

Abstract
Chronic airway inflammation is one of the main features of asthma. Release of mediators from infiltrating inflammatory cells in the airway mucosa has been proposed to contribute directly or indirectly to changes in airway structure and function. The airway smooth muscle, which has been regarded as a contractile component of the airways responding to various mediators and neurotransmitters, has recently been recognised as a rich source of pro-inflammatory cytokines, chemokines and growth factors. In this review, we discuss the role of airway smooth muscle cells in the regulation and perpetuation of airway inflammation that contribute to the pathogenesis of asthma.
Keywords: airway smooth muscle, chemokine, cytokine, growth factor, inflammation
Introduction
Inflammation of the airway wall is a central characteristic of asthma [1]. Airborne allergens often lead to an accumulation of eosinophils, lymphocytes (predominantly CD4 type), mast cells and macrophages, resulting in an inflammatory reaction in the mucosa. Neutrophil numbers can increase during an exacerbation. The release of mediators from these inflammatory cells has been proposed to contribute directly or indirectly to changes in airway structure and function.
Important structural changes of inflamed airways include epithelial cell shedding, basement membrane thickening, goblet cell hyperplasia (increase in cell number) and hypertrophy (increase in cell size), as well as an increase in airway smooth muscle (ASM) content [2]. These structural changes consequently form the basis for airway remodelling, a phenomenon believed to have profound consequences for airway function [3]. Bronchial vascular remodelling, with an increase in size and number of blood vessels as well as vascular hyperaemia have been proposed as contributing factors in airway wall remodelling in patients with chronic asthma.
The ASM has been typically described as a contractile tissue, responding to pro-inflammatory mediators and neurotransmitters by contracting, and responding to bronchodilators by relaxing. It has recently been recognised, however, that the synthetic function of ASM cells may be related to the perpetuation and intensity of airway wall inflammation. A number of recent studies have shown that ASM cells are also a rich source of biologically active cytokines, chemokines and growth factors, which may modulate airway inflammation through chemotactic, autocrine or paracrine effects. The expression of adhesion molecules and the release of cyclooxygenase (COX)-derived products by ASM cells may also influence inflammatory processes in the airways. In this review, we discuss the role of ASM cells in the regulation of airway inflammation and its contribution to subsequent airway wall remodelling.
Inflammatory mediators in the airways
Inflammatory mediators may be generated by resident cells within the airways and lungs, as well as by cells that have migrated into the airway from the circulation. The release of pro-inflammatory mediators can induce airway hyper-reactivity and airway wall remodelling [4,5,6]. Additionally, these mediators may further help in the recruitment and activation of other inflammatory cells, thus augmenting the inflammatory cascade. Potential sources of pro-inflammatory mediators in inflamed airways include eosinophils, epithelial cells, lymphocytes, mast cells, macrophages, neutrophils and platelets, as summarised in Table 1. The effects of a number of these individual mediators on ASM cell synthetic functions have recently been described. It seems unlikely that one particular mediator is solely responsible for the perpetuation of airway inflammation; perhaps a network of mediators contributes to the inflammatory processes.
Table 1.
Cellular sources of inflammatory mediators in inflamed airways
| Source | Inflammatory mediators | Reference |
| Eosinophil | MBP, EDN, ECP, EPO | [9,34,78] |
| LTC4, PAF, O2-, MMP-9 | ||
| IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-8, TGF-β, GM-CSF, TNF-α, MIP-1α, TIMP-1, PDGF | ||
| Epithelium | IL-1β, IL-6, IL-8, GM-CSF, ET-1, FGF, RANTES, MCP, MIP-1α, TIMP-1, TNF-a, TGF-β, PDGF | [9,48] |
| Macrophage | IL-1, IL-6, IL-10, GM-CSF, TNF-α, prostaglandins, TXs, LTs, PAF, FGF, ET-1, TGF-β, PDGF, MCP | [9,48] |
| Mast cell | IL-1, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-13, GM-CSF, TNF-α, TGF-β, histamine, tryptase, chymase, bradykinin, prostaglandins, PAF, LTs | [9,34] |
| Neutrophil | Myeloperoxidase, lysozyme, LTA4, LTB4, IL-1β, IL-6, IL-8, prostaglandins, PAF, TXA2, TNF-α, TGF-β, elastase, collagenase, MMP-9 | [34] |
| Th1 lymphocyte | IL-2, IL-3, GM-CSF, IFN-γ, TNF-α, TNF-β | [9] |
| Th2 lymphocyte | IL-3, IL-4, IL-5, IL-6, IL-10, IL-13, GM-CSF | [9] |
| Plasma | 5-HT, angiotensin II | [79,80] |
ECP, Eosinophil cationic protein; EDN, eosinophil derived neurotoxin; EPO, eosinophil peroxidase; ET, endothelin; FGF, fibroblast growth factor; GM-CSF, granulocyte/macrophage-colony stimulating factor; 5-HT, 5-hydroxytryptamine; IFN, interferon; IL, interleukin; LT, leukotriene; MBP, major basic protein; MCP, monocyte chemotactic protein; MIP, macrophage inflammatory protein; MMP-9, matrix metalloprotease-9; PAF, platelet activating factor; PDGF, platelet-derived growth factor; RANTES, regulated on activation, normal T cell expressed and secreted; TGF-β, transforming growth factor-β; TIMP-1, tissue inhibitor metalloprotease; TNF-α, tumour necrosis factor-α; TX, thromboxane.
Cytokine production by human ASM
Chronic airway inflammation is orchestrated and regulated by a complex network of cytokines, and these cytokines have numerous and divergent biological effects. ASM cells have been shown to be capable of producing a number of cytokines (Th1 type: IL-2, granulocyte/ macrophage-colony stimulating factor [GM-CSF], IFN-γ, IL-12; and Th2 type: IL-5, IL-6, GM-CSF), which then have the potential to influence airway inflammation and the development of airway remodelling (Table 2).
Table 2.
Airway smooth muscle (ASM) cell-derived inflammatory cytokines, their target cells and effects
| Cytokine | Target | Effect | Reference |
| IL-1 | ASM | IL-6, IL-11, LIF, GM-CSF, MCP, eotaxin, PDGF, PGE secretion↑ | [11,13,21,26,31,32] |
| Eosinophil | Recruitment | ||
| IL-2 | T cell | Proliferation↑ | [9] |
| Eosinophil | Recruitment | ||
| IL-5 | Eosinophil, mast cell, T cells | Recruitment, activation↑, survival↑ | [7,9] |
| ASM | Responsiveness↑ | ||
| IL-6 | mθ | Proliferation↓ | [9] |
| Goblet cell | Proliferation↑, mucus secretion↑ | ||
| B cell | Differentiation to plasma cell | ||
| Cytotoxic T cell | Differentiation | ||
| Fibroblast | Activation | ||
| IL-11 | ASM | Proliferation↑ | [81,82] |
| mθ, monocyte | Secretion↓ TIMP-1 | ||
| IL-12 | T cell | Proliferation↑ | [9] |
| IFN-γ | Th2 cells | Proliferation↓ | [9,62] |
| Th0 cells | → Th1 | ||
| B cells | IL-4-driven immunoglobulin isotype switching to IgE synthesis↓, proliferation↓ | ||
| Eosinophil, mast cell | MCP, PGE secretion↑ | ||
| ASM | MHC II expression | ||
| Activation, secretion↑ | |||
| mθ | ICAM-1↑ | ||
| LIF | Monocytes | IL-8 secretion↑ | [83,84] |
| Neurons | Phenotype regulation, tachykinin release↑ | ||
| mθ | Differentiation | ||
↑, Increase; ↓, decrease; →, unchanged. GM-CSF, Granulocyte/macrophage-colony stimulating factor; ICAM-1, intercellular adhesion molecule-1; IFN, interferon; IL, Interleukin; LIF, leukaemia inhibitory factor; MCP, monocyte chemotactic protein; MHC, major histocompatibility complex; mθ, macrophage; PDGF, platelet-derived growth factor; PGE, prostaglandin E; Th, T helper cells; TIMP-1, tissue inhibitor metalloprotease.
Secretion of Th1-type cytokines by ASM cells
In a study by Hakonarson et al., it was demonstrated that sensitised ASM cells could express the Th1-type cytokines IL-2, IL-12 and IFN-γ hours after the initial upregulation of Th2-type cytokines [7]. ASM cell-derived IL-2 and IFN-γ may play a protective role in the airway considering the results published by Hakonarson et al., whereby exogenous IL-2 or INF-γ attenuated atopic serum-induced ASM hyper-responsiveness to acetylcholine. IFN-γ may also play a protective role in atopic asthma by functionally antagonising IL-4-driven immunoglobulin isotype switching to IgE synthesis.
Inhibition of the proliferation of Th2 cells, mast cells and eosinophils or promotion of the differentiation of Th0 cells into those expressing a Th1 phenotype may also be considered protective in asthmatic airways. Low levels of IFN-γ have been detected in the bronchoalveolar lavage (BAL) fluid of patients with stable asthma, whereas the levels of mRNA for IFN-γ were not elevated in BAL fluid from patients with mild asthma [8]. This observation supports the notion that the pro-asthmatic state reflects an imbalance between Th1-type and Th2-type cytokine production. In patients with acute severe asthma, however, serum levels of IFN-γ were found to be elevated [9].
Secretion of Th2-type cytokines by ASM cells
The pro-inflammatory cytokines IL-1β and tumour necrosis factor-α (TNF-α) are found in exaggerated quantities in the BAL fluid from symptomatic asthmatics and can cause airway hyper-responsiveness and eosinophilia [10]. Cultured human ASM cells stimulated with IL-1β or TNF-α release IL-6 and GM-CSF [11,12,13,14,15,16].
IL-6 is a pleiotropic cytokine with a number of pro-inflammatory properties that could be relevant to the development and perpetuation of airway inflammation during asthma. These properties include mucus hypersecretion, the terminal differentiation of B cells into antibody-producing cells, upregulation of IL-4-dependent IgE production and stimulation of cytotoxic T-cell differentiation, as well as differentiation of immature mast cells. Possible anti-inflammatory properties of IL-6 include the inhibition of macrophage production of inflammatory cytokines and reduced airway responsiveness to methacholine.
GM-CSF has been implicated in the activation, proliferation and subsequent survival of infiltrating inflammatory cells such as neutrophils and eosinophils. Elevated levels of GM-CSF have been found in airway biopsies from asthma patients, and its overexpression is associated with pulmonary eosinophilia and fibrosis. Increased levels of IL-6 and GM-CSF have also been detected in the BAL fluid of asthmatic subjects [10]. IL-6 and GM-CSF expression by human ASM cells can be decreased by treatment with the glucocorticosteroid dexamethasone [11,12,13], suggesting that these cells may be an important target cell for the anti-inflammatory effects of steroids in asthma therapy [17].
BAL fluid samples isolated from atopic asthmatic patients also reveal significantly increased levels of IL-5 [18]. This cytokine is predominantly produced by infiltrating T cells in asthmatic airways, and possibly by mast cells [8], and is involved in the recruitment and subsequent activation of mast cells and eosinophils that are characteristic of asthmatic airway inflammation. IL-5 promotes mobilisation of eosinophils from the bone marrow. A recent study, however, has shown that human bronchial smooth muscle cells in culture, when passively sensitised with serum from patients with atopic asthma, can also express and secrete IL-5. Treatment of naive ASM with exogenous IL-5 potentiated its responsiveness to acetylcholine, suggesting that this Th2 cytokine may be involved in the pathobiology of asthma [7]. It should be borne in mind, however, that the concentrations of IL-5 in these experiments were significantly higher than the concentrations of IL-5 secreted by the sensitised ASM cells into the culture medium.
Passive sensitisation of ASM cells in culture also induced the synthesis and release of GM-CSF, IL-1β, IL-6 and IL-8 [7,15,19,20]. The release and subsequent autocrine action of IL-1β is of particular interest considering its pro-inflammatory effects mentioned earlier. The timing and order of secretion of Th1 and Th2 cytokines by ASM cells may be an important intrinsic regulatory mechanism. The question remains whether this mechanism is defective in ASM cells in asthmatic airways, thereby leading to an exaggerated Th2 response.
Secretion of other cytokines by ASM cells
IL-11 and leukaemia inhibitory factor (LIF) are classified as IL-6-type cytokines and they are produced by fibroblasts and epithelial cells of the airways. IL-11 has a variety of biological properties including the stimulation of tissue inhibitor of metalloproteinase-1, inhibition of macrophage/ monocyte-derived cytokine production and inhibition of nitric oxide (NO) production. A reduction in NO production in the asthmatic airway could have deleterious consequences for airway calibre considering the fact that endogenous NO is partly responsible for maintaining ASM tone. On the contrary, a reduction in NO production (by inducible NO synthase) in the asthmatic airway could reduce tissue damage and inflammation, depending on the relative amount of NO produced. LIF is a multifunctional cytokine with the ability to regulate macrophage differentiation. It also acts as a potent regulator of neuronal phenotype by modulating sympathetic neurones to adopt a cholinergic phenotype. LIF can also enhance neuronal tachykinin production and differentially regulate the expression of neural muscarinic receptors.
All of these mechanisms can alter airway function and, therefore, may become relevant to the pathogenesis of inflammatory airway diseases. A study by Elias et al. showed that transforming growth factor-β1 (TGF-β1) and/or IL-1α could stimulate human ASM cells to express and release IL-11, IL-6 and LIF. Respiratory syncytial virus and para-influenza virus type 3 were also potent stimulators of IL-11 by human ASM cells [21]. These viruses are known to be important triggers of asthma exacerbations [22].
Chemokine production by human ASM
A complex network of chemokines also regulates chronic airway inflammation. Chemokines are 8–10 kDa proteins that have been divided into subfamilies on the basis of the position of their cysteine residues located near the N-terminus of the protein. These mediators have numerous and divergent biological effects including leukocyte trafficking, degranulation of cells, angiogenesis, haematopoiesis and immune responses [23]. Chemokines produced and secreted by ASM cells may amplify the chemokine signal generated by the infiltrating inflammatory cells in the airway, thereby augmenting the recruitment of eosinophils, neutrophils, monocytes and lymphocytes to the airway (Table 3). The accumulation of these inflammatory cells subsequently contributes to the development of airway hyper-responsiveness, local inflammation and tissue injury through the release of granular enzymes and other cytokines. Eosinophils are also known to produce growth factors such as TGF-β1 and platelet-derived growth factor (PDGF). These growth factors can induce proliferation of fibroblasts and smooth muscle cells in vitro, possibly leading to the observed increase in smooth muscle mass in the asthmatic airway.
Table 3.
Airway smooth muscle cell-derived chemokines, their target cells and effects
| Chemokine | Target | Effect | Ref |
| IL-8 | Neutrophil | Recruitment, activation↑ | [34] |
| Eosinophil, T cell | Recruitment | ||
| Eotaxin | Eosinophil, T cells, basophil | Recruitment | [23,24,29,34,85,86] |
| Mast cell | Induces growth | ||
| RANTES | Eosinophil, T cells, monocytes | Recruitment | [23,24,29,34] |
| GM-CSF | Eosinophil, mq, monocyte | Recruitment, activation↑, proliferation↑ | [34] |
| Neutrophil | Survival↑ | ||
| MCP | Monocytes, lymphocytes, eosinophil, basophil, | Recruitment, activation↑ | [23,24,29] |
| dendritic cells, NK cells | |||
| mθ | IL-1 secretion↑ | ||
| MIP-1a | Eosinophil, NK, T cells, monocytes, mast cell, basophil | Recruitment, activation↑ | [23,24] |
↑, Increase; GM-CSF, granulocyte/macrophage-colony stimulating factor; IL, interleukin; MCP, monocyte chemotactic protein; MIP, macrophage inflammatory protein; mθ, macrophage; NK, natural killer; RANTES, regulated on activation, normal T cell expressed and secreted.
CC chemokines
The CC chemokines (or beta subfamily) have two juxtaposed cysteine residues. RANTES (regulated on activation, normal T cell expressed and secreted) is a potent chemoattractant for monocytes, T lymphocytes and eosinophils [23], and is produced by inflammatory cells and epithelial cells of the airways. Increased levels of RANTES in the BAL fluid and the bronchial mucosa of allergic asthmatic patients have been measured [24]. Several studies have demonstrated that human ASM cells are also capable of expressing and secreting biologically active RANTES following stimulation with TNF-α or IL-1β [14,25,26]. Stimulation of human ASM cells with IFN-γ in combination with TNF-α and/or IL-1β potentiated this effect, possibly via upregulation of IFN-γ receptor expression [25]. Treatment of the cells with dexamethasone inhibited expression of RANTES mRNA and inhibited secretion of RANTES protein. The Th2-type cytokine IL-10, however, failed to attenuate RANTES mRNA expression but did inhibit secretion of RANTES induced by a combination of IFN-γ and TNF-α . IL-10 could not inhibit TNF-α-induced RANTES secretion from cultured ASM cells [25,26].
The spectrum of target cells for the monocyte chemotactic proteins (MCPs) MCP-1, MCP-2, MCP-3, MCP-4 and MCP-5 includes monocytes, lymphocytes, eosinophils, basophils, dendritic cells and natural killer cells. Cellular sources of MCPs include lymphocytes, monocytes, alveolar macrophages and bronchial epithelial cells. Increased levels of mRNA and protein encoding MCPs have been detected in BAL fluid and bronchial biopsies of patients with asthma [24,27,28,29,30]. Work published by two groups describes the expression and secretion of MCP-1, MCP-2 and MCP-3 by human ASM cells treated with the pro-inflammatory cytokines TNF-α, IFN-γ, IL-1β or IL-1α, although induction patterns between chemokine mRNA expression after stimulation with the individual cytokines differed [26,31]. Furthermore, Pype et al. show that dexamethasone inhibited MCP mRNA expression and protein secretion in human ASM cells, whereas IL-10 had no effect on the expression [26]. No real data have so far been published showing the secretion of MCP-4, MCP-5 or the weak eosinophil attractant macrophage inflammatory protein-1α by ASM cells.
Eotaxin is a potent chemoattractant for eosinophils, basophils and Th2-like T lymphocytes. It cooperates with IL-5 in vivo to induce eosinophil recruitment; IL-5 promotes mobilisation of eosinophils from the bone marrow, whereas eotaxin recruits eosinophils in the tissue. Moreover, eotaxin has the ability to induce mast cell growth. Eotaxin is highly expressed by epithelial cells and inflammatory cells in asthmatic airways, and has been measured in increased quantities in BAL fluid from asthmatic subjects [23,24,29]. Two recent reports demonstrated that human ASM cells expressed eotaxin mRNA and protein following TNF-α and/or IL-1β stimulation [32,33]. Neither dexamethasone nor IL-10 inhibited the expression of mRNA encoding eotaxin, although IL-10 did inhibit the release of eotaxin protein into the culture medium, suggesting inhibition at a translational and/or post-translational level [32].
CXC chemokines
The CXC chemokines (or alpha subfamily) have two cysteine residues separated by an amino acid residue located near the N-terminus of the protein. IL-8 is an example of a CXC chemokine, and it is a potent chemoattractant and activator for neutrophils as well as a chemoattractant for eosinophils [6,34]. IL-8 is produced by inflammatory cells and epithelial cells in the airways and has been found to be elevated in the BAL fluid from asthma patients [35]. There are several studies demonstrating that human ASM cells stimulated with TNF-α, IL-1β or IL-1α can express and secrete IL-8 in vitro [15,31,36]. The study carried out by Herrick et al. also showed that atopic/asthmatic serum stimulates ASM cells to express mRNA encoding IL-8 [15]. Pang and Knox showed that bradykinin also stimulates the production of IL-8 in human ASM cells [37]. Both dexamethasone and IL-10 can inhibit the release of IL-8 protein into the culture medium [36].
Growth factor production by human ASM
ASM cells are also a potential source of growth factors that have been implicated in airway wall thickening and may indirectly influence airway inflammation (Table 4). Fibroblast growth factor-2 (FGF-2) is produced by fibroblasts and vascular smooth muscle cells in vitro and is described as mitogenic for cells of mesenchymal origin [38,39,40]. Increased concentrations of FGF-2 have been measured in the BAL fluid from asthmatic patients [6]. Rödel et al. suggest that this increase in FGF-2 in the airways may be the result of chronic Chlamydia pneumoniae infections, supported by their in vitro experiments showing that C. pneumoniae infection of human ASM cells significantly increased production of FGF-2 and IL-6 [41]. FGF-2 is released after host-cell lysis and may then act as a paracrine growth factor for neighbouring ASM cells, as well as upregulating the expression of interstitial collagenase, which mediates extracellular matrix (ECM) turnover. This turnover may support the proliferation of ASM cells in vivo, leading ultimately to airway remodelling.
Table 4.
Airway smooth muscle (ASM) cell-derived growth factors, their target cells and effects
| Growth factor | Target | Effect | Reference |
| FGF-2 | ASM | Proliferation↑ | [38,39] |
| ECM | Collagenase↑ | ||
| Fibroblast | Proliferation↑ | ||
| PDGF | ASM | Proliferation↑, IL-6 secretion↑ | [38,47,48,72] |
| Fibroblast | Recruitment and proliferation↑ | ||
| Epithelial cell | Proliferation↑ | ||
| IGF-2 | Fibroblast | Collagen↑ | [38,49] |
| ASM | Proliferation↑ | ||
| TGF-β1 | ASM | Proliferation, hypertrophy↑ | [21,38,43,44,48] |
| IL-11, IL-6, LIF secretion↑ | |||
| β-Adrenergic receptor expression↓ | |||
| Monocyte | Recruitment, NO production↓ | ||
| Neutrophil | Recruitment | ||
| Fibroblast | Recruitment, proliferation, collagen↑, fibronectin↑ | ||
| T cells | Recruitment | ||
| Endothelial cell | E-Selectin↓ | ||
| Eosinophil | Survival↓, degranulation↓ | ||
| VEGF | Endothelial cell | Proliferation↑, migration, tube formation | [50] |
↑, Increase; ↓, decrease; ECM, extracellular matrix; FGF-2, fibroblast growth factor-2; IGF-2, insulin-like growth factor-2; IL, interleukin; LIF, leukaemia inhibitory factor; NO, nitric oxide; PDGF, platelet-derived growth factor; TGF-β, transforming growth factor-β ; VEGF, vascular endothelial growth factor.
TGF-β1 can be produced in the lung by a variety of cells, including macrophages, platelets, eosinophils, mast cells, activated T lymphocytes and epithelial cells, and it is detected in exaggerated quantities in asthmatic BAL fluid before and after antigen challenge [6]. It is an extremely potent stimulus for the synthesis of ECM components such as collagen and fibronectin leading to tissue fibrosis and it can also inhibit ASM NO production, potentially resulting in a loss of control of airway calibre. The modulation of smooth muscle cell β-adrenergic receptor number and function by TGF-β1 can attenuate the effects of endogenous catecholamines or therapeutically applied β-adrenergic agonists.
In some studies, TGF-β1 expression correlates with basement membrane thickness and fibroblast number and/or disease severity. Black et al. showed that ASM cells secreted latent TGF-β1 into the culture medium [42]. Results from our laboratory demonstrated that human bronchial ASM cells express and secrete significant amounts of TGF-β1 in response to the potent vasoconstrictor angiotensin II [43]. The production of TGF-β1 coincided with ASM cellular hypertrophy, suggesting an autocrine effect of TGF-β1 on ASM cell phenotype. Work by Cohen et al. demonstrates that TGF-β1 can also modulate epidermal growth factor-induced DNA biosynthesis in human tracheal ASM cells [44]. Several studies show, however, that exogenous TGF-β1 can also stimulate bovine ASM cell mitogenesis [45].
Activated macrophages, eosinophils, epithelial cells, fibroblasts and smooth muscle cells produce PDGF. De et al. reported that ASM cells in culture could also express PDGF following IL-1β stimulation [46]. PDGF is a highly potent mitogen for ASM cells and fibroblasts [47,48], and has been shown to act as a chemoattractant for fibroblasts as well as a stimulator for collagenase production [38]. Vignola et al., however, showed that PDGF-AA, PDGF-BB and PDGF-AB levels in BAL between controls and asthmatics were not significantly different [48], inferring that PDGF might not play an important role in the remodelling of asthmatic airways, although it may be involved in fibrotic diseases of the lung.
Insulin-like growth factor (IGF)-2 and IGF binding protein-2 have also been detected in the conditioned medium of confluent ASM cell cultures [49]. IGF is a mitogen for these cells, and IGF binding protein-2 modulates the bioavailability of IGF by binding to it and thereby decreasing its mitogenic potency. IGF expression is not increased in the airways of asthmatics. In our laboratory, we more recently found that bronchial ASM cells are capable of expressing and releasing vascular endothelial growth factor, an angiogenic peptide, following stimulation with TNF-α, angiotensin II or endothelin-1 (unpublished data). These results suggest that ASM cells may be involved in the regulation of vascular remodelling in the airway wall during inflammation [50]. Furthermore, these growth factors may form a link between airway inflammation and airway and vascular remodelling. Subsequent to hyperplasia and/or hypertrophy, ASM cells remaining in the synthetic/secretory phenotype may further undergo increased production of inflammatory mediators.
COX products in the airway
COX is the enzyme that converts arachidonic acid to prostaglandins (PGs), prostacyclin (PGI2) and thromboxane (TX) A2. COX-1 is the constitutively expressed isoform involved in the production of PGs under physiological conditions. The inducible isoform, COX-2, is expressed in response to pro-inflammatory stimuli, suggesting it may play a role in the pathophysiology of asthma. Several studies have shown that ASM cells can express COX-2 and release PGE2, and to a lesser extent PGI2, and also the pro-inflammatory TXB2, PGF2α and PGD2 in response to pro-inflammatory cytokines [51,52]. The relative contribution of the individual COX products on airway inflammation depends ultimately on the presence of their respective receptors on target tissues (Table 5).
Table 5.
Airway smooth muscle (ASM) cell-derived arachidonic acid metabolites, their target cells and effects
| Cyclooxygenase product | Target | Effect | Reference |
| PGD | ASM | Contraction | [9,87,88] |
| Goblet cell | Mucous secretion↑ | ||
| PGE | ASM | Contraction, proliferation↓ GM-CSF secretion↓ | [9,52,56] |
| Goblet cell | Mucous secretion↑ | ||
| Mast cell | Secretion↓ | ||
| Eosinophil | Recruitment and survival↓ | ||
| T cell | IL-2 secretion↓ | ||
| PGF | ASM | Contraction | [9,87] |
| Goblet cell | Mucous secretion↑ | ||
| Thromboxane | ASM | Contraction and proliferation↑ | [57,89] |
↑, Increase; ↓, decrease; GM-CSF, granulocyte/macrophage-colony stimulating factor; IL, interleukin; PG = prostaglandin.
PGE2 production by ASM cells can be upregulated in the presence of pro-inflammatory mediators such as bradykinin, IL-1β and TNF-α [51,52], but also by β-adrenoceptor agonists, IFN-γ and agents that elevate cAMP levels [53]. Important anti-inflammatory effects of ASM cell-derived PGE2 include inhibition of mast cell mediator release, eosinophil chemotaxis and survival, IL-2 and IgE production by lymphocytes [54], inhibition of ASM cell mitogenesis [55,56] and inhibition of GM-CSF release by ASM cells [52]. These studies support the notion that a negative feedback mechanism exists to limit the inflammatory response. However, PGs also have the ability to induce bronchoconstriction, to increase mucous secretion from bronchial wall explants and to enhance airway responsiveness. TXB2, a pro-inflammatory and bronchoconstricting mediator, can also be expressed by ASM cells [51]. It has been demonstrated to have mitogenic activity on ASM cells and it can trigger cysteinyl-leukotriene synthesis [57]. TX has also been implicated in airway hyper-responsiveness [58].
Cytokine-induced COX-2 activity can be inhibited by the anti-inflammatory steroid dexamethasone, and by non-steroidal anti-inflammatory drugs [51]. The potential therapeutic effects are complicated, however, because the consequences of COX-2 induction and PG production may be beneficial or deleterious. PGE2 is an important anti-inflammatory mediator and, at low concentrations, it acts as a bronchodilator, whereas higher concentrations can lead to bronchoconstriction via the TX receptor.
Lipoxygenase products
5-lipoxygenase (5-LO) is the enzyme that converts arachidonic acid to leukotriene (LT) A4, which is quickly converted to LTC4, LTB4, LTD4 and LTE4. The cysteinyl LTC4, LTD4 and LTE4 are known to mediate bronchoconstriction. Expression of mRNA for enzymes of the 5-LO pathway (5-LO, epoxide hydrolase, LTC4 synthase and γ-glutamyl transpeptidase) by ASM has also been reported following exposure of ASM cells to atopic serum or IL-1β [59]. Products of 5-LO can cause tissue oedema and migration of eosinophils, and it can stimulate airway secretions. LTD4 can also stimulate ASM cell proliferation [60]. Whether, under pathological conditions, ASM cells generate relevant amounts of LTs remains to be shown.
T-cell interactions with ASM cells
It is well known that adhesion of lymphocytes to endothelial cells, mediated by adhesion molecules and integrins, is necessary for their migration from the blood circulation to areas of tissue injury [6]. The subsequent interactions of the T lymphocytes with ASM cells in the bronchial mucosa have also been investigated [61,62]. These studies showed that ASM cells constitutively express high levels of CD44, the principal cell surface receptor for hyaluronate, and express intercellular adhesion molecule-1 and vascular cell-adhesion molecule-1 when stimulated with TNF-α.
Interactions between ASM cells and activated T lymphocytes, possibly via specific adhesion molecules, have been shown to stimulate ASM cell DNA synthesis and the subsequent ASM cell hyperplasia involved in airway wall remodelling in inflamed airways. Adherence of anti-CD3-stimulated peripheral blood T cells to ASM cells markedly upregulated intercellular adhesion molecule-1 expression as well as the expression of MHC class II antigens. IFN-γ stimulation also induced the expression of MHC class II antigens by ASM cells. These studies suggest that ASM cells may also act as antigen-presenting cells for pre-activated T lymphocytes in the asthmatic airways. However, the ASM cells were unable to support the proliferation of resting CD4+ T cells by presenting alloantigen [61,62].
Therapeutic intervention for airway inflammation
A large number of cells in the airways, such as eosinophils, mast cells, lymphocytes, neutrophils and ASM cells, contribute to the pathogenesis of inflammatory airway diseases. Here we specifically discuss potential anti-inflammatory interventions that target ASM-driven inflammation.
As mentioned earlier, ASM cells are potential targets for glucocorticosteroid therapy. Other workers and ourselves have recently demonstrated that the expression and secretion of pro-inflammatory cytokines and chemokines by ASM cells in vitro can be inhibited by glucocorticosteroid treatment [11,12,17,25,26,32,36,40]. COX-2 induction and the resulting production of arachidonic acid metabo-lites could similarly be inhibited by treatment with dexa-methasone [13,51]. Suppression of the COX-2 pathway, on the contrary, may result in deleterious consequences considering the bronchoprotective properties of PGE2 in asthmatic airways.
Mechanistically, it has been proposed that glucocorticoid receptors interact with transcription factors such as activator protein-1 and nuclear factor-κB, that are activated by inflammatory signals. Protein-protein complexes thus formed prevent DNA binding and subsequent transcription of pro-inflammatory cytokines that amplify inflammation, chemokines involved in recruitment of eosinophils, inflammatory enzymes that synthesise mediators and adhesion molecules involved in the trafficking of inflammatory cells to sites of inflammation. Corticosteroids may also control airway inflammation by increasing the transcription of anti-inflammatory genes like IL-10, IL-12 or IL-1 receptor antagonist, the gene products of which appear to be the most potent anti-inflammatory drugs for use in the treatment of airway inflammation [63].
Stewart et al. have demonstrated that pretreatment with dexamethasone, methylprednisolone and hydrocortisone can inhibit serum-induced, FGF-2-induced and thrombin-induced human ASM cell proliferation [40]. Beclomethasone and cortisol were also found to inhibit bovine ASM cell proliferation in culture [64]. In view of the complexity of the mechanisms involved in airway inflammation, however, a treatment for the inhibition or reversal of airway wall remodelling has yet to be fully validated [65]. In addition, inhibition of metalloproteinases and growth factors during glucocorticosteroid therapy may eventually lead to the persistence of chronic inflammation by preventing proper wound healing, and thus may indirectly enhance airway remodelling.
Novel therapies aimed at reducing the effects of (ASM-derived) chemokines and IL-5 include chemokine receptor antagonists and IL-5 antagonists [23,66]. A methionine extension on the amino terminus of RANTES and the modified form of macrophage inflammatory protein-4, met-chemokine β7, have been shown to interfere with CCR1 and CCR3 chemokine receptors, thereby inhibiting eosinophil chemotaxis in animal models of airway inflammation and allergy. However, a multi-mechanistic approach would probably be advantageous for the treatment of airway inflammation due to the large number of chemokines and the promiscuous binding pattern for multiple receptors (redundancy), making it unlikely that a single chemokine or chemokine receptor approach would be beneficial [23].
Similar to the use of chemokine receptor antagonists, the IL-5 antagonist approach should result in decreased eosinophilia in inflamed airways. Anti-IL-5 antibodies were effective in animal models to abrogate eosinophilia, suggesting that the IL-5 receptor is a potential drug target [66]. A 19-amino acid peptide that binds to the IL-5 receptor alpha/beta heterodimer complex, with an affinity equal to that of IL-5, has been shown to be a potent and specific antagonist of IL-5 activity in a human eosinophil adhesion assay [67]. However, this IL-5 antagonist has not yet been tested in vivo.
Our increasing knowledge of the intercellular communication between structural and infiltrating inflammatory cells in the airways and in view of the role of various cytokines, chemokines and other inflammatory mediators, provides an insight into the complex inflammatory processes and that may subsequently help us to identify novel therapeutic targets.
Consequences for ongoing airway inflammation
Allergic airway inflammation develops following the uptake and processing of inhaled allergens by antigen-presenting cells such as dendritic cells and macrophages. The subsequent interactions between these cells, T lymphocytes and resident cells leads to a cascade of events contributing to chronic inflammation, bronchospasm, mucus secretion, oedema and airway remodelling.
The contemporary viewpoint is that the pro-asthmatic state reflects an imbalance between Th1-type and Th2-type cytokine production and action, with an upregulated Th2 cytokine response and a downregulated Th1 cytokine response [68]. It is postulated that the release of pre-formed cytokines by mast cells is the initial trigger for the early infiltration of inflammatory cells (including T cells) into the airways, and that their subsequent activation and the release of pro-inflammatory mediators induces airway hyper-reactivity and recruitment of further inflammatory cells. The recent data showing that ASM cells exposed to an inflammatory environment can express and secrete chemokines, Th1-type and Th2-type cytokines and growth factors thus provide evidence that these structural cells could regulate airway inflammation by influencing the local environment within the airway wall.
Prolonged survival of infiltrating inflammatory cells is thought to be a result of delayed apoptosis (programmed cell death), a mechanism that would normally limit tissue injury during inflammation and promote resolution rather than progression of inflammation. Both GM-CSF and IL-5 can reduce eosinophil apoptosis, resulting in persistence of the inflammatory infiltrate and even more tissue damage. The numerous chemokines secreted by ASM cells amplify this effect by recruiting more inflammatory cells to the airway wall (Fig. 1).
Figure 1.
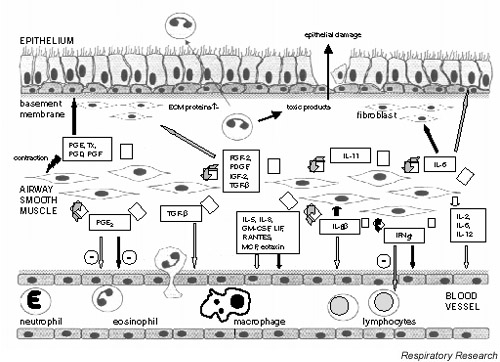
Schematic diagram depicting the role of airway smooth muscle (ASM) cell-derived mediators in airway inflammation. The release of cytokines, chemokines or growth factors (open arrows) can result in ASM as well as inflammatory cell proliferation (dark grey arrows), or the recruitment (light grey arrows) or activation (black arrows) of various cells in the airways. ECM, Extracellular membrane; FGF-2, fibroblast growth factor-2; GM-CSF, granulocyte/macrophage-colony stimulating factor; IGF-2, insulin-like growth factor-2; IFN, interferon; IL, interleukin; LIF, leukaemia inhibitory factor; MCP, monocyte chemotactic protein; PDGF, platelet-derived growth factor; PG, prostaglandin; RANTES, regulated on activation, normal T cell expressed and secreted; TGF-β, transforming growth factor-β; TX, thromboxane.
TGF-β1, however, can inhibit eosinophil survival and degranulation, and may therefore play a role in the resolution of inflammation by stimulating the development of post-inflammatory repair processes in the airways [69]. The pro-inflammatory cytokine-induced release of PGE2 by ASM cells may also limit the inflammatory response through a mechanism whereby the secretion of GM-CSF and possibly other cytokines is inhibited [52]. It should be noted that mast cell products may have anti-inflammatory properties. Heparin and heparan sulphate can modulate cell differentiation, ASM cell proliferation and inflammation [56,65].
Consequences for airway remodelling
The continuous process of healing and repair, due to chronic airway inflammation, can lead to airway wall remodelling [4,70,71]. The release of cytokines, chemokines and growth factors by ASM cells exposed to an inflammatory environment can result in ASM cell and goblet cell hyperplasia and/or hypertrophy [21,38,39,43, 44,47,48,71,72]. Proliferation of fibroblasts and the subsequent deposition of ECM components also contribute to airway wall thickening [71]. Components of the ECM can form a reservoir for cytokines and growth factors. Upregulation of tissue inhibitor of metalloproteinase expression can inhibit the degradation of ECM components, resulting in an amplification of this effect. The increase in airway wall thickness can increase bronchial hyper-responsiveness and profoundly affect airway narrowing, with a subsequent increase in resistance to airflow caused by smooth muscle shortening [3].
Consequences for ASM physiology
The recent study by Hakonarson et al. [7] showed that cytokine exposure can influence ASM contractile function. They demonstrated that passive sensitisation of ASM strips augments constrictor responses and reduces relaxation responsiveness. These effects are ablated by exposure to the Th1-type cytokines, IL-2 and IFN-γ. Furthermore, exposure of naïve ASM strips to IL-5 and GM-CSF (Th2-type cytokines) increased muscarinic responsiveness and impaired ASM relaxation to the β-agonist isoproterenol. This study suggests that ASM contractile function can be modulated in an autocrine fashion during an inflammatory episode.
Work from Stephens et al. shows that ragweed pollen-sensitised canine bronchial smooth muscle displayed altered contractile phenotypes when compared with controls, with increased muscle shortening at maximum velocity. ASM cells obtained from asthmatic patients also showed increased shortening compared with controls [73]. Such increased muscle shortening could be attributed to enhanced activity of actin-activated myosin Mg2+-ATPase.
Increased quantity and activity of myosin light chain kinase, thereby increasing the actomyosin cycling rate, was also reported [74]. TGF-β exposure has been reported to activate myosin light chain kinase, indicating that stimuli present in inflamed airways may be able to influence actin-myosin cycling [75]. Hautmann et al. showed that TGF-β exposure also increases smooth muscle α-actin, smooth muscle myosin heavy chain and h1-calponin mRNA in ASM cells in vitro [76].
A recent review by Solway discusses in more detail the mechanisms whereby a variety of inflammatory stimuli, such as TNF-α, lysophosphatidic acid and smooth muscle mitogens, could enhance the actomyosin cycling rate in ASM cells, thereby influencing ASM physiology in chronically inflamed asthmatic airways [77].
Concluding remarks
Chronic persistent asthma is still a major health problem causing morbidity and mortality despite the widespread use of anti-inflammatory drugs, suggesting a clear need to develop new therapeutic strategies; in particular, to tackle structural and functional changes in the airway wall. The expression and secretion of cytokines, chemokines and growth factors by ASM cells in vitro support the notion that ASM cells are actively involved in the inflammatory response in the airway. These biologically active multipo-tent mediators can act to alter ASM contractility and proliferative responses as well as exaggerating or dampening the inflammatory response by amplifying signals generated by infiltrating inflammatory cells. The interaction between structural ASM cells and cells recruited from the circulation should be further explored to understand this complex cellular cross-talk and to further define the contributions of the cellular and molecular events involved in the pathogenesis of airway diseases.
Whether the endogenous generation of inflammatory mediators by ASM cells enhances ASM hyper-responsiveness, alters ASM function or supports airway remodelling in vivo remains to be elucidated. Although the emerging data from various laboratories implicates a regulatory role for the ASM in airway inflammatory responses, there is a clear need to explore the synthetic properties of these cells. Determination of secretory profiles of chemokines, cytokines and growth factors by ASM cells would give us an insight into the complex mechanisms contributing to chronic airway inflammation and eventually help us identify potential targets for future drug therapy.
Abbreviations
ASM = airway smooth muscle; BAL = bronchoalveolar lavage; COX = cyclooxygenase; ECM = extracellular matrix; FGF = fibroblast growth factor; GM-CSF = granulocyte/macrophage-colony stimulating factor; IFN = interferon; IGF = insulin-like growth factor; IL = interleukin; LIF = leukaemia inhibitory factor; 5-LO = 5-lipoxygenase; LT = leukotriene; MCP = monocyte chemotactic protein; MHC = major histocompatibility complex; NO = nitric oxide; PDGF = platelet-derived growth factor; PG = prostaglandin; RANTES = regulated on activation, normal T cell expressed and secreted; TGF = transforming growth factor; Th = T helper cells; TNF-α = tumour necrosis factor-α; TX = thromboxane.
Acknowledgments
Acknowledgements
The authors acknowledge the financial support from the Netherlands Asthma Foundation (grant number NAF 95.46). They thank Dr Johan de Jongste for critically reading the manuscript.
References
- Djukanovic R, Roche WR, Wilson JW, Beasley CR, Twentyman OP, Howarth RH, Holgate ST. Mucosal inflammation in asthma. Am Rev Respir Dis. 1990;142:434–457. doi: 10.1164/ajrccm/142.2.434. [DOI] [PubMed] [Google Scholar]
- Heard BE, Hossain S. Hyperplasia of bronchial muscle in asthma. J Pathol. 1973;110:310–331. doi: 10.1002/path.1711010212. [DOI] [PubMed] [Google Scholar]
- James AL. Relationship between airway wall thickness and airway hyperresponsiveness. In Airway Wall Remodelling in Asthma Edited by Stewart AG London: CRC; 1997. pp. 29–63.
- Vignola AM, Chanez P, Bonsignore G, Godard P, Bousquet J. Structural consequences of airway inflammation in asthma. J Allergy Clin Immunol. 2000;105:S514–S517. doi: 10.1067/mai.2000.103049. [DOI] [PubMed] [Google Scholar]
- Elias JA, Zhu Z, Chupp G, Homer RJ. Airway remodeling in asthma. J Clin Invest. 1999;104:1001–1006. doi: 10.1172/JCI8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradding P, Redington AE, Holgate ST. Airway wall remodelling in the pathogenesis of asthma: Cytokine expression in the airways. In Airway Wall Remodelling in Asthma Edited by Stewart AG London: CRC; 1997. pp. 29–63.
- Hakonarson H, Maskeri N, Carter C, Grunstein MM. Regulation of TH1- and TH2-type cytokine expression and action in atopic asthmatic sensitized airway smooth muscle. J Clin Invest. 1999;103:1077–1087. doi: 10.1172/JCI5809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Durham SR, Corrigan CJ, Hamid Q, Kay AB. Phenotype of cells expressing mRNA for TH2-type (interleukin 4 and interleukin 5) and TH1-type (interleukin 2 and interferon gamma) cytokines in bronchoalveolar lavage and bronchial biopsies from atopic asthmatic and normal control subjects. Am J Respir Cell Mol Biol. 1995;12:477–487. doi: 10.1165/ajrcmb.12.5.7742012. [DOI] [PubMed] [Google Scholar]
- Wilson JW, Li X. Inflammation and cytokines in airway wall remodelling. In Airway Wall Remodelling in Asthma Edited by Stewart AG London: CRC; 1997. pp. 65–109.
- Broide DH, Lotz M, Cuomo AJ, Coburn DA, Federman EC, Wasserman SI. Cytokines in symptomatic asthma airways. J Allergy Clin Immunol. 1992;89:958–967. doi: 10.1016/0091-6749(92)90218-q. [DOI] [PubMed] [Google Scholar]
- Hallsworth M. Cultured human airway smooth muscle cells stimulated by IL-1beta enhance eosinophil survival. Am J Respir Cell Mol Biol. 1998;19:910–919. doi: 10.1165/ajrcmb.19.6.3275. [DOI] [PubMed] [Google Scholar]
- McKay S, Hirst SJ, Haas MB, de Jongste JC, Hoogsteden HC, Saxena PR, Sharma HS. Tumor necrosis factor-alpha enhances mRNA expression and secretion of interleukin-6 in cultured human airway smooth muscle cells. Am J Respir Cell Mol Biol. 2000;23:103–111. doi: 10.1165/ajrcmb.23.1.3765. [DOI] [PubMed] [Google Scholar]
- Saunders MA, Mitchell JA, Seldon PM, Yacoub MH, Barnes PJ, Giembycz MA, Belvisi MG. Release of granulocyte-macrophage colony stimulating factor by human cultured airway smooth muscle cells: suppression by dexamethasone. Br J Pharmacol. 1997;120:545–546. doi: 10.1038/sj.bjp.0700998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammit AJ, Hoffman RK, Amrani Y, Lazaar AL, Hay DW, Torphy TJ, Penn RB, Panettieri RA. Tumor necrosis factor-alpha-induced secretion of RANTES and interleukin-6 from human airway smooth-muscle cells. Modulation by cyclic adenosine monophosphate. Am J Respir Cell Mol Biol. 2000;23:794–802. doi: 10.1165/ajrcmb.23.6.4184. [DOI] [PubMed] [Google Scholar]
- Herrick DJ, Hakonarson H, Grunstein MM. Induced expression of the IL-6 and IL-8 cytokines in human bronchial smooth muscle cells exposed to asthmatic serum or cytokines [abstract]. Am J Respir Crit Care Med. 1996;153:A164. [Google Scholar]
- McKay S, De Jongste JC, Hoogsteden HC, Saxena PR, Sharma HS. Interleukin-1b induced expression of transcription factors precedes upregulation of interleukin-6 in cultured human airway smooth muscle cells [abstract]. Eur Respir J. 1999;12:57s. [Google Scholar]
- Hirst SJ, Lee TH. Airway smooth muscle as a target of glucocorticoid action in the treatment of asthma. Am J Resp Crit Care Med. 1998;158:S201–S206. doi: 10.1164/ajrccm.158.supplement_2.13tac190. [DOI] [PubMed] [Google Scholar]
- Teran LM, Carroll MP, Shute JK, Holgate ST. Interleukin 5 release into asthmatic airways 4 and 24 hours after endobronchial allergen challenge: its relationship with eosinophil recruitment. Cytokine. 1999;11:518–522. doi: 10.1006/cyto.1998.0457. [DOI] [PubMed] [Google Scholar]
- Hakonarson H, Herrick DJ, Serrano PG, Grunstein MM. Mechanism of cytokine-induced modulation of beta-adrenoceptor responsiveness in airway smooth muscle. J Clin Invest. 1996;97:2593–2600. doi: 10.1172/JCI118708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakonarson H, Herrick DJ, Serrano PG, Grunstein MM. Autocrine role of interleukin-1 beta in altered responsiveness of atopic asthmatic sensitized airway smooth muscle. J Clin Invest. 1997;99:117–124. doi: 10.1172/JCI119122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JA, Wu Y, Zheng T, Panettieri R. Cytokine- and virus-stimulated airway smooth muscle cells produce IL-11 and other IL-6-type cytokines. Am J Physiol. 1997;273:L648–L655. doi: 10.1152/ajplung.1997.273.3.L648. [DOI] [PubMed] [Google Scholar]
- Johnston SL, Pattemore PK, Sanderson G, Smith S, Lampe F, Josephs L, Symington O, O'Toole S, Myint SH, Tyrrell DA, Holgate ST. A community study of the role of virus infections in exacerbations of asthma in 9–11 year old children. Br Med J. 1995;310:1225–1228. doi: 10.1136/bmj.310.6989.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangur V, Oppenheim JJ. Are chemokines essential or secondary participants in allergic responses? Ann Allergy Asthma Immunol. 2000;84:569–579. doi: 10.1016/S1081-1206(10)62403-9. [DOI] [PubMed] [Google Scholar]
- Teran LM. CCL chemokines and asthma. Immunol Today. 2000;21:235–242. doi: 10.1016/S0167-5699(00)01634-0. [DOI] [PubMed] [Google Scholar]
- John M, Hirst SJ, Jose PJ, Robichaud A, Berkman N, Witt C, Twort CH, Barnes PJ, Chung KF. Human airway smooth muscle cells express and release RANTES in response to T helper 1 cytokines: regulation by T helper 2 cytokines and corticosteroids. J Immunol. 1997;158:1841–1847. [PubMed] [Google Scholar]
- Pype JL, Dupont LJ, Menten P, Van Coillie E, Opdenakker G, Van Damme J, Chung KF, Demedts MG, Verleden GM. Expression of monocyte chemotactic protein (MCP)-1, MCP-2, and MCP-3 by human airway smooth-muscle cells. Modulation by corticosteroids and T-helper 2 cytokines. Am J Respir Cell Mol Biol. 1999;21:528–536. doi: 10.1165/ajrcmb.21.4.3660. [DOI] [PubMed] [Google Scholar]
- Sousa AR, Lane SJ, Nakhosteen JA, Yoshimura T, Lee TH, Poston RN. Increased expression of the monocyte chemoattractant protein-1 in bronchial tissue from asthmatic subjects. Am J Respir Cell Mol Biol. 1994;10:142–147. doi: 10.1165/ajrcmb.10.2.8110469. [DOI] [PubMed] [Google Scholar]
- Powell N, Humbert M, Durham SR, Assoufi B, Kay AB, Corrigan CJ. Increased expression of mRNA encoding RANTES and MCP-3 in the bronchial mucosa in atopic asthma. Eur Respir J. 1996;9:2454–2460. doi: 10.1183/09031936.96.09122454. [DOI] [PubMed] [Google Scholar]
- Ying S, Meng Q, Zeibecoglou K, Robinson DS, Macfarlane A, Humbert M, Kay AB. Eosinophil chemotactic chemokines (eotaxin, eotaxin-2, RANTES, monocyte chemoattractant protein-3 (MCP-3), and MCP-4), and C-C chemokine receptor 3 expression in bronchial biopsies from atopic and nonatopic (Intrinsic) asthmatics. J Immunol. 1999;163:6321–6329. [PubMed] [Google Scholar]
- Alam R, York J, Boyars M, Stafford S, Grant JA, Lee J, Forsythe P, Sim T, Ida N. Increased MCP-1, RANTES and MIP-1alpha in bronchoalveolar lavage fluid of allergic asthmatic patients. Am J Respir Crit Care Med. 1996;153:1398–1404. doi: 10.1164/ajrccm.153.4.8616572. [DOI] [PubMed] [Google Scholar]
- Watson ML, Grix SP, Jordan NJ, Place GA, Dodd S, Leithead J, Poll CT, Yoshimura T, Westwick J. Interleukin 8 and monocyte chemoattractant protein 1 production by cultured human airway smooth muscle cells. Cytokine. 1998;10:346–352. doi: 10.1006/cyto.1997.0350. [DOI] [PubMed] [Google Scholar]
- Chung KF, Patel HJ, Fadlon EJ, Rousell J, Haddad EB, Jose PJ, Mitchell J, Belvisi M. Induction of eotaxin expression and release from human airway smooth muscle cells by IL-1beta and TNFalpha: effects of IL-10 and corticosteroids. Br J Pharmacol. 1999;127:1145–1150. doi: 10.1038/sj.bjp.0702660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaffar O, Hamid Q, Renzi PM, Allakhverdi Z, Molet S, Hogg JC, Shore SA, Luster AD, Lamkhioued B. Constitutive and cytokine-stimulated expression of eotaxin by human airway smooth muscle cells. Am J Respir Crit Care Med. 1999;159:1933–1942. doi: 10.1164/ajrccm.159.6.9805039. [DOI] [PubMed] [Google Scholar]
- Sampson AP. The role of eosinophils and neutrophils in inflammation. Clin Exp Allergy. 2000;30(suppl 1):22–27. doi: 10.1046/j.1365-2222.2000.00092.x. [DOI] [PubMed] [Google Scholar]
- Folkard SG, Westwick J, Millar AB. Production of interleukin-8, RANTES and MCP-1 in intrinsic and extrinsic asthmatics. Eur Respir J. 1997;10:2097–2104. doi: 10.1183/09031936.97.10092097. [DOI] [PubMed] [Google Scholar]
- John M, Au BT, Jose PJ, Lim S, Saunders M, Barnes PJ, Mitchell JA, Belvisi MG, Chung KF. Expression and release of interleukin-8 by human airway smooth muscle cells: inhibition by Th-2 cytokines and corticosteroids. Am J Respir Cell Mol Biol. 1998;18:84–90. doi: 10.1165/ajrcmb.18.1.2813. [DOI] [PubMed] [Google Scholar]
- Pang L, Knox AJ. Bradykinin stimulates IL-8 production in cultured human airway smooth muscle cells: role of cyclooxygenase products. J Immunol. 1998;161:2509–2515. [PubMed] [Google Scholar]
- Li X, Wilson JW. Fibrogenic cytokines in airway fibrosis. Airway Wall Remodelling in Asthma Edited by Stewart AG London: CRC; 1997. pp. 111–138.
- Hawker KM, Johnson PR, Hughes JM, Black JL. Interleukin-4 inhibits mitogen-induced proliferation of human airway smooth muscle cells in culture. Am J Physiol. 1998;275:L469–L477. doi: 10.1152/ajplung.1998.275.3.L469. [DOI] [PubMed] [Google Scholar]
- Stewart AG, Fernandes D, Tomlinson PR. The effect of glucocorticoids on proliferation of human cultured airway smooth muscle. Br J Pharmacol. 1995;116:3219–3226. doi: 10.1111/j.1476-5381.1995.tb15127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rödel J, Woytas M, Groh A, Schmidt KH, Hartmann M, Lehmann M, Straube E. Production of basic fibroblast growth factor and interleukin 6 by human smooth muscle cells following infection with Chlamydia pneumoniae. Infect Immun. 2000;68:3635–3641. doi: 10.1128/IAI.68.6.3635-3641.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black PN, Young PG, Skinner SJ. Response of airway smooth muscle cells to TGF-beta 1: effects on growth and synthesis of glycosaminoglycans. Am J Physiol. 1996;271:L910–L917. doi: 10.1152/ajplung.1996.271.6.L910. [DOI] [PubMed] [Google Scholar]
- McKay S, de Jongste JC, Saxena PR, Sharma HS. Angiotensin II induces hypertrophy of human airway smooth muscle cells: expression of transcription factors and transforming growth factor-beta1. Am J Respir Cell Mol Biol. 1998;18:823–833. doi: 10.1165/ajrcmb.18.6.2924. [DOI] [PubMed] [Google Scholar]
- Cohen MD, Ciocca V, Panettieri RA. TGF-beta 1 modulates human airway smooth-muscle cell proliferation induced by mitogens. Am J Respir Cell Mol Biol. 1997;16:85–90. doi: 10.1165/ajrcmb.16.1.8998083. [DOI] [PubMed] [Google Scholar]
- Kilfeather SA, Okona-Mensah KB, Tagoe S, Costello J, Page C. Stimulation of airway smooth muscle cell mitogenesis by transforming growth factor beta and inhibition by heparin and smooth muscle explant-conditioned media [abstract]. Eur Respir J. 1995;8:547s. [Google Scholar]
- De S, Zelazny ET, Souhrada JF, Souhrada M. IL-1 beta and IL-6 induce hyperplasia and hypertrophy of cultured guinea pig airway smooth muscle cells. J Appl Physiol. 1995;78:1555–1563. doi: 10.1152/jappl.1995.78.4.1555. [DOI] [PubMed] [Google Scholar]
- Hirst SJ, Barnes PJ, Twort CHC. PDGF isoform-induced proliferation and receptor expression in human cultured airway smooth muscle cells. Am J Physiol. 1996;270:L415–L428. doi: 10.1152/ajplung.1996.270.3.L415. [DOI] [PubMed] [Google Scholar]
- Vignola AM, Chiappara G, Chanez P, Merendino AM, Pace E, Spatafora M, Bousquet J, Bonsignore G. Growth factors in asthma. Monaldi Arch Chest Dis. 1997;52:159–169. [PubMed] [Google Scholar]
- Noveral JP, Bhala A, Hintz RL, Grunstein MM, Cohen P. Insulin like growth factor axis in airway smooth muscle cells. Am J Physiol. 1994;267:L761–L765. doi: 10.1152/ajplung.1994.267.6.L761. [DOI] [PubMed] [Google Scholar]
- Hoshino M, Takahashi M, Aoike N. Expression of vascular endothelial growth factor, basic fibroblast growth factor, and angiogenin immunoreactivity in asthmatic airways and its relationship to angiogenesis. J Allergy Clin Immunol. 2001;107:295–301. doi: 10.1067/mai.2001.111928. [DOI] [PubMed] [Google Scholar]
- Pang L, Knox AJ. Effect of interleukin-1 beta, tumour necrosis factor-alpha and interferon-gamma on the induction of cyclo-oxygenase-2 in cultured human airway smooth muscle cells. Br J Pharmacol. 1997;121:579–587. doi: 10.1038/sj.bjp.0701152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzeri N, Belvisi MG, Patel HJ, Yacoub MH, Chung KF, Mitchell JA. Effects of prostaglandin E2 and cAMP elevating drugs on GM-CSF release by cultured human airway smooth muscle cells: relevance to asthma therapy. Am J Respir Mol Cell Biol. 2001;24:44–48. doi: 10.1165/ajrcmb.24.1.4027. [DOI] [PubMed] [Google Scholar]
- Barry T, Delamere F, Holland E, Pavord I, Knox A. Production of PGE2 by bovine cultured airway smooth muscle cells: regulation by cAMP. J Appl Physiol. 1995;78:623–628. doi: 10.1152/jappl.1995.78.2.623. [DOI] [PubMed] [Google Scholar]
- Johnson SR, Knox AJ. Synthetic functions of airway smooth muscle in asthma. Trends Pharmacol Sci. 1997;18:288–292. doi: 10.1016/S0165-6147(97)01092-4. [DOI] [PubMed] [Google Scholar]
- Belvisi MG, Saunders M, Yacoub M, Mitchell JA. Expression of cyclo-oxygenase-2 in human airway smooth muscle is associated with profound reductions in cell growth. Br J Pharmacol. 1998;125:1102–1108. doi: 10.1038/sj.bjp.0702104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PRA, Armour CL, Carey D, Black JL. Heparin and PGE-2 inhibit DNA synthesis in human airway smooth muscle cells in culture. Am J Physiol. 1995;269:514–519. doi: 10.1152/ajplung.1995.269.4.L514. [DOI] [PubMed] [Google Scholar]
- Noveral JP, Grunstein MM. Role and mechanism of thromboxane-induced proliferation of cultured airway smooth muscle cells. Am J Physiol. 1992;263:L555–L561. doi: 10.1152/ajplung.1992.263.5.L555. [DOI] [PubMed] [Google Scholar]
- Jongejan RC, de Jongste JC, Raatgeep HC, Stijnen T, Bonta IL, Kerrebijn KF. Effects of inflammatory mediators on the responsiveness of isolated human airways to methacholine. Am Rev Respir Dis. 1990;142:1129–1132. doi: 10.1164/ajrccm/142.5.1129. [DOI] [PubMed] [Google Scholar]
- Hakonarson H, Gandhi S, Strusberg L, Carter S, Grunstein MM. Altered 5-lipoxygenase, epoxide hydrolase, LTC4 synthase, gamma-glutamyl transpeptidase, and leukotriene B4 and cys-LT1 receptor expression in atopic asthmatic sensitised airway smooth muscle [abstract]. Am J Respir Crit Care Med. 1999;159:A401. [Google Scholar]
- Cohen P, Noveral JP, Bhala A, Nunn SE, Herrick DJ, Grunstein MM. Leukotriene D4 facilitates airway smooth muscle cell proliferation via modulation of the IGF axis. Am J Physiol. 1995;269:L151–L157. doi: 10.1152/ajplung.1995.269.2.L151. [DOI] [PubMed] [Google Scholar]
- Lazaar AL, Albelda SM, Pilewski JM, Brennan B, Pure E, Panettieri RA., Jr T lymphocytes adhere to airway smooth muscle cells via integrins and CD44 and induce smooth muscle cell DNA synthesis. J Exp Med. 1994;180:807–816. doi: 10.1084/jem.180.3.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaar AL, Reitz HE, Panettieri RA, Jr, Peters SP, Pure E. Antigen receptor-stimulated peripheral blood and bronchoalveolar lavage-derived T cells induce MHC class II and ICAM-1 expression on human airway smooth muscle. Am J Respir Cell Mol Biol. 1997;16:38–45. doi: 10.1165/ajrcmb.16.1.8998077. [DOI] [PubMed] [Google Scholar]
- Barnes PJ, Pedersen S, Busse WW. Efficacy and safety of inhaled corticosteroids: new developments. Am J Respir Crit Care Med. 1998;157:s1–s53. doi: 10.1164/ajrccm.157.3.157315. [DOI] [PubMed] [Google Scholar]
- Young PG, Skinner JM, Black PN. Effects of glucocorticoids and beta-adrenoceptor agonists on the proliferation of airway smooth muscle. Eur J Pharmacol. 1995;273:137–143. doi: 10.1016/0014-2999(94)00679-2. [DOI] [PubMed] [Google Scholar]
- Bousquet J, Jeffery PK, Busse WW, Johnson M, Vignola AM. Asthma. From bronchoconstriction to airways inflammation and remodeling. Am J Respir Crit Care Med. 2000;161:1720–1745. doi: 10.1164/ajrccm.161.5.9903102. [DOI] [PubMed] [Google Scholar]
- Singh AD, Sanderson CJ. Anti-interleukin-5 strategies as a potential treatment for asthma. Thorax. 1997;52:483–485. doi: 10.1136/thx.52.5.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England BP, Balasubramanian P, Uings I, Bethell S, Chen MJ, Schatz PJ, Yin Q, Chen YF, Whitehorn EA, Tsavaler A, Martens CL, Barrett RW, McKinnon M. A potent dimeric peptide antagonist of interleukin-5 that binds two interleukin-5 receptor alpha chains. Proc Natl Acad Sci USA. 2000;97:6862–6867. doi: 10.1073/pnas.110053997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzarella G, Bianco A, Catena E, De Palma R, Abbate GF. Th1/Th2 lymphocyte polarization in asthma. Allergy. 2000;55 (suppl 61):6–9. doi: 10.1034/j.1398-9995.2000.00511.x. [DOI] [PubMed] [Google Scholar]
- Atsuta J, Fujisawa T, Iguchi K, Terada A. Inhibitory effect of TGF-beta1 on cytokine-enhanced eosinophil survival. Arerugi. 1994;43:1194–1200. [PubMed] [Google Scholar]
- Fahy JV, Corry DB, Boushey HA. Airway inflammation and remodeling in asthma. Curr Opin Pulmon Med. 2000;6:15–20. doi: 10.1097/00063198-200001000-00004. [DOI] [PubMed] [Google Scholar]
- Redington AE. Fibrosis and airway remodelling. Clin Exp Allergy. 2000;30:42–45. doi: 10.1046/j.1365-2222.2000.00096.x. [DOI] [PubMed] [Google Scholar]
- Mckay S, de Jongste JC, Hoogsteden HC, Saxena PR, Sharma HS. Enhanced expression and secretion of interleukin-6 in human airway smooth muscle cells treated with platelet derived growth factor-AB [abstract]. Am J Resp Crit Care Med. 2000;161:A261. [Google Scholar]
- Stephens NL, Li W, Wang Y, Ma X. The contractile apparatus of airway smooth muscle. Am J Respir Crit Care Med. 1998;158:S80–S94. doi: 10.1164/ajrccm.158.supplement_2.13tac300. [DOI] [PubMed] [Google Scholar]
- Jiang H, Rao K, Halayko AJ, Liu X, Stephens NL. Ragweed sensitization-induced increase of myosin light chain kinase content in canine airway smooth muscle. Am J Respir Cell Mol Biol. 1992;7:567–573. doi: 10.1165/ajrcmb/7.6.567. [DOI] [PubMed] [Google Scholar]
- Takeda M, Homma T, Breyer MD, Horiba N, Hoover RL, Kawamoto S, Ichikawa I, Kon V. Volume and agonist-induced regulation of myosin light-chain phosphorylation in glomerular mesangial cells. Am J Physiol. 1993;264:L421–L426. doi: 10.1152/ajprenal.1993.264.3.F421. [DOI] [PubMed] [Google Scholar]
- Hautmann MB, Madsen CS, Owens GK. A transforming growth factor-beta control element drives TGF-beta-induced stimulation of smooth muscle alpha-actin gene expression in concert with two CArG elements. J Biol Chem. 1997;272:10948–10956. doi: 10.1074/jbc.272.16.10948. [DOI] [PubMed] [Google Scholar]
- Solway J. What makes the airways contract abnormally? Is it inflammation? Am J Respir Crit Care Med. 2000;161:S164–S167. doi: 10.1164/ajrccm.161.supplement_2.a1q4-3. [DOI] [PubMed] [Google Scholar]
- Ohno I, Nitta Y, Yamauchi K, Hoshi H, Honma M, Woolley K, O'Byrne P, Dolovich J, Jordana M, Tamura G, Tanno Y, Shirato K. Eosinophils as a potential source of platelet-derived growth factor B-chain (PDGF-B) in nasal polyposis and bronchial asthma. Am J Respir Cell Mol Biol. 1995;13:639–647. doi: 10.1165/ajrcmb.13.6.7576701. [DOI] [PubMed] [Google Scholar]
- Millar EA, Angus RM, Hulks G, Morton JJ, Connell JMC, Thomson NC. Activity of the renin-angiotensin system in acute severe asthma and the effect of angiotensin II on lung function. Thorax. 1994;49:492–495. doi: 10.1136/thx.49.5.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechin F, van der Dijs B, Orozco B, Lechin M, Lechin AE. Increased levels of free serotonin in plasma of symptomatic asthmatic patients. Ann Allergy Asthma Immunol. 1996;77:245–253. doi: 10.1016/S1081-1206(10)63263-2. [DOI] [PubMed] [Google Scholar]
- Trepicchio WL, Bozza M, Pedneaullt G, Dorner AJ. Recombinant human IL-11 attenuates the inflammatory response through downregulation of proinflammatory cytokine release and nitric oxide production. J Immunol. 1996;157:3627–3634. [PubMed] [Google Scholar]
- Redlich AA, Gao X, Rockwell S, Kelley M, Elias JA. IL-11 enhances survival and decreases TNF production after radiation-induced thoracic injury. J Immunol. 1996;157:1705–1710. [PubMed] [Google Scholar]
- Hilton DJ. LIF: lots of interesting functions. Trends Biochem Sci. 1992;17:72–76. doi: 10.1016/0968-0004(92)90505-4. [DOI] [PubMed] [Google Scholar]
- Shadiack AM, Hart RP, Carlson CD, Jonakait MG. Interleukin-1 induces substance P in sympathetic ganglia through the induction of leukemia inhibitory factor (LIF). J Neurosci. 1993;13:2601–2609. doi: 10.1523/JNEUROSCI.13-06-02601.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conroy DM, Humbles AA, Rankin SM, Palframan RT, Collins PD, Griffiths-Johnson DA, Jose PJ, Williams TJ. The role of the eosinophil-selective chemokine, eotaxin, in allergic and non-allergic airways inflammation. Mem Inst Oswaldo Cruz. 1997;92:183–191. doi: 10.1590/s0074-02761997000800024. [DOI] [PubMed] [Google Scholar]
- Rankin SM, Conroy DM, Williams TJ. Eotaxin and eosinophil recruitment: implications for human disease. Mol Med Today. 2000;6:20–27. doi: 10.1016/S1357-4310(99)01635-4. [DOI] [PubMed] [Google Scholar]
- Marom Z, Shelhamer JH, Kaliner M. The effect of arachidonic acid, monohydroxyeicosatetraenoic acid, and prostaglandins on the release of mucous glycoproteins from human airways in vitro. J Clin Invest. 1981;67:1695–1702. doi: 10.1172/JCI110207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson WR. Eicosanoids and platelet-activating factor in allergic respiratory diseases. Am Rev Respir Dis. 1991;143:S86–S90. doi: 10.1164/ajrccm/143.5_Pt_2.S86. [DOI] [PubMed] [Google Scholar]
- Armour CL, Johnson PR, Alfredson ML, Black JL. Characterization of contractile prostanoid receptors on human airway smooth muscle. Eur J Pharmacol. 1989;165:215–222. doi: 10.1016/0014-2999(89)90715-2. [DOI] [PubMed] [Google Scholar]