

Abstract
Neuropeptides are an important class of signaling molecules in the nervous and neuroendocrine system, but they are challenging to study due to their low concentration in vivo in the presence of numerous interfering artifacts. Often the limitation of mass spectrometry analyses of neuropeptides in complex tissue extracts is not due to neuropeptides being below the detection limit but due to ions not being selected for tandem mass spectrometry during the liquid chromatography elution time and therefore not being identified. In this study, a data independent acquisition (DIA) method was developed to improve the coverage of neuropeptides in neural tissue from the model organism C. borealis. The optimal mass-to-charge ratio range and isolation window were determined and subsequently used to detect more neuropeptides in extracts from the brain and pericardial organs than the conventional data dependent acquisition method. The DIA method led to the detection of almost twice as many neuropeptides in the brain and approximately 1.5-fold more neuropeptides in the pericardial organs. The technical and biological reproducibility were also explored and found to be improved over the original method, with 56% of neuropeptides detected in 3 out of 3 replicate injections and 62% in 3 out of 3 biological replicates. Furthermore, 68 putative novel neuropeptides were detected and identified with de novo sequencing. The quantitative accuracy of the method was also explored. The developed method is anticipated to be useful for gaining a deeper profiling of neuropeptides, especially those in low abundance, in a variety of sample types.
Graphical Abstract
Neuropeptides are important signaling molecules in the nervous and neuroendocrine system that act both locally and long-range to modulate and regulate various biological activities.1 These short-chain amino acid sequences represent a diverse class of biological molecules responsible for the modulation of various functions such as feeding2,3 and adaptation to external stresses.4,5 Dysregulation of the expression of neuropeptides can contribute to neurological diseases by altering the behavior of neurons, and so a more comprehensive characterization of neuropeptides is essential to the treatment of these diseases.6 However, the complexity and wide variety in characteristics of neuropeptides, in addition to their low abundance and propensity toward rapid degradation, make them difficult to study.
In order to characterize both sequence information and function of neuropeptides, it is necessary to use a model organism that simplifies the experimental matrix and enables samples to be reliably collected. The Jonah crab, Cancer borealis, provides a useful model for mass spectrometric investigation into the neuropeptidome and its changes in response to external stimuli, as its simple yet elegant nervous system has already been well-characterized with electrophysiological studies,7–9 and many neuropeptides in crustaceans are direct homologues to mammalian neuropeptides (e.g., the allatostatin family is homologous to the mammalian galanin family10 and RFamide and tachykinin families are conserved across both invertebrates and vertebrates). Furthermore, despite not yet having a completely sequenced genome, an extensive database of neuropeptides has been compiled through a combination of multifaceted mass spectrometry (MS) and in silico prediction approaches.11,12
Despite these advantages, neuropeptides are difficult to study, even in model organisms such as crustaceans. Neuropeptides are highly variable, and there are numerous neuropeptide isoforms belonging to each family, often differing only by a single amino acid. These isoforms can have identical or drastically different effects in the nervous and neuroendocrine systems. For example, the C-type allatostatin (AST-C) neuropeptides I, II, and III have all been evaluated in the modulation of lobster heart rhythm in a recent study, and AST-C I and AST-C III were shown to have similar, though not identical, functionality, while AST-C II had largely contrasting effects, despite the high degree of sequence similarity.13 Moreover, the same neuropeptide can have varying effects in different parts of the nervous/neuroendocrine system or at different concentrations, such as pyrokinin neuropeptide PevPK2, whose effect on cardiac amplitude and frequency changes with varying dosage concentrations.14
MS has been demonstrated to be effective at discriminating between similar isoforms of neuropeptides and providing relative changes in abundance between samples, and many studies have been successfully implemented to both profile neuropeptides in crustacean species and assess their changes in response to various stimuli.15–18 However, current methodologies are still limited in the depth at which they can profile the neuropeptidome and reproducibly detect low-abundance neuropeptides. Though these challenges stem from several factors, a major influence is inherent limitations in the way the samples are analyzed via MS. Up until now, data dependent acquisition (DDA) has been predominantly used to select ions for tandem MS (MS/MS) fragmentation, in which only the highest-abundance ions are selected and fragmented for subsequent identification. As a result, the method is largely biased toward high-abundance species, often selecting interfering artifacts such as lipids or protein fragments over low-abundance yet biologically active neuropeptides, limiting the total number of neuropeptides detected. Furthermore, slight variations in relative abundance of ions, either due to differing conditions, natural biological variability, or even slight differences between repeat injections, alter which ions are selected for MS/MS, limiting the reproducibility of the method.
Alternative methods have been investigated for addressing these limitations, such as using a targeted approach, in which ions are only fragmented if they are included on a list.19 This method works well if all neuropeptides of interest are known before analyzing the samples. However, this method is not suitable for discovery-based neuropeptidomic studies and largely limits the breadth of analysis. Data independent acquisition (DIA) is emerging as a promising alternative strategy that provides balance between the sensitivity and selectivity of targeted MS methods while retaining the breadth of coverage offered by DDA methods.20–28 In DIA methods, a large window of ions is simultaneously fragmented across an entire mass range, enabling theoretically comprehensive fragmentation of all ions in a sample in a reproducible and nonspecific manner. While the method is easy to implement on the instrument, data analysis has been difficult due to the lack of precursor ion information. The most common method of analysis has been to use spectral libraries that typically are generated by first analyzing the samples using a DDA method.29 While this helps to reduce run-to-run variability, relying on DDA for spectral libraries retains the bias toward high-abundance neuropeptides.
Recently, an alternative method of analysis has emerged in which no spectral libraries are required for data analysis. These methods use sophisticated computational algorithms to parse complex DIA spectra that typically contain fragments from several ions. The two most prominent software for this are DIA-Umpire and PECAN. DIA-Umpire uses deconvolution of MS/MS spectra over time to map fragments to individual precursors, thereby creating “pseudo MS/MS spectra” that are similar to typical DDA MS/MS spectra.30 PECAN is part of the open-source data analysis platform Skyline and uses a dot-product scoring method for database searching.31 These methods offer the ability to perform discovery-based analyses in a reproducible, unbiased manner.
This study sought to develop a DIA method to improve the coverage of neuropeptides detected in C. borealis neural tissue in a manner that is reproducible both at the technical and biological level and does not rely on the generation of spectral libraries. The resulting method almost doubled the number of neuropeptides detected that are present in the database and demonstrated improved reproducibility when directly compared to the conventional DDA method. The method also demonstrated improved results compared to previous profiling of the C. borealis neuropeptidome, despite the previous study combining multiple MS methods.11 Furthermore, 68 putative novel neuropeptides not currently present in the database were detected and identified with de novo sequencing. The method was also evaluated for quantitative accuracy and found to be acceptable for recognizing large changes in abundance between samples.
METHODS
Instrumental Analysis.
See the Supporting Information for details of chemicals and materials, animal dissection, tissue collection, and sample preparation. Samples were reconstituted in 15 μL of 0.1% FA in water (Optima-grade) for MS analysis, with the exception of the brain and pericardial organ (PO) samples for method optimization, which were reconstituted in 90 μL so that all methods could be tested on injections from the same vial. Samples were analyzed on a Thermo Scientific Q Exactive instrument (Thermo Scientific, Bremen, Germany) coupled to a nano-ESI source connected to a Waters nanoAcquity LC system (Waters Corp, Milford, MA, USA). LC separation was carried out using a 15 cm self-packed C18 column with a 75 μm internal diameter and 1.7 μm particle size. Water with 0.1% FA and acetonitrile with 0.1% FA were used as mobile phases A and B, respectively. The flow rate was set to 0.300 μL/min. Two μL of sample was injected onto the column and separated over a 120 min gradient as follows: 0–1 min 3–10% B; 1–90 min 10–35% B; 90–92 min 35–95% B; 92–102 min 95% B; 102–105 min 95–3% B; 105–120 min 3% B.
For MS analysis, 6 different acquisition methods were used: 2 DDA methods and 4 DIA methods. All MS acquisition methods used positive electrospray ionization (ESI) with a collision energy of 30 eV, with 70,000 resolution for MS scans and 17,500 resolution for MS/MS scans. The MS/MS scan range was adjusted depending on the parent mass or isolation window, with 50 m/z being the first fixed mass. The DDA methods were set to top 10 and top 20 for precursor ion selection, with an isolation window of 2 m/z and dynamic exclusion set to 40 s. The MS scan range was 300–2000 m/z. The DIA methods varied in their MS scan ranges and isolation widths: scan range of 250 to 850 m/z with a 20 m/z isolation width; scan range of 250 to 930 m/z with an isolation width of 20 m/z for 250 to 510 m/z and 60 m/z for 510 to 930 m/z; scan range of 250 to 1000 m/z with an isolation width of 30 m/z for 250 to 700 m/z and 60 m/z for 700 to 1000 m/z; scan range of 400 to 800 m/z with an isolation width of 20 m/z.
Data Analysis.
Resulting MS spectra from the DIA methods were first converted to the mzXML file format using MSConvert32 and processed with DIA-Umpire30 using the default parameters. The output mgf files from DIA-Umpire were converted to mzXML files again using MSConvert and processed in PEAKS 7.0 (Bioinformatics Solutions Inc., Waterloo, ON, CAN).33 The resulting DDA files were directly processed in PEAKS with no initial processing. The parameters used for PEAKS de novo sequencing and database searching were as follows: no enzyme cleavage specified, instrument orbi–orbi, HCD fragmentation, and precursor correction enabled. Modifications were set to include amidation, pyroglutamate, dehydration, and oxidation. All other parameters were set to the default. An in-house crustacean neuropeptide database was used for searches. For quantification, abundances were obtained by taking the area under the curve of MS1 peaks from the raw file with no normalization used. For identification of putative neuropeptide sequences, de novo sequences exported from PEAKS were filtered using PepExplorer for sequence similarity to the neuropeptide database and then manually filtered for characteristic sequence motifs.34 Only neuropeptides with an average local confidence (ALC) score greater than or equal to 75% were included.
RESULTS AND DISCUSSION
Optimization of Method Isolation Window and m/z Range.
DIA performs its acquisition in a cyclic manner in which it selects a window of ions at a time for fragmentation, sequentially working through an entire mass range, then cycles back, and scans the range again, with intermittent MS scans within each loop. Therefore, the key factors affecting the detection efficacy of the method are the size of the mass range, the size of each isolation window, and the total number of scans in each loop. As the number of scans per loop is dependent upon the scan range and window size, this number was kept constant at 20 MS/MS scans in order to simplify the comparison of methods. The mass-to-charge ratio (m/z) scan range and isolation window were varied based on the frequency of m/z values commonly detected for neuropeptide samples. Histogram plots were constructed for several neuropeptide samples previously analyzed, and it was found that most detected neuropeptides fall within the m/z range of 450 to 800, with a small number appearing outside this window and substantially decreasing in frequency further from this number. Very few neuropeptides were detected with m/z values greater than 1000, and so this number was the upper limit tested with the method. The window sizes were determined based on the mass range, with narrower windows for m/z values with a higher density and wider windows for lower density ranges.
A total of 4 DIA methods were tested for two neuropeptide sample types. The samples used were C. borealis brain and PO extracts, which typically have different neuropeptide compositions. The first method used a variable window optimized based on masses of commonly detected neuropeptides, mass range 250 to 930 m/z, window size 20 m/z for lower mass range (250 to 510 m/z), and window size 60 m/z for upper mass range (510 to 930 m/z). The second method also used a variable window but with a larger mass range that encompassed close to the entire range of m/z values detected for neuropeptide samples, 250 to 1000 m/z, with a window size of 30 m/z for the lower mass range (250 to 700 m/z) and 60 m/z for the upper mass range (700 to 1000 m/z). The third method used a uniform window size of 30 m/z with a slightly smaller m/z range of 250 to 850 m/z. The fourth method also used a uniform window size, but slightly smaller at 20 m/z, with the m/z range only encompassing the high-density m/z values detected for neuropeptide samples (400 to 800 m/z). For comparison, a conventional top-10 DDA method was also tested that reflected what has previously been optimized for neuropeptide samples. Additionally, a top-20 DDA method was also tested in order to have a more direct comparison with similar duty cycles between methods. A total of 20 isolation windows were used for method optimization because it coincides with the duty cycle typically employed on common mass spectrometers, thus enabling the method to be easily transferrable across instruments.
Overall, the results from the 4 DIA methods were largely similar to each other but showed a noticeable improvement over the DDA methods (Figure 1). The methods were evaluated based on the number of neuropeptides detected, the number detected with full sequence coverage, and the average score of detected neuropeptides. The total number of detected neuropeptides improved substantially from DDA to DIA. The DDA methods resulted in the detection of 100 to 150 neuropeptides, while the DIA methods resulted in the detection of over 200 neuropeptides (with the exception of one of the DIA methods for the brain sample that only detected 151 neuropeptides). Furthermore, the number of neuropeptides with 100% sequence coverage improved substantially with 3 of the 4 DIA methods. The DIA method encompassing the scan range 250 to 930 m/z did not detect as many neuropeptides as the other methods and had a much lower number with full sequence coverage, which is likely attributed to its bias toward low m/z values. The method incorporated a small isolation width at low m/z values and a large isolation width for large m/z values, which likely resulted in more fragment peptides being detected and fewer intact neuropeptides, resulting in a lower number of neuropeptides detected with full sequence coverage. The fourth method (scan range 400 to 800 m/z) showed the greatest improvement in detected neuropeptides (total and with full sequence coverage) for the PO samples and showed approximately the same results as the third method (scan range 250 to 850 m/z) for the brain samples. In order to ensure that the detected neuropeptides were reflecting quality identifications, the average PEAKS scores of identified neuropeptides were also compared between the methods, and all were in close proximity to each other, between 65 and 90 −10logP. Though all of the scores were similar, the fourth DIA method (scan range 400 to 800 m/z) had the highest average scores for both the brain and PO samples (85.33 and 89.35, respectively). Figure 2 shows three representative raw MS/MS spectra with annotated fragmentation peaks. While there appears to be some coisolation, as anticipated with DIA, the spectra are still very clear, and most of the backbone fragmentation can be identified without excessive interference from coisolating peaks. Based on these observations, it was concluded that the DIA method encompassing the scan range 400 to 800 m/z was the most appropriate to use for the data, and it was the one selected for all subsequent analyses. If the resulting MS/MS spectra acquired from DIA-MS analysis showed extensive coisolation or the detected neuropeptides were low-scoring, a follow-up targeted DDA method could be employed in order to confirm the identification of those neuropeptides. However, the MS/MS spectra displayed clear fragment peaks and most peptides were high-scoring, so the incorporation of a follow-up DDA analysis does not appear to be necessary and would not be worth the increased analysis time required.
Figure 1.

Comparison of the number of neuropeptides detected in the brain (blue) and in pericardial organs (POs) (red) using each acquisition method. The solid fills indicate the total number of neuropeptides detected, and the patterns indicate the number of neuropeptides detected with 100% sequence coverage.
Figure 2.

MS/MS spectra of three detected neuropeptides using the data independent acquisition (DIA) method, including (a) an orcokinin neuropeptide detected in the brain, (b) a tachykinin neuropeptide detected in the brain, and (c) an allatostatin B-type neuropeptide detected in the pericardial organs.
Comparison of Neuropeptides Detected in DIA and DDA Methods.
In order to ensure that the chosen DIA method was reliably detecting neuropeptides in each sample, the neuropeptides identified with the method were compared to those detected with the conventional top-10 DDA method. The total numbers of detected neuropeptides with each method were compared, as shown in Figure 3. As shown in the figure, there is a large amount of overlap in neuropeptides detected between the two methods, which indicates that the DIA method is comprehensive, not complementary to the DDA method.
Figure 3.
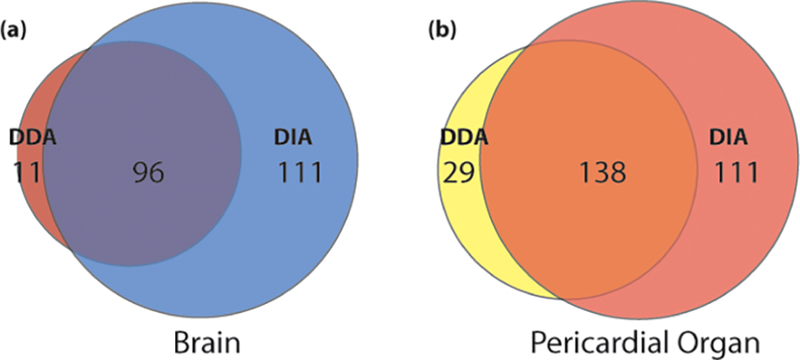
Overlap of neuropeptides detected using data dependent acquisition (DDA) and data independent acquisition (DIA) for (a) brain and (b) pericardial organ (PO) tissue extracts.
However, while there was very good overlap between the two methods, there was still a small fraction that were detected by DDA but not DIA. To make sure that there was no bias against certain types of neuropeptides, the neuropeptides not in common between the two methods were compared. The average score of neuropeptides detected with only DDA was 55.93 and 42.55 in the brain and POs, respectively, while the average score detected with only DIA was 62.98 and 71.26 in the brain and POs, respectively, indicating that the additional neuropeptides detected by DIA were not just low-scoring peptides likely to be false-positives. The average mass of neuropeptides detected with only DDA was 1194.33 and 1688.56 in the brain and POs, respectively, and the average m/z value detected with only DIA was 2832.625 and 2266.798 in the brain and POs, respectively. These numbers initially suggest that DDA and DIA have differential bias toward neuropeptide masses. However, there were more low-molecular weight neuropeptides detected in both tissues with DIA only than with DDA only, which shows that DDA is not more likely to detect low-molecular weight neuropeptides than DIA.
The potential bias toward specific neuropeptide families or sequence motifs was also investigated. For each neuropeptide detected in DDA only, there were more neuropeptides from the same family detected in DIA only. For example, in the brain sample, 7 RFamide neuropeptides were only detected in DDA, but 27 RFamide neuropeptides were only detected in DIA. In addition, there were several neuropeptide families detected only in DIA, but no families detected exclusively in DDA. In summary, the DIA method appears to generally detect more neuropeptides than DDA without any apparent systematic bias. Those neuropeptides detected only in DDA are likely due to technical variation between analyses and not due to limitations of the method.
Evaluation of Experimental and Biological Reproducibility.
In addition to detecting a wider range and greater number of neuropeptides, DIA is useful for improving run-to-run reproducibility both within and between experiments. In order to assess this, the experimental (or technical) reproducibility of the DIA method was compared to that of the top-10 DDA method. Replicate injections of the brain extract sample were compared to determine the overlap in detected neuropeptides, as shown in Figure S1 (see the Supporting Information). As can be seen, the DIA method showed much greater overlap between replicate injections, with 56% detected in every injection, compared to 32% detected in every injection with DDA.
The biological reproducibility of the DIA method was also explored. Neural tissue samples often contain a large amount of interfering biological matrices such as lipids, protein fragments, etc. These background interfering compounds can be present at different levels in different animals of the same species, which can affect which ions are selected for MS/MS fragmentation in samples from different animals. As a result, differences in identified neuropeptides may be incorrectly attributed to different neuropeptide compositions in different samples, which may not necessarily reflect the actual sample composition. It was suspected that using a DIA method would enable consistent identification of the same neuropeptides across different biological samples, with exceptions reflecting the true natural variability of individual animals. To test this, brains from 3 individual animals were extracted and analyzed separately using the DIA method, and the resulting detected neuropeptides were compared. Figure S2 (see the Supporting Information) shows the overlap in neuropeptides detected in the three biological replicates. As can be seen, the results indicate excellent reproducibility at the biological level, with 62% of neuropeptides detected in every biological replicate.
Evaluation of Quantitative Accuracy and Dynamic Range.
The data indicate that the method is very useful for improving the coverage of the neuropeptidome. To further evaluate its potential utility in neuropeptide analysis, the quantitative accuracy of the method was investigated. Pooled extracts from 15 C. borealis thoracic ganglia (TGs) were aliquoted into separate vials with concentration ratios of 0.5:1:1:2:5. The purpose of this experiment was to compare the abundance of neuropeptides across samples of different concentrations. TGs were used for the analysis because they are a larger tissue than the brain or POs and so offer a greater number of neuropeptides to be used for comparison. The samples were analyzed using the DIA method and quantified at the MS1 level by calculating the area under the curve of each MS1 peak. Ratios were calculated by dividing the peak area of each neuropeptide in each respective sample. The area of neuropeptides in the fifth sample was divided by those in the first sample to give a ratio of 1:10 without overloading the LC column. Figure S3 (see the Supporting Information) shows boxplots of the results obtained for each ratio. The experimental errors in the ratio for 1:1, 2:1, 5:1, and 10:1 were 18%, 23.5%, 32.8%, and 21.4%, respectively, indicating that this quantitation can be used to discriminate up and down regulation in the samples at large fold-changes. The quantitative accuracy can likely be substantially improved by the incorporation of chemical labeling, especially isotopic labeling where quantitation is performed at the MS1 level. However, labeling and combining samples increase spectral complexity, which would limit the ability of the DIA method to perform confident identifications. As a result, incorporating chemical labeling would require additional method optimization and may be explored in future studies.
Detection of Putative Novel Neuropeptides with De Novo Sequencing.
A key advantage of using DIA software that converts DIA spectra into pseudo DDA spectra is that it does not require spectral libraries to process the data, which enables the possibility of discovery of novel peptides via de novo sequencing. As the C. borealis genome is not yet completely sequenced, ensuring a complete neuropeptide profile relies on de novo sequencing for detection of putative novel neuropeptides. The DIA method has been shown to be useful for obtaining a deeper neuropeptidomic profile of neural tissue samples, and so it was expected that the method could also detect neuropeptides not previously identified by MS analysis and are therefore unknown. Putative, potentially interesting/novel neuropeptides were determined by looking for common sequence motif characteristics of neuropeptide families.
In the brain sample, 38 putative neuropeptides from 6 different families were detected, including orcomyotropin, orcokinin, pyrokinin, RFamide, tachykinin, and YRamide. There were 31 neuropeptides from 3 families in the PO sample, including B-type allatostatin, RFamide, and RYamide. Table S1 lists all putative novel neuropeptides detected in the brain samples, and Table S2 lists all putative novel neuro-peptides that have been detected in PO samples (see the Supporting Information). These results are consistent with what would be expected for each tissue. The brain is connected to the crustacean stomatogastric nervous system. Orcokinins are responsible for influencing hindgut contractions and modulating stomatogastric nervous system outputs, and therefore these neuropeptides tend to have a greater presence in the brain and stomatogastric nervous system.35 Orcomyotropin neuropeptides also modulate hindgut contractions, and pyrokinin neuropeptides have a role in the gastric mill rhythm, and so it is expected that neuropeptides belonging to these families would also be identified in the brain.35 B-type allatostatins function both locally and as secreted circulating hormones, and so they are commonly found in neurosecretory tissue such as the POs.35 Neuropeptides ending in −RFamide can be classified into a variety of subfamilies with various functions, and it is therefore not surprising that a large number of neuropeptides with this sequence motif were detected in both tissues.35 One neuropeptide from this family, SENRDFLRFamide, was identified in both tissues, indicating that it may function both locally in the central nervous system and long-range as a circulating hormone. Figure 4 shows three representative spectra of neuropeptides detected and identified with de novo sequencing with their key fragment ions annotated. Spectra of all de novo sequenced neuropeptides listed in Tables S1 and S2 are shown in Figures S4–S72 (Supporting Information). As with the spectra in Figure 2 for database-matched neuropeptides, there appears to be some coisolation with other ions, which is to be expected with DIA analysis because the isolation window is large. Each neuro-peptide was searched for in the comprehensive neuropeptide database NeuroPep,36 and no matches were found, indicating that these neuropeptides have not yet been identified in any species. The sequences did not match parts of longer sequences either, indicating that they are not simply degradation products from larger neuropeptides. Most of the putative novel neuropeptides listed possess the classic neuropeptide motif. The rest of the neuropeptides listed possess a similar motif but with the variation of one amino acid or variation in the presence of C-terminal amidation, indicating the possibility of a mutation that may alter the biological activity. Many other peptides were also sequenced with PEAKS software and did not possess similarity to common neuropeptide motifs. These peptides were not included in the list of putative neuropeptides, as they are likely degradation products from large proteins, but they may still be biologically active as nonclassical neuropeptides.37 Further studies of the precursor proteins and functional assessment of the peptides are necessary to properly classify them as nonclassical neuropeptides.37
Figure 4.

MS/MS spectra of three putative neuropeptides detected with the data independent acquisition (DIA) method, including (a) a tachykinin neuropeptide detected in the brain, (b) a pyrokinin neuropeptide detected in the brain, and (c) an RFamide neuropeptide detected in the pericardial organs.
CONCLUSIONS
In this study, DIA was explored as an alternative method to DDA to address current limitations in neuropeptide research. DIA’s key advantage of unbiased precursor selection for MS/MS fragmentation analysis allowed for a greatly improved coverage of the crustacean neuropeptidome without sacrificing the quality of neuropeptide identifications, and the inclusion of most DDA-detected neuropeptides indicated that DIA can be used alone without being accompanied by DDA analyses. The Thermo Q Exactive Obitrap MS used in this study enabled a duty cycle time of 3.6 s, which was less than half the width of most peaks in the chromatogram. The breadth and depth of coverage may be increased by additional optimization on more sophisticated instruments with faster scan times, which would improve the duty cycle, but the method will still likely be viable on instruments with slower scan times as long as the duty cycle time does not exceed the width of the peaks in the chromatogram. With a faster scan time, more MS/MS acquisitions could be made in the same amount of time, enabling either a larger m/z range or narrower m/z windows to be obtained. However, the duty cycle was not found to be a limitation of DIA compared to DDA, as with identical duty cycles, DIA still outperformed DDA. The depth of coverage could also be improved upon by implementing a multidimensional separation platform, such as by the incorporation of ion mobility MS for gas-phase separation, or incorporating an orthogonal front-end separation such as capillary electrophoresis.38–41 The additional level of separation would reduce the number of cofragmented ions in each spectra, potentially improving the number of and quality of identifications.
The consistent fragmentation of all ions within a given m/z range allowed for reproducible identifications between both technical replicates and biological replicates, making the data more rigorous and reproducible for neuropeptidome profiling. Furthermore, through the use of PEAKS de novo sequencing, the method was able to identify putative neuropeptides that have previously gone undetected, likely due to their low in vivo abundance compared to other neuropeptides. The 68 novel neuropeptides identified here indicate the power of DIA for providing a more complete assessment of a biological sample without depending upon spectral libraries. Additional studies will need to be carried out in order to validate these neuropeptides, such as by performing a targeted analysis and matching to a synthetic standard. Functional studies can also be implemented in order to assess the biological activity of the neuropeptides. The quantitative accuracy of the method was also evaluated and found to be satisfactory for obtaining general trends in up or down regulations. However, the accuracy of quantitation is limited and can likely be improved with the use of an internal standard or more sophisticated computational methods, such as those available by software tools.30,31 In general, DIA quantification would greatly benefit from the development of labeling methods using chemical tags, which would both improve quantitative accuracy and increase the multiplexing capabilities of the method.42–47 We anticipate that this DIA method will serve as a useful tool for profiling neuropeptides and other endogenous peptides in a variety of samples.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Science Foundation (CHE-1710140) and National Institutes of Health through grants R01DK071801 and R01NS029436. K.D. acknowledges a predoctoral fellowship supported by the National Institutes of Health, under Ruth L. Kirschstein National Research Service Award T32 HL 007936 from the National Heart Lung and Blood Institute to the University of Wisconsin–Madison Cardiovascular Research Center and the National Institutes of Health-General Medical Sciences F31 National Research Service Award (1F31GM126870–01A1) for funding. The Orbitrap instruments were purchased through the support of an NIH shared instrument grant (S10RR029531) and Office of the Vice Chancellor for Research and Graduate Education at the University of Wisconsin–Madison. L.L. acknowledges a Vilas Distinguished Achievement Professorship and Charles Melbourne Johnson Distinguished Chair Professorship with funding provided by the Wisconsin Alumni Research Foundation and University of Wisconsin–Madison School of Pharmacy.
ABBREVIATIONS
- MS
mass spectrometry
- DIA
data independent acquisition
- DDA
data dependent acquisition
- MS/MS
tandem MS
- PO
pericardial organ
- TG
thoracic ganglion
- m/z
mass to charge ratio
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.8b05734.
Details of animal dissection, tissue collection, and sample preparation; tables of putative neuropeptides detected with de novo sequencing in brain and pericardial organs; figures showing MS/MS spectra of all de novo sequences (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).DeLaney K; Buchberger AR; Atkinson L; Grunder S; Mousley A; Li LJ Exp. Biol 2018, 221 (3), jeb151167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Tachibana T; Tsutsui K Front. Neurosci 2016, 10, 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Audsley N; Weaver RJ Gen. Comp. Endocrinol 2009, 162 (1), 93–104. [DOI] [PubMed] [Google Scholar]
- (4).Li W; Papilloud A; Lozano-Montes L; Zhao N; Ye X; Zhang X; Sandi C; Rainer G Proteomics 2018, 18 (7), No. 1700408. [DOI] [PubMed] [Google Scholar]
- (5).Bowers ME; Choi DC; Ressler KJ Physiol. Behav 2012, 107 (5), 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Beal MF; Martin JB Ann. Neurol 1986, 20 (5), 547–565. [DOI] [PubMed] [Google Scholar]
- (7).Follmann R; Goldsmith CJ; Stein WJ Comp. Neurol 2017, 525 (8), 1827–1843. [DOI] [PubMed] [Google Scholar]
- (8).White RS; Spencer RM; Nusbaum MP; Blitz DM J. Neurophysiol 2017, 118 (5), 2806–2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Otopalik AG; Goeritz ML; Sutton AC; Brookings T; Guerini C; Marder E eLife 2017, 6, e22352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Birgül N; Weise C; Kreienkamp HJ; Richter D EMBO J 1999, 18 (21), 5892–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ma MM; Wang JH; Chen RB; Li LJ J. Proteome Res 2009, 8 (5), 2426–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Christie AE; Pascual MG Gen. Comp. Endocrinol 2016, 237, 53–67. [DOI] [PubMed] [Google Scholar]
- (13).Dickinson PS; Armstrong MK; Dickinson ES; Fernandez R; Miller A; Pong S; Powers BW; Pupo-Wiss A; Stanhope ME; Walsh PJ; Wiwatpanit T; Christie AE J. Neurophysiol 2018, 119 (5), 1767–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Dickinson PS; Sreekrishnan A; Kwiatkowski MA; Christie AE J. Exp. Biol 2015, 218, 2892–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zhang Y; Buchberger A; Muthuvel G; Li L Proteomics 2015, 15, 3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhang Y; DeLaney K; Hui L; Wang J; Sturm RM; Li LJ Am. Soc. Mass Spectrom 2018, 29 (5), 948–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Jia C; Lietz CB; Ye H; Hui L; Yu Q; Yoo S; Li LJ Proteomics 2013, 91, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ye H; Hui LM; Kellersberger K; Li LJ J. Am. Soc. Mass Spectrom 2013, 24 (1), 134–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Schmerberg CM; Liang ZD; Li LJ ACS Chem. Neurosci 2015, 6 (1), 174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Yan Z; Yan R Anal. Chem 2018, 90 (7), 4380–4388. [DOI] [PubMed] [Google Scholar]
- (21).Pan KT; Chen CC; Urlaub H; Khoo KH Anal. Chem 2017, 89 (8), 4532–4539. [DOI] [PubMed] [Google Scholar]
- (22).Lin CH; Krisp C; Packer NH; Molloy MP J. Proteomics 2018, 172, 68–75. [DOI] [PubMed] [Google Scholar]
- (23).Egertson JD; MacLean B; Johnson R; Xuan Y; MacCoss MJ Nat. Protoc 2015, 10 (6), 887–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Searle BC; Pino LK; Egertson JD; Ting YS; Lawrence RT; MacCoss MJ; Villeń J Nat. Commun 2018, 9 (1), 5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Collins BC; Hunter CL; Liu Y; Schilling B; Rosenberger G; Bader SL; Chan DW; Gibson BW; Gingras AC; Held JM; Hirayama-Kurogi M; Hou G; Krisp C; Larsen B; Lin L; Liu S; Molloy MP; Moritz RL; Ohtsuki S; Schlapbach R; Selevsek N; Thomas SN; Tzeng SC; Zhang H; Aebersold R Nat. Commun 2017, 8 (1), 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Rosenberger G; Liu Y; Röst HL; Ludwig C; Buil A; Bensimon A; Soste M; Spector TD; Dermitzakis ET; Collins BC; Malmström L; Aebersold R Nat. Biotechnol 2017, 35 (8), 781–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Panchaud A; Scherl A; Shaffer SA; von Haller PD; Kulasekara HD; Miller SI; Goodlett DR Anal. Chem 2009, 81 (15), 6481–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Jung S; Smith JJ; von Haller PD; Dilworth DJ; Sitko KA; Miller LR; Saleem RA; Goodlett DR; Aitchison JD Mol. Cell. Proteomics 2013, 12 (5), 1421–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Hu A; Noble WS; Wolf-Yadlin A F1000Research 2016, 5, 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Tsou CC; Avtonomov D; Larsen B; Tucholska M; Choi H; Gingras AC; Nesvizhskii AI Nat. Methods 2015, 12 (3), 258–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ting YS; Egertson JD; Bollinger JG; Searle BC; Payne SH; Noble WS; MacCoss MJ Nat. Methods 2017, 14 (9), 903–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kessner D; Chambers M; Burke R; Agus D; Mallick P Bioinformatics 2008, 24 (21), 2534–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zhang J; Xin L; Shan B; Chen W; Xie M; Yuen D; Zhang W; Zhang Z; Lajoie GA; Ma B Mol. Cell. Proteomics 2012, 11 (4), No. M111.010587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Leprevost FV; Valente RH; Lima DB; Perales J; Melani R; Yates JR 3rd; Barbosa VC; Junqueira M; Carvalho PC Mol. Cell. Proteomics 2014, 13 (9), 2480–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Christie AE; Stemmler EA; Dickinson PS Cell. Mol. Life Sci 2010, 67 (24), 4135–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wang Y; Wang M; Yin S; Jang R; Wang J; Xue Z; Xu T Database 2015, 2015, No. bav038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Fricker LD Mol. BioSyst 2010, 6 (8), 1355–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Lietz C; Yu Q; Li LJ Am. Soc. Mass Spectrom 2014, 25 (12), 2009–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lubeckyj RA; McCool EN; Shen X; Kou Q; Liu X; Sun L Anal. Chem 2017, 89 (22), 12059–12067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kaszycki JL; Bowman AP; Shvartsburg AA J. Am. Soc. Mass Spectrom 2016, 27 (5), 795–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Zhang Z; Jia C; Li LJ Sep Sci 2012, 35 (14), 1779–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Xiang F; Ye H; Chen RB; Fu Q; Li LJ Anal. Chem 2010, 82 (7), 2817–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Frost DC; Greer T; Xiang F; Liang ZD; Li LJ Rapid Commun. Mass Spectrom 2015, 29 (12), 1115–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Frost DC; Greer T; Li L Anal. Chem 2015, 87 (3), 1646–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Greer T; Lietz CB; Xiang F; Li LJ J. Am. Soc. Mass Spectrom 2015, 26 (1), 107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Bai B; Tan H; Pagala VR; High AA; Ichhaporia VP; Hendershot L; Peng J Methods Enzymol 2017, 585, 377–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Thompson A; Schafer J; Kuhn K; Kienle S; Schwarz J; Schmidt G; Neumann T; Hamon C Anal. Chem 2003, 75 (8), 1895–904. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.