

Abstract
Regulator of G protein signaling 2 (RGS2−/−) deficient mice feature an increased resting blood pressure and an excessive pressor response to stress. We measured renal sympathetic nerve activity (RSNA) directly to test the hypothesis that RSNA is increased in RGS2−/− mice, compared to RGS2+/+ mice. Seventeen mice (RGS2−/−, n = 9; RGS2+/+, n = 8) were anesthetized with isoflurane. We cannulated the left jugular vein for drug administration. Renal sympathetic nerve activity (RSNA) was recorded using bipolar electrodes. Arterial blood pressure (BP) from the femoral artery, ECG (needle electrodes), and RSNA were recorded (sample rate 10 kHz) simultaneously. RSNA was analysed offline using a modified wavelet de-noising technique and the classical discriminator method. RSNA detected during phenylephrine bolus injections or after the animals death was subtracted from baseline values. Mean arterial blood pressure, norepinephrine plasma levels, the responsiveness to vasoactive drugs, and the sympathetic baroreflex gain were similar in anesthetized RGS2+/+ and RGS2−/− animals. RSNA was lower in RGS2−/− mice compared to wild-type controls (wavelet: spike rate in Hz: RGS2+/+ 25.5 ± 5.1; RGS2−/− 17.4 ± 4.0; discriminator method: RGS2+/+ 41.4 ± 5.7, RGS2−/− 22.0 ± 4.3, p < 0.05). Thus, the expected result proved not to be the case. Our data suggest a mismatch between sympathetic nerve traffic and plasma norepinephrine concentrations. This observation may depend on altered coupling between electrical nerve activity and norepinephrine release and/or a changed norepinephrine uptake in RGS2−/− mice.
Keywords: Cardiovascular physiology, Hypertension, RGS2-deficient mice, Renal sympathetic nerve activity
1. Introduction
Many great transmembrane receptors belong to the superfamily of G protein coupled receptors (GPCR) and about half of all medicines modulate GPCRs activity (Miura et al., 2003). GPCRs in the central nervous system and in peripheral tissues have a critical role in blood pressure regulation. In the central nervous system, various GPCRs modulate sympathetic activity (Vizi, 2000). In the kidney, GPCRs regulate sodium excretion (DiBona, 2002). Vascular tone is also regulated by GPCRs (Heximer et al., 2003; Tang et al., 2003). Therefore, mechanisms modulating G protein-mediated responses have an important bearing on the cardiovascular system and could be also a target for therapeutic interventions. The regulator of G protein signaling (RGS) 2 was first identified as belonging to the GTPase activating proteins (GAPs). The RGS proteins were subsequently shown to regulate G protein-effector interactions (Obst et al., 2005; Abramow-Newerly et al., 2006). RGS2 preferentially alters Gq-mediated signaling (Heximer et al., 1997) by accelerating the inactivation of G proteins (Riddle et al., 2006). Thus, RGS2 attenuates the responses to angiotensin (Ang) II type-1 and alpha-1 adrenoreceptor stimulation. RGS2-deficient (RGS2−/−) mice feature an increased resting blood pressure and an excessive pressor response to stress (Kehrl and Sinnarajah, 2002; Gross et al., 2005). Sympathetic activation is implicated by the observation that urinary norepinephrine excretion rate and the depressor response to alpha-1 adrenoceptor blockade were increased in these animals (Gross et al., 2005). We measured renal sympathetic nerve activity (RSNA) directly to test the hypothesis that RSNA is increased in RGS2−/− mice, compared to RGS2+/+ mice.
2. Methods
2.1. Animals
We studied 7 adult male RGS2−/− mice weighing 27 ± 1 g and 8 male wild-type RGS2+/+ mice weighing 30 ± 1 g. In a subset of RGS2−/− (n = 4, 30 ± 1 g) and RGS2+/+ (n = 4, 29± 1 g), we measured plasma catecholamine concentrations. All animals were obtained from Washington University School of Medicine, Department of Cell Biology and Physiology, St. Louis, Missouri, USA. The genetic background of the mouse strain was C57Bl6. The animals were allowed free access to standard chow (0.25% sodium, SNIFF Spezialitäten GmbH, Soest, Germany) and drinking water. The protocol was approved by the local council on animal care in accordance with the guidelines of the American Physiological Society.
2.2. Instrumentation
We anesthetized mice with isoflurane through a nose cone (CuraMed Pharma, Karlsruhe, Germany). Body temperature was maintained within normal range using a heating table. Mice ventilated spontaneously. We cannulated the right jugular vein and the right femoral artery with polyethylene tubing for drug administration and for arterial blood pressure measurements (MIO-0501, Föhr Medical Instruments, See- heim, Germany), respectively. The left kidney was exposed through a left flank incision and with blunt dissection retroperitoneally. A branch of the renal nerve running parallel to the renal artery was carefully isolated. A bipolar stainless steel wire electrode pair (wire 0.003 in. bare diameter and 0.0045 in. coated diameter, part number 316SS3T, MedWire Corp, NY, USA) was hooked onto the nerve. When the signal quality was optimal, the electrodes were secured with silicone adhesive gel (QuickSeal, World Precision Instruments, Sarasota, Florida, USA). Therefore, nerve activity was recorded from the whole nerve in contrast to single fiber recordings. The nerve signal was band pass filtered (100–3000 Hz) and amplified (gain 10000) by an isolated differential amplifier (ISO-80, World Precision Instruments, Sarasota, Florida, USA). At the end of the experiment, animals were euthanized to record post mortal signal to correct for background noise.
2.3. Data acquisition and analysis
Arterial blood pressure (femoral artery), ECG (MVU- 0607, Föhr Medical Instruments, Seeheim, Germany) and RSNA were recorded using a computerized data-acquisition system. Signals were recorded using a WINDAQ data acquisition system (DI410, DATAQ, Acron, OH) with 14 Bit resolution at 10000 Hz sample frequency. Data were processed off-line using customized software written in PV-Wave language (Visual Numerics Inc., Houston, TX, USA) and Matlab environment (The MathWorks, Inc., Natick, MA, USA).
We applied three different methods to determine renal sympathetic nerve activity. The area under the curve was calculated as the integrated absolute values related to one second of the nerve signal after subtracting the offset. As a second technique we used the classical discriminator method. The frequency of spikes exceeding a preselected threshold voltage just above noise level was counted per second. The third technique was a modified wavelet denoising technique. Wavelet decomposition effectively filters the nerve signal into several frequency sub-bands while preserving its temporal structure and seems suitable for unsupervised de-noising and detection of single channel, multiunit data. In particular, wavelet-based processing has been demonstrated to be effective in the detection of human sympathetic action potentials (Diedrich et al., 2003), suggesting that wavelet detection methods could be applied to the mouse sympathetic nerve activity as described in more detail elsewhere (Brychta et al., 2007).
2.4. Experimental protocol
After surgery the mice were allowed 30–45 min for stabilization. Thereafter arterial pressure and RSNA were recorded. The reliability of the nerve preparation was tested by incremental intravenous bolus injections of phenylephrine hydrochloride (1.5, 3, and 6 μg/kg in 2–8 μl of saline) and sodium nitroprusside (1.5, 3.5, and 7 μg/kg in 1–4 μl of glucose 5%). Injections were separated by 5 min to reach stable baseline values before the next bolus. A representative tracing is shown in Fig. 1. Only nerve recordings with the characteristic baroreflex mediated reduction of RSNA in response to phenylephrine and with the characteristic increase of RSNA in response to nitroprusside were included in the study.
Fig. 1.
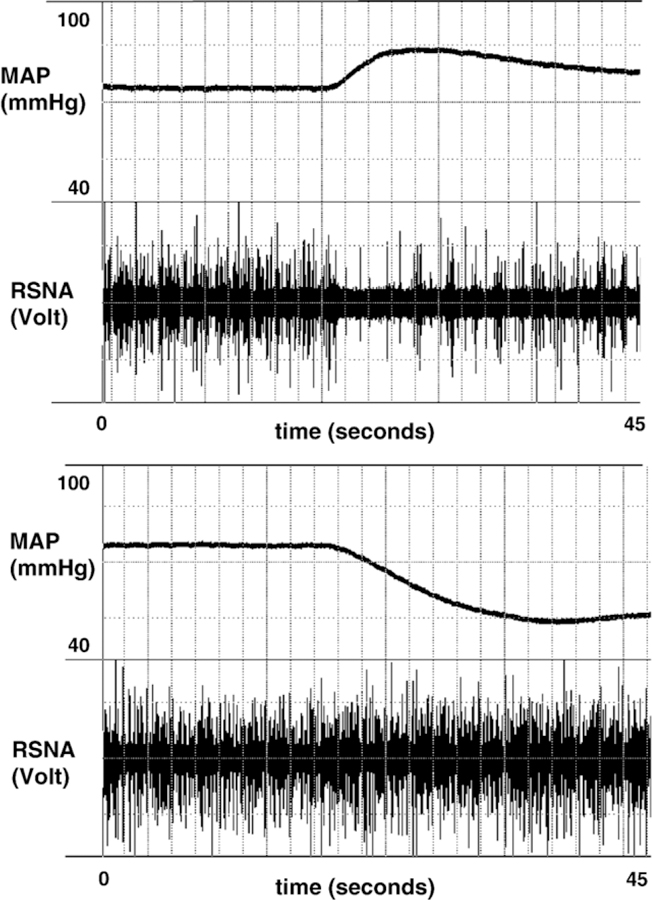
Recordings of renal sympathetic nerve activity (RSNA) and mean arterial pressure (MAP) during bolus injections of phenylephrine (6 μg/kg, upper panel) and nitroprusside (7 μg/kg, lower panel) in an isoflurane- anesthetized RGS+/+ mouse. Note the baroreflex mediated decrease of RSNA in response to the blood pressure increase during phenylephrine and the increase of RSNA in response to the blood pressure decrease during nitroprusside.
2.5. Baroreflex control of RSNA
The baroreflex gain was calculated as the percentage of the maximum change in RSNA (Δspike rate in %) divided by the maximum change in mean arterial blood pressure (ΔMAP in mm Hg) during phenylephrine bolus injections.
2.6. Plasma catecholamine level
Epinephrine and norepinephrine were extracted from plasma of isoflurane anaesthetized mice in a microtiter plate and coated with boronate-affinity gel. The catecholamines were acylated and released from the boronate-affinity gel. The solution of acylated epinephrine and norepinephrine was then analysed by a conventional ELISA assay (Dr. K. Burling, Mouse Biochemistry Laboratory, Dept. Clinical Biochemistry, Addenbrooke`s Hospital, Cambridge, UK).
2.7. Statistics
Data are presented as means ± SEM. Statistically significant differences between mean group values were evaluated by the Mann–Whitney U-test. P values b 0.05 were considered statistically significant.
3. Results
3.1. Blood pressure and catecholamines
The values are displayed in Fig. 2. Mean arterial blood pressure (Fig. 2, top) was 71 ± 4 mm Hg in anaesthetized RGS2−/− and 76 ± 3 mm Hg in RGS2+/+ mice, which is comparable to MAP values in C57Bl6 mouse strains during isoflurane anesthesia (Zuurbier et al., 2002). Plasma norepinephrine (Fig. 2, middle) and epinephrine concentrations (Fig. 2, bottom) were similar in anesthetized RGS2−/− and RGS2+/+ mice.
Fig. 2.
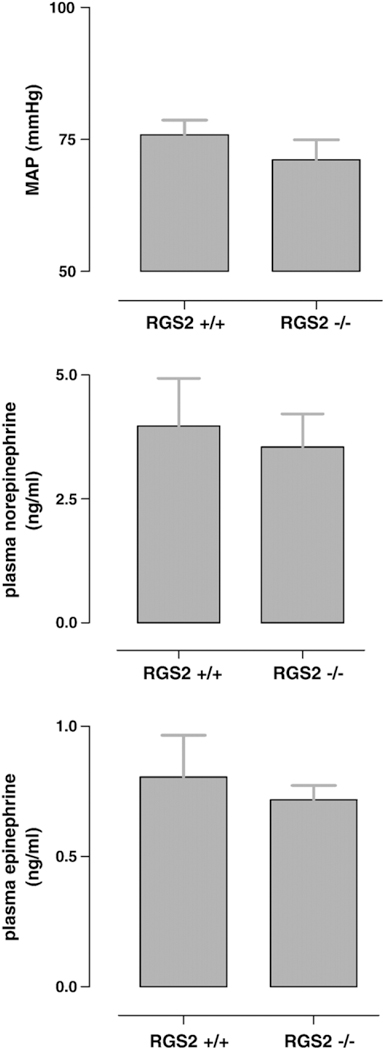
Mean arterial blood pressure values (MAP, upper panel), plasma norepinephrine values (middle panel), and plasma epinephrine values for RGS−/− and RGS+/+ mice are shown. No significant differences were found. Data are presented as mean SEM.
3.2. Basal sympathetic nerve activity
Fig. 3 shows the individual data of RSNA for RGS2- deficient mice compared to wild-type mice as scatter plots. RSNA, calculated as the area under the curve by merely rectifying the nerve signal, was 117 ± 41 a.u./s in RGS2−/− and 202 ± 32 a.u./s in RGS2+/+ mice (Fig. 3, top, n.s.). RSNA, calculated as spike rate in Hz by use of the modified wavelet de-noising technique (Fig. 3, middle) and by the classical discriminator method (Fig. 3, bottom), was lower in RGS2−/− mice compared to wild-type mice (wavelet: spike rate in Hz: RGS2+/+= 25.5 ± 5.1; RGS2−/− = 17.4 ± 4.0; discriminator method: RGS2+/+= 41.4 ± 5.7, RGS2−/− = 22.0 ± 4.3, p b<0.05).
Fig. 3.
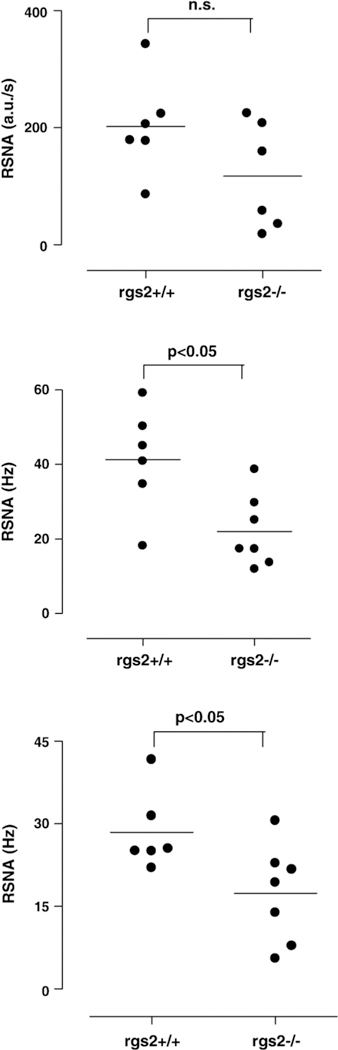
The individual data of renal sympathetic nerve activity (RSNA) for RGS2-deficient (RGS−/−) mice compared to wild-type mice (RGS+/+) determined by three different methods are shown as scatter plots. Top- RSNA calculated as the area under the curve by merely rectifying the nerve signal related to one second in arbitrary units (a.u./s). Middle-the frequency of spikes in Hz calculated using the wavelet de-noising technique. Bottom-classical discriminator method. The frequency of spikes exceeding a preselected threshold voltage was counted per second. Bars represent the mean values. RSNA was lower in RGS2-/- mice compared with RGS2+/+ mice independent of the used method (*p < 0.05, Mann-Whitney U-test).
3.3. Responses to phenylephrine and nitroprusside
The sensitivities to phenylephrine and to nitroprusside are shown in Fig. 4. The responses to phenylephrine (Fig. 4, top) were similar in RGS2+/+ mice compared to RGS2−/− mice, as were those to nitroprusside (Fig. 4, bottom). The phenylephrine dose to increase mean arterial blood pressure (MAP) 12.5 mm Hg was 3.6 μg/kg in RGS2+/+ mice and 4.5 μg/kg in RGS2−/− mice (n.s.). The nitroprusside dose that decreased MAP 12.5 mm Hg was
Fig. 4.
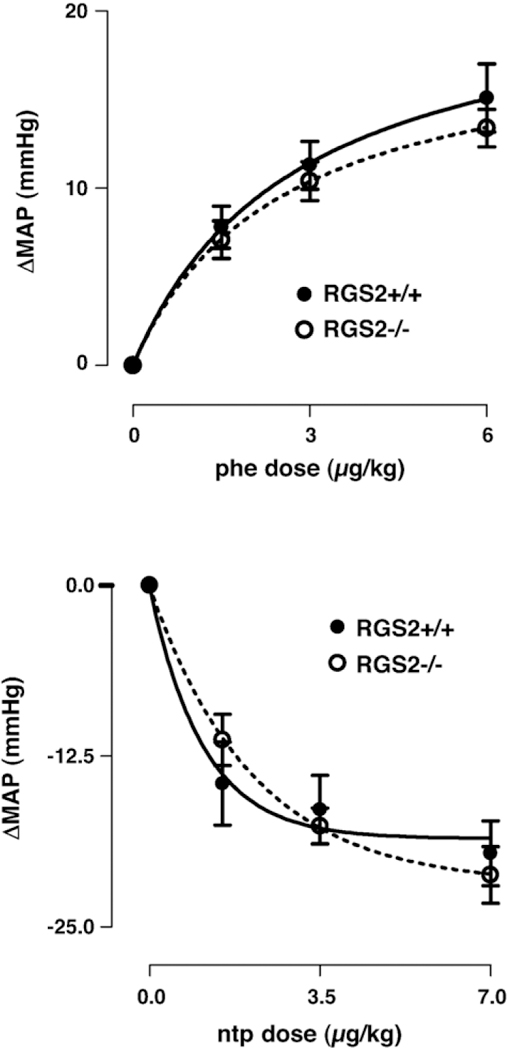
Changes of mean arterial blood pressure (ΔMAP) with incremental bolus injections of phenylephrine (phe, 1.5, 3, 6 μg/kg, upper panel) and nitroprusside (ntp, 1.5, 3.5, 7 μg/kg, lower panel) in RGS2-deficient mice (RGS2−/−) compared with RGS2+/+ mice. The sensitivity to vasoactive drugs was similar in both strains.
1.3 μg/kg in RGS2+/+ mice and 1.7 μg/kg in RGS2−/− mice (n.s.).
3.4. Baroreflex control of RSNA
Maximum gain was 2.4 ± 0.5%/mm Hg in RGS2+/+ mice and 2.1 ± 0.3%/mm Hg in RGS2−/− mice (n.s.).
4. Discussion
We observed that, in anesthetized RGS2−/− and RGS2+/+ mice, mean arterial blood pressure and circulating plasma catecholamines were not different, although RSNA was reduced in RGS2−/− mice, compared to controls. In earlier studies, we found that in conscious RGS2−/− mice, blood pressure is moderately increased, compared to RGS2+/+ mice and urinary catecholamines are increased in this strain. Furthermore, the difference in blood pressure can be abrogated by alpha-1 agonist blockade. Thus, our current hypothesis was that RSNA is increased in RGS2−/− mice, compared to RGS2+/+ mice. The expected result proved not to be the case.
We believe that the unexpected reduction in RSNA in our RGS2−/− mice is an important observation. Under the conditions of our study that was conducted under isoflurane anesthesia, the blood pressure values between strains were no longer different and the circulating catecholamines were similar. We interpret our result as indicating that with isoflurane anesthesia, the blood pressures are no longer different because of baroreceptor resetting in RGS2−/− mice leftward, compared to controls. The observation supports the idea that RGS2−/− mice are more stress sensitive than control animals. In our earlier studies, we measured catecholamines in urine in conscious RGS2−/− and RGS2+/+ mice. Urinary norepinephrine measurements were increased in awake RGS2−/− mice. In the present study we measured plasma norepinephrine, which was similar in both strains. Nevertheless, the present results could be explained by a reduction in the humoral levels of these compounds by the same mechanism that reduced sympathetic outflow, namely an anesthesia induced reduction in centrally generated sympathetic activity. Our earlier interpretation remains basically unaltered. Sympathetic support of blood pressure is increased in RGS2−/− mice compared to RGS2+/+ mice. In this study, the isoflurane anesthesia unmasked a suppressive potential that was not present in the conscious state.
Our data raise the speculation that RSNA and circulating catecholamines are disparate in their behaviors and that RGS2 deficiency has unmasked that state-of-affairs. RGS2 may be involved in the regulation of transmitter release and reuptake. RGS2 proteins may change presynaptic G- protein signaling as well as ion channel function and thereby adjust the amount of norepinephrine released per action potential (Abramow-Newerly et al., 2006). We propose that in RGS2−/− mice, the amount of norepinephrine released per action potential may be increased.
There is evidence for a possible mismatch between electrical sympathetic nerve activity, catecholamine levels, and vasoconstriction (Jacob et al., 2000; Thompson et al., 1995). Clinical examples are the inherited dopamine beta- hydroxylase deficiency (Thompson et al., 1995) or pharmacological inhibition of the norepinephrine transporter (NET) (Schroeder et al., 2002; Eisenhofer et al., 1995). With pharmacological NET inhibition, sympathetic nerve traffic is profoundly reduced whereas norepinephrine concentrations are normal or even increased. Thus, NET inhibition recapitulates the peculiar sympathetic abnormalities in RGS2−/− mice. One potential limitation of our study was the comparison of absolute levels of sympathetic nerve activity measured in arbitrary units per second or in spikes per second, which depends on recording conditions, the number and size of the nerve fibers, and the proximity of the active fibers to the electrode (Ma et al., 2002). Hence, comparing separate groups of animals may be influenced by those factors. However, we obtained the nerve signals under the exact same circumstances and used three different methods of nerve signal analysis after subtraction of the individual noise level. We admit that the anesthetic has complicated our results. The technical problems we face are immense. We know that the recovery time from abdominal surgery in terms of cardiovascular regulation exceeds 7 days in mice (Gross et al., 2002). To maintain an RSNA electrode for such a period of time is impossible. Similar studies in rats require 24 h recovery (Brockway et al., 1991). However, the 25 g mouse cannot be compared to the 250 g rat. We are improving our technical approaches; however, conscious mouse measurements of nerve activity are currently not possible.
Even more important could be the simultaneous recording from different target organs, which allows deeper insight into the differential regulation within the sympathetic nervous system (Kamiya et al., 2005; Ramchandra et al., 2006).
Despite these issues, we believe that our observations in anesthetized animals might be helpful. We found differences in sympathetic outflow in RGS2−/− compared to RGS2+/+ mice. We abide by the conclusion that the hypertension in conscious RGS2−/− mice is at least in part mediated through alpha-adrenoreceptor stimulation. Under isoflurane anesthesia, the remaining detectable signal is a diminished RSNA traffic in these mice. An interaction of RGS2 with the release or reuptake of catecholamines and with the central and peripheral sympathetic nervous system continues to be particularly important in this regard. For example, the reduction in sympathetic nerve traffic in our study did not indicate reduction in sympathetic contribution to blood pressure. RGS2 remains a putative blood pressure-lowering target.
Acknowledgment
The “Deutsche Forschungsgemeinschaft (DFG)” and the “Nationale Genomforschungsnetz (KGCV1 in NGFN2, 01GS0416)” supported the study. We thank Ilona Kamer (Max Delbrück Center for Molecular Medicine, Berlin, Germany) for technical assistance.
References
- Abramow-Newerly M, Roy AA, Nunn C, Chidiac P, 2006. RGS proteins have a signalling complex: interactions between RGS proteins and GPCRs, effectors, and auxiliary proteins. Cell. Signal 18, 579–591. [DOI] [PubMed] [Google Scholar]
- Brockway BP, Mills PA, Azar SH, 1991. A new method for continuous chronic measurement and recording of blood pressure, heart rate and activity in the rat via radio-telemetry. Clin. Exp. Hypertens., A 13, 885–895. [DOI] [PubMed] [Google Scholar]
- Brychta RJ, Tuntrakool S, Appalsamy M, Keller NR, Finney C, Robertson D, Shiavi R, Diedrich A, 2007. Wavelet methods for spike detection in mouse renal sympathetic nerve activity. IEEE Trans Biomed. Eng 54, 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiBona GF, 2002. Sympathetic nervous system and the kidney in hypertension. Curr. Opin. Nephrol. Hypertens 11, 197–200. [DOI] [PubMed] [Google Scholar]
- Diedrich A, Charoensuk W, Brychta RJ, Ertl AC, Shiavi R, 2003. Analysis of raw microneurographic recordings based on wavelet de- noising technique and classification algorithm: wavelet analysis in microneurography. IEEE Trans. Biomed. Eng 50, 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhofer G, Friberg P, Goldstein DS, Esler M, 1995. Differential actions of desipramine on sympathoadrenal release of noradrenaline and adrenaline. Br. J. Clin. Pharmacol 40, 263–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross V, Plehm R, Tank J, Jordan J, Diedrich A, Obst M, Luft FC, 2002. Heart rate variability and baroreflex function in AT2 receptor- disrupted mice. Hypertension 40, 207–213. [DOI] [PubMed] [Google Scholar]
- Gross V, Tank J, Obst M, Plehm R, Blumer KJ, Diedrich A, Jordan J, Luft FC, 2005. Autonomic nervous system and blood pressure regulation in RGS2-deficient mice. Am. J. Physiol., Regul. Integr. Comp. Physiol 288, R1134–R1142. [DOI] [PubMed] [Google Scholar]
- Heximer SP, Watson N, Linder ME, Blumer KJ, Hepler JR, 1997. RGS2/G0S8 is a selective inhibitor of Gqalpha function. Proc. Natl. Acad. Sci. U. S. A 94, 14389–14393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heximer SP, Knutsen RH, Sun X, Kaltenbronn KM, Rhee MH, Peng N, Oliveira-dos-Santos A, Penninger JM, Muslin AJ, Steinberg TH, Weiss JM, Mecham RP, Blumer KJ, 2003. Hypertension and prolonged vasoconstrictor signaling in RGS2- deficient mice. J. Clin. Invest 111, 445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob G, Costa F, Shannon J, Robertson D, Biaggioni I, 2000. Dissociation between neural and vascular responses to sympathetic stimulation: contribution of local adrenergic receptor function. Hypertension 35, 76–81. [DOI] [PubMed] [Google Scholar]
- Kamiya A, Kawada T, Yamamoto K, Michikami D, Ariumi H, Miyamoto T, Uemura K, Sugimachi M, Sunagawa K, 2005. Muscle sympathetic nerve activity averaged over 1 minute parallels renal and cardiac sympathetic nerve activity in response to a forced baroreceptor pressure change. Circulation 112, 384–386. [DOI] [PubMed] [Google Scholar]
- Kehrl JH, Sinnarajah S, 2002. RGS2: a multifunctional regulator of G-protein signaling. Int. J. Biochem. Cell Biol 34, 432–438. [DOI] [PubMed] [Google Scholar]
- Ma X, Abboud FM, Chapleau MW, 2002. Analysis of afferent, central, and efferent components of the baroreceptor reflex in mice. Am. J. Physiol., Regul. Integr. Comp. Physiol 283, R1033–R1040. [DOI] [PubMed] [Google Scholar]
- Miura S, Saku K, Karnik SS, 2003. Molecular analysis of the structure and function of the angiotensin II type 1 receptor. Hypertens. Res 26, 937–943. [DOI] [PubMed] [Google Scholar]
- Obst M, Tank J, Plehm R, Blumer KJ, Diedrich A, Jordan J, Luft FC, Gross V, 2005. NO-dependent blood pressure regulation in RGS2-deficient mice. Am. J. Physiol., Regul. Integr. Comp. Physiol 290 (4), R1012–R1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramchandra R, Barrett CJ, Guild SJ, Malpas SC, 2006. Evidence of differential control of renal and lumbar sympathetic nerve activity in conscious rabbits. Am. J. Physiol., Regul. Integr. Comp. Physiol 290 (3), R701–R708. [DOI] [PubMed] [Google Scholar]
- Riddle EL, Rana BK, Murthy KK, Rao F, Eskin E, O’Connor DT, Insel PA, 2006. Polymorphisms and haplotypes of the regulator of G protein signaling-2 gene in normotensives and hypertensives. Hypertension 47 (3), 415–420. [DOI] [PubMed] [Google Scholar]
- Schroeder C, Tank J, Boschmann M, Diedrich A, Sharma AM, Biaggioni I, Luft FC, Jordan J, 2002. Selective norepinephrine reuptake inhibition as a human model of orthostatic intolerance. Circulation 105, 347–353. [DOI] [PubMed] [Google Scholar]
- Tang M, Wang G, Lu P, Karas RH, Aronovitz M, Heximer SP, Kaltenbronn KM, Blumer KJ, Siderovski DP, Zhu Y, Mendel-sohn ME, 2003. Regulator of G-protein signaling-2 mediates vascular smooth muscle relaxation and blood pressure. Nat. Med 9, 1506–1512. [DOI] [PubMed] [Google Scholar]
- Thompson JM, O’Callaghan CJ, Kingwell BA, Lambert GW, Jennings LG, Esler MD, 1995. Total norepinephrine spillover, muscle sympathetic nerve activity and heart-rate spectral analysis in a patient with dopamine beta-hydroxylase deficiency. J. Auton. Nerv. Syst 55, 198–206. [DOI] [PubMed] [Google Scholar]
- Vizi ES, 2000. Role of high-affinity receptors and membrane transporters in nonsynaptic communication and drug action in the central nervous system. Pharmacol. Rev 52, 63–89. [PubMed] [Google Scholar]
- Zuurbier CJ, Emons VM, Ince C, 2002. Hemodynamics of anesthetized ventilated mouse models: aspects of anesthetics, fluid support, and strain. Am. J. Physiol.: Heart Circ. Physiol 282, H2099–H2105. [DOI] [PubMed] [Google Scholar]