

Abstract
SIX1 and SIX2 encode closely related transcription factors of which disruptions have been associated with distinct craniofacial syndromes, with mutations in SIX1 associated with branchiootic syndrome 3 (BOS3) and heterozygous deletions of SIX2 associated with frontonasal dysplasia defects. Whereas mice deficient in Six1 recapitulated most of the developmental defects associated with BOS3, mice lacking Six2 function had no obvious frontonasal defects. We show that Six1 and Six2 exhibit partly overlapping patterns of expression in the developing mouse embryonic frontonasal, maxillary, and mandibular processes. We found that Six1–/–Six2–/– double-mutant mice were born with severe craniofacial deformity not seen in the Six1–/– or Six2–/– single mutants, including skull bone agenesis, midline facial cleft, and syngnathia. Moreover, whereas Six1–/– mice exhibited partial transformation of maxillary zygomatic bone into a mandibular condyle-like structure, Six1–/–Six2+/– mice exhibit significantly increased penetrance of the maxillary malformation. In addition to ectopic Dlx5 expression at the maxillary-mandibular junction as recently reported in E10.5 Six1–/– embryos, the E10.5 Six1–/–Six2+/– embryos showed ectopic expression of Bmp4, Msx1, and Msx2 messenger RNAs in the maxillary-mandibular junction. Genetically inactivating 1 allele of either Ednra or Bmp4 significantly reduced the penetrance of maxillary malformation in both Six1–/– and Six1–/–Six2+/– embryos, indicating that Six1 and Six2 regulate both endothelin and bone morphogenetic protein-4 signaling pathways to pattern the facial structures. Furthermore, we show that neural crest–specific inactivation of Six1 in Six2–/– embryos resulted in midline facial cleft and frontal bone agenesis. We show that Six1–/–Six2–/– embryos exhibit significantly reduced expression of key frontonasal development genes Alx1 and Alx3 as well as increased apoptosis in the developing frontonasal mesenchyme. Together, these results indicate that Six1 and Six2 function partly redundantly to control multiple craniofacial developmental processes and play a crucial neural crest cell–autonomous role in frontonasal morphogenesis.
Keywords: BMP4, frontonasal dysplasia, median facial cleft, neural crest, transcription factor, Alx1
Introduction
Frontonasal dysplasia (FND) consists of a group of disorders characterized by ocular hypertelorism, midline facial cleft affecting the nose and/or upper lip and palate, notching or clefting of the alae nasi, and it is sometimes associated with anterior cranium bifidum and other malformations (Wu et al. 2007; Kayserili et al. 2009; Twigg et al. 2009). Although it has long been recognized that FND results from abnormal development of the embryonic frontonasal prominence, which forms from cranial neural crest cells populating in between the forebrain and surface ectoderm and ultimately gives rise to the forehead, nose, philtrum, and premaxillary component of the upper jaw, the causes and molecular mechanisms of FND pathogenesis are not well understood (Farlie et al. 2016). Mutations in 5 genes, including ALX1, ALX3, ALX4, EFNB1, and ZSWIM6, have been identified in a small number of patients with FND disorders (Twigg et al. 2004; Wieland et al. 2004; Kayserili et al. 2009; Twigg et al. 2009; Uz et al. 2010; Smith et al. 2014; Farlie et al. 2016). Remarkably, loss of function of each of the ALX family genes, which encode homeodomain-containing transcription factors, has been associated with distinct autosomal recessive FND syndromes (Kayserili et al. 2009; Twigg et al. 2009; Uz et al. 2010; Farlie et al. 2016). Gene knockout studies in mice showed that Alx1 function is required for survival of embryonic forebrain mesenchyme cells, whereas Alx3 and Alx4 play redundant roles in survival of embryonic nasal mesenchyme cells (Zhao et al. 1996; Beverdam et al. 2001). Loss of function of the X-linked EFNB1 gene caused severe hypertelorism with a grooved nasal tip and craniosynostosis in heterozygous females, whereas hemizygous males showed mildly affected hypertelorism (Twigg et al. 2004; Twigg et al. 2013), due to mosaicism in EFNB1 expression caused by random X-inactivation in heterozygous females (Davy et al. 2006; Twigg et al. 2013). A single recurrent missense mutation in ZSWIM6 has been associated with severe frontonasal and limb malformations (Smith et al. 2014), but the mechanism involving ZSWIM6 in frontonasal and limb development is unknown.
Recently, heterozygous microdeletions of the SIX2 gene and flanking noncoding sequences have been associated with an FND syndrome in patients of 2 unrelated families, characterized by frontal bossing, a large anterior fontanelle, hypertelorism, a broad nasal bridge, and conductive hearing loss (Hufnagel et al. 2016; Henn et al. 2018). Although Six2 messenger RNAs (mRNAs) are highly expressed in frontonasal mesenchyme in mouse embryos, Six2-null mice exhibit kidney dysgenesis and premature fusion of the bones in the cranial base but no obvious defects in frontonasal development (Oliver et al. 1995; Self et al. 2006; He et al. 2010). However, mice homozygous for an X-irradiation–induced mutation, Brachyrrhine (Br), which was mapped to the Six2 locus and caused significantly reduced Six2 mRNA expression in developing kidney and craniofacial tissues, exhibited a large median facial cleft (Fogelgren et al. 2008). Six2 belongs to the SIX gene family that consists of 6 members (Six1–6), each coding for a DNA-binding protein containing a homeodomain and an adjacent SIX domain (Kumar 2009). In both mouse and human genomes, the Six2 and Six3 genes are physically linked immediate neighbors (Fogelgren et al. 2008; Hufnagel et al. 2016; O’Brien et al. 2018). Recent genome sequencing revealed that the Br mutation caused inversion of the Six2/Six3 locus, resulting in aberrant Six3 gene expression in developing craniofacial and kidney tissues in addition to loss of Six2 expression. Thus, whether and how Six2 regulates frontonasal development require furthers investigation.
In this study, we investigated whether Six2 function in mouse craniofacial development is compensated by Six1, which shares the highest sequence similarity with Six2 and is also highly expressed in developing frontonasal and other craniofacial tissues (Oliver et al. 1995; Laclef et al. 2003). Mutations in SIX1 have been associated with autosomal dominant branchiootic syndrome 3 (BOS3), characterized by preauricular pits, hearing loss, branchial cleft fistula or cyst, and renal dysplasia (Ruf et al. 2004; Sanggaard et al. 2007). Mice lacking Six1 function display malformation of several craniofacial structures, including the nasal cavity, ear, and maxillary and mandibular skeletons, in addition to kidney agenesis (Laclef et al. 2003; Li et al. 2003; Xu et al. 2003; Ozaki et al. 2004; Tavares et al. 2017). We report here that mice lacking both Six1 and Six2 function exhibit more severe defects in multiple craniofacial tissues than in the single mutant mice and that Six1 and Six2 have a crucial but overlapping neural crest cell–autonomous function in frontonasal development.
Materials and Methods
Mouse Strains and Mouse Breeding
This study used mice carrying previously reported Bmp4null (Liu et al. 2004), Six1Cre/hpAP (Guo et al. 2011), Six1flox (Le Grand et al. 2012), and Six2GCE (Kobayashi et al. 2008) alleles as well as the EIIa-Cre and Wnt1Cre transgenic mouse lines (Lakso et al. 1996; Danielian et al. 1998). The Ednra+/– mice, heterozygous for the Ednratm1b(EUCOMM)Hmgu allele, were obtained from The Jackson Laboratory (JAX Stock 027942). Six1Cre/hpAP is functionally a Six1 null allele, whereas Six2GCE is functionally a Six2 null allele (Kobayashi et al. 2008; Guo et al. 2011). Six1Cre/hpAP heterozygous mice were crossed with Six2GCE heterozygous mice to generate Six1+/–Six2+/– mice, which were subsequently crossed to the ICR strain to establish the Six1+/–Six2+/– colony. The Six1flox allele (Le Grand et al. 2012) is referred to as Six1c in this report. Six1c/c and Six1c/+Wnt1Cre mice were separately crossed with Six2GCE/+ mice to generate Six1c/+Six2+/– and Six1c/+Six2+/–Wnt1Cre mice, which were subsequently used to generate Six1c/cSix2–/–Wnt1- Cre compound mutant pups.
All animal work procedures were performed following recommendations in the Guide for Care and Use of Laboratory Animals by the National Institutes of Health and approved by the Institutional Animal Care and Use Committee (IACUC) at Cincinnati Children’s Hospital Medical Center. This study conformed with ARRIVE guidelines for preclinical animal studies.
Histology, In Situ Hybridization, and Skeletal Preparations
Embryos were collected and processed for histology or in situ hybridization as described previously (Li et al. 2017). Skeletal preparations of newborn and late-term fetuses were processed and stained with alizarin red and Alcian blue as described previously (Ovchinnikov 2009).
Immunofluorescence Detection
Immunofluorescent staining was performed using paraffin sections following standard protocols (Xu et al. 2014). The following primary antibodies were used: rabbit antiactive capase 3 (cat. 559565, 1:300; BD Biosciences), rabbit anti-SIX1 (D4A8K, cat. 12891S, 1:200; Cell Signaling Technology), rabbit anti-SIX2 (cat. 11562-1-AP, 1:200; Proteintech), and guinea pig anti-SIX3 (cat. 200-201-A26S, 1:100; Rockland).
Quantitative Real-Time Polymerase Chain Reaction Analysis
Quantitative real-time polymerase chain reaction (RT-qPCR) was performed as previously described (Li et al. 2017). The relative levels of mRNAs in each sample were normalized to that of Hprt mRNAs. Student’s t test was used to analyze differential expression data. The gene-specific PCR primers are Alx1F (ACGATACGGCCAAATACAGC) and Alx1R (TAAG GTGTCATGCAGGAGGA), Alx3F (AGCGTTATGGGAA GATGCAG) and Alx3R (ATGCCCTCTGGAGACATGAG), and Alx4F (CTGCGCATCTCTACTTGCAG) and Alx4R (CACCCAGGTTGCTCTCTTTG).
Results
Patterns of Expression of Six1 and Six2 Exhibit Extensive Overlap in Developing Frontonasal, Maxillary, and Mandibular Processes
To investigate whether Six1 might complement Six2 function in frontonasal development, we compared expression patterns of Six1 and Six2 mRNAs and proteins in developing mouse embryos. Whole-mount in situ hybridization analysis showed that both Six1 and Six2 mRNAs were highly expressed in the nasal and maxillary processes at E10.5 (Fig. 1A, G). Immunofluorescent analyses showed that Six1 protein was strongly expressed in medial nasal, maxillary, and mandibular mesenchyme, as well as in the nasal pit epithelium and Rathke’s pouch (Fig. 1B–E). Strong Six2 protein expression was detected in the medial nasal and maxillary mesenchyme, whereas lower levels of Six2 protein were detected in mandibular mesenchyme and nasal epithelium (Fig. 1H–K). We validated specificity of the antibodies by directly comparing immunofluorescent staining of wild-type and Six2–/– embryo samples and found that the anti-SIX2 antibody did not detect any specific signal in Six2–/– embryos, whereas the patterns of anti-SIX1 immunostaining of the developing facial tissues were unaltered (Fig. 1F, L; compare with Fig. 1E, K, respectively). These data indicate that Six1 and Six2 expression exhibits extensive overlap in the developing facial tissues, particularly in medial nasal and maxillary processes. Since Br/Br mutant mouse embryos showed altered pattern of Six3 expression in addition to disruption of Six2 expression (O’Brien et al. 2018), we also examined Six3 expression in Six2–/– mutant embryos and control littermates. Our results show that Six3 expression in the developing facial tissues was unaltered in Six2–/– mutant embryos (Appendix Fig. 1), indicating that the alteration in Six3 expression in Br/Br mutant embryos was not due to loss of Six2 function.
Figure 1.

Expression of Six1 and Six2 in the developing frontonasal, maxillary, and mandibular processes. (A) Whole-mount view of the pattern of Six1 messenger RNA (mRNA) expression in E10.5 wild-type (WT) embryo. (B–E) Representative sagittal (B, C) and frontal (D, E) sections of E10.5 WT embryos showing immunofluorescent staining (green) using the anti-SIX1 antibody. DAPI counterstaining is shown in blue. (F) Representative frontal section of E10.5 Six2–/– embryo showing immunofluorescent staining (green) using the anti-SIX1 antibody. (G) Whole-mount view of the pattern of Six2 mRNA expression in E10.5 WT embryo. (H–K) Representative sagittal (H, I) and frontal (J, K) sections of E10.5 WT embryos showing immunofluorescent staining (green) using the anti-SIX2 antibody. (L) Immunofluorescent staining of frontal sections of E10.5 Six2–/– embryos using the same anti-SIX2 antibody. Multiple serial sections from 3 wild-type and 3 Six2–/– embryos were used for immunofluorescent staining using each antibody. hm, head mesenchyme; mdp, mandibular process; mnp, medial nasal process; mxp, maxillary process; np, nasal pit; Rp, Rathke’s pouch. Scale bar in panel B, 100 µm.
Mice Lacking Both Six1 and Six2 Exhibit Severe Craniofacial Malformations Not Seen in Six1–/– or Six2–/– Mutants
No overt craniofacial defects were detected in Six1+/–Six2+/– double heterozygous mice. We intercrossed Six1+/–Six2+/– mice and analyzed craniofacial phenotypes of compound mutant pups. At E18.5, Six1–/– pups (n = 28) exhibited a domed head, smaller mandible, and a characteristic curvature between the forehead and snout (Fig. 2B, B′). Six2–/– pups (n = 9) showed shortened snout (Fig. 2C, C′). In contrast, Six1–/–Six2–/– pups showed severe craniofacial defects, including midline facial cleft, widely open eyes lacking upper eyelid, absence of external ear, and severely shortened mandible (Fig. 2D, D′) (n = 8). Skeletal preparations showed that, whereas Six1–/– pups had defects in nasal and mandibular structures and Six2–/– pups showed premature fusion of the presphenoid and basisphenoid bones, the Six1–/–Six2–/– mutants exhibited agenesis of frontal and parietal bones of the skull, significantly reduced nasal bones, bony fusion of the maxilla and mandible (syngnathia), midline cleft of the nasal capsule, and absence of the anterior cranial base, with the basioccipital bone aberrantly fused anteriorly with the merged pre/basisphenoid and laterally with the exoccipital bones (Fig. 2E–H′). These results indicate that Six1 and Six2 play crucial and partly redundant roles in the development of multiple craniofacial structures.
Figure 2.
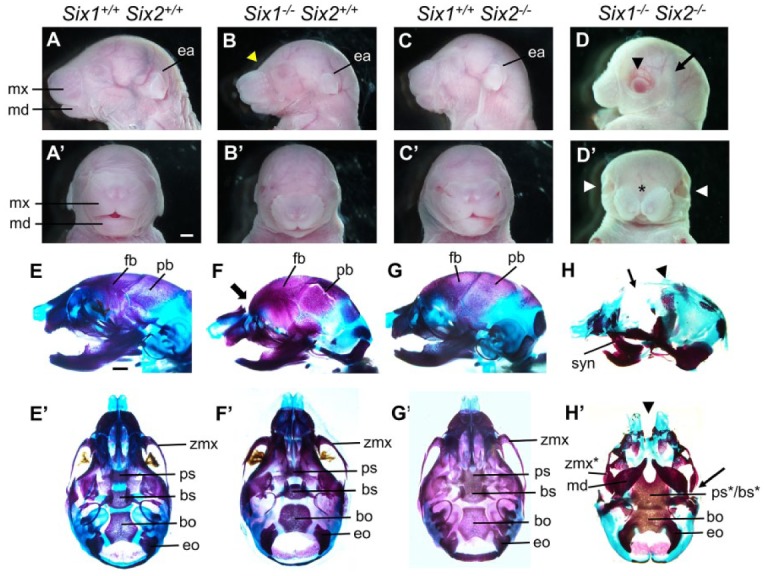
Craniofacial skeletal defects in the Six1 and Six2 compound mutant mice. (A–D′) Lateral view (A–D) and frontal view (A′–D′) of E18.5 heads of the Six1+/+Six2+/+ (A, A′), Six1–/–Six2+/+ (B, B′), Six1+/+Six2–/– (C, C′), and Six1–/–Six2–/– (D, D′) embryos. The yellow arrowhead in B points to the abnormal shape of the forehead. The black arrowhead in D and the white arrowheads in D′ point to widely open eyes. Asterisk in D′ marks midline facial cleft. Scale bar in A’, 1 mm. (E–H′) Lateral view (E–H) and ventral view (E′–H′) of alizarin red– and Alcian blue–stained E18.5 head skeletons of the Six1+/+Six2+/+ (E, E′), Six1–/–Six2+/+ (F, F′), Six1+/+Six2–/– (G, G′), and Six1–/–Six2–/– (H, H′) embryos. The thick arrow in F points to the abnormal shape of the forehead. The thin arrow in H points to lack of frontal bones, and the arrowhead points to the severely reduced parietal bones. The arrowhead in H′ points to the midline facial cleft, and the arrow points to abnormal zygomatic bone. Scale bar in E, 1 mm. bo, basioccipital; bs, basisphenoid; ea, ear; eo, exoccipital; fb, frontal bone; md, mandible; mx, maxilla; pb, parietal bone; ps, presphenoid; syn, syngnathia; zmx, zygomatic process of maxilla. The asterisks in the ps*/bs* and zmx* labels in Panel H point to severe malformation of those structures in the double homozygous mutants.
Six2 Acts in Concert with Six1 to Pattern the Maxillary/Mandibular Junction
Tavares et al. (2017) recently reported that mice homozygous for one of the Six1 knockout alleles, Six1tm1Kwk (Ozaki et al. 2004), exhibited a maxillary defect in which the zygomatic bone was transformed to a rod-like structure resembling the mandibular condyle. However, craniofacial skeletal preparations reported for mice homozygous for other Six1 knockout alleles did not show this maxillary defect (Laclef et al. 2003; Guo et al. 2011). Since we detected strong expression of both Six1 and Six2 mRNAs and proteins in the developing maxillary mesenchyme (Fig. 1), we carefully examined maxillary phenotypes in Six1–/– and Six1–/–Six2+/– mutant pups. We found that 11 of 28 (39.3%) Six1–/– pups showed partial mandibular transformation of the zygomatic bone, with 6 of them showing the defect unilaterally (Fig. 3B; Table). Notably, 16 of 18 (88.9%) Six1–/–Six2+/– pups showed the zygomatic bone defect, with 11 being bilateral (Fig. 3C; Table). These data, together with the syngnathia phenotype in Six1–/–Six2–/– mutants, indicate that Six1 and Six2 regulate maxillomandibular patterning in a dose-dependent manner.
Figure 3.
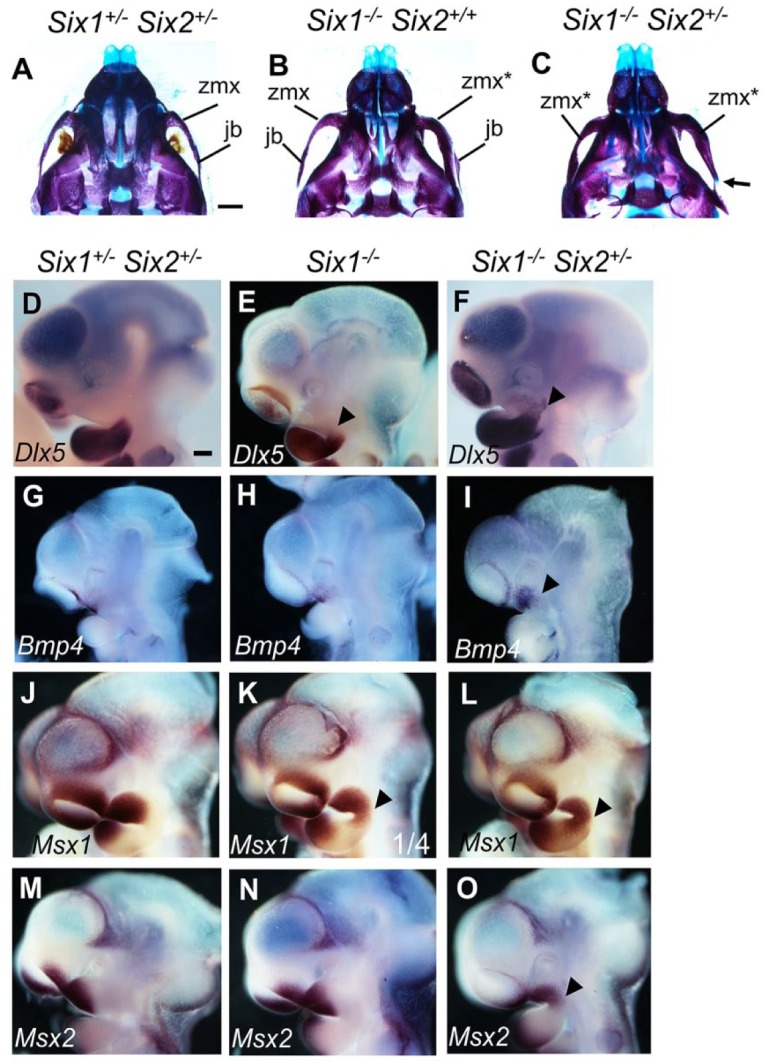
Maxillary defects in the Six1 and Six2 compound mutant mice. (A–C) Ventral view of alizarin red– and Alcian blue–stained E18.5 head skeletons of the Six1+/–Six2+/– (A), Six1–/–Six2+/+ (B), and Six1–/–Six2+/– (C) embryos. Arrow in C points to the ectopic cartilage at the proximal end of the zygomatic bone. Scale bar in A, 1 mm. The zmx* label in Panels B and C points to malformed zmx. (D–O) Lateral view of patterns of Dlx5 (D–F), Bmp4 (G–I), Msx1 (J–L), and Msx2 (M–O) messenger RNA expression in the E10.5 Six1+/–Six2+/– (D, G, J, M), Six1–/– (E, H, K, N), and Six1–/–Six2+/– (F, I, L, O) embryos. At least 3 embryos of each genotype were analyzed using each probe. Arrowheads point to ectopic expression of Dlx5 (E, F), Bmp4 (I), Msx1 (K, L), and Msx2 (O) in the maxillary-mandibular junction. Scale bar in D, 200 μm. jb, jugal bone; zmx, zygomatic process of maxilla.
Table.
Comparison of the Maxillary Defect in E18.5 Six1–/–, Six1–/–Six2+/–, Six1–/–Ednra+/–, Six1–/–Six2+/–Ednra+/–, Six1–/–Bmp4+/–, and Six1–/–Six2+/–Bmp4+/– Mutant Mice.
| Genotype | Total Number | Unilateral Maxillary Defect | Bilateral Maxillary Defect | Percentage with Maxillary Defecta |
|---|---|---|---|---|
| Six1 –/– | 28 | 6 | 5 | 39.3 |
| Six1 –/– Six2 +/– | 18 | 5 | 11 | 88.9 |
| Six1 –/– Ednra +/– | 8 | 1 | 0 | 12.5 |
| Six1 –/– Six2 +/– Ednra +/– | 9 | 5 | 0 | 55.6 |
| Six1 –/– Bmp4 +/– | 9 | 1 | 0 | 11.1 |
| Six1 –/– Six2 +/– Bmp4 +/– | 11 | 3 | 1 | 36.4 |
Student’s t test analyses showed significant increase in the frequency of the maxillary defect from Six1–/– to Six1–/–Six2+/– mice (P < 0.05) and significant reduction in frequency of the phenotype from Six1–/– to Six1–/–Ednra+/– and Six1–/–Bmp4+/– mice, respectively (P < 0.05). The frequency of the maxillary defect also significantly decreased from Six1–/–Six2+/– to Six1–/–Six2+/–Ednra+/– and Six1–/–Six2+/–Bmp4+/– mice, respectively (P < 0.05).
Tavares et al. (2017) showed that Six1–/– mutant embryos had ectopic expression of Edn1 in the mandibular arch epithelium and ectopic expression of the EDNRA signaling target gene Dlx5 in the maxillomandibular junction at E10.5. We also detected ectopic Dlx5 mRNA expression in the maxillomandibular junction in E10.5 Six1–/– embryos and found that Six1–/–Six2+/– embryos exhibited stronger ectopic Dlx5 expression in the maxillary prominences (Fig. 3D–F). Furthermore, inactivating 1 Ednra allele significantly reduced the percentage of Six1–/– and Six1–/–Six2+/– mutant pups with the maxillary bone defect: only 1 of 8 Six1–/–Ednra+/– and 5 of 9 Six1–/–Six2+/–Ednra+/– mutant pups showed a unilateral transformation of the zygomatic bone, and none of the pups of these genotypes showed bilateral maxillary defects (Table).
The incomplete rescue of the maxillary defect in Six1–/–Six2+/– pups by Ednra heterozygosity, together with the finding that Six1–/–Six2–/– mutants exhibited much more severe craniofacial defects than that previously reported of mice with ectopic activation of Edn1-EDNRA signaling throughout the cranial neural crest lineage (Tavares et al. 2017), prompted us to investigate whether Six1 and Six2 regulate additional signaling pathways important in maxillomandibular patterning. At E10.5, Bmp4 mRNAs were expressed in the distal maxillary and mandibular epithelium, whereas no morphogenetic protein (BMP) target genes Msx1 and Msx2 were expressed in distal regions of the maxillary and mandibular mesenchyme in control embryos (Fig. 3G, J, M). Whereas Six1–/– embryos showed similar patterns of expression of Bmp4 and Msx2, respectively, in the maxillary and mandibular prominences as in control embryos (Fig. 3H, N), 1 of 4 E10.5 Six1–/– embryos showed ectopic expression of Msx1 mRNAs at the maxillomandibular junction (Fig. 3K). Furthermore, expression of Bmp4 was increased in the maxillary region, and expression of both Msx1 and Msx2 mRNAs was expanded to the maxillomandibular junction in all E10.5 Six1–/–Six2+/– embryos examined (n = 4 for each of the Bmp4 and Msx1 probes and n = 3 for Msx2 probe) (Fig. 3I, L, O). Moreover, inactivation of 1 Bmp4 allele dramatically reduced the percentage of Six1–/– and Six1–/–Six2+/– mutant pups with the maxillary defect: only 1 of 9 (11.1%) Six1–/–Bmp4+/– and 4 of 11 (36.4) Six1–/–Six2+/–Bmp4+/– mutant pups showed the zygomatic bone defect (Table). These data indicate that Six1 and Six2 control maxillomandibular patterning through regulating both Edn1 and Bmp4 signaling pathways.
Six1 and Six2 Function Is Required in the Neural Crest Lineage for Frontonasal Development
Most of the craniofacial bones affected in the Six1–/–Six2–/– mutants form from ossification of neural crest–derived mesenchyme (Chai and Maxson 2006). We investigated whether Six1 is required cell-autonomously in neural crest cells for craniofacial development by analyzing Six1c/cWnt1Cre conditional mutants. All Six1c/c Wnt1Cre pups were born with a shortened mandible (n = 11) (Fig. 4D, E; compare with control samples in Fig. 4A, B). Notably, the skull, maxillary, and nasal skeletal elements appeared normal in Six1c/cWnt1Cre pups (Fig. 4B, C, E, F). We next analyzed Six1c/cSix2–/–Wnt1Cre mutant embryos. In contrast to Six1c/cWnt1Cre and Six2–/– pups, all Six1c/cSix2–/–Wnt1Cre mutant pups examined (n = 8) showed a midline facial cleft and agenesis of the frontal bones (Fig. 4G–I). In contrast to the Six1–/–Six2–/– pups (Fig. 2D, D′, H, H′), Six1c/cSix2–/–Wnt1Cre pups had well-formed parietal and mandibular bones comparable to Six1c/cWnt1Cre pups (Fig. 4H) and no maxillomandibular fusion (Fig. 4H, I). Together, these results indicate that Six1 expression in the neural crest–derived craniofacial mesenchyme is required for mandibular morphogenesis and has a crucial but overlapping function with Six2 in regulating frontonasal and maxillary development.
Figure 4.
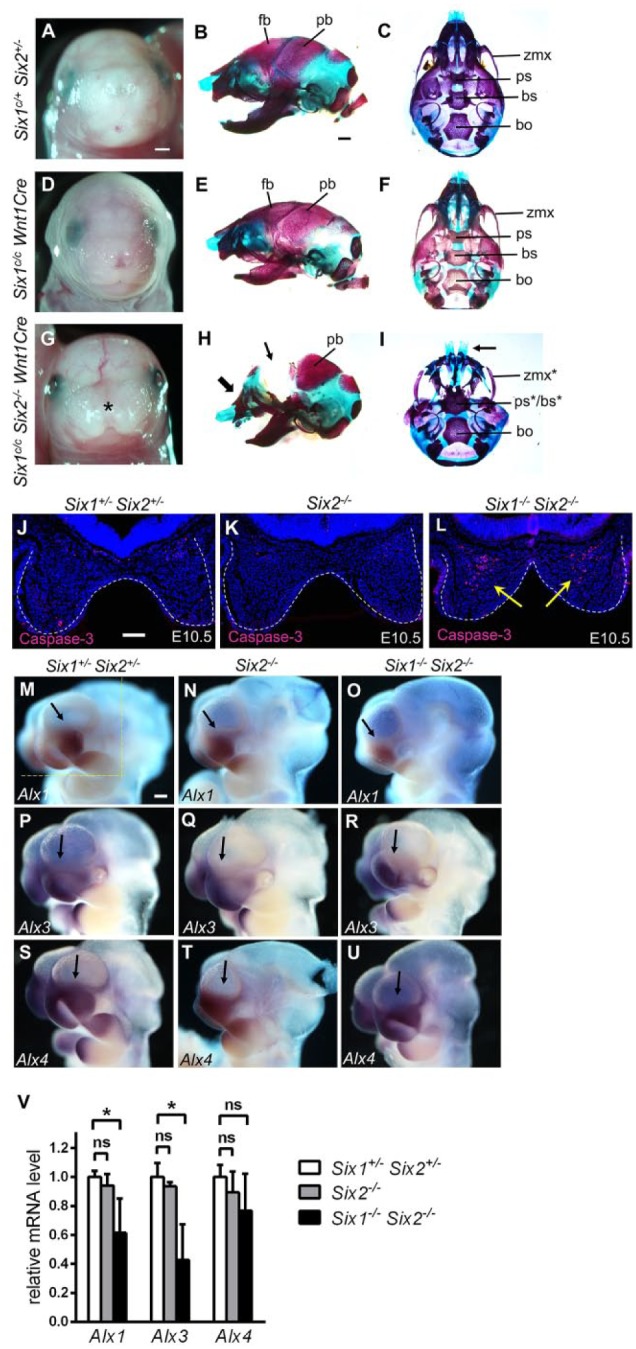
Analyses of craniofacial defects in Six1c/cSix2–/–Wnt1Cre embryos and mechanisms underlying frontonasal defects in Six1–/–Six2–/– embryos. (A, D, G) Frontal view of E18.5 heads of the Six1c/+Six2+/– (A), Six1c/cWnt1Cre (D), and Six1c/cSix2–/–Wnt1Cre (G) embryos. (B, E, H) Lateral view of alizarin red– and Alcian blue–stained E18.5 head skeletons of the Six1c/+Six2+/– (B), Six1c/cWnt1Cre (E), and Six1c/cSix2–/–Wnt1Cre (H) embryos. (C, F, I) Ventral view of E18.5 head skeletons of the Six1c/+Six2+/– (C), Six1c/cWnt1Cre (F), and Six1c/cSix2–/–Wnt1Cre (I) embryos. Asterisk in G indicates midline facial cleft in the Six1c/cSix2–/–Wnt1Cre embryo. The thick arrow in H points to the abnormal shape of the nasal region, whereas the thin arrow points to the deficiency in frontal bones in the Six1c/cSix2–/–Wnt1Cre mutant. The arrow in I points to the midline facial cleft in Six1c/cSix2–/–Wnt1Cre mutant. The asterisk in the ps*/bs* and zmx* labels in Panel I points to malformation of those structures. Scale bar in A and B, 1 mm. (J–L) Representative frontal sections of E10.5 Six1+/–Six2+/– (J), Six2–/– (K), and Six1–/– Six2–/– (L) embryos showing immunofluorescent staining of active caspase-3 (red). White dashes outline the medial nasal processes. Arrows in L point to a large number of active caspase-3–positive cells in the medial nasal mesenchyme in the Six1–/– Six2–/– embryo. Scale bar in J, 100 µm. Multiple serial sections from 3 embryos of each genotype were analyzed. (M–U) Lateral view of patterns of Alx1 (M–O), Alx3 (P–R), and Alx4 (S–U) messenger RNA (mRNA) expression in the E10.5 Six1+/–Six2+/– (M, P, S), Six2–/– (N, Q, T), and Six1–/–Six2–/– (O, R, U) embryos. Arrow points to the lateral nasal process in each sample. At least 2 embryos of each genotype were analyzed using each probe. Scale bar in M, 200 µm. (V) Quantitative real-time polymerase chain reaction (RT-qPCR) analysis of the levels of expression of Alx1, Alx3, and Alx4 mRNAs in the E10.5 Six1+/–Six2+/–, Six2–/–, and Six1–/–Six2–/–embryos (n = 3 each). RT-qPCR analyses were performed using total RNAs extracted from microdissected facial tissues from the region of E10.5 embryos as marked by the yellow-dashed lines in panel M. The asterisk indicates significantly different expression levels (P < 0.05 by Student’s t test), whereas ns indicates no significant difference (P > 0.05 by Student’s t test). bo, basioccipital; bs, basisphenoid; fb, frontal bone; pb, parietal bone; ps, presphenoid; zmx, zygomatic process of maxilla.
We further investigated the cellular basis of midline facial clefting in Six1–/–Six2–/– embryos. In contrast to Six1+/–Six2+/– and Six2–/– littermates, Six1–/–Six2–/– embryos showed a large amount of active caspase-3–positive cells in the medial nasal mesenchyme at E10.5 (Fig. 4J–L), indicating that loss of both Six1 and Six2 function causes abnormal apoptosis in developing medial nasal processes.
The midline facial defects in Six1–/–Six2–/– mutant mice are reminiscent of the Alx1+/–Alx4–/– and Alx3–/–Alx4–/– mutant mice reported previously (Qu et al. 1999; Beverdam et al. 2001). Through in situ hybridization and RT-qPCR analyses, we found that expression of Alx1 and Alx3, but not that of Alx4, was significantly reduced in the developing nasal processes in Six1–/–Six2–/– embryos in comparison with the single mutant and control littermates at E10.5 (Fig. 4M–V). These data indicate that Six1 and Six2 function in a gene regulatory network with the Alx family transcription factors to control frontonasal development.
Discussion
Whereas studies of Six1 mutant mice have confirmed critical roles for Six1 in the development of inner ear, kidney, and other organs affected in BOS3 patients (Xu et al. 2003; Bosman et al. 2009), no obvious frontonasal defects have been reported in mice heterozygous or homozygous for Six2 null alleles. In this report, we show that Six1 and Six2 exhibit partly overlapping patterns of expression in the developing frontonasal, maxillary, and mandibular processes (Fig. 1) and that Six1–/–Six2–/– double-mutant pups exhibit severe craniofacial defects, including midline facial cleft and agenesis of the frontal and parietal bones. These results identify a crucial but overlapping function of Six1 and Six2 in frontonasal development.
Six2 is strongly expressed in the neural crest–derived frontonasal mesenchyme, whereas Six1 is expressed not only in frontonasal mesenchyme but also in developing nasal and olfactory epithelium during mouse facial development. Our analyses of Six1c/cSix2–/–Wnt1Cre mutant mice indicate that a neural crest cell–autonomous function of Six1 and Six2 is required for frontonasal development. The midline facial cleft phenotype of Six1–/–Six2–/– mutant pups resembles the facial defects in Alx1+/–Alx4–/– and Alx3–/–Alx4–/– mice (Qu et al. 1999; Beverdam et al. 2001). Both Alx3–/–Alx4–/– and Six1–/–Six2–/– mutant embryos exhibited increased apoptosis of the developing nasal mesenchyme, whereas Alx1–/– embryos showed increased apoptosis of early embryonic forebrain mesenchyme (Zhao et al. 1996; Beverdam et al. 2001; Fig. 4 in this study). Remarkably, expression of both Alx1 and Alx3 in the developing nasal processes was significantly reduced in Six1–/–Six2–/– embryos (Fig. 4M–V). These data suggest that Six1 and Six2 regulate frontonasal development at least in part through regulating expression of the Alx family genes and that the pathogenic mechanism underlying frontonasal dysplasia in patients with deletion of SIX2 is related to that in patients with loss of ALX gene function.
In addition to validating a crucial role of Six2 in frontonasal development, we found that Six2 complements Six1 function in patterning the jaws. Partial transformation of the maxillary zygomatic bone to a mandibular condyle-like structure has only been reported in 1 of 3 previously characterized Six1 null mutant mouse lines (Laclef et al. 2003; Guo et al. 2011; Tavares et al. 2017). In this report, we found that 39.3% of mice homozygous for the Six1Cre/hpAP allele (Guo et al. 2011) in the ICR background exhibited the zygomatic bone defect. The lower frequency of maxillary-to-mandibular transformation in this line of Six1 mutant mice than that observed by Tavares et al. (2017) in the Six1tm1Kwk mutant line (Ozaki et al. 2004) could be due to differences in the gene-targeted alleles and/or genetic background modification. We found that inactivating 1 allele of Six2 increased the frequency of the maxillary defect in the Six1–/– mice in the ICR background to 88.9%. Moreover, the Six1–/–Six2–/– double homozygous mutant pups showed much more severe maxillary and mandibular defects than in Six1–/– and Six1–/–Six2+/– mutants. These data indicate that Six2 complements Six1 function in maxillary and mandibular development. Furthermore, we found that the Six1–/–Six2+/– mutant embryos had ectopic expression of Bmp4, Msx1, and Msx2 in the proximal maxillary processes and that inactivating 1 allele of Bmp4 significantly rescued maxillary bone formation in both Six1–/– and Six1–/–Six2+/– mutants. Thus, in addition to the previously identified role of Six1 in regulating Edn1-EDNRA signaling (Tavares et al. 2017), our results indicate that Six1 and Six2 act together to pattern the mammalian jaws through regulating BMP signaling as well. Related to this finding, Garcez et al. (2014) showed previously that double-stranded RNA-mediated silencing of Six1, Six2, and Six4 mRNAs in the cranial neural crest cells abolished formation of craniofacial skeletal structures in chick embryos and that overexpression of Noggin, a BMP antagonist, in the cranial neural crest cells nearly completely restored facial skeleton formation in Six1/2/4 triple-knockdown chick embryos. Together, these results indicate that Six1 and Six2 play an evolutionarily conserved role in patterning the facial skeleton through controlling BMP signaling.
Author Contributions
Z. Liu and C. Li contributed to design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; J. Xu and H. Liu contributed to data acquisition, critically revised the manuscript; Y. Lan and X. Wang contributed to design, data analysis, interpretation, and critically revised the manuscript; X. Li and P. Maire contributed to design, critically revised the manuscript; R. Jiang contributed to conception, design, data analysis, and interpretation, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
Supplemental material, DS_10.1177_0022034519835204 for Crucial and Overlapping Roles of Six1 and Six2 in Craniofacial Development by Z. Liu, C. Li, J. Xu, Y. Lan, H. Liu, X. Li, P. Maire, X. Wang and R. Jiang in Journal of Dental Research
Footnotes
A supplemental appendix to this article is available online.
This work was supported by National Institutes of Health/National Institute of Dental and Craniofacial Research (NIH/NIDCR) grants DE018401 and DE027046 to RJ. ZL was supported partly by the China Scholarship Council scholarship.
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Beverdam A, Brouwer A, Reijnen M, Korving J, Meijlink F. 2001. Severe nasal clefting and abnormal embryonic apoptosis in Alx3/Alx4 double mutant mice. Development. 128(20):3975–3986. [DOI] [PubMed] [Google Scholar]
- Bosman EA, Quint E, Fuchs H, Hrabé de Angelis M, Steel KP. 2009. Catweasel mice: a novel role for Six1 in sensory patch development and a model for branchio-oto-renal syndrome. Dev Biol. 328(2):285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y, Maxson RE., Jr. 2006. Recent advances in craniofacial morphogenesis. Dev Dyn. 235(9):2353–2375. [DOI] [PubMed] [Google Scholar]
- Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. 1998. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol. 8(24):1323–1326. [DOI] [PubMed] [Google Scholar]
- Davy A, Bush JO, Soriano P. 2006. Inhibition of gap junction communication at ectopic Eph/ephrin boundaries underlies craniofrontonasal syndrome. PLoS Biol. 4(10):e315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farlie PG, Baker NL, Yap P, Tan TY. 2016. Frontonasal dysplasia: towards an understanding of molecular and developmental aetiology. Mol Syndromol. 7(6):312–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogelgren B, Kuroyama MC, McBratney-Owen B, Spence AA, Malahn LE, Anawati MK, Cabatbat C, Alarcon VB, Marikawa Y, Lozanoff S. 2008. Misexpression of Six2 is associated with heritable frontonasal dysplasia and renal hypoplasia in 3H1 Br mice. Dev Dyn. 237(7):1767–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcez RC, Le Douarin NM, Creuzet SE. 2014. Combinatorial activity of six1-2-4 genes in cephalic neural crest cells controls craniofacial and brain development. Cell Mol Life Sci. 71(11):2149–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Sun Y, Zhou B, Adam RM, Li X, Pu WT, Morrow BE, Moon A, Li X. 2011. A Tbx1-Six1/Eya1-Fgf8 genetic pathway controls mammalian cardiovascular and craniofacial morphogenesis. J Clin Invest. 121(4):1585–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G, Tavella S, Hanley KP, Self M, Oliver G, Grifone R, Hanley N, Ward C, Bobola N. 2010. Inactivation of Six2 in mouse identifies a novel genetic mechanism controlling development and growth of the cranial base. Dev Biol. 344(2):720–730. [DOI] [PubMed] [Google Scholar]
- Henn A, Weng H, Novak S, Rettenberger G, Gerhardinger A, Rossier E, Zirn B. 2018. SIX2 gene haploinsufficiency leads to a recognizable phenotype with ptosis, frontonasal dysplasia, and conductive hearing loss. Clin Dysmorphol. 27(2):27–30. [DOI] [PubMed] [Google Scholar]
- Hufnagel RB, Zimmerman SL, Krueger LA, Bender PL, Ahmed ZM, Saal HM. 2016. A new frontonasal dysplasia syndrome associated with deletion of the SIX2 gene. Am J Med Genet A. 170(2):487–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayserili H, Uz E, Niessen C, Vargel I, Alanay Y, Tuncbilek G, Yigit G, Uyguner O, Candan S, Okur H, et al. 2009. ALX4 dysfunction disrupts craniofacial and epidermal development. Hum Mol Gen. 18(22):4357–4366. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Valerius MT, Mugford JW, Carroll TJ, Self M, Oliver G, McMahon AP. 2008. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell. 3(2):169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar JP. 2009. The sine oculis homeobox (six) family of transcription factors as regulators of development and disease. Cell Mol Life Sci. 66(4):565–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laclef C, Souil E, Demignon J, Maire P. 2003. Thymus, kidney and craniofacial abnormalities in Six1 deficient mice. Mech Dev. 120(6):669–679. [DOI] [PubMed] [Google Scholar]
- Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, Alt FW, Westphal H. 1996. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci U S A. 93(12):5860–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Grand F, Grifone R, Mourikis P, Houbron C, Gigaud C, Pujol J, Maillet M, Pagès G, Rudnicki M, Tajbakhsh S, et al. 2012. Six1 regulates stem cell repair potential and self-renewal during skeletal muscle regeneration. J Cell Biol. 198(5):815–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Lan Y, Krumlauf R, Jiang R. 2017. Modulating Wnt signaling rescues palate morphogenesis in Pax9 mutant mice. J Dent Res. 96(11):1273–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Ohgi KA, Zhang J, Krones A, Bush KT, Glass CK, Nigam SK, Aggarwal AK, Maas R, Rose DW, et al. 2003. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature. 426(6964):247–254. [DOI] [PubMed] [Google Scholar]
- Liu W, Selever J, Wang D, Lu MF, Moses KA, Schwartz RJ, Martin JF. 2004. Bmp4 signaling is required for outflow-tract septation and branchial-arch artery remodeling. Proc Natl Acad Sci U S A. 101(13):4489–4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien LL, Guo Q, Bahrami-Samani E, Park JS, Hasso SM, Lee YJ, Fang A, Kim AD, Guo J, Hong TM, et al. 2018. Transcriptional regulatory control of mammalian nephron progenitors revealed by multi-factor cistromic analysis and genetic studies. PLoS Genet. 14(1):e1007181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver G, Wehr R, Jenkins NA, Copeland NG, Cheyette BN, Hartenstein V, Zipursky SL, Gruss P. 1995. Homeobox genes and connective tissue patterning. Development. 121(3):693–705. [DOI] [PubMed] [Google Scholar]
- Ovchinnikov D. 2009. Alcian blue/alizarin red staining of cartilage and bone in mouse. Cold Spring Harb Protoc. 2009(3):pdb.prot5170. [DOI] [PubMed] [Google Scholar]
- Ozaki H, Nakamura K, Funahashi JI, Ikeda K, Yamada G, Tokano H, Okamura HO, Kitamura K, Muto S, Kotaki H, et al. 2004. Six1 controls patterning of the mouse otic vesicle. Development. 131(3):551–562. [DOI] [PubMed] [Google Scholar]
- Qu SH, Tucker SC, Zhao Q, Wisdom R. 1999. Physical and genetic interactions between Alx4 and Cart1. Development. 126(2):359–369. [DOI] [PubMed] [Google Scholar]
- Ruf RG, Xu PX, Silvius D, Otto EA, Beekmann F, Muerb UT, Kumar S, Neuhaus TJ, Kemper MJ, Raymond RM, et al. 2004. SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes. Proc Natl Acad Sci U S A. 101(21):8090–8095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanggaard KM, Rendtorff ND, Kjaer KW, Eiberg H, Johnsen T, Gimsing S, Dyrmose J, Nielsen KO, Lage K, Tranebjærg L. 2007. Branchio-oto-renal syndrome: detection of EYA1 and SIX1 mutations in five out of six Danish families by combining linkage, MLPA and sequencing analyses. Eur J Hum Genet. 15(11):1121–1131. [DOI] [PubMed] [Google Scholar]
- Self M, Lagutin OV, Bowling B, Hendrix J, Cai Y, Dressler GR, Oliver G. 2006. Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J. 25(21):5214–5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JD, Hing AV, Clarke CM, Johnson NM, Perez FA, Park SS, Horst JA, Mecham B, Maves L, Nickerson DA, et al. 2014. Exome sequencing identifies a recurrent de novo ZSWIM6 mutation associated with acromelic frontonasal dysostosis. Am J Hum Genet. 95(2):235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares AL, Cox TC, Maxson RM, Ford HL, Clouthier DE. 2017. Negative regulation of endothelin signaling by SIX1 is required for proper maxillary development. Development. 144(11):2021–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twigg SR, Babbs C, van den Elzen ME, Goriely A, Taylor S, McGowan SJ, Giannoulatou E, Lonie L, Ragoussis J, Akha ES, et al. 2013. Cellular interference in craniofrontonasal syndrome: males mosaic for mutations in the X-linked EFNB1 gene are more severely affected than true hemizygotes. Hum Mol Gen. 22(8):1654–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twigg SR, Kan R, Babbs C, Bochukova EG, Robertson SP, Wall SA, Morriss-Kay GM, Wilkie AO. 2004. Mutations of ephrin-B1 (EFNB1), a marker of tissue boundary formation, cause craniofrontonasal syndrome. Proc Natl Acad Sci U S A. 101(23):8652–8657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twigg SR, Versnel SL, Nürnberg G, Lees MM, Bhat M, Hammond P, Hennekam RC, Hoogeboom AJ, Hurst JA, Johnson D, et al. 2009. Frontorhiny, a distinctive presentation of frontonasal dysplasia caused by recessive mutations in the ALX3 homeobox gene. Am J Hum Genet. 84(5):698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uz E, Alanay Y, Aktas D, Vargel I, Gucer S, Tuncbilek G, von Eggeling F, Yilmaz E, Deren O, Posorski N, et al. 2010. Disruption of ALX1 causes extreme microphthalmia and severe facial clefting: expanding the spectrum of autosomal-recessive ALX-related frontonasal dysplasia. Am J Hum Genet. 86(5):789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland I, Jakubiczka S, Muschke P, Cohen M, Thiele H, Gerlach KL, Adams RH, Wieacker P. 2004. Mutations of the ephrin-B1 gene cause craniofrontonasal syndrome. Am J Hum Genet. 74(6):1209–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu E, Vargevik K, Slavotinek AM. 2007. Subtypes of frontonasal dysplasia are useful in determining clinical prognosis. Am J Med Genet A. 143(24):3069–3078. [DOI] [PubMed] [Google Scholar]
- Xu J, Liu H, Park JS, Lan Y, Jiang R. 2014. Osr1 acts downstream of and interacts synergistically with Six2 to maintain nephron progenitor cells during kidney organogenesis. Development. 141(7):1442–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu PX, Zheng W, Huang L, Maire P, Laclef C, Silvius D. 2003. Six1 is required for the early organogenesis of mammalian kidney. Development. 130(14):3085–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Behringer RR, de Crombrugghe B. 1996. Prenatal folic acid treatment suppresses acrania and meroanencephaly in mice mutant for the Cart1 homeobox gene. Nat Genet. 13(3):275–283. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, DS_10.1177_0022034519835204 for Crucial and Overlapping Roles of Six1 and Six2 in Craniofacial Development by Z. Liu, C. Li, J. Xu, Y. Lan, H. Liu, X. Li, P. Maire, X. Wang and R. Jiang in Journal of Dental Research