

Abstract
Hydrogen deuterium exchange mass spectrometry (H/DX MS) provides a quantitative comparison of the relative rates of exchange of amide protons for solvent deuterons. In turn, the rate of amide exchange depends on a complex combination of the stability of local secondary structure, solvent accessibility, and dynamics. H/DX MS has, therefore, been widely used to probe structure and function of soluble proteins, but its application to membrane proteins was limited previously to detergent solubilized samples. The large excess of lipids from model membranes, or from membrane fractions derived from in vivo samples, presents challenges with mass spectrometry. The lipid nanodisc platform, consisting of apolipoprotein A-derived membrane scaffold proteins, provides a native like membrane environment in which to capture analyte membrane proteins with a well defined, and low, ratio of lipid to protein. Membrane proteins in lipid nanodiscs are amenable to H/DX MS, and this is expected to lead to a rapid increase in the number of membrane proteins subjected to this analysis. Here we review the few literature examples of the application of H/DX MS to membrane proteins in nanodiscs. The incremental improvements in the experimental workflow of the H/DX MS are described and potential applications of this approach to study membrane proteins are described.
Keywords: Conformational dynamics, Conformational exchange, H/DX mass spectrometry, Membrane protein, Nanodisc
1. Introduction
Lipid nanodisc technology has matured dramatically since its initial introduction, and nanodiscs have enabled the study of membrane proteins by many biochemical and biophysical techniques that were previously considered to be inapplicable. Many recent reviews amplify this maturity, and nanodiscs have been exploited for structural, thermodynamic, and functional studies (Bayburt and Sligar, 2010; Denisov and Sligar, 2017; 2016; Rouck et al., 2017). Arguably, the use of lipid nanodiscs to enable structural and functional studies of membrane proteins might appear to be nearly ‘routine,’ on the basis of this issue of The Chemistry and Physics of Lipids dedicated to the topic of nanodiscs. However, the application of nanodiscs is not always routine and some limits to their utility exist even with some mature technologies. A mature technology that has only been used in combination with lipid nanodiscs in a few cases is hydrogen deuterium exchange mass spectrometry (H/DX MS). H/DX MS provides a probe of peptide amide structure, dynamics, and solvent accessibility and it has been applied extensively for soluble proteins for a few decades (Busenlehner and Armstrong, 2005; Johnson and Walsh, 1994; Nemirovskiy et al., 1999; Oganesyan et al., 2018; Pirrone et al., 2015a). In contrast, the use of H/DX MS to study membrane proteins remains sparse, and was largely limited to detergent-solubilized samples until recently (Busenlehner et al., 2008; 2006). With only modest additional experimental workup, H/DX MS can be applied to lipid nanodiscs, which provide a platform with lipid membrane properties in the absence of other proteins that normally inhabit cellular membranes. Based on our own experience with this combination of tools, and very recent work from other labs, the time is right for a review of the systems for which H/DX MS and lipid nanodiscs have been exploited, and of the novel insights obtained. We limit this review to examples with lipid nanodiscs that utilize the apo-lipoprotein A-derived membrane scaffold proteins, originally designed by Sligar and co-workers (Bayburt and Sligar, 2010; Denisov and Sligar, 2017; 2016; Rouck et al., 2017), but other membrane mimics might be used as well (Duc et al., 2015; Hall et al., 2018; Pirrone et al., 2015b; Reading et al., 2017; Schmidt and Sturgis, 2018). Because both lipid nanodiscs and H/DX MS have been reviewed extensively as separate topics, we include only brief overviews of each. This review emphasizes examples of their successful combination.
The aim of this highly focused review is to recruit additional investigators to this small, but rapidly growing, community. First we provide a brief overview of the H/DX MS method, followed by examples of its application for proteins in lipid nanodiscs. Some of the published work is from our lab but we have included all other examples that we were able to find. In addition to a review of these published results, we include a prospective of this field based on potential methodological improvements that others and we have recently incorporated.
1.1. Overview of Lipid Nanodiscs
The nanodiscs that are the focus of this review are self assembled proteolipid particles with membrane scaffold proteins (MSP) that encapsulate a well-defined number of phospholipid molecules in a bilayer. Two MSPs per nanodisc particle form helical belts around the periphery of the lipid bilayers. The size of the discs and number of encapsulated lipid molecules are dictated by the specific MSP, which may be chosen from a number of engineered variants of Apo lipoprotein A. Several reviews describe these MSPs in detail. Interestingly, the Apo lipoprotein A from which MSPs are derived is involved in the formation of spherical LDL particles from nascent discoidal particles in vivo. It should be emphasized that each target protein-MSP combination requires optimization of lipid:MSP:protein ratios and reconstitution conditions, as reviewed previously. This can require significant investment of time, but once a protocol is optimized generation of protein-nanodiscs is highly reproducible. Of course, after experiments with one type of nanodisc are completed, if a change in MSP or lipid is desired then re-optimization will be required.
Despite these challenges, lipid nanodiscs provide a way to maintain monomeric, monodisperse, preparations of purified membrane proteins in a lipid bilayer environment, without the need for detergents to prevent aggregation. Furthermore, they can, in principle, allow for the systematic study of the effects of lipid composition on membrane protein structure and dynamics. Liposomes provide an alternative membrane mimic for H/DX MS of membrane proteins, with some potential advantages, but they can also be polydisperse and the higher ratios of lipid:protein can add to difficulties in chromatography required for peptide resolution.
1.2. Overview of the H/DX MS Theory
The static conformations of a protein obtained by X-ray crystallography and electron-microscopy are rarely sufficient to capture all of the relevant mechanistic features for a given system. In solution, a protein can sample a vast conformational landscape and a firm understanding of a protein’s conformational dynamics is critical to understanding its function. Despite advances in crystallography, it is still difficult to perform crystallographic analyses for many membrane proteins (Carpenter et al., 2008; Moraes et al., 2014; Parker and Newstead, 2016). H/DX MS in combination with nanodisc technology is an alternative approach that allows one to characterize a protein’s spatial and temporal conformational dynamics. While NMR is heavily utilized for a wide range of protein dynamics experiments, the protein/complex size limitations, isotope costs, and large sample requirement make this approach rather intractable for many challenging, large membrane protein/nanodisc systems. Many older elegant NMR studies demonstrated the utility of mapping H/DX of individual amides to probe structure of soluble proteins (Jeng et al., 1990; Krishna et al., 2004), but the requirement for high sample concentrations and the presence of lipids has made H/DX NMR studies intractable for membrane proteins. In contrast, the exchange of protons for deuterons is easily monitored by mass spectrometry, and can be localized to specific peptides within the primary structure, upon proteolytic digestion. There is no theoretical protein size limitation for H/DX MS (spectra are somewhat more crowded), and typical experiments with several time points only require 100s of picomoles to a few nanomoles of the target protein. Despite the utility of H/DX as a tool to probe dynamics and structure, the technique is most powerful when combined with other methods. H/DX is not capable of generating de novo structural models, nor can it unambiguously distinguish between changes in secondary structure vs. changes in solvation due to environmental changes. Thus, H/DX should be viewed as a powerful, but complementary, probe of behavior.
Exposure of a protein to a D2O buffer induces exchange of amide backbone hydrogens for deuterium. Note that the experiment can also be done in reverse, where a fully labeled protein is exposed to an H2O buffer. In either case, deuterium exchange is typically measured at the peptide level by monitoring the shift in the mass envelope for each peptide over time. Regions with defined secondary structural elements, such as α-helices and β-sheets, are stabilized by hydrogen bonding networks involving the backbone amide groups, so they tend to exchange more slowly than unstructured regions. HDX will also report on sequestration of regions of a protein in a solvent-inaccessible hydrophobic core, as deuteration will require both exposure of the region to solvent and fluctuations in hydrogen bonding. In some special cases, a bimodal isotopic distribution (two mass envelopes) is observed for a single peptide. Regions that display this type of behavior are associated with slow, correlated protein motions. We briefly expand on the two modes of uptake but urge the reader to explore more in-depth reviews (Ferraro et al., 2004; Konermann et al., 2014; Weis et al., 2006).
Linderstrøm-Lang and colleagues first described H/DX and their kinetic model is shown in figure 1 (Berger and Linderstrom-Lang, 1957; Englander et al., 1997). In their model, amides equilibrate between exchange-incompetent (N-Hclosed) and exchange-competent (N-Hopen) states, and the relative population of each is influenced by the rate of opening (kop) and closing (kcl) motions. When a local unfolding event exposes a backbone amide, thereby rendering it “exchange-competent”, deuterium uptake can occur and is influenced by the intrinsic rate of exchange (kch). Two conformational regimes are frequently distinguished in the H/DX literature, known as ‘EX1’ and ‘EX2’ exchange kinetics. Both modes arise from the relationship between the rate of re-folding (kcl) and the rate of deuterium exchange (kch). The majority of peptides identified in H/DX MS studies exhibit EX2 behavior, wherein the rate of re-folding is much faster than the rate of exchange (kcl >> kch). In this case, several opening and closing events can take place before exchange occurs. A single population is observed. EX1 kinetics are less common and occur under the opposite conditions (kcl << kch) where the rate-limiting step is the rate of opening (kop). In this case, once the amide is exposed, deuterium exchange occurs before the re-folding event can occur. Multiple populations are observed and the rate of uptake in this case is equal to the rate of opening (kop).
Figure 1.
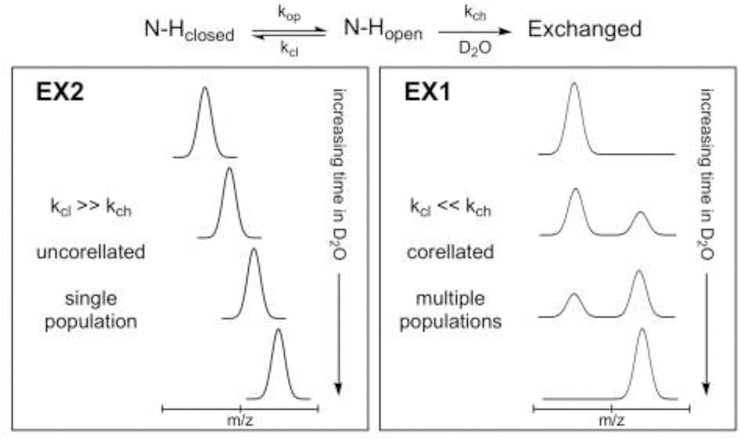
Protein unfolding and deuterium labeling in H/DX MS. The overall kinetic scheme is shown at the top and the resulting extremes of the two kinetic limits, EX2 and EX1, shown below. Figure adapted from (Weis et al., 2006).
1.3. Overview of H/DX MS Methodology
Below, we detail the experimental workflow for a standard continuous labeling experiment (Engen and Wales, 2015). In the H/DX analysis of soluble proteins, the target protein is diluted into D2O buffer (pD ≈ pH + 0.4, (Glasoe and Long, 1960)) and incubated for varying times ranging from a few seconds to many hours. These time points are selected based on the inherent dynamics of the protein and the regions of interest to give an overall profile. The intrinsic rate of exchange is temperature and pH dependent, so maintenance of these conditions is critical (Englander, 2006; Marcsisin and Engen, 2010). After discrete times following introduction of D2O, the exchange processes are quenched by rapid acidification and the temperature is decreased (pH 2.5 and 0 °C). The quench conditions need to be maintained for the remainder of the experiment to minimize “back exchange” (deuterated amides begin to exchange with the protonated quench and LC buffer). Samples are then digested typically with pepsin in solution or chromatographed through an immobilized pepsin column. Other proteolytic enzymes that function at low pH can be used for alternative peptide coverage. The resulting peptides are analyzed by LC-MS. Briefly, digested samples are separated on a reverse-phase column (typically C18) with an acetonitrile gradient at low pH (0.1% formic acid). Near-freezing conditions must be maintained during chromatography and digestion/separation time should be minimized to limit back-exchange. Results are often displayed in a butterfly plot as shown in Figure 4.
Figure 4.
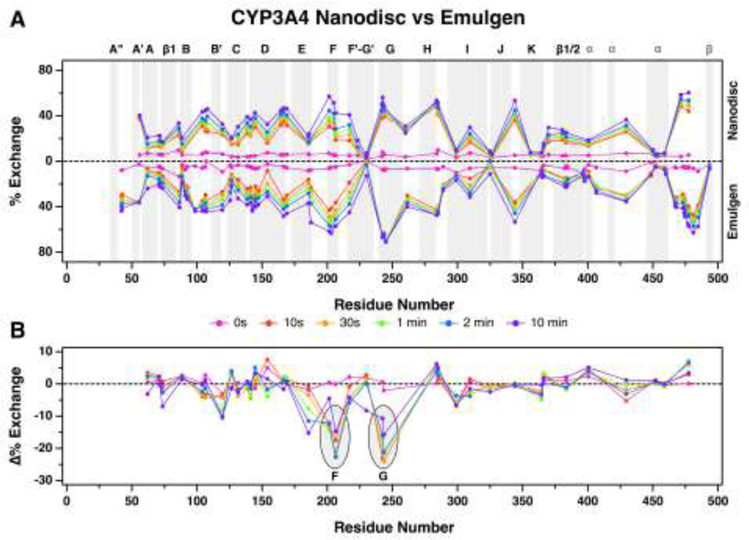
HDX comparison of ligand-free CYP3A4 nanodiscs versus detergent solubilized CYP3A4. (A) The butterfly plot shows deuterium exchange profiles of ligand-free CYP3A4 nanodiscs (top) vs detergent solubilized CYP3A4 (bottom). Percent uptake for each time point is plotted at the midpoint of the primary sequence for each peptide. For example, uptake for the peptide spanning residues 33 to 51 is plotted at 42. The alpha-helices and beta-sheets are highlighted in gray and labeled above the plot. Generic “α” and “β” labels in gray are given to helices and sheets that are not named in this article, respectively. (B) A plot of the differences between matching peptides of CYP3A4 in nanodiscs and detergent solubilized CYP3A4 at each time point. The difference plot highlights regions of CYP3A4 nanodiscs that are more (positive value) or less (negative value) solvent accessible compared to detergent solubilized CYP3A4. Differences are not plotted when matching peptides do not exist between states. For example, no difference is calculated for the peptide spanning residues 33−51 because there was no peptide observed in that region for CYP3A4 nanodiscs. Figure from (Treuheit et al., 2016). Copyright 2016 American Chemical Society.
An alternative experiment is “pulsed” H/DX MS, which provides kinetic information on a protein undergoing changes in conformation or dynamics (Deng et al., 1999). The above protocol is only slightly modified. After a kinetic reaction is started, samples are taken at desired time points and deuterated for a fixed period of time. The resulting peptide profiles show the rate or points in a reaction at which a protein is changing conformation.
2. Initial H/DX MS studies on Nanodiscs
The advantages of the lipid nanodiscs for providing a membrane mimic and for ensuring that the samples are monodisperse are bundled with the disadvantages of having significant lipid present, which can significantly decrease the sensitivity of most MS analyses. As a result, the successful combination of H/DX MS with nanodisc-captured proteins requires modifications to typical H/DX MS protocols used with soluble proteins.
2.1. H/DX MS Debut to the Nanodisc World
Hebling et al. described the experimental workflow that has been the basis for most H/DX studies of protein in nanodiscs (Hebling et al., 2010). In their experiment, the deuterium exchange and the subsequent quench processes were performed following standard HDX procedures. Because of the nature of the sample (a fully formed nanodisc composed of lipid, belt-like scaffold proteins, and the target membrane protein), additional considerations had to be addressed in the remainder of the experiment (pepsin digest and final peptide separation before analysis on the MS). An obvious requirement in any H/DX MS experiment is the generation of several peptides that span the length of the protein. They found that detergent-facilitated nanodisc disassembly in the pepsin digest increased the number of recovered peptides by 4-fold compared to whole nanodisc digestion. Further, removal of lipid by addition of ZrO2 beads in the final minute of pepsin digest increased column lifetime and retention time reproducibility in the chromatographic peptide separation step. Sample cleanup with a centrifugal filter was useful to quickly remove the lipid saturated ZrO2 beads, the immobilized pepsin beads, and any precipitate caused by the nanodisc disassembly. Finally, they reported that separation of the peptides for MS analysis via UPLC was faster and provided greater resolution compared to HPLC. The workflow is summarized in Figure 2.
Figure 2.
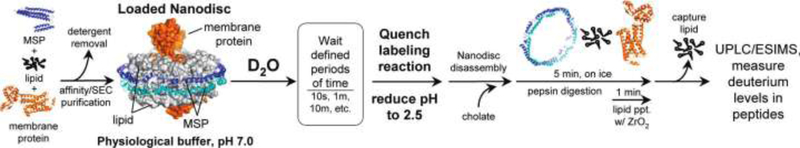
The integrated nanodisc-HX MS workflow. Loaded nanodiscs were assembled from a mixture of membrane scaffold protein (MSP), lipids, and the target membrane protein solubilized with detergent. Nanodiscs self-assemble as detergent was removed. Loaded nanodiscs were purified with size exclusion chromatography (SEC) and exposed to deuterated buffer for various times before quenching the exchange reaction. Cholate was immediately added to the quenched reaction to begin disassembly of the nanodiscs. Protein was digested with pepsin for 5 min on ice. In the last minute of digestion, ZrO2 resin was added to the digestion mixture to selectively remove phospholipid. Filtration removed immobilized-pepsin beads and the ZrO2 resin. UPLC/ESIMS were used to measure the incorporation of deuterium. Figure from (Hebling et al., 2010). Copyright 2010 American Chemical Society.
The authors then tested the robustness of their modified H/DX protocol with nanodiscs containing γ-glutamyl carboxylase (GGCX), a 94 kDa protein with five transmembrane domains. They obtained 45% sequence coverage of GGCX with 71 unique peptides. Data analyses for both the empty nanodisc and the GGCX nanodisc were reported in two different follow-up papers.
2.2. Characterization of scaffold protein in empty nanodiscs via H/DX MS
Morgan et al. performed an in-depth comparison of the conformational dynamics of the membrane scaffold protein, MSP1D1, in solution vs. in DOPC nanodiscs (Morgan et al., 2011). In general, MSP1D1 in a nanodisc exchanged deuterons more slowly than those in solution, which is consistent with the higher helical content in the lipid-associated scaffold compared to the lipid-free scaffold protein measured by circular dichroism. Despite the high helical content, uptake neared completion within 2 hours in both conditions. An interesting result was the presence of several peptides displaying EX1 kinetic profiles with half-lives on the order of 1–5 minutes (Figure 3). Regions displaying bimodal uptake kinetics were confined to the N-terminal half of the protein in the lipid-free state but expand to include the C-terminus in the lipid-associated state. The authors propose a flexing motion in the nanodisc or unraveling in the helices as possible explanations for the cooperative unfolding events. The presence of EX1 kinetics and the fact that exchange neared completion within 2 hours suggest that nanodiscs are dynamic bodies capable of accommodating flexible proteins within their membrane. Collectively, the work contributed to the characterization of nanodiscs, demonstrated the feasibility of the modified H/DX workflow, and provided reference MSP1D1 uptake data for future H/DX studies.
Figure 3.
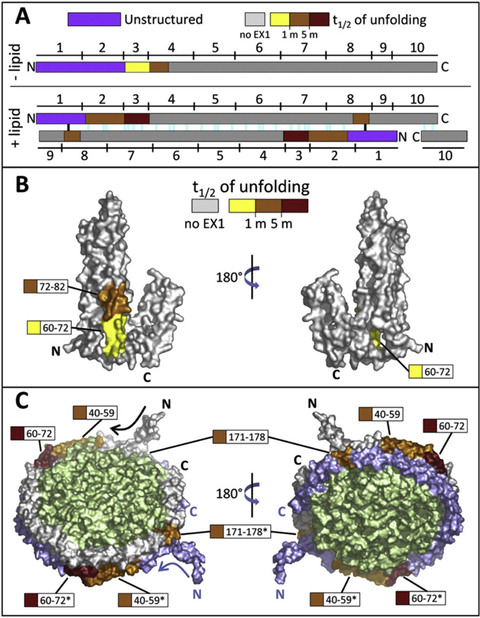
Location and magnitude of EX1-related cooperative unfolding in MSP1D1. A, Diagram showing the spatial relationship between unstructured regions (purple) and EX1 unfolding regions (colored according to the half-life of unfolding, as shown) in lipid-free apoA-1 (top) and apoA-1 in discoidal HDL particle model (bottom). The 10 amphipathic helices (denoted by numbers above and below the colored bars) and predicted salt bridges (colored lines in the +lipid panel, bottom) for apoA-1 are shown. Two of the salt bridges (black) are most likely not present because of the unstructured nature of MSP1D1, which differs from apoA-1 in the N terminus. B, Locations of EX1 unfolding plotted on a space-filling model of lipid-free apoA-1 (Ajees et al., 2006), PDB 2A01. Regions that exhibited EX1 unfolding are colored with respect to the magnitude of the half-life of unfolding. Regions colored gray did not undergo EX1 unfolding. C, Regions of EX1 unfolding plotted on a space-filling model of apoA-1 in the discoidal double belt model (Segrest et al., 1999). Regions that exhibited EX1 kinetics are colored as in panel B. The top copy of MSP is shown in gray and the bottom copy in blue (regions from the bottom copy are shown with asterisks). The lipid bilayer is shown in green. Figure from (Morgan et al., 2011). Copyright 2011 The American Society for Biochemistry and Molecular Biology, Inc.
3. Applications of H/DX MS to membrane embedded nanodiscs
To date, H/DX MS has been utilized to characterize protein dynamics for nine different ‘analyte’ proteins that have diverse structures and functions in addition to their degree of membrane insertion. Six of the proteins span the transmembrane and the dynamics have been analyzed for γ-glutamyl carboxylase (GGCX, a carboxylase studied by C. H. Parker et al., 2014), Leucine transporter (LeuT, a nonpolar amino acid studied by Adhikary et al., 2017), and P-glycoprotein (P-gp, a drug efflux transporter studied by Li et al., 2018), Xylose transporter (XylE), Lactose permease (LacY) and Glycerol-3-phosphate antiporter (GlpT) (the latter three of which were studied by Martens et al., 2018). The remaining two analyte proteins studied by H/DX MS are peripheral membrane proteins and include Cytochrome P450 3A4 (CYP3A4, a drug metabolizing enzyme studied by Treuheit et al., 2016) and monoacylglycerol lipase (MGL, an esterase involved in endocannabinoid signal regulation studied by Nasr et al., 2013). The last protein is a soluble cytosolic protein that translocates to the membrane surface in a calcium-dependent manner (Droege et al., 2017). Here we summarize some of the key findings from each study.
3.1. H/DX MS to characterize membrane topology
Some H/DX MS experiments can provide information regarding membrane orientation as well as depth of insertion for a target analyte protein if sufficient global sequence coverage is achieved. The technique can be especially useful when no crystal structure exists, as was done by Parker et al. in their study of GGCX, an integral membrane protein that consists of 758 amino acids with 5 transmembrane domains and is predicted to have solvent exposed regions on either side of the membrane (C. H. Parker et al., 2014). In their work, a 42% overall sequence coverage was attained. For the majority of peptides, deuterium uptake was consistent with predicted topology maps for the protein. More specifically, regions predicted to interact with the membrane showed little to no exchange for the duration of the experiment. Additionally, regions predicted to have secondary structure showed little uptake compared to regions predicted to lack structure. The authors note that further improvements in sample preparation, chromatographic separation, MS sensitivity, and ion mobility mass spectrometry can increase peptide coverage and thus provide additional information. Recent improvements will be discussed in section 4. Regardless of the low sequence coverage, Parker et al. made a major contribution to the understanding of spatial and temporal conformational fluctuations in GGCX.
The structures of the remaining proteins have been extensively characterized and at least one crystal structure has been obtained for each protein. The proteins in the crystal structure lack a native membrane and in some cases are heavily truncated or engineered to promote a more soluble version of the protein amenable for crystallography. The data from each H/DX study were remarkably consistent with structural information provided by the crystal structures, with some significant differences in each case. Additional evidence for membrane interaction was provided when comparing uptake in the nanodisc-incorporated protein with the same protein in detergent or detergent-free solution. For example, a comparison of the detergent solubilized form of CYP3A4 and P-gp with the nanodisc-incorporated equivalents was reported by Treuheit et al. and Li et al., respectively (Li et al., 2018; Treuheit et al., 2016). The detergent did not perfectly mimic the environment provided by the lipid bilayer (example figure shown in Figure 4). Similarly, Nasr et al. made comparisons with MGL in solution with MGL–incorporated nanodiscs (Nasr et al., 2013). In general, membrane embedded regions of each protein tended to have slower rates of H/DX in the nanodisc compared to the protein in solution or solubilized by detergent, demonstrating the ability of nanodisc to provide a membrane capable of shielding hydrophobic regions of the protein from solvent.
3.2. Membrane association
Nanodiscs are useful in monitoring the details of protein-membrane association events, for proteins that transiently bind at and dissociate from the surface as part of their function. Droege et al. investigated the calcium-dependent membrane association of the soluble cytosolic protein, 15-LOX (Droege et al., 2017). The conformational dynamics and structural changes after membrane association as well as the changes induced upon binding of the endogenous substrate, arachadonic acid, were unknown. The work helped identify regions involved in anchoring the enzyme to the membrane surface and provided insight to the ongoing debate in the field regarding the extent to which large structural rearrangements were required for membrane association. The results demonstrated that large conformational changes are not required for membrane association and the work provided evidence for substrate-dependent effects on inter-domain communication.
3.3. Effects of lipid composition
Protein incorporation into nanodiscs with different lipid composition is routine. Martens et al. assessed the effects of lipid composition on the conformational landscape for three secondary transporters in E. coli (Martens et al., 2018). The enzymes have a conserved network of charged residues believed to act as a switch between the structurally different inward and outward facing conformations of the transporters. In their work, they compared two different mixtures of lipids for their protein-incorporated nanodiscs to further investigate the role membrane charge has on the equilibrium between the closed and open states of the transporters. Interestingly, one peptide mixture shifted the equilibrium of each transporter to favor an inward facing conformation demonstrating the importance of lipid composition and their ability to fine-tune the conformational landscape for membrane proteins. The work demonstrates the importance of membrane mimic systems such as nanodiscs.
3.4. H/DX MS as a probe of ligand-induced changes in protein dynamics
H/DX MS is well suited to probe interactions with small-molecule ligands and their target proteins, especially in cases where the protein/ligand complex of interest cannot be crystalized. Interpretation of ligand-dependent changes in H/DX requires careful consideration for a number of reasons. At first glance, one might be tempted to assign a binding site as the region where the largest decrease in deuterium uptake occurs for the ligand-bound state compared to the ligand-free state. While in some cases this holds true, one must consider the possibility that ligand might interact with side-chains in the binding site but may not necessarily alter the hydrogen-bonding network for the amide groups that H/DX MS probes. Nasr et al. observe such a case where no difference in H/DX occurs in peptides that include a residue that is covalently modified upon incubation with an irreversible carbamylate inhibitor, AM6580 (Nasr et al., 2013). However, they note a decrease in exchange in the surrounding regions. Thus, H/DX may not be altered for peptides that directly interact with a ligand, but ligands can induce changes in H/DX of peptides remote from the exact binding site. To further complicate the matter, ligand binding can alter H/DX in regions within and beyond the ligand-binding site. Treuheit et al. note ligand-dependent changes throughout CYP3A4 when they incubate the protein with ketoconazole, a potent inhibitor (Treuheit et al., 2016). In contrast to the case with MGL, addition of ketoconazole induces global conformational fluctuations where both increases and decreases in H/DX are observed. Thus, although identification of a ligand-binding site may prove inconclusive in some cases, characterization of global conformational changes upon binding of an allosteric effector can readily be studied by H/DX MS, as observed with the nonmembrane protein Thrombin (Handley et al 2015). In this regard, H/DX may be uniquely useful for identifying the peptide networks through which allosteric signal are transmitted. Due to these complexities, if a ligand binding site is suspected due to localized ligand-induced changes in H/DX, other orthogonal methods should be utilized, such as reactive analogs that yield covalent adducts or mutagenesis. Of course, a crystal structure of the ligand complex, presumably in the absence of lipid membrane, would be the best corroboration.
3.5. H/DX MS to map protein-protein interfaces
Protein-protein interface mapping is a common goal of H/DX MS experiments. Unfortunately, the same disadvantages described in the previous small-molecule section can apply. However, an advantage when interrogating protein-protein interactions is that one can measure uptake in both proteins in some cases. Parker et al. used H/DX MS to characterize changes in H/DX upon binding of pCON, an 18 amino acid consensus propeptide present in vitamin K-dependent (VKD) proteins recognized by GGCX (C. H. Parker et al., 2014). Previous reports hypothesize VKD protein binding induces conformational changes in GGCX such that the target glutamic acid is accessible for carboxylation at the GGCX active site. They were able to observe a sustained decrease in deuterium uptake (over ~4 hours) at the predicted binding site. Interestingly, they observed protection of the same order when pCON was incubated with the GGCX nanodisc compared to immediate uptake when incubated alone. Their work provided valuable supporting evidence for identification of the VKD protein-binding site of GGCX, a protein that has eluded crystallographic analyses. A potential limitation for monitoring multiple proteins in a single nanodisc is the possible overlap in the chromatography of peptides, but in some cases this can be managed by using ion mobility for a second dimension of separation. Thus, the ability to monitor multiple proteins simultaneously in a single nanodisc may be limited by the available mass spectrometers.
For the current status of MSP proteins, it is unlikely that large protein complexes with more than two target membrane proteins could be studied. In this regard liposomes may have the advantage of accommodating larger protein complexes in the membrane. However, in liposomes the detailed stoichiometry of proteins within a complex may be unknown or heterogeneous, so H/DX data for protein complexes in liposomes could report on the average H/D exchange behavior for a complex population. In contrast, although the nanodiscs may limit studies of protein-protein interaction to small or low stoichiometry complexes, they may be more homogeneous and monodisperse. So, the advantages of liposomes vs. nanodiscs would need to be considered before initiating H/DX experiments to map protein binding sites.
3.6. H/DX MS to probe slow conformational effects
H/DX MS can be particularly useful to characterize two or more disparate protein conformations in slow exchange, as described for ‘EX1 kinetics’ above. Two examples include the integral proteins LeuT and P-gp, which were studied by Adhikary et al. and Li et al., respectively (Adhikary et al., 2017; Li et al., 2018). Both LeuT and P-gp exist in a dynamic equilibrium between macroscopic inward facing and outward facing conformations, or ensembles.
P-gp is a large, ATP-dependent, promiscuous drug transporter that consists of 12 transmembrane helices (TMHs) and two nucleotide binding domains (NBDs) (Subramanian et al., 2018). The size of P-gp alone presents a challenge for H/DX, due to the need to separate the abundant peptides generated. The mechanism of drug transport is thought to include a conformational switch from an inward facing state that engages hydrophobic drugs in the membrane, to an outward facing conformation in the presence of nucleotide, from which drugs are presumed to dissociate to the external milieu. The outward conformation can be trapped for characterization by addition of nucleotide and vanadate. Structures of the inward and nucleotide-bound outward facing conformations of P-gp have been captured via X-ray crystallography and cryo-EM (Aller et al., 2009; Esser et al., 2017; Frank et al., 2016; Kim and Chen, 2018; Moeller et al., 2015). Li et al. incorporated P-gp into DMPC nanodiscs and compared the H/DX of the inward facing nucleotide-free state with the ‘vanadate-trapped’ state. In this initial work, the overall peptide coverage for P-gp in nanodiscs was 37%, where ~68% of the NBD peptides were recovered and only ~14.5% of the TMHs were found. The NBDs of the vanadate-trapped state were not as protected from H/DX as would be expected from the two state inward-facing vs. outward facing paradigm. This finding supported results from other methods that suggested the vanadate-trapped state may be heterogeneous, or the NBDs are not tightly complexed (Liu et al., 2018; Wen et al., 2013). Furthermore, many peptides in the NBDs exhibited EX1 kinetics on a very wide range of time scales, up to several hours. The broad spectrum of EX1 kinetics indicates that the two state paradigm is incomplete and the conformational landscape is more complex, as might be expected for a substrate promiscuous transporter.
The importance of EX1 kinetics was also suggested by H/DX with the LeuT neurotransmitter/Na+ symporter. Adhikary et al. incorporated wild type and a mutant LeuT into POPC:POPG nanodiscs. The LeuT transporter plays a critical role in the Na+-dependent reuptake of neurotransporters after release into the synaptic cleft. As with P-gp, the protein exists in a dynamic equilibrium between inward facing and outward facing conformations, or ensembles. The mutant Y268A switches the bias of the ensembles toward the inward facing population and provided a benchmark for this ensemble. EX1 kinetics with very slow exchange were also observed. Interestingly LeuT is not as promiscuous as P-gp so the link of conformational heterogeneity to substrate promiscuity, and the functional role of slow conformational exchange, are not obvious.
An interesting and important aspect of the EX1 kinetics observed with P-gp and LeuT is that they occur on a much wider range of time scales, including some very slow processes, than the EX1 kinetics observed in the scaffold protein in empty nanodiscs. This indicates that the dynamics of the nanodiscs are not coupled directly to, and do not drive, all of the conformational relaxations of these analyte protein transporters. P-gp and LeuT slowly exchange conformations independent of the fluctuations of the nanodisc. It would be interesting to determine whether P-gp or LeuT decrease or quench the EX1 kinetics of the MSP1D1 scaffold proteins, but we have not analyzed this possibility in detail.
3.7. Potential uses of H/DX MS on nanodiscs
In addition to the examples already described in the literature, there are other potential uses of H/DX MS that could be explored in the future. Here we provide a prospective of possible applications of H/DX MS with proteins in nanodiscs.
One area in which H/DX MS could be useful would be in mapping interactions between two proteins embedded in a membrane. Although we describe systems where a single target protein is embedded in each nanodisc, we note that nanodiscs with multiple proteins embedded have been isolated (Denisov et al., 2007; Grinkova et al., 2010; McDougle et al., 2013; Raschle et al., 2015). A key finding in two of the studies described here is that uptake in the detergent solubilized protein differs from the nanodisc equivalent and serves as a reminder that detergent solubilization is not a perfect mimic of a lipid bilayer. For this reason, H/DX MS can be advantageous when interrogating protein-protein interface mapping particularly for different membrane proteins.
Another interesting and fundamental utility of nanodiscs would be to study the effects of lipid composition of the membrane on conformational dynamics of analyte proteins embedded in them. It is widely appreciated that lipid composition affects functional properties of many membrane proteins including conformational dynamics, stability, ligand binding kinetics etc., and this has been demonstrated in a few cases with nanodiscs (Dijkman and Watts, 2015; Henrich et al., 2016; McClary et al., 2016; Rues et al., 2016; Shenkarev et al., 2014; Wang et al., 2015). It is logical to presume, therefore, that the conformational dynamics of membrane proteins is also affected, and H/DX MS is poised to document the relationships between membrane charge and fluidity on protein dynamics.
4. Advances in H/DX MS workflow including a prospective on potential improvements
Here we describe several potential improvements to the H/DX workflow for nanodiscs from the reports already described, advancements in non-nanodisc fields that could be applied, and our own unpublished observations of P-gp to provide some general comments/observations to aid others in similar endeavors. Some of these observations have not been rigorously tested in published work but are intuitively sensible. As a result, they should not be considered as ‘guarantees’ of improved results, but rather possible solutions to problems that we have encountered. Sample preparation, injection, and analysis are all highly laborious steps that the H/DX community is constantly striving to streamline. As samples are typically manually generated and injected, there is increased risk of variability and therefore improvements in reproducibility have been key to robust interpretations. However, the largest concern for the small nanodisc community has been improving sequence coverage of large membrane proteins that are less amenable to quick digestion and whose peptides overlap with other contaminants not inherent to other H/DX users. The foundations of peptide coverage are robust proteolytic digestion, resolution of peptides from each other and contaminants, and adequate signal intensity. However, these elements are intertwined and limited by time constraints dictated by the need to limit back-exchange after quenching, therefore a careful balance must be considered for each proteins system.
4.1. Selection of quench conditions and proteolysis optimization
We have found that optimization of quench/digestion conditions for P-gp has dramatically increased coverage in the nanodisc context. In our published work (Li et al., 2018), we obtained 38% total coverage of P-gp in nanodiscs, with 14% coverage of the TMDs. While the ample coverage of the NBDs provided insight into conformational changes with nucleotide binding, the low TMD coverage limited interpretations of the likely shifting transmembrane helices (as seen in crystal and EM structures). We have since dramatically improved overall coverage to 64%, with 42% coverage of the TMDs, so that we can probe more extensive conformational changes in or near the membrane (Figure 5).
Figure 5.
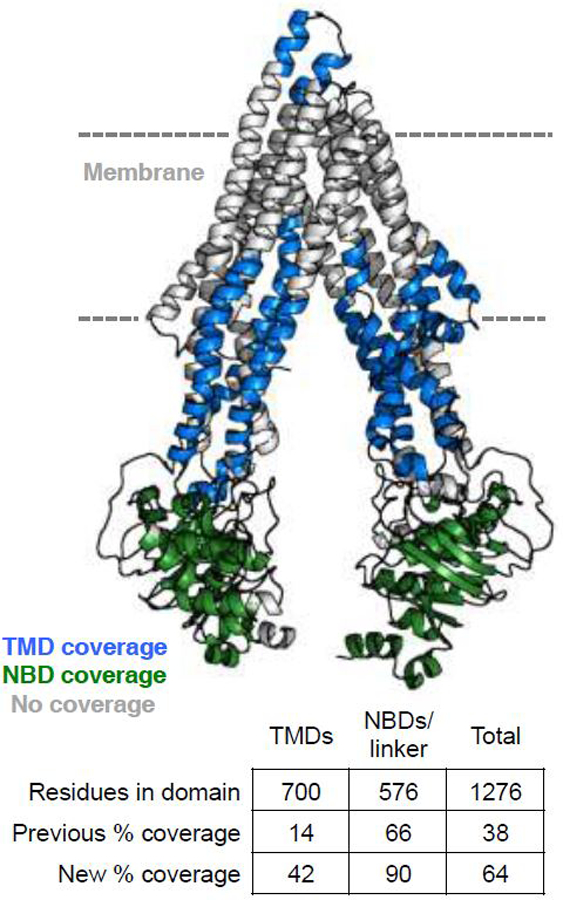
Improved peptide coverage for P-gp in nanodiscs. Coverage for the trans-membrane and nucleotide-binding domains are mapped onto the 5KPI structure of mouse P-gp.
Routine H/DX MS of smaller soluble proteins requires little optimization of quench conditions and typically yields high levels of peptide coverage. Larger, stable proteins (e.g. antibodies) often require chaotropic agents (urea, guanidinium) in the quench buffer to promote unfolding and subsequent accessibility to pepsin (Hamuro and Coales, 2018). However, these agents also limit pepsin activity and are problematic in cases where they trigger aggregation rather than aid in protein unfolding. We have found that the “less is more” approach, reported for other membrane protein mass spectrometry studies (Zhang, 2015; Zhang et al., 2010), improves coverage for P-gp in nanodiscs; specifically no denaturing agent and only detergent and low levels of reducing agent can be used.
We have altered our choice of detergent for disassembly of P-gp nanodiscs. We had previously used cholate, common in other studies discussed here, but comparison with non-ionic DDM yielded increased coverage. The likely reasons for improvement are threefold: (1) P-gp “prefers” the environment of DDM compared to other detergents during purification and experiments. (2) The less harsh properties of DDM vs. cholate presumably have a less inhibitory effect on pepsin. (3) DDM is less of an ion suppressant during mass spectrometry, therefore peptides eluting with detergent have improved signal.
For increased digestion, we have also turned to the use of soluble pepsin over beads with immobilized pepsin. For bulky protein complexes, proteins in liposomes, and large proteins in nanodiscs, accessibility for pepsin becomes a concern. While immobilized pepsin eliminates one source of overlapping peptides in mass spectra (discussed in next section), the tradeoff for increased digestion with soluble pepsin can be favorable. Self-cleavage of pepsin is minimal and we have found that even in the presence of 20-fold molar excess of pepsin, the signal of pepsin peptides is far below that of the most abundant P-gp peptides. Undigested pepsin elutes far from peptides of interest. Finally, pepsin quantity and digestion time should be balanced with gradient elution time and resolution/sensitivity limitations of the mass spectrometer being used. In summary, several digestions protocols with different types and amounts of pepsin should be compared to optimize the experiment, and the optimal conditions can vary with target protein.
4.2. Removal of contaminants
H/DX studies in lipid nanodiscs inherently introduce the contaminants of lipids and scaffold protein at a molar excess over the analyte protein. As mentioned earlier, addition of solid ZrO2 beads to the protein digest mixture in the final minute facilitates lipid removal. The quick, accurate, and reproducible delivery of ZrO2 beads to the digestion mixture can be difficult as this is a manual process. Adhikary et al. simplify the process by preparing a 300 mg/mL solution of ZrO2 beads equilibrated in 0.8% formic acid. We recommend optimizing amount and duration of incubation to maximize lipid removal and minimize peptide adsorption.
The digestion of large membrane proteins can generate hundreds of peptides that require a high level of separation in order to analyze isotopic profiles for each one. The process becomes increasingly difficult the larger an analyte protein becomes as the likelihood of overlap between analyte peptides and scaffold protein peptides increases. Adhikary et al. lessen the complexity by selectively removing a biotinylated form of the scaffold protein with Neutravidin Ultralink beads (Pierce) before the digestion step. Similarly, in non-nanodisc studies, others have utilized affinity tag strategies to selectively remove non-analyte proteins necessary for the deuteration experiment (i.e. protein-protein interactions) just prior to proteolysis (Jensen et al., 2013). This overall approach could theoretically be applied to any protein-protein interaction in nanodiscs.
4.3. Mass spectrometer options and internal standards
Although many mass spectrometers are inherently suitable for collection of H/DX data, there are considerations for sensitivity and resolution that can be more crucial for membrane proteins in nanodiscs.
Any system that involves multiple proteins (e.g. nanodiscs) or very large proteins will generate abundant, overlapping peptides in the mass spectrum. Therefore, resolution of peptides becomes critical for peptide coverage of the analyte protein. Separation of peptides by reverse-phase chromatography is limited in time by the need to limit back-exchange of deuterated amides with the solvent. Ion mobility after chromatography can substantially improve peptide coverage (Iacob et al., 2008) and has proven useful in our studies through the use of a Waters Synapt-G2 with ion mobility (Treuheit et al. and Li et al.). However, a high level of sensitivity is required for initial tandem-MS identification of peptides. Because transmembrane regions of membrane proteins are less efficiently digested, peptides from these regions tend to have weaker signals, necessitating an even higher level of sensitivity for tandem-MS. Therefore, we have found it to be advantageous to identify analyte peptides by tandem-MS using a highly sensitive Thermo LTQ-Orbitrap coupled with the same LC system as used for HDX samples. With a precise map of peptide chromatographic retention times, we then utilize the resolution provided by the Synapt-G2 to analyze deuterated samples.
The additional steps in nanodisc assembly over other routine protein purifications can result in increased prep-to-prep variability. Ideally, H/DX samples are prepared on the same day with identical buffer components and injected on the same day (separate from sample prep) to minimize further experimental inconsistencies. However, it is often unfeasible for larger experiments to stay in these reproducible confines. The use of an internal peptide standard introduced to each sample prior to deuteration has provided a simple method to account for day-to-day variability in the LC/MS injection process (Li et al., 2018). This allows one to more quantitatively compare datasets and identify possible prep-to-prep irregularities by ruling out errors in the later experimental steps.
Finally, very recent advances are being made to automate preparation of H/DX samples and their injection onto LC/MS systems. Anderson et al. advanced the H/DX workflow by automating the lipid removal step with ZrO2 (Anderson et al., 2018). In their study, they use a fully automated HDX PAL robotic arm (LEAP Technologies, Carrboro, NC) to test a fully automated H/DX MS workflow on a transmembrane protein expressed in liposomes. The workflow has the potential to work for a nanodisc system and we anticipate future publications using the automated workflow. In theory, increased automation will dramatically improve reproducibility for all H/DX studies.
HIGHLITES.
H/DX mass spectrometry enables interrogation of the backbone dynamics and solvent accessibility membrane
proteins in a native-like lipid environment.
Several improvements in experimental workflow of H/DX MS with proteins in nanodiscs have led to increased
peptide coverage, allowing interrogation of even some membrane spanning regions.
H/DX MS can be used to probe membrane topology, ligand binding, and protein-protein interactions of
membrane proteins.
The existing literature examples of H/DX MS of membrane proteins in lipid nanodiscs reveal a wide range of
H/DX behavior including combinations of EX1 and EX2 kinetics.
Acknowledgments
Funding: Preparation of this review was supported by NIH R01GM130810 (WMA) and NIH R01GM121603 (WMA).
Abbreviations
- pCON
Consensus propeptide
- CYP3A4
Cytochrome P450 3A4
- DMPC
1,2-dimyristoyl-sn-glycero-3-phosphocholine
- GGCX
γ-glutamyl carboxylase
- GlpT
Glycerol-3-phosphate antiporter
- H/DX MS
Hydrogen deuterium exchange mass spectrometry
- LacY
Lactose permease
- LeuT
Leucine transporter
- MGL
Monoacylglycerol lipase
- MSP
membrane scaffold proteins
- NBD
Nucleotide binding domain
- P-gp
P-glycoprotein
- POPC
1-palmitoyl-2-oleoyl-glycero-3-phosphocholine
- POPG
1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1’-rac-glycerol)
- TMD
Transmembrane domain
- VKD
vitamin K-dependent
- XylE
Xylose transporter
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adhikary S, Deredge DJ, Nagarajan A, Forrest LR, Wintrode PL, Singh SK, 2017. Conformational dynamics of a neurotransmitter:sodium symporter in a lipid bilayer. Proceedings of the National Academy of Sciences 114, E1786–E1795. doi: 10.1073/pnas.1613293114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajees AA, Anantharamaiah GM, Mishra VK, Hussain MM, Murthy HMK, 2006. Crystal structure of human apolipoprotein A-I: insights into its protective effect against cardiovascular diseases. Proc. Natl. Acad. Sci. U.S.A 103, 2126–2131. doi: 10.1073/pnas.0506877103 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, Chang G, 2009. Structure of P-Glycoprotein Reveals a Molecular Basis for Poly-Specific Drug Binding. Science 323, 1718–1722. doi: 10.1074/jbc.M609899200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KW, Gallagher ES, Hudgens JW, 2018. Automated Removal of Phospholipids from Membrane Proteins for H/D Exchange Mass Spectrometry Workflows. Anal. Chem 90, 6409–6412. doi: 10.1021/acs.analchem.8b00429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayburt TH, Sligar SG, 2010. Membrane protein assembly into Nanodiscs. FEBS Lett [DOI] [PMC free article] [PubMed]
- Berger A, Linderstrom-Lang K, 1957. Deuterium exchange of poly-DL-alanine in aqueous solution. Archives of Biochemistry and Biophysics 69, 106–118. [DOI] [PubMed] [Google Scholar]
- Busenlehner LS, Armstrong RN, 2005. Insights into enzyme structure and dynamics elucidated by amide H/D exchange mass spectrometry. Archives of Biochemistry and Biophysics 433, 34–46. doi: 10.1016/j.abb.2004.09.002 [DOI] [PubMed] [Google Scholar]
- Busenlehner LS, Brändén G, Namslauer I, Brzezinski P, Armstrong RN, 2008. Structural elements involved in proton translocation by cytochrome c oxidase as revealed by backbone amide hydrogen-deuterium exchange of the E286H mutant. Biochemistry 47, 73–83. doi: 10.1021/bi701643a [DOI] [PubMed] [Google Scholar]
- Busenlehner LS, Salomonsson L, Brzezinski P, Armstrong RN, 2006. Mapping protein dynamics in catalytic intermediates of the redox-driven proton pump cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A 103, 15398–15403. doi: 10.1073/pnas.0601451103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter EP, Beis K, Cameron AD, Iwata S, 2008. Overcoming the challenges of membrane protein crystallography. Curr. Opin. Struct. Biol 18, 581–586. doi: 10.1016/j.sbi.2008.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Zhang Z, Smith DL, 1999. Comparison of continuous and pulsed labeling amide hydrogen exchange/mass spectrometry for studies of protein dynamics. J Am Soc Mass Spectrom 10, 675–684. doi: 10.1016/S1044-0305(99)00038-0 [DOI] [PubMed] [Google Scholar]
- Denisov IG, Baas BJ, Grinkova YV, Sligar SG, 2007. Cooperativity in cytochrome P450 3A4: linkages in substrate binding, spin state, uncoupling, and product formation. J. Biol. Chem 282, 7066–7076. doi: 10.1074/jbc.M609589200 [DOI] [PubMed] [Google Scholar]
- Denisov IG, Sligar SG, 2017. Nanodiscs in Membrane Biochemistry and Biophysics. Chem. Rev 117, 4669–4713. doi: 10.1021/acs.chemrev.6b00690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denisov IG, Sligar SG, 2016. Nanodiscs for structural and functional studies of membrane proteins. Nat. Struct. Mol. Biol 23, 481–486. doi: 10.1038/nsmb.3195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkman PM, Watts A, 2015. Lipid modulation of early G protein-coupled receptor signalling events. Biochim. Biophys. Acta 1848, 2889–2897. doi: 10.1016/j.bbamem.2015.08.004 [DOI] [PubMed] [Google Scholar]
- Droege KD, Keithly ME, Sanders CR, Armstrong RN, Thompson MK, 2017. Structural Dynamics of 15-Lipoxygenase-2 via Hydrogen-Deuterium Exchange. Biochemistry 56, 5065–5074. doi: 10.1021/acs.biochem.7b00559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duc NM, Du Y, Zhang C, Lee SY, Thorsen TS, Kobilka BK, Chung KY, 2015. Effective application of bicelles for conformational analysis of G protein-coupled receptors by hydrogen/deuterium exchange mass spectrometry. J Am Soc Mass Spectrom 26, 808–817. doi: 10.1007/s13361-015-1083-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engen JR, Wales TE, 2015. Analytical Aspects of Hydrogen Exchange Mass Spectrometry. Annu Rev Anal Chem (Palo Alto Calif) 8, 127–148. doi: 10.1146/annurev-anchem-062011-143113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander SW, 2006. Hydrogen exchange and mass spectrometry: A historical perspective. J Am Soc Mass Spectrom 17, 1481–1489. doi: 10.1016/j.jasms.2006.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander SW, Mayne L, Bai Y, Sosnick TR, 1997. Hydrogen exchange: the modern legacy of Linderstrøm-Lang., Protein science : a publication of the Protein Society doi: 10.1002/pro.5560060517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser L, Zhou F, Pluchino KM, Shiloach J, Ma J, Tang W-K, Gutierrez C, Zhang A, Shukla S, Madigan JP, Zhou T, Kwong PD, Ambudkar SV, Gottesman MM, Xia D, 2017. Structures of the Multidrug Transporter P-glycoprotein Reveal Asymmetric ATP Binding and the Mechanism of Polyspecificity. J. Biol. Chem 292, 446–461. doi: 10.1074/jbc.M116.755884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro DM, Lazo ND, Robertson AD, 2004. EX1 hydrogen exchange and protein folding. Biochemistry 43, 587–594. doi: 10.1021/bi035943y [DOI] [PubMed] [Google Scholar]
- Frank GA, Shukla S, Rao P, Borgnia MJ, Bartesaghi A, Merk A, Mobin A, Esser L, Earl LA, Gottesman MM, Xia D, Ambudkar SV, Subramaniam S, 2016. Cryo-EM Analysis of the Conformational Landscape of Human P-glycoprotein (ABCB1) During its Catalytic Cycle. Mol. Pharmacol 90, 35–41. doi: 10.1073/pnas.1309275110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasoe PK, Long FA, 1960. Use Of Glass Electrodes To Measure Acidities In Deuterium Oxide 1,2. J. Phys. Chem 64, 188–190. doi: 10.1021/j100830a521 [DOI] [Google Scholar]
- Grinkova YV, Denisov IG, Sligar SG, 2010. Biochemical and Biophysical Research Communications. Biochemical and Biophysical Research Communications 398, 194–198. doi: 10.1016/j.bbrc.2010.06.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall SCL, Tognoloni C, Price GJ, Klumperman B, Edler KJ, Dafforn TR, Arnold T, 2018. Influence of Poly(styrene-co-maleic acid) Copolymer Structure on the Properties and Self-Assembly of SMALP Nanodiscs. Biomacromolecules 19, 761–772. doi: 10.1021/acs.biomac.7b01539 [DOI] [PubMed] [Google Scholar]
- Hamuro Y, Coales SJ, 2018. Optimization of Feasibility Stage for Hydrogen/Deuterium Exchange Mass Spectrometry. J Am Soc Mass Spectrom 29, 623–629. doi: 10.1007/s13361-017-1860-3. [DOI] [PubMed] [Google Scholar]
- Handley LD, Treuheit NA, Venkatesh VJ, Komives EA 2015. Thrombomodulin Binding Selects the Catalytically Active Form of Thrombin. Biochemistry 54(43):6650–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebling CM, Morgan CR, Stafford DW, Jorgenson JW, Rand KD, Engen JR, 2010. Conformational Analysis of Membrane Proteins in Phospholipid Bilayer Nanodiscs by Hydrogen Exchange Mass Spectrometry. Anal. Chem 82, 5415–5419. doi: 10.1021/ac100962c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrich E, Ma Y, Engels I, Münch D, Otten C, Schneider T, Henrichfreise B, Sahl H-G, Dötsch V, Bernhard F, 2016. Lipid Requirements for the Enzymatic Activity of MraY Translocases and in Vitro Reconstitution of the Lipid II Synthesis Pathway. J. Biol. Chem 291, 2535–2546. doi: 10.1074/jbc.M115.664292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacob RE, Murphy JP, Engen JR, 2008. Ion mobility adds an additional dimension to mass spectrometric analysis of solution-phase hydrogen/deuterium exchange. Rapid Commun. Mass Spectrom 22, 2898–2904. doi: 10.1002/rcm.3688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeng MF, Englander SW, Elöve GA, Wand AJ, Roder H, 1990. Structural description of acid-denatured cytochrome c by hydrogen exchange and 2D NMR. Biochemistry 29, 10433–10437. [DOI] [PubMed] [Google Scholar]
- Jensen PF, Jorgensen TJD, Koefoed K, Nygaard F, Sen JW, 2013. Affinity Capture of Biotinylated Proteins at Acidic Conditions to Facilitate Hydrogen/Deuterium Exchange Mass Spectrometry Analysis of Multimeric Protein Complexes. Anal. Chem 85, 7052–7059. doi: 10.1021/ac303442y [DOI] [PubMed] [Google Scholar]
- Johnson RS, Walsh KA, 1994. Mass spectrometric measurement of protein amide hydrogen exchange rates of apo- and holo-myoglobin. Protein Sci 3, 2411–2418. doi: 10.1002/pro.5560031224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Chen J, 2018. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 359, 915–919. doi: 10.1016/j.jsb.2006.06.006 [DOI] [PubMed] [Google Scholar]
- Konermann L, Rodriguez AD, Sowole MA, 2014. Type 1 and Type 2 scenarios in hydrogen exchange mass spectrometry studies on protein-ligand complexes. Analyst 139, 6078–6087. doi: 10.1039/c4an01307g [DOI] [PubMed] [Google Scholar]
- Krishna MMG, Hoang L, Lin Y, Englander SW, 2004. Hydrogen exchange methods to study protein folding. Methods 34, 51–64. doi: 10.1016/j.ymeth.2004.03.005 [DOI] [PubMed] [Google Scholar]
- Li MJ, Guttman M, Atkins WM, 2018. Conformational dynamics of P-glycoprotein in lipid nanodiscs and detergent micelles reveal complex motions on a wide time scale. J. Biol. Chem 293, 6297–6307. doi: 10.1074/jbc.RA118.002190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Liu Y, He L, Zhao Y, Zhang XC, 2018. Biophysics Reports. Biophysics Reports 1–13. doi: 10.1007/s41048-018-0057-z29577065 [DOI] [Google Scholar]
- Marcsisin SR, Engen JR, 2010. Hydrogen exchange mass spectrometry: what is it and what can it tell us? Anal Bioanal Chem 397, 967–972. doi: 10.1007/s00216-010-3556-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens C, Shekhar M, Borysik AJ, Lau AM, Reading E, Tajkhorshid E, Booth PJ, Politis A, 2018. Direct protein-lipid interactions shape the conformational landscape of secondary transporters. Nat Commun 9, 4151. doi: 10.1038/s41467-018-06704-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClary WD, Sumida JP, Scian M, Paço L, Atkins WM, 2016. Membrane Fluidity Modulates Thermal Stability and Ligand Binding of Cytochrome P4503A4 in Lipid Nanodiscs. Biochemistry 55, 6258–6268. doi: 10.1021/acs.biochem.6b00715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDougle DR, Palaria A, Magnetta E, Meling DD, Das A, 2013. Functional studies of N-terminally modified CYP2J2 epoxygenase in model lipid bilayers. Protein Science 22, 964–979. doi: 10.1002/pro.2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller A, Lee SC, Tao H, Speir JA, Chang G, Urbatsch IL, Potter CS, Carragher B, Zhang Q, 2015. Distinct Conformational Spectrum of Homologous Multidrug ABC Transporters. Structure 23, 450–460. doi: 10.1016/j.str.2014.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraes I, Evans G, Sanchez-Weatherby J, Newstead S, Stewart PDS, 2014. Membrane protein structure determination - the next generation. Biochim. Biophys. Acta 1838, 78–87. doi: 10.1016/j.bbamem.2013.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan CR, Hebling CM, Rand KD, Stafford DW, Jorgenson JW, Engen JR, 2011. Conformational transitions in the membrane scaffold protein of phospholipid bilayer nanodiscs. Mol. Cell Proteomics 10, M111.010876. doi: 10.1074/mcp.M111.010876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasr ML, Shi X, Bowman AL, Johnson M, Zvonok N, Janero DR, Vemuri VK, Wales TE, Engen JR, Makriyannis A, 2013. Membrane phospholipid bilayer as a determinant of monoacylglycerol lipase kinetic profile and conformational repertoire. Protein Science 22, 774–787. doi: 10.1002/pro.2257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemirovskiy O, Giblin DE, Gross ML, 1999. Electrospray ionization mass spectrometry and hydrogen/deuterium exchange for probing the interaction of calmodulin with calcium. J Am Soc Mass Spectrom 10, 711–718. doi: 10.1016/S1044-0305(99)00036-7 [DOI] [PubMed] [Google Scholar]
- Oganesyan I, Lento C, Wilson DJ, 2018. Contemporary hydrogen deuterium exchange mass spectrometry. Methods 144, 27–42. doi: 10.1016/j.ymeth.2018.04.023 [DOI] [PubMed] [Google Scholar]
- Parker CH, Morgan CR, Rand KD, Engen JR, Jorgenson JW, Stafford DW, 2014. A Conformational Investigation of Propeptide Binding to the Integral Membrane Protein γ-Glutamyl Carboxylase Using Nanodisc Hydrogen Exchange Mass Spectrometry. Biochemistry 53, 1511–1520. doi: 10.1021/bi401536m [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JL, Newstead S, 2016. Membrane Protein Crystallisation: Current Trends and Future Perspectives. Adv. Exp. Med. Biol 922, 61–72. doi: 10.1007/978-3-319-35072-1_5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirrone GF, Iacob RE, Engen JR, 2015a. Applications of hydrogen/deuterium exchange MS from 2012 to 2014. Anal. Chem 87, 99–118. doi: 10.1021/ac5040242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirrone GF, Vernon BC, Kent MS, Engen JR, 2015b. Hydrogen Exchange Mass Spectrometry of Proteins at Langmuir Monolayers. Anal. Chem 87, 7022–7029. doi: 10.1021/acs.analchem.5b01724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschle T, Lin C, Jungmann R, Shih WM, Wagner G, 2015. Controlled Co-reconstitution of Multiple Membrane Proteins in Lipid Bilayer Nanodiscs Using DNA as a Scaffold. ACS Chem. Biol 10, 2448–2454. doi: 10.1021/acschembio.5b00627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reading E, Hall Z, Martens C, Haghighi T, Findlay H, Ahdash Z, Politis A, Booth PJ, 2017. Interrogating Membrane Protein Conformational Dynamics within Native Lipid Compositions. Angew. Chem. Int. Ed. Engl 56, 15654–15657. doi: 10.1002/anie.201709657 [DOI] [PubMed] [Google Scholar]
- Rouck JE, Krapf JE, Roy J, Huff HC, Das A, 2017. Recent advances in nanodisc technology for membrane protein studies (2012–2017). FEBS Lett 591, 2057–2088. doi: 10.1002/1873-3468.12706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rues R-B, Dötsch V, Bernhard F, 2016. Co-translational formation and pharmacological characterization of beta1-adrenergic receptor/nanodisc complexes with different lipid environments. Biochim. Biophys. Acta 1858, 1306–1316. doi: 10.1016/j.bbamem.2016.02.031 [DOI] [PubMed] [Google Scholar]
- Schmidt V, Sturgis JN, 2018. Modifying styrene-maleic acid co-polymer for studying lipid nanodiscs. Biochim Biophys Acta Biomembr 1860, 777–783. doi: 10.1016/j.bbamem.2017.12.012 [DOI] [PubMed] [Google Scholar]
- Segrest JP, Jones MK, Klon AE, Sheldahl CJ, Hellinger M, De Loof H, Harvey SC, 1999. A Detailed Molecular Belt Model for Apolipoprotein A-I in Discoidal High Density Lipoprotein. J. Biol. Chem 274, 31755–31758. doi: 10.1021/bi961876e [DOI] [PubMed] [Google Scholar]
- Shenkarev ZO, Lyukmanova EN, Paramonov AS, Panteleev PV, Balandin SV, Shulepko MA, Mineev KS, Ovchinnikova TV, Kirpichnikov MP, Arseniev AS, 2014. Lipid-protein nanodiscs offer new perspectives for structural and functional studies of water-soluble membrane-active peptides. Acta Naturae 6, 84–94. [PMC free article] [PubMed] [Google Scholar]
- Subramanian N, Condic-Jurkic K, O’Mara ML, 2018. Neurochemistry International 1–7. doi: 10.1016/j.neuint.2016.05.005 [DOI] [PubMed] [Google Scholar]
- Treuheit NA, Redhair M, Kwon H, McClary WD, Guttman M, Sumida JP, Atkins WM, 2016. Membrane Interactions, Ligand-dependent Dynamics, and Stability of Cytochrome P4503A4 in Lipid Nanodiscs. Biochemistry doi: 10.1021/acs.biochem.5b01313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Mu Z, Li Y, Bi Y, Wang Y, 2015. Smaller Nanodiscs are Suitable for Studying Protein Lipid Interactions by Solution NMR. The Protein Journal 34, 205–211. doi: 10.1007/s10930-015-9613-2 [DOI] [PubMed] [Google Scholar]
- Weis DD, Wales TE, Engen JR, Hotchko M, 2006. Identification and Characterization of EX1 Kinetics in H/D Exchange Mass Spectrometry by Peak Width Analysis. Journal of the American [DOI] [PubMed]
- Wen P-C, Verhalen B, Wilkens S, Mchaourab HS, Tajkhorshid E, 2013. On the Origin of Large Flexibility of P-glycoprotein in the Inward-facing State. J. Biol. Chem 288, 19211–19220. doi: 10.1042/BJ20050047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, 2015. Less is More: Membrane Protein Digestion Beyond Urea-Trypsin Solution for Next-level Proteomics. Mol. Cell Proteomics 14, 2441–2453. doi: 10.1074/mcp.R114.042572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chien EYT, Chalmers MJ, Pascal BD, Gatchalian J, Stevens RC, Griffin PR, 2010. Dynamics of the β 2-Adrenergic G-Protein Coupled Receptor Revealed by Hydrogen−Deuterium Exchange. Anal. Chem 82, 1100–1108. doi: 10.1021/ac902484p [DOI] [PMC free article] [PubMed] [Google Scholar]