

Abstract
The neural crest—a key innovation of the vertebrates—gives rise to diverse cell types including melanocytes, neurons and glia of the peripheral nervous system, and chondrocytes of the jaw and skull. Proper development of the cephalic region is dependent on the tightly-regulated specification and migration of cranial neural crest cells (NCCs). The core PCP proteins Frizzled and Disheveled have previously been implicated in NCC migration. Here we investigate the functions of the core PCP proteins Prickle1a and Prickle1b in zebrafish cranial NCC development. Using analysis of pk1a and pk1b mutant embryos, we uncover similar roles for both genes in facilitating cranial NCC migration. Disruption of either gene causes pre-migratory NCCs to cluster together at the dorsal aspect of the neural tube, where they adopt aberrant polarity and movement. Critically, in investigating Pk1-deficient cells that fail to migrate ventrolaterally, we have also uncovered roles for pk1a and pk1b in the epithelial-to-mesenchymal transition (EMT) of pre-migratory NCCs that precedes their collective migration to the periphery. Normally, during EMT, pre-migratory NCCs transition from a neuroepithelial to a bleb-based and subsequently, mesenchymal morphology capable of directed migration. When either Pk1a or Pk1b is disrupted, NCCs continue to perform blebbing behaviors characteristic of pre-migratory cells over extended time periods, indicating a block in a key transition during EMT. Although some Pk1-deficient NCCs transition successfully to mesenchymal, migratory morphologies, they fail to separate from neighboring NCCs. Additionally, Pk1b-deficient NCCs show elevated levels of E-Cadherin and reduced levels of N-Cadherin, suggesting that Prickle1 molecules regulate Cadherin levels to ensure the completion of EMT and the commencement of cranial NCC migration. We conclude that Pk1 plays crucial roles in cranial NCCs both during EMT and migration. These roles are dependent on the regulation of E-Cad and N-Cad.
Keywords: Zebrafish, Cranial Neural Crest, Prickle, Planar Cell Polarity, EMT, Cadherin
Graphical Abstract

Introduction
The neural crest is an embryonic, multipotent cell population that arises from the lateral edges of the developing neural plate, at the interface between neural and non-neural ectoderm (reviewed in Hall, 2000; Le Douarin and Kalcheim, 1999; Theveneau and Mayor, 2012). It is capable of giving rise to a large array of cell types that contribute to the vertebrate body plan (reviewed in Donoghue et al., 2008) including melanocytes, the neurons and glia of the entire peripheral nervous system, and in the cranial region only, the chondrocytes that form the bony elements of the jaw and skull. As a defining feature of the vertebrate phylum, neural crest has been key to the evolution of the complex vertebrate cephalic region (Gans and Northcutt, 1983).
Induction, specification, and onset of migration of neural crest cells (NCCs) occur in concert with neurulation. In teleosts such as zebrafish, neurulation processes differ markedly from those in other vertebrate models. Specifically, the zebrafish neural ectoderm does not form an epithelial neural plate that folds to form a central lumen, as in amniotes and amphibians. Rather, the zebrafish neural plate cells begin to converge at about 10 hours post fertilization (hpf), to produce a multilayered neural keel and subsequently a neural rod, with the first NCCs specified as early as 12 hpf at the lateral edges of the neural ectoderm. As neurulation continues, neuroepithelial cells undergo polarized cell divisions to establish the midline by 18 hpf, allowing the rod to cavitate and produce the neural tube (reviewed in Clarke, 2009). Throughout neurulation NCCs continue to emerge from the dorsal-most neural tissue and migrate away (Jimenez et al., 2016; Schilling and Kimmel, 1994).
NCCs travel large distances through the developing embryo and display a migratory potential that has been likened to that of metastatic cancer cells (reviewed in Gallik et al., 2017; Maguire et al., 2015). Like cancer cells, before NCCs migrate to colonize the embryo, they undergo an epithelial-to-mesenchymal transition (EMT) during which they lose their epithelial character (Kaufman et al., 2016). In a variety of vertebrate models, including zebrafish, NCCs reduce their previously-high levels of E-Cadherin during EMT and up-regulate N-Cadherin to become highly-protrusive, migratory mesenchymal cells (Acloque et al., 2009; Hay, 2005; Scarpa et al., 2015; Wheelock et al., 2008). Interestingly, despite the well-established down-regulation of E-Cadherin in migratory NCCs, Xenopus E-Cadherin is nevertheless required for NCC migration (Huang et al., 2016). During EMT, NCCs have been reported to show changes in expression levels of other Cadherin molecules as well, including Cadherin-6, Cadherin-7, and Cadherin-11 (Acloque et al., 2009; Berndt et al., 2008; Clay and Halloran, 2014; reviewed in Taneyhill and Schiffmacher, 2017). In tandem, NCCs alter the expression of polarity molecules that contribute to their high directionality: in both Xenopus and zebrafish embryos, presumptive NCCs lose apico-basal polarity, and subsequently activate non-canonical Wnt/PCP signaling molecules (Berndt et al., 2008; reviewed in Gallik et al., 2017; Lee et al., 2006; Mayor and Theveneau, 2014; Sauka-Spengler and Bronner-Fraser, 2008; Scarpa et al., 2015; Thiery and Sleeman, 2006; Thompson and Williams, 2008). These dynamic molecular changes are tightly associated with the changes in cell morphology and behavior that accompany the onset of NCC migration.
Recently, the classical understanding of the process of EMT that precedes a variety of cell migration, wound healing, and metastasis processes, has come under greater scrutiny. Classical studies have treated the EMT transition as a binary state change from a tightly-packed, highly-adhesive epithelial morphology to a more dispersed, highly-protrusive, migratory mesenchymal one. By contrast, more recent studies from different cell types across multiple model organisms have revealed a range of transient cell states that span the ‘spectrum’ or ‘continuum’ from epithelial to mesenchymal morphologies (reviewed in Campbell and Casanova, 2016; Nieto et al., 2016). For instance, metastatic carcinoma cells that show hybrid characteristics during the process of EMT have been described as occupying an intermediate ‘metastable’ state, owing to their transitory morphology (reviewed in Lee et al., 2006; Savagner, 2010). Similarly, zebrafish cranial NCCs have also been reported to adopt transitional morphologies during EMT between the fully-neuroepithelial morphology and the migratory, mesenchymal morphology (Berndt et al., 2008; Clay and Halloran, 2014). First, presumptive NCCs in the neuroepithelium detach from their apical surfaces. These pre-migratory NCCs at the dorsal aspect of the neural tube change morphologically from elongated, tightly-packed cells to loose, rounded cells capable of bleb-based protrusions. Subsequently, NCCs adopt highly mesenchymal, filopodial- and lamellipodial-based morphologies capable of migration in streams (Berndt et al., 2008; Clay and Halloran, 2014). Since ‘EMT’ has often been used interchangeably with the term ‘delamination’, an appreciation for the particular state changes that need to occur during the EMT has been lacking, causing difficulty in ascribing particular phenotypes to specific phases of neural crest development.
Many collective cell migration processes in vertebrates, including NCC migration, depend on Planar Cell Polarity (PCP) molecules (reviewed in Davey and Moens, 2017; Roszko et al., 2009). The non-canonical Wnt/PCP pathway influences cranial NCC migration in both Xenopus and zebrafish by mediating contact inhibition of locomotion (CIL) (Carmona-Fontaine et al., 2008; De Calisto et al., 2005; Matthews et al., 2008; Scarpa et al., 2015; Theveneau et al., 2010; Theveneau et al., 2013). Specifically, highly-protrusive migratory NCCs contact one another, activate the core PCP proteins Frizzled and Disheveled at the points of contact, and reorient their protrusions to migrate away from each other (Carmona-Fontaine et al., 2008; Scarpa et al., 2015; Szabo et al., 2016; Theveneau et al., 2013).
The role of the core PCP protein Prickle (Pk) in cranial neural crest development has not previously been comprehensively investigated. However, several characteristics make it an attractive candidate for study. In Drosophila, Pk is required to amplify cellular asymmetries of other core PCP proteins, and differential expression of Pk isoforms is critical for the establishment of tissue polarity (Ambegaonkar and Irvine, 2015; Merkel et al., 2014; Sharp and Axelrod, 2016; Tree et al., 2002; reviewed in Zallen, 2007). Additionally, in vertebrates, multiple orthologs of Drosophila Pk control the localization and dynamics of other core PCP proteins in morphogenesis and collective cell migration (reviewed in Davey and Moens, 2017; Jussila and Ciruna, 2017). Like other core PCP proteins, Pk1 molecules play roles in convergent extension during gastrulation and neurulation (Ciruna et al., 2006; Sepich et al., 2011; Veeman et al., 2003; Yin et al., 2008), in organizing the sensory cells of the inner ear (reviewed in May-Simera and Kelley, 2012; Rida and Chen, 2009), in the polarization of ciliated epithelia (Butler and Wallingford, 2015), and in the migration of facial branchiomotor neurons (Carreira-Barbosa et al., 2003; Mapp et al., 2011; Mapp et al., 2010).
In this study, we present evidence for roles of the zebrafish Prickle1 paralogs, Prickle1b (Pk1b) and Prickle1a (Pk1a), in both EMT and NCC migration. Our investigation has utilized both our previously-described pk1bfh12/fh122 mutant (Mapp et al., 2011), and a new pk1ach105/ch105 mutant. Time-lapse imaging analysis reveals that disruption of either pk1b or pk1a causes pre-migratory NCCs to cluster with neighboring NCCs at the dorsal aspect of the neural tube, adopt aberrant cell polarity, and migrate in an inappropriate directions along the anteroposterior (AP) axis of the dorsal neural tube. Unlike wild-type pre-migratory NCCs that rapidly transition from a neuroepithelial morphology to a transient blebbing state to a mesenchymal morphology, we find that dorsal pre-migratory NCCs in Pk1-deficient specimens maintain blebbing behaviors over extended periods. Those Pk1-deficient NCCs that do successfully transition to the mesenchymal state, fail to separate from neighboring NCCs, consistent with defects in CIL. Further, the two pk1 paralogs function redundantly in EMT and migration. Finally, we find that Pk1b regulates E-Cadherin both in pre-migratory and migratory NCCs, and N-Cadherin in migratory cells. Our findings demonstrate that the core PCP Pk1 molecules are required not only in regulating migration, but also in the process of EMT that precedes and is required for the normal migration of the cranial neural crest.
Materials and methods
Zebrafish husbandry
Zebrafish (Danio rerio) were maintained using standard procedures and used in accord with IACUC-approved protocols. Embryos were maintained in E3 solution (in mM: 5.0 NaCl, 0.17 KCl, 0.33 CaCl, 0.33 MgSO4) at 24°C–28.5°C and staged as described (Kimmel et al., 1995). Embryos were obtained from crosses of adult fish stocks of mutants and/or transgenics. Specimens analyzed at stages later than 24 hours post-fertilization (hpf) were treated with 0.2 mM 1-phenyl 2-thiourea (PTU; Sigma) from 24 hpf onwards to inhibit melanin synthesis. Transgenic zebrafish lines Tg(−4.9sox10:EGFP)ba2 (hereafter Tg(sox10:EGFP) (Carney et al., 2006) and Tg(−7.2sox10:mRFP)vu234 (hereafter Tg(sox10:mRFP)) (Kirby and Hutson, 2010) and the mutant line pk1bfh122 (Mapp et al., 2011) have been previously described.
In situ hybridization
Detection of pk1b (Rohrschneider et al., 2007), pk1a (Carreira-Barbosa et al., 2003; Veeman et al., 2003), crestin (Solomon et al., 2003), and dlx2 (Akimenko et al., 1994) by in situ hybridization was carried out as previously described (Prince et al., 1998) using NBT/BCIP as the enzyme substrate. Embryos were cleared in 70% dimethylformamide overnight at 4°C, washed twice with 100% Methanol for 30 minutes at room temperature, and then mounted in 80% glycerol in PBS+0.1%Tween-20. Images were acquired using a a Leica DFC490 camera on a Zeiss Axioskop microscope.
Generation of the pk1ach105 mutant
The CHOPCHOP website (Montague et al., 2014) was used to select genomic target sites on the zebrafish pk1a locus. Two gRNAs targeting exon 2 were designed and one gRNA was selected based on higher efficacy as determined using a T7 endonuclease I assay (below). The following sequence containing an SP6 promoter (underlined), genomic target site (gRNA sequence in lowercase, with PAM sequence italicized) and an optimized single guide (sg) RNA scaffold, modified for efficient transcription with an extended stem loop designed to improve interaction with the Cas9 protein (Chen et al., 2013) was purchased as a gBlock from Integrated DNA Technologies and used as a template for transcribing sgRNA: 5’-AAAAATTTAGGTGACACTATAgtgatggagctggagaatcggTTTAAGAGCTATGCTGGAAACAGCATAGCAAGTTTAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTTT-3’ sgRNA and Cas9 RNA were transcribed, purified and injected as previously described (Dalgin and Prince, 2015). The sgRNA (final stock concentration: 100–200 ng/μl) and Cas9 mRNA (final stock concentration: 600 ng/μl) were transcribed using the MEGAscript SP6 Kit (Ambion), treated with TURBO DNase from the Ambion kit, extracted using an equal volume of phenol/chloroform, and precipitated using 3 volumes of 100% ethanol. RNA was resuspended in water, and 70 pg sgRNA and 500 pg Cas9 RNA were injected into one-cell stage embryos. Genomic DNA was extracted from 10–15 48 hpf embryos. DNA was resuspended in 10 l TE buffer and stored at 4°C. A PCR reaction using 2 μl of genomic DNA was assembled using primers that amplified a 230 bp genomic region flanking the pk1a target site (forward primer 5’-GTAAGTGTGTGGCGGTA-3’, reverse primer 5’-CCATACCTGCTCTGGTCTGAGT-3’). The T7 Endonuclease Assay I was performed on PCR amplicons as a measure of gRNA efficiency as previously described (Dalgin and Prince, 2015). PCR products were subcloned into the pGEM-T Easy vector (Promega A1360) according to manufacturer’s protocols, and clones were sequenced to identify insertion/deletion mutant alleles.
Injected F0 specimens were raised to adulthood and outcrossed to wild-type AB embryos. Genomic DNA was extracted from 10–15 F1 offspring embryos per cross, PCR amplified as described above, and subcloned to identify INDELs. Sequence information from F1 offspring identified two founder F0 fish with putative germline mutations. F1 offspring from these founders were then raised to adult stages, and DNA isolated from fin clips used to genotype putative mutants as described (Jing, 2012). Of these, multiple F1 fish were heterozygous for specific individual mutations, with one F1 fish identified as heterozygous for a putative null pk1a genomic allele, hereafter designated pk1ach105. The pk1ach105 allele has a 19 bp deletion, causing a frame shift and predicted STOP at amino acid 24 (Fig. 1B). A subsequent F2 generation was raised by out-crossing the F1 mutant fish to a Tg(sox10:EGFP) fish. Fin clips from F3 and F4 progeny of heterozygous adult fish were used to genotype and identify heterozygous pk1ach105/+ and homozygous pk1ach105/ch105 mutants using PCR with primer sequences 5’-GTAAGTGTGTGGTGGCGGTA-3’ and 5’-TCAGTCGTTTAAGGCGGGTC-3’. The resulting PCR products were then run on a 4% agarose gel. Heterozygous, homozygous and wild-type fish were distinguished by the presence of two bands 19 bp apart in the first case, versus different, single bands in the latter two cases.
Figure 1: Zebrafish pk1b and pk1a are expressed in domains that partially overlap with expression of the pan-neural crest marker crestin.

(A-C) In situ hybridizations for the pan-neural crest marker crestin (A), prickle1b (B) and prickle1a (C), dorsal views at 24 hpf, show that both pk1b and pk1a are expressed in tissue lateral to the neural tube. Low levels of expression are also seen in the neuroepithelium. Solid black arrows indicate gene expression in the cranial NCC stream leading to pharyngeal arch 1 (PA1), solid white arrow indicates pk1b expression in the FBMNs, r4 is indicated. (D) Schematic of protein domain structure for both Prickle1b and Prickle1a. Arrows indicate lesions in pk1ach105 and pk1bfh122 mutants. The first codon, and the C-terminal CAAX domain are also indicated. (E) Amino-acid sequence of Pk1a N-terminus; the pk1ach105 mutant encodes a truncated protein with a premature STOP codon.
Morpholino design and microinjection
Two morpholinos (MOs) designed to block pk1b splicing (Gene Tools, LLC) were used as previously described (Rohrschneider et al., 2007) at a standard concentration of 2 ng/nl. The first MO was targeted to intron3-exon4, with a sequence of 5’-GGCAGTAGCGAATCTGTGTTGAAGC-3’, and the second MO was targeted to exon6-intron6 with a sequence of 5’-TTAATGAAACTCACCAATATTCTCT-3’. MOs were solubilized in water (Sigma-Aldrich) for a stock concentration of 20 mg/ml. Tg(sox10:EGFP) and Tg(sox10:mRFP) embryos were microinjected with MOs at the one-cell stage. The vangl2-MO (5’-GTACTGCGACTCGTTATCCATGTC-3’) was used as previously described (Jessen et al., 2002) at a concentration of 1 ng/nl.
Image Acquisition
For assays in fixed specimens, embryos were fixed in 4% paraformaldehyde (PFA, diluted in 1× phosphate buffered saline; PBS) overnight. Following overnight fixation, embryos were washed in 1× PBS five times for 10 minutes each. For long-term storage of embryos, embryos were washed in 30%, 60% and 100% methanol (diluted in 1× PBS) and stored in 100% methanol at −20°C. If stored in 100% methanol, embryos were progressively washed in 60%, 30% methanol as well as 1× PBS + 0.1%Tween-20 before being transferred to 80% glycerol. The embryos were cleared in 80% glycerol, deyolked using fine forceps and flat-mounted in glycerol in dorsal view. For transverse section analysis, embryos in PBS were embedded in 3% low-melt agarose (MidSci IB70051 St. Louis, Missouri), and 100 m sections cut using a Vibratome 1000 plus sectioning system, then mounted in glycerol for subsequent imaging. Images were collected using an upright Zeiss LSM710 confocal microscope using either a Plan-Apochromat 20×/0.8 (working distance: 0.55mm) objective or a Plan Apochromat 40×/1.0 water-immersion (working distance: 2.5mm) objective. Green fluorescent proteins (GFP) and dyes (Alexa Fluor 488) were excited by a 488nm laser. Red fluorescent proteins (mRFP, RFP) and dyes (Alexa Fluor 564) were excited by a 543nm laser. DAPI dye and cyan fluorescent protein (CFP) were excited using a 405nm laser. For a single fluorophore or a combination of fluorophores, spectral unmixing was used to define emission fluorescence range. For each sample, transmitted-light images were also collected. Images were acquired and saved as .czi files using Zen (Zeiss) software.
Live imaging was performed at room temperature by mounting 16 hpf embryos in 1% low-melt agarose (MidSci IB70051 St. Louis, Missouri) prepared in E3 medium with anesthetic (MS-222, tricaine methanesulfonate, Sigma-Aldrich; 0.20mg/mL in E3 solution). Embryos were staged before and after experimentation. A Plan-Apochromat 40×/1.0 water-immersion objective (working distance: 0.55mm) on a Zeiss LSM710 confocal microscope was used over a period of two hours with images captured at a time interval between 1 and 2 mins, with images collected with transmitted light (brightfield) acquired before and after experimentation to provide a reference for drift correction in image processing of migrating cells relative to static cells. Longer time-lapses were performed at room temperature by mounting 12 hpf embryos in 0.5% low-melt agarose and imaged in the same fashion as above with a time interval of 5 mins. Transmitted light (brightfield) images were acquired at each time point to allow for drift correction.
mRNA generation and microinjection
Capped mRNA was generated using the MEGAscript SP6 or T7 Kit (Ambion). mRNAs at concentrations of Tol2 Transposase (150 ng/μl) (Kawakami and Shima, 1999), EGFP-XCentrin (150 ng/μl), Cherry-XCentrin (150 ng/μl) (Sepich et al., 2011), H2B-CFP (300 ng/μl), and mRFP (200 ng/μl) (last two generously provided by Gokhan Dalgin, University of Chicago) (Dalgin and Prince, 2015) were kept on ice and microinjected into the yolk-cell interphase of one-cell stage embryos.
NCC aspect ratio measurements
Measurements of the width and length ratio of NCCs were performed in wild-type embryos and mutant embryos in the Tg(sox10:EGFP) background. Embryos were co-injected with RNA encoding mRFP, and width and length was measured using the mRFP label in sox10-positive cells.
Quantification of NCC contacts over time
To quantify breakage of contacts between NCCs over time intervals of 0–10 min, 10–20 min, 20–30 min and 30–40 min, we counted the total number of pairs of NCCs in contact within a cluster at t=0 such that a single cell could be in pairs with multiple cells in contact, and then counted the pairs that broke at each time interval. Dividing the second measure by the first, we generated a ratiometric measure of ‘pair breakage’ within a cluster. To measure the relative proportions of individuals NCCs and NCC clusters of varying sizes, we measured the number of cells that persisted in a given configuration (from an individual cell to cells in increasing sizes of clusters) over non-overlapping 20-minute time windows.
Transient transgenesis
To label cranial NCCs mosaically, a DNA construct encoding I-SceI −4.9sox10:LifeAct-GFP (gift from Michael Granato, University of Pennsylvania) was microinjected as described (Banerjee et al., 2011) into one-cell stage Tg(sox10:mRFP) embryos at a concentration of 100 ng/μl together with the meganuclease I-SceI (NEB) at a concentration of 1U/μl in 1× phosphate-buffered saline. Cells were imaged in live embryos, mounted as described above, and imaged at approximately 16 hpf for at least 20 minutes using a Plan-Apochromat 40×/1.0 water-immersion objective (working distance: 0.55mm) on a Zeiss LSM710 confocal microscope. Transmitted light (brightfield) images were also collected to categorize cells as pre-migratory or migratory based on their position relative to the apparent edge of the neural tube.
Alcian labeling
Cartilage was labeled with 0.02% Alcian Green, as previously described (Schilling and Kimmel, 1997). The pharyngeal apparatus was dissected and flat mounted. Samples were then imaged with a Leica DFC490 camera on a Zeiss Axioskop microscope.
Immunohistochemistry
Embryos were fixed in 4% paraformaldehyde (PFA; Sigma), and immunohistochemistry was performed as previously described (Prince et al., 1998) using the following primary antibodies: rabbit polyclonal anti-Cdh1 (E-Cadherin; 1:100; GeneTex GTX100443), rabbit polyclonal Cdh2 (N-Cadherin; 1:200; GeneTex GTX125885), rabbit polyclonal anti-Sox10 (1:250; Genetex GTX128374) and rabbit polyclonal anti-Caspase-3 (1:100; Millipore #AB3623). The following secondary antibodies were used: goat-anti mouse highly cross-adsorbed Alexa Fluor Plus 488 (Molecular Probes A32723), goat anti-rabbit highly cross-adsorbed Alexa Fluor 488 (Molecular Probes A11034), goat anti-rabbit cross-adsorbed Alexa Fluor 546 conjugate (Molecular Probes A11010), and anti-GFP Alexa Fluor 488 conjugate (Molecular Probes A21311). Embryos were then cleared in 80% glycerol, deyolked and flat-mounted.
Subcellular localization measurements
To quantify subcellular localization of E-Cad, fluorescence intensity in Tg(sox10:mRFP) embryos immunolabeled with E-Cad, and counterstained with DAPI was measured at the plasma membrane with average ratios calculated and intensities normalized with respect to background levels of E-Cad. To determine subcellular localization of N-Cad, Tg(sox10:mRFP) embryos were immunolabeled for N-Cad and counterstained with DAPI and fluorescence intensities measured in the same manner as for E-Cad.
To determine subcellular localization of the intracellular microtubule organizing center (MTOC) in NCCs, either Tg(sox10:mRFP) embryos were injected with RNA coding for EGFP-XCentrin as described (Sepich et al., 2011) as well as RNA coding for H2B-CFP, or Tg(sox10:EGFP) embryos were injected with RNA coding for Cherry-XCentrin (Sepich et al., 2011) as well as RNA coding for H2B-CFP. Injected embryos were fixed at 24 hpf. To quantify angles of the MTOC relative to the nucleus and the AP axis, bisecting lines were drawn from the MTOC to the nucleus, the angle relative to the AP axis for each cell was determined, and measurement of the angle of the MTOC with respect to the nucleus and the AP body axis was performed using Fiji and subsequently analyzed using MATLAB.
Quantification and statistical analysis
Fiji (NIH) was used to process and analyze data, and Prism (GraphPad) and Microsoft Excel were used for statistical analyses. Cell counts for z-stacks of confocal images with optical sections between 2 and 4 μm were performed using the Fiji cell counter plugin. Cell aspect ratio and area quantifications were performed using the Fiji Cell Magic Wand plugin. Statistical significance was determined using two-way ANOVA or the student t-test (* indicates p<0.05, ** indicates p<0.01, *** indicates p<0.001, **** indicates p<0.0001; non-significance indicates p>0.05).
Polar plots for directionality trajectories were made by importing Excel-generated measurements of displacement vectors into MATLAB to allow for generation of polar plots. Polarity measurements were analyzed using Fiji; statistical measurements and polar histograms were generated using MATLAB. Statistical significance (p<0.05) for polar plots was determined using the Watson-Williams F-test.
Manual tracking of migrating cells was performed using the Fiji MTrackJ plugin by following cells through multiple stacks. Persistence measurements were made using tracking data imported into Excel and using the program DiPer, an open-source set of Excel macros, as previously described (Gorelik and Gautreau, 2014). Collected time-lapse data were registered using Fiji and exported as TIF images and AVI videos.
For cluster rearrangement analysis, a MATLAB script (available upon request) was used to threshold and segment cells, enabling counts on pair breaks within clusters. The data on pair-breaks within clusters were imported into Prism and tabulated. All figures were created in Illustrator and InDesign (Adobe).
Results
1. Disruption of pk1b and pk1a function alters cranial NCC disposition
Both zebrafish pk1b and pk1a paralogs are expressed in the developing nervous system (Carreira-Barbosa et al., 2003; Rohrschneider et al., 2007; Roszko et al., 2009; Veeman et al., 2003). Our previous in situ hybridization analysis has demonstrated that pk1b is expressed in the developing hindbrain by the 16 hpf stage, with elevated levels of expression in migrating facial branchiomotor neurons (FBMNs) (Rohrschneider et al., 2007). Similarly, pk1a expression has also been reported in the hindbrain (Carreira-Barbosa et al., 2003; Roszko et al., 2009). In these reports, expression can additionally be noted in domains immediately lateral to the developing hindbrain, suggesting that the pk1 paralogs might also be expressed in cranial neural crest cells. To investigate cranial NCC expression, we performed in situ hybridization with pk1b and pk1a probes at 24 hpf, and compared the expression patterns with those of the pan-neural crest marker crestin (Fig. 1A–C). While the genes are expressed at low levels, expression of both pk1b and pk1a is detectable in domains lateral to the anterior hindbrain and midbrain that overlap with expression of crestin. These results indicate that pk1b and pk1a are expressed at an appropriate time and place to play a role in the development of cranial neural crest.
To investigate the functions of pk1b and pk1a in developing cranial NCCs, we have utilized zebrafish embryos mutant for each gene. We previously described the pk1bfh122/fh122 mutant, which is characterized by a complete block to collective cell migration of FBMNs through the hindbrain (Mapp et al., 2011). Importantly, Pk1b-morpholino (MO) knockdown precisely phenocopies the pk1bfh122/fh122 mutant FBMN defect, implying that this C-terminal point mutation (Fig. 1D) is a strong hypomorph (Mapp et al., 2011). To allow analysis of Pk1a function, we used the CRISPR/Cas system to generate a novel mutant, pk1ach105/ch105. The ch105 allele is a predicted null, with a premature stop codon at amino acid 24, truncating the protein close to its N-terminus (Fig. 1D, E). In contrast to previous reports of specimens in which pk1a function was disrupted using morpholino knockdown (Carreira-Barbosa et al., 2003; Veeman et al., 2003), pk1ach105/ch105 mutant embryos do not show obvious gastrulation defects, except when combined with the knockdown of Pk1b. Although homozygous pk1ach105/ch105 mutant embryos do not survive at Mendelian ratios, they can nevertheless occasionally be raised to fertile adulthood.
To investigate cranial NCC behaviors in the pk1 mutants, we made use of the Tg(sox10:EGFP) line (Carney et al., 2006), which expresses EGFP in all NCCs beginning at approximately 12 hpf. In the cranial region, streams of NCCs derive from specific anteroposterior (AP) regions of the developing midbrain and hindbrain and migrate out to populate the pharyngeal arches and frontonasal process (Couly et al., 2002; Couly et al., 1993; Lumsden et al., 1991; Schilling and Kimmel, 1994). In wild-type Tg(sox10:EGFP) specimens counterstained with the nuclear marker DAPI at 18 hpf, streams of cranial NCCs were detected migrating ventrolaterally towards the developing pharyngeal arches (PAs) and the fronto-nasal process (Fig. 2A). In both the pk1bfh122/fh122 and pk1ach105/ch105 single homozygous mutants, NCCs were found in groups of closely-apposed cells located at the dorsal-most aspect of the midbrain and anterior hindbrain, showing a markedly different organization to the more dispersed, individual NCCs found in wild-type specimens (compare Fig. 2B, C with 2A). We next used transverse section analysis to investigate the precise localization of the cranial NCCs. Sections of wild type specimens (Fig. 2D, G) revealed a few NCCs located just dorsal to the neural tube, with others in the process of migrating ventrolaterally around the neural tube, and the majority located lateral to the neural tube within the surrounding head mesenchyme (arrows). By contrast, in pk1bfh122/fh122 embryos (Fig. 2E, H) and pk1ach105/ch105 embryos (Fig. 2F, I) fewer NCCs had migrated out into the lateral head mesenchyme, and groups of closely-apposed NCCs or ‘clusters’ of cells, were present immediately adjacent to the dorsal neural tube.
Figure 2: Disruption of pk1b or pk1a function disrupts cranial NCC disposition.
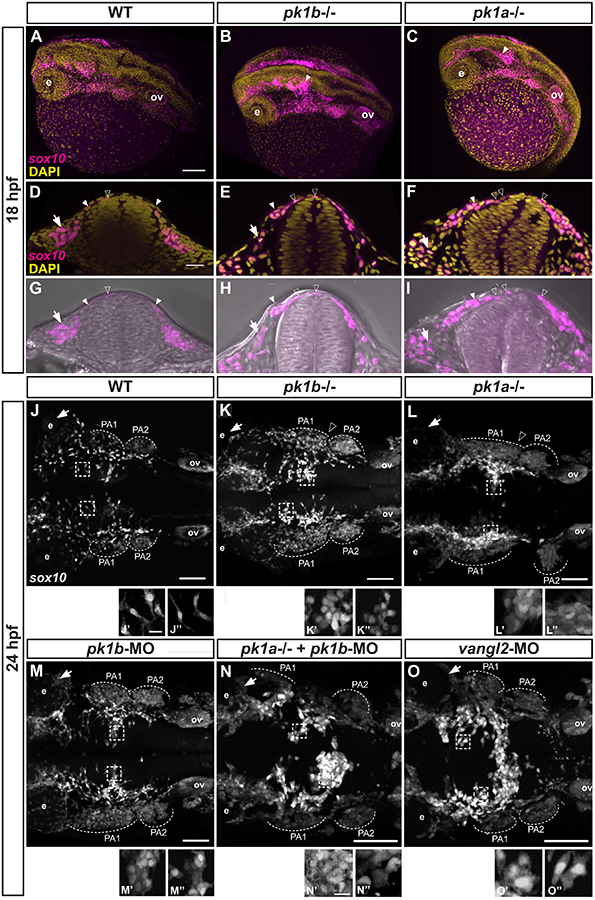
(A-C) Maximal projections of confocal images in dorsolateral view of fixed Tg(sox10:EGFP) 18 hpf embryos (EGFP indicated in magenta), counterstained with DAPI (yellow), show the 3D formation of NCC streams migrating ventrolaterally. The pk1bfh122/fh122 (B) and pk1ach105/ch105 (C) mutant embryos exhibit dorsal clusters of NCCs along the AP axis, as indicated by solid white arrowheads; ov = otic vesicle, e = eye, scale bar = 100μm. (D-I) Single confocal z-slices of transverse sections through the anterior hindbrain of wild-type (D, G), pk1bfh122/fh122 (E, H) and pk1ach105/ch105 (F, I) Tg(sox10:EGFP) 18 hpf embryos counterstained with DAPI (yellow; D-F) or together with bright-field views (G-I). Open arrowheads indicate nuclei of dorsally located NCCs (EGFP signal in magenta), closed arrowheads indicate NCCs in process of ventrolateral migration around the neural keel, arrows indicate NCCs that have migrated out into the surrounding head mesenchyme, scale bar = 25μm. (J-O) Maximal projections of confocal images in dorsal view from fixed, deyolked, flat-mounted Tg(sox10:EGFP) embryos at 24 hpf. The scale (bar = 100μm) is identical for all specimens shown except for pk1ach105/ch105 + pk1bmorphant, and vangl2-morphant embryos, which showed a shortened AP body axis and a wider mediolateral body axis due to convergence defects. As compared to WT embryos (n=13 embryos) (J), pk1bfh122/fh122 (n=12 embryos) (K) and pk1ach105/ch105 (n=9 embryos) (L) mutant embryos exhibit the maintenance of distinct dorsally-localized NCC clusters. pk1b-morphant (n=18 embryos) (M) embryos phenocopy the pk1bfh122/fh122 mutant embryos (K). The pk1ach105/ch105 + pk1b-morphant (n=8 embryos) (N), and vangl2-morphant (n=7 embryos) (O) show a more severe phenotype of dorsal NCC clustering across the midline. (J’-O’’) High magnification insets from associated micrographs (boxed) show that in contrast to WT specimens (J’, J’’) where NCCs exhibit bipolar, protrusive morphologies, pk1bfh122/fh122 (K’, K’’) pk1ach105/ch105 (L’, L’’), pk1b-morphant (M’, M’’), pk1ach105/ch105 + pk1b-morphant (N’, N’’), and vangl2-morphant (O’, O’’) NCCs show less protrusive, more rounded morphologies that exhibit close contacts with neighboring NCCs. The scale (bar = 10μm) is identical for each inset. PA1=pharyngeal arch 1, PA2=pharyngeal arch 2, PA3=pharyngeal arch 3, ov=otic vesicle.
To investigate the cell morphology of the NCCs in more detail, we next imaged the cranial region of wild-type and pk1 mutant embryos in dorsal view at 24 hpf. In wild-type specimens, although the majority of dorsal NCCs were individual, we also observed a few small clusters of cells (Fig. 2J). Both individual and clustered wild-type NCCs displayed highly protrusive morphologies with filopodia and lamellipodia (Fig. 2J’–J’’). By contrast, pk1bfh122/fh122 and pk1ach105/ch105 mutants showed both a higher frequency of clusters and larger NCC clusters lying dorsally at midbrain and hindbrain levels, consistent with our observations at the 18 hpf stage, with the cells displaying rounded, non-protrusive morphologies (Fig. 2K’–K’’, L’–L’’). We confirmed that morpholino knockdown of Pk1b, using our previously described reagents (Mapp et al., 2011), fully phenocopied the pk1bfh122/fh122 mutant (Fig. 2M–M’’).
To assay embryos deficient in both Pk1b and Pk1a, we imaged pk1ach105/ch105 embryos injected with morpholinos targeted against Pk1b, and found a more severe phenotype, with larger dorsal NCC clusters than in the single homozygous mutants alone, as well as gross morphological defects consistent with disruptions in convergent extension (Fig. 2N–N’’). These double-deficient embryos also began to die shortly after the 24 hpf stage. We then compared the phenotype of embryos deficient in both Pk1b and Pk1a to the phenotype caused by knockdown of another core PCP molecule, Vangl2, which is also known to be important in convergent extension (Ferrante et al., 2009; Jessen et al., 2002). We found that knockdown of Vangl2 also caused a severe dorsal NCC clustering phenotype, with NCCs meeting across the dorsal midline of the neural tube at approximately the level of r3 (Fig. 2O–O’’), as well as gross morphological defects. These results are consistent with previous reports that Vangl2 is required for proper convergence of neuroepithelial cells towards the midline during neurulation (Ciruna et al., 2006; Tawk et al., 2007). As the resultant altered neural keel morphology makes it difficult to ascribe NCC phenotypes to deficits in NCCs themselves versus earlier defects in neurulation, we elected not to study vangl2 morphants or embryos deficient for both pk1 paralogs in any additional detail. However, the lack of convergence defects both in single homozygous pk1bfh122/fh122 and pk1ach105/ch105 mutants, as well as in double-heterozygous embryos lacking one copy each of pk1b and pk1a, provided us a unique opportunity to study the roles of the Pk1 paralogs specifically in the developing neural crest.
To investigate the unusual NCC clusters in pk1 mutants in more detail, we mapped their spatial localization. For this analysis we defined ‘clusters’ as groups of three or more cells in close apposition. We allocated the clusters to three regions, corresponding to different axial positions as indicated in Fig. 3A: Region I corresponds to anterior midbrain-derived NCCs that contribute to the fronto-nasal process; region II corresponds to posterior midbrain and rhombomere 1, 2 and 3 (r1-to-r3)-derived NCCs from the anterior hindbrain that contribute to pharyngeal arch (PA)1; region III corresponds to central hindbrain (r3–r5)-derived NCCs that migrate anterior of the otic vesicle to contribute to PA2. Interestingly, despite a widespread distribution of dorsally-located NCC clusters along the AP axis in both mutant conditions, a slight spatial bias was detectable, with clusters most frequently located towards the anterior. Moreover, NCC clusters that spanned the space between the origins of two separate streams of migratory NCCs occurred more frequently in the pk1 mutants than in wild-type specimens (Fig. 3A).
Figure 3: Additional analysis of cranial neural crest cell clusters in wild-type and pk1 mutant embryos.
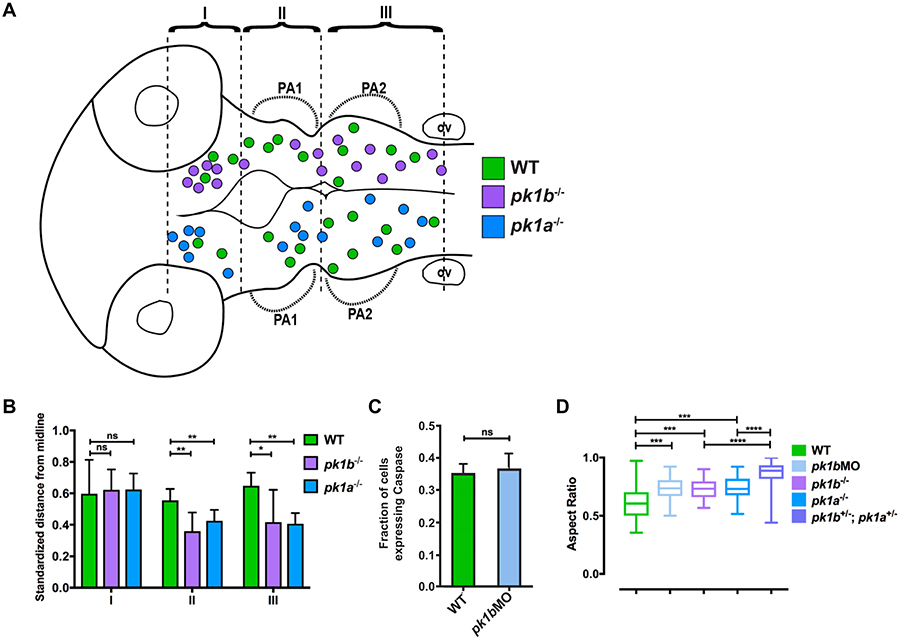
(A-B) The spatial distribution of NCC clusters across multiple 24 hpf embryos shows that both pk1bfh122/fh122 and pk1ach105/ch105 clusters show anterior and medial biases. (A) The centroids of cell clusters, defined as two or more cells in contact with one another, across multiple pk1bfh122/fh122 specimens (n=5 embryos) are plotted above the midline and those of pk1ach105/ch105 specimens (n=5 embryos) are plotted below the midline, with WT clusters on both sides for comparison (green filled circles, n=5 embryos). There is a bias for both pk1bfh122/fh122 and pk1ach105/ch105 clusters in region 1 (R1), as well as a tendency to misalign such that the cluster cannot be ascribed to distinct streams for either PA1 or PA2. (B) Normalized distances of centroids of clusters from the midline show that in both RII and RIII there is a tendency for pk1bfh122/fh122 and pk1ach105/ch105 NCCs to be located medially as compared to WT NCCs. The bias was found to be statistically significant for regions II and III using a two-way ANOVA test (*, p<0.05, **, p<0.01) for either mutant conditions as compared to WT. There was no statistically significant difference between either mutant condition and WT in Region I (p>0.05). (C) Comparison of the fraction of NCCs in a given z-slice that expresses Caspase-3 in 24 hpf WT (n=78 cells, 3 embryos) versus 24 hpf pk1bmorphant (n=172 cells, 5 embryos) specimens. No statistically-significant difference was found between WT and pk1b-morphant NCCs (p>0.05). (D) Measurements of aspect ratio (the ratio of width/length of NCCs) for WT (n=34 cells), pk1b-morphant (n=48 cells), pk1bfh122/fh122 (n=40 cells), pk1ach105/ch105 (n=30 cells), and double-heterozygous pk1ach105/+; pk1bfh122/+ (n=94 cells) 24 hpf specimens were performed by measuring the width versus length ratio of NCCs either in Tg(sox10:mRFP) embryos, which label the membranes of all NCCs, or in Tg(sox10:EGFP) embryos co-injected with RNA encoding mRFP. WT NCCs had lower aspect ratios in comparison to each Pk1-deficient condition. Statistical significance was calculated in each pairwise case using unpaired t-tests (***, p<0.001; ****, p<0.0001). Double-heterozygous pk1ach105/+; pk1bfh122/+ embryos showed a significantly higher aspect ratio than either pk1bfh122/fh122 or pk1ach105/ch105 alone (p<0.0001 in both cases).
To query whether there was a mediolateral bias in cluster location, we measured the distances between the centroids of clusters and the midline, and expressed the distances as a ratio of the longest possible distance between any NCC and the midline at that specific AP position. This analysis confirmed that for NCCs migrating towards PA 1 and 2 there is a statistically significant bias for cell clusters to be located closer to the midline in both pk1bfh122/fh122 and pk1ach105/ch105 mutant conditions as compared to wild-type. However, no such bias was found for the midbrain-derived cells migrating towards the fronto-nasal process (Fig. 3B). The medial bias of Pk1-deficient NCCs in clusters indicates that Pk1-deficient cells do not undergo the normal wild-type lateral migration, and suggests that these Pk1-deficient NCCs may instead remain pre-migratory.
As we found that many NCCs in Pk1-deficient embryos showed unusual dorsal clustering, we evaluated whether the broader dorsoventral distribution of NCCs migrating ventrolaterally towards the pharyngeal region was also disrupted. To query the dorsoventral localization of NCCs, we again used confocal imaging, at two different stages, 16 and 24 hpf. In each case, we counted the sox10:EGFP-positive NCCs located in both the dorsal-most 4 μm and the ventral-most 4 μm of the EGFP-positive domain, across 5 embryos for each condition, in a region of interest spanning the midbrain and anterior half of the hindbrain (regions I, II, III as defined above). As shown in Table 1, in 16 hpf wild-type embryos an average of 53% of all EGFP-positive NCCs were present in the dorsal-most domain and only 25.6% in the ventral-most domain. However, by 24 hpf wild-type embryos showed 11.8% of NCCs in the dorsal-most domain and 54.2% in the ventral-most domain, indicating that the majority of wild-type NCCs had completed ventrolateral migration to the pharyngeal domain. In contrast, in both pk1bfh122/fh122and pk1ach105/ch105 embryos, the majority of NCCs were present in the dorsal-most domain, both at 16 hpf (61.2% and 62.2%, respectively) and at 24 hpf (55.6% and 59.4%). Although the percentage of NCCs in dorsal-most domains decreased slightly in Pk1-deficient embryos between 16 and 24 hpf, a large proportion of NCCs remained dorsally localized, suggesting that unlike wild-type NCCs, most Pk1-deficient NCCs fail to undergo normal ventrolateral migration by 24 hpf.
Table 1: Aberrant dorsoventral distribution of cranial neural crest cells in Pk1-deficient specimens.
The dorsoventral locations of NCCs were established using confocal z-stacks of Tg(sox10:EGFP) specimens in dorsal view (Fig. 2). EGFP-positive cells, on both left and right sides of the midline, were counted within a region of interest spanning the midbrain and anterior hindbrain (regions I, II, III, see Fig. 3A). The percentage of the total number of EGFP-positive cells in the entire dorsoventral range at 16 hpf and 24 hpf was calculated for the most dorsal and most ventral domains across wild-type, pk1bfh122/fh122, and pk1ach105/ch105 conditions (5 embryos in each condition and at each stage, with mean percentages and standard error of the mean shown). At 16 hpf, the dorsoventral range is ~40μm, with the dorsal-most domain in the range defined as 0–4 μm, and the ventral-most domain in the range defined as 36–40 μm (the first z-slice showing EGFP-positive cells is denoted by 0μm). At 24 hpf, the dorsoventral range is ~64 μm, with the dorsal-most domain in the range defined as 0–4 μm, and the ventral-most domain in the range defined as 60–64 μm. The total number of cells counted, across 5 embryos for each condition, is indicated.
| 16 hpf | 24 hpf | |||||
|---|---|---|---|---|---|---|
| % of total no. of cells (Mean ± SEM) | Total no. of cells | % of total no. of cells (Mean ± SEM) | Total no. of cells | |||
| Dorsal-most | Ventral-most | Dorsal-most | Ventral-most | |||
| WT | 53.0 ± 3.2 | 25.6 ± 1.9 | 960 | 11.8 ± 2.4 | 54.2 ± 3.7 | 1430 |
| Pklb−/− | 61.2 ± 3.4 | 9.6 ± 2.0 | 973 | 55.6 ± 2.3 | 20.8 ± 3.7 | 1392 |
| pkla−/− | 64.2 ± 3.1 | 12.8 ± 1.5 | 1063 | 59.4 ± 4.3 | 18.6 ± 4.6 | 1470 |
We also investigated whether the dorsally located clusters of NCCs might be failing to migrate because they were undergoing apoptosis. To address this, we performed immunolabeling for Caspase-3, a member of the caspase family of proteins required for apoptosis (Li and Yuan, 2008), in both wild-type and pk1bmorphant embryos at 24 hpf. We found no significant difference in the proportion of NCCs that expressed Caspase-3 in wild-type versus pk1b-morphant conditions (Fig. 3C), indicating that the formation of dorsal NCC clusters in Pk1-deficient embryos is not related to NCCs undergoing cell death.
We qualitatively observed that wild-type NCCs were more elongated and protrusive than Pk1-deficient NCCs, which were more rounded and less protrusive. To confirm the differences in cell shape, we measured the aspect (width:length) ratios of NCCs in wild-type and mutant conditions, with a ratio tending to 1 indicating greater roundedness. Wild-type NCCs showed elongated shapes with a mean aspect ratio of 0.61. In contrast, pk1b-MO, pk1bfh122/fh122, and pk1ach105/ch105 NCCs exhibited significantly higher mean aspect ratios of 0.73, 0.73, and 0.74, respectively, (p<0.001 for each). Interestingly, pk1bfh122/+ ; pk1ach105/+ double-heterozygous embryos exhibited a higher mean aspect ratio, of 0.86, than either wild-type or single homozygous mutant NCCs (p<0.0001) (Fig. 3D). The unusually rounded morphology of Pk1-deficient NCCs indicates that Pk1 may play a role in the acquisition of elongated, mesenchymal morphology.
In summary, we conclude that in the absence of Pk1b or Pk1a function, NCCs cluster at dorsal positions in the embryo, adopt aberrant rounded morphologies, and frequently fail to migrate our into the pharyngeal region by 24 hpf.
2. Cell fates of NCCs contributing to the pharyngeal elements are unchanged in Pk1-deficient specimens
In wild-type 24 hpf embryos, the most ventrally-located migrating NCCs are destined to populate the pharyngeal arches and ultimately differentiate into cartilage cells that establish the pharyngeal apparatus (Schilling and Kimmel, 1994). Our analysis of NCC dorsoventral localization (Table 1) indicates a significant reduction in the number of ventrally-localized cells in Pk1-deficient embryos relative to wild-types at 24 hpf. To evaluate whether this change in cell position is accompanied by an alteration of cell fate, we performed in situ hybridization with the pharyngeal NCC marker dlx2. In 24 hpf wild-type embryos, dlx2 expression can be distinguished in individual streams of cranial NCC cells (Fig. 4A). We found that dlx2 expression was maintained in both pk1bfh122/fh122 and pk1ach105/ch105 mutants (Fig. 4B, C), with a pattern essentially indistinguishable from that of wild-types, despite the aberrant distribution of NCCs migrating towards the pharyngeal arches in Pk1-deficient embryos. To investigate potential subsequent disruptions to the pharyngeal apparatus, we stained cartilage cells at 6 dpf (days post fertilization) with Alcian Green. We found that the size and organization of the cartilage elements that comprise the pharyngeal apparatus were indistinguishable between wild-type specimens (Fig. 4D) and either pk1bfh122/fh122 or pk1ach105/ch105 mutants (Fig. 4E, F).
Figure. 4: pk1b and pk1a mutants do not show alterations in pharyngeal cartilage fates.

(A-C) In situ hybridizations for the pharyngeal neural crest marker dlx2 in WT (A), pk1bfh122/fh122 (B) and pk1ach105/ch105 (C) 24 hpf embryos in lateral view show no significant differences between conditions. Solid arrowheads indicate streams of NCCs. (D-F) Alcian Green labeling of cartilage elements of the pharyngeal apparatus in WT (D), pk1bfh122/fh122 (E) and pk1ach105/ch105 (F) 6 dpf larvae, show no significant changes in size and organization of elements between conditions. m=Meckel’s cartilage; pq=palatoquadrate; ch=ceratohyal; cb=ceratobranchial; cb(i) indicates first branchial arch, cb(iv) indicates fourth branchial arch, with solid arrows indicating the corresponding ceratobranchial cartilage elements.
In summary, despite significant early deficits in cranial NCC organization in Pk1-deficient embryos, we found no evidence of subsequent changes in NCC fate or in the later morphological organization of NCC derived cartilages. These results suggest that the cranial neural crest cells of pk1bfh122/fh122 or pk1ach105/ch105 mutants have a significant capacity to recover from early defects, consistent with the ability of both mutants to survive to viable fertile adulthood. However, as noted in the previous section, embryos deficient for both Pk1b and Pk1a die shortly after 24 hpf, precluding analysis of NCC fates.
3. Loss of pk1b or pk1a function disrupts ventrolateral migration of cranial neural crest cells
Based on our observations of abberant dorsoventral localization of NCCs, coupled with clustering of NCCs at the dorsal aspect of the cranial neural tube, we hypothesized that in Pk1-deficient embryos, NCCs fail to migrate ventrolaterally into streams. We addressed this using time-lapse confocal imaging of Tg(sox10:EGFP) embryos over a two-hour period, from 16 hpf to 18 hpf, of active NCC migration in the cranial region. For this analysis, we imaged the region corresponding to the first pharyngeal arch (PA1) stream of NCCs, deriving from the midbrain and r1–r2 (indicated by the dashed box in Fig. 5A) in dorsal view. At 16 hpf the neural keel has not yet formed a midline, which occurs through alignment of neuroepithelial cell end feet by 18 hpf (Jimenez et al., 2016; Tawk et al., 2007). At approximately 16 hpf, cranial neural crest cells from the dorsal neural keel complete EMT and actively migrate laterally and then ventrally around the neural keel towards the pharyngeal region. As shown for the embryo imaged in Supplemental Movie 1 and analyzed in Fig. 5B, wild-type NCCs form dynamic, transient contacts with neighboring NCCs and move laterally towards the “edges” of the neuroepithelium as seen from a dorsal view (line in Supplemental Movie 1, and dashed line in Fig. 5B) between 16 and 18 hpf. To assay the overall movement of these NCCs, we plotted displacement trajectories using the first and last position of individual cells after correcting for drift and embryo growth. We found that approximately 89% of wild-type NCCs (n=59 cells, 3 embryos, representative embryo shown in Supplemental Movie 1 and Fig. 5B) showed net lateral movement, as expected, over the two-hour time period.
Figure 5: Loss of function of either pk1b or pk1a causes aberrant movement of cranial neural crest cells.

(A) Schematic dorsal view of a 16 hpf embryo with optic cup, pharyngeal arches (PA), and otic vesicle (ov) indicated; dashed box indicates region that was time-lapse imaged. (B-F) Displacement tracks of motile NCCs in dorsal view, anterior to left, from 16–18 hpf. The x and y components of the displacement tracks were drift-corrected relative to bright-field images of the embryos to account for growth and movement of the embryo. In contrast to WT embryos (B) where the net displacement of NCCs is lateral, the net displacement of NCCs in pk1b-morphant (C), pk1bfh122/fh122 (D) and pk1ach105/ch105 (E) mutant embryos is in the anterior direction. NCCs in double-heterozygous pk1ach105/+; pk1bfh122/+ specimens (F) also move in net anterior directions. Scale bar = 25 μm for all conditions. (G-I) To show inter-embryo variation, 10 individual NCC displacement trajectories, for each of 3 embryos, were plotted at the origin, with both left and right sides of the embryo in the case of WT (G) embryos transposed on to one side for clarity. Trajectories from different embryos are colored in red, blue and green. (G) WT NCCs move in the lateral direction as is expected for the first stage of normal ventrolateral NCC migration, (H, I) pk1bfh122/fh122 and pk1ach105/ch105 mutant NCCs display a bias for anterior directionalities.
Because loss of Pk1b or Pk1a function caused aberrant NCC cluster formation, we postulated that it would also result in a loss of overall NCC motility. Unexpectedly, we found that pk1b-morphant cells did not lose motility. Rather 72% of pk1b-MO NCCs (n=47 cells, 3 embryos) showed net movement in the anterior direction, with NCCs typically retaining contacts with neighboring cells as they moved in a cluster (Supplemental Movie 2, Fig. 5C). A similar anterior bias of NCC motility was noted in pk1bfh122/fh122 embryos (Fig. 5D), with 77% of NCCs showing net movement in the anterior direction (n=65 cells, 3 embryos). In both pk1b-MO and pk1bfh122/fh122 specimens, clusters of NCCs showed less dynamic and protrusive behaviors than wild-type NCCs (Supplemental Movies 2, 3, Fig. 5C, D). pk1ach105/ch105 embryos exhibited a similar phenotype (Supplemental Movie 4, Fig. 5E), with 75% of NCCs showing net movement in the anterior direction (n=47 cells, 3 embryos). We also assayed double-heterozygous pk1bfh122/+ ; pk1ach105/+ embryos, which do not show gross morphological defects, unlike pk1ach105/ch105 +pk1b-MO embryos. We found that 86% of pk1bfh122/+ ; pk1ach105/+NCCs (n=38 cells, 3 embryos) also moved in net anterior directions (Supplemental Movie 5, Fig. 5F), a higher proportion than in either Pk1b-deficient or Pk1a-deficient embryos. We conclude that Pk1-deficient specimens do not demonstrate a loss of NCC motility, but instead show an altered direction of NCC movement, with cells moving towards the anterior rather than laterally and subsequently ventrally. We further conclude that the Pk1 paralogs function partially redundantly, such that double-heterozygous embryos show a more severe phenotype than either single homozygous mutant despite the same number of alleles being disrupted, likely because of the dosage sensitivity of the combined action of the genes. There is precedent for PCP genes acting in a highly dosage-sensitive manner—notably in members of the Vangl family in mouse embryos (Torban et al., 2008)—such that a genetic interaction between two homologs manifests as a more severe phenotype in a double-heterozygous embryo than a single homozygous mutant for either homolog.
To highlight the degree of inter-embryo variation in NCC motility, we plotted the displacement trajectories of 10 NCCs from three individual embryos of each of the wild-type, pk1bfh122/fh122, and pk1ach105/ch105 conditions, with NCCs from different embryos allocated a different color (Fig. 3G–I). This analysis reaffirmed that wild-type NCCs move in lateral directions. Further, in mutant conditions the trend for anterior movement was highly consistent. We conclude that both Pk1b and Pk1a are required for lateral NCC migration. When two pk1 alleles are disrupted (i.e. homozygous mutation of one pk1 paralog, or disruption of a single copy of both pk1 paralogs), cranial NCCs form clusters that do not migrate laterally but instead preferentially move towards the anterior.
In summary, the rounded morphologies of Pk1-deficient NCCs are correlated with the inability of NCCs to migrate in the normal ventrolateral direction, moving in anterior directions instead. We conclude that although Pk1b and Pk1a are not required for NCCs to be motile, they are required for NCCs to move specifically in the ventrolateral direction.
4. Loss of Pk1 function disrupts NCC polarity
The finding that Pk1-deficient NCCs preferentially move in an anterior direction led us to postulate that the dorsally-located pre-migratory NCCs adopt aberrant polarities, rather than losing polarity entirely as might be expected in a PCP-deficient condition. Previously, it has been reported that PCP signals control the position of the intracellular microtubule organizing center (MTOC), and that cells polarize MTOCs in the direction of motility (Sepich et al., 2011). To assay the polarity of NCCs at the dorsal aspect of the neural tube, in both wild-type and Pk1-deficient conditions, we measured the angle of the MTOC relative to the primary body axis (Fig. 6A, B).
Figure 6: Disruption of pk1b or pk1a causes NCCs to adopt aberrant polarity.
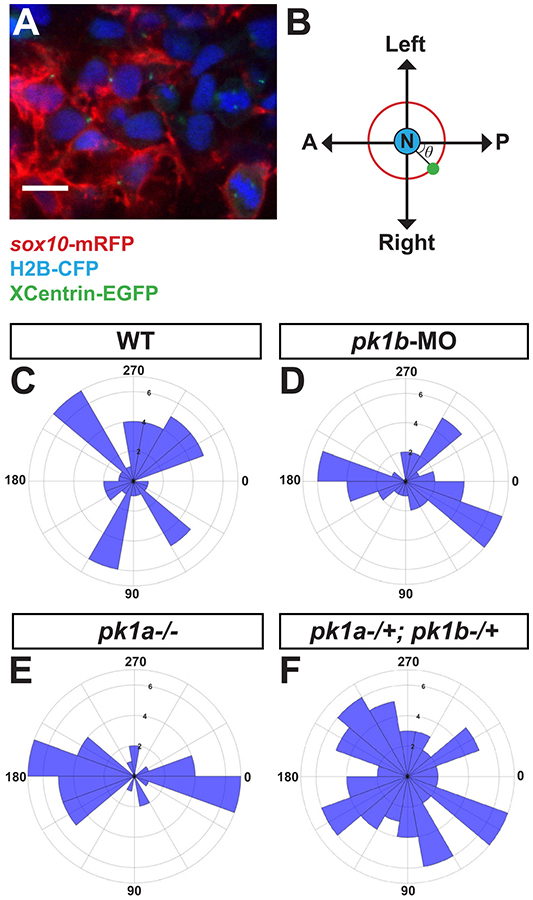
To assay the polarity of NCCs, MTOCs in NCCs were labeled by injecting either XCentrin-EGFP RNA in one-cell stage Tg(sox10:mRFP) or Cherry-XCentrin RNA in one-cell stage Tg(sox10:EGFP) embryos that were also injected with H2B-CFP to label nuclei. Fixed, deyolked, flat-mounted 24 hpf embryos were imaged in dorsal view (A). Scale bar = 10 μm. The angle θ of the MTOC relative to the nucleus and the AP body axis (B) was measured for each cell and quantified using Fiji. (C-F) Polar histograms generated using MATLAB show the polarity of assayed NCCs. In all cases, the Watson-Williams F-test was used to measure statistical significance between conditions as well as with a randomly-distributed polar histogram. In WT embryos, NCCs (n=48 cells, 5 embryos) were polarized along the mediolateral axis (C), with WT cells showing a significant difference from a random distribution (***, p<0.001). However, in pk1b-morphant specimens (n=42 cells, 3 embryos) (D) and pk1ach105/ch105 specimens (n=40 cells, 3 embryos) (E) NCCs were polarized along the AP axis. As compared to WT NCCs, the distributions of both pk1b-morphant and pk1ach105/ch105 NCCs were significantly altered, and also showed a significant difference from random distributions (***, p<0.001 for each case). However, double-heterozygous pk1ach105/+; pk1bfh122/+ NCCs (n=71 cells, 4 embryos) (F) showed no statistically significant difference as compared to a random distribution (p>0.05), indicating that disrupting one copy each of the pk1a and pk1b genes is sufficient to randomize NCC polarity.
In accord with previous findings, we found that wild-type NCCs at 24 hpf on either side of the midline polarized laterally in the direction of normal migration (Fig. 6C). In contrast, pk1b-morphant (Fig. 6D, p<0.001) and pk1ach105/ch105 (Fig. 6E, p<0.001) NCCs on either side of the midline polarized along the AP axis. However, NCCs in double-heterozygous pk1bfh122/+ ; pk1ach105/+ embryos displayed a randomized polarity (Fig. 6F, p>0.05), despite our previous finding that these NCCs moved as clusters in anterior directions similar to the single pk1 mutants (Fig. 5F). These findings suggest that whereas Pk1b-deficient, and Pk1a-deficient NCCs polarize to correspond to the aberrant direction of migration, in double-heterozygous NCCs, MTOC position is decoupled from the direction of migration. Interestingly, this finding argues against a direct correspondence between cell polarity and the direction of cell migration.
We conclude that while both Pk1b and Pk1a are required to establish normal NCC polarity, the absence of either alone is insufficient to cause a complete loss of polarity. However, the double-heterozygous condition is sufficient for loss of polarity, indicating that the two Pk1 paralogs function redundantly and again likely reflecting the importance of gene dosage. Importantly, the altered polarity of Pk1-deficient NCCs occurs in conjunction with aberrant morphologies and increased clustering.
5. Dorsal clusters of pre-migratory NCCs are established and maintained in Pk1-deficient embryos
Having observed dorsally clustered NCCs in embryos lacking either Pk1b or Pk1a function, we investigated at what stage of NCC development the clusters formed and probed their characteristics in more detail. We hypothesized that the clustering defect arose at stages prior to the migration of cranial NCCs in well-defined streams, and that wild-type cells, even early in neural crest development, are able to resolve contacts with neighboring NCCs at faster rates than Pk1-deficient cells. To test these hypotheses, we performed time-lapse imaging of Tg(sox10:EGFP) embryos beginning at 12 hpf and extending for at least 4 hours. We noted that even as early as 12 hpf, some NCCs are present ventrolaterally, a finding which is consistent with previous reports of an early population of NCCs originating from the non-neural ectoderm adjacent to the neural keel (Jimenez et al., 2016; Schilling and Kimmel, 1994), and one that explains why even in conditions causing severe neural crest defects, some cells are observed at ventrolateral positions in the presumptive pharyngeal arches (Fig. 2J–M). Importantly, we found that in wild-type embryos, NCCs in contact with one another at dorsal-most positions tended to move rapidly away from one another, with most cell contacts resolving within 20–30 mins (Fig. 7A–A’’’’, with NCCs in a given group of cells pseudocolored to indicate pairs in contact). However, in both pk1bfh122/fh122 (Fig. 7B–B’’’’) and pk1ach105/ch105 (Fig. 7C–C’’’’) embryos, NCCs in clusters frequently failed to break contacts, despite moving relative to other cells in the cluster.
Figure 7: Loss of Pk1 function causes dorsal NCC clusters to form and be maintained in early stages of neural crest development.
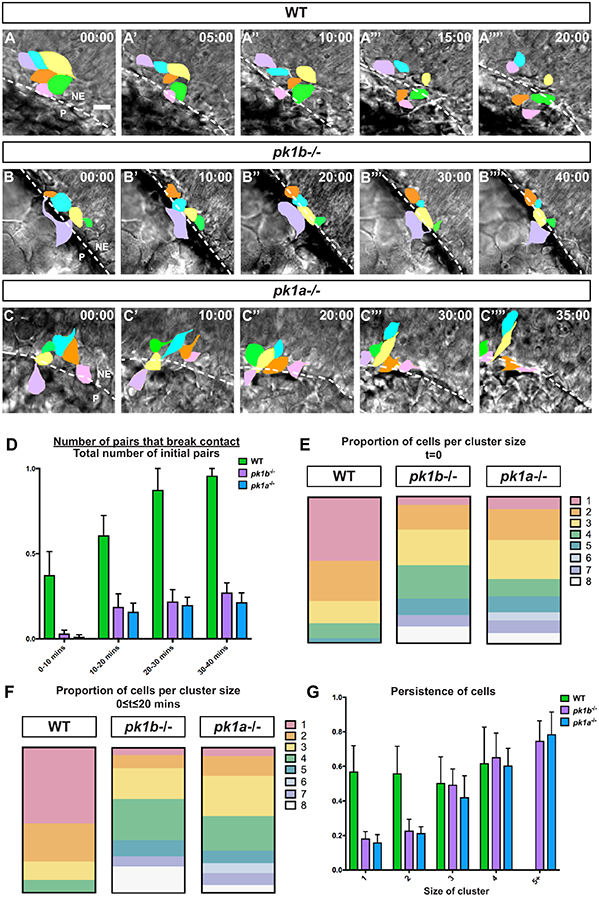
Confocal time-lapse imaging of dorsally-mounted Tg(sox10:EGFP) embryos was started at 12 hpf when the neural keel is still developing, with both EGFP and DIC (brightfield) images collected every 5 minutes. (A-C) Reorganization of EGFP+ NCCs (pseudocolored) in clusters of NCCs at t=0 to t=40 mins; dotted line indicates border of the neuroepithelium. The neuroepithelium (NE) is located at top right, and the periphery (P) is located at bottom left, in all panels. Scale bar=10 μm. (A) In WT embryos, many of the NCCs in contact at t=0 break contact by 20 minutes, with almost all contacts breaking by 30 mins and 40 mins. (B, C) In pk1bfh122/fh122 and pk1ach105/ch105 specimens NCC clusters largely remained in contact for 40 mins. (D) To quantify breakage of contacts between NCCs over the time intervals 0–10 min, 10–20 min, 20–30 min and 30–40 min, a ratiometric measure of ‘pair breakage’ within a cluster was used (Methods). In WT embryos, ~61% of pairs of cells broke contacts between 0 and 20 minutes (n=43 pairs, 3 embryos), with a large majority of pairs losing contact by 20–30 minutes. In contrast, pairs of NCCs in both pk1bfh122/fh122 (n=61 pairs, 3 embryos) and pk1ach105/ch105 embryos (n=58 pairs, 3 embryos) did not break over extended periods of time, with only ~27% of pairs in pk1bfh122/fh122 clusters breaking contact and ~21% of pairs in pk1ach105/ch105 clusters breaking contact by 30–40 minutes. (E) To measure the relative proportions of individual cells and cell clusters of varying sizes, the organization of cells at 12 hpf in Tg(sox10:EGFP) embryos was quantified. In WT embryos (n=37 cells, 3 embryos), 43.9% of NCCs were found as individuals, 27.6% were in pairs, 15.3% in clusters of 3 cells, 10.2% in clusters of 4 cells, and 3.1% in clusters of 5 cells. In pk1bfh122/fh122 specimens (n=53 cells, 3 embryos), 5.9% of NCCs were found as individuals, and 17.0% as pairs. Most NCCs were found in cluster sizes of 3 (24.2%) or 4 (22.9%), with clusters consisting of as many as 8 cells. In pk1ach105/ch105 specimens (n=46 cells, 3 embryos), 8.6% of NCCs were found as individuals, and 21.1% as pairs. Most NCCs were found as pairs or in cluster sizes of 3 cells (26.6%), with appreciable percentages of cluster sizes of 4 (11.7%) and 5 (10.9%) and with clusters consisting of as many as 8 cells. (F) As another measure of the relative proportions of individual cells and clusters of varying sizes, the number of cells that persisted in a given configuration (from an individual cell to cells in increasing sizes of clusters) was measured over non-overlapping 20-minute time windows. In WT embryos (n=117 cells, 3 embryos), 53.8% of NCCs remain individual over 20 minutes, whereas 26.5% were in pairs, 11.1% in clusters of 3 cells, and 8.5% in clusters of 4 cells. In pk1bfh122/fh122 embryos (n=99 cells, 3 embryos), 5.05% of NCCs were found as individual cells over the 20-minute time window. Most NCCs were found in cluster sizes of 3 (21.2%) or 4 (28.3%), with NCCs found in clusters consisting of as many as 8 cells. In pk1ach105/ch105 embryos (n=148 cells, 3 embryos), 6.1% of NCCs were individual, with most NCCs found in cluster sizes of 3 (27.7%) or 4 (23.6%) and clusters consisting of as many as 8 cells. (G) To assay how persistence—a measure of the total length of a trajectory of a cell as a ratio of the displacement of the cell with a value of 1.0 indicating a targeted route from starting point to end point—varied as a function of cluster size, the persistence of individual cells and clusters of sizes 2 or more was measured for each condition. Persistence is agnostic to the ‘correct’ direction of the cells, and is a measure of the targeted directionality of NCCs, independent of their trajectories. In WT embryos (n=42 cells, 3 embryos), the persistence of individual NCCs or clusters of 2, 3 or 4 was similar. In both pk1bfh122/fh122 (n=49 cells, 3 embryos) and pk1ach105/ch105 embryos (n=64 cells, 3 embryos), the persistence of individual cells and cells in pairs was lower than the persistence of clusters consisting of 3 or more NCCs, or of WT NCCs. Clusters of 3 or more NCCs in both pk1-mutants showed high levels of persistence, with statistically insignificant (p>0.5) differences in persistence as compared to WT cells. Both pk1-mutants showed small increases in persistence as the size of the cluster increased, with no statistically significant difference between clusters of 5 or more cells.
To quantify the breakage of cell contacts, we measured the total number of pairs of NCCs in contact with one another in any given cluster. Here, we defined clusters as two or more cells in contact with one another. We then binned pairs of cells that broke contacts with one another in ten-minute time intervals relative to the total number of pairs at the beginning of observation of a given cluster (new contacts formed that were not present at time t=0 were not counted in the analysis). We found that in wild-type embryos, ~61% of cell pairs broke contacts between 0 and 20 minutes (n=43 pairs, 3 embryos), with 88% of cell pairs breaking contact by 30 minutes (Fig. 7D). In contrast, pairs of NCCs in both pk1bfh122/fh122 (n=61 pairs, 3 embryos) and pk1ach105/ch105 embryos (n=58 pairs, 3 embryos) typically did not break contact over extended periods of time, with only 27% and 21% of cell pairs breaking contact in pk1bfh122 fh122 and pk1ach105/ch105 embryos, respectively, by 30 minutes. Since Pk1-deficient NCC clusters formed well before dorsally-located NCCs begin migration, we conclude that the Pk1 paralogs are required as early as 12 hpf.
We also hypothesized that NCCs in pk1-mutants were present in larger cell clusters at 12 hpf than NCCs in wild-type embryos. To investigate the configuration of NCCs at 12 hpf, we counted the number of cells in any given configuration (as single cells, or in clusters of two or more cells). We found that 44% of NCCs in wild-type specimens were present as single cells, 28% in pairs, 15% in clusters of three cells, 10% in clusters of 4 cells, and 3% in clusters of 5 cells (n=37 cells, 3 embryos, Fig. 7E). By contrast, only 6% of NCCs in pk1bfh122/fh122 specimens (n=53 cells, 3 embryos) and 9% of NCCs in pk1ach105/ch105 specimens (n=46 cells, 3 embryos) were present as individual NCCs, with far greater percentages of NCCs in clusters of two or more cells, consistent with our hypothesis (Fig. 7E). Notably, in Pk1-deficient specimens, clusters consisting of as many as 8 cells were observed.
We further hypothesized that since pk1-mutant NCCs were present in higher-order clusters than wild-type NCCs, pk1-mutant clusters were also likely to be maintained for extended time periods relative to wild-type cells, leading in turn to the defects we observed at later stages. To test this hypothesis, we quantified the clusters of NCCs that maintained contact over non-overlapping time intervals of 20 minutes (Fig. 7F; see Methods). This analysis showed that in wild-type embryos 54% of NCCs were largely individual (n=117 cells, 3 embryos), with transient contacts lasting less than 20 minutes. In contrast, only 5% of NCCs in pk1bfh122/fh122 embryos (n=99 cells, 3 embryos) and 6% of NCCs in pk1ach105/ch105 embryos (n=148 cells, 3 embryos) remained as individuals in the same time frame (Fig. 7F). Wild-type embryos did include some NCCs that maintained contact with other NCCs for at least 20 minutes. 26% of these wild-type cells were pairs of cells, 13% were in clusters of 3 cells and 9% in clusters of 4 cells. In contrast, both pk1bfh122/fh122 and pk1ach105/ch105 embryos maintained appreciable percentages of clusters as large as 8 cells, with most clusters comprising 3 cells (21% in pk1bfh122/fh122 and 28% in pk1ach105/ch105 embryos) or 4 cells (28% in pk1bfh122/fh122 and 24% in pk1ach105/ch105 embryos) (Fig. 7F). Together, these data indicate that while NCCs establish contact with other NCCs in clusters as early as 12 hpf in both wild-type and Pk1-deficient specimens, Pk1-deficient embryos form and maintain larger NCC clusters than wild-type embryos.
Since Pk1-deficient NCCs in clusters tended to exhibit high levels of motility—albeit in the anterior direction as opposed to the wild-type lateral direction—we postulated that the persistence (a measure of the degree of directional movement calculated as the ratio of the total distance traversed by a cell relative to its displacement) of NCC motility would be dependent on the number of cells in the cluster. We found that wild-type NCCs, in any configuration, showed a high level of persistence over a 20-minute time period. In pk1 mutants, individual NCCs and pairs of NCCs showed lower levels of persistence than their corresponding wild-type configurations (Fig. 7G). However, consistent with our hypothesis, in either pk1 mutant, clusters of three or more NCCs showed high levels of persistence similar to those of wild-type NCCs, albeit in an anterior direction (Fig. 7G). We conclude that Pk1-deficient NCCs in clusters of three or more cells demonstrate a high level of targeted movement in an aberrant (anterior) direction, whereas individual cells or pairs of cells display a low level of targeted movement in any direction. In both cases, Pk1-deficient NCCs fail to migrate ventrolaterally.
6. Pk1 regulates the transition from bleb-based to mesenchymal morphologies in EMT, in addition to regulating the breakage of cell contacts during migration.
We have shown that Pk1-deficient cranial NCCs cluster aberrantly at the dorsal surface of the neuroepithelium as early as 12 hpf, a stage that precedes the vast majority of ventrolateral migration of NCCs (Jimenez et al., 2016; Schilling and Kimmel, 1994). These findings led us to hypothesize that NCC defects in Pk1-deficient embryos might result from a previously undocumented role for PCP in regulating the morphological changes that NCCs must undergo before they migrate: during the epithelial-tomesenchymal transition. The process of EMT occurs continuously at early stages of NCC development, as waves of pre-migratory NCCs emerge from the dorsal neuroepithelium, and prepare to migrate in a lateral and subsequently ventral direction, around the developing neural keel, to reach their final ventral positions. To test our hypothesis, we performed a detailed investigation of the morphological states of pre-migratory and migratory NCCs, comparing wild-type with Pk1-deficient embryos.
To observe NCC morphological transitions, we used a transient-transgenesis approach to drive expression of a LifeAct-GFP transgene from sox10 regulatory sequences, allowing visualization of the actin-rich protrusions of both pre-migratory and migratory NCCs in dorsal views at 16 hpf. We defined pre-migratory NCCs as sox10-positive cells localized at the dorsal aspect of the neural tube, which in a dorsal view do not appear to have traversed the apparent “edges” of the neuroepithelium, and migratory NCCs as sox10-positive cells that have moved laterally beyond the edge of the neuroepithelium to actively migrate ventrally.
In wild-type specimens, pre-migratory NCCs undergo a series of morphological transitions following the detachment of neuroepithelial precursor cells from the apical midline (Supplemental Movies 6–9), consistent with previous reports (Berndt et al., 2008; Clay and Halloran, 2014). Following apical detachment (Supplemental Movie 6), NCCs adopt rounded morphologies, making short, transient protrusions (Fig. 8A, Supplemental Movie 7), and forming blebs (Fig. 8A’, 8A’’, Supplemental Movie 8), which have previously been described as rounded, actin-rich extensions created through membrane invagination (Goudarzi et al., 2017). Subsequently, wild-type NCCs transition rapidly to the migratory state, which involves highly-protrusive, mesenchymal morphologies with multiple, long, dynamic protrusions, including filopodia and lamellipodia (Fig. 8A’’’, 8A’’’’, Supplemental Movie 9).
Figure 8: Loss of Pk1 function causes defects in both pre-migratory NCCs in the transitional states of EMT, as well as in migratory ‘escaper’ NCCs.

To query the role of Pk1 molecules in the morphological transitions of EMT, as well as during active migration, NCCs were labeled in a mosaic fashion by injecting DNA encoding LifeAct-GFP under the control of a sox10 promoter into one-cell stage embryos. (A-H) Confocal time-lapse imaging of LifeAct-GFP positive cells for at least 20 minutes in 16 hpf embryos revealed F-actin rich protrusions and distinct morphologies of NCCs. Scale bar=10 μm. (A-D’’’’) Frames from confocal time-lapses of pre-migratory NCCs. (A-A’’’’) WT NCCs display short protrusions even before displaying rounded bleb-based protrusions. WT NCCs adopt the bleb-based morphology before transitioning to morphologies with longer filopodial protrusions, with NCCs moving through these transitional morphologies in EMT over the time-span of approximately 20 minutes. (B-B’’’’, C-C’’’’, D-D’’’’) Over the same time frame as WT NCCs, pk1b-morphant, pk1bfh122/fh122, and pk1ach105/ch105 NCCs show bleb-based protrusions that are actively maintained, on occasion changing the location of the bleb along the edges of a NCC. (B’-B’’’) White arrowheads indicate bleb-based protrusions. (E-H) Confocal time-lapse images of NCCs in the mandibular stream actively migrating towards the first pharyngeal arch. (E-E’’’’) Actively-migrating WT cells that are highly filopodial make transient contacts with neighboring cells, often making, breaking and re-establishing a contact in the form of a thick actin-rich protrusion with the same neighboring cell over a time span of at least 20 minutes. White arrowheads indicate a protrusion contacting a neighboring NCC. (F-F’’’’, G-G’’’’, H-H’’’’) pk1b-morphant, pk1bfh122/fh122, and pk1ach105/ch105 migratory NCCs are highly protrusive but display an inability to separate from neighboring NCCs and maintain contact, often over extended time periods. (I) Bar graph showing the percentage of pre-migratory NCCs that successfully transitioned from a bleb-based to a highly-protrusive mesenchymal-like morphology relative to those that remained in the persistently-blebbing state over the time span of 20 minutes. 89% of WT NCCs (n=18 cells, 8 embryos), 16% of pk1b-morphant NCCs (n=17 cells, 8 embryos), 6% of pk1bfh122/fh122 NCCs (n=32, 8 embryos), and 17% of pk1ach105/ch105 NCCs (n=12 cells, 5 embryos) transitioned successfully. (J) Bar graph showing the percentage of migratory NCCs that established and severed contact with a neighboring NCC relative to NCCs that failed to sever contact with a neighboring NCC over the time span of 20 minutes. 93% of WT NCCs (n=28, 6 embryos) NCCs, 15% of pk1b-morphant NCCs (n=41 cells, 10 embryos), 9% of pk1bfh122/fh122 NCCs (n=22 cells, 7 embryos), and 15% of pk1ach105/ch105 NCCs (n=13 cells, 4 embryos) severed contact with neighboring NCCs.
Using the LifeAct-GFP label, we found no defect in apical detachment of NCCs in Pk1-deficient embryos as compared to wild-types. By contrast, unlike wild-type NCCs, which transition from a bleb-based morphology to mesenchymal morphology over the span of 20 minutes (Fig. 8A–A’’’’), pk1b-morphant (Fig. 8B–B’’’’), pk1bfh122/fh122 (Fig. 8C–C’’’’), and pk1ach105/ch105 (Fig. 8D–D’’’’) NCCs typically maintained blebbing behaviors over the entire time span. In fact, quantifications of the cell behaviors showed that whereas 89% of wild-type NCCs transitioned from pre-migratory to migratory states over a 20-minute time interval, only 16% of pk1b-MO, 6% of pk1bfh122/fh122, and 17% of pk1ach105/ch105 cells made the same transition, with the vast majority of cells instead remaining in a persistently-blebbing state over a 20-minute time interval (Fig. 8I). These results indicate that the defects observed in pk1-deficient specimens reflect a failure of NCCs to fully transition to the mesenchymal state: many NCCs remain trapped in the transitional blebbing state. Thus, Pk1 molecules play a central role in the regulation of key changes in NCC morphology and behavior during EMT.
Although most Pk1-deficient NCCs fail to complete EMT in 20 minutes, we nevertheless found that some NCCs are present migrating ventrolaterally in Pk1-deficient embryos (Table 1). These cells likely include the population of NCCs previously reported to lie lateral to the neuroepithelium as early at 12 hpf (Jimenez et al., 2016); this subset of NCCs localizes lateral to the basal edge of the neuroepithelium without migrating there from the dorsal aspect of the neuroepithelium. While our data are consistent with the presence of an early lateral population of Tg(sox10:EGFP) cells, we also find that a subset of Pk1-deficient NCCs arising from more dorsal locations (Supplemental Movies 2–4) ‘escape’ the neuroepithelium to successfully migrate ventrolaterally into the pharynx.
To observe the behaviors of migratory NCCs, we again imaged NCCs expressing LifeAct-GFP in live embryos. Our data showed that while migrating wild-type NCCs made contacts with neighboring labeled cells, the contacts were transitory and dynamic: over a 30-minute time frame NCCs broke contacts and created new contacts, even with the same cell (Fig. 8E–E’’’’). In contrast, in pk1b-MO (Fig. 8F–F’’’’), pk1bfh122/fh122 (Fig. 8G–G’’’’), and pk1ach105/ch105 (Fig. 8H–H’’’’) embryos, we found that despite ‘escaper’ migratory NCCs adopting highly-protrusive mesenchymal morphologies, they were unable to separate from neighboring NCCs over extended time periods. This indicated that the clustering phenotype in Pk1-deficient embryos is not entirely exclusive to dorsal, pre-migratory NCCs. Quantifications of the cell behaviors showed that whereas 93% of labeled migratory NCCs in wild-type specimens established and severed contacts with neighboring cells over a period of 20 minutes, only 15% of migratory NCCs in pk1b-morphant embryos, 9% in pk1bfh122/fh122 embryos, and 15% in pk1ach105/ch105 embryos severed contacts, with the majority of paired NCCs instead retaining their contacts over this time frame and beyond (Fig. 8J).
In summary, whereas wild-type NCCs show dynamic, transitory contacts with neighboring NCCs during their migration, Pk1-deficient NCCs instead show prolonged contact with neighboring NCCs, even during active migration. This role for Pk1 molecules during migration is consistent with the previously-established role for core PCP molecules in contact inhibition of locomotion of zebrafish NCCs (Carmona-Fontaine et al., 2008; Matthews et al., 2008). Together, our data show that the core PCP Pk1 molecules are required both during EMT, and in the migration of cranial neural crest.
7. Pk1b regulates E-Cad and N-Cad in NCCs
Our results indicate that cranial NCCs require Pk1 proteins for a key transition during EMT and for normal ventrolateral migration towards the pharyngeal region. Interestingly, recent work has established that a crucial Cadherin switch must occur during EMT in order for CIL to be activated in the zebrafish NCCs. Scarpa et al. (2015) showed that zebrafish E-Cad (Cdh1) is highly expressed in epithelial NCC progenitors and subsequently downregulated in N-Cad (Cdh2)-expressing mesenchymal NCCs, and that over-expression of E-Cad is sufficient to block normal NCC migration. Therefore, we postulated that the inability of Pk1-deficient NCCs to transition from a blebbing to a mesenchymal state could be due to inappropriate levels of E-Cad and N-Cad.
As E-Cad over-expression causes a phenotype that shares properties with our pk1-deficient phenotypes we hypothesized that E-Cad levels would be elevated in the absence of Pk1b function. By performing immunohistochemistry at 16 hpf, we found that E-Cad puncta are present in both pre-migratory and migratory wild-type NCC membranes (Fig. 9A–A’’’, C–C’’’). The presence of E-Cad expression in migratory as well as pre-migratory NCCs, albeit at lower levels, is consistent with previous reports that ECad is required in migratory NCCs (Huang et al., 2016). Quantification of normalized fluorescence intensity (Fig. 9E) confirmed that E-Cad levels are higher in wild-type pre-migratory NCCs than in wild-type migratory NCCs (compare Fig. 9A–A’’’ and Fig. 9C–C’’’, p<0.0001), consistent with the previously reported down-regulation of E-Cad during EMT. Morpholino knockdown of Pk1b function resulted in elevated levels of E-Cad in both pre-migratory and migratory NCCs (Fig. 9B–B’’’, 9D–D’’’). Quantification of normalized fluorescence intensity revealed a statistically significant increase in E-Cad levels in Pk1b-deficient specimens as compared to wild-type specimens in both pre-migratory and migratory NCCs (Fig. 9E, p<0.001 in both cases). Intriguingly, Pk1b-deficient migratory NCCs also show significantly lower levels of E-Cad than Pk1b-deficient pre-migratory NCCs (Fig. 9E, p<0.0001; compare Fig 9D with Fig. 9B), suggesting that Pk1b-deficient ‘escaper’ cells require decreased E-Cad levels. Thus, the failure of Pk1b-deficient NCCs to complete EMT may be a consequence of inappropriate E-Cad levels.
Figure 9: Pk1b regulates E-Cad levels in pre-migratory and migratory NCCs and N-Cad levels in migratory NCCs.

To investigate the effect of Pk1b-knockdown on the levels of E-Cad (Cdh1) and N-Cad (Cdh2), immunolabeling at 16 hpf embryos was used to assay both pre-migratory and migratory NCCs, which were defined based on their z-position relative to the most dorsally located NCCs. Pre-migratory NCCs were defined as those located within 7 μm of the most dorsal position; the migratory cells imaged were located between 16 and 37 μm of the most dorsal position. (A-D) Tg(sox10:mRFP) embryos were immunolabeled for E-Cad (Cdh1) (A’-D’), and counterstained with DAPI to mark nuclei (A’’’-D’’’). Scale bar=10 μm. White arrowheads indicate levels of E-Cad in NCCs with labeled membranes and nuclei. Merged channels are also shown (A’’-D’’’). (A-A’’’) In pre-migratory NCCs, E-Cad is present at high levels in WT specimens (A’), with puncta localizing largely on the membrane (A’’). (B-B’’’) In pk1b-morphant specimens, E-Cad levels are elevated (B’) with more puncta localizing on the membrane (B’’) as compared to WT (compare A’’ and B’’). (C-C’’’) In migratory NCCs, E-Cad is present at reduced levels (C’, C’’) as compared to pre-migratory NCCs (compare C’’ and A’’). (D-D’’’). In pk1bmorphant specimens, E-Cad levels are elevated (D’) with more puncta localizing on the membrane (D’’) as compared to WT (compare C’’ and D’’). (E) To quantify the levels of E-Cad in different conditions, fluorescence pixel intensity was measured. Each cell was normalized by subtracting background pixel intensity and dividing the remainder by the area of the cell. High levels of E-Cad were found in pre-migratory WT cells (n=92 cells, 4 embryos). E-Cad levels in pk1b-deficient NCCs (n=73 cells, 4 embryos) were significantly elevated as compared to WT NCCs (***, p=0.0002). E-Cad levels in migratory WT cells (n=100 cells, 4 embryos), were significantly reduced as compared to levels in pre-migratory WT cells (****, p<0.0001), however pk1b-morphant migratory NCCs (n=65, 4 embryos) showed significantly higher levels of E-Cad as compared to WT migratory NCCs (***, p=0.0006). Similarly, (F-I) Tg(sox10:mRFP) embryos were immunolabeled for N-Cad (Cdh2) and counterstained with DAPI to mark nuclei (F’’-I’’’). White arrowheads indicate levels of N-Cad in NCCs with labeled membranes and nuclei. Merged channels are also shown (F’’-I’’). (F-F’’’) In pre-migratory NCCs, N-Cad (F’) is highly localized at the membrane in WT specimens (F’’). (G-G’’’) In pk1b-morphant specimens, N-Cad levels (G’) showed no discernable difference in localization on the membrane (G’’) as compared to WT (compare G’’ and F’’). (H-H’’’) In migratory NCCs, N-Cad is present at increased levels as compared to pre-migratory NCCs (compare H’’ and F’’). (I-I’’’) However, in pk1b-morphant specimens, N-Cad levels in migratory NCCs are decreased (I’) with less N-Cad localization on the membrane (I’’) as compared to WT (compare I’’ and H’’). (J) To quantify the levels of N-Cad in different conditions, fluorescence pixel intensity was measured and normalized in the same manner as with E-Cad. N-Cad was present on the membrane in pre-migratory WT NCCs (n=164 cells, 5 embryos). N-Cad levels in pk1bmorphant NCCs (n=204 cells, 4 embryos) showed no difference to WT NCCs (p>0.05). N-Cad levels in migratory WT cells (n=192 cells, 5 embryos), were significantly elevated as compared to levels in pre-migratory WT NCCs (****, p<0.0001), however pk1b-morphant migratory NCCs (n=189, 4 embryos) showed significantly lower levels of N-Cad as compared to WT migratory NCCs (**, p=0.0027). N-Cad levels in WT pre-migratory, pk1b-morphant pre-migratory, and pk1b-morphant NCCs showed no significant difference from each other (p>0.05 for each combination).
Since up-regulation of N-Cad is normally required in migratory NCCs (Carmona-Fontaine et al., 2008; Theveneau et al., 2010; Theveneau et al., 2013), we further hypothesized that N-Cad levels might be reduced in the absence of Pk1b function. By performing immunohistochemistry, we found that N-Cad is also present on both pre-migratory and migratory NCC membranes in wild-type specimens. Reciprocal to E-Cad, N-Cad levels were elevated in wild-type migratory NCCs as compared to wild-type pre-migratory NCCs (compare Fig. 9F–F’’’ and Fig. 9H–H’’’, p<0.0001), consistent with previous reports. Pk1b-deficient embryos showed no discernable change in N-Cad levels in pre-migratory NCCs relative to unmanipulated controls (Fig. 9G–G’’’, p>0.5). However, Pk1b-deficient migratory NCCs showed reduced levels of N-Cad at the membrane relative to wild-type specimens (Fig. 9I–I’’’), consistent with our hypothesis. Quantification of normalized fluorescence intensity confirmed a statistically significant reduction in N-Cad levels in pk1b-MO specimens, as compared to wild-type specimens, in migratory (p<0.01), but not pre-migratory, NCCs (Fig. 9J).
In summary, in Pk1b-deficient specimens, E-Cad levels in both pre-migratory and migratory NCCs are elevated relative to unmanipulated wild-type controls. However, the Pk1b-deficient embryos do still show a downregulation of E-Cad in migratory relative to pre-migratory NCCs. Further, in Pk1b-deficient embryos, N-Cad levels are unaffected in pre-migratory NCCs, but are reduced in migratory NCCs relative to wild-type controls. Thus, Pk1b function is required for appropriate regulation of E-Cad and N-Cad levels at multiple stages during zebrafish neural crest development. We conclude that the important role played by Pk1 proteins in promoting EMT and subsequent migration of cranial neural crest cells, is at least in part mediated though the ability to regulate levels of E-Cad and N-Cad and thus facilitate the transition from epithelial to mesenchymal cell behavior.
Discussion
Through analysis of the pk1bfh122/fh122 and pk1ach105/ch105 mutants, we have uncovered broad roles for zebrafish Prickle1 in the developing cranial neural crest. The similar roles for Pk1b and Pk1a suggest that the two proteins share very similar functions. Importantly, a central function of vertebrate PCP molecules is the regulation of embryonic convergence movements that are necessary for both gastrulation and neurulation to proceed normally. Thus, the absence of convergence defects in either of the pk1bfh122/fh122 and pk1ach105/ch105 mutants likely reflects functional redundancy between these paralogous genes. Consistent with this model we have found that Pk1b-knockdown in pk1ach105/ch105 mutants does indeed cause morphological defects similar to those resulting from loss of function of other core PCP molecules, such as Vangl2 (Ciruna et al., 2006; Jessen et al., 2002; Veeman et al., 2003). In contrast to the results we present here, previous reports (Carreira-Barbosa et al., 2003; Ciruna et al., 2006; Veeman et al., 2003) have suggested that Pk1a function is necessary for convergence movements. While it is formally possible that our pk1ach105/ch105 mutant does not represent a genetic null, we consider this unlikely given the N-terminal location of the genetic lesion. Rather, we note that the previous analyses of Pk1a-function relied exclusively on morpholino knockdown, and some aspects of the phenotypes could thus reflect off-target effects of the reagent. Alternatively, the discrepancy between the morpholino knockdown and the pk1ach105/ch105 mutant phenotype may be a consequence of genetic compensation, as recently described for other zebrafish mutants (Rossi et al., 2015). Importantly, by generating the pk1ach105/ch105 mutation, we have established a useful new tool that enables detailed investigation of the range of functions of zebrafish Pk1 proteins. In this study, the functional redundancy of the pk1 paralogs has proven to be a significant advantage of the zebrafish model, as it has allowed the nuanced roles of the Pk1 molecules in NCC development, including during EMT, to be teased apart from their earlier roles in convergence. Functional redundancy also allows both pk1bfh122/fh122 mutant fish and pk1ach105/ch105 mutant fish to reach fertile adulthood, whereas pk1ach105/ch105 mutants injected with morpholinos targeted against Pk1b begin to die shortly after 24 hpf. As also discussed below, we conclude that developing zebrafish embryos are able to recover from the early loss of a single pk1 gene, whereas depletion of both pk1 paralogs proves lethal.
By analysing the pk1bfh122/fh122 and pk1ach105/ch105 mutants using in vivo imaging we have uncovered roles for zebrafish Prickle1 both before and during the migration of the developing cranial neural crest. Specifically, we have uncovered equivalent roles for the two zebrafish pk1 paralogs during both EMT and directional migration of the NCCs. We find that Pk1 function is required for ventrolateral migration of the cranial crest, consistent with the previously-established roles for other core PCP molecules in contact inhibition of locomotion (CIL) of both Xenopus and zebrafish NCCs (Carmona-Fontaine et al., 2008; De Calisto et al., 2005; Matthews et al., 2008; Theveneau et al., 2010; Theveneau et al., 2013). However, by investigating the complex morphological transitions that precede migration, we demonstrate a broader role for Prickle1 before crest migration commences. We show that NCCs transition from neuroepithelial cells to a transient, blebbing, motile state before adopting a mesenchymal morphology capable of ventrolateral migration. These findings are consistent with previous descriptions of EMT as a series of progressive state changes rather than a binary switch (reviewed in Campbell and Casanova, 2016; Nieto et al., 2016), as well as with previous descriptions of NCC behaviors in zebrafish (Berndt et al., 2008). Critically, we find that pre-migratory NCCs in embryos deficient in either Pk1b or Pk1a complete apical detachment, indicating that they initiate NCC development as normal, but fail to transition from blebbing to mesenchymal morphologies. To our knowledge, this is the first report of a function for Prickle1—or indeed any PCP molecule—in controlling the key epithelial-to-mesenchymal transition that is necessary for neural crest cells to migrate out into the periphery.
We find that the NCC blebbing morphology in particular is critical to the understanding of EMT. Previous work in other systems has shown that in some contexts, bleb-based cells are capable of migration. For instance, some modes of amoeboid migration (reviewed in Lammermann and Sixt, 2009) and zebrafish primordial germ cells (PGCs), which migrate through the developing embryo (Goudarzi et al., 2017), use blebs as the predominant type of protrusion. Consistent with these findings, we have shown that Pk1-deficient blebbing cranial NCCs are motile, undergoing aberrant migration towards the anterior of the embryo, but they are not capable of migration in the appropriate ventrolateral direction unless they transition to mesenchymal states. Similarly, Scarpa et al. (2015) have demonstrated that when EMT is blocked using ectopic E-Cadherin expression, zebrafish NCCs again remain motile, moving at speeds equivalent to those of unmanipulated NCCs, yet they are incapable of normal ventrolateral migration. These findings are consistent with recent understanding of EMT in several contexts, particularly in metastatic cancer cells, where cells that express attributes of both epithelial and mesenchymal cell types have been denoted as intermediate, ‘metastable’, states (reviewed in Lee et al., 2006). We propose that the transient bleb-based morphology of NCCs represents such an intermediate, ‘metastable’ state in EMT, and that Prickle1 regulates the transition from the metastable state to the more stable, mesenchymal state. Again, this is consistent with recent understanding that EMT is a multi-state process (reviewed in Campbell and Casanova, 2016; Nieto et al., 2016). Importantly, our findings also suggest that NCCs may use the same planar cell polarity proteins in sequential stages of neural crest development, to control both EMT and CIL behaviors.
The most obvious phenotypic consequence of zebrafish Pk1-deficency is the clustering of NCCs at the dorsal aspect of the developing neural tube at stages when NCCs should be completing EMT and commencing their ventrolateral migration to the periphery. Similar NCC clustering phenotypes have previously been reported in embryos deficient for molecules that are required for EMT. For example, as discussed above, NCC clustering occurs when EMT is blocked by ectopic expression of E-Cad (Scarpa et al., 2015). In Xenopus embryos, the Smad-interacting protein-1 (Sip-1)—known to repress E-Cad expression in cancer cells—is required for NCCs to complete EMT, disperse, and migrate out from dorsal positions of the neural tube (Rogers et al., 2013). The metalloproteinase MMP14, through control of both E-Cad and N-Cad, also regulates both EMT and migration (Garmon et al., 2018), at least in part through a change in apicobasal polarity (Andrieu et al., 2018). Transcription factors such as Ets (Theveneau et al., 2007) and the Wnt signaling effector Axud1 (Simoes-Costa et al., 2015) are also required for the successful separation of NCCs from the neuroepithelium, with the latter additionally playing an earlier role in NCC specification. Deficiency of transcription factor Ovo1 also leads to NCC clustering phenotypes, although in this case only pigment cell progenitors are affected. Notably, Ovo1 is also a Wnt target, and functions to regulate intracellular movements of N-Cad (Piloto and Schilling, 2010). Completion of EMT also requires the dynamic regulation of canonical Wnt signaling through the secreted molecule Draxin (Hutchins and Bronner, 2018); both loss of and ectopic expression of Draxin prevent NCCs from adopting mesenchymal morphologies capable of migration. Further, the dynamic regulation of Cadherins other than E-Cad and N-Cad is also critical for EMT. The proteolytic cleavage of Cadherin-6B, for instance, is required for cranial NCCs to undergo EMT (Schiffmacher et al., 2014); one of these fragments is critical to up-regulate EMT effectors, such as Snail1 and Snail2 (Schiffmacher et al., 2016). In summary, a range of molecules and pathways play important roles in NCC EMT, and a common theme in the NCC clustering phenotypes associated with blocks in EMT is misregulation of Cadherin molecules.
Our findings suggest that Cadherin regulation similarly underlies the roles of the core PCP Pk1 molecules in the zebrafish NCC. Specifically, we suggest that the link between Pk1 and EMT behavior lies at the level of E-Cad and N-Cad, possibly in the form of a feedback loop. Scarpa et al. (2015) showed that the Xenopus E-Cad/N-Cad switch is required for CIL; cell contacts subsequently trigger non-canonical Wnt/PCP signaling (Carmona-Fontaine et al., 2008; Theveneau et al., 2013). Although down-regulation of E-Cad has long been considered a hallmark of the end of EMT and the beginning of NCC migration, ECad is nevertheless not only present, but also required, in migrating cranial NCCs of Xenopus, chick, and mouse (reviewed in Cousin, 2017). Consistent with findings in other species, we find that in unmanipulated zebrafish embryos E-Cad is indeed detectable in migrating NCCs at low levels. Notably, we find that E-Cad levels are elevated in Pk1b-deficient specimens, both in pre-migratory and migratory NCCs, and that N-Cad levels are reduced in migratory NCCs. We suggest that the previously-proposed activation of PCP signaling in response to decreased levels of E-cad and increased levels of N-Cad may be part of a broader feedback loop, such that Pk1 (a core PCP molecule) is in turn necessary to regulate both E-Cad levels and N-Cad. Consistent with this model, our analysis has shown that disruption of pk1b is sufficient to disrupt the typical reduction of E-Cad levels, likely leading to NCCs becoming trapped in the transitional blebbing morphology rather than completing EMT. According to this model, once EMT is completed, Pk1 is necessary for increase of N-Cad levels, and the continued reduction of E-Cad, such that cells make only transient contacts with neighbors, and migrate in streams, consistent with CIL behavior (Carmona-Fontaine et al., 2008; Theveneau et al., 2010; Theveneau et al., 2013).
One of our more striking findings is that even when ventrolateral migration is impaired in Pk1-deficient specimens, NCCs maintain the ability to move in a directed fashion, albeit in an aberrant anterior direction. This finding implies that when core PCP function is disrupted, and NCCs do not complete EMT, they nevertheless remain able to respond to directional cue(s) that exist in the embryo. Szabo et al. (2016) have shown that the collective migration of Xenopus NCCs depends upon their initial confinement by the extracellular matrix molecule Versican, which also functions as a guidance cue, allowing NCCs to migrate collectively in streams and respect exclusionary boundaries between streams. Several other guidance cues, including VEGF (McLennan et al., 2015; McLennan et al., 2010) and Sdf1 (Kasemeier-Kulesa et al., 2010; Olesnicky Killian et al., 2009; Saito et al., 2012; Theveneau et al., 2013), are also important for cranial NCC migration (reviewed in Kulesa et al., 2010). Although we find that clustered NCCs can move collectively in Pk1b or Pk1a-deficient embryos—perhaps in response to local environmental signals—these NCCs largely retain a location immediately dorsal to the neural tube, failing to migrate away laterally and instead moving towards the anterior and crossing the exclusionary boundaries that would typically exist between normal cranial crest streams. Interestingly, despite the aberrant cranial NCC migration we have documented early in the NCC development process, we have not found evidence of altered cranial crest cell fates, nor do we find any obvious defects in later cranial morphology in Pk1b or Pk1a-deficient zebrafish larvae. Rather, the normal organization of pharyngeal cartilages in 6 dpf larvae mutant for a single pk1 gene, coupled with the viability of each mutant, demonstrates a remarkable ability of single pk1 mutant cranial NCC derivatives to recover from their early deficits. Consistent with the hypothesis that this recovery reflects functional redundancy of the two zebrafish Pk1 paralogs, it has recently been reported that the single mouse Prickle1 gene is required for the migration and differentiation of NCCs into osteoblasts of the skull frontal bone (Wan et al., 2018).
We also find that pre-migratory NCCs deficient in either Pk1b or Pk1a display defects in polarity such that they orient along the AP axis. A recent study found that in both in vivo mouse mammary gland epithelia and in vitro 3D mammary gland cultured cells, EMT was accompanied by a change in polarity, and that polarity reversal promoted the scattering of mesenchymal cells (Burute et al., 2017). Moreover, previous work has shown that in zebrafish, PCP proteins polarize neuroepithelial cells along the AP axis during neurulation, with GFP-tagged Drosophila Prickle protein localizing specifically on the anterior side of these cells (Ciruna et al., 2006). Considering our data in light of these previous studies, it may be tempting to speculate that when key transitions during EMT are blocked, NCCs move in aberrant directions because of a failure to switch from a neuroepithelial-like apicobasal polarity to a mesenchymal “front-back” polarity. However, our findings in double-heterozygous specimens, where NCCs lack clear polarity yet continue to show anteriorly-directed migrations, argue that anterior movement can be decoupled from polarity in NCCs. Future investigation of how the dynamics of Pk1b and Pk1a localization correlate with polarization of NCCs and their progenitors will be important to dissect the dynamic role(s) of Prickle1.
While previous work has implicated Frizzled and Disheveled in NCC migration, our work implicates Pk1 in the process of NCC EMT as well as in subsequent NCC migration. This suggests that the entire core PCP suite may be required during EMT. It is probable that the core PCP factors act in concert at the same developmental stages when we have established Pk1 functions, especially given the previously-elucidated role of PCP-signaling in CIL. However, it is also formally possible that Pk1 functions at an earlier stage, to set up the asymmetries of other core PCP proteins, similar to how the Drosophila Prickle and Spiny legs isoforms differentially amplify the asymmetries of core PCP proteins (Gubb et al., 1999; Merkel et al., 2014; Sharp and Axelrod, 2016; reviewed in Strutt and Strutt, 2009), as well as establish the polarity of a microtubule network that determines the direction of tissue polarity in Drosophila (Olofsson et al., 2014). Approaches to visualize endogenous levels of zebrafish Pk1 proteins, as well as the other core PCP proteins and previously-implicated downstream molecules, coupled with detailed temporal analysis during EMT processes, would help to clarify regulatory interactions in the core PCP module during different stages of NCC development.
Overall, our study has revealed that zebrafish Pk1 is required during cranial neural crest EMT and migration. Together with previously-published work, our findings reveal broad functions for the core PCP molecule Prickle1 in neural crest development. We conclude that PCP molecules act as a key source of regulation and feedback for the complex cellular dynamics, including EMT processes, that function to allow proper development of the cranial neural crest.
Supplementary Material
Movie 1: Migration behavior of wild-type cranial NCCs Time-lapse maximum projection confocal movie of wild-type Tg(sox10:EGFP) embryo from ~16 hpf to 18 hpf. Z-stacks were taken at 4 min 30 sec intervals. Anterior to left, solid line indicates left-side edge of the neuroepithelium as assayed by DIC imaging. NCCs move from the dorsal aspect of the neuroepithelium (above the line), laterally towards the periphery (below the line). Time stamp at top left follows hh:mm format.
Movie 2: Migration behavior of pk1b-morphant cranial NCCs Time-lapse maximum projection confocal movie of pk1b-MO Tg(sox10:EGFP) embryo from ~16 hpf to 18 hpf. Z-stacks were taken at 5 min intervals. Anterior to left, solid lines indicate lateral edges of the neuroepithelium as assayed by DIC imaging. NCCs remain dorsal to the neuroepithelium, at center above solid line. Time stamp at top left follows hh:mm format.
Movie 3: Migration behavior of pk1b-mutant cranial NCCs Time-lapse maximum projection confocal movie of pk1bfh122/fh122 Tg(sox10:EGFP) embryo from ~16 hpf to 18 hpf. Z-stacks were taken at 3 min 30 sec intervals. Anterior to left, solid lines indicate lateral edges of the neuroepithelium as assayed by DIC imaging. NCCs remain dorsal to the neuroepithelium, at center between solid lines. Time stamp at top left follows hh:mm format.
Movie 4: Migration behavior of pk1a-mutant cranial NCCs Time-lapse maximum projection confocal movie of pk1ach105/ch105 Tg(sox10:EGFP) embryo from ~16 hpf to 18 hpf. Z-stacks were taken at 2 min intervals. Anterior to left, solid lines indicate lateral edges of the neuroepithelium as assayed by DIC imaging. NCCs remain dorsal to neuroepithelium, at center between solid lines. Time stamp at top left follows hh:mm format.
Movie 5: Migration behavior of double-heterozygous pk1b/pk1a-mutant cranial NCCs Time-lapse maximum projection confocal movie of double-heterozygous pk1bfh122/+;pk1ach105/+ Tg(sox10:EGFP) embryo from ~16 hpf to 18 hpf. Z-stacks were taken at 2 min 30 sec intervals. Anterior to left, solid lines indicate lateral edges of the neuroepithelium as assayed by DIC imaging. NCCs remain dorsal to neuroepithelium, at center between solid lines. Time stamp at top left follows hh:mm format.
Movie 6: NCC undergoing apical detachment Time-lapse maximum projection confocal movie of 16 hpf wild-type embryo injected with sox10:LifeAct-GFP. Z-stacks were taken at 1 min 8 sec intervals. An apically-delaminating NCC rounds up within the neuroepithelium; the midline is to the left and the edge of the neuroepithelium to the right. Time stamp at top left in minutes, with two decimal places. Scale bar=10 μm
Movie 7: NCC transitioning to blebbing behavior Time-lapse maximum projection confocal movie of 16 hpf wild-type embryo injected with sox10:LifeAct-GFP. Z-stacks were taken at 33 sec intervals. A dorsally located NCC transitions from a rounded morphology, to a one with small protrusions, edge of neuroepithelium to the right. Time stamp at top left in minutes, with two decimal places. Scale bar=10 μm.
Movie 8: NCC in the blebbing state Time-lapse maximum projection confocal movie of 16 hpf wild-type embryo injected with sox10:LifeAct-GFP. Z-stacks were taken at 1 min 18 sec intervals. A dorsally located NCC shows blebbing behaviors, edge of neuroepithelium to right. Time stamp at top left in minutes, with two decimal places. Scale bar=10 μm.
Movie 9: NCC transitioning from blebbing to mesenchymal state Time-lapse maximum projection confocal movie of 16 hpf wild-type embryo injected with sox10:LifeAct-GFP. Z-stacks were taken at 1 min 9 sec intervals. A NCC with small protrusions transitions to a highly-protrusive mesenchymal cell, which is crossing the edge of the neuroepithelium. Time stamp at top left in minutes with two decimal places. Scale bar=10 μm.
Highlights.
Pk1a & Pk1b are required for normal polarity and migration of cranial neural crest
Pk1-deficient neural crest cells adopt rounded morphologies and cluster dorsally
Pk1 regulates the epithelial-to-mesenchymal transition of cranial neural crest
Pk1b regulates Cadherin levels in the epithelial-to-mesenchymal transition
Acknowledgements
We are grateful to numerous colleagues for sharing genetic tools and reagents that made this study possible. The Tg(−4.9sox10:EGFP)ba2 fish line was provided by Tom Schilling with permission from Rob Kelsh and the Tg(−7.2sox10:mRFP)vu234 fish line was provided by Bruce Appel. The −4.9sox10:LifeAct-GFP construct was provided by Michael Granato, the Cherry-XCentrin and EGFPXCentrin plasmids by Diane Sepich, the pk1a probe by Diane Slusarski, and the Vangl2 morpholino reagent by Anand Chandrasekhar. We thank Sarah Wanner and Crystal Love for general technical guidance, Rachel Warga for advice on Cadherin antibody immunolabeling, Gokhan Dalgin for advice on CRISPR/Cas9 mutagenesis and for sharing reagents, and Anita Ng for expert fish care. We are grateful to Mary Halloran, Ankur Saxena, and members of the Ho and Prince labs for helpful discussions, and to Ana Beiriger, Alana Beadell, Edwin (Chip) Ferguson, and Cliff Ragsdale for comments on the manuscript. This work was funded in part by the Chicago Biomedical Consortium with support from the Searle Funds at The Chicago Community Trust, and by the National Center for Advancing Translational Sciences of the National Institutes of Health through Grant Number UL1 TR000430. K.A was supported by the George A. Hines Scholarship fund, and M.R. was supported by NIH T32 HD055164. This work also benefitted from the resources of the ZFIN database (zfin.org).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, and Nieto MA (2009). Epithelial mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest 119, 1438–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimenko MA, Ekker M, Wegner J, Lin W, and Westerfield M (1994). Combinatorial expression of three zebrafish genes related to distal-less: part of a homeobox gene code for the head. J Neurosci 14, 3475–3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambegaonkar AA, and Irvine KD (2015). Coordination of planar cell polarity pathways through Spiny-legs. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrieu C, Montigny A, Alfandari D, and Theveneau E (2018). Basolateral localization of MMP14 drives apicobasal polarity change during EMT independently of its catalytic activity. bioRxiv, 402180. [Google Scholar]
- Banerjee S, Gordon L, Donn TM, Berti C, Moens CB, Burden SJ, and Granato M (2011). A novel role for MuSK and non-canonical Wnt signaling during segmental neural crest cell migration. Development 138, 3287–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndt JD, Clay MR, Langenberg T, and Halloran MC (2008). Rho-kinase and myosin II affect dynamic neural crest cell behaviors during epithelial to mesenchymal transition in vivo. Dev Biol 324, 236–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burute M, Prioux M, Blin G, Truchet S, Letort G, Tseng Q, Bessy T, Lowell S, Young J, Filhol O, et al. (2017). Polarity Reversal by Centrosome Repositioning Primes Cell Scattering during Epithelial-to-Mesenchymal Transition. Dev Cell 40, 168–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MT, and Wallingford JB (2015). Control of vertebrate core planar cell polarity protein localization and dynamics by Prickle 2. Development 142, 3429–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell K, and Casanova J (2016). A common framework for EMT and collective cell migration. Development 143, 4291–4300. [DOI] [PubMed] [Google Scholar]
- Carmona-Fontaine C, Matthews HK, Kuriyama S, Moreno M, Dunn GA, Parsons M, Stern CD, and Mayor R (2008). Contact inhibition of locomotion in vivo controls neural crest directional migration. Nature 456, 957–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney TJ, Dutton KA, Greenhill E, Delfino-Machin M, Dufourcq P, Blader P, and Kelsh RN (2006). A direct role for Sox10 in specification of neural crest-derived sensory neurons. Development 133, 4619–4630. [DOI] [PubMed] [Google Scholar]
- Carreira-Barbosa F, Concha ML, Takeuchi M, Ueno N, Wilson SW, and Tada M (2003). Prickle 1 regulates cell movements during gastrulation and neuronal migration in zebrafish. Development 130, 4037–4046. [DOI] [PubMed] [Google Scholar]
- Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li GW, Park J, Blackburn EH, Weissman JS, Qi LS, et al. (2013). Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155, 1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciruna B, Jenny A, Lee D, Mlodzik M, and Schier AF (2006). Planar cell polarity signalling couples cell division and morphogenesis during neurulation. Nature 439, 220–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke J (2009). Role of polarized cell divisions in zebrafish neural tube formation. Curr Opin Neurobiol 19, 134–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay MR, and Halloran MC (2014). Cadherin 6 promotes neural crest cell detachment via F-actin regulation and influences active Rho distribution during epithelial-to-mesenchymal transition. Development 141, 2506–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couly G, Creuzet S, Bennaceur S, Vincent C, and Le Douarin NM (2002). Interactions between Hox-negative cephalic neural crest cells and the foregut endoderm in patterning the facial skeleton in the vertebrate head. Development 129, 1061–1073. [DOI] [PubMed] [Google Scholar]
- Couly GF, Coltey PM, and Le Douarin NM (1993). The triple origin of skull in higher vertebrates: a study in quail-chick chimeras. Development 117, 409–429. [DOI] [PubMed] [Google Scholar]
- Cousin H (2017). Cadherins function during the collective cell migration of Xenopus Cranial Neural Crest cells: revisiting the role of E-cadherin. Mech Dev 148, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalgin G, and Prince VE (2015). Differential levels of Neurod establish zebrafish endocrine pancreas cell fates. Dev Biol 402, 81–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey CF, and Moens CB (2017). Planar cell polarity in moving cells: think globally, act locally. Development 144, 187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Calisto J, Araya C, Marchant L, Riaz CF, and Mayor R (2005). Essential role of non-canonical Wnt signalling in neural crest migration. Development 132, 2587–2597. [DOI] [PubMed] [Google Scholar]
- Donoghue PC, Graham A, and Kelsh RN (2008). The origin and evolution of the neural crest. Bioessays 30, 530–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante MI, Romio L, Castro S, Collins JE, Goulding DA, Stemple DL, Woolf AS, and Wilson SW (2009). Convergent extension movements and ciliary function are mediated by ofd1, a zebrafish orthologue of the human oral-facial-digital type 1 syndrome gene. Hum Mol Genet 18, 289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallik KL, Treffy RW, Nacke LM, Ahsan K, Rocha M, Green-Saxena A, and Saxena A (2017). Neural crest and cancer: Divergent travelers on similar paths. Mech Dev 148, 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gans C, and Northcutt RG (1983). Neural crest and the origin of vertebrates: a new head. Science 220, 268–273. [DOI] [PubMed] [Google Scholar]
- Garmon T, Wittling M, and Nie S (2018). MMP14 Regulates Cranial Neural Crest Epithelial-to-Mesenchymal Transition and Migration. Dev Dyn 247, 1083–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelik R, and Gautreau A (2014). Quantitative and unbiased analysis of directional persistence in cell migration. Nat Protoc 9, 1931–1943. [DOI] [PubMed] [Google Scholar]
- Goudarzi M, Tarbashevich K, Mildner K, Begemann I, Garcia J, Paksa A, Reichman-Fried M, Mahabaleshwar H, Blaser H, Hartwig J, et al. (2017). Bleb Expansion in Migrating Cells Depends on Supply of Membrane from Cell Surface Invaginations. Dev Cell 43, 577–587e575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubb D, Green C, Huen D, Coulson D, Johnson G, Tree D, Collier S, and Roote J (1999). The balance between isoforms of the prickle LIM domain protein is critical for planar polarity in Drosophila imaginal discs. Genes Dev 13, 2315–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BK (2000). The neural crest as a fourth germ layer and vertebrates as quadroblastic not triploblastic. Evol Dev 2, 3–5. [DOI] [PubMed] [Google Scholar]
- Hay ED (2005). The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Dev Dyn 233, 706–720. [DOI] [PubMed] [Google Scholar]
- Huang C, Kratzer MC, Wedlich D, and Kashef J (2016). E-cadherin is required for cranial neural crest migration in Xenopus laevis. Dev Biol 411, 159–171. [DOI] [PubMed] [Google Scholar]
- Hutchins EJ, and Bronner ME (2018). Draxin acts as a molecular rheostat of canonical Wnt signaling to control cranial neural crest EMT. J Cell Biol 217, 3683–3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen JR, Topczewski J, Bingham S, Sepich DS, Marlow F, Chandrasekhar A, and Solnica-Krezel L (2002). Zebrafish trilobite identifies new roles for Strabismus in gastrulation and neuronal movements. Nat Cell Biol 4, 610–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez L, Wang J, Morrison MA, Whatcott C, Soh KK, Warner S, Bearss D, Jette CA, and Stewart RA (2016). Phenotypic chemical screening using a zebrafish neural crest EMT reporter identifies retinoic acid as an inhibitor of epithelial morphogenesis. Dis Model Mech 9, 389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing L (2012). Genotyping for Single Zebrafish (Fin Clip) or Zebrafish Embryo. Bio-protocol 2, e182. [Google Scholar]
- Jussila M, and Ciruna B (2017). Zebrafish models of non-canonical Wnt/planar cell polarity signalling: fishing for valuable insight into vertebrate polarized cell behavior. Wiley Interdiscip Rev Dev Biol 6. [DOI] [PubMed] [Google Scholar]
- Kasemeier-Kulesa JC, McLennan R, Romine MH, Kulesa PM, and Lefcort F (2010). CXCR4 controls ventral migration of sympathetic precursor cells. J Neurosci 30, 13078–13088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman CK, Mosimann C, Fan ZP, Yang S, Thomas AJ, Ablain J, Tan JL, Fogley RD, van Rooijen E, Hagedorn EJ, et al. (2016). A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science 351, aad2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami K, and Shima A (1999). Identification of the Tol2 transposase of the medaka fish Oryzias latipes that catalyzes excision of a nonautonomous Tol2 element in zebrafish Danio rerio. Gene 240, 239–244. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, and Schilling TF (1995). Stages of embryonic development of the zebrafish. Dev Dyn 203, 253–310. [DOI] [PubMed] [Google Scholar]
- Kirby ML, and Hutson MR (2010). Factors controlling cardiac neural crest cell migration. Cell Adh Migr 4, 609–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulesa PM, Bailey CM, Kasemeier-Kulesa JC, and McLennan R (2010). Cranial neural crest migration: new rules for an old road. Dev Biol 344, 543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammermann T, and Sixt M (2009). Mechanical modes of ‘amoeboid’ cell migration. Curr Opin Cell Biol 21, 636–644. [DOI] [PubMed] [Google Scholar]
- Le Douarin N, and Kalcheim C (1999). The neural crest, 2nd edn (Cambridge, UK; New York, NY, USA: Cambridge University Press; ). [Google Scholar]
- Lee JM, Dedhar S, Kalluri R, and Thompson EW (2006). The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol 172, 973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, and Yuan J (2008). Caspases in apoptosis and beyond. Oncogene 27, 6194–6206. [DOI] [PubMed] [Google Scholar]
- Lumsden A, Sprawson N, and Graham A (1991). Segmental origin and migration of neural crest cells in the hindbrain region of the chick embryo. Development 113, 1281–1291. [DOI] [PubMed] [Google Scholar]
- Maguire LH, Thomas AR, and Goldstein AM (2015). Tumors of the neural crest: Common themes in development and cancer. Dev Dyn 244, 311–322. [DOI] [PubMed] [Google Scholar]
- Mapp OM, Walsh GS, Moens CB, Tada M, and Prince VE (2011). Zebrafish Prickle1b mediates facial branchiomotor neuron migration via a farnesylation-dependent nuclear activity. Development 138, 2121–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mapp OM, Wanner SJ, Rohrschneider MR, and Prince VE (2010). Prickle1b mediates interpretation of migratory cues during zebrafish facial branchiomotor neuron migration. Dev Dyn 239, 1596–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews HK, Marchant L, Carmona-Fontaine C, Kuriyama S, Larrain J, Holt MR, Parsons M, and Mayor R (2008). Directional migration of neural crest cells in vivo is regulated by Syndecan-4/Rac1 and non-canonical Wnt signaling/RhoA. Development 135, 1771–1780. [DOI] [PubMed] [Google Scholar]
- May-Simera H, and Kelley MW (2012). Planar cell polarity in the inner ear. Curr Top Dev Biol 101, 111–140. [DOI] [PubMed] [Google Scholar]
- Mayor R, and Theveneau E (2014). The role of the non-canonical Wnt-planar cell polarity pathway in neural crest migration. Biochem J 457, 19–26. [DOI] [PubMed] [Google Scholar]
- McLennan R, Schumacher LJ, Morrison JA, Teddy JM, Ridenour DA, Box AC, Semerad CL, Li H, McDowell W, Kay D, et al. (2015). VEGF signals induce trailblazer cell identity that drives neural crest migration. Dev Biol 407, 12–25. [DOI] [PubMed] [Google Scholar]
- McLennan R, Teddy JM, Kasemeier-Kulesa JC, Romine MH, and Kulesa PM (2010). Vascular endothelial growth factor (VEGF) regulates cranial neural crest migration in vivo. Dev Biol 339, 114–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkel M, Sagner A, Gruber FS, Etournay R, Blasse C, Myers E, Eaton S, and Julicher F (2014). The balance of prickle/spiny-legs isoforms controls the amount of coupling between core and fat PCP systems. Curr Biol 24, 2111–2123. [DOI] [PubMed] [Google Scholar]
- Montague TG, Cruz JM, Gagnon JA, Church GM, and Valen E (2014). CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res 42, W401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto MA, Huang RY, Jackson RA, and Thiery JP (2016). Emt: 2016. Cell 166, 21–45. [DOI] [PubMed] [Google Scholar]
- Olesnicky Killian EC, Birkholz DA, and Artinger KB (2009). A role for chemokine signaling in neural crest cell migration and craniofacial development. Dev Biol 333, 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olofsson J, Sharp KA, Matis M, Cho B, and Axelrod JD (2014). Prickle/spiny-legs isoforms control the polarity of the apical microtubule network in planar cell polarity. Development 141, 2866–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piloto S, and Schilling TF (2010). Ovo1 links Wnt signaling with N-cadherin localization during neural crest migration. Development 137, 1981–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince VE, Moens CB, Kimmel CB, and Ho RK (1998). Zebrafish hox genes: expression in the hindbrain region of wild-type and mutants of the segmentation gene, valentino. Development 125, 393–406. [DOI] [PubMed] [Google Scholar]
- Rida PC, and Chen P (2009). Line up and listen: Planar cell polarity regulation in the mammalian inner ear. Semin Cell Dev Biol 20, 978–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers CD, Saxena A, and Bronner ME (2013). Sip1 mediates an E-cadherin-to-N-cadherin switch during cranial neural crest EMT. J Cell Biol 203, 835–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrschneider MR, Elsen GE, and Prince VE (2007). Zebrafish Hoxb1a regulates multiple downstream genes including prickle1b. Dev Biol 309, 358–372. [DOI] [PubMed] [Google Scholar]
- Rossi A, Kontarakis Z, Gerri C, Nolte H, Holper S, Kruger M, and Stainier DY (2015). Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 524, 230–233. [DOI] [PubMed] [Google Scholar]
- Roszko I, Sawada A, and Solnica-Krezel L (2009). Regulation of convergence and extension movements during vertebrate gastrulation by the Wnt/PCP pathway. Semin Cell Dev Biol 20, 986–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito D, Takase Y, Murai H, and Takahashi Y (2012). The dorsal aorta initiates a molecular cascade that instructs sympatho-adrenal specification. Science 336, 1578–1581. [DOI] [PubMed] [Google Scholar]
- Sauka-Spengler T, and Bronner-Fraser M (2008). A gene regulatory network orchestrates neural crest formation. Nat Rev Mol Cell Biol 9, 557–568. [DOI] [PubMed] [Google Scholar]
- Savagner P (2010). The epithelial-mesenchymal transition (EMT) phenomenon. Ann Oncol 21 Suppl 7, vii89–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpa E, Szabo A, Bibonne A, Theveneau E, Parsons M, and Mayor R (2015). Cadherin Switch during EMT in Neural Crest Cells Leads to Contact Inhibition of Locomotion via Repolarization of Forces. Dev Cell 34, 421–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffmacher AT, Padmanabhan R, Jhingory S, and Taneyhill LA (2014). Cadherin-6B is proteolytically processed during epithelial-to-mesenchymal transitions of the cranial neural crest. Mol Biol Cell 25, 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffmacher AT, Xie V, and Taneyhill LA (2016). Cadherin-6B proteolysis promotes the neural crest cell epithelial-to-mesenchymal transition through transcriptional regulation. J Cell Biol 215, 735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling TF, and Kimmel CB (1994). Segment and cell type lineage restrictions during pharyngeal arch development in the zebrafish embryo. Development 120, 483–494. [DOI] [PubMed] [Google Scholar]
- Schilling TF, and Kimmel CB (1997). Musculoskeletal patterning in the pharyngeal segments of the zebrafish embryo. Development 124, 2945–2960. [DOI] [PubMed] [Google Scholar]
- Sepich DS, Usmani M, Pawlicki S, and Solnica-Krezel L (2011). Wnt/PCP signaling controls intracellular position of MTOCs during gastrulation convergence and extension movements. Development 138, 543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp KA, and Axelrod JD (2016). Prickle isoforms control the direction of tissue polarity by microtubule independent and dependent mechanisms. Biol Open 5, 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoes-Costa M, Stone M, and Bronner ME (2015). Axud1 Integrates Wnt Signaling and Transcriptional Inputs to Drive Neural Crest Formation. Dev Cell 34, 544–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon KS, Kudoh T, Dawid IB, and Fritz A (2003). Zebrafish foxi1 mediates otic placode formation and jaw development. Development 130, 929–940. [DOI] [PubMed] [Google Scholar]
- Strutt H, and Strutt D (2009). Asymmetric localisation of planar polarity proteins: Mechanisms and consequences. Semin Cell Dev Biol 20, 957–963. [DOI] [PubMed] [Google Scholar]
- Szabo A, Melchionda M, Nastasi G, Woods ML, Campo S, Perris R, and Mayor R (2016). In vivo confinement promotes collective migration of neural crest cells. J Cell Biol 213, 543–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneyhill LA, and Schiffmacher AT (2017). Should I stay or should I go? Cadherin function and regulation in the neural crest. Genesis 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawk M, Araya C, Lyons DA, Reugels AM, Girdler GC, Bayley PR, Hyde DR, Tada M, and Clarke JD (2007). A mirror-symmetric cell division that orchestrates neuroepithelial morphogenesis. Nature 446, 797–800. [DOI] [PubMed] [Google Scholar]
- Theveneau E, Duband JL, and Altabef M (2007). Ets-1 confers cranial features on neural crest delamination. PLoS One 2, e1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theveneau E, Marchant L, Kuriyama S, Gull M, Moepps B, Parsons M, and Mayor R (2010). Collective chemotaxis requires contact-dependent cell polarity. Dev Cell 19, 39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theveneau E, and Mayor R (2012). Neural crest delamination and migration: from epithelium-to mesenchyme transition to collective cell migration. Dev Biol 366, 34–54. [DOI] [PubMed] [Google Scholar]
- Theveneau E, Steventon B, Scarpa E, Garcia S, Trepat X, Streit A, and Mayor R (2013). Chase-and-run between adjacent cell populations promotes directional collective migration. Nat Cell Biol 15, 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP, and Sleeman JP (2006). Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol 7, 131–142. [DOI] [PubMed] [Google Scholar]
- Thompson EW, and Williams ED (2008). EMT and MET in carcinoma--clinical observations, regulatory pathways and new models. Clin Exp Metastasis 25, 591–592. [DOI] [PubMed] [Google Scholar]
- Torban E, Patenaude AM, Leclerc S, Rakowiecki S, Gauthier S, Andelfinger G, Epstein DJ, and Gros P (2008). Genetic interaction between members of the Vangl family causes neural tube defects in mice. Proc Natl Acad Sci U S A 105, 3449–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tree DR, Shulman JM, Rousset R, Scott MP, Gubb D, and Axelrod JD (2002). Prickle mediates feedback amplification to generate asymmetric planar cell polarity signaling. Cell 109, 371–381. [DOI] [PubMed] [Google Scholar]
- Veeman MT, Slusarski DC, Kaykas A, Louie SH, and Moon RT (2003). Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr Biol 13, 680–685. [DOI] [PubMed] [Google Scholar]
- Wan Y, Lantz B, Cusack BJ, and Szabo-Rogers HL (2018). Prickle1 regulates differentiation of frontal bone osteoblasts. Sci Rep 8, 18021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheelock MJ, Shintani Y, Maeda M, Fukumoto Y, and Johnson KR (2008). Cadherin switching. J Cell Sci 121, 727–735. [DOI] [PubMed] [Google Scholar]
- Yin C, Kiskowski M, Pouille PA, Farge E, and Solnica-Krezel L (2008). Cooperation of polarized cell intercalations drives convergence and extension of presomitic mesoderm during zebrafish gastrulation. J Cell Biol 180, 221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallen JA (2007). Planar polarity and tissue morphogenesis. Cell 129, 1051–1063. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie 1: Migration behavior of wild-type cranial NCCs Time-lapse maximum projection confocal movie of wild-type Tg(sox10:EGFP) embryo from ~16 hpf to 18 hpf. Z-stacks were taken at 4 min 30 sec intervals. Anterior to left, solid line indicates left-side edge of the neuroepithelium as assayed by DIC imaging. NCCs move from the dorsal aspect of the neuroepithelium (above the line), laterally towards the periphery (below the line). Time stamp at top left follows hh:mm format.
Movie 2: Migration behavior of pk1b-morphant cranial NCCs Time-lapse maximum projection confocal movie of pk1b-MO Tg(sox10:EGFP) embryo from ~16 hpf to 18 hpf. Z-stacks were taken at 5 min intervals. Anterior to left, solid lines indicate lateral edges of the neuroepithelium as assayed by DIC imaging. NCCs remain dorsal to the neuroepithelium, at center above solid line. Time stamp at top left follows hh:mm format.
Movie 3: Migration behavior of pk1b-mutant cranial NCCs Time-lapse maximum projection confocal movie of pk1bfh122/fh122 Tg(sox10:EGFP) embryo from ~16 hpf to 18 hpf. Z-stacks were taken at 3 min 30 sec intervals. Anterior to left, solid lines indicate lateral edges of the neuroepithelium as assayed by DIC imaging. NCCs remain dorsal to the neuroepithelium, at center between solid lines. Time stamp at top left follows hh:mm format.
Movie 4: Migration behavior of pk1a-mutant cranial NCCs Time-lapse maximum projection confocal movie of pk1ach105/ch105 Tg(sox10:EGFP) embryo from ~16 hpf to 18 hpf. Z-stacks were taken at 2 min intervals. Anterior to left, solid lines indicate lateral edges of the neuroepithelium as assayed by DIC imaging. NCCs remain dorsal to neuroepithelium, at center between solid lines. Time stamp at top left follows hh:mm format.
Movie 5: Migration behavior of double-heterozygous pk1b/pk1a-mutant cranial NCCs Time-lapse maximum projection confocal movie of double-heterozygous pk1bfh122/+;pk1ach105/+ Tg(sox10:EGFP) embryo from ~16 hpf to 18 hpf. Z-stacks were taken at 2 min 30 sec intervals. Anterior to left, solid lines indicate lateral edges of the neuroepithelium as assayed by DIC imaging. NCCs remain dorsal to neuroepithelium, at center between solid lines. Time stamp at top left follows hh:mm format.
Movie 6: NCC undergoing apical detachment Time-lapse maximum projection confocal movie of 16 hpf wild-type embryo injected with sox10:LifeAct-GFP. Z-stacks were taken at 1 min 8 sec intervals. An apically-delaminating NCC rounds up within the neuroepithelium; the midline is to the left and the edge of the neuroepithelium to the right. Time stamp at top left in minutes, with two decimal places. Scale bar=10 μm
Movie 7: NCC transitioning to blebbing behavior Time-lapse maximum projection confocal movie of 16 hpf wild-type embryo injected with sox10:LifeAct-GFP. Z-stacks were taken at 33 sec intervals. A dorsally located NCC transitions from a rounded morphology, to a one with small protrusions, edge of neuroepithelium to the right. Time stamp at top left in minutes, with two decimal places. Scale bar=10 μm.
Movie 8: NCC in the blebbing state Time-lapse maximum projection confocal movie of 16 hpf wild-type embryo injected with sox10:LifeAct-GFP. Z-stacks were taken at 1 min 18 sec intervals. A dorsally located NCC shows blebbing behaviors, edge of neuroepithelium to right. Time stamp at top left in minutes, with two decimal places. Scale bar=10 μm.
Movie 9: NCC transitioning from blebbing to mesenchymal state Time-lapse maximum projection confocal movie of 16 hpf wild-type embryo injected with sox10:LifeAct-GFP. Z-stacks were taken at 1 min 9 sec intervals. A NCC with small protrusions transitions to a highly-protrusive mesenchymal cell, which is crossing the edge of the neuroepithelium. Time stamp at top left in minutes with two decimal places. Scale bar=10 μm.