

Abstract
Purpose:
Variants in PRPF31, a splicing factor, are a common cause of autosomal dominant retinitis pigmentosa (RP). Deleterious variants are thought to cause disease by haploinsufficiency. In anticipation of upcoming replacement gene therapy trials, we present the phenotype and clinical progression of a large cohort of patients with PRPF31-mediated RP.
Design:
Cross-sectional with retrospective review
Methods:
A total of 26 patients with RP and 5 asymptomatic individuals, all with deleterious variants in PRPF31 (from 13 families), were selected from our database of patients followed longitudinally. Ages ranged from 9–77 years (mean 47 years old), with an average follow up time of 16 years. All patients underwent ophthalmic examination including psychophysical tests, electrophysiology, and imaging. All available records were reviewed retrospectively. Additionally, all patients were contacted, and all available patients (n=7) were examined in an additional prospective follow up visit.
Results:
Age of onset ranged from 6 to 71 years of age, without apparent relationship to specific variant. Two adults (ages 42 and 77) and 3 teenaged children were found to harbor a mutation with no evidence of RP. In those with RP, visual field area (spot size III) declined exponentially at a rate of 8.1% per year of disease duration (p<0.001, 95% CI 5.6–10.6), electroretinogram (ERG) cone amplitude declined exponentially at a rate of 7.3% per year of disease duration (p<0.001, 95% CI 5.4–9.1), and ellipsoid zone (EZ) area declined exponentially at a rate of 5.4% per year of disease duration (p<0.001, 95% CI 3.7–7.1).
Conclusions:
PRPF31-mediated retinitis pigmentosa is characterized by a variable age of onset. Once disease develops, it follows a predictable exponential time course.
Introduction
Retinitis pigmentosa (RP) is a genetically heterogenous form of inherited retinal degeneration characterized by night blindness and progressive loss of peripheral vision, eventually leading to blindness.1 It occurs in roughly 1 in 3,000–5,000 individuals, without any apparent link to geographic origin or racial background.2 On exam, patients are generally found to have atrophy of the outer retina, optic nerve pallor, attenuated retinal vessels, and bone spicule pigment. RP can be inherited in an autosomal dominant, autosomal recessive, or X-linked manner.
Over 22 genes causing autosomal dominant, non-syndromic RP have been identified to date, with most of these genes directly responsible for proteins involved in phototransduction and other processes which take place specifically within the outer retina.2–4
Interestingly, a subset of patients with non-syndromic autosomal dominant RP have been found to have mutations in spliceosomal proteins, which are necessary in every cell of the body.5 The spliceosome is a ribonucleoprotein necessary for removing introns from nascent RNA.6 It is also responsible for alternative splicing, the process by which RNA is spliced to generate different proteins from one sequence of coding DNA.7
PRPF31 mutations are the most prevalent of the splicing factor mutations, accounting for 2.5–10% of autosomal dominant retinitis pigmentosa. 8–10 PRPF31 mutations have been postulated to cause retinal pathology by haploinsufficiency.11 Because PRPF31 is more highly expressed in the retina than in other parts of the body, it is thought that the retina may rely more heavily on alternative splicing than other tissues and thus is affected by a relative insufficiency that other tissues can tolerate.12, 13 Defective PRPF31 proteins have been found to undergo nonsense-mediated decay, resulting in an insufficiency of the protein within the retina.14 Biochemical evidence supports this disease mechanism.15, 16
Previous studies have shown PRPF31-mediated retinitis pigmentosa to be associated with reduced phenotypic penetrance.11 Retrospective data on natural progression suggests that the rate of change in visual field area and cone ERG amplitude is between 7 and 10% per year with considerable variation among patients. However, previous data sets are limited by the use of age as the dependent variable, as PRPF31-mediated retinitis pigmentosa is characterized by a variable age of onset.17 As it may be a candidate for gene therapy,18 more information is needed about the natural history of PRPF31-mediated retinitis pigmentosa. In this study, we seek to clarify the disease course of PRPF31-mediated retinitis pigmentosa in a large cohort of patients.
Methods
All procedures were approved by the institutional review board of UT Southwestern Medical Center. Informed consent was obtained, and all research was conducted in accordance with the Declaration of Helsinki. Thirty-one subjects with PRPF31 mutations were identified from the Southwest Eye Registry, a pre-existing database with phenotype and genotype data from patients followed in research studies at the Retina Foundation of the Southwest. All available records were reviewed retrospectively. Additionally, all patients were contacted, and available patients (n=7) were examined in an additional prospective follow up visit.
Genomic DNA was extracted from either whole blood as reported previously19 or saliva collected with Oragene collection kits (DNA Genotek, Inc., Kanata, ON, Canada) and extracted according to the manufacturer’s recommended protocol. The affected proband from each family was tested for possible mutations in known adRP genes with fluorescent dideoxy sequencing as previously described.9, 19–23 Additional affected and asymptomatic family members underwent known mutation analysis for the identified familial variant.
Best-corrected visual acuity was measured with the E-ETDRS or with a Snellen chart, with results converted to ETDRS scores24 for statistical analysis. Static fields were obtained using the 30–2 grid on the Humphrey visual field analyzer. Kinetic fields with Goldmann equivalent stimuli V4e, III4e, and I4e were obtained on a semi-automated perimeter (Octopus 900; Haag-Streit AG, Köniz, Switzerland).
The eye with the better visual acuity, or the left eye if there was no difference between the two eyes, was dilated with eye drops (tropicamide 1% and phenylephrine 2.5%) and patched. After 30 minutes of dark adaptation, full-field electroretinography (ERG) recordings were obtained (Diagnosys LLC, Lowell, Massachusetts, USA) for all patients using the International Society for Clinical Electrophysiology of Vision (ISCEV) standard protocol.25 Subsequently, morphologic examinations including fundus photography, fundus autofluorescence, and spectral-domain optical coherence tomography (SD-OCT) (Spectralis HRA-OCT, version 5.3.3.0; Heidelberg Engineering, Heidelberg, Germany) was performed. Horizontal and vertical SD line scans were obtained and then the ellipsoid zone (EZ) width was manually measured. EZ area was obtained using the formula for an ellipse given height and width.26–29
For statistical analysis, patient-reported disease duration was determined by subtracting the age at which the patient first noticed the onset of either reduced night vision or constriction of visual fields from the patient’s age at the visit.
As patient-reported disease duration is a subjective measurement, we sought to determine if disease duration can be estimated using functional and structural data. Patients were divided into two groups: group A (n=15), the “reliable reporters” group, presenting in childhood or with early stage retinitis pigmentosa (measurable ERG rod response and spot size III visual field diameter >30 degrees), and group B (n=11), the “less reliable reporters group,” presenting in adulthood with intermediate or advanced disease (defined as visual field <30 degrees and no measurable ERG rod response).
Disease duration at initial visit was estimated for patients in group B using linear regressions of ERG data and EZ width data from group A. The two estimated durations were subsequently averaged. Two patients had to be excluded from analysis in group B because they had absent ERG cone responses and no measurable EZ width at their initial visit, and thus no way to estimate disease duration. For the remaining 9 patients in group B, duration at each follow up visit was offset by the duration at initial visit, and the time interval between visits was kept intact.
Results
We enrolled 31 subjects out of 35 subjects identified in the registry (4 patients had undergone genetic testing but were unavailable for follow-up visits). Ages ranged from 9 to 77, mean 47 years old; 15 subjects were female and 16 were male. Thirteen mutations were represented, from fourteen different families. Of the 31 individuals with PRPF31 mutations identified by genetic analysis, 26 were affected by retinitis pigmentosa and 5 were asymptomatic with no signs of retinal degeneration at their most recent visit. Seventeen patients had multiple visits, each separated by at least 6 months (mean ± standard deviation 16 ± 15). Baseline patient characteristics are presented in Table 1.
Table 1:
Baseline Characteristics
| Age at onset | Age at initial visit | Visual Acuity (ETDRS) | Visual Field Diameter | ||||
|---|---|---|---|---|---|---|---|
| ID | Mutation | OD | OS | OD | OS | ||
| 1 | c.527+1G>T | 48 | 48 | 85 | 84 | 25 | 25 |
| 2 | c.527+1G>T | 13 | 13 | 85 | 84 | 30 | 30 |
| 3 | c.527+3A>G | 29 | 52 | 61 | 63 | 25 | 25 |
| 4 | c.527+3A>G | 28 | 30 | 77 | 64 | >30 | >30 |
| 5 | c.527+3A>G | 27 | 70 | 23 | 51 | 10 | 10 |
| 6 | c.1060C>T (p.Arg354X) | 12 | 24 | 74 | 69 | 10 | 10 |
| 7 | c.1073+1G>A | 40 | 41 | 65 | 65 | 20 | 20 |
| 8 | c.1073+1G>A | n/a | 42 | 82 | 89 | >30 | >30 |
| 9 | c.1073+1G>A | n/a | 12 | 92 | 91 | >30 | >30 |
| 10 | c.1073+1G>A | n/a | 9 | 90 | 90 | >30 | >30 |
| 11 | c.1073+1G>A | n/a | 14 | 90 | 91 | >30 | >30 |
| 12 | c.1073+1G>A | 28 | 36 | 90 | 90 | >30 | >30 |
| 13 | c.−3_7del (p.Met1?) | 17 | 58 | 71 | 71 | 15 | 15 |
| 14 | c.−3_7del (p.Met1?) | 10 | 10 | 75 | 75 | ||
| 15 | c.−3_7del (p.Met1?) | 29 | 62 | 63 | 53 | 10 | 10 |
| 16 | c.550_552del (p.Leu184del) | 71 | 71 | 88 | 82 | >30 | >30 |
| 17 | c.758_767del (p.Gly253fs*317) | 19 | 31 | 30 | 65 | 10 | 20 |
| 18 | c.1084delA (p.Met362X) | 6 | 44 | 44 | 42 | 5 | 5 |
| 19 | c.895T>C (p.Cys299Arg) | 63 | 63 | 75 | 75 | >30 | >30 |
| 20 | c.895T>C (p.Cys299Arg) | 41 | 65 | 58 | 61 | 10 | |
| 21 | c.895T>C (p.Cys299Arg) | 21 | 44 | 65 | 70 | ||
| 22 | exon1indel: 149 bp deleted/640 bp inserted | 46 | 46 | 55 | 55 | ||
| 23 | exon1indel: 149 bp deleted/640 bp inserted | 25 | 50 | 70 | 75 | 10 | 10 |
| 24 | exon1indel: 149 bp deleted/640 bp inserted | 14 | 52 | 63 | 69 | 10 | 10 |
| 25 | exon1indel: 149 bp deleted/640 bp inserted | n/a | 77 | 87 | 89 | >30 | >30 |
| 26 | exon1indel: 149 bp deleted/640 bp inserted | 16 | 37 | 79 | 68 | 15 | 15 |
| 27 | c.390delC (p.Asn131fsX197) | 10 | 21 | 85 | 85 | ||
| 28 | c.390delC (p.Asn131fsX197) | 17 | 48 | 65 | 65 | ||
| 29 | c.390delC (p.Asn131fsX197) | 10 | 16 | 85 | 85 | ||
| 30 | c.525_526insAG | 16 | 47 | 69 | 49 | 15 | 10 |
| 31 | c.220C>T (p.Gln74X) | 7 | 9 | 65 | 75 | >30 | |
Of the 5 asymptomatic individuals known to have deleterious variants of PRPF31, two were adults, ages 42 and 77 at their most recent visits, and three were teenaged children. Both asymptomatic adults had first degree relatives affected at variable ages. Notably, the 77-year-old had a daughter who developed symptoms at age 6. The 42-year-old had multiple affected relatives, including his mother and a sibling affected in her teenaged years. Extensive examination, including ERG, showed no evidence of retinal degeneration in the asymptomatic patients. See Figure 1 for a comparison of the asymptomatic 42-year-old individual with his affected 53-year-old aunt.
FIGURE 1.
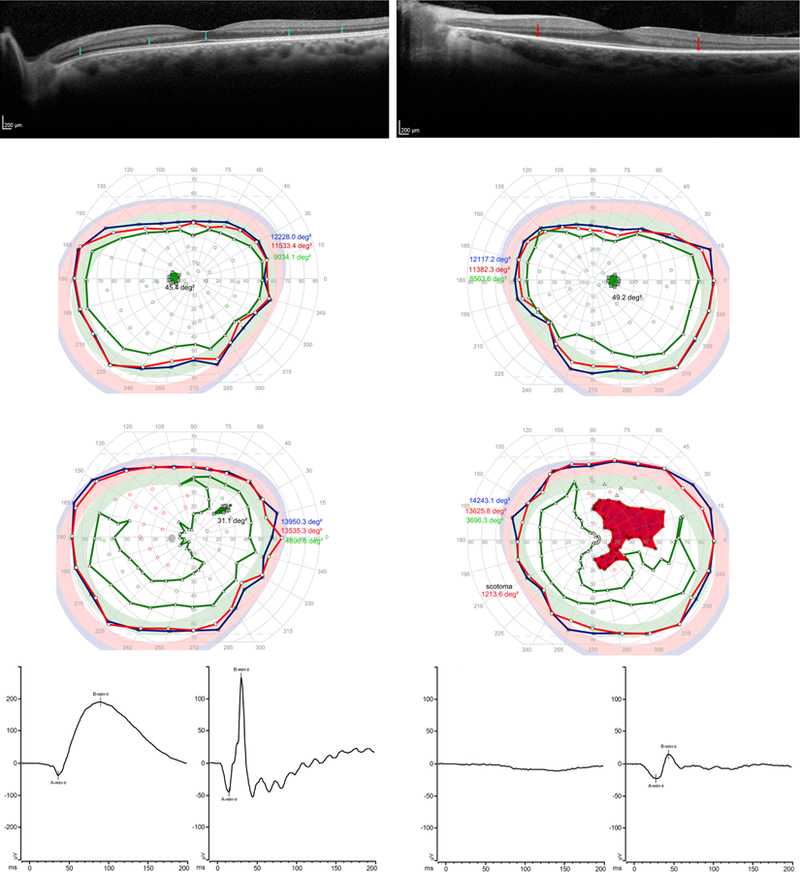
Comparison of an unaffected individual found to have a deleterious variant in PRPF31 (P8, age 42, left) with his affected aunt (P12, age 53, right). (Top left) For the asymptomatic individual, note that the ellipsoid zone, indicated by blue arrows, is intact throughout the SD-OCT scan. (Top right) For the symptomatic patient, the ellipsoid zone, with its endpoints indicated by red arrows, is shortened. (Middle left) For the asymptomatic individual, visual field area is within normal limits for Goldmann equivalent stimulus size V (blue), size III (red) and size I (green). (Middle right) For the 53-year-old with RP, visual fields are within normal limits for stimulus size V (blue.) There is a large scotoma for spot size III (red) in the right eye. Visual fields are reduced to a crescent shaped pattern for spot size I (green) in both eyes, with a small scotoma for spot size I in the right eye. These visual field changes are typical for early-intermediate PRPF31-mediated RP. (Bottom left) ERG rod (bottom left corner) and cone (bottom left middle) response for the asymptomatic individual. ERG rod peak-to-peak amplitude is 229.0 (normal 72–243) and implicit time is 89.0 ms (normal 69.4– 88.2). ERG cone response for the asymptomatic patient peak-to-peak amplitude 178.7 (normal 42–168) and implicit time is 30.0 (normal 24.−31.7). (Bottom right) ERG rod responses (bottom right middle) are absent for the symptomatic individual, but cone amplitude (bottom right corner) is relatively preserved (peak to peak amplitude 38.4, implicit time 44), as is characteristic until advanced stages of the disease.
The age at which symptomatic patients first noticed either night blindness or field loss varied from ages 6–71, with most patients reporting disease onset within the first 3 decades of life. A subset of 5 patients noticed symptoms in early childhood, and a subset of 5 patients noticed symptoms after age 40 (Figure 2). There was no apparent relationship between the specific type of PRPF31 mutation and age of onset (Supplemental Figure 1).
FIGURE 2.
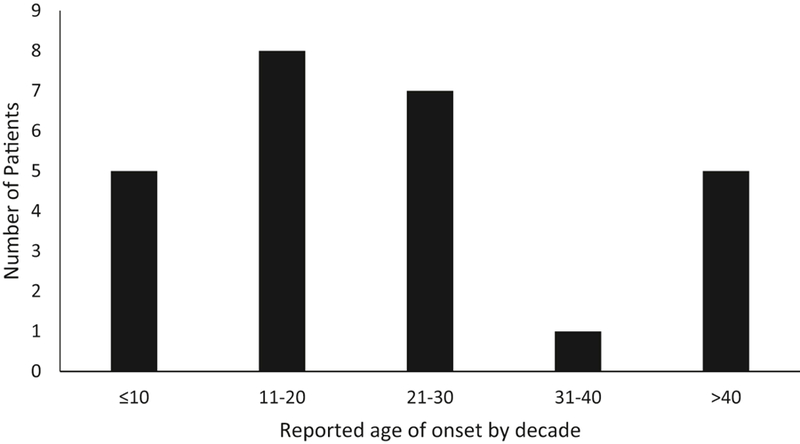
Patient reported age of onset, by decade. Most patients first report symptoms starting in their teens or twenties, with a subset noticing symptoms later in life. The subjective nature of age of onset does not fully account for the variability in patient reported age of onset, as several patients who noticed symptoms late in life had early stage disease at initial presentation.
Fundus appearance of all affected individuals was consistent with retinitis pigmentosa, with waxy pallor of the optic nerve, retinal vessel attenuation, and bone spicule pigment (Figure 3). Patient-reported disease duration of the affected individuals in our cohort varied between 0 and 49 years (mean 21 ± 15 years).
FIGURE 3.
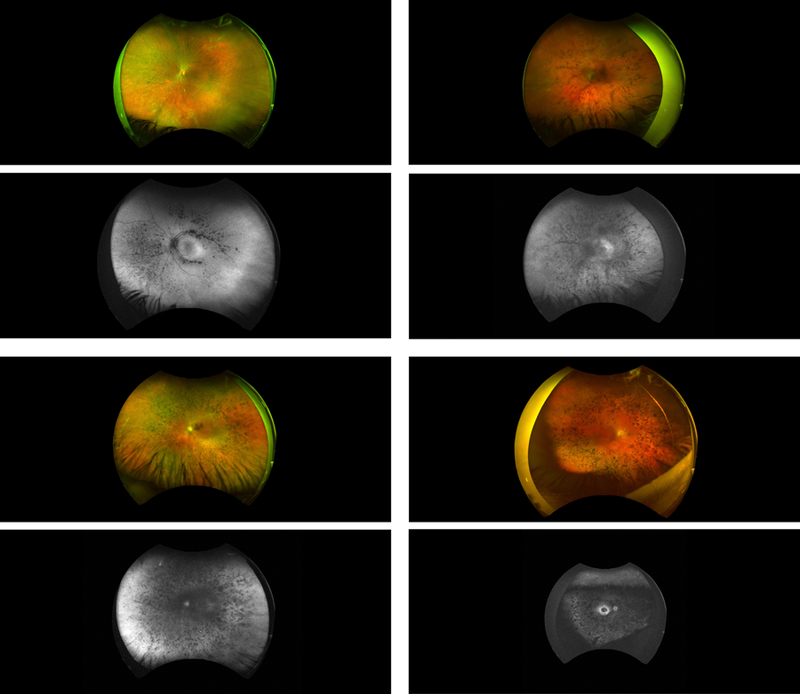
Representative wide field color fundus images and autofluorescence images for several patients. Note the bone spicule pigment, optic nerve pallor, and attenuation of the retinal vessels. (Top left) Fundus photograph and autofluorescence for P4, female, 40 years old, 12 years reported disease duration. (Bottom left) P17, female, age 67, 29 years reported disease duration. (Top right) P26, female, age 52, 36 years reported disease duration. (Bottom right) P15, female, age 67, 38 years reported disease duration.
Fifteen patients were included in group A, the “reliable reporters group,” with childhood onset or early stage disease at initial presentation. Visual field area, ERG cone response amplitude, and EZ width were found to decline exponentially (Figure 4). Visual acuity was poorly correlated with disease duration.
FIGURE 4.
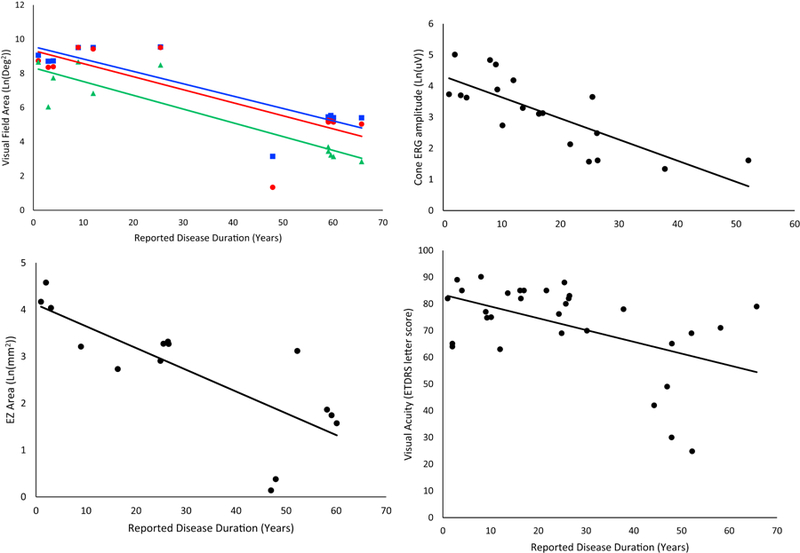
Rates of change for patients in group A, the “reliable reporters.” All available data is represented in the following figures, including data from multiple visits for some patients. The eye with better visual acuity for each patient, or the left eye, if the visual acuity is the same in both eyes, was used in this analysis. Acuity declined linearly and is shown on an ETDRS number scale. All other metrics declined exponentially and are shown transformed linearly by natural log. (Top left) Visual field area declined exponentially for all stimuli. Goldmann equivalent V (blue square) declined at a rate of 7.2% per year of disease duration (95% CI 4.2–10.2, R2=0.74, p=0.00032). Field area for equivalent III (red circle) declined at a rate of 7.6% per year of disease duration (95% CI 3.5–11.8, R2=0.63, p=0.002). Field area for equivalent I (green triangle), the smallest stimulus, declined at a rate of 8% per year of disease duration (95% CI 5.2–10.9, R2=0.82, p=0.0001). (Top right) Cone ERG amplitude, shown as a function of natural log, declined exponentially at a rate of 6.8% per year (95% CI 4.0–9.6, R2=0.61, p=8×10−5). (Bottom left) EZ area, shown as a function of natural log, declined exponentially at a rate of 4.6% per year (95% CI 2.4–6.9, R2=0.61, p=0.0006). (Bottom right) Acuity was poorly correlated with disease duration but demonstrated linear kinetics with a decline of 0.44% per year (95% CI 0.15–0.72, R2=0.25, p=0.004).
Eleven patients, all of whom had visual fields <20 degrees diameter as well as absent ERG rod response at their initial visit, were included in group B, the “less reliable reporters group.” An indicator variable revealed that there was a statistically significant difference in the relationship between patient reported disease duration and ocular function between groups A and B. Thus, patient reported disease duration is unreliable for group B. The same analysis was conducted using age in place of patient reported disease duration. Age was also found to be an unreliable measure. Disease duration for patients in group B was then estimated as a function of EZ width and ERG cone response from group A. The two measurements were demonstrated to have a high level of agreement, and both indicated a longer disease duration than the patient reported. The indicator variable was not statistically significant when reported (A) and estimated (B) duration were assessed as a function of visual acuity, fields, cone response, and EZ width, suggesting that the estimated disease duration is an effective means to correct for unreliability in patient-reported disease duration. Thus, the following results assess ocular parameters as a function of a combination of patient-reported disease duration (for patients in group A, n=15) and estimated disease duration (for patients in group B, n=9).
Kinetic visual fields were measured for 12 patients, and serial data was available for 6 patients. Visual field area declined exponentially for all stimuli. For spot size V, area declined at a rate of 8.0% per year of disease duration (95% CI 5.6–10.0, p<0.001). For spot size III, visual field area declined at a rate of 8.1% per year of disease duration (95% CI 5.6–10.6, p<0.001). For spot size I, visual field area declined at a rate of 8.4% (95% CI 6.2–10.7, p<0.001) per year of disease duration (Figure 5).
FIGURE 5.
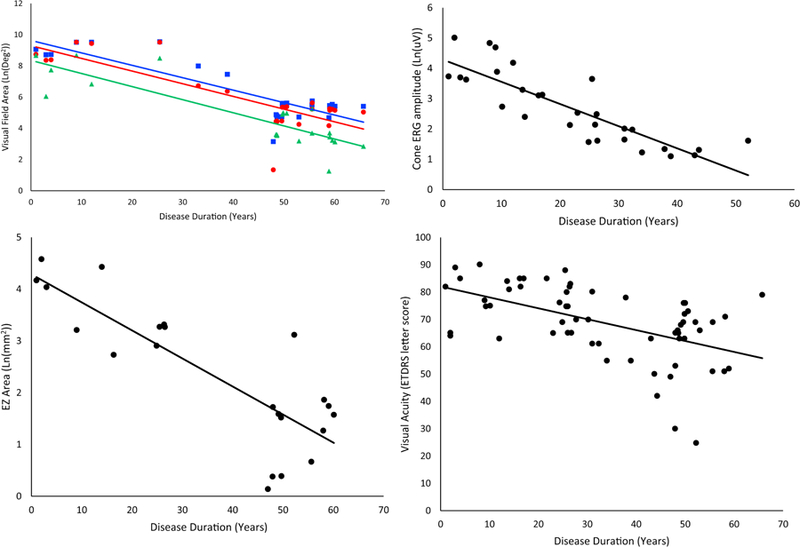
Rates of change as reported for patients in group A and as estimated for patients in group B. All available data is represented in the following figures, including data from multiple visits for some patients. The eye with better visual acuity for each patient, or the left eye, if the visual acuity is the same in both eyes, was used in this analysis. (Top left) Visual field area declined exponentially for all stimuli and is represented here transformed linearly by natural log. Goldmann equivalent V (blue square) declined at a rate of 8.0% per year (95% CI 5.6–10.0, R2=0.74, p=3×10−8). Equivalent III (red circle) declined at a rate of 8.1% per year (95% CI 5.6–10.6, R2=0.67, p=7×10−7). Equivalent I4e (green triangle), the smallest stimulus, declined at a rate of 8.4% per year (95% CI 6.2–10.7, R2=0.76, p=1×10−7). (Top right) Cone ERG amplitude, shown as a function of natural log, declined exponentially at a rate of 7.3% per year (95% CI 5.4–9.1, R2=0.72, p=5×10−9). (Bottom left) EZ area, shown as a function of natural log, declined at a rate of 5.4% per year (95% CI 3.7–7.1, R2=0.68, p=2×10−6) (Bottom right) Visual acuity, represented on an ETDRS scale, where points are assigned for letters guessed correctly; 85 is equivalent to 20/20 vision, 70 is equivalent to 20/40, and 50 is 20/100. Acuity declined linearly at a rate of 0.4% per year (95% CI 0.23–0.57, R2=0.27, p=2×10−5), which is the equivalent of seeing one less letter on the chart every 2 years.
Regarding the rod and cone ERG waveforms, 21 patients had no detectable rod b-wave at their initial visit, while 5 patients had rod b-wave amplitude severely reduced in amplitude. The 5 asymptomatic patients had rod responses within normal limits. Only 6 patients had a non-recordable cone response on their initial visit, suggesting that rod function deteriorates before cone function. For those symptomatic individuals with detectable cone ERG responses on initial presentation (n=19), the ERG cone response was found to decline exponentially at a rate of 7.3% per year of disease duration (95% CI 5.4–9.1, p<0.001) (Figure 5).
Excluding asymptomatic individuals, EZ area was measurable for 15 patients, and serial data available for 8 patients. EZ area was found to decline exponentially at a rate of 5.4% per year of disease duration (95% CI 3.7–7.1, p<0.001) (Figure 5).
Visual acuity was measured for all 26 symptomatic patients, and it was checked on multiple occasions separated by at least 6 months for 13 patients. It varied for individual patients and scores would often increase and decrease by 2 or more Snellen lines several times during a patient’s disease course. Although it was poorly correlated with disease duration, there appeared to be an overall decline of visual acuity of approximately 0.4% per year (95% CI= 0.23–0.57, p<0.001) after symptom onset (Figure 5), which translates into correctly identifying one fewer letter on an ETDRS chart, with score dropping one point, (where a score of 85=20/20 and 50=20/100) roughly every two years.
Discussion
In this study, we demonstrate that PRPF31-mediated RP is characterized by a variable age of onset, but disease progresses at a constant, measurable rate once it develops.
Many factors, such as allelic heterogeneity and genetic modifiers, may contribute to the wide age range at which patients with PRPF31 mutations develop symptoms. Other forms of neurodegeneration mediated by haploinsufficiency, for instance, frontotemporal dementia linked to progranulin mutations, have been also been associated with a highly variable age of onset.30 It has been hypothesized that haploinsufficiency itself leads to these differences, as some wild type alleles may be better able to compensate for the defective allele than others.11, 12, 31, 32
Other genetic modifiers may also play a role in the phenotypic presentation of PRPF31-mediated RP. It has been suggested that CNOT3, a transcriptional regulator, may play a protective role in asymptomatic individuals with PRPF31 mutations by directly modulating PRPF31 expression.11, 33, 34
The variable age of onset necessitates the use of a different metric, other than the patient’s age, for tracking visual function over time. Our results support the use of disease duration as a means of tracking clinical progression. Additionally, our results support the use of ERG and EZ width data to perform this estimation, as an indicator variable was statistically insignificant between groups A and B when reported duration was used for A and estimated duration was used for B.
In this study, visual field area, ERG cone response, and EZ area all declined exponentially, following a pattern of first order kinetics consistent with that observed in retinitis pigmentosa in general.35–37 Visual acuity was only weakly correlated with a linear pattern of decline. It was noted that individuals had inconsistent visual acuity at repeat visits. Many factors such as cataracts, surgeries, and fluctuations in macular edema may contribute to variable visual acuity, making it a less reliable endpoint than the others considered in this study. The rates of change from this study differ slightly from those previously reported for autosomal dominant RP,26, 35, 37, 38 and from those determined previously for PRPF31.17 However, the difference is likely accounted for by our use of disease duration as a more accurate metric than age.
Our finding of exponential kinetics for visual fields, ERG responses, and EZ area support the one hit model of cell death proposed by Clark et al, in which any individual photoreceptor has a constant or decreasing probability of apoptosis over time.39, 40 There may be some triggering event, such as trauma, systemic infection, or UV exposure, which sets off this cascade of random cell death, resulting in a “critical age” at which retinal deterioration begins.36 It is likely that this model applies to other forms of RP, with an exponential decline in function after a certain age,41 and PRPF31 represents an extreme example of the model.
Supplementary Material
SUPPLEMENTAL FIGURE 1. Age of onset by genetic variant. Genetic variants are grouped together by color, with asymptomatic individuals represented by white bars outlined with the color assigned to their variant. Patient reported age of onset varies widely, even within families. The numbers at the bottom indicate patient number, as outlined in the baseline characteristics table. Variants are as follows: P1–2 c.527+1G>T; P3–5 c.527+3A>G; P7–12 c.1073+1G>A; P13–15 c.−3_7del (p.Met1?); P16 c.550_552del (p.Leu184del); P17 c.758_767del (p.Gly253fs*317); P18 c.1084delA (p.Met362X); P19–22 c.895T>C (p.Cys299Arg); P22–26 exon1indel: 149 bp deleted/640 bp inserted; P27–29 c.390delC (p.Asn131fsX197); P30 c.525_526insAG; P31 c.220C>T (p.Gln74X)
{kind=link}
Acknowledgements
Supported by National Eye Institute/NIH EY09076, The Foundation Fighting Blindness, UT Southwestern Medical Center, the Max and Minnie Tomerline Voelcker Fund, NIH grant EY007142, and a grant from the William Stamps Farish Fund. David G Birch is a consultant for Nightstar, Inc., Lexington, MA; AGTC, Alachua, FL; Nacuity Pharmaceuticals, Ft. Worth, TX; Editas Medicine, Inc.; Ionis Pharmaceuticals, Inc., Carlesbad, CA; Acucela Inc., Seattle, WA. The following authors have no financial interests to disclose: Kelly Kiser, Kaylie Webb-Jones, Sara Bowne, Lori Sullivan, Stephen Daiger. All authors attest that they meet the current ICMJE criteria for authorship. We thank the family members for their participation in this study and the members of Retina Foundation of the Southwest Rose Silverthorne Retinal Degenerations lab for support and advice.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet 2006;368(9549):1795–809. [DOI] [PubMed] [Google Scholar]
- 2.Daiger SP, Sullivan LS, J Bowne SJ. Genes and mutations causing retinitis pigmentosa. Clin Genet 2013;84(2):132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dias MF, Joo K, Kemp JA, et al. Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Prog Retin Eye Res 2018;63:107–131. [DOI] [PubMed] [Google Scholar]
- 4.Daiger SP, Bowne SJ, Sullivan LS. Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmol 2007;125(2):151–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruzickova S, Stanek D. Mutations in spliceosomal proteins and retina degeneration. RNA Biol 2017;14(5):544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell 2009;136(4):701–18. [DOI] [PubMed] [Google Scholar]
- 7.Wang ET, Sandberg R, Luo S, et al. Alternative isoform regulation in human tissue transcriptomes. Nature 2008;456(7221):470–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Cauwenbergh C, Coppieters F, Roels D, et al. Mutations in Splicing Factor Genes Are a Major Cause of Autosomal Dominant Retinitis Pigmentosa in Belgian Families. PLoS One 2017;12(1):e0170038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sullivan LS, Bowne SJ, Seaman CR, et al. Genomic rearrangements of the PRPF31 gene account for 2.5% of autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci 2006;47(10):4579–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinez-Gimeno M, Gamundi MJ, Hernan I, et al. Mutations in the pre-mRNA splicing-factor genes PRPF3, PRPF8, and PRPF31 in Spanish families with autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci 2003;44(5):2171–7. [DOI] [PubMed] [Google Scholar]
- 11.Rose AM, Bhattacharya SS. Variant haploinsufficiency and phenotypic non-penetrance in PRPF31-associated retinitis pigmentosa. Clin Genet 2016;90(2):118–26. [DOI] [PubMed] [Google Scholar]
- 12.Tanackovic G, Ransijn A, Thibault P, et al. PRPF mutations are associated with generalized defects in spliceosome formation and pre-mRNA splicing in patients with retinitis pigmentosa. Hum Mol Genet 2011;20(11):2116–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 2008;40(12):1413–5. [DOI] [PubMed] [Google Scholar]
- 14.Rio Frio T, Wade NM, Ransijn A, Berson EL, Beckmann JS, Rivolta C. Premature termination codons in PRPF31 cause retinitis pigmentosa via haploinsufficiency due to nonsense-mediated mRNA decay. J Clin Invest 2008;118(4):1519–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deery EC, Vithana EN, Newbold RJ, et al. Disease mechanism for retinitis pigmentosa (RP11) caused by mutations in the splicing factor gene PRPF31. Hum Mol Genet 2002;11(25):3209–19. [DOI] [PubMed] [Google Scholar]
- 16.Bujakowska K, Maubaret C, Chakarova CF, et al. Study of gene-targeted mouse models of splicing factor gene Prpf31 implicated in human autosomal dominant retinitis pigmentosa (RP). Invest Ophthalmol Vis Sci 2009;50(12):5927–33. [DOI] [PubMed] [Google Scholar]
- 17.Hafler BP, Comander J, Weigel DiFranco C, Place EM, Pierce EA. Course of Ocular Function in PRPF31 Retinitis Pigmentosa. Semin Ophthalmol 2016;31(1–2):49–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pensado A, Diaz-Corrales FJ, De la Cerda B, et al. Span poly-L-arginine nanoparticles are efficient non-viral vectors for PRPF31 gene delivery: An approach of gene therapy to treat retinitis pigmentosa. Nanomedicine 2016;12(8):2251–2260. [DOI] [PubMed] [Google Scholar]
- 19.Sullivan LS, Bowne SJ, Birch DG, et al. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: a screen of known genes in 200 families. Invest Ophthalmol Vis Sci 2006;47(7):3052–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bowne SJ, Sullivan LS, Avery CE, et al. Mutations in the small nuclear riboprotein 200 kDa gene (SNRNP200) cause 1.6% of autosomal dominant retinitis pigmentosa. Mol Vis 2013;19:2407–17. [PMC free article] [PubMed] [Google Scholar]
- 21.Bowne SJ, Sullivan LS, Gire AI, et al. Mutations in the TOPORS gene cause 1% of autosomal dominant retinitis pigmentosa. Mol Vis 2008;14:922–7. [PMC free article] [PubMed] [Google Scholar]
- 22.Churchill JD, Bowne SJ, Sullivan LS, et al. Mutations in the X-linked retinitis pigmentosa genes RPGR and RP2 found in 8.5% of families with a provisional diagnosis of autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci 2013;54(2):1411–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gire AI, Sullivan LS, Bowne SJ, et al. The Gly56Arg mutation in NR2E3 accounts for 1–2% of autosomal dominant retinitis pigmentosa. Mol Vis 2007;13:1970–5. [PubMed] [Google Scholar]
- 24.Gregori NZ, Feuer W, Rosenfeld PJ. Novel method for analyzing snellen visual acuity measurements. Retina 2010;30(7):1046–50. [DOI] [PubMed] [Google Scholar]
- 25.Marmor MF, Fulton AB, Holder GE, Miyake Y, Brigell M, Bach M. ISCEV Standard for full-field clinical electroretinography (2008 update). Doc Ophthalmol 2009;118(1):69–77. [DOI] [PubMed] [Google Scholar]
- 26.Cai CX, Locke KG, Ramachandran R, Birch DG, Hood DC. A comparison of progressive loss of the ellipsoid zone (EZ) band in autosomal dominant and x-linked retinitis pigmentosa. Invest Ophthalmol Vis Sci 2014;55(11):7417–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hood DC, Ramachandran R, Holopigian K, Lazow M, Birch DG, Greenstein VC. Method for deriving visual field boundaries from OCT scans of patients with retinitis pigmentosa. Biomed Opt Express 2011;2(5):1106–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Birch DG, Locke KG, Felius J, et al. Rates of decline in regions of the visual field defined by frequency-domain optical coherence tomography in patients with RPGR-mediated X-linked retinitis pigmentosa. Ophthalmology 2015;122(4):833–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Birch DG, Locke KG, Wen Y, Locke KI, Hoffman DR, Hood DC. Spectral-domain optical coherence tomography measures of outer segment layer progression in patients with X-linked retinitis pigmentosa. JAMA Ophthalmol 2013;131(9):1143–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petkau TL, Leavitt BR. Progranulin in neurodegenerative disease. Trends Neurosci 2014;37(7):388–98. [DOI] [PubMed] [Google Scholar]
- 31.Rivolta C, McGee TL, Rio Frio T, Jensen RV, Berson EL, Dryja TP. Variation in retinitis pigmentosa- 11 (PRPF31 or RP11) gene expression between symptomatic and asymptomatic patients with dominant RP11 mutations. Hum Mutat 2006;27(7):644–53. [DOI] [PubMed] [Google Scholar]
- 32.Vithana EN, Abu-Safieh L, Pelosini L, et al. Expression of PRPF31 mRNA in patients with autosomal dominant retinitis pigmentosa: a molecular clue for incomplete penetrance? Invest Ophthalmol Vis Sci 2003;44(10):4204–9. [DOI] [PubMed] [Google Scholar]
- 33.Venturini G, Rose AM, Shah AZ, Bhattacharya SS, Rivolta C. CNOT3 is a modifier of PRPF31 mutations in retinitis pigmentosa with incomplete penetrance. PLoS Genet 2012;8(11):e1003040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rose AM, Shah AZ, Venturini G, Rivolta C, Rose GE, Bhattacharya SS. Dominant PRPF31 mutations are hypostatic to a recessive CNOT3 polymorphism in retinitis pigmentosa: a novel phenomenon of “linked trans-acting epistasis”. Ann Hum Genet 2014;78(1):62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berson EL, Sandberg MA, Rosner B, Birch DG, Hanson AH. Natural course of retinitis pigmentosa over a three-year interval. Am J Ophthalmol 1985;99(3):240–51. [DOI] [PubMed] [Google Scholar]
- 36.Massof RW, Dagnelie G, Benzschawel T, Palmer RW, Finkel stein D. First order dynamics of visual field loss in retinitis pigmentosa. Clinical Vision Sciences 1990;5(1):1–26. [Google Scholar]
- 37.Birch DG, Anderson JL, Fish GE. Yearly rates of rod and cone functional loss in retinitis pigmentosa and cone-rod dystrophy. Ophthalmology 1999;106(2):258–68. [DOI] [PubMed] [Google Scholar]
- 38.Berson EL, Rosner B, Weigel-DiFranco C, Dryja TP, Sandberg MA. Disease progression in patients with dominant retinitis pigmentosa and rhodopsin mutations. Invest Ophthalmol Vis Sci 2002;43(9):3027–36. [PubMed] [Google Scholar]
- 39.Clarke G, Collins RA, Leavitt BR, et al. A one-hit model of cell death in inherited neuronal degenerations. Nature 2000;406(6792):195–9. [DOI] [PubMed] [Google Scholar]
- 40.Burns J, Clarke G, Lumsden CJ. Photoreceptor death: spatiotemporal patterns arising from one- hit death kinetics and a diffusible cell death factor. Bull Math Biol 2002;64(6):1117–45. [DOI] [PubMed] [Google Scholar]
- 41.McGuigan DB, Heon E, Cideciyan AV, et al. EYS Mutations Causing Autosomal Recessive Retinitis Pigmentosa: Changes of Retinal Structure and Function with Disease Progression. Genes (Basel) 2017;8(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTAL FIGURE 1. Age of onset by genetic variant. Genetic variants are grouped together by color, with asymptomatic individuals represented by white bars outlined with the color assigned to their variant. Patient reported age of onset varies widely, even within families. The numbers at the bottom indicate patient number, as outlined in the baseline characteristics table. Variants are as follows: P1–2 c.527+1G>T; P3–5 c.527+3A>G; P7–12 c.1073+1G>A; P13–15 c.−3_7del (p.Met1?); P16 c.550_552del (p.Leu184del); P17 c.758_767del (p.Gly253fs*317); P18 c.1084delA (p.Met362X); P19–22 c.895T>C (p.Cys299Arg); P22–26 exon1indel: 149 bp deleted/640 bp inserted; P27–29 c.390delC (p.Asn131fsX197); P30 c.525_526insAG; P31 c.220C>T (p.Gln74X)