

Abstract
Conventional parallel group randomized controlled clinical trials (RCT) in Alzheimer’s disease (AD) are too large, long, expensive and insensitive to clinical change to meet the urgent need for an effective treatment. While providing good evidence for a treatment’s benefit, parallel group RCTs in AD must have very large samples and broad measures of change to accommodate the marked heterogeneity of demographics, genetics, symptoms, pathophysiologies, comorbidities and rates of progression. Multi-crossover, placebo-controlled, double-blind RCTs, including those with sample sizes as small as a single subject (“N-of-1”), are robust designs wherein subjects serve as their own controls in repeated blocks of randomly sequenced crossover treatments. Heterogeneities are inherently controlled and the sensitivity, specificity and clinical relevance of outcomes can be enhanced further by including personalized outcome measures for each individual. Multi-crossover N-of-1 RCTs enable unbiased estimation of efficacy for single subjects, and joint analysis of multiple N-of-1 trials enables estimation of efficacy for populations with much smaller sample sizes than those needed in conventional parallel group studies. It is important to identify carryover effects and natural history time trends to achieve unbiased estimation and testing of the treatment effect. We assert that in AD, multi-crossover RCTs offer many advantages over standard parallel group trials for drugs or other treatments with suitable mechanisms of action, pharmacokinetics and pharmacodynamics; despite the fact that they are almost never conducted. They may be especially useful for therapies directed at symptomatic improvement of cognitive and neuropsychiatric symptoms, but also can be used in early phase studies of disease modifying treatments with biomarker outcome measures.
Introduction
The US Congress’s National Alzheimer’s Project Act and nations around the world have the ambitious goal to prevent and effectively treat Alzheimer’s disease (AD) by 2025.1 Progress towards this goal however remains at a standstill. The most recent drug approval for AD was memantine in 2002/3 (Europe/US) and this very modestly beneficial medicine along with the more widely prescribed, but similarly modest, earlier-approved cholinesterase inhibitors constitute the current pharmacopeia indicated for AD.2 In the last 15 years many promising drugs for both symptom alleviation and disease modification have failed, some spectacularly so in late phase trials.3–8
AD is a complex neurodegenerative disease driven by heterogeneous pathophysiologies in vicious cycles of proteinopathies, inflammation, metabolic and neurovascular derangements leading to synaptic dysfunction and neuronal death.9–14 Insidious, progressive dementia is the clinical manifestation of these processes whose cumulative effects evolve over years before and after onset of clinical symptoms.15 The complexity of AD’s biology and protean clinical features present many challenges, but also many entry points for affecting the disease and its symptoms.
Progress towards achieving an effective prevention or treatment for AD is, at least in part, mired in lumbering clinical trials. Today’s trials often are built on narrow mechanistic concepts of AD, failing to accommodate the heterogeneous pathophysiological and co-morbid drivers of the dementia syndrome. Commonly used outcome measures are broad and noisy. Meaningful changes in AD’s variable symptoms and rates of progression can get washed out amidst “good days and bad days” with infrequently acquired cognitive assessments. There continue to be few biomarkers or other surrogate markers that are validated, sensitive, specific and informative for diagnosis, staging and progression, disease mechanisms or even therapeutic target engagement.16–19 Overcoming these limitations to demonstrate effectiveness in conventional parallel group randomized controlled trials (RCT) usually requires sample sizes of hundreds to thousands and long commitments from patients to detect a difference from control conditions, especially for disease modifying treatments. Such trials cost hundreds of millions of dollars.
In conventional parallel group RCTs, eligible participants are randomly assigned to one of two or more comparison groups and the primary and secondary endpoints are compared between groups. The well-designed study provides a good estimate of a drug’s effect of interest in the average patient – whether positive or null -- and is the most common design by far for evidence in the regulatory approval process for common diseases. The relevance of results to real world patients however, may be constrained by often narrow eligibility criteria and the exclusion of subjects with common co-morbidities, concurrent medications, too mild or too severe disease or other factors that might coarsen group responses. It has been noted that the average RCT excludes three quarters of potential participants and ultimately enrolls only 10% of all candidates with the disease of interest.20 Such stringency in eligibility is particularly common in today’s clinical trials research in AD. Highly focused eligibility criteria may boost the signal to noise ratio for a medicine if responders are correctly targeted, but their external validity can be questioned for the large percentage of patients not enrolled. Paradoxically, they may miss detecting a signal because subgroups for which the medicine might have helped were unwittingly excluded. Sample sizes also need to be very large when treatment effects are modest. This makes them almost impossible in rarer AD-related dementias such as dementias with Lewy Bodies or frontotemporal lobar degenerations.
Challenges of recruiting and screening thousands of participants to fill the large, hyper-selective samples of today’s pivotal clinical trials in AD and retaining them for years is among the most difficult, time consuming and expensive aspect of a study.21 This is especially true in the current era of trials that require PET scans for amyloid or tau to determine eligibility. Hundreds of diverse sites around the world, each recruiting a handful or two of subjects to achieve sample size, are logistically managed by a complex multivendor infrastructure struggling to maintain a semblance of uniformity of protocol. To this burden is added the “broken” funding, regulatory, and training envrionment.16
Definition of Multi-Crossover “N-of-1” Randomized Controlled Trials
An open label single patient “trial” is the general practice of clinical medicine. A practitioner reasons that a particular medicine will benefit their particular patient, prescribes it, and then patient and provider gauge the response after an adequate period of time. The patient serves as their own control. Of course, such trials are rarely conducted systematically and are confounded by many biases, especially placebo effects for both patient and provider. As case reports in the medical literature, they constitute anecdotes at a low level of medical evidence.
More rigorous formulations of single patient trials, so-called N-of-1 trials, have been conducted and discussed since the 1950s.22–27 As defined by the Consolidated Standards of Reporting Trials (CONSORT) extension for reporting N-of-1 trials (CENT) 2015 statement,28 an N-of-1 trial is “an experimental clinical design to determine the effect of an intervention in a single study participant,” in which they undergo several blocks of crossovers between the experimental treatment and control condition and the relative response in each block is assessed. When rigorously conducted with appropriately selected treatments, pre-specified doses, durations and placebo washouts of each treatment, blinding, repeated blocks, randomized sequences of treatments in each block and systematically applied outcome measures, a multi-crossover N-of-1 RCT is a robust design that provides an unbiased estimate of effect for a single participant. Some consider such a study as Level 1 evidence, on par with systematic reviews of conventional parallel group RCTs.29 Guyatt and others have also shown how multi-crossover N-of-1 RCTs can be valuable in clinical practice to demonstrate benefit of a prescribed treatment for a given patient, or in the absence of benefit, to de-prescribe a medicine and spare side effects, iatrogenic risk and expense.24, 30, 31
When N-of-1 RCTs employ the same treatment, trial design and outcome measures in multiple subjects, the data can be combined easily and analyzed to obtain an estimate of the treatment’s effect in the condition more generally. With subjects serving as their own controls, many sources of bias (heterogeneity, co-morbidities) that mandate large sample sizes in parallel group RCTs are automatically controlled and statistical power is increased greatly.
Design of Multi-Crossover Randomized Controlled Trials
The essential design elements of multi-crossover RCTs are repetition, randomized block sequence assignment, blinding, and systematic outcomes measurement.32 The treatment, its doses, and the durations of each treatment and washout periods are pre-determined based on the drug’s pharmacokinetic and pharmacodynamic properties and natural history of the disease. A trial consists of a series of blocks (e.g., experimental treatment “A” versus placebo treatment “B”), whose sequence (AB or BA) is randomly assigned, with or without counterbalancing. Multi-crossover RCTs are described as especially practical for drugs that provide symptomatic benefit in a chronic condition, have rapid onsets of action with short half-lives and quick cessation of biological activity after withdrawal to avoid carryover confounding.33 However, they may be useful even in the presence of carryover effects, with accommodations in placebo washout durations and analyses.
Repetition and Block Sequence Assignment
Parallel group RCTs seek comparability of groups by randomized assignment to experimental treatment or control/placebo conditions. Multi-crossover RCTs seek comparability through repeated exposures to experimental and control/placebo treatment conditions in randomized sequence. A single crossover, AB or BA, compares two treatments in one or more subjects but is vulnerable to both random and systematic errors. Examples of random errors include fluctuations in the measurement of the outcome, as well as other unmeasured factors such as diet and sleep, seasonal changes or other health conditions that arise. Repeating blocks attenuates the effects of random error. Systematic errors include predictable features of the disease and the drug, such as disease progression, carryover effects or treatment-by-time interactions. Blocks that are short in length relative to natural progression and carry-over of the drug temper the effects of systematic error.
As seen in Figure 1, a trial with two treatments has two types of blocks, meaning that a three-block N-of-1 trial has eight possible treatment sequences. But even with randomization of block type, care must be taken to protect against bias of time-dependent or non-linear confounders. For instance, a regularly ordered design such as AB-AB-AB might be affected by carry-over effects or other factors. On the other hand, a partially counterbalanced design such as AB-BA-AB offers some protection against this. In multi-crossover RCTs with fewer block repetitions, i.e., three or four, random assignment to one of the sequences with counterbalancing may be preferred. With more block repetitions and a large sample size, randomization alone would suffice. More repetitions provide greater statistical power for determining the superiority of one treatment condition over another of course, but their implementation must be weighed against the practical matters of trial duration and complexity, participant retention, cost, and natural time trends in the disease course.
Figure 1:
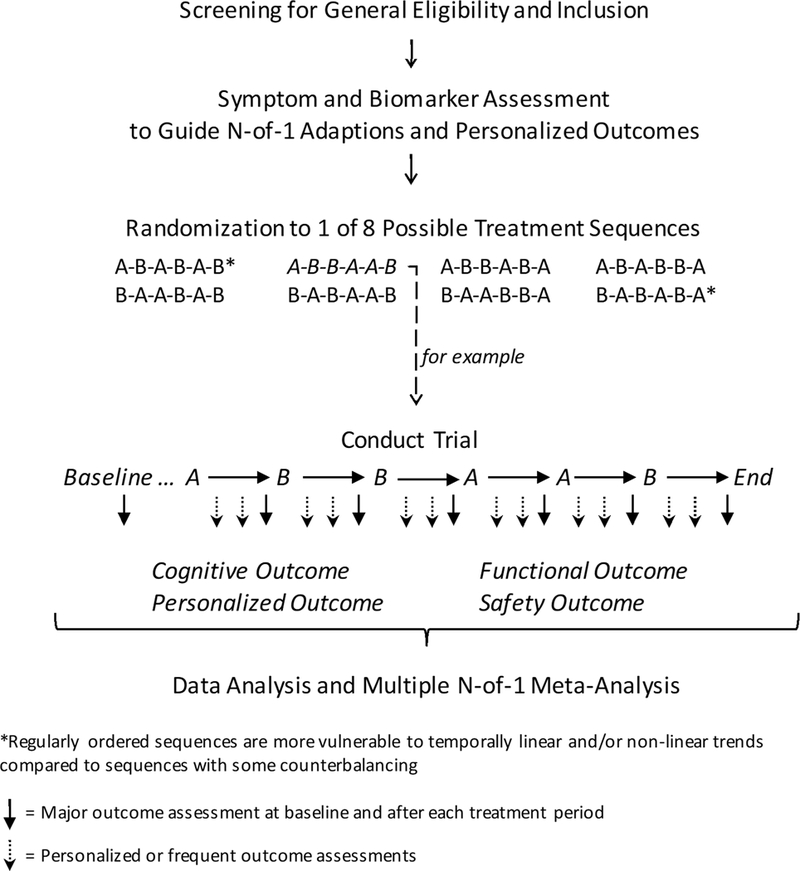
Three-block Multi-crossover N-of-1 Randomized controlled Trial of Active Treatment “A” vs. Comparator placebo “B” in Alzheimer’s Disease
Blinding
Placebo effects for study participants and outcomes ascertainment bias for investigators are major biasing factors in all clinical trials, but especially so for dementias in which inexact and subjective cognitive, psychological and daily functions are used as primary outcomes. For multi-crossover N-of-1 RCTs, as for conventional parallel group RCTs, it is important that participants and study personnel are blinded to treatment. This may not be possible for some interventions, e.g., behavioral interventions, but some blinding, at least for staff members measuring outcomes, is usually possible. Randomized sequences of treatments within blocks may make blinding more likely.
Outcome Measures
Multi-crossover N-of-1 RCTs can employ a full range of measures, from simple subjective rating scales and questionnaires to standardized clinician rating instruments, computerized cognitive or neurological assessments, wearable and passive monitoring technologies, biochemical and imaging biomarkers and health care utilization statistics. Of special importance are assessment strategies and tools that are robust for repeated measures. A run-in period allows the participant to become fully familiar with an assessment prior to baseline assessment and allows appraisal of the inherent variability of the measure in the participant prior to baseline assessment. Validated cognitive tests with alternative but equivalent versions of stimuli can be used, e.g., different word lists for memory testing. Or, a study can incorporate very frequent assessments or even continuous monitoring using innovative wearable or other passive ecological assessment technologies (see below).
A major advantage of multi-crossover N-of-1 trials is their adaptability for personalized outcomes, while rigorously controlling for subject-specific features. Along with standardized outcome measures, a multi-crossover N-of-1 RCT can readily customize assessments, enriching with measures of the participant’s most distressing symptoms or troublesome functional deficits based on their illness, severity stage or personal goals.
Adaptive Extensions in the Multi-Crossover N-of-1 Design
A natural extension of the N-of-1 design is to utilize a stopping rule for each subject that would pre-define criteria for stopping the trial in that subject as soon as sufficiently strong evidence in favor of treatments A or B or of their practical equivalence emerges. Like those used in standard parallel comparison RCTs, this stopping rule would preserve the type I error for each subject and would guarantee pre-selected operating characteristics. This approach recognizes heterogeneity across patients, even if not understood a priori through patient features, and allows the data to automatically select subgroups that respond to particular drugs or not. At a second level, another stopping rule could monitor the experience of each subject and enable the overall trial of multiple N-of-1 trials to stop if there is sufficient population-level evidence in favor of A or B or their equivalence. Development of these designs remains for future research.
Analysis of Multi-Crossover Randomized Controlled Trials
The basic data of a multi-crossover N-of-1 RCT are the measurements obtained in two or more different treatment conditions occurring repeatedly over time. The principal goal is to compare outcomes for each condition within a randomized block structure while accommodating repetition within subjects and possible effects of carryover or disease progression. Statistical methods used in multi-crossover N-of-1 RCTs include visual inspection of outcomes, aggregated t-tests, mixed effects longitudinal modeling and Bayesian analysis. These are well considered by Schmid, Duan and the DeCIDE Methods N-of-1 Guidance Panel.34 Similar, but more complex mixed effects longitudinal models could accommodate carryover effects and time trends in settings in which the block lengths are longer than temporal effects and so they are not washed out in the comparison of treatments within blocks.
The benefit of a treatment for a single patient can be inferred from a multi-crossover N-of-1 RCT; however, the generalizability of observations in any single subject for others is limited. Analyzing multiple multi-crossover N-of-1 RCTs together is an efficient approach to estimate treatment benefit at the population level. Multiple multi-crossover N-of-1 RCTs of similar design and outcome measures can be analyzed to reveal the characteristics of patients most likely to respond to the treatment and experience side effects. Such analyses can adaptively instruct subsequent trial designs. Approaches for combining individual treatment effects from multi-crossover N-of-1 RCTs include summary and individual participant data meta-analysis, repeated-measure models, Bayesian hierarchical models and single-period, single-pair, and averaged outcome crossover models.35–38 One recommended approach uses multi-level random effects models in which intra-individual and inter-individual variances of block-specific treatment effect estimates are included, along with patient-level characteristics such as demographics, co-morbidities, or other factors that may be important. This approach can generate an overall mean treatment effect for the population studied. Network meta-analysis models also have been proposed and are flexible in their inclusion of all the pairwise comparisons into a single model for simultaneous estimation, even when specific designs or even treatment comparators differ between trials.34, 39, 40 A simple approach for continuous data is to reduce the data to the within-block differences between treatments and to model them as the sum of a subject specific term (also called the treatment by patient interaction) and an error term; the differences are assumed independent across blocks within subjects and between subjects. The subject specific treatment effects are assumed to follow a normal distribution with a certain between-subject variance.41 Which approach to use will depend on many factors, but especially important are the number of trials to be combined, the extent of missing data, and the particular characteristics of the multi-crossover N-of-1 trials, such as number of repeated blocks and within patient heterogeneity of outcome measures and between patient variances.
Multi-Crossover N-of-1 Randomized Controlled Trials in AD Research
To our knowledge, there have been only two multi-crossover RCTs conducted in the Alzheimer’s field, both several decades ago. Jotkowitz found no benefit of physostigmine in a single-blind trial in which the drug was administered to ten patients in multiple crossovers over a period of ten months.42 Molloy et al. described three randomly ordered double-blinded crossover cycles of tetrahydroaminoacridine vs. placebo in 34 AD subjects and found no drug benefit for cognition, function or behavior in the 22 completers.43 An inquiry in PubMed.com (03/10/18) using key words of Alzheimer’s and either “N-of-1” or “crossover” yielded no additional studies in which N-of-1 or group RCTs with more than a single crossover could be discerned. In a search of Clinicatrials.gov (11/24/17), of 912 drug, dietary supplement or biological intervention clinical trials in ADRD since 1993, 648 are parallel group RCTs and 66 list crossover as their intervention model, while 198 are single group, uncontrolled, open label or otherwise indeterminate. None could be discerned to use a multi-crossover design.
Efficiency Considerations for Multiple Multi-Crossover N-of-1 versus Parallel Group RCTs in AD Research
Multi-crossover N-of-1 RCTs can provide very large increases in statistical power for demonstrating treatment effects in cognition, although at the expense of a longer participation for each subject and the assumption of stable treatment effect over time. To model the relative efficiencies of multi-crossover N-of-1 versus parallel group RCTs for symptomatic benefit in AD, we used data from a recent single crossover study of metformin versus placebo.44 In this pilot study of MCI and early dementia due to AD, participants were randomly assigned to receive either placebo or drug for eight weeks and then crossed over to drug or placebo for eight weeks respectively. Using the data for change in the Montreal Cognitive Assessment (MoCA)45 over eight weeks in the placebo group to estimate within and between subject variance terms, if the true treatment effect were to increase MoCA by 1 point, the 80% powered parallel group study requires 575 subjects and 16 weeks, while an N-of-1 study with 3 crossover blocks requires 39 subjects (total of 39×2×3=234 tests and 48 weeks). So, the multi-crossover RCT requires 6% of the subjects and 41% of tests but 300% of the study time per participant. If the true treatment effect is to increase MoCA by 2 points in 30% of people, the average effect is 0.6 points and the 80% powered parallel group study requires 1591 subjects, while the N-of-1 with 3 blocks requires 104 subjects (total of 104×2×3=624 tests). The multi-crossover design requires 6.5% of the subjects and 39% of tests.
Multi-Crossover N-of-1 RCTs for Symptomatic Treatment in AD
Cognitive and functional symptoms in AD are progressive, but slowly so.46–50 The relative stability, at least over reasonably short periods of time such as a year, make AD an appropriate condition for evaluating symptomatic treatments in multi-crossover RCTs.
“Cognitive enhancer,” “nootropic,” and “smart drug” are terms for drugs with cognitive function as their major target. Cholinesterase inhibitors and memantine, various psychostimulants, and a host of unproven dietary supplements and other drugs are in common use for symptomatic treatment in AD and other cognitive disorders.51–54 There are also many investigational drugs in various phases of development for symptoms of AD and other cognitive impairment disorders.17 Some examples include α−7 nicotinic receptor modulators, 5HT2A and 5HT6 serotonin receptor antagonists, inhibitors of various phosphodiesterases (PDE), p38MAPK inhibitors, AMPA receptor modulators, glycine transporter-1 inhibitors, GABA neurotransmission inhibitors, voltage-gated sodium channel modulators, and H3 histamine receptor antagonists. Aside from drugs, there is also interest in neurostimulation, exercise and other behavioral and somatic interventions for cognitive enhancement. Multi-crossover RCTs may be an efficient way to identify the most promising treatments in relatively small sample size studies, winnowing out those with no beneficial effects or de-risking future development of those showing promise in larger RCTs for regulatory approval.
Thoughtful consideration of carryover and potential disease-modifying as well as symptomatic effects of the drug, and natural disease progression in statistical analyses are critical. Treatment periods must be tailored to a drug’s pharmacokinetics and pharmacodynamics. For instance, an intervention like methylphenidate has a relatively rapid onset and cessation of action for symptoms. A trial of methylphenidate might be designed with weekly crossovers of drug and placebo in three or more randomly sequenced blocks, with the entire trial taking six weeks. A different example might be a PDE4 inhibitor, which augments the cAMP - protein kinase A - cAMP response element-binding protein (CREB) pathway to enhance memory consolidation via gene transcriptional changes and synaptic modification. Such drugs might be expected to have slower onset with greater carryover. A symptomatic multi-crossover trial for a PDE4 inhibitor might be designed with three blocks of two months of active treatment alternating with two or three months of placebo washout in each block, and thus take twelve to fifteen months to complete.
Personalized Extensions in the Multi-Crossover N-of-1 Design
Because participants serve as their own controls, multi-crossover N-of-1 RCTs allow latitude and creativity in outcome measures. Common cognitive batteries (e.g. RBANS55) can be complemented with custom-selected measures based on a participant’s disease severity or cognitive profile. “Designer” outcome measures can be employed to target cognitive, behavioral and functional impairments that are most meaningful to the individual. With mild cognitive impairment for instance, a driving reaction time and navigation game may be used with someone for whom driving is interesting or valued. Bridge may be an enjoyable leisure activity for another, and so daily performance in tablet-based games might reveal treatment-associated changes over the different periods.
Along with standard functional assessment instruments (e.g., FAQ56 or ADCS-ADL57) at the end of each treatment period, a multi-crossover N-of-1 RCT for AD might incorporate innovative daily personal functional outcome measures spanning widely differing levels of severity or interests. Kaye and colleagues have compellingly argued for high frequency ecological outcome measures with actimetric, biometric and other pervasive computing assessment technologies to gauge disease progression and treatment effects.58, 59 These are especially well-suited for multi-crossover RCTs. For instance, passive monitoring of duration and type of computer usage or even keystrokes can be informative as measures of memory, attention, speed of processing, motor coordination and dexterity, as well as apathy and social engagement.
Neuropsychiatric symptoms are common contributors to dysfunction and distress in AD, including apathy, anxiety, agitation, depression, psychosis and sleep disturbances.60 They are complex, with heterogeneous neurobiological, cognitive and environmental/psychosocial contributions and diverse manifestations. For example, agitation may be manifest as pacing in one person, constantly packing a suitcase in another, or attempting to leave the house in another. These could be tracked with well-established instruments like the Neuropsychiatric Inventory61 or Cohen Mansfield Agitation Inventory,62 but adding more personalized outcome measures such as actigraphy, sensors on a suitcase or sensors on doors in the house might offer greater sensitivity, specificity and objectivity for measuring symptom frequency, intensity and change with intervention. Apathy might manifest as physical inertia in one person, lack of interest in family and friends in another or lack of interest in sports in another. N-of-1 RCTs for apathy could include personal outcomes like actigraphy, phone usage records for outgoing phone calls or tracking sports-oriented TV and website usage, along with standard apathy rating instruments.
Personalized assessments can sensitively demonstrate meaningful effects of a treatment at the individual level. Multiple N-of-1 trials with diverse outcome measures could then be combined through various approaches to demonstrate efficacy in the disease more generally. At the simplest level, data from binary classification of personalized outcomes - improved/not improved - in each block for each participant could be combined, or more sophisticated meta-analytic approaches for Z-scores, such as multi-level random effects models, can be applied in which responses occurring at different levels of a data hierarchy are used in a single model.
Potential Role of Multi-Crossover N-of-1 RCTs for Disease Modification Effects
Prevention or slowing of clinical dementia in AD with disease modifying treatments are not practicable outcomes for N-of-1 RCTs with multiple crossovers and relatively short durations for each block. This is due both to AD’s slow and variable progression as well as the types of biological interventions that seek to permanently alter disease activity. Theoretically, one could design a study in which the duration of both treatments A and B in a block are each a year long, and AD progression rate is measured for cognition and function, hippocampal or cortical atrophy, or topographical spread of tau pathology with tau PET imaging. However, a three-block trial would take six years to complete and be complicated by the non-linear progression of AD (i.e., different treatment effects at different points in time), as well as other random and non-random factors that are difficult to control for with repetitions and/or analytical solutions, including carryover, unpredictable illnesses, major life events, or aging itself.
On the other hand, multi-crossover N-of-1 RCTs with repeatable biomarker outcomes can be useful in gauging target engagement of potential disease-modifying drugs. Functional MRI measures of perfusion, activation and network connectivity or EEG-based measures of event-related potentials and spectra may be dynamically responsive to a disease-modifying treatment. Emerging ultra-sensitive immunoassays in blood may be useful for measuring CNS-specific proteins reflecting AD pathology.63 For instance, repeated lowering of plasma levels of tau or phospho-tau with active drug but not placebo would provide evidence of a drug’s ability to modify disease pathology in a single participant. On the other hand, some biomarkers would be impractical or difficult in a multi-crossover trial, like cerebrospinal fluid biomarkers obtained via lumbar punctures with their procedural risks and low acceptability, or PET scans of tau, amyloid-β or inflammation with their radiation exposure and expense.
Conclusions
The need for new, effective treatments for AD is urgent. Reducing the number of participants required for a well-powered study using novel clinical trial designs is an efficient way to accelerate progress, as recruitment and screening are among the most time-intensive and costly activities in a clinical trial program. Multi-crossover N-of-1 RCTs are robust designs in which heterogeneities are automatically controlled. When standardized outcome measures are used, data can be combined to estimate group level effect sizes. The design also allows flexibility for adaptive modifications and personalized outcomes that may enhance sensitivity, specificity and clinical relevance of a treatment’s effects. Such designs can be especially useful in Phase II for new and re-purposed drugs, with subsequent “graduation” of the drug to more conventional, larger trial designs for regulatory agency marketing approval. While biomarkers or other surrogate endpoints would be typically used in the early phase trials, Phase III or other pivotal trials for approval would likely use clinical endpoints. From the participant’s perspective, the design is attractive as exposure to the experimental treatment is guaranteed and not subject to chance via randomization. At the end of the trial, the data for treatment benefit, or lack thereof, for each individual can be shared, perhaps providing guidance for their ongoing care. However, the overall time and effort commitment to the trial for each subject will usually be greater than a conventional parallel group study.
While our focus here has been AD, we advocate that this design has wide applicability in other chronic and/or slowly progressive neurological disorders, including epilepsy, migraine, long-term residual deficits post-stroke and traumatic brain injury, Parkinson’s disease and others (see Table 3, Lillie et al27). The suitability of a multi-crossover trial design for a given treatment depends on a variety of factors, particularly the drug’s pharmacokinetic and pharmacodynamic properties, the precision and sensitivity of outcome measures, and the natural course of AD progression. For many investigational treatments in AD, these factors can be managed by design with adequate placebo washout periods and biostatistical methods that accommodate carryover, frequent outcome measures and changing baselines. Multi-crossover N-of-1 RCTs can play a valuable role in building the evidence base for investigational drugs and other treatments for AD.
Acknowledgment
The authors acknowledge support of the Challenger Foundation and the Massachusetts Alzheimer’s Disease Research Center (NIH P50 AG005134) in the preparation of this manuscript.
Footnotes
Potential Conflicts of Interest
Nothing to report.
References
- 1.Snyder HM, Hendrix J, Bain LJ, Carrillo MC. Alzheimer’s disease research in the context of the national plan to address Alzheimer’s disease. Mol Aspects Med 2015. Jun-Oct;43–44:16–24. [DOI] [PubMed] [Google Scholar]
- 2.Tricco AC, Ashoor HM, Soobiah C, et al. Comparative Effectiveness and Safety of Cognitive Enhancers for Treating Alzheimer’s Disease: Systematic Review and Network Metaanalysis. J Am Geriatr Soc 2017. September 29. [DOI] [PubMed] [Google Scholar]
- 3.Jones RW. Dimebon disappointment. Alzheimers Res Ther 2010. September 13;2(5):25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vandenberghe R, Rinne JO, Boada M, et al. Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alzheimers Res Ther 2016. May 12;8(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doody RS, Raman R, Farlow M, et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med 2013. July 25;369(4):341–50. [DOI] [PubMed] [Google Scholar]
- 6.Lilly announces detailed results of solanezumab Phase 3 EXPEDITION 3 study at the Clinical Trials on Alzheimer’s Disease (CTAD) 2016 Meeting 2016. . https://wwwprnewswirecom/news-releases/lilly-announces-detailed-results-of-solanezumab-phase-3-expedition3-study-at-the-clinical-trials-on-alzheimers-disease-ctad-2016-meeting-300375751html.Accessed 12/10/17.
- 7. Merck Announces EPOCH Study of Verubecestat for the Treatment of People with Mild to Moderate Alzheimer’s Disease to Stop for Lack of Efficacy. 2017 http://investorsmerckcom/news/press-release-details/2017/Merck-Announces-EPOCH-Study-of-Verubecestat-for-the-Treatment-of-People-with-Mild-to-Moderate-Alzheimers-Disease-to-Stop-for-Lack-of-Efficacy/defaultaspx; Accessed 12/10/17.
- 8.Cummings J, Lee G, Mortsdorf T, Ritter A, Zhong K. Alzheimer’s disease drug development pipeline: 2017. Alzheimers Dement (N Y) 2017. September;3(3):367–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mufson EJ, Ikonomovic MD, Counts SE, et al. Molecular and cellular pathophysiology of preclinical Alzheimer’s disease. Behav Brain Res 2016. September 15;311:54–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci 2015. June;16(6):358–72. [DOI] [PubMed] [Google Scholar]
- 11.Chiti F, Dobson CM. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu Rev Biochem 2017. June 20;86:27–68. [DOI] [PubMed] [Google Scholar]
- 12.Niedzielska E, Smaga I, Gawlik M, et al. Oxidative Stress in Neurodegenerative Diseases. Mol Neurobiol 2016. August;53(6):4094–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Santos CY, Snyder PJ, Wu WC, Zhang M, Echeverria A, Alber J. Pathophysiologic relationship between Alzheimer’s disease, cerebrovascular disease, and cardiovascular risk: A review and synthesis. Alzheimers Dement (Amst) 2017;7:69–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arnold SE, Arvanitakis Z, Macauley-Rambach SL, et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol 2018. January 29. [DOI] [PMC free article] [PubMed]
- 15.Jack CR Jr. , Holtzman DM. Biomarker modeling of Alzheimer’s disease. Neuron 2013. December 18;80(6):1347–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cummings J, Aisen P, Barton R, et al. Re-Engineering Alzheimer Clinical Trials: Global Alzheimer’s Platform Network. J Prev Alzheimers Dis 2016. June;3(2):114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cummings J, Aisen PS, DuBois B, et al. Drug development in Alzheimer’s disease: the path to 2025. Alzheimers Res Ther 2016. September 20;8:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cummings J Lessons Learned from Alzheimer Disease: Clinical Trials with Negative Outcomes. Clin Transl Sci 2017. August 02. [DOI] [PMC free article] [PubMed]
- 19.Sugino H, Watanabe A, Amada N, et al. Global Trends in Alzheimer Disease Clinical Development: Increasing the Probability of Success. Clin Ther 2015. August;37(8):1632–42. [DOI] [PubMed] [Google Scholar]
- 20.Rothwell PM. External validity of randomised controlled trials: “to whom do the results of this trial apply?”. Lancet 2005. January 1–7;365(9453):82–93. [DOI] [PubMed] [Google Scholar]
- 21.Fargo KN, Carrillo MC, Weiner MW, Potter WZ, Khachaturian Z. The crisis in recruitment for clinical trials in Alzheimer’s and dementia: An action plan for solutions. Alzheimers Dement 2016. November;12(11):1113–5. [DOI] [PubMed] [Google Scholar]
- 22.Hogben L, Sim M. The self-controlled and self-recorded clinical trial for low-grade morbidity. Br J Prev Soc Med 1953. October;7(4):163–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hare EH. Comparative efficacy of hypnotics a self-controlled, self-recorded clinical trial in neurotic patients. Br J Prev Soc Med 1955. July;9(3):140–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guyatt G, Sackett D, Taylor DW, Chong J, Roberts R, Pugsley S. Determining optimal therapy--randomized trials in individual patients. N Engl J Med 1986. April 03;314(14):889–92. [DOI] [PubMed] [Google Scholar]
- 25.Nikles CJ, Mitchell GK, Del Mar CB, Clavarino A, McNairn N. An n-of-1 trial service in clinical practice: testing the effectiveness of stimulants for attention-deficit/hyperactivity disorder. Pediatrics 2006. June;117(6):2040–6. [DOI] [PubMed] [Google Scholar]
- 26.Mirza RD, Punja S, Vohra S, Guyatt G. The history and development of N-of-1 trials. J R Soc Med 2017. August;110(8):330–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lillie EO, Patay B, Diamant J, Issell B, Topol EJ, Schork NJ. The n-of-1 clinical trial: the ultimate strategy for individualizing medicine? Per Med 2011. March;8(2):161–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vohra S, Shamseer L, Sampson M, et al. CONSORT extension for reporting N-of-1 trials (CENT) 2015 Statement. BMJ 2015. May 14;350:h1738. [DOI] [PubMed] [Google Scholar]
- 29.Howick J, Chalmers I, Glasziou P, et al. The 2011 Oxford CEBM Evidence Levels of Evidence (Introductory Document). Oxford Centre for Evidence-Based Medicine 2011:http://www.cebm.net/index.aspx?o=5653.
- 30.Guyatt G N of 1 randomized trials: a commentary. J Clin Epidemiol 2016. August;76:4–5. [DOI] [PubMed] [Google Scholar]
- 31.Berlin JA. N-of-1 clinical trials should be incorporated into clinical practice. J Clin Epidemiol 2010. December;63(12):1283–4. [DOI] [PubMed] [Google Scholar]
- 32.Kravitz RL, Duan N, Panel DMCN-o-G. Design and Implementation of N-of-1 Trials: A User’s Guide. AHRQ Publication No. 13(14)-EHC122-EF. In: Quality AfHRa, editor. Rockville, MD: Agency for Healthcare Research and Quality; 2014. p. http://www.effectivehealthcare.ahrq.gov/N-1-Trials.cfm. [Google Scholar]
- 33.Guyatt GH, Heyting A, Jaeschke R, Keller J, Adachi JD, Roberts RS. N of 1 randomized trials for investigating new drugs. Control Clin Trials 1990. April;11(2):88–100. [DOI] [PubMed] [Google Scholar]
- 34.Schmid CH, Duan N. Statistical design and analytic considerations for N-of-1 trials. Design and Implementation of N-of-1 Trials: A User’s Guide Rockville, MD: Agency for Healthcare Research and Quality; 2014. p. 33–52. [Google Scholar]
- 35.Zucker DR, Schmid CH, McIntosh MW, D’Agostino RB, Selker HP, Lau J. Combining single patient (N-of-1) trials to estimate population treatment effects and to evaluate individual patient responses to treatment. J Clin Epidemiol 1997. April;50(4):401–10. [DOI] [PubMed] [Google Scholar]
- 36.Duan N, Kravitz RL, Schmid CH. Single-patient (n-of-1) trials: a pragmatic clinical decision methodology for patient-centered comparative effectiveness research. J Clin Epidemiol 2013. August;66(8 Suppl):S21–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huber AM, Tomlinson GA, Koren G, Feldman BM. Amitriptyline to relieve pain in juvenile idiopathic arthritis: a pilot study using Bayesian metaanalysis of multiple N-of-1 clinical trials. J Rheumatol 2007. May;34(5):1125–32. [PubMed] [Google Scholar]
- 38.Zucker DR, Ruthazer R, Schmid CH. Individual (N-of-1) trials can be combined to give population comparative treatment effect estimates: methodologic considerations. J Clin Epidemiol 2010. December;63(12):1312–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.White IR, Barrett JK, Jackson D, Higgins JP. Consistency and inconsistency in network meta-analysis: model estimation using multivariate meta-regression. Res Synth Methods 2012. June;3(2):111–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Higgins JP, Jackson D, Barrett JK, Lu G, Ades AE, White IR. Consistency and inconsistency in network meta-analysis: concepts and models for multi-arm studies. Res Synth Methods 2012. June;3(2):98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Senn S Sample size considerations for n-of-1 trials. Stat Methods Med Res 2017. January 1:962280217726801. [DOI] [PubMed] [Google Scholar]
- 42.Jotkowitz S Lack of clinical efficacy of chronic oral physostigmine in Alzheimer’s disease. Ann Neurol 1983. December;14(6):690–1. [DOI] [PubMed] [Google Scholar]
- 43.Molloy DW, Guyatt GH, Wilson DB, Duke R, Rees L, Singer J. Effect of tetrahydroaminoacridine on cognition, function and behaviour in Alzheimer’s disease. CMAJ 1991. January 01;144(1):29–34. [PMC free article] [PubMed] [Google Scholar]
- 44.Koenig AM, Mechanic-Hamilton D, Xie SX, et al. Effects of the Insulin Sensitizer Metformin in Alzheimer Disease: Pilot Data From a Randomized Placebo-controlled Crossover Study. Alzheimer Dis Assoc Disord 2017. Apr-Jun;31(2):107–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nasreddine ZS, Phillips NA, Bedirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005. April;53(4):695–9. [DOI] [PubMed] [Google Scholar]
- 46.Qian J, Wolters FJ, Beiser A, et al. APOE-related risk of mild cognitive impairment and dementia for prevention trials: An analysis of four cohorts. PLoS Med 2017. March;14(3):e1002254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Amariglio RE, Donohue MC, Marshall GA, et al. Tracking early decline in cognitive function in older individuals at risk for Alzheimer disease dementia: the Alzheimer’s Disease Cooperative Study Cognitive Function Instrument. JAMA Neurol 2015. April;72(4):446–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Storandt M, Balota DA, Aschenbrenner AJ, Morris JC. Clinical and psychological characteristics of the initial cohort of the Dominantly Inherited Alzheimer Network (DIAN). Neuropsychology 2014. January;28(1):19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilson RS, Beckett LA, Bennett DA, Albert MS, Evans DA. Change in cognitive function in older persons from a community population: relation to age and Alzheimer disease. Arch Neurol 1999. October;56(10):1274–9. [DOI] [PubMed] [Google Scholar]
- 50.Monsell SE, Liu D, Weintraub S, Kukull WA. Comparing measures of decline to dementia in amnestic MCI subjects in the National Alzheimer’s Coordinating Center (NACC) Uniform Data Set. Int Psychogeriatr 2012. October;24(10):1553–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Froestl W, Pfeifer A, Muhs A. Cognitive enhancers (nootropics). Part 3: drugs interacting with targets other than receptors or enzymes. disease-modifying drugs. J Alzheimers Dis 2013;34(1):1–114. [DOI] [PubMed] [Google Scholar]
- 52.Froestl W, Muhs A, Pfeifer A. Cognitive enhancers (nootropics). Part 2: drugs interacting with enzymes. J Alzheimers Dis 2013;33(3):547–658. [DOI] [PubMed] [Google Scholar]
- 53.Froestl W, Muhs A, Pfeifer A. Cognitive enhancers (nootropics). Part 1: drugs interacting with receptors. J Alzheimers Dis 2012;32(4):793–887. [DOI] [PubMed] [Google Scholar]
- 54.Zanforlin E, Zagotto G, Ribaudo G. An Overview of New Possible Treatments of Alzheimer’s Disease, Based on Natural Products and Semi-Synthetic Compounds. Curr Med Chem 2017. November 17;24(34):3749–73. [DOI] [PubMed] [Google Scholar]
- 55.Randolph C, Tierney MC, Mohr E, Chase TN. The Repeatable Battery for the Assessment of Neuropsychological Status (RBANS): preliminary clinical validity. J Clin Exp Neuropsychol 1998. June;20(3):310–9. [DOI] [PubMed] [Google Scholar]
- 56.Pfeffer RI, Kurosaki TT, Harrah CH Jr., Chance JM, Filos S. Measurement of functional activities in older adults in the community. J Gerontol 1982. May;37(3):323–9. [DOI] [PubMed] [Google Scholar]
- 57.Galasko D, Bennett D, Sano M, et al. An inventory to assess activities of daily living for clinical trials in Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. Alzheimer Dis Assoc Disord 1997;11 Suppl 2:S33–9. [PubMed] [Google Scholar]
- 58.Lyons BE, Austin D, Seelye A, et al. Pervasive Computing Technologies to Continuously Assess Alzheimer’s Disease Progression and Intervention Efficacy. Front Aging Neurosci 2015;7:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Arneric SP, Cedarbaum JM, Khozin S, et al. Biometric monitoring devices for assessing end points in clinical trials: developing an ecosystem. Nat Rev Drug Discov 2017. October;16(10):736. [DOI] [PubMed] [Google Scholar]
- 60.Rosenberg PB, Nowrangi MA, Lyketsos CG. Neuropsychiatric symptoms in Alzheimer’s disease: What might be associated brain circuits? Mol Aspects Med 2015. Jun-Oct;43–44:25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cummings JL. The Neuropsychiatric Inventory: assessing psychopathology in dementia patients. Neurology 1997. May;48(5 Suppl 6):S10–6. [DOI] [PubMed] [Google Scholar]
- 62.Cohen-Mansfield J Agitated behaviors in the elderly. II. Preliminary results in the cognitively deteriorated. J Am Geriatr Soc 1986. October;34(10):722–7. [DOI] [PubMed] [Google Scholar]
- 63.Blennow K A Review of Fluid Biomarkers for Alzheimer’s Disease: Moving from CSF to Blood. Neurol Ther 2017. July;6(Suppl 1):15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]