 mLs) and once with brine (25 mLs). The organic layer was dried with magnesium sulfate and evaporated under reduced pressure. The crude solids were purified by flash chromatography (5–20% EtOAc/Hexanes) to afford pure products.
N-(2-(1H-indol-3-yl)ethyl)-4,5-dichloro-1H-pyrrole-2-carboxamide (1):
Compound was synthesized using the methods previously reported by Richards et al. Spectral data were consistent with previous reports.14
N-(2-(1H-indol-3-yl)ethyl)-4,5-dibromo-1H-pyrrole-2-carboxamide (2):
Compound was synthesized using the methods previously reported by Richards et al. Spectral data were consistent with previous reports.14
N-(2-(1H-indol-3-yl)ethyl)-4-bromo-1H-pyrrole-2-carboxamide (3):
Compound was synthesized using the methods previously reported by Richards et al. Spectral data were consistent with previous reports.14
N-(2-(1H-indol-3-yl)ethyl)-4-bromo-1-methyl-1H-pyrrole-2-carboxamide (4):
Compound was synthesized using the methods previously reported by Richards et al. Spectral data were consistent with previous reports.14
N-(2-(1H-indol-3-yl)ethyl)-1-methyl-1H-pyrrole-2-carboxamide (5):
Compound was synthesized using the methods previously reported by Richards et al. Spectral data were consistent with previous reports.14
N-(2-(1H-indol-3-yl)ethyl)-1H-pyrrole-2-carboxamide (6):
Compound was synthesized using the methods previously reported by Richards et al. Spectral data were consistent with previous reports.14
2,2,2-trichloro-1-(4,5-dibromo-1H-pyrrol-2-yl)ethan-1-one (7):
Compound was synthesized using the methods previously reported by Richards et al. Spectral data were consistent with previous reports.14
N-((1H-indol-3-yl)methyl)-4,5-dibromo-1H-pyrrole-2-carboxamide (8):
Compound was synthesized using the general procedure for condensing primary amines with 2,2,2-trichloroacetyl pyrroles with (1H-indol-3-yl)methanamine (323 mg, 2.14 mmols) and 7 (396 mg, 1.07 mmols) as condensation partners to afford 8 as a tan solid (372 mg, 77%). 1H NMR (400 MHz, DMSO-d6) δ 12.69 (s, 1H), 10.94 (s, 1H), 8.41 (s, 1H), 7.60 (d, J = 7.9 Hz, 1H), 7.36 (d, J = 8.1 Hz, 1H), 7.28 (s, 1H), 7.07 (t, J = 7.5 Hz, 1H), 7.01 – 6.88 (m, 2H), 4.57 (d, J = 5.5 Hz, 2H); 13C NMR (101 MHz, dmso) δ 158.54, 136.20, 128.26, 126.46, 123.82, 121.20, 118.81, 118.56, 112.71, 112.25, 111.44, 104.21, 97.86, 33.91 ppm; UV (λmax nm) 288; IR vmax (cm−1) 3392, 1504, 804, 749, 492; HRMS (ESI) calcd for C14H11Br2N3O [M+H]− 393.9196, found 393.9192.
4,5-dibromo-N-(2-(1-butyl-1H-indol-3-yl)ethyl)-1H-pyrrole-2-carboxamide (9):
Compound was synthesized using the general procedure for condensing primary amines with 2,2,2-trichloroacetyl pyrroles with (1H-indol-3-yl)propylamine (150 mg, 0.86 mmols) and 7 (287 mg, 0.78 mmols) as condensation partners to afford 9 as a tan solid (186 mg, 38%). 1H NMR (400 MHz, DMSO-d6) δ 12.70 (s, 1H), 8.26 (s, 1H), 7.58 (d, J = 7.8 Hz, 1H), 7.39 (d, J = 8.1 Hz, 1H), 7.16 (s, 1H), 7.14 – 7.07 (m, 1H), 7.05 – 6.96 (m, 1H), 6.92 (s, 1H), 4.07 (t, J = 6.5 Hz, 2H), 3.62 – 3.39 (m, 2H), 2.91 (t, J = 6.9 Hz, 2H), 1.82 – 1.49 (m, 2H), 1.19 (dd, J = 14.7, 7.3 Hz, 2H), 0.84 (t, J = 7.2 Hz, 3H); 13C NMR (101 MHz, acetone) δ 160.09, 137.48, 129.43, 129.00, 126.97, 122.00, 119.62, 119.26, 113.08, 113.05, 112.35, 110.34, 99.44, 46.24, 40.79, 33.20, 26.21, 20.74, 14.01 ppm; UV (λmax nm) 296; IR vmax (cm−1) 3117, 1611, 1557, 742, 492; HRMS (ESI) calcd for C19H21Br2N3O [M+H]− 463.9978, found 463.9976.
N-(2-(1H-imidazol-4-yl)ethyl)-4,5-dibromo-1H-pyrrole-2-carboxamide (10):
Compound was synthesized using the methods previously reported by Richards et al. Spectral data were consistent with previous reports.14
N-benzyl-4,5-dibromo-1H-pyrrole-2-carboxamide (11):
Compound was synthesized using the general procedure for condensing primary amines with 2,2,2-trichloroacetyl pyrroles with benzylamine (89 μL, 0.81 mmols) and 7 (300 mg, 0.81 mmols) as condensation partners to afford 11 as a white solid (162 mg, 56%). 1H NMR (300 MHz, DMSO-d6) δ 12.73 (s, 1H), 8.68 (s, 1H), 7.40 – 7.17 (m, 5H), 6.98 (d, J = 2.8 Hz, 1H), 4.42 (d, J = 5.8 Hz, 2H); 13C NMR (75 MHz, dmso) δ 158.93, 139.51, 128.36, 128.06, 127.21, 126.86, 112.73, 104.78, 97.92, 41.99 ppm; UV (λmax nm) 286; IR vmax (cm−1) 3187, 1602, 1558, 1232, 711; HRMS (ESI) calcd for C12H10Br2N2O [M+H]− 354.9087, found 354.9088.
4,5-dibromo-N-(2-(pyridin-2-yl)ethyl)-1H-pyrrole-2-carboxamide (12):
Compound was synthesized using the general procedure for condensing primary amines with 2,2,2-trichloroacetyl pyrroles with 2-(pyridin-2-yl)ethan-1-amine (97 μL, 0.81 mmols) and 7 (300 mg, 0.81 mmols) as condensation partners to afford 12 as a white solid (97 mg, 32%). 1H NMR (300 MHz, DMSO-d6) δ 12.67 (s, 1H), 8.50 (d, J = 6.4 Hz, 1H), 8.23 (t, J = 5.4 Hz, 1H), 7.69 (td, J = 7.6, 1.9 Hz, 1H), 7.23 (dd, J = 14.0, 7.2 Hz, 2H), 6.87 (s, 1H), 3.56 (q, J = 6.7 Hz, 2H), 2.95 (t, J = 7.3 Hz, 2H); 13C NMR (101 MHz, dmso) δ 159.05, 158.91, 149.11, 136.55, 128.25, 123.23, 121.60, 112.49, 104.52, 97.84, 38.65, 37.50 ppm; UV (λmax nm) 288; IR vmax (cm−1) 3108, 1626, 1566, 1327, 765; HRMS (ESI) calcd for C12H11Br2N3O [M+H]− 369.9196, found 369.9197.
4,5-dibromo-N-(2,3-dihydro-1H-inden-2-yl)-1H-pyrrole-2-carboxamide (13):
Compound was synthesized using the general procedure for condensing primary amines with 2,2,2-trichloroacetyl pyrroles with 2-aminoindane (211 μL, 1.62 mmols) and 7 (300 mg, 0.81 mmols) as condensation partners to afford 13 as a gray solid (102 mg, 33%). 1H NMR (300 MHz, DMSO-d6) δ 12.68 (s, 1H), 8.29 (d, J = 6.2 Hz, 1H), 7.20 (d, J = 20.0 Hz, 4H), 6.99 (s, 1H), 4.85 – 4.38 (m, 1H), 3.22 (dd, J = 16.3, 7.3 Hz, 2H), 2.88 (dd, J = 16.0, 5.5 Hz, 2H); 13C NMR (101 MHz, dmso) δ 158.80, 141.22, 128.21, 126.46, 124.52, 112.97, 104.50, 97.84, 50.12, 39.07 ppm; UV (λmax nm) 288; IR vmax (cm−1) 3133, 1635, 1503, 811, 740; HRMS (ESI) calcd for C14H12Br2N2O [M+H]− 380.9243, found 380.9245.
N-((1H-indol-2-yl)methyl)-4,5-dibromo-1H-pyrrole-2-carboxamide (14):
Compound was synthesized using the general procedure for condensing primary amines with 2,2,2-trichloroacetyl pyrroles with (1H-indol-2-yl)methanamine (237 mg, 1.62 mmols) and 7 (300 mg, 0.81 mmols) as condensation partners to afford 14 as a yellow solid (239 mg, 74%). 1H NMR (300 MHz, DMSO-d6) δ 12.76 (s, 1H), 10.96 (s, 1H), 8.64 (s, 1H), 7.45 (d, J = 7.7 Hz, 1H), 7.33 (d, J = 8.2 Hz, 1H), 7.07 – 6.99 (m, 2H), 6.98 – 6.89 (m, 1H), 6.28 (s, 1H), 4.57 (s, 2H); 13C NMR (101 MHz, dmso) δ 158.98, 137.01, 136.18, 128.08, 127.98, 120.72, 119.60, 118.92, 113.08, 111.14, 104.82, 99.20, 98.01, 36.25 ppm; UV (λmax nm) 294; IR vmax (cm−1) 3314, 1685, 1521, 1229, 684; HRMS (ESI) calcd for C15H14ClN3O [M+H]− 393.9196, found 393.9198.
N-((1H-indol-6-yl)methyl)-4,5-dibromo-1H-pyrrole-2-carboxamide (15):
Compound was synthesized using the general procedure for condensing primary amines with 2,2,2-trichloroacetyl pyrroles with (1H-indol-6-yl)methanamine (236 mg, 1.62 mmols) and 7 (300 mg, 0.81 mmols) as condensation partners to afford 14 as a white solid (268 mg, 83%). 1H NMR (300 MHz, DMSO-d6) δ 12.73 (s, 1H), 11.01 (s, 1H), 8.66 (s, 1H), 7.47 (d, J = 8.1 Hz, 1H), 7.30 (s, 2H), 7.00 (s, 1H), 6.94 (d, J = 9.0 Hz, 1H), 6.38 (s, 1H), 4.51 (d, J = 5.9 Hz, 2H); 13C NMR (101 MHz, dmso) δ 158.85, 136.06, 132.19, 128.30, 126.64, 125.30, 119.90, 118.85, 112.73, 110.00, 104.68, 100.94, 97.97, 42.54 ppm; UV (λmax nm) 292; IR vmax (cm−1) 3403, 1637, 1512, 867, 769; HRMS (ESI) calcd for C15H14ClN3O [M+H]− 393.9196, found 393.9198.
4,5-dibromo-N-(2-(5-hydroxy-1H-indol-3-yl)ethyl)-1H-pyrrole-2-carboxamide (16):
Compound was synthesized using the general procedure for condensing primary amines with 2,2,2-trichloroacetyl pyrroles with one exception: final product was purified by flash chromatograpy (0.5 – 5% MeOH-NH3/DCM) in lieu of EtOAc/Hexanes. 5-hydroxytryptamine (117 mg, 0.54 mmols) and 7 (200 mg, 0.54 mmols) were condensed to afford 16 as a tan solid (48 mg, 21%). 1H NMR (400 MHz, DMSO-d6) δ 12.68 (s, 1H), 10.50 (s, 1H), 8.61 (s, 1H), 8.25 (s, 1H), 7.13 (d, J = 8.5 Hz, 1H), 7.06 (s, 1H), 6.93 (s, 1H), 6.87 (s, 1H), 6.61 (d, J = 8.6 Hz, 1H), 3.53 – 3.42 (m, 2H), 2.82 (t, J = 7.0 Hz, 2H); 13C NMR (101 MHz, dmso) δ 158.86, 150.24, 130.85, 128.44, 127.93, 123.16, 112.46, 111.73, 111.35, 110.79, 104.40, 102.29, 97.83, 39.44, 25.46 ppm; UV (λmax nm) 296; IR vmax (cm−1) 3161, 1569, 1178, 829, 614; HRMS (ESI) calcd for C15H13Br2N3O2 [M+H]− 423.9301, found 423.9304.
4,5-dibromo-N-(2-(5-methoxy-1H-indol-3-yl)ethyl)-1H-pyrrole-2-carboxamide (17):
Compound was synthesized using the general procedure for condensing primary amines with 2,2,2-trichloroacetyl pyrroles with 5-methoxytryptamine (318 mg, 1.62 mmols) and 7 (300 mg, 0.81 mmols) as condensation partners to afford 14 as a pink solid (207 mg, 58%). 1H NMR (300 MHz, DMSO-d6) δ 12.69 (s, 1H), 10.66 (s, 1H), 8.26 (s, 1H), 7.22 (d, J = 8.6 Hz, 1H), 7.12 (s, 1H), 7.04 (s, 1H), 6.91 (s, 1H), 6.71 (d, J = 8.7 Hz, 1H), 3.53 – 3.39 (m, 2H), 2.87 (t, J = 7.1 Hz, 2H); 13C NMR (101 MHz, dmso) δ 158.94, 153.04, 131.43, 128.46, 127.65, 123.43, 112.50, 112.09, 111.69, 111.18, 104.42, 100.16, 97.85, 55.28, 39.72, 25.35 ppm; UV (λmax nm) 296; IR vmax (cm−1) 3340, 1606, 1467, 1167, 923; HRMS (ESI) calcd for C16H15Br2N3O2 [M+H]− 437.9458, found 437.9460.
2-(benzo[b]thiophen-3-yl)ethan-1-amine (18):
Compound was synthesized using the methods previously reported by Szostak. Spectral data were consistent with previous reports.15
N-(2-(benzo[b]thiophen-3-yl)ethyl)-4,5-dibromo-1H-pyrrole-2-carboxamide (19):
Compound was synthesized using the general procedure for condensing primary amines with 2,2,2-trichloroacetyl pyrroles with 18 (100 mg, 0.56 mmols) and 7 (209 mg, 0.56 mmols) as condensation partners to afford 19 as a yellow solid (105 mg, 44%). 1H NMR (300 MHz, DMSO-d6) δ 12.71 (s, 1H), 8.35 (s, 1H), 7.95 (dd, J = 16.0, 7.2 Hz, 2H), 7.48 (s, 1H), 7.45 – 7.33 (m, 2H), 6.89 (s, 1H), 3.54 (q, J = 6.8 Hz, 2H), 3.05 (t, J = 7.2 Hz, 2H); 13C NMR (101 MHz, dmso) δ 159.03, 139.73, 138.81, 133.73, 128.24, 124.35, 124.11, 122.98, 122.86, 121.77, 112.52, 104.59, 97.85, 38.50, 28.34 ppm; UV (λmax nm) 286; IR vmax (cm−1) 3410, 3117, 2921, 1629, 739; HRMS (ESI) calcd for C15H12Br2N2OS [M+H]− 424.8964, found 424.8969.
(E)-6-fluoro-3-(2-nitrovinyl)-1H-indole (20):
Compound was synthesized using the methods previously reported by Muratore. Spectral data were consistent with previous reports.18
(E)-7-methyl-3-(2-nitrovinyl)-1H-indole (21):
Compound was synthesized using the general procedure for synthesis of 3-(2-nitrovinyl) indoles with 7-methyl indole (800 mg, 6.09 mmols) as the starting material to afford 21 as an orange solid (730 mg, 59%). 1H NMR (400 MHz, DMSO-d6) δ 12.22 (s, 1H), 8.35 (d, J = 13.4 Hz, 1H), 8.19 (d, J = 2.7 Hz, 1H), 7.93 (d, J = 13.4 Hz, 1H), 7.68 (d, J = 7.8 Hz, 1H), 7.07 (t, J = 7.5 Hz, 1H), 7.00 (d, J = 7.1 Hz, 1H), 2.44 (s, 3H); 13C NMR (101 MHz, dmso) δ 137.17, 135.99, 134.82, 131.02, 124.53, 123.99, 122.21, 122.09, 117.98, 108.68, 16.62 ppm; UV (λmax nm) 396; IR vmax (cm−1) 3104, 1609, 1521, 1201, 793; HRMS (ESI) calcd for C11H10N2O2 [M+H]− 201.0669, found 201.0669.
2-(6-fluoro-1H-indol-3-yl)ethan-1-amine (22):
Compound was synthesized using the methods previously reported by Muratore and taken to the next reaction without further purification.18
2-(7-methyl-1H-indol-3-yl)ethan-1-amine (23):
Compound was synthesized using the methods previously reported by Blough and taken on to the next reaction without further purification.19
4,5-dibromo-N-(2-(6-fluoro-1H-indol-3-yl)ethyl)-1H-pyrrole-2-carboxamide (24):
Compound was synthesized using the general procedure for condensing primary amines with 2,2,2-trichloroacetyl pyrroles with 22 (204 mg, 1.14 mmols) and 7 (300 mg, 0.81 mmols) as condensation partners to afford 24 as a brown solid (129 mg, 37%). 1H NMR (300 MHz, Chloroform-d) δ 11.25 (s, 1H), 8.11 (s, 1H), 7.50 (dd, J = 8.6, 5.3 Hz, 1H), 7.09 – 7.00 (m, 2H), 6.90 (td, J = 9.1, 2.1 Hz, 1H), 6.37 (s, 1H), 5.97 (s, 1H), 3.76 (q, J = 6.4 Hz, 2H), 3.03 (t, J = 6.5 Hz, 2H); 13C NMR (101 MHz, cd3od) δ 161.64, 159.89, 137.95, 128.85, 125.40, 123.79, 120.13, 114.21, 113.48, 108.09, 105.88, 99.94, 98.19, 41.34, 26.33 ppm; UV (λmax nm) 288; IR vmax (cm−1) 2924, 1613, 1556, 1454, 799; HRMS (ESI) calcd for C15H12Br2FN3O [M+H]− 425.9258, found 425.9262.
4,5-dibromo-N-(2-(7-methyl-1H-indol-3-yl)ethyl)-1H-pyrrole-2-carboxamide (25):
Compound was synthesized using the general procedure for condensing primary amines with 2,2,2-trichloroacetyl pyrroles with 23 (201 mg, 1.14 mmols) and 7 (300 mg, 0.81 mmols) as condensation partners to afford 25 as a tan solid (141 mg, 41%). 1H NMR (400 MHz, DMSO-d6) δ 12.70 (s, 1H), 10.79 (s, 1H), 8.27 (s, 1H), 7.40 (d, J = 7.4 Hz, 1H), 7.15 (s, 1H), 6.93 (s, 1H), 6.88 (t, J = 9.1 Hz, 2H), 3.50 (q, J = 6.3 Hz, 2H), 2.91 (t, J = 7.2 Hz, 2H), 2.44 (s, 3H); 13C NMR (101 MHz, dmso) δ 158.85, 135.79, 128.42, 126.92, 122.39, 121.45, 120.46, 118.49, 115.90, 112.43, 112.19, 104.34, 97.79, 39.57, 25.42, 16.79 ppm; UV (λmax nm) 286; IR vmax (cm−1) 3392, 3085, 1559, 777, 448; HRMS (ESI) calcd for C16H15Br2N3O [M+H]− 421.9509, found 421.9514.
1-methyl-1H-indole (26a):
Compound was synthesized via the general procedure for N-alkylation of indole with methyl iodide as the electrophile. Spectral data were consistent with previous reports.20
1-butyl-1H-indole (26b):
Compound was synthesized via the general procedure for N-alkylation of indole with butyl iodide (4.84 mLs, 42.7 mmols) as the electrophile to afford 26b as a yellow oil (1.36 g, 92%). 1H NMR (400 MHz, Chloroform-d) δ 7.90 (d, J = 8.5 Hz, 1H), 7.57 (d, J = 8.2 Hz, 1H), 7.46 (t, J = 7.6 Hz, 1H), 7.37 (t, J = 7.4 Hz, 1H), 7.28 (d, J = 3.1 Hz, 1H), 6.74 (d, J = 3.1 Hz, 1H), 4.27 (t, J = 7.1 Hz, 2H), 2.01 (p, J = 7.2 Hz, 2H), 1.59 – 1.51 (m, 2H), 1.17 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, cdcl3) δ 136.08, 128.71, 127.79, 121.34, 120.99, 119.22, 109.44, 100.92, 46.06, 32.37, 20.22, 13.75 ppm; UV (λmax nm) 298; IR vmax (cm−1) 2928, 1510, 1313, 734, 425; HRMS (ESI) calcd for C12H15N [M+H]+ 174.1277, found 174.1277.
1-benzyl-1H-indole (26c):
Compound was synthesized using the methods previously reported by Yu. Spectral data were consistent with previous reports.21
(E)-1-methyl-3-(2-nitrovinyl)-1H-indole (27a):
Compound was synthesized using the general procedure for synthesis of 3-(2-nitrovinyl) indoles with 26a (536 mg, 4.08 mmols) as the starting material to afford 27a as an orange solid (115 mg, 14%). 1H NMR (400 MHz, DMSO-d6) δ 8.34 (d, J = 13.4 Hz, 1H), 8.16 (s, 1H), 8.03 – 7.89 (m, 2H), 7.56 (d, J = 8.0 Hz, 1H), 7.33 (t, J = 7.6 Hz, 1H), 7.27 (t, J = 7.5 Hz, 1H), 3.84 (s, 3H); 13C NMR (101 MHz, dmso) δ 139.48, 138.27, 134.20, 131.00, 125.06, 123.42, 122.24, 120.69, 111.26, 107.25, 33.33 ppm; UV (λmax nm) 394; IR vmax (cm−1) 2918, 1614, 1296, 948, 740; HRMS (ESI) calcd for C11H10N2O2 [M+H]− 201.0669, found 201.0672
(E)-1-butyl-3-(2-nitrovinyl)-1H-indole (27b):
Compound was synthesized using the general procedure for synthesis of 3-(2-nitrovinyl) indoles with 26b (1.21 g, 6.98 mmols) as the starting material to afford 27b as a brown oil (545 mg, 32%). 1H NMR (400 MHz, Chloroform-d) δ 8.26 (d, J = 14.6 Hz, 1H), 7.78 – 7.71 (m, 2H), 7.55 (s, 1H), 7.44 (d, J = 7.9 Hz, 1H), 7.40 – 7.35 (m, 1H), 7.35 – 7.29 (m, 1H), 4.14 (t, J = 7.1 Hz, 2H), 1.84 (dq, J = 12.5, 5.7, 5.0 Hz, 2H), 1.37 (h, J = 7.3 Hz, 2H), 0.98 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, cdcl3) δ 137.74, 136.29, 133.63, 131.51, 125.51, 123.67, 122.32, 120.58, 110.83, 107.91, 46.75, 31.67, 19.96, 13.54 ppm; UV (λmax nm) 394; IR vmax (cm−1) 2929, 1615, 1294, 1174, 740; HRMS (ESI) calcd for C14H16N2O2 [M+H]+ 245.1284, found 245.1285.
(E)-1-benzyl-3-(2-nitrovinyl)-1H-indole (27c):
Compound was synthesized using the general procedure for synthesis of 3-(2-nitrovinyl) indoles with 26c (770 mg, 3.71 mmols) as the starting material to afford 27c as an orange solid (390 mg, 38%). 1H NMR (400 MHz, Chloroform-d) δ 8.23 (d, J = 13.4 Hz, 1H), 7.75 (d, J = 13.3 Hz, 2H), 7.53 (s, 1H), 7.37 (td, J = 14.0, 12.6, 9.3 Hz, 5H), 7.19 (d, J = 6.0 Hz, 2H), 5.33 (s, 2H); 13C NMR (101 MHz, cdcl3) δ 137.99, 136.27, 135.38, 133.43, 132.14, 129.11, 128.39, 127.17, 125.68, 124.04, 122.63, 120.72, 111.19, 108.55, 50.73 ppm; UV (λmax nm) 394; IR vmax (cm−1) 3105, 1616, 1488, 1247, 701; HRMS (ESI) calcd for C17H14N2O2 [M+H] 277.0982, found 277.0982.
(E)-7-methyl-3-(2-nitrovinyl)-1H-indole (28a):
Compound was synthesized using the general procedure for the reduction of N-alkylated 3-nitrovinyl indoles and taken to the next reaction without further purification.
2-(1-butyl-1H-indol-3-yl)ethan-1-amine (28b):
Compound was synthesized using the general procedure for the reduction of N-alkylated 3-nitrovinyl indoles and taken to the next reaction without further purification.
2-(1-benzyl-1H-indol-3-yl)ethan-1-amine (28c):
Compound was synthesized using the general procedure for the reduction of N-alkylated 3-nitrovinyl indoles and taken to the next reaction without further purification.
4,5-dibromo-N-(2-(1-methyl-1H-indol-3-yl)ethyl)-1H-pyrrole-2-carboxamide (29a):
Compound was synthesized using the general procedure for condensing primary amines with 2,2,2-trichloroacetyl pyrroles with 28a (330 mg, 1.89 mmols) and 7 (701 mg, 1.89 mmols) as condensation partners to afford 29a as a white solid (476 mg, 59%). 1H NMR (400 MHz, DMSO-d6) δ 12.75 (s, 1H), 8.33 (s, 1H), 7.60 (d, J = 7.8 Hz, 1H), 7.35 (d, J = 8.1 Hz, 1H), 7.21 – 7.07 (m, 2H), 7.08 – 6.91 (m, 2H), 3.70 (s, 3H), 3.58 – 3.44 (m, 2H), 2.94 (t, J = 6.4 Hz, 2H); 13C NMR (101 MHz, dmso) δ 158.99, 136.70, 128.45, 127.62, 127.13, 121.17, 118.59, 118.43, 112.57, 111.22, 109.60, 104.48, 97.92, 39.77, 32.26, 25.28 ppm; UV (λmax nm) 298; IR vmax (cm−1) 3113, 1639, 1516, 824, 736; HRMS (ESI) calcd for C16H15Br2N3O [M+H]− 421.9509, found 421.9521.
4,5-dibromo-N-(2-(1-butyl-1H-indol-3-yl)ethyl)-1H-pyrrole-2-carboxamide (29b):
Compound was synthesized using the general procedure for condensing primary amines with 2,2,2-trichloroacetyl pyrroles with 28b (204 mg, 0.95 mmols) and 7 (350 mg, 0.95 mmols) as condensation partners to afford 29b as a tan solid (168 mg, 38%). 1H NMR (400 MHz, DMSO-d6) δ 12.70 (s, 1H), 8.26 (s, 1H), 7.58 (d, J = 7.8 Hz, 1H), 7.39 (d, J = 8.1 Hz, 1H), 7.16 (s, 1H), 7.14 – 7.07 (m, 1H), 7.05 – 6.96 (m, 1H), 6.92 (s, 1H), 4.07 (t, J = 6.5 Hz, 2H), 3.62 – 3.39 (m, 2H), 2.91 (t, J = 6.9 Hz, 2H), 1.82 – 1.49 (m, 2H), 1.19 (dd, J = 14.7, 7.3 Hz, 2H), 0.84 (t, J = 7.2 Hz, 3H); 13C NMR (101 MHz, acetone) δ 160.09, 137.48, 129.43, 129.00, 126.97, 122.00, 119.62, 119.26, 113.08, 113.05, 112.35, 110.34, 99.44, 46.24, 40.79, 33.20, 26.21, 20.74, 14.01 ppm; UV (λmax nm) 296; IR vmax (cm−1) 3111, 1638, 1506, 745, 494; HRMS (ESI) calcd for C19H21Br2N3O [M+H]− 463.9978, found 463.9976.
N-(2-(1-benzyl-1H-indol-3-yl)ethyl)-4,5-dibromo-1H-pyrrole-2-carboxamide (29c):
Compound was synthesized using the general procedure for condensing primary amines with 2,2,2-trichloroacetyl pyrroles with 28c (400 mg, 1.59 mmols) and 7 (300 mg, 0.81 mmols) as condensation partners to afford 29c as a tan solid (117 mg, 15%). 1H NMR (300 MHz, DMSO-d6) δ 12.69 (s, 1H), 8.25 (s, 1H), 7.60 (d, J = 7.8 Hz, 1H), 7.40 (d, J = 8.1 Hz, 1H), 7.32 (s, 1H), 7.24 (t, J = 8.0 Hz, 3H), 7.16 (d, J = 7.7 Hz, 2H), 7.12 – 7.05 (m, 1H), 7.03 – 6.97 (m, 1H), 6.90 (s, 1H), 5.35 (s, 2H), 3.54 – 3.46 (m, 2H), 2.92 (t, J = 7.3 Hz, 2H); 13C NMR (101 MHz, dmso) δ 158.91, 138.40, 136.10, 128.51, 128.40, 127.86, 127.26, 127.04, 126.75, 121.28, 118.74, 118.63, 112.44, 111.71, 110.11, 104.43, 97.84, 48.94, 39.43, 25.16 ppm; UV (λmax nm) 290; IR vmax (cm−1) 3109, 1638, 824, 746, 490; HRMS (ESI) calcd for C22H19Br2N3O [M+H]− 497.9822, found 497.9821.
N-(2-(1H-indol-3-yl)ethyl)thiophene-2-carboxamide (30):
Tryptamine (152 mg, 0.984 mmols) and TEA (0.13 mLs, 0.984 mmols) were dissolved in 3 mLs anhydrous DCM and stirred for 10 mins at RT. To the stirring solution was added thiophene-2-carbonyl chloride (100 μL, 0.984 mmols) dropwise over five minutes. The reaction was stirred for an additional hour at RT and then poured into 75 mLs EtOAc and 25 mLs of deionized water, the aqueous layer was discarded and the organic was further washed thrice with water, twice with 25mLs 1N HCl, and once with 25 mLs brine. The organic layer was then dried with anhydrous magnesium sulfate and evaporated under reduced pressure. The crude solid was purified by flash chromatography (5–20% EtOAc/Hexanes) to afford 30 as a brown solid (192 mg, 80%).1H NMR (300 MHz, DMSO-d6) δ 10.83 (s, 1H), 8.65 (s, 1H), 7.74 (d, J = 4.0 Hz, 2H), 7.58 (d, J = 7.8 Hz, 1H), 7.34 (d, J = 8.0 Hz, 1H), 7.18 (s, 1H), 7.16 – 7.11 (m, 1H), 7.07 (t, J = 7.4 Hz, 1H), 6.98 (t, J = 7.3 Hz, 1H), 3.57 – 3.47 (q, 2H), 2.94 (t, J = 7.5 Hz, 2H); 13C NMR (101 MHz, cd3od) δ 164.44, 140.40, 138.12, 131.36, 129.42, 128.78, 128.68, 123.41, 122.31, 119.60, 119.34, 113.30, 112.21, 42.03, 26.38 ppm; UV (λmax nm) 292; IR vmax (cm−1) 3401, 1551, 741, 717, 504; HRMS (ESI) calcd for C15H14N2OS [M+H]+ 271.0899, found 271.0899.
N-(2-(1H-indol-3-yl)ethyl)-4-chloro-1H-pyrrole-2-carboxamide (31):
Compound was synthesized using the general procedure for condensing primary amines with 2,2,2-trichloroacetyl pyrroles with tryptamine (81 mg, 0.41 mmols) and 2,2,2-trichloro-1-(4-chloro-1H-pyrrol-2-yl)ethan-1-one (50 mg, 0.20 mmols) as condensation partners to afford 31 as a tan solid (46 mg, 79%). 1H NMR (300 MHz, DMSO-d6) δ 11.76 (s, 1H), 10.82 (s, 1H), 8.24 (s, 1H), 7.58 (d, J = 7.8 Hz, 1H), 7.34 (d, J = 8.0 Hz, 1H), 7.17 (s, 1H), 7.07 (t, J = 7.5 Hz, 1H), 7.03 – 6.92 (m, 2H), 6.77 (s, 1H), 3.49 (q, J = 6.8 Hz, 2H), 2.91 (t, J = 7.3 Hz, 2H); 13C NMR (101 MHz, cd3od) δ 162.67, 138.08, 128.72, 126.77, 123.39, 122.31, 120.07, 119.58, 119.31, 113.71, 113.31, 112.19, 110.69, 41.45, 26.50 ppm; UV (λmax nm) 288; R vmax (cm−1) 2922, 1610, 1516, 1454, 740; HRMS (ESI) calcd for C15H14ClN3O [M+H]− 286.0752, found 286.0752.
N-(2-(1H-indol-3-yl)ethyl)-4,5-dibromofuran-2-carboxamide (32):
Compound was synthesized using the general procedure for coupling carboxylic acids to primary amines using PyBOP. Tryptamine (712 mg, 4.44 mmols) and 4,5-dibromofuran-2-carboxylic acid (400 mg, 1.48 mmols) were coupled to afford 32 as a white solid (290 mg, 48%). 1H NMR (400 MHz, Chloroform-d) δ 8.51 (s, 1H), 7.62 (d, J = 7.8 Hz, 1H), 7.38 (d, J = 8.1 Hz, 1H), 7.22 (t, J = 7.2 Hz, 1H), 7.14 (t, J = 7.4 Hz, 1H), 7.10 (s, 1H), 7.02 (d, J = 1.8 Hz, 1H), 6.59 (s, 1H), 3.74 (q, J = 6.7 Hz, 2H), 3.06 (t, J = 6.8 Hz, 2H); 13C NMR (101 MHz, cdcl3) δ 156.64, 149.34, 136.51, 127.24, 125.21, 122.28, 122.22, 119.52, 118.61, 118.53, 112.40, 111.45, 104.32, 39.85, 25.37 ppm; UV (λmax nm) 286; IR vmax (cm−1) 3286, 1643, 1479, 978, 738; HRMS (ESI) calcd for C15H12Br2N2O2 [M+H]+ 410.9338, found 410.9329.
N-(2-(1H-indol-3-yl)ethyl)-4,5-dibromothiophene-2-carboxamide (33):
Compound was synthesized using the general procedure for coupling carboxylic acids to primary amines using PyBOP. Tryptamine (672 mg, 4.19 mmols) and 4,5-dibromothiophene-2-carboxylic acid (400 mg, 1.39 mmols) were coupled to afford 33 as a white solid (387 mg, 65%). 1H NMR (400 MHz, DMSO-d6) δ 10.83 (s, 1H), 8.80 (s, 1H), 7.76 (s, 1H), 7.55 (d, J = 7.8 Hz, 1H), 7.35 (d, J = 8.0 Hz, 1H), 7.17 (s, 1H), 7.07 (t, J = 7.5 Hz, 1H), 6.98 (t, J = 7.4 Hz, 1H), 3.51 (q, J = 6.7 Hz, 2H), 2.94 (t, J = 7.3 Hz, 2H); 13C NMR (101 MHz, dmso) δ 159.17, 141.61, 136.26, 129.95, 127.20, 122.74, 120.95, 118.27, 118.20, 116.21, 113.72, 111.54, 111.41, 40.22, 25.00 ppm; UV (λmax nm) 294; IR vmax (cm−1) 3285, 2921, 1620, 1540, 738; HRMS (ESI) calcd for C15H12Br2N2OS [M+H]− 424.8964, found 424.8966.
N-(2-(1H-indol-3-yl)ethyl)-5-bromofuran-2-carboxamide (34):
Compound was synthesized using the general procedure for coupling carboxylic acids to primary amines using PyBOP with modifications to the purification procedure. Tryptamine (671 mg, 4.19 mmols) and 5-bromofuran-2-carboxylic acid (400 mg, 2.09 mmols) were coupled under standard conditions. In lieu of flash chromatography, the crude product was recrystallized from chloroform to afford 34 as a white solid (202 mg, 29%). 1H NMR (400 MHz, DMSO-d6) δ 10.82 (s, 1H), 8.57 (s, 1H), 7.57 (d, J = 7.9 Hz, 1H), 7.34 (d, J = 7.6 Hz, 1H), 7.20 – 7.16 (m, 1H), 7.12 (d, J = 3.5 Hz, 1H), 7.06 (t, J = 7.5 Hz, 1H), 6.98 (t, J = 7.4 Hz, 1H), 6.74 (dd, J = 3.5, 1.1 Hz, 1H), 3.49 (q, J = 6.7 Hz, 2H), 2.92 (t, J = 7.5 Hz, 2H); 13C NMR (101 MHz, dmso) δ 156.60, 149.95, 136.23, 127.22, 124.10, 122.60, 120.92, 118.26, 118.21, 115.57, 113.91, 111.66, 111.36, 39.45, 25.16 ppm; UV (λmax nm) 286; IR vmax (cm−1) 3385, 1596, 1120, 797, 582; HRMS (ESI) calcd for C15H13BrN2O2 [M+H]− 331.0087, found 331.0090.
N-(2-(1H-indol-3-yl)ethyl)-3-bromothiophene-2-carboxamide (35):
Compound was synthesized using the general procedure for coupling carboxylic acids to primary amines using PyBOP with modifications to the purification procedure. Tryptamine (619 mg, 3.86 mmols) and 3-bromothiophene-2-carboxylic acid (400 mg, 1.93 mmols) were coupled under standard conditions. Flash chromatography (1% MeOH-NH3/DCM) followed by trituration of the solid in diethyl ether gave 35 as a white solid (249 mg, 36%). 1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 8.20 (s, 1H), 7.79 (d, J = 5.2 Hz, 1H), 7.59 (d, J = 7.8 Hz, 1H), 7.34 (d, J = 8.1 Hz, 1H), 7.21 (d, J = 2.3 Hz, 1H), 7.17 (d, J = 5.2 Hz, 1H), 7.11 – 7.03 (m, 1H), 6.99 (td, J = 7.5, 7.1, 1.0 Hz, 1H), 3.60 – 3.46 (m, 2H), 2.95 (t, J = 7.4 Hz, 2H); 13C NMR (101 MHz, dmso) δ 160.12, 136.29, 133.67, 131.61, 129.74, 127.20, 122.87, 120.98, 118.33, 118.28, 111.48, 111.40, 109.48, 40.31, 24.94 ppm; UV (λmax nm) 290; IR vmax (cm−1) 3275, 1605, 1536, 729, 544; HRMS (ESI) calcd for C15H13BrN2OS [M+H]− 346.9859, found 346.9863.
N-(2-(1H-indol-3-yl)ethyl)-5,6-dibromopicolinamide (36):
Compound was synthesized using the general procedure for coupling carboxylic acids to primary amines using PyBOP. Tryptamine (684 mg, 4.27 mmols) and 5,6-dibromopicolinic acid (400 mg, 1.42 mmols) were coupled to afford 36 as a white solid (274 mg, 46%). 1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 8.86 (s, 1H), 8.36 (d, J = 8.1 Hz, 1H), 7.93 (d, J = 8.1 Hz, 1H), 7.62 (d, J = 7.7 Hz, 1H), 7.34 (d, J = 8.0 Hz, 1H), 7.18 (s, 1H), 7.06 (t, J = 7.5 Hz, 1H), 6.98 (t, J = 7.4 Hz, 1H), 3.57 (q, J = 6.7 Hz, 2H), 3.02 – 2.89 (m, 2H); 13C NMR (101 MHz, dmso) δ 161.89, 149.71, 143.70, 141.51, 136.26, 127.22, 126.05, 122.69, 122.60, 120.95, 118.39, 118.23, 111.65, 111.36, 39.99, 25.14 ppm; UV (λmax nm) 288; IR vmax (cm−1) 3256, 1672, 1524, 1339, 744; HRMS (ESI) calcd for C16H13Br2N3O [M+H]+ 421.9498, found 421.9493.
N-(2-(1H-indol-3-yl)ethyl)-6-bromopicolinamide (37):
Compound was synthesized using the general procedure for coupling carboxylic acids to primary amines using PyBOP. Tryptamine (476 mg, 2.97 mmols) and 6-bromopicolinic acid (300 mg, 1.49 mmols) were coupled to afford 37 as a white solid (251 mg, 49%). 1H NMR (400 MHz, DMSO-d6) δ 10.83 (s, 1H), 8.81 (s, 1H), 8.06 (d, J = 7.5 Hz, 1H), 7.94 (t, J = 7.7 Hz, 1H), 7.85 (d, J = 7.9 Hz, 1H), 7.63 (d, J = 7.8 Hz, 1H), 7.34 (d, J = 8.6 Hz, 1H), 7.19 (s, 1H), 7.07 (t, J = 7.5 Hz, 1H), 6.98 (t, J = 7.4 Hz, 1H), 3.59 (q, J = 6.7 Hz, 2H), 2.96 (t, J = 7.5 Hz, 2H); 13C NMR (75 MHz, acetone) δ 163.21, 152.64, 141.30, 141.03, 137.58, 131.43, 128.50, 123.17, 122.14, 122.04, 119.45, 119.40, 113.20, 112.10, 40.96, 29.84, 26.28 ppm; UV (λmax nm) 290; IR vmax (cm−1) 3372, 1666, 1552, 1408, 746; HRMS (ESI) calcd for C16H14BrN3O [M+H]+ 344.0393, found 344.0391.
N-(2-(1H-indol-3-yl)ethyl)-5-bromonicotinamide (38):
Tryptamine (581 mg, 3.63 mmols) and trimethylamine (762 μL, 5.44 mmols) were dissolved in anhydrous DMF under argon and stirred for 15 mins and 5-bromonicotinoyl chloride (400 mg, 1.81 mmols) was added in a single portion. The reaction was allowed to stir at RT for 2 hours and was then poured into 75 mLs EtOAc / 25 mLs deionized water. The aqueous layer was discarded and the organic layer was washed twice with water (25 mLs) and once with brine (25 mLs). The organic layer was dried with anhydrous magnesium sulfate and evaporated under reduced pressure. The crude solid was triturated in diethyl ether to afford pure 38 as a tan solid (372 mg, 60%). 1H NMR (400 MHz, DMSO-d6) δ 10.85 (s, 1H), 8.96 (s, 1H), 8.92 (s, 1H), 8.85 (s, 1H), 8.40 (s, 1H), 7.57 (d, J = 7.8 Hz, 1H), 7.34 (d, J = 8.1 Hz, 1H), 7.20 (s, 1H), 7.07 (t, J = 7.5 Hz, 1H), 6.98 (t, J = 7.4 Hz, 1H), 3.55 (q, J = 6.7 Hz, 2H), 2.96 (t, J = 7.3 Hz, 2H); 13C NMR (101 MHz, dmso) δ 163.70, 152.81, 147.44, 137.69, 136.69, 132.14, 127.69, 123.21, 121.38, 120.48, 118.69, 118.66, 112.10, 111.86, 40.84, 25.37 ppm; UV (λmax nm) 286; IR vmax (cm−1) 3392, 1628, 1545, 739, 692; HRMS (ESI) calcd for C16H14BrN3O [M+H]− 342.0247, found 342.0248.
N-(2-(1H-indol-3-yl)ethyl)picolinamide (39):
Compound was synthesized using the general procedure for coupling carboxylic acids to primary amines using PyBOP. Tryptamine (1.56 g, 9.74 mmols) and picolinic acid (400 mg, 3.25 mmols) were coupled to afford 39 as a tan solid (475 mg, 55%). 1H NMR (300 MHz, Chloroform-d) δ 8.74 (s, 1H), 8.49 (d, J = 4.7 Hz, 1H), 8.36 – 8.27 (m, 1H), 8.24 (d, J = 7.8 Hz, 1H), 7.85 – 7.74 (m, 1H), 7.68 (d, J = 7.7 Hz, 1H), 7.36 (dd, J = 7.5, 4.9 Hz, 2H), 7.24 – 7.17 (m, 1H), 7.17 – 7.08 (m, 1H), 7.03 (s, 1H), 3.85 (q, J = 7.0 Hz, 2H), 3.12 (t, J = 7.0 Hz, 2H); 13C NMR (75 MHz, cdcl3) δ 164.51, 149.87, 148.08, 137.35, 136.49, 127.32, 126.13, 122.27, 122.11, 121.93, 119.22, 118.72, 112.67, 111.37, 39.86, 25.55 ppm; UV (λmax nm) 286; IR vmax (cm−1) 3244, 1662, 1522, 1430, 747; HRMS (ESI) calcd for C16H15N3O [M+H]+ 266.1287, found 266.1282.
2,2,2-trichloro-1-(4,5-dibromo-1-methyl-1H-pyrrol-2-yl)ethan-1-one (40a):
Compound was synthesized using the methods previously reported by Richards. Spectral data were consistent with previous reports.14
2,2,2-trichloro-1-(4,5-dibromo-1-butyl-1H-pyrrol-2-yl)ethan-1-one (40b):
7 (500 mg, 1.35 mmols) was dissolved in 15 mLs DMF and cooled to 0°C under argon. Sodium hydride (59 mg, 1.48 mmols) was added in portions to the solution which was then stirred at 0°C for 30 mins. Butyl iodide (460 μL, 4.05 mmols) was then added to the solution and stirred at RT for 18 hours. The reaction was poured into 75 mLs EtOAc and 25 mLs of deionized water, the aqueous layer was discarded and the organic was further washed thrice with water, twice with 25 mLs 1N HCl, and once with 25 mLs brine. The organic layer was then dried with anhydrous magnesium sulfate and evaporated under reduced pressure. The crude solid was purified by flash chromatography (5–20% EtOAc/Hexanes) to afford 40b as a brown oil (299 mg, 52%). 1H NMR (300 MHz, Chloroform-d) δ 7.58 (s, 1H), 4.60 – 4.30 (m, 2H), 1.68 (p, J = 7.6 Hz, 2H), 1.39 (dt, J = 14.9, 7.4 Hz, 2H), 1.02 – 0.93 (m, 3H); 13C NMR (101 MHz, cdcl3) δ 171.25, 125.09, 122.64, 119.02, 100.45, 95.57, 50.00, 32.33, 19.87, 13.83 ppm; UV (λmax nm) 324; IR vmax (cm−1) 2959, 1675, 1360, 830, 683; HRMS (ESI) calcd for C10H10Br2Cl3NO [M+H]− 421.8122, found 421.8130.
2,2,2-trichloro-1-(4,5-dibromo-1-butyl-1H-pyrrol-2-yl)ethan-1-one (40c):
Compound was synthesized using the methods previously reported by Richards. Spectral data were consistent with previous reports.14
N-(2-(1H-indol-3-yl)ethyl)-4,5-dibromo-1-methyl-1H-pyrrole-2-carboxamide (41a):
Compound was synthesized using the methods previously reported by Richards. Spectral data were consistent with previous reports.14
N-(2-(1H-indol-3-yl)ethyl)-4,5-dibromo-1-butyl-1H-pyrrole-2-carboxamide (41b):
Compound was synthesized using the general procedure for condensing primary amines with 2,2,2-trichloroacetyl pyrroles with tryptamine (179 mg, 1.11 mmols) and 40b (238 mg, 0.56 mmols) as condensation partners to afford 41b as a tan solid (176 mg, 68%). 1H NMR (400 MHz, DMSO-d6) δ 10.83 (s, 1H), 8.34 (s, 1H), 7.56 (d, J = 7.8 Hz, 1H), 7.36 (d, J = 8.0 Hz, 1H), 7.18 (s, 1H), 7.08 (t, J = 7.5 Hz, 1H), 7.03 – 6.92 (m, 2H), 4.42 (t, J = 7.3 Hz, 2H), 3.49 (q, J = 6.8 Hz, 2H), 2.93 (t, J = 7.3 Hz, 2H), 1.59 (p, J = 7.6 Hz, 2H), 1.25 (dq, J = 14.3, 7.1 Hz, 2H), 0.86 (s, 3H); 13C NMR (75 MHz, cdcl3) δ 160.45, 136.43, 127.25, 127.18, 122.21, 122.16, 119.43, 118.58, 113.74, 112.50, 111.41, 110.53, 98.01, 48.03, 39.83, 33.00, 25.30, 19.73, 13.77 ppm; UV (λmax nm) 286; IR vmax (cm−1) 3281, 1630, 1421, 807, 736; HRMS (ESI) calcd for C19H21Br2N3O [M+H]− 463.9978, found 463.9980.
N-(2-(1H-indol-3-yl)ethyl)-1-benzyl-4,5-dibromo-1H-pyrrole-2-carboxamide (41c):
Compound was synthesized using the general procedure for condensing primary amines with 2,2,2-trichloroacetyl pyrroles with tryptamine (170 mg, 1.06 mmols) and 40c (245 mg, 0.53 mmols) as condensation partners to afford 41c as a tan solid (143 mg, 53%). 1H NMR (300 MHz, DMSO-d6) δ 10.80 (s, 1H), 8.42 (s, 1H), 7.52 (d, J = 7.9 Hz, 1H), 7.29 (dt, J = 14.8, 8.4 Hz, 4H), 7.04 (dq, J = 21.3, 7.3 Hz, 6H), 5.76 (s, 2H), 3.46 – 3.39 (m, 2H), 2.86 (t, J = 7.2 Hz, 2H); 13C NMR (101 MHz, dmso) δ 159.65, 137.66, 136.25, 128.53, 128.53, 128.07, 127.24, 127.18, 126.35, 122.70, 120.95, 118.27, 114.55, 111.72, 111.41, 110.35, 97.90, 50.07, 39.73, 25.10 ppm; UV (λmax nm) 290; IR vmax (cm−1) 3398, 1626, 1327, 772, 692; HRMS (ESI) calcd for C22H19Br2N3O [M+H]− 497.9822, found 497.9827.
2,2,2-trichloro-1-(4,5-diiodo-1H-pyrrol-2-yl)ethan-1-one (42):
Compound was synthesized using the methods previously reported by Essa. Spectral data were consistent with previous reports.17
2,2,2-trichloro-1-(4-iodo-1H-pyrrol-2-yl)ethan-1-one (43):
Compound was synthesized using the methods previously reported by Essa and was isolated as a side product in this procedure along with the major product 42. Spectral data were consistent with previous reports.17
N-(2-(1H-indol-3-yl)ethyl)-4,5-diiodo-1H-pyrrole-2-carboxamide (44):
Compound was synthesized using the general procedure for condensing primary amines with 2,2,2-trichloroacetyl pyrroles with tryptamine (376 mg, 2.34 mmols) and 42 (545 mg, 1.17 mmols) as condensation partners to afford 44 as a white solid (121 mg, 21%). 1H NMR (400 MHz, DMSO-d6) δ 12.32 (s, 1H), 10.81 (s, 1H), 8.19 (s, 1H), 7.57 (d, J = 7.8 Hz, 1H), 7.34 (d, J = 8.0 Hz, 1H), 7.15 (s, 1H), 7.06 (t, J = 7.5 Hz, 1H), 6.97 (t, J = 7.3 Hz, 1H), 6.90 (s, 1H), 3.48 (q, J = 6.5 Hz, 2H), 2.90 (t, J = 7.2 Hz, 2H); 13C NMR (101 MHz, cd3od) δ 161.37, 138.12, 133.82, 128.74, 123.44, 122.30, 119.85, 119.58, 119.31, 113.29, 112.20, 82.15, 76.24, 41.46, 26.46 ppm; UV (λmax nm) 288; IR vmax (cm−1) 3556, 2396, 1597, 1451, 747; HRMS (ESI) calcd for C15H13I2N3O [M+H]− 503.9075, found 503.9075.
N-(2-(1H-indol-3-yl)ethyl)-4-iodo-1H-pyrrole-2-carboxamide (45):
Compound was synthesized using the general procedure for condensing primary amines with 2,2,2-trichloroacetyl pyrroles with tryptamine (516 mg, 3.22 mmols) and 43 (545 mg, 1.61 mmols) as condensation partners to afford 45 as a tan solid (172 mg, 28%). 1H NMR (400 MHz, DMSO-d6) δ 11.80 (s, 1H), 10.81 (s, 1H), 8.22 (s, 1H), 7.58 (d, J = 7.8 Hz, 1H), 7.35 (d, J = 8.8 Hz, 1H), 7.17 (s, 1H), 7.07 (t, J = 7.5 Hz, 1H), 7.03 – 6.94 (m, 2H), 6.91 (s, 1H), 3.50 (q, J = 6.6 Hz, 2H), 2.92 (t, J = 7.4 Hz, 2H); 13C NMR (101 MHz, cd3od) δ 162.28, 138.06, 129.08, 128.71, 127.78, 123.39, 122.29, 119.58, 119.30, 118.13, 113.30, 112.18, 60.99, 41.44, 26.49 ppm; UV (λmax nm) 284; IR vmax (cm−1) 3341, 1633, 1548, 752, 592; HRMS (ESI) calcd for C15H14IN3O [M+H]− 378.0108, found 378.0108.
N-((1H-indol-3-yl)methyl)-6-bromopicolinamide (46):
Compound was synthesized using the general procedure for coupling carboxylic acids to primary amines using PyBOP. (1H-indol-3-yl)methanamine (362 mg, 2.47 mmols) and 6-bromopicolinic acid (250 mg, 1.24 mmols) were coupled to afford 46 as a red solid (22 mg, 6.9%). 1H NMR (400 MHz, DMSO-d6) δ 10.95 (s, 1H), 8.82 (s, 1H), 8.08 (d, J = 7.5 Hz, 1H), 7.93 (t, J = 7.9 Hz, 1H), 7.82 (d, J = 7.9 Hz, 1H), 7.67 (d, J = 7.9 Hz, 1H), 7.35 (d, J = 8.2 Hz, 1H), 7.31 (s, 1H), 7.07 (t, J = 7.5 Hz, 1H), 6.97 (t, J = 7.5 Hz, 1H), 4.64 (d, J = 6.0 Hz, 2H); 13C NMR (101 MHz, cdcl3) δ 162.75, 151.07, 140.61, 139.68, 136.48, 130.72, 126.64, 123.64, 122.39, 121.38, 119.88, 118.93, 112.26, 111.48, 35.16 ppm; UV (λmax nm) 286; IR vmax (cm−1) 3434, 1665, 1515, 759, 492; HRMS (ESI) calcd for C15H12BrN3O [M+Na]+ 352.0056, found 352.0049.
N-((1H-indol-3-yl)methyl)-4,5-diiodo-1H-pyrrole-2-carboxamide (47):
Compound was synthesized using the general procedure for condensing primary amines with 2,2,2-trichloroacetyl pyrroles with (1H-indol-3-yl)methanamine (182 mg, 1.25 mmols) and 42 (290 mg, 0.62 mmols) as condensation partners to afford 47 as a pink solid (146 mg, 48%). 1H NMR (400 MHz, DMSO-d6) δ 12.31 (s, 1H), 10.92 (s, 1H), 8.31 (s, 1H), 7.59 (d, J = 7.8 Hz, 1H), 7.35 (d, J = 8.1 Hz, 1H), 7.27 (s, 1H), 7.07 (t, J = 7.5 Hz, 1H), 6.96 (t, J = 7.5 Hz, 1H), 6.92 (s, 1H), 4.55 (d, J = 5.3 Hz, 2H) 13C NMR (101 MHz, dmso) δ 158.25, 136.31, 132.93, 126.47, 123.89, 121.13, 118.74, 118.50, 118.24, 112.31, 111.41, 83.00, 76.03, 33.94 ppm; UV (λmax nm) 290; IR vmax (cm−1) 3386, 3145, 1643, 1543, 809; HRMS (ESI) calcd for C14H11I2N3O [M+H]− 489.8918, found 489.8915.
Supplementary Material
4. Acknowledgements
The authors would like to thank the National Institutes of Health for support (GM055769 and AI136904)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests
Dr. Melander is a co-founder and board of directors member of Agile Sciences, a biotechnology company seeking to commercialize antibiotic adjuvants.
6. Notes and References
- 1.O’Neill J, The Review on Antimicrobial resistance 2014.
- 2.Clatworthy AE; Pierson E; Hung DT, Targeting virulence: a new paradigm for antimicrobial therapy. Nat Chem Biol 2007, 3 (9), 541–8. [DOI] [PubMed] [Google Scholar]
- 3.Garonzik SM; Li J; Thamlikitkul V; Paterson DL; Shoham S; Jacob J; Silveira FP; Forrest A; Nation RL, Population pharmacokinetics of colistin methanesulfonate and formed colistin in critically ill patients from a multicenter study provide dosing suggestions for various categories of patients. Antimicrob. Agents Chemother. 2011, 55 (7), 3284–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.CLSI, Performance Standards for Antimicrobial Susceptibility Testing. 28th ed.; Clinical Laboratory Standards Institute: Wayne, PA, 2018. [Google Scholar]
- 5.Wanty C; Anandan A; Piek S; Walshe J; Ganguly J; Carlson RW; Stubbs KA; Kahler CM; Vrielink A, The Structure of the Neisserial Lipooligosaccharide Phosphoethanolamine Transferase A (LptA) Required for Resistance to Polymyxin. J. Mol. Biol 2013, 425 (18), 3389–3402. [DOI] [PubMed] [Google Scholar]
- 6.Pelletier MR; Casella LG; Jones JW; Adams MD; Zurawski DV; Hazlett KR; Doi Y; Ernst RK, Unique structural modifications are present in the lipopolysaccharide from colistin-resistant strains of Acinetobacter baumannii. Antimicrob Agents Chemother 2013, 57 (10), 4831–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Snitkin ES; Zelazny AM; Gupta J; Program NCS; Palmore TN; Murray PR; Segre JA, Genomic insights into the fate of colistin resistance and Acinetobacter baumannii during patient treatment. Genome research 2013, 23 (7), 1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y-Y; Wang Y; Walsh TR; Yi L-X; Zhang R; Spencer J; Doi Y; Tian G; Dong B; Huang X; Yu L-F; Gu D; Ren H; Chen X; Lv L; He D; Zhou H; Liang Z; Liu J-H; Shen J, Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. The Lancet Infectious Diseases 2016, 16 (2), 161–168. [DOI] [PubMed] [Google Scholar]
- 9.Wang X; Wang Y; Zhou Y; Li J; Yin W; Wang S; Zhang S; Shen J; Shen Z; Wang Y, Emergence of a novel mobile colistin resistance gene, mcr-8, in NDM-producing Klebsiella pneumoniae. Emerging Microbes & Infections 2018, 7 (1), 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harris TL; Worthington RJ; Hittle LE; Zurawski DV; Ernst RK; Melander C, Small molecule downregulation of PmrAB reverses lipid A modification and breaks colistin resistance. ACS chemical biology 2014, 9 (1), 122–7. [DOI] [PubMed] [Google Scholar]
- 11.Brackett CM; Furlani RE; Anderson RG; Krishnamurthy A; Melander RJ; Moskowitz SM; Ernst RK; Melander C, Second generation modifiers of colistin resistance show enhanced activity and lower inherent toxicity. Tetrahedron 2016, 72 (25), 3549–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barker WT; Martin SE; Chandler CE; Nguyen TV; Harris TL; Goodell C; Melander RJ; Doi Y; Ernst RK; Melander C, Small molecule adjuvants that suppress both chromosomal and mcr-1 encoded colistin-resistance and amplify colistin efficacy in polymyxin-susceptible bacteria. Bioorg Med Chem 2017, 25 (20), 5749–5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu YY; Chandler CE; Leung LM; McElheny CL; Mettus RT; Shanks RMQ; Liu JH; Goodlett DR; Ernst RK; Doi Y, Structural Modification of Lipopolysaccharide Conferred by mcr-1 in Gram-Negative ESKAPE Pathogens. Antimicrob. Agents Chemother. 2017, 61 (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richards JJ; Ballard TE; Huigens RW III; Melander C, Synthesis and screening of an oroidin library against Pseudomonas aeruginosa biofilms. ChemBioChem 2008, 9 (8), 1267–1279. [DOI] [PubMed] [Google Scholar]
- 15.Szostak M; Sautier B; Spain M; Procter DJ, Electron Transfer Reduction of Nitriles Using SmI2–Et3N–H2O: Synthetic Utility and Mechanism. Organic Letters 2014, 16 (4), 1092–1095. [DOI] [PubMed] [Google Scholar]
- 16.Holloway CA; Muratore ME; Storer R. l.; Dixon DJ, Direct Enantioselective Brønsted Acid Catalyzed N-Acyliminium Cyclization Cascades of Tryptamines and Ketoacids. Organic Letters 2010, 12 (21), 4720–4723. [DOI] [PubMed] [Google Scholar]
- 17.Essa AH; Lerrick RI; Tuna F; Harrington RW; Clegg W; Hall MJ, Reduction of 2,2,2-trichloro-1-arylethanones by RMgX: mechanistic investigation and the synthesis of substituted α,α-dichloroketones. Chemical Communications 2013, 49 (27), 2756–2758. [DOI] [PubMed] [Google Scholar]
- 18.Muratore ME; Holloway CA; Pilling AW; Storer RI; Trevitt G; Dixon DJ, Enantioselective Brønsted Acid-Catalyzed N-Acyliminium Cyclization Cascades. Journal of the American Chemical Society 2009, 131 (31), 10796–10797. [DOI] [PubMed] [Google Scholar]
- 19.Blough BE; Landavazo A; Partilla JS; Decker AM; Page KM; Baumann MH; Rothman RB, Alpha-ethyltryptamines as dual dopamine–serotonin releasers. Bioorganic & Medicinal Chemistry Letters 2014, 24 (19), 4754–4758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poot AJ; van Ameijde J; Slijper M; van den Berg A; Hilhorst R; Ruijtenbeek R; Rijkers DTS; Liskamp RMJ, Development of Selective Bisubstrate-Based Inhibitors Against Protein Kinase C (PKC) Isozymes By Using Dynamic Peptide Microarrays. ChemBioChem 2009, 10 (12), 2042–2051. [DOI] [PubMed] [Google Scholar]
- 21.Yu H; Yu Z, Direct Alkenylation of Indoles with α-Oxo Ketene Dithioacetals: Efficient Synthesis of Indole Alkaloids Meridianin Derivatives. Angewandte Chemie International Edition 2009, 48 (16), 2929–2933. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.