

Abstract
Ichthyosis follicularis, a distinct cutaneous entity reported in combination with atrichia, and photophobia has been associated with mutations in MBTPS2. We sought the genetic cause of a novel syndrome of ichthyosis follicularis, bilateral severe sensorineural hearing loss and punctate palmoplantar keratoderma in two families. We performed whole exome sequencing on three patients from two families. The pathogenicity and consequences of mutations were studied in Xenopus oocyte expression system and by molecular dynamics simulation analysis. Compound heterozygous mutations in the GJB2 gene were discovered: a pathogenic c.526A>G; p.Asn176Asp, and a common frameshift mutation, c.35delG; p.Gly12Valfs*2. The p.Asn176Asp mutation was demonstrated to significantly reduce the cell-cell gap junction channel activity and increase the non-junctional hemichannel activity in the Xenopus oocyte expression system. Molecular dynamics simulation analyses of the mutant Cx26 protein revealed significant changes in the structural characteristics and electrostatic potential of the Cx26, either in hemichannel or gap junction conformation. Thus, association of a new entity of an autosomal recessive disorder of ichthyosis follicularis, bilateral severe sensorineural hearing loss and palmoplantar keratoderma with mutations in GJB2, expanding the phenotypic spectrum of the GJB2-associated disorders. The findings attest to the complexity of the clinical consequences of different mutations in GJB2.
Keywords: Connexin 26, GJB2 mutations, deafness, sensorineural hearing loss, ichthyosis follicularis
INTRODUCTION
Gap junction channels, critical for cell-cell communication, are composed of connexins (Cx), a family of proteins with four transmembrane domains. There are as many as 21 human connexins, and at least 10 of them have been associated with genetic disorders in humans, including skin findings (Table 1) (Srinivas, Verselis, & White, 2018). The individual connexins oligomerize within the endoplasmic reticulum-Golgi pathway to form connexons, hemichannels that contain six connexin monomers (Laird, 2006). The individual connexons are transported to the plasma membrane where they partner with a hemichannel in the adjacent cell to complete the gap junction channel formation. These channels couple the adjacent cells both electrically and metabolically allowing trans-plasma membrane flow of molecules, including calcium, ATP and NAD+, thereby eliciting signaling cascades resulting in diverse biological responses (Evans, De Vuyst, & Leybaert, 2006).
Table 1.
The phenotypic spectrum of cutaneous findings in the diseases with GJB2 mutations.*
| NO. | Phenotype | OMIM # | Inheritance | SNHL | Spinyhyperkeratosis† | Knuckle pads | PPK† | Leukonychia | Alopecia | Keratitis | Pseudoainhum | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Germline | ||||||||||||
| 1 | Current Family | - | AR | Y | Y(Gen) | N | N | Y | N | N | N | |
| 2 | Bart-Pumphrey syndrome | 149200 | AD | Y | N | Y | Y | Y | Y | N | N | Richard et al.(Richard, Brown, Ishida-Yamamoto, & Krol, 2004) |
| 3 | Deafness, autosomal dominant 3A | 601544 | AD | Y | N | N | N | N | N | N | N | Kelsell et al.(Kelsell et al., 1997) |
| 4 | Deafness, autosomal recessive 1A | 220290 | AR | Y | N | N | N | N | N | N | N | Brown et al.(Brown et al., 1996) |
| 5 | Hystrix-like ichthyosis with deafness | 602540 | AD | Y | Y(Loc) | N | N | Y | Y | Y | N | van Geel et al.(van Geel et al., 2002) |
| 6 | Keratitis-ichthyosis-deafness syndrome | 148210 | AD | Y | N | N | Y | Y | Y | Y | N | Richard et al.(Richard et al., 2002) |
| 7 | Keratoderma, palmoplantar, with deafness | 148350 | AD | Y | N | N | Y | Y | Y | Y | N | Brown et al.52 |
| 8 | Vohwinkel syndrome | 124500 | AD | Y | N | Y | Y | N | Y | Y | Y | Maestrini et al.(Maestrini et al., 1999) |
| Mosaic | ||||||||||||
| 9 | Porokeratotic eccrine ostial and dermal duct nevus (PEODDN) | - | Mosaic | N | Y (Loc) | N | N | N | N/A | N | N | Easton et al.(Easton et al., 2012) |
| 10 | Spiny Hyperkeratosis without Eccrine Involvement | - | Mosaic | N | Y (Loc) | N | N | N | N | N | N | Eskin-Schwartz et al.(Eskin-Schwartz et al., 2016) |
The clinical findings are according to https://www.omim.org and literature review.
Gen, generalized; Loc, localized; PPK, palmoplantar keratoderma; SNHL, sensorineural hearing loss.
The gap junction channels formed by different connexins are functionally distinct with respect to their gating, conductance and permeability characteristics. The complexity of these channels is emphasized by the fact that hemichannels and complete gap junction channels can be either homotypic or heterotypic (Karademir, Aoyama, Yue, Chen, & Bai, 2016; Koval, Molina, & Burt, 2014). Consequently, genetic mutations in a single connexin gene may impact formation of many types of channels, thus contributing to the phenotypic spectrum of diseases that result from such mutations.
Mutations in the connexin 26 (Cx26) have been associated with a spectrum of phenotypic findings, and the carrier frequencies of mutations in the GJB2 gene, which encodes Cx26, can be as high as 4% of unaffected individuals in certain populations (Srinivas et al., 2018). The most common clinical finding associated with GJB2 mutations is autosomal recessive or autosomal dominant non-syndromic deafness which account for about half of mild to very severe cases of all genetic deafness and hearing loss (Leclere, Le Gac, Le Marechal, Ferec, & Marianowski, 2017; Snoeckx et al., 2005). In addition to non-syndromic hearing loss, there are forms of syndromic deafness associated with skin manifestations caused by mutations in GJB2 (Table 1) (Lilly, Sellitto, Milstone, & White, 2016). In addition to these disorders caused by germline mutations in GJB2, cases diagnosed as porokeratotic eccrine ostial and dermal duct nevus (PEODDN) have been described in association with somatic GJB2 mutations (Table 1) (Bandyopadhyay, Saha, Das, & Das, 2015; Levinsohn, McNiff, Antaya, & Choate, 2015; Marsden, Fleming, & Dawber, 1979). These cutaneous lesions are considered to represent epidermal nevi that follow the Blaschko lines, found in most cases in limited distribution on the hands and feet, although more widespread distribution has been noted in some patients. These lesions have been suggested to be hyperkeratotic epidermal nevi originating from the eccrine duct. However, in one case, no eccrine gland association was found by histopathological observations, and this condition, also associated with somatic mosaicism with a GJB2 mutation, was diagnosed as spiny hyperkeratosis, a rare clinical phenotype characterized by non-follicular hyperkeratotic projections (Eskin-Schwartz et al., 2016).
Ichthyosis follicularis manifests with hyperkeratotic papules centering on hair follicles in association with a number of other cutaneous findings. In particular, it has been reported in association with alopecia and photophobia (IFAP syndrome; OMIM 308205), in some cases in association with BRESHECK syndrome, characterized by brain anomalies, retardation, ectodermal dysplasia, hearing loss, skeletal abnormalities, Hirschsprung disease, and dysmorphic features (OMIM 308205). These conditions are inherited in an X-linked recessive manner due to mutations in MBTPS2 (Oeffner et al., 2009).
In this study, we have examined two families consisting of a total of four individuals manifesting with a new phenotype of ichthyosis follicularis, congenital sensorineural deafness and palmoplantar keratoderma (PPK), associated with compound heterozygous germline mutations in the GJB2 gene.
MATERIALS AND METHODS
Patients data:
This study was approved by the institutional review board of the Pasteur Institute of Iran, and all subjects and parents of underaged patients gave written informed consent to participate in research and to publish their images. In this study, 92 extended families affected by nonsyndromic and syndromic forms of ichthyosis, diagnosed in various medical centers in Iran, were evaluated. The families were personally examined by a dermatologist (HM) and by medical geneticists (LY, AK and HV), who are authors, and the diagnostic clinical features and demographic data were carefully recorded. Criteria for inclusion were clinical presentation of ichthyosis supported in some of the patients by histopathology.
Mutation Detection and Bioinformatics
Whole exome sequencing (WES):
DNA was extracted from peripheral blood samples of three affected family members, IV-1, III-10, and III-11 by salting out method. DNA concentration was measured using a Qubit 3.0 fluorometer (Life Technologies, Carlsbad, CA, USA). The qualified genomic DNA samples were randomly fragmented by Covaris and the size of the library fragments was distributed predominantly between 150 to 200 bp. Adapters were then ligated to both ends of the resulting fragments. The adapter-ligated templates were purified by the Agencourt AMPure SPRI beads, and fragments with insert size about 176 bp were excised. Extracted DNA was amplified by ligation-mediated PCR (LM-PCR), purified, and hybridized to the SureSelect Biotinylated RNA Library (BAITS) for enrichment. The hybridized fragments were bound to the strepavidin beads whereas non-hybridized fragments were washed out after 24 h. Captured LM-PCR products were subjected to Agilent 2100 Bioanalyzer to estimate the magnitude of enrichment. Each captured library was then loaded on HiSeq2000 platform, and high-throughput sequencing for each captured library was performed to ensure that each sample met the desired average sequencing depth.
Raw image files were processed by Illumina Software 1.7 for base-calling with default parameters and the sequences of each individual were generated as 90/100 bp pair-end reads. The bioinformatics analysis was applied to the sequencing data (“raw data”) which was generated from the Illumina pipeline. First, the adapter sequence in the raw data was removed, and low quality reads which have too many undefined nucleotides or low base quality were discarded. This step produced the “clean data”. Secondly, Burrows-Wheeler Aligner (BWA) (Li & Durbin, 2010) was used for alignment. Read groups were added, duplicates were marked and the reads were sorted using Picard (http://broadinstitute.github.io/picard). After these processes, the final BAM files were used for variant calling using the GATK haplotype caller (McKenna et al., 2010) to identify both single nucleotide variants (SNVs) and indels. Candidate variants were then annotated using ANNOVAR software (Wang, Li, & Hakonarson, 2010). ANNOVAR MetaSVM scores, representing metascores across several tools, including SIFT, PolyPhen2, MutationTaster and MutationAssessor were used to identify variants as deleterious.
Variant interpretation:
SNVs and CNVs were examined using the annotated CSV files of WES from the three affected family members, IV-1, IV-10, and IV-11. The sequence variants were filtered from the VCF files for missense, nonsense, and splice site-affecting variants. Indel variants were filtered for exonic in-frame insertions and deletions, frameshift mutations, and gained or lost start or stop codon. Additionally, only variants with 1000 Genomes database total frequencies of <0.05 (see Figure 2a), or those without frequency data available, were examined. Candidate genes using these filtering parameters were then overlapped between the three affected individuals in the family to examine potential common disease-causing variants. Considering the given phenotype and segregation analysis allowed us to identify two pathogenic variants in GJB2.
Figure 2.

Mutation detection and expression of the mutant protein in skin. (a) Whole exome sequencing, followed by filtering with the steps indicated, resulted in identification of compound heterozygous p.Asn176Asp and c.35delG mutations in the GJB2 gene in the affected individuals. (b) The sequence variants were confirmed by Sanger sequencing. The reference sequence of unrelated healthy controls is shown below each mutation. (c) Molecular modeling of Cx26 places the asparagine 176 residue at the rim of the extracellular part of the protein at the interface of docking of two hemichannels to form a gap junction. (d) Immunofluorescence staining with an anti-Cx26 antibody revealed the presence of the mutant protein in the patient’s skin.
Gap Junction and Hemichannel Functional Analyses in Xenopus oocyte System
In vitro transcription, oocyte microinjection, and pairing:
Wild-type Cx26 was cloned into the pCS2+ expression vector for functional studies in Xenopus oocytes (Mese, Londin, Mui, Brink, & White, 2004; Turner & Weintraub, 1994). The Cx26-N176D mutation was amplified from the patient’s genomic DNA with Cx26 primers containing BamH1 and Xba1 linkers (Rouan et al., 2001). Cx26-N176D was cloned into pBlueScript II (Agilent Technologies, Santa Clara, CA) and sequenced on both strands prior to subcloning into pCS2+. Constructs were linearized with Not1 and transcribed using the SP6 mMessage mMachine (Ambion, Austin, TX). Xenopus laevis oocytes were purchased (Xenopus 1, Dexter, MI) and cultured in modified Barth’s (MB) medium (Mhaske et al., 2013). Oocytes were injected with 10 ng of antisense Xenopus Cx38 oligonucleotide (Barrio et al., 1991; Bruzzone, Haefliger, Gimlich, & Paul, 1993), followed by connexin transcripts (5 ng/cell) alone or in combination. Water injected oocytes served as a negative control.
Recording of hemichannel currents:
Hemichannel currents were recorded 24 hours after cRNA injection using a GeneClamp 500 amplifier controlled by a PC-compatible computer through a Digidata 1440A interface using pClamp 8.0 software (Axon Instruments, Foster City, CA). Electrodes (1.5 mm diameter glass, World Precision Instruments, Sarasota, FL) were pulled to a resistance of 1-2 MΩ (Narishige, Tokyo, Japan) and filled with 3 M KCl, 10 mM EGTA, and 10 mM HEPES, pH 7.4. Oocytes were recorded in MB medium without added calcium (Gerido, DeRosa, Richard, & White, 2007). Hemichannel current-voltage (I-V) curves were obtained by clamping cells at −40 mV and subjecting them to 5 second depolarizing voltage steps ranging from −30 to +60 mV in 10 mV increments.
Recording of junctional conductance:
Gap junctional conductance (Gj) was measured by initially clamping both cells in a pair at −40 mV (a transjunctional potential (Vj) of zero). One cell was subjected to alternating pulses of 00B120 mV and the current produced by the change in voltage was recorded in the second cell, which was equal in magnitude to the junctional current (Ij). Conductance was calculated by dividing Ij by the voltage difference, Gj = Ij/(V1-V2) (Spray, Harris, & Bennett, 1981).
Western blotting:
Oocytes extracts were prepared as previously described(White, Bruzzone, Goodenough, & Paul, 1992), separated on 12% SDS gels and transferred to nitrocellulose membranes. Blots were blocked with 5% milk and 0.1% Tween20 in TBS, probed with polyclonal antibodies against Cx26 (Life Technologies, Carlsbad CA), followed by horseradish peroxidase conjugated secondary antibodies (Jackson Laboratories and GE Healthcare). A monoclonal β-actin antibody (Abcam, Cambridge, MA) was used as a loading control.
Structural Bioinformatics Analysis of Conformational and Electrostatic Potential of Cx26-Asn176Asp
Preparation of crystal structure and induction of mutations:
Initial structure of hexamer of Cx26 hemichannel was obtained from RCSB.org (PDBID:2ZW3) (Maeda et al., 2009). Missing residues and hydrogen atoms within the structure were filled using Modeller 9.16 (Eswar et al., 2007) and Reduce 3.23 (Word, Lovell, Richardson, & Richardson, 1999), respectively. In addition to the wild-type structure, three mutant conformations were generated (p.N176D, p.G45E and p.W44C) by inducing point mutations using psfgen plugin of Visual Molecular Graphics (VMD 1.9.3) (Humphrey, Dalke, & Schulten, 1996). The p.G45E and p.W44C mutants were previously reported as a cause of syndromic and non-syndromic deafness (Minami et al., 2013). All structures were energetically minimized to 10,000 steps to eliminate bad contacts by conjugate gradient algorithm using Nanoscale Molecular Dynamics program (NAMD) 2.12 (Phillips et al., 2005).
Calculation of electrostatic potentials:
Atomic charges and radius parameters of CHARMM force field were assigned to the structures by PDB2PQR software (Dolinsky, Nielsen, McCammon, & Baker, 2004). Electrostatic potentials of conformations were then calculated using Adaptive Poisson-Boltzmann Solver (APBS) software package (Holst, Baker, & Wang, 2000) and visualized with VMD 1.9.3.
Protein docking:
Easy interface of HADDOCK webserver (de Vries, van Dijk, & Bonvin, 2010) was used to predict possible modes of channel formation between the hemichannels in wild type and mutant conformations. Since the dimerization of connexin hemichannels require their head-to-head interactions, some residues within the surface of hemichannel were considered as the active residue which contribute directly in the process of channel formation (N54-C60, C169-V178 from each monomer); other residues around the active zones were automatically selected as passive ones. Top cluster with lowest HADDOCK score (highest affinity) was selected as the best mode of dimers in each wild type and mutant conformation for further complementary analysis. R statistical language was used for all analysis. VMD 1.9.3 was used for structural representation and intermolecular hydrogen bonds were measured with H-Bonds plugins of VMD 1.9.3.
Histopathology and Immunofluorescence:
Skin biopsies were obtained from a lesional area together with adjacent normal appearing skin of the proband and processed for histopathology with standard techniques. Paraffin sections (5 μm) were stained with Hematoxylin and Eosin. Parallel sections (3 μm) were stained with mouse anti-human connexin 26 antibody (Cx-12H10(13–1800), Life Technologies, Carlsbad, CA), followed by a fluorescein conjugated goat anti-mouse secondary antibody (IRDye 800CW, LI-COR Biosciences, Lincoln, NE). The images were visualized by EVOSTM Auto Imaging System, (Thermo Fisher Scientific, Waltham, MA).
RESULTS
Clinical features and histopathology
Two families with a total of four similarly affected individuals, residing in the same geographical area in Northern part of Iran, were subjected to study.
Family 1.
The proband (IV-1), a nine-year-old male, manifested with painful diffuse skin lesions present since first month of his life. The parents were unrelated, healthy, and belonged to Iranian-Tabari ethnicity. On skin examination, xerosis, numerous discreet widespread spiny follicular papules, consistent with the diagnosis of ichthyosis follicularis were noted on his non-glabrous skin (Figure 1). Furthermore, focal hyperkeratosis of the palms and soles with multiple punctate palmoplantar pits were noted and diagnosed as PPK. Hypertrophic nail cuticle, and leukonychia totalis were also noted. No other abnormality was detected in the hair, nails, or teeth. His parents reported a modest improvement in cutaneous follicular lesions after administration of oral acitretin. The parents noticed severe hearing loss at the age of 7 months. Audiometric examination revealed profound Sensorineural Hearing Loss (SNHL), which ultimately led to cochlear implant at the age of 5 years. Developmental milestones and intelligence quotient (IQ) were within normal range.
Figure 1.

Pedigree structures, cutaneous features and histopathology of skin in the families with autosomal recessive follicular hyperkeratosis, PPK and bilateral sensorineural deafness. (a) Family pedigrees with autosomal recessive inheritance. Note the clinically unaffected parents of the affected individuals, consistent with autosomal recessive inheritance. (b-d) Skin examination of the proband in Family 1 (identified by an arrow in a1) revealed multiple discrete hyperkeratotic projections centering on hair follicles in a wide-spread distribution with scalp findings. (e) focal keratoderma of the soles with punctate pits consistent with PPK in patient no. 9 of Family 2. (f) On the backs of the hands of the proband (IV-1) these lesions formed well demarcated plaques separated by normal appearing skin. Note the white nails on the hands as part of leukonychia totalis hyperkeratosis plugs (f1), and hypertrophic cuticle (f2). (g, h) Palm of the proband (IV-1) with hyperkeratosis and accentuated creases which contain punctate pits (h).
(j) Histopathology of a skin lesion delineated in (i) revealed a parakeratotic column of hyperkeratotic skin invaginating into epidermis (j1). The granular layer is absent and the underlying epidermis is pale consisting of cells with perinuclear halo (j2).
Family 2:
Two affected siblings, a 32-year-old male and 28-year-old female (III-10 and III-11, respectively), with the clinical findings similar to Family 1, including SNHL, generalized spiny follicular papules (ichthyosis follicularis) limited to their non-glabrous skin, and PPK, were presented to our dermatology clinic. The spiny papules appeared within the first few weeks after birth, and they gradually became erythematous in both patients. They also reported the presence of hypertrophic nail cuticles (similar to the first case in Family 1). They habitually used to manipulate cuticles resulting in paronychia and secondary nail plate changes. Their mother reported that the skin manifestations improved gradually during aging, and a modest improvement was seen during oral acitretin treatment. SNHL was suspected at the age of 4 and 1.5 years in the male and female patients, respectively. No problem with hair was noted and developmental milestones and IQ were within the normal range.
Histopathology of a skin lesion in the proband of Family 1 revealed a parakeratotic column of hyperkeratosis invaginating into the epidermis with adjacent basket weave hyperkeratosis (Figure 1j1). The bottom of the parakeratotic lesion was devoid of granular layer, and the epidermis below the hyperkeratotic column was pale with keratinocytes with distinct perinuclear halo. There was a mild perivascular inflammatory cell infiltrate in the upper papillary dermis, and the reticular dermis appeared normal with compact collagen fibers.
Immunostaining of the peri-lesional skin in the proband revealed a characteristic punctate pattern with an anti-Cx26 antibody, indicating the presence of Cx26 (Figure 2d). The staining pattern was indistinguishable from that in control skin (not shown).
Identification of compound heterozygous GJB2 mutations
DNA samples from three affected individuals (nos. IV-1, III-9, and III-10) were examined by WES on an Illumina platform. Initially, ~77,000 sequence variants were identified in each of the three affected individuals. Following a filtering process, as described in Materials and Methods and summarized in Figure 2a, two allelic heterozygous mutations in the GJB2 gene common in the three patients were identified (Figure 2b). One of the variants was NM_004004.5, GJB2:c.526A>G; p.Asn176Asp (rs781767722), which was submitted to ClinVar (https://submit.ncbi.nlm.nih.gov/subs/clinvar_file/SUB4534284/; submission ID: SUB4621706). This sequence variant was predicted to be pathogenic by predictive software programs (SIFT, PolyPhen2, MutationTaster, and MutationAssessor). This variant was heterozygous in the clinically normal mother of the patient no. 1, and its frequency in publicly available variant database ExAC in heterozygous state was 1 out of 121,028 alleles (8.263e-6). This variant was not reported as homozygous in any of the population databases examined, including 1000 Genome Project, and Greater Middle East (GME) Variome Project, or in the literature. Amino acid 176, along with a chain of six residues, including Asn54, Leu56, Gln57, Lys168, Thr177, and Asp179, is evolutionarily highly conserved in the extracellular domain responsible for the docking of two connexons (Zonta et al., 2015). Among those six conserved residues, only a heterozygous variant in Asn54 has been reported in association of palmoplantar keratoderma, sensorineural hearing loss, knuckle pads in Bart-Pumphrey syndrome (OMIM# 149200) (Akiyama et al., 2007). So far, there are no homozygous variants associated with these critical residues with skin manifestation, and to the best of our knowledge this is the first reported variant in critical residues of the second extracellular domain (E2) of Cx26.
In addition, a heterozygous frameshift mutation, NM_004004.5, GJB2:c.35delG; p.Gly12Valfs*2, was identified in the patients as well as in the clinically normal father of patient no. 1 (Figure 2b). This is a common mutation associated with non-syndromic hearing loss and when in homozygous state, is predicted to result in loss of functional Cx26 due to frameshift and premature termination codon at the early part of the protein. Collectively, two GJB2 mutations as compound heterozygous were identified in the patients in two families, and clinically normal parents of the Patient 1 were heterozygous carriers of the p.Asn176Asp and c.35delG mutations, respectively, as determined by Sanger sequencing (Figures 2b), consistent with autosomal recessive inheritance pattern.
Consequences of the p.Asn176Asp mutation to gap junction and hemichannel functional activities
Cx26-N176D has reduced cell-cell gap junction channel activity (Figure 3a):
Figure 3.
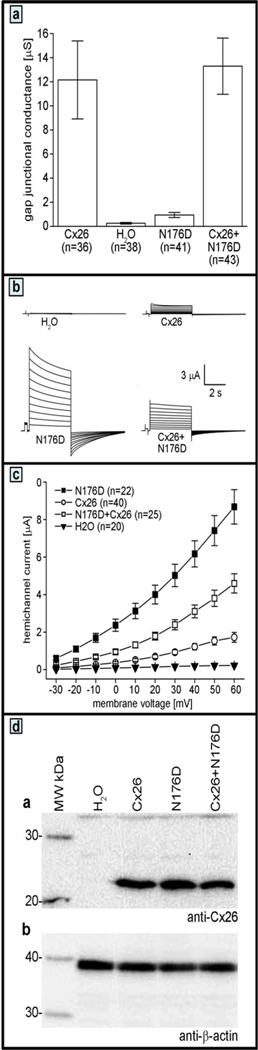
Assay of gap junction conductance and hemichannel current of the mutant connexin 26. (a) Cx26-N176D has reduced gap junction channel activity. Xenopus oocyte pairs expressing Cx26 formed gap junction channels, whereas water injected control cell pairs displayed negligible conductance. Oocytes expressing Cx26-N176D had significantly reduced conductance compared to wild-type Cx26. Co-expression of Cx26- N176D with wild-type Cx26 did not reduce conductance compared to Cx26 alone. Data are the mean ± SE. (b) Cx26-N176D induced large increases in hemichannel currents in Xenopus oocytes. Single cells were clamped at a holding potential of −40 mV and subjected to voltage pulses ranging from −30 to +60 mV in 10 mV steps. H2O injected cells displayed negligible membrane currents. The Cx26-N176D mutant showed a large increase in the magnitude of these outward currents when compared to the either H2O or wild-type Cx26 injected cells. (c) Steady-state currents from each pulse were plotted as a function of membrane voltage. Steady state currents in H2O injected control cells (filled triangles) were negligible at all membrane voltages. Cx26 currents (open circles) were similar to those observed in control cells at lower voltages, but increased at higher membrane voltage. Cx26-N176D (filled squares) expressing cells exhibited significantly increased steady-state currents compared to either control or Cx26 oocytes. Co-expression of Cx26-N176D and wild-type Cx26 produced currents that were intermediate in magnitude. Data are the mean ± SE. (d) Wild-type and mutant connexins are equivalently expressed in Xenopus oocytes. Equal amounts of membrane extracts were first probed with an antibody that recognized Cx26. H2O injected cells did not express Cx26. Wild-type Cx26 and Cx26-N176D expressed alone, or in combination, were readily detected in lanes corresponding to each injection condition with similar band intensities. To confirm equal sample loading, blots were stripped and reprobed with an antibody against β-actin, which was present at comparable levels in all lanes.
Wild-type Cx26 and the corresponding mutant connexin, designated as Cx26-N176D, were expressed in Xenopus oocyte pairs, and gap junctional conductance, Gj, was measured. Control oocyte pairs injected with water showed negligible conductance (Gj = 0.26 µS), while cells with wild-type Cx26 channels had an average Gj of 12.2 µS (Figure 3a). Cell pairs expressing Cx26-N176D had conductance levels that were reduced by 93% (0.94 µS, Student’s t-test p<0.05) compared to wild-type Cx26. Cell pairs co-injected with equal amounts of wild-type Cx26 and Cx26-N176D RNA were well coupled, with a Gj of 13.3 µS (p>0.05, compared to wild-type Cx26 alone) (Figure 3a). Thus, Cx26-N176D had reduced gap junction channel activity when expressed alone, and failed to inhibit co-expressed wild-type Cx26, consistent with recessive mode of inheritance.
Cx26-N176D has increased hemichannel activity in single Xenopus oocytes (Figure 3b, c):
Wild-type Cx26 and Cx26-N176D were expressed in Xenopus oocytes either alone or in combination. Single cells were subjected to depolarizing voltage pulses and the resulting membrane currents were recorded. Control oocytes injected with H2O showed negligible current flow for voltage steps between −30 and +60 mV. Hemichannel activity of wild-type human Cx26 has been previously reported (Gerido et al., 2007; Gonzalez, Gomez-Hernandez, & Barrio, 2006; Lee, Derosa, & White, 2009; Ripps, Qian, & Zakevicius, 2004; Sanchez, Mese, Srinivas, White, & Verselis, 2010), and was characterized by outward currents that increased with greater depolarization. The Cx26-N176D mutant showed a large increase in the magnitude of these outward currents when compared to either H2O or wild-type Cx26 injected cells.
Mean steady state currents were plotted as a function of the membrane potential to quantify differences in the recorded hemichannel activity (Figure 3c). Control cells injected with H2O showed negligible currents at all tested voltages. In comparison, wild-type Cx26 injected cells displayed larger outward currents that increased at greater depolarizing voltages. Cx26-N176D expressing cells produced significantly larger outward currents than wild-type Cx26 injected cells. At +60 mV, Cx26-N176D produced currents that were five times larger than wild-type and thirty-nine times larger than control cells, differences that were statistically significant (p <0.05, one-way ANOVA). Cells co-expressing equal amounts of Cx26-N176D with wild-type Cx26 displayed currents that were intermediate between the values of mutant or wild-type Cx26 alone at all tested potentials (Figure 3c). This significantly increased membrane current suggested the presence of increased hemichannel activity resulting from the Cx26-N176D mutation.
Cx26-N176D and wild-type Cx26 have equivalent levels of protein expression (Figure 3d):
Synthesis of wild-type and mutant Cx26 was examined by Western blot analysis. Immunoblotting for Cx26 revealed ~26kDa bands in lanes corresponding to oocytes injected with either Cx26 or Cx26-N176D alone or in combination. No Cx26 was detected in H2O injected control cells. When the blot was re-probed for β-actin, it was detected at similar intensity in all samples, confirming equal protein loading.
Electrostatic potential of mutant Cx26 p.Asn176Asp hemichannel
Molecular modeling of Cx26 proteins indicated that the p.Asn176Asp mutation is located at the rim of the extracellular part of Cx26 hemichannel which is directly involved in gap junction channel formation (Figure 4a). To assess the effects of p.Asn176Asp mutation on hemichannel ion conductance and permeation activity of Cx26, electrostatic surface potential and electrostatic field of Cx26 hemichannel were calculated in normal and mutant conformations (Figure 4a). As shown in this figure, the substitution of polar Asn with negatively charged Asp disturbed and elevated the electrostatic field which contributes to the alteration in ion transduction through hemichannel in the p.Asn176Asp mutant conformation. This result is consistent with those obtained in the Xenopus oocyte system which showed elevation of hemichannel activity.
Figure 4.

Molecular architecture of hemichannel and gap junction conformations in wild-type and mutant Cx26. (a) The location of p.Asn176Asp mutation on the top of Cx26 hemichannel is indicated in a ribbon grey representation (left panel). Computed electrostatic potentials and electrostatic fields projected on the surface of the Cx26 in wild-type (middle panel), and p.Asn176Asp (right panel) forms; positive and negative potentials are shown in blue and red, respectively (color scale is 1 to +1 kTe−1). (b) Modeled structure of Cx26 gap junction channels in wild-type (left) and mutant p.Asn176Asp (right) conformations predicted by HADDOCK (left panel); Electrostatic field-lines with positive and negative electrostatic potentials are shown in blue and red, respectively (right panel) (color scale is −1 to +1 kTe−1).
For comparison, two other cases of Cx26 mutants, p.Trp44Cys and p.Gly45Glu were selected to compare the possible differences between their hemichannel electrostatic potential (Supp. Figure S1a–d) (Denoyelle et al., 1998; Jonard et al., 2008). The Trp44Cys is an established mutation underlying non-syndromic deafness without skin manifestations (Denoyelle et al., 1998), while mutation p.Gly45Glu is associated with PEODDN, a skin disorder resulting from somatic mutation in GJB2 with skin histopathy very similar to that shown in our cases (Levinsohn et al., 2015). Thus, these two mutations in GJB2 represent independently the major features of our patients, i.e., deafness and ichthyosis. The electrostatic field increased in the case of the p.Gly45Glu conformation (Supp. Figure S1c) but remained unchanged in the p.Trp44Cys form (Supp. Figure S1d), indicating similar increased pattern of p.Gly45Glu as in p.Asn176Asp.
Effect of Cx26 p.Asn176Asp mutation on channel formation
Since p.Asn176Asp is located at the rim of the extracellular part of the hemichannel, we next evaluated the impact of this mutation on dimerization affinity and channel formation capability of Cx26. Figure 4b shows the results of protein docking in p.Asn176Asp conformation in comparison with wild type Cx26. The interaction affinity (denoted as HADDOCK score) between hemichannels in p.Asn176Asp dimers was reduced in comparison with wild type (199.5 ± 21.1 vs. –235.6 ± 14.5), indicating that this mutation interferes with the dimerization and channel formation ability (Figure 4b and Supp. Supp. Table S1). The negatively charged repulsion force also suggested lower affinity of dimerization in this case as shown by negative repulsion in the electrostatic field (Figure 4b). Moreover, intermolecular hydrogen bonds were reduced in mutant channel (Supp. Table S2). In contrast, the interaction affinity in p.Gly45Glu conformation was increased, suggesting higher affinity to form channels, compared with the wild type structures (Supp. Figure S1e and Supp. Table S1). Moreover, the higher electrostatic field inside the p.Gly45Glu channel (red lines within the channel lumen in Supp. Figure S1e) suggested an increase in transcellular transfer of ions which was not detected in the case of p.Asn176Asp channel. Thus, although extracellular ion transfer was increased in both p.Gly45Glu and p.Asn176Asp mutants hemichannel conformations, the mechanisms of actions in the channel forms were completely different.
DISCUSSION
This study expands the phenotypic spectrum of GJB2-associated disorders in a family with four affected individuals with congenital bilateral deafness and distinct skin manifestations. The cutaneous findings consisted of widespread distribution of discrete hyperkeratotic papules, which were extremely painful upon touch, inherited in an autosomal recessive pattern. Clinical diagnosis of the skin lesions, confirmed by histopathology, was consistent with ichthyosis follicularis, a condition previously associated with atrichia and photophobia with or without brain anomalies. Ichthyosis follicularis has been associated with mutations in MBTPS2 in an X-linked recessive pattern. In our cases, the clinical phenotype of the affected individuals is clearly associated with compound heterozygous GJB2 mutations inherited in an autosomal recessive fashion. Specifically, the affected individuals were compound heterozygous of a novel, previously unreported missense mutation, p.Asn176Asp, and a common deletion mutation, c.35delG, in GJB2 encoding Cx26.
It should be noted that the histopathologic findings were similar to that in PEODDN, a condition associated with GJB2 mutations. However, all the published mutations reported in PEODDN are somatic, and the corresponding germline mutations have been suggested to be lethal (Bandyopadhyay et al., 2015; Eskin-Schwartz et al., 2016; Levinsohn et al., 2015; Marsden et al., 1979). Also, the clinical presentations of the patients with PEODDN, even in the localized areas as expected by somatic mosaicism, are quite different, and our cases may represent a new entity of ichthyosis follicularis with severe sensorineural deafness and PPK. In the differential diagnosis, follicular or spiny hyperkeratosis is a consideration as reported by Eskin-Schwartz et al., 2016 (Eskin-Schwartz et al., 2016), but the GJB2 mutation was somatic.
The functional consequences of the missense mutation p.Asn176Asp were examined by recording the currency of hemichannels and the junctional conductance of mutant Cx26 in the Xenopus oocyte system. The current in hemichannel made of mutant Cx26 showed markedly increased activity, and the hemichannels heterozygous for p.Asn176Asp and wild-type Cx26 showed intermediate levels, as compared to wild-type Cx26. However, the gap junction conductance of channels composed of mutant p.Asn176Asp protein was reduced to less than 10% of that noted with wild-type Cx26 containing channels. Interestingly, however, the gap junction conductance compromised by p.Asn176Asp in Cx26 could be restored to the normal level by adding an equal amount of wild-type Cx26. This observation apparently explains why the mother of the proband in Family 1, who was heterozygous for p.Asn176Asp, was clinically normal. Similarly, the mutation c.35delG when heterozygous in the father of an affected individual, did not cause any clinical phenotype. However, this mutation in combination with non-functional Cx26 containing the p.Asn176Asp mutation, rendering it functionally hemizygous, was able to elicit congenital bilateral deafness in the affected individuals.
Interestingly, the membrane currents elicited by hemichannels made of mutant p.Asn176Asp protein were five times greater than those produced by wild-type Cx26. Some of the specific somatic mutations in Cx26 that have been linked to PEODDN, have also been found as germline mutations in patients with keratitis-ichthyosis-deafness (KID) syndrome which is inherited in an autosomal dominant manner (Easton et al., 2012; Lazic, Li, Frank, Uitto, & Zhou, 2012; Levinsohn et al., 2015). It has been shown that increased mutant Cx26 hemichannel activity is a common feature of KID mutations and is likely to cause the epidermal phenotype of KID syndrome (Donnelly et al., 2012; Gerido et al., 2007; Lee et al., 2009; Mhaske et al., 2013; Sanchez et al., 2010; Sanchez, Villone, Srinivas, & Verselis, 2013; Stong, Chang, Ahmad, & Lin, 2006). Although the specific mutation p.Asn176Asp found in our patients has not been linked to KID syndrome, it is possible that its increased hemichannel activity contributes to the skin pathology, as there is a growing recognition of the role for dysregulated hemichannels in many of the other genetic connexin skin disorders (Lilly et al., 2016; Srinivas et al., 2018). These mutations attest to the complexity of the clinical phenotypes as a consequence of different mutations in GJB2.
The consequences of the p.Asn176Asp mutation on the structural and electrostatic characteristics of the Cx26 protein either in hemichannel or gap junction conformation were explored by computational analysis of Cx26 protein conformations. In agreement with the electrophysiology (Xenopus oocyte system) results, the computational analysis of the Cx26 hemichannel structures suggested that the increase in hemichannel activity in the p.Asn176Asp mutant is due to the elevation of electrostatic field within the hemichannel lumen. This alteration increased the permeability of ions between cytosol and the extracellular region of the cells. Similar changes could be detected in the case of previously reported p.Gly45Glu mutation in hemichannel form (Jonard et al., 2008). In contrast, there was no significant changes in the case of a previously reported mutation in non-syndromic deafness, p.Trp44Cys conformation, indicating the involvement of different mechanisms in this Cx26-related disease category. Moreover, the results on protein docking suggested that the reduction of gap junction conductance, as demonstrated in Xenopus oocytes, is due to the decrease in formation of gap junction channels by the mutant hemichannels. This could be explained by the lower affinity of hemichannels due to the high electrostatic repulsion of Asp residues at the rim of the extracellular part of each hemichannel during the channel formation. In contrast, the affinity of hemichannels to form gap junctions was increased in the case of p.Gly45Glu mutants, suggesting that regardless of the same consequences of these two missense mutations on hemichannel conformations, the interfering mechanism in the form of channel is different.
In conclusion, structural analysis indicated that Asn176Asp mutation interferes the Cx26 function at two levels: (a) reducing the possibility of channel formation between the cells compared to the wild-type, and (b) increasing the extracellular ion transfer in the hemichannel conformation which was not detected in the case of other mutations such as p.Gly45Glu and p.Trp 44Cys.
Supplementary Material
ACKNOWLEDGMENTS
Carol Kelly assisted in manuscript preparation. Drs. Sylvia Hsu, Jason B. Lee, John McGrath and Eli Sprecher, provided expert assistance in interpretation of the clinical findings and histopathology. Ali Jazayeri contributed to data analysis. This work was supported in part by R01 EY013163 and R01 EY026911 (TWW). This manuscript is in partial fulfilment of the PhD Thesis of Leila Youssefian.
Abbreviations:
- CNV
copy number variation
- Cx
connexin
- BPS
Bart-Pumphrey syndrome
- PEODDN
porokeratotic eccrine ostial and dermal duct nevus
- KID
keratitis ichthyosis-deafness
- SNV
single nucleotide variants
- SNHL
sensorineural hearing loss
- PPK
palmoplantar keratoderma
- WES
whole exome sequencing
Footnotes
Publisher's Disclaimer: This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record.
Conflict of Interest: The authors have no conflict of interest to declare.
Declaration of Interests The authors declare no competing interests.
Patient consent Obtained.
Ethics approval Institutional Review Board of the Pasteur Institute of Iran.
Supplemental Data
Supplemental data include one Figure and two Tables.
REFERENCES
- Akiyama M, Sakai K, Arita K, Nomura Y, Ito K, Kodama K, … Shimizu H (2007). A novel GJB2 mutation p.Asn54His in a patient with palmoplantar keratoderma, sensorineural hearing loss and knuckle pads. J Invest Dermatol, 127(6), 1540–1543. 10.1038/sj.jid.5700711 [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay D, Saha A, Das D, & Das A (2015). Porokeratotic eccrine ostial and dermal duct nevus. Indian Dermatol Online J, 6(2), 117–119. 10.4103/2229-5178.153016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrio LC, Suchyna T, Bargiello T, Xu LX, Roginski RS, Bennett MV, & Nicholson BJ (1991). Gap junctions formed by connexins 26 and 32 alone and in combination are differently affected by applied voltage. Proc Natl Acad Sci U S A, 88(19), 8410–8414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruzzone R, Haefliger JA, Gimlich RL, & Paul DL (1993). Connexin40, a component of gap junctions in vascular endothelium, is restricted in its ability to interact with other connexins. Mol Biol Cell, 4(1), 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries SJ, van Dijk M, & Bonvin AM (2010). The HADDOCK web server for data-driven biomolecular docking. Nat Protoc, 5(5), 883–897. 10.1038/nprot.2010.32 [DOI] [PubMed] [Google Scholar]
- Denoyelle F, Lina-Granade G, Plauchu H, Bruzzone R, Chaib H, Levi-Acobas F, … Petit C (1998). Connexin 26 gene linked to a dominant deafness. Nature, 393(6683), 319–320. 10.1038/30639 [DOI] [PubMed] [Google Scholar]
- Dolinsky TJ, Nielsen JE, McCammon JA, & Baker NA (2004). PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res, 32(Web Server issue), W665–667. 10.1093/nar/gkh381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly S, English G, de Zwart-Storm EA, Lang S, van Steensel MA, & Martin PE (2012). Differential susceptibility of Cx26 mutations associated with epidermal dysplasias to peptidoglycan derived from Staphylococcus aureus and Staphylococcus epidermidis. Exp Dermatol, 21(8), 592–598. 10.1111/j.1600-0625.2012.01521.x [DOI] [PubMed] [Google Scholar]
- Easton JA, Donnelly S, Kamps MA, Steijlen PM, Martin PE, Tadini G, … van Steensel MA (2012). Porokeratotic eccrine nevus may be caused by somatic connexin26 mutations. J Invest Dermatol, 132(9), 2184–2191. 10.1038/jid.2012.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskin-Schwartz M, Metzger Y, Peled A, Weissglas-Volkov D, Malchin N, Gat A, … Sarig O (2016). Somatic mosaicism for a “lethal” GJB2 mutation results in a patterned form of spiny hyperkeratosis without eccrine involvement. Pediatr Dermatol, 33(3), 322–326. 10.1111/pde.12848 [DOI] [PubMed] [Google Scholar]
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, … Sali A (2007). Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci, Chapter 2, Unit 2 9 10.1002/0471140864.ps0209s50 [DOI] [PubMed] [Google Scholar]
- Evans WH, De Vuyst E, & Leybaert L (2006). The gap junction cellular internet: connexin hemichannels enter the signalling limelight. Biochem J, 397(1), 1–14. 10.1042/BJ20060175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerido DA, DeRosa AM, Richard G, & White TW (2007). Aberrant hemichannel properties of Cx26 mutations causing skin disease and deafness. Am J Physiol Cell Physiol, 293(1), C337–345. 10.1152/ajpcell.00626.2006 [DOI] [PubMed] [Google Scholar]
- Gonzalez D, Gomez-Hernandez JM, & Barrio LC (2006). Species specificity of mammalian connexin-26 to form open voltage-gated hemichannels. FASEB J, 20(13), 2329–2338. 10.1096/fj.06-5828com [DOI] [PubMed] [Google Scholar]
- Holst M, Baker NA, & Wang F (2000). Adaptive multilevel finite element solution of the Poisson-Boltzmann Equation I. Nat Protoc, 5, 883–897. [Google Scholar]
- Humphrey W, Dalke A, & Schulten K (1996). VMD: visual molecular dynamics. J Mol Graph, 14(1), 33–38, 27–38. doi:0263785596000185 [pii] [DOI] [PubMed] [Google Scholar]
- Jonard L, Feldmann D, Parsy C, Freitag S, Sinico M, Koval C, … Hadj-Rabia S (2008). A familial case of Keratitis-Ichthyosis-Deafness (KID) syndrome with the GJB2 mutation G45E. Eur J Med Genet, 51(1), 35–43. 10.1016/j.ejmg.2007.09.005 [DOI] [PubMed] [Google Scholar]
- Karademir LB, Aoyama H, Yue B, Chen H, & Bai D (2016). Engineered Cx26 variants established functional heterotypic Cx26/Cx43 and Cx26/Cx40 gap junction channels. Biochem J, 473(10), 1391–1403. 10.1042/BCJ20160200 [DOI] [PubMed] [Google Scholar]
- Koval M, Molina SA, & Burt JM (2014). Mix and match: investigating heteromeric and heterotypic gap junction channels in model systems and native tissues. FEBS Lett, 588(8), 1193–1204. 10.1016/j.febslet.2014.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird DW (2006). Life cycle of connexins in health and disease. Biochem J, 394(Pt 3), 527–543. 10.1042/BJ20051922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazic T, Li Q, Frank M, Uitto J, & Zhou LH (2012). Extending the phenotypic spectrum of keratitis-ichthyosis-deafness syndrome: Report of a patient with GJB2 (G12R) connexin 26 mutation and unusual clinical findings. Pediatr Dermatol, 29, 349–357. 10.1111/j.1525-1470.2011.01425.x [DOI] [PubMed] [Google Scholar]
- Leclere JC, Le Gac MS, Le Marechal C, Ferec C, & Marianowski R (2017). GJB2 mutations: Genotypic and phenotypic correlation in a cohort of 690 hearing-impaired patients, toward a new mutation? Int J Pediatr Otorhinolaryngol, 102, 80–85. 10.1016/j.ijporl.2017.09.011 [DOI] [PubMed] [Google Scholar]
- Lee JR, Derosa AM, & White TW (2009). Connexin mutations causing skin disease and deafness increase hemichannel activity and cell death when expressed in Xenopus oocytes. J Invest Dermatol, 129(4), 870–878. 10.1038/jid.2008.335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinsohn JL, McNiff JM, Antaya RJ, & Choate KA (2015). A somatic p.G45E GJB2 mutation causing porokeratotic eccrine ostial and dermal duct nevus. JAMA Dermatol, 151(6), 638–641. 10.1001/jamadermatol.2014.5069 [DOI] [PubMed] [Google Scholar]
- Li H, & Durbin R (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics, 26(5), 589–595. 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilly E, Sellitto C, Milstone LM, & White TW (2016). Connexin channels in congenital skin disorders. Semin Cell Dev Biol, 50, 4–12. 10.1016/j.semcdb.2015.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Nakagawa S, Suga M, Yamashita E, Oshima A, Fujiyoshi Y, & Tsukihara T (2009). Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature, 458(7238), 597–602. 10.1038/nature07869 [DOI] [PubMed] [Google Scholar]
- Marsden RA, Fleming K, & Dawber RP (1979). Comedo naevus of the palm--a sweat duct naevus? Br J Dermatol, 101(6), 717–722. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, … DePristo MA (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res, 20(9), 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mese G, Londin E, Mui R, Brink PR, & White TW (2004). Altered gating properties of functional Cx26 mutants associated with recessive non-syndromic hearing loss. Hum Genet, 115(3), 191–199. 10.1007/s00439-004-1142-6 [DOI] [PubMed] [Google Scholar]
- Mhaske PV, Levit NA, Li L, Wang HZ, Lee JR, Shuja Z, … White TW (2013). The human Cx26-D50A and Cx26-A88V mutations causing keratitis-ichthyosis-deafness syndrome display increased hemichannel activity. Am J Physiol Cell Physiol, 304(12), C1150–1158. 10.1152/ajpcell.00374.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami SB, Mutai H, Nakano A, Arimoto Y, Taiji H, Morimoto N, … Matsunaga T (2013). GJB2-associated hearing loss undetected by hearing screening of newborns. Gene, 532(1), 41–45. 10.1016/j.gene.2013.08.094 [DOI] [PubMed] [Google Scholar]
- Oeffner F, Fischer G, Happle R, Konig A, Betz RC, Bornholdt D, … Grzeschik KH (2009). IFAP syndrome is caused by deficiency in MBTPS2, an intramembrane zinc metalloprotease essential for cholesterol homeostasis and ER stress response. Am J Hum Genet, 84(4), 459–467. 10.1016/j.ajhg.2009.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, … Schulten K (2005). Scalable molecular dynamics with NAMD. J Comput Chem, 26(16), 1781–1802. 10.1002/jcc.20289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripps H, Qian H, & Zakevicius J (2004). Properties of connexin26 hemichannels expressed in Xenopus oocytes. Cell Mol Neurobiol, 24(5), 647–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouan F, White TW, Brown N, Taylor AM, Lucke TW, Paul DL, … Richard G (2001). trans-dominant inhibition of connexin-43 by mutant connexin-26: implications for dominant connexin disorders affecting epidermal differentiation. J Cell Sci, 114(Pt 11), 2105–2113. [DOI] [PubMed] [Google Scholar]
- Sanchez HA, Mese G, Srinivas M, White TW, & Verselis VK (2010). Differentially altered Ca2+ regulation and Ca2+ permeability in Cx26 hemichannels formed by the A40V and G45E mutations that cause keratitis ichthyosis deafness syndrome. J Gen Physiol, 136(1), 47–62. 10.1085/jgp.201010433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez HA, Villone K, Srinivas M, & Verselis VK (2013). The D50N mutation and syndromic deafness: altered Cx26 hemichannel properties caused by effects on the pore and intersubunit interactions. J Gen Physiol, 142(1), 3–22. 10.1085/jgp.201310962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snoeckx RL, Huygen PLM, Feldmann D, Marlin S, Denoyelle F, Waligora J, … Van Camp G (2005). GJB2 mutations and degree of hearing loss: A multicenter study. Am J Hum Genet, 77, 945–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spray DC, Harris AL, & Bennett MV (1981). Equilibrium properties of a voltage-dependent junctional conductance. J Gen Physiol, 77(1), 77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas M, Verselis VK, & White TW (2018). Human diseases associated with connexin mutations. Biochimica et Biophysica Acta, 1860, 192–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stong BC, Chang Q, Ahmad S, & Lin X (2006). A novel mechanism for connexin 26 mutation linked deafness: cell death caused by leaky gap junction hemichannels. Laryngoscope, 116(12), 2205–2210. 10.1097/01.mlg.0000241944.77192.d2 [DOI] [PubMed] [Google Scholar]
- Turner DL, & Weintraub H (1994). Expression of achaete-scute homolog 3 in Xenopus embryos converts ectodermal cells to a neural fate. Genes Dev, 8(12), 1434–1447. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, & Hakonarson H (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res, 38(16), e164 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TW, Bruzzone R, Goodenough DA, & Paul DL (1992). Mouse Cx50, a functional member of the connexin family of gap junction proteins, is the lens fiber protein MP70. Mol Biol Cell, 3(7), 711–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Word JM, Lovell SC, Richardson JS, & Richardson DC (1999). Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation. J Mol Biol, 285(4), 1735–1747. 10.1006/jmbi.1998.2401 [DOI] [PubMed] [Google Scholar]
- Zonta F, Girotto G, Buratto D, Crispino G, Morgan A, Abdulhadi K, … Mammano F (2015). The p.Cys169Tyr variant of connexin 26 is not a polymorphism. Hum Mol Genet, 24(9), 2641–2648. 10.1093/hmg/ddv026 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.