

Abstract
Idelalisib (ILB) is a selective phosphatidylinositol-3-kinase delta inhibitor approved for the treatment of hematological malignancies. However, ILB frequently causes hepatotoxicity and the exact mechanism remains unclear. The current study profiled the metabolites of ILB in mouse liver, urine and feces. The major metabolites found in the liver were oxidized metabolite GS-563117 (M1) and ILB-glutathione (GSH) adduct (M2). These metabolic pathways were confirmed by analysis of urine and feces from mice treated with ILB. Identification of ILB-GSH adduct (M2) suggests the formation of reactive metabolites of ILB. We also found that M1 can produce reactive metabolites and form M1-GSH adducts. The GSH-conjugates identified in mouse liver were also found in the incubations of ILB and M1 with human liver microsomes. Furthermore, we illustrated that CYP3A4 and 2C9 are the key enzymes contributing to the bioactivation pathway of ILB and M1. In summary, our work revealed that both ILB and its major metabolite M1 can undergo bioactivation to produce reactive metabolites in the liver. Further studies are required to determine whether these metabolic pathways contribute to ILB hepatotoxicity.
Keywords: idelalisib, bioactivation, metabolomics, hepatotoxicity, CYP
TOC graphic
INTRODUCTION
Idelalisib (ILB) is a selective phosphatidylinositol-3-kinase delta (PI3Kδ) inhibitor that inhibits the binding of adenosine-5’-triphosphate to the catalytic subunit of PI3Kδ.1–3 ILB has been approved by the US Food and Drug Administration (FDA) for the treatment of relapsed chronic lymphocytic leukaemia (CLL, in combination with rituximab), relapsed follicular B-cell non-Hodgkin lymphoma, and relapsed small lymphocytic lymphoma. The overall response rate is 57% and the median duration response is 12.5 months using ILB as a monotherapy for the treatment of previously treated indolent non-Hodgkin lymphoma.4 As a combination therapy with rituximab, ILB improved overall response rate and overall survival at 12 months in patients with relapsed CLL.5
Although ILB is highly effective, various side effects of ILB have been observed in clinical practice, including enterocolitis, transaminitis, hematopoietic laboratory abnormality, and pneumonitis. In a phase 2 study of ILB for CLL treatment, a high incidence of hepatotoxicity was observed, as 54% of patients showed more than 3-fold increase in alanine aminotransferase.6 In addition, ILB has been given a black box warning by the FDA for its fatal side effects including hepatotoxicity. The detailed mechanisms of ILB hepatotoxicity remain unknown.
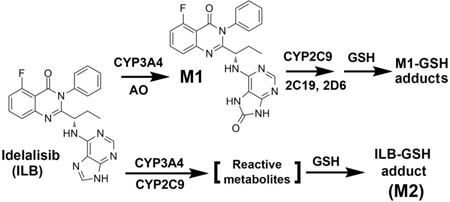
Drug metabolism and bioactivation is often associated with adverse drug reactions.7,8 The metabolism of ILB has been studied in vitro and in vivo.9–13 In a mass balance study using 14C-ILB, approximately 78% of the total radioactive dose administered was recovered in feces, whereas 14% was recovered in urine.10 The major metabolite of ILB in urine, feces ans1s1d plasma is GS-563117 (M1, Table S1), which is primarily formed by aldehyde oxidase (AO) with lesser involvement of cytochrome P450 (CYP) 3A.12 In addition, three glucuronides of ILB were detected in human urine.13 However, limited information is available for the metabolism and disposition of ILB in the liver.
The current study profiled the metabolites of ILB in mouse liver, urine, feces and human live microsomes (HLM) using a metabolomic approach. GS-563117 (M1) and ILB-glutathione (GSH) adduct (M2) were identified as the major metabolites of ILB in mouse liver. We also identified M1-GSH adducts in the liver. Identification of ILB-GSH and M1-GSH adducts suggests that reactive metabolites are produced in ILB metabolism in the liver. Furthermore, we demonstrated that CYP3A4 and 2C9 are the major enzymes contributing to the bioactivation of ILB and its major metabolite M1.
EXPERIMENTAL PROCEDURES
Chemicals and Reagents.
ILB ((S)-2-(1-((9H-purin-6-yl)amino)propyl)-5-fluoro-3-phenylquinazolin-4(3H)-one) was purchased from Ark Pharm (Arlington Heights, IL). M1 was purchased from TLC Pharmaceutical Standards (Aurora, Ontario, Canada). β-nicotinamide adenine dinucleotide phosphate (NADPH), GSH, ketoconazole (KCZ), fluoxetine hydrochloride, dimethyl sulfoxide (DMSO), corn oil, cysteine, cysteine-glycine and N-acetyl cysteine were purchased from Sigma-Aldrich (St. Louis, MO). HLM and the recombinant human CYPs (EasyCYP Bactosomes) were purchased from XenoTech (Lenexa, KS). All solvents for ultra-performance liquid chromatography and quadrupole time-of-flight mass spectrometry (UPLC-QTOFMS) analysis were of the highest grade commercially available.
Animals and Treatments.
BALB/c mice (male, 6–8 weeks old) were treated with ILB (100 mg/kg or 36 mg/kg, p.o.) or vehicle (n = 4). One hour later, blood was collected from the retro-orbital plexus into the BD Microtainer SST serum tubes (Franklin Lakes, NJ), and serum was harvested by centrifugation. Then, all mice were sacrificed and liver tissues were harvested. All the liver tissues were flash-frozen in liquid nitrogen and stored at −80 °C until further analysis. Another two groups of mice were treated with ILB (100 mg/kg, p.o.) or vehicle (n = 4), and housed separately in metabolic cages for 12 h to collect urine and feces. The study protocol was approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh.
Sample Preparation for Metabolite Analysis.
The methods for metabolite analysis in the liver, urine, feces and serum have been reported in previous papers.14, 15 Briefly, urinary samples were prepared by mixing 40 μL of urine with 160 μL acetonitrile followed by centrifugation at 15,000 rpm for 10 min. Feces were homogenized in water (100 mg feces in 1 mL water). Two hundred microliters of acetonitrile was added to 100 μL of the resulting suspension, and vortexed for 1 min. Subsequently, the resulting mixture was centrifuged at 15,000 rpm for 10 min. Twenty microliters of serum sample was vortexed with 80 μL methanol for 1 min followed by centrifugation at 15,000 rpm for 10 min. Liver samples were homogenized in water (50 mg tissue in 200 μL water) using PowerGen homogenizer (Fisher Scientific, Waltham, MA), and then 200 μL of methanol was added to 100 μL of the liver homogenate. The resulting mixtures were vortexed for 1 min, and centrifuged at 15,000 rpm for 10 min. Each supernatant from all samples mentioned above was transferred to an auto sampler vial, and injected into the UPLC-QTOFMS system (Waters, Milford, MA) for metabolite analysis.
Metabolism of ILB and M1 in HLM and mouse liver microsomes (MLM).
Incubations were carried out in 1 × PBS (137 mM NaCl, 2.7 mM KCl, and 11.9 mM phosphate buffer, pH 7.4), containing 0.2 mg HLM and 30 μM ILB or M1 in a final volume of 190 μL. The reactions were initiated by adding 10 μL of 20 mM NADPH (final concentration 1.0 mM) and continued at 37 °C for 45 min with gentle shaking. Incubations in the absence of NADPH were used as the control. To investigate possible reactive metabolites, 2.5 mM of the following trapping agents GSH, cysteine, cysteine-glycine or N-acetyl cysteine were used in the incubations with HLM. Incubations without NADPH or GSH were used as controls. In addition, 2.5 mM GSH was co-incubated with ILB or M1 in MLM. The reactions were terminated by adding 200 μL methanol. The mixture was vortexed for 30 s and centrifuged at 15,000 rpm for 10 min. Two microliters or 10 μL (for trapping assay) aliquot of the supernatant was injected into the UPLC-QTOFMS system for metabolite analysis. All incubations were performed in triplicate.
Metabolism of ILB or M1 by Recombinant CYPs.
Incubations were performed in 1 × PBS (pH 7.4), containing 30 μM ILB or M1, 2.5 mM GSH, 10 pmol of each cDNA-expressed CYP (control, CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and CYP3A4), and 1 mM NADPH in a final volume of 100 μL. After incubation at 37 °C for 45 min, the reactions were terminated with 100 μL of methanol. The mixture was then vortexed for 30 s and centrifuged at 15,000 rpm for 10 min. The supernatant was dried and reconstituted in 60 μL of acetonitrile/water (1:1, v/v). Co-incubations of KCZ (1 μM) or fluoxetine (200 μM) with CYP3A4, CYP2C9 or HLM, was performed to confirm the role of CYP3A4 and CYP2C9 in ILB metabolism. Ten microliters aliquot of the supernatant was injected into the UPLC-QTOFMS system for metabolite analysis. All incubations were performed in triplicate.
UPLC-QTOFMS Analysis.
Chromatographic separation of metabolites was performed on an Acquity UPLC BEH C18 column (2.1 × 100 mm, 1.7 μm; Waters Corporation, Milford, MA). The mobile phase A (MPA) was 0.1% formic acid in water, and the mobile phase B (MPB) was 0.1% formic acid in acetonitrile. For the liver, urine, feces and serum samples, the gradient began at 2% MPB then held for 1 min, followed by 11 min linear gradient to 95% MPB, held for 8 min, and then decreased to 2% MPB for column equilibration. For the samples from HLM and CYP incubation, the gradient began at 2% MPB and held for 1 min, followed by 8 min linear gradient to 95% MPB, held for 4.5 min, and then decreased to 2% MPB for column equilibration. The flow rate of mobile phase was 0.5 mL/min and the column temperature was maintained at 50 °C. The QTOFMS system was operated in a positive high resolution mode with electrospray ionization. The source and desolvation temperatures were set at 150 and 500 °C, respectively. Nitrogen was applied as the cone gas (50 L/h) and desolvation gas (800 L/h). Argon was applied as collision gas. The capillary and cone voltages were set at 0.8 kV and 40 V. QTOFMS was calibrated with sodium formate and monitored by the intermittent injection of lock mass leucine encephalin (m/z = 556.2771) in real time. Product ion scans with collision energy ramping from 10 to 45 eV were used for structural elucidations of ILB and its metabolites.
Data Analysis.
Mass spectra were acquired by MassLynx 4.1 in centroid format from m/z 50 to 1000. A multivariate data matrix containing sample identity, ion characteristics (retention time and m/z) and ion abundance was generated by Markerlynx XS through deisotoping, filtering, peak recognition and integration. The data matrix was further exported into SIMCA-P software (Version 13, Umetrics, Kinnelon, NJ) and transformed by mean-centering and Pareto-scaling to increase the importance of low abundance ions without significant amplification of noise. Orthogonal partial least-squares discriminant analysis (OPLS-DA) was conducted to maximize the class discrimination. An S-plot was generated and the variables that significantly contributed to the discrimination between groups were considered as potential metabolites and subjected to further analysis.
Statistical Analysis.
All data are expressed as means ± SEM. Statistical analysis was performed with two-tailed Student’s t-test and p value < 0.05 was considered as statistically significant.
RESULTS
Profiling ILB Metabolites.
The OPLS-DA score plot revealed two clusters corresponding to the control and ILB groups (Figure 1A). The S-plot displayed the ion contribution to the group separation. The top ranking ions appearing in the upper-right quadrant of the S-plot was identified as ILB and its metabolites (Figure 1B). According to peak areas, the most abundant metabolites were identified as M1, followed by M2 (Figure 1C). This trend is the same in the liver from mice treated with a lower dose of ILB (Figure S1). In addition, the metabolites in feces and urine were also profiled by the metabolomic approach (Figure S2 and S3). Overall, 20 ILB metabolites were identified (Table S1), including 7 novel metabolites (M2, M3, M6, M15, M16, M19 and M20).
Figure 1.
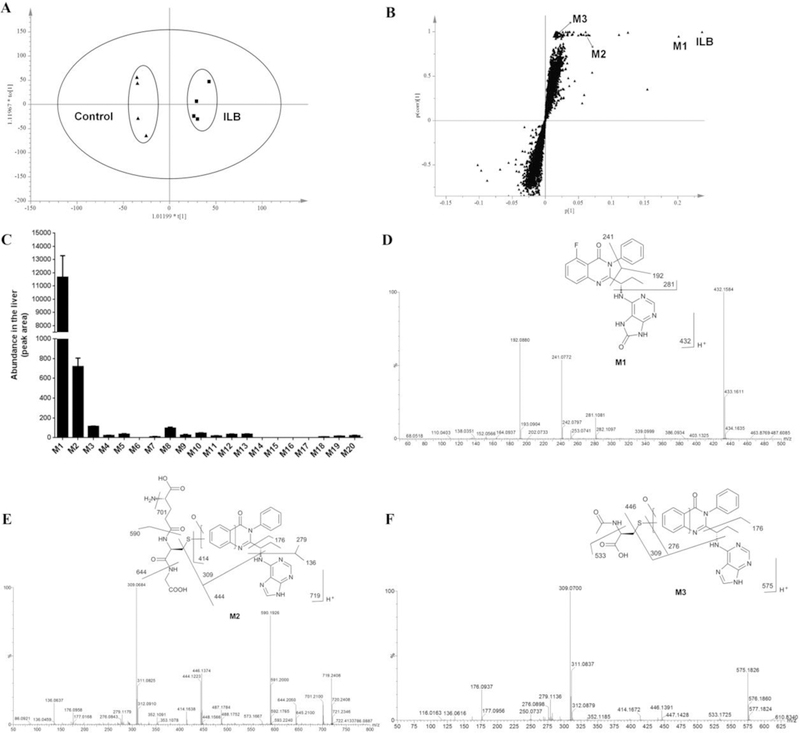
Metabolomic analysis of the liver from mice treated with vehicle or ILB (100 mg/kg). All samples were analyzed by UPLC-QTOFMS. (A) Separation of liver samples from control and ILB group in an OPLS-DA score plot. (B) A loading S-plot generated by OPLS-DA analysis. The top ranking ions were identified as ILB and its metabolites. (C) Relative abundance of ILB metabolites in the liver. The data are expressed as means ± SEM (n = 4). (D) MS/MS spectrum of M1. (E) MS/MS spectrum of M2. (F) MS/MS spectrum of M3.
Identification of M1, an Oxidized Metabolite of ILB.
M1 was the most abundant metabolite found in the liver (Figure 1C), feces (Figure S2), urine (Figure S3) and serum (data not shown). Based on the protonated molecule [M+H] at m/z = 432.1584 Da, M1 was identified as an oxidized metabolite of ILB. The major MS/MS fragmental ions of M1 were at m/z 281, 241 and 192 (Figure 1D). The ion at m/z 192 indicated that the hydroxyl group was introduced at the adenine moiety. All the above information implied that the structure of M1 was in agreement with the reported metabolite GS-563117.10 And this was further confirmed by comparison with the authentic standard of GS-563117 (M1) via retention time and MS/MS fragments (data not shown).
Identification of M2, an ILB-GSH Adduct.
M2 was the second abundant metabolite of ILB found in the liver. M2 was eluted at 4.48 min, and had a protonated molecule [M+H] at m/z = 719.2408 Da. The structural elucidation of M2 was determined by MS/MS fragmentations and interpreted in the inlaid structural diagram (Figure 1E). The MS/MS fragmental ions at m/z 701 (-NH3), 644 (-Gly), 590 (-Glu), 444 and 414 (-GSH) suggest the conjugation of GSH. The MS/MS fragmental ions at m/z 279, 176 and 136 corresponded to the hydroxylated ILB moiety. The MS/MS fragmental ions at m/z 309 determined that the conjugation position was at the quinazoline moiety. The structure of M2 was further confirmed by comparison with the metabolite formed in the incubation of ILB in HLM with GSH (Figure S4).
Identification of M3, a Degraded Metabolite of ILB-GSH Adduct (M2).
M3 was the third abundant metabolite in the liver. The accurate mass of M3 was 575.1826 Da, which indicated the loss of a fluorine atom and the addition of C5H8NO4S to the ILB structure. The MS/MS results showed the major fragmental ions at m/z 533, 446, 414, 309, 279, 276, 176 and 136, which were interpreted in the inlaid structure (Figure 1F). Compared with the MS/MS fragments of M2, the components of C5H8NO4S were elucidated as one oxygen atom and one acetyl cysteine moiety. Incubation of ILB in HLM with N-acetyl cysteine confirmed the structure of M3 by comparison of the retention time and MS/MS data of the corresponding metabolite found in the liver (Figure S4).
Identification of Other Degraded Metabolites of ILB-GSH Adduct (M2).
In addition to M3, the metabolites M4, M6, and M7 were also identified as degraded products of M2. M4 had a protonated molecule [M+H] at m/z = 533.1695 Da, suggesting that a cysteine molecule was attached to the ILB structure. The MS/MS of M4 produced the major fragmental ions at m/z 444, 309, 276, 176 and 136 Da (Figure S5A). Similar to M2, the MS/MS fragmental ions at m/z 309 and 276 determined the conjugated position of the cysteine to ILB. M6 had a protonated molecule [M+H] at m/z = 590.1959 Da. The accurate mass implied the loss of the glutamic acid (−129) moiety of M2. The MS/MS of M6 produced the major fragmental ions at m/z 487, 444, 414, 309, 276, 176 and 136 (Figure S5B). M4 could be considered as the hydrolytic specie of M6. The structures of M4 and M6 were further verified by the incubation of ILB with cysteine and cysteine-glycine, respectively in HLM (Figure S4). Based on the accurate mass and MS/MS fragmental ions, the structure of M7 was elucidated as the addition of a methylthio group along with oxidative defluorination to ILB. We proposed that M7 is the further metabolite of M4 through P-elimination followed by methylation.
Identification of M19 and M20, Two M1-GSH Adducts.
The m/z values of M20 and M19 were rationalized by the addition of a GSH-moiety to M1 and oxygenated M1, respectively. M19 had a protonated molecule [M+H] at m/z = 735.2325 Da. The structure of M19 was interpreted in the inlaid structural diagram (Figure S6A). The MS/MS fragmental ions at m/z 660 (-Gly), 606 (-Glu), 460 and 430 (-GSH) indicated the structure of GSH. The MS/MS fragmental ions at m/z 309 determined that the GSH conjugation position of M19 was similar to that of M2. The accurate mass of M20 is 737.2250 Da, which implies a GSH molecule is directly conjugated to the M1 structure. The structural elucidation is showed in Figure S6B. The GSH moiety was verified by the MS/MS fragmental ions at m/z 608, 505 and 464, whereas the fragmental ions at m/z 241 and 281 indicated the upper quinazoline part of M1. The MS/MS fragmental ions at m/z 224 determined that the GSH conjugation position of M20 was at the adenine moiety. Furthermore, both of M19 and M20 were identified in the incubation of M1 with HLM and GSH (Figure S7). The formation of M19 and M20 were NADPH dependent, and the production of M19 from the HLM incubation was much higher than that of M20 (Figure S6C).
Identification of Degraded Metabolites of M1-GSH Adducts (M19 and M20).
The metabolites M15-M18 were identified as the degraded metabolites of M19 and M20. M15 had a protonated molecule [M+H] at m/z = 591.1805 Da. The structure of M15 was interpreted in the inlaid structural diagram (Figure S5C). The major MS/MS fragmental ions were at m/z 549, 462, 430, 309, 276, 192 and 152. The ion at m/z 192 indicated the oxidation formed at the adenine moiety. The major MS/MS fragmental ions of M16 were at m/z 460, 309, 276, 192 and 152, and the whole structure was interpreted in Figure S5D. The structure of the cysteine adduct M18 was interpreted in the inlaid structural diagram in Figure S5E. The conjugation position of the cysteine to the adenine group was determined by the representative MS/MS fragmental ions at m/z 311 and 224, which is similar to M20.
Incubation of M1 in HLM with the trapping agent cysteine or N-acetyl cysteine further confirmed the structures of M15, M16 and M18 (Figure S7). Two N-acetyl cysteine adducts (M15 and M21) were identified in vitro. Theoretically, M21 should be formed in vivo, but the abundance may be too low to be detected. Through a similar metabolic pathway of M7, M17 is metabolized from M16 via de-alkylation followed by methylation. M3 and M15 is the acetylation product of M4 and M16, respectively, which is probably catalyzed by the N-acetyltransferase.16, 17 In agreement with the in vitro data of levels of M19 and M20, M4 and M16 were much more abundant than M18 in mouse feces and urine treated with ILB (Figure S2C and S3C). All these results suggested that the reactive species of ILB and M1 were mostly formed at quinazoline moiety compared with the adenine part.
Comparison of ILB and M1 Metabolism and Bioactivation between HLM and MLM.
In the incubations of ILB with HLM and MLM, the productions of M1, M2, and M5 in MLM were higher than that in HLM (Figure S8A). On the contrary, the productions of M19 and M20 were higher in the incubations of Ml with HLM than that in MLM (Figure S8B). These data suggest that there are species differences in the metabolism and bioactivation of ILB and Ml.
Role of CYPs in M2 Formation.
M2 was recaptured in the incubation of ILB with GSH in HLM. The formation of M2 was NADPH dependent (Figure 2A). Further studies using human cDNA-expressed CYPs revealed that the formation of M2 was mediated by CYP2C9 and CYP3A4 (Figure 2B). Co-incubation of ILB with KCZ and/or fluoxetine verified the roles of CYP3A4 and CYP2C9. In HLM, the formation of M2 was suppressed 59% and 57% by 1 μM of KCZ and 200 μM of fluoxetine, respectively, while the two inhibitors together decreased the formation of M2 to 19% (Figure 2C). The incubation of KCZ or fluoxetine with recombinant CYPs gave similar results, as the formation of M2 was decreased significantly when compared with that of the control group (Figure 2D).
Figure 2.
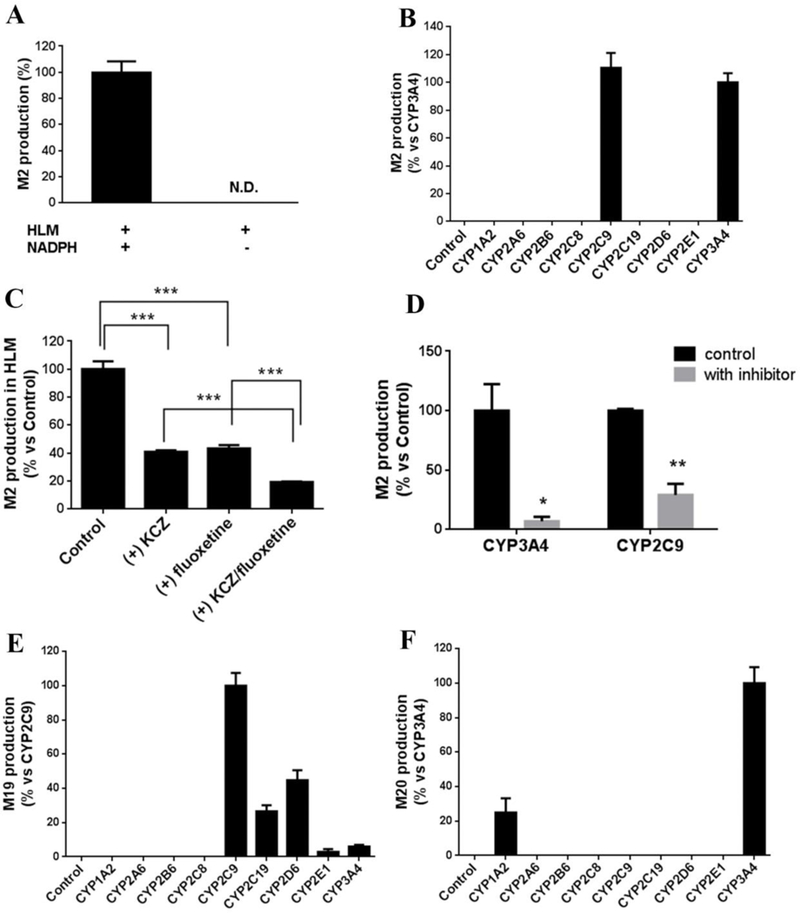
Roles of CYPs in bioactivation of ILB (formation of M2) and M1 (formation of M19 and M20). (A) The formation of M2 is NADPH dependent in the incubation with human liver microsomes (HLM). N.D. not detected. (B) Role of CYPs in M2 formation. The relative abundance of M2 from the incubation with CYP3A4 was set as 100%. (C) Effect of KCZ and fluoxetine on M2 formation in the incubation with HLM. KCZ and fluoxetine were used as an inhibitor of CYP3A4 and CYP2C9, respectively. (D) Effect of KCZ and fluoxetine on M2 formation in the incubation with cDNA-expressed CYP3A4 and CYP2C9, respectively. The relative abundance of M2 in control groups was set as 100%. (E) Role of CYPs in M19 formation. The relative abundance of M19 from the incubation with CYP2C9 was set as 100%. (F) Role of CYPs in M20 formation. The relative abundance of M20 from the incubation with CYP3A4 was set as 100%. All the data are expressed as means ± SEM (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001.
Role of CYPs in the Formation of M1 and its GSH adducts (M19 and M20).
M1 was the predominant metabolite in the liver. A previous report showed that M1 was formed mainly through aldehyde oxidase (AO), with a lesser involvement of CYP3A4.12 Our data agreed with this finding that CYP3A4 is involved in Ml formation (Figure S9). We further profiled that role of CYPs in Ml bioactivation. Incubation of Ml with CYPs and GSH revealed that the formation of M19 was majorly mediated by CYP2C9, followed by CYP2C19 and 2D6 (Figure 2E). M20 was produced majorly through CYP3A4, followed by 1A2 (Figure 2F).
DISCUSSION
The current study employed a metabolomic approach to profile the metabolism and bioactivation of ILB in mouse liver, urine and feces. Overall, 20 metabolites of ILB were identified, including 7 new metabolites (Figure 3 and Table S1). The data from the current work can be used to guide the future studies on drug-drug interactions and hepatotoxicity of ILB.
Figure 3.
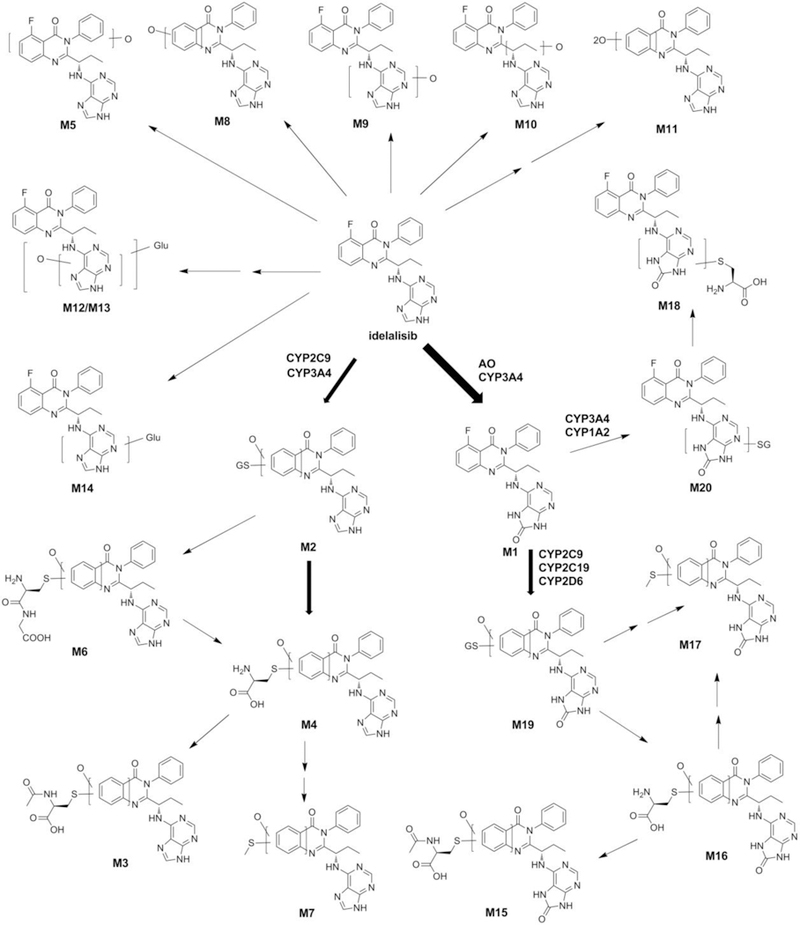
The metabolic map of ILB. These metabolites were summarized from the analysis of liver, fecal, urinary and serum samples of mice treated with ILB. The structures were determined based on the exact mass (mass error less than 10 ppm) and MS/MS fragments. The major metabolites of ILB in the liver were elucidated as M1, followed by CYP3A4/2C9-mediated generation of M2. M2, M3, M6, M15, M16, M19 and M20 were identified as novel metabolites of ILB. AO, aldehyde oxidase; Glu, glucuronic acid; GS, GSH.
We found that M1 was the major metabolite of ILB in mouse liver. M1 (GS-563117) has been identified as a mechanism-based CYP3A inhibitor, and its half-maximal inhibitory concentration (5.1 μM) is clinically relevant.11, 12 Mechanism-based CYP inhibitors usually undergo the formation of reactive metabolites that covalently modify the CYP proteins or the heme moiety.18 In agreement with this knowledge, we recaptured the reactive metabolites of M1 in HLM with the trapping agent GSH. We further demonstrated that CYP2C9, 2C19, 2D6 and 3A4 were involved in M1 bioactivation. Although the levels of M19 and M20 were low in the liver, the high levels of their degraded metabolites (M16 and M18) indicated that a considerable amount of reactive metabolites of Ml were formed in the liver.
The ILB-GSH adduct M2 is the second abundant metabolite in mouse liver, which indicates that a high amount of ILB undergoes bioactivation in mice. The high levels of the downstream metabolites of M2, including M3, M4 and M6 in feces and urine, also suggest that a large amount of reactive metabolites of ILB are generated in mouse liver. We further illustrated that CYP3A4 and 2C9 were the two major enzymes contributing to the formation of M2. We propose that the CYP3A4- and 2C9-mediated bioactivation of ILB produced epoxides and then reacted with GSH to form M2 (Figure S10A).
Species differences are often observed in drug metabolism. It has been shown that GSH adducts of diclofenac formed in HLM are much more abundant than that in rat and monkey liver microsomes. 19 Our study on ILB metabolism found that the formation of ILB-GSH adduct (M2) in MLM was higher than that in HLM. On the contrary, we found that M1-GSH adducts (M19 and M20) formed in HLM were much more abundant than that in MLM. Because M1 is the dominant pathway in ILB metabolism,12 further in vivo studies are needed to evaluate the species differences in M1 metabolism, bioactivation and toxicity.
Inferred from the mechanism of gefitinib bioactivation,20 we proposed that the formation ILB and M1 GSH adducts is through the epoxides pathway (Figure S10). For M2, oxidation of ILB by CYP2C9 and 3A4 at the quinazoline ring formed an epoxide. There might be 2 positions for the GSH addition, showed as M2–1 and M2–2. GSH could react with epoxide followed by oxidative defluorination to form M2–1, or substitute the fluorine atom followed by protonation to form M2–2. In addition, M2–2 might also be produced through aromatic hydroxylation para to the N-atom followed by quinonimine formation via CYPs, and further substitution of the fluorine atom by GSH. M19 was formed from M1 by CYP2C9, 2C19, 2D6, and 3A4, with 2 possible positions of GSH (M19–1 and M19–2). M20 formation might have gone through an epoxide intermediate followed by dehydration occurring at the adenine ring of M1. Oxidation by CYP3A4 and CYP1A2 may form 2 possible epoxides on the pyrimidine ring, which reacts with GSH to produce 4 adducts. Further dehydration led to the formation of a double bond, to increase the structural stability, and finally formed the conjugated molecules M20–1, M20–2, M20–3, and M20–4. However, other positions of the double bond formed by dehydration should not be disregarded.
The formation of reactive metabolites by metabolic activation is considered as one of the causes of drug toxicity.7, 8 Reactive metabolites, like quinones, epoxides, aldehydes and iminium ions, may modify hepatic proteins and cause liver toxicity.21–23 Further studies are needed to address whether these reactive metabolites of ILB and M1 are involved in ILB hepatotoxicity, especially in the conditions of GSH depletion and CYP induction. Because CYP2C9, 2C19 and 3A4 are highly inducible, their inducers are likely to increase the production of the reactive metabolites of ILB and M1, and then augment ILB hepatotoxicity. Pregnane X receptor (PXR) transcriptionally regulates the expression of CYP2C9, 2C19 and 3A4.24, 25 It is possible that PXR ligands can induce the expression of CYP2C9, 2C19 and 3A4 and increase the metabolism of ILB and M1 to form their reactive epoxides. Therefore, further studies are also suggested to investigate the effect of CYP2C9, 2C19, and 3A4 inducers on ILB bioactivation and hepatotoxicity.
In summary, the metabolism of ILB in mice was profiled using a metabolomic approach. The most abundant metabolite of ILB in the liver was M1 (GS-563117), which can further undergo bioactivation through CYP2C9, 2C19 and 3A4. The second abundant metabolite of ILB in the liver was a GSH adduct, which was produced through CYP3A4- and 2C9-mediated bioactivation of ILB. Due to the high abundance of M1 and M2 in the liver, further studies are suggested to determine the role of these two metabolic pathways in ILB-induced liver injury.
Supplementary Material
Acknowledgement
We thank Dr. Jane Maddigan for proofreading our manuscript.
Funding information
This work was supported in part by the National Institute of Diabetes and Digestive and Kidney Diseases (DK090305) and the National Institute of Allergy And Infectious Diseases (AI131983).
Abbreviations
- ILB
Idelalisib
- PI3Kδ
phosphatidylinositol-3-kinase delta
- GSH
glutathione
- PXR
pregnane X receptor
- FDA
food and drug administration
- CLL
chronic lymphocytic leukaemia
- CYP
cytochrome P450
- NADPH
β-nicotinamide adenine dinucleotide phosphate
- HLM
human liver microsomes
- MLM
mouse liver microsomes
- UPLC-QTOFMS
ultra-performance liquid chromatography and quadrupole time-of-flight mass spectrometry
- AO
aldehyde oxidase
- PBS
phosphate-buffered saline
- KCZ
ketoconazole
- OPLS-DA
orthogonal partial least-squares discriminant analysis
Footnotes
Supporting Information
Metabolomic analysis of mouse feces and urine from the control and ILB-treated groups; the relative abundance of ILB metabolites in feces and urine; identification of M2, M3, M4, M6, M15, M16, M18, M19, M20 and M21; comparison of the metabolism of ILB and M1 between HLM and MLM; role of CYPs in M1 formation; proposed mechanisms for the formation of GSH adducts.
REFERENCES
- (1).Herman SE, Lapalombella R, Gordon AL, Ramanunni A, Blum KA, Jones J, Zhang X, Lannutti BJ, Puri KD, Muthusamy N, Byrd JC, and Johnson AJ (2011) The role of phosphatidylinositol 3-kinase-delta in the immunomodulatory effects of lenalidomide in chronic lymphocytic leukemia. Blood 117, 4323–4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lannutti BJ, Meadows SA, Herman SE, Kashishian A, Steiner B, Johnson AJ, Byrd JC, Tyner JW, Loriaux MM, Deininger M, Druker BJ, Puri KD, Ulrich RG, and Giese NA (2011) CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood 117, 591–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Hoellenriegel J, Meadows SA, Sivina M, Wierda WG, Kantarjian H, Keating MJ, Giese N, O’Brien S, Yu A, Miller LL, Lannutti BJ, and Burger JA (2011) The phosphoinositide 3’-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood 118, 3603–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Gopal AK, Kahl BS, de Vos S, Wagner-Johnston ND, Schuster SJ, Jurczak WJ, Flinn IW, Flowers CR, Martin P, Viardot A, Blum KA, Goy AH, Davies AJ, Zinzani PL, Dreyling M, Johnson D, Miller LL, Holes L, Li D, Dansey RD, Godfrey WR, and Salles GA (2014) PI3Kdelta inhibition by idelalisib in patients with relapsed indolent lymphoma. N. Engl. J. Med. 370, 1008–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, Barrientos JC, Zelenetz AD, Kipps TJ, Flinn I, Ghia P, Eradat H, Ervin T, Lamanna N, Coiffier B, Pettitt AR, Ma S, Stilgenbauer S, Cramer P, Aiello M, Johnson DM, Miller LL, Li D, Jahn TM, Dansey RD, Hallek M, and O’Brien SM (2014) Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 370, 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lampson BL, Kasar SN, Matos TR, Morgan EA, Rassenti L, Davids MS, Fisher DC, Freedman AS, Jacobson CA, Armand P, Abramson JS, Arnason JE, Kipps TJ, Fein J, Fernandes S, Hanna J, Ritz J, Kim HT, and Brown JR (2016) Idelalisib given front-line for treatment of chronic lymphocytic leukemia causes frequent immune-mediated hepatotoxicity. Blood 128 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Walgren JL, Mitchell MD, and Thompson DC (2005) Role of metabolism in drug-induced idiosyncratic hepatotoxicity. Crit. Rev. Tocicol. 35, 325–361. [DOI] [PubMed] [Google Scholar]
- (8).Baillie TA (2006) Future of toxicology-metabolic activation and drug design: challenges and opportunities in chemical toxicology. Chem. Res. Toxicol. 19, 889–893. [DOI] [PubMed] [Google Scholar]
- (9).Chen H, Evarts J, Webb H, and Ulrich R (2012) Biotransformation of treatment of patients with hematologic malignancies. FASEB J. 26, 850.810–850.810. [Google Scholar]
- (10).Robeson M, Zhou H, Kwan E, and Ramanathan S (2013) Pharmacokinetics, metabolism and excretion of idelalisib. Blood 122, 5570–5570. [Google Scholar]
- (11).Jin F, Robeson M, Zhou H, Moyer C, Wilbert S, Murray B, and Ramanathan S (2015) Clinical drug interaction profile of idelalisib in healthy subjects. J. Clin. Pharmacol. 55, 909–919. [DOI] [PubMed] [Google Scholar]
- (12).Ramanathan S, Jin F, Sharma S, and Kearney BP (2016) Clinical pharmacokinetic and pharmacodynamic profile of idelalisib. Clin Pharmacokinet. 55, 33–45. [DOI] [PubMed] [Google Scholar]
- (13).Chen H, Webb H, Evarts J, and Ulrich R (2011) UGT-catalyzed glucuronidation: an important elimination mechanism of GS-1101 (CAL-101), a potent and selective inhibitor of the p110 delta isoform of phosphoinositide 3-kinase (PI3K delta) for treatment of patients with hematologic malignancies. 17th North American Regional International society for the study of xenobiotics Meeting, p88. [Google Scholar]
- (14).Li F, Lu J, and Ma X (2011) Metabolomic screening and identification of the bioactivation pathways of ritonavir. Chem. Res. Toxicol.24, 2109–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Liu K, Zhu J, Huang Y, Li C, Lu J, Sachar M, Li S, and Ma X (2017) Metabolism of KO143, an ABCG2 inhibitor. Drug Metab. Pharmacok. 32, 193–200. [DOI] [PubMed] [Google Scholar]
- (16).Duffel MW, and Jakoby WB (1982) Cysteine S-conjugate N-acetyltransferase from rat kidney microsomes. Mol. Pharmacol. 21, 444–448. [PubMed] [Google Scholar]
- (17).Green RM, and Elce JS (1975) Acetylation of S-substituted cysteines by a rat liver and kidney microsomal N-acetyltransferase. Biochem. J. 147, 283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Lin JH, and Lu AY (1998) Inhibition and induction of cytochrome P450 and the clinical implications. Clin. Pharmacokinet. 35, 361–390. [DOI] [PubMed] [Google Scholar]
- (19).Yan Z, Li J, Huebert N, Caldwell GW, Du Y, and Zhong H (2005) Detection of a novel reactive metabolite of diclofenac: evidence for CYP2C9-mediated bioactivation via arene oxides. Drug Metab. Dispos. 33, 706–713. [DOI] [PubMed] [Google Scholar]
- (20).Li X, Kamenecka TM, and Cameron MD (2009) Bioactivation of the epidermal growth factor receptor inhibitor gefitinib: implications for pulmonary and hepatic toxicities. Chem. Res. Toxicol. 22, 1736–1742. [DOI] [PubMed] [Google Scholar]
- (21).Evans DC, Watt AP, Nicoll-Griffith DA, and Baillie TA (2004) Drug-protein adducts: an industry perspective on minimizing the potential for drug bioactivation in drug discovery and development. Chem. Res. Toxicol. 17, 3–16. [DOI] [PubMed] [Google Scholar]
- (22).LoPachin RM, and Gavin T (2014) Molecular mechanisms of aldehyde toxicity: a chemical perspective. Chem. Res. Toxicol. 27, 1081–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wang J, Davis M, Li F, Azam F, Scatina J, and Talaat R (2004) A novel approach for predicting acyl glucuronide reactivity via Schiff base formation: development of rapidly formed peptide adducts for LC/MS/MS measurements. Chem. Res. Toxicol. 17, 1206–1216. [DOI] [PubMed] [Google Scholar]
- (24).Willson TM, and Kliewer SA (2002) PXR, CAR and drug metabolism. Nat. Rev. Drug Discov. 1, 259–266. [DOI] [PubMed] [Google Scholar]
- (25).Gerbal-Chaloin S, Pascussi JM, Pichard-Garcia L, Daujat M, Waechter F, Fabre JM, Carrere N, and Maurel P (2001) Induction of CYP2C genes in human hepatocytes in primary culture. DrugMetab. Dispos. 29, 242–251. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.