

Abstract
Objective
To study genetic causes of the low frequency of Huntington disease (HD) in the Finnish population, we determined HTT haplogroups in the population and patients with HD and analyzed intergenerational Cytosine-Adenosine-Guanosine (CAG) stability.
Methods
A national cohort of patients with HD was used to identify families with mutant HTT (mHTT). HTT haplogroups were determined in 225 archival samples from patients and from 292 population samples. CAG repeats were phased with HTT haplotypes using data from parent-offspring pairs and other mHTT carriers in the family.
Results
The allele frequencies of HTT haplotypes in the Finnish population differed from those in 411 non-Finnish European subjects (p < 0.00001). The frequency of haplogroup A was lower than that in Europeans and haplogroup C was higher. Haplogroup A alleles were significantly more common in patients than in controls. Among patients with HD haplotypes A1 and A2 were more frequent than among the controls (p = 0.003). The mean size of the CAG repeat change was +1.38 units in paternal transmissions being larger than that (−0.17) in maternal transmissions (p = 0.008). CAG repeats on haplogroup A increased by 3.18 CAG units in paternal transmissions, but only by 0.11 units in maternal transmissions (p = 0.008), whereas haplogroup C repeat lengths decreased in both paternal and maternal transmissions.
Conclusions
The low frequency of HD in Finland is partly explained by the low frequency of the HD-associated haplogroup A in the Finnish population. There were remarkable differences in intergenerational CAG repeat dynamics that depended on HTT haplotype and parent gender.
Huntington disease (HD) is caused by an expansion of a Cytosine-Adenosine-Guanosine (CAG) repeat in the HTT gene.1 The expansion is driven by paternal transmissions, where the repeat instability correlates with repeat length.2–5 The prevalence of HD varies markedly between populations. The disease is most common among white populations, whereas the prevalence is only a 10th or less in Asian populations.6 Among whites, the Finns and Icelanders have low prevalence, whereas the prevalence rates in other Scandinavian countries are similar to those in other whites.7,8
Shortly after the identification of the HTT gene, it was established that the risk of CAG expansion is associated with certain chromosome 4 haplotypes.9 Indeed, differences in the frequency of HD between populations correlate with differences in the frequency of HTT haplogroups defined by single-nucleotide polymorphisms (SNPs). Haplogroup A is the most frequent haplogroup in white patients with HD,10 while haplogroup C is the most common one in white general populations. On the other hand, East Asian populations lack haplotypes A1 and A2 and HD cases harbor haplogroup C.11 HTT haplogroups have been suggested to differ in the rate of new mutations,11 implying differences in the intergenerational stability of the CAG repeats.
We have previously estimated that the frequency of haplogroup A is lower in the Finnish general population than that in most European populations.12 Here we report the frequencies of HTT haplogroups in Finnish patients with HD and in the general population and assess intergenerational stability of CAG repeat expansion across HTT haplogroups.
Methods
Patients and controls
A previously reported national cohort of patients with HD12 was used to identify families with mutant HTT (mHTT). Archival deoxyribonucleic acid (DNA) samples (N = 225) remaining from diagnostic or predictive HTT analyses were obtained from the 2 national laboratories that perform HD diagnostics. Family relationships were determined using data from the Population Register Centre, a governmental authority keeping records of citizens, the archives of Family Federation of Finland, a nongovernmental nonprofit welfare organization that has provided genetic counselling to Finnish families with HD, and hospital records. Phasing of CAG repeat and HTT haplotype was conducted using data from parent-offspring pairs and other mHTT carriers in the family. As controls, we had 292 healthy Finnish blood donors with a mean age of 40.5 years and range 19–64 years.13
The study has been approved by the ethics committee of Hospital District of Southwestern Finland (Dnro ETMK 19/180/2010) and received the national study permits from the National Institute for Health and Welfare (THL, Dnro THL/1456/5.05.00/2010) and the National Supervisory Authority for Welfare and Health, Valvira (Dnro 1195/06.01.03.01/2012). The study involved no contact with patients. Hence, no informed consent was stipulated.
Molecular methods
HTT haplogroups A, B, and C were defined using 2 intragenic SNPs, rs762855 and rs4690073, and haplotype A variants were further defined using 4 additional intragenic SNPs, rs2857936, rs363096, rs2276881, and rs362307.10 The SNPs were determined with restriction fragment analysis (FastDigest; Thermo Fisher Scientific, Waltham, MA), allele-specific amplification using locked nucleic acid (LNA) primers (Exiqon, Vedbaek, Denmark), or by sequencing (ABI3500xL Genetic Analyzer, Applied Biosystems, Foster City, CA) as appropriate (table e-1, links.lww.com/NXG/A153). The PCRs were performed using Phire Hot Start II DNA Polymerase (Thermo Fisher Scientific) according to the standard procedures. The LNA amplifications were conducted in duplicates or confirmed by sequencing. Additional SNPs (table e-1, links.lww.com/NXG/A153) were analyzed in samples, where haplogroup A variant remained undetermined.10,14 Haplotypes were manually annotated and then phased to CAG repeat length by familial relationship or by homozygosity of haplotype.
Statistical analysis
Exact test of population differentiation as implemented in Arlequin 3.515 was used to compare frequencies of haplogroups between cases and controls. Chi-square test or t test was used to compare 2 groups as appropriate.
Data availability statement
Access to data was regulated by the Finnish law, THL, and Valvira. Permission to conduct this study prohibits disclosing data to third parties without explicit permission from THL and Valvira. Those fulfilling the requirements for viewing confidential data as required by the Finnish law and who receive the permission from THL and Valvira are able to access the data.
Results
Frequencies
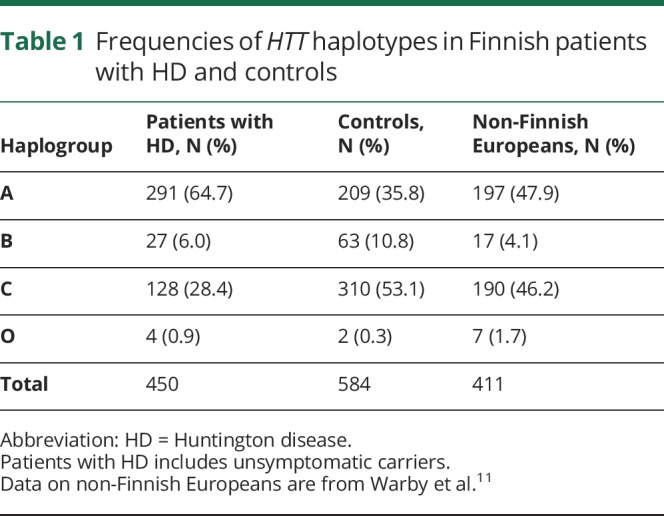
The allele frequencies of HTT haplotypes in the Finnish population differed from those in 411 non-Finnish European subjects11 (p < 0.00001, exact test of population differentiation). The frequency of haplogroup A was lower (p = 0.0003, χ2 test) than that in Europeans, and haplogroup C was higher (p = 0.046, χ2 test), but there was no difference in the proportion of haplotypes A1 and A2 out of all haplogroup A alleles.
The allele frequencies of HTT haplotypes among Finnish patients with HD and unsymptomatic mHTT carriers differed from those in the controls (p < 0.00001, exact test of population differentiation). Haplogroup A alleles were significantly more common in patients than in controls, while haplogroup B and C alleles were more common in population controls (table 1). Frequencies of haplotypes A1–A5 differed between controls and patients with HD (p < 0.00001, exact test of population differentiation). Among patients with HD (including unsymptomatic mHTT carriers), haplotypes A1 and A2 were more frequent and haplotypes A4 and A5 less frequent than among the controls (p = 0.003, exact test of population differentiation).
Table 1.
Frequencies of HTT haplotypes in Finnish patients with HD and controls
Transmissions
We were able to analyze the change in the size of mHTT CAG repeat in 65 transmissions (table 2). The mean size of the change was +1.38 CAG units in paternal transmissions being larger than that in maternal transmissions (−0.17 CAG units) (p = 0.008, t test). mHTT haplotype SNPs and CAG repeat could be phased in 50 transmissions (haplogroup A, 38; haplogroup C, 10; other haplogroup, 2; figure 1). mHTT CAG repeats in haplogroup A (42.6 CAG units) were shorter than those in haplogroup C (48.4 CAG units). In haplogroup C, the mHTT CAG repeats decreased in paternal as well as maternal transmissions (p = 0.74 for gender difference), whereas the mean increase of the repeats in haplogroup A was considerable in paternal transmissions (3.18 CAG units) but meager in maternal transmissions (0.11 CAG units) (p = 0.008 for difference) (figure 2). The gender difference in mHTT CAG change was significant in haplotype A1 inheritances (p = 0.022), but not in haplotype A2 or A3 inheritance.
Table 2.
The change in the size of mHTT CAG repeat in 65 transmissions
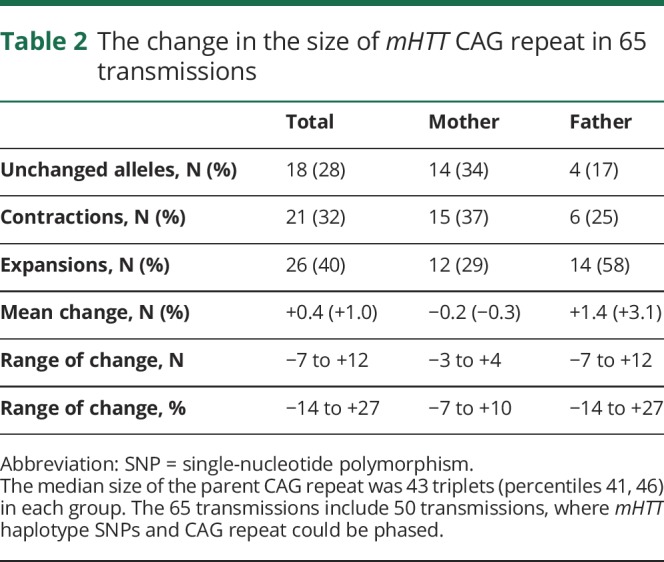
Figure 1. Proportions of changes in mHTT CAG repeat tracts in transmission events in 48 Finnish families with Huntington disease.

Two families belonging to haplogroups other than A or C are not included. (A) Changes in haplogroups A and C. (B) Changes in haplogroup A haplotypes.
Figure 2. Mean change in mHTT CAG repeat length in maternal and paternal transmission events in 48 Finnish families with Huntington disease.

Two families belonging to haplogroups other than A or C are not included. Changes are shown for haplogroup A haplotypes and for haplogroup C.
Discussion
We found remarkable differences in intergenerational CAG repeat dynamics that depended on HTT haplotype and parent gender. HTT haplotype A1 was not only associated with HD, but also showed a predilection to CAG repeat expansion in paternal transmissions. Repeat tracts on haplotypes A2 and A3 showed propensity to expand, whereas those on haplogroup C showed a preference for contraction irrespective of parental gender.
Previous studies have shown that paternal transmission drives intergenerational CAG expansion in HTT.2–5 We identified 65 Finnish parent-child pairs, where paternal transmission was associated with repeat expansion (+1.38 CAG units), whereas maternal transmission mainly resulted in repeat contraction (−0.17 CAG units). Strikingly similar figures have been obtained in 337 parent-offspring transmissions in Dutch patients, where paternal transmission was associated with CAG repeat expansion (+1.76 CAG units) and maternal transmissions resulted in contraction (−0.07 CAG units).3 These findings suggest that intergenerational stability of mHTT in Finnish HD families does not differ from that in other white populations. Interestingly, the intergenerational stability of the CAG repeat differed between mHTT haplotypes. On average, repeat tracts on haplotype A1 gained length in paternal transmissions, while maternally inherited tracts decreased in length. Haplotype A1 seems to be a major contributor to paternal anticipation in HD, as 48% of all the pathogenic expansions were on haplotype A1.
We found that repeat tracts on haplogroup C were larger than those on haplogroup A and that repeat tracts on haplogroup C showed a tendency to contract. Indeed, the ratio between the number of contraction events and the number of expansion events was 2.0 in haplogroup C transmissions, while the corresponding ratio was 0.4 in haplogroup A transmissions (figure 1A). The skew toward contraction suggests that HTT haplogroup C harbors DNA sequence elements that favor contraction in previously elongated repeat tracts. These findings should be interpreted with caution, however, as HTT haplotype and CAG repeat could be phased in only 10 haplogroup C samples. The haplogroup C samples showed a tendency for contraction in both paternal and maternal transmissions, which resembles the findings in some Cretan HD families.16 The repeat tracts are stable or contract in transmission in these families and, clinically, the families do not show anticipation.
Remarkable ethnic differences in the prevalence of HD have suggested a genetic contribution. The frequency is highest among European populations and a recent systematic review compiled 14 studies and found that the median prevalence is 5.7/100,000 (range 0.96–11.8).6 The lowest prevalence rates included in the review were 0.96/100,000 in Iceland17 and 2.14/100,000 in Finland.12 In the European population, CAG expansions occur predominantly on haplotypes A1 and A2, which are present in 20% of the individuals with <27 CAG repeats.10,11 We found that haplogroup frequencies in the Finnish general population differ from those among non-Finnish Europeans. In particular, haplogroup A was low, and thus the low frequency of the susceptibility haplogroups partly explains the low HD prevalence in the Finnish population. Similar to other white populations, the majority of HD cases in Finland harbored haplotype A1 or A2 and the 2 haplotypes constituted similar proportions of haplogroup A chromosomes.
In addition to HTT haplotype, other possible contributors to variation in prevalence rates include differences in the rate of new mutations and founder effect. A recent analysis has suggested that a higher frequency of haplotype A1 in the general population yield a higher rate of new HD mutations.18 Hence, the relatively low rate of new mutations would be the mechanism producing low prevalence of HD in the Finnish population by virtue of the low proportion of HTT haplotypes A1 and A2. In Finland, the haplotype distribution may be because of small founder population and geographic, cultural, and linguistic isolation have shaped the gene pool of the Finnish population.19 These events have yielded Finnish disease heritage, where certain Mendelian diseases are enriched in the population, while some others are lacking or occur at a low prevalence, such as HD.12 The population genomics also shows some ancient features that are undetectable in most other regions of Europe.20
Taking into account the low frequency of HD in Finland, our sample set of 225 is rather comprehensive. The archival nature of the samples posed some limitations, as lack of samples precluded phasing in many families. However, homozygosity of the HTT haplotype or results from 79 out of the 225 samples enabled us to phase HTT haplotype and CAG repeat in 50 transmissions.
Here we found that the intergenerational stability of the CAG repeat differed between mHTT haplotypes. Repeat tracts on haplotype A1 gained length only in paternal transmissions, while paternally as well as maternally inherited tracts decreased in length on haplogroup C. In addition, we showed that the frequency of the HD-associated HTT haplogroup A was lower in the Finnish population than that in the non-Finnish European population and that the frequency of haplogroup A was higher in patients with HD than that in the population. These findings at least partly explain the low prevalence of HD in Finland.
Disclosure
S. Ylönen has nothing to declare. J.O.T. Sipilä has received honoraria (Merck, Pfizer, Sanofi Genzyme), has received a consultancy fee (Rinnekoti Foundation), has received travel grants and congress sponsorship (Orion Corporation, Merck Serono, Sanquin, Lundbeck, Novartis), and holds shares (Orion Corporation). M. Hietala has nothing to declare. K. Majamaa has received travel support from MSD Finland and honoraria from Sanofi Genzyme and UCB. This study was not industry-sponsored. Go to Neurology.org/NG for full disclosure.
Acknowledgment
The expert technical assistance of Ms. Anja Heikkinen is gratefully acknowledged. The authors would like to thank Dr. Annukka M. Tuiskula, PhD, and Mr. Antti-Jussi Kortevaara for valuable assistance in obtaining the DNA samples archived by HUSLAB and the staff of the genetic laboratory of the Hospital District of Southwest Finland for gathering and dispatching the samples stored there.
Glossary
- CAG
cytosine-adenosine-guanosine
- DNA
deoxyribonucleic acid
- HD
Huntington's disease
- LNA
locked nucleic acid
- mHTT
mutant HTT
- SNP
single-nucleotide polymorphism
- THL
National Institute for Health and Welfare
Appendix. Authors
Study funding
This study was supported in part by grants from the Sigrid Juselius Foundation, Turku University Foundation, and the Finnish Parkinson Foundation.
References
- 1.MacDonald ME, Ambrose CM, Duyao MA, et al. . A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell 1993;72:971–983. [DOI] [PubMed] [Google Scholar]
- 2.Nørremølle A, Sørensen SA, Fenger K, Hasholt L. Correlation between magnitude of CAG repeat length alterations and length of the paternal repeat in paternally inherited Huntington's disease. Clin Genet 1995;47:113–117. [DOI] [PubMed] [Google Scholar]
- 3.Aziz NA, van Belzen MJ, Coops ID, Belfroid RDM, Roos RAC. Parent-of-origin differences of mutant HTT CAG repeat instability in Huntington's disease. Eur J Med Genet 2011;54:e413–e418. [DOI] [PubMed] [Google Scholar]
- 4.Ramos EM, Cerqueira J, Lemos C, Pinto-Basto J, Alonso I, Sequeiros J. Intergenerational instability in Huntington disease: extreme repeat changes among 134 transmissions. Mov Disord 2012;27:583–585. [DOI] [PubMed] [Google Scholar]
- 5.Semaka A, Kay C, Doty C, et al. . CAG size-specific risk estimates for intermediate allele repeat instability in Huntington disease. J Med Genet 2013;50:696–703. [DOI] [PubMed] [Google Scholar]
- 6.Baig SS, Strong M, Quarrell OW. The global prevalence of Huntington's disease: a systematic review and discussion. Neurodegener Dis Manag 2016;6:331–343. [DOI] [PubMed] [Google Scholar]
- 7.Gilling M, Budtz-Jørgensen E, Boonen SE, et al. . The Danish HD Registry-a nationwide family registry of HD families in Denmark. Clin Genet 2017;92:338–341. [DOI] [PubMed] [Google Scholar]
- 8.Roos AK, Wiklund L, Laurell K. Discrepancy in prevalence of Huntington's disease in two Swedish regions. Acta Neurol Scand 2017;136:511–515. [DOI] [PubMed] [Google Scholar]
- 9.Andrew SE, Hayden MR. Origins and evolution of Huntington disease chromosomes. Neurodegeneration 1995;4:239–244. [DOI] [PubMed] [Google Scholar]
- 10.Warby SC, Montpetit A, Hayden AR, et al. . CAG expansion in the Huntington disease gene is associated with a specific and targetable predisposing haplogroup. Am J Hum Genet 2009;84:351–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Warby SC, Visscher H, Collins JA, et al. . HTT haplotypes contribute to differences in Huntington disease prevalence between Europe and East Asia. Eur J Hum Genet 2011;19:561–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sipilä JO, Hietala M, Siitonen A, Päivärinta M, Majamaa K. Epidemiology of Huntington's disease in Finland. Parkinsonism Relat Disord 2015;21:46–49. [DOI] [PubMed] [Google Scholar]
- 13.Meinilä M, Finnilä S, Majamaa K. Evidence for mtDNA admixture between the Finns and the Saami. Hum Hered 2001;52:160–170. [DOI] [PubMed] [Google Scholar]
- 14.Kay C, Collins JA, Skotte NH, et al. . Huntingtin haplotypes provide prioritized target panels for allele-specific silencing in Huntington disease patients of European ancestry. Mol Ther 2015;23:1759–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online 2007;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- 16.Tsagournissakis M, Fesdjian OC, Shashidharan P, Plaitakis A. Stability of the Huntington disease (CAG)n repeat in a late onset form occurring on the island of Crete. Hum Mol Genet 1996;4:2239–2243. [DOI] [PubMed] [Google Scholar]
- 17.Sveinsson O, Halldórsson S, Olafsson E. An unusually low prevalence of Huntington's disease in Iceland. Eur Neurol 2012;68:48–51. [DOI] [PubMed] [Google Scholar]
- 18.Kay C, Collins JA, Wright GEB, et al. . The molecular epidemiology of Huntington disease is related to intermediate allele frequency and haplotype in the general population. Am J Med Genet B Neuropsychiatr Genet 2018;177:346–357. [DOI] [PubMed] [Google Scholar]
- 19.Norio R. Finnish Disease Heritage I: characteristics, causes, background. Hum Genet 2003;112:441–456. [DOI] [PubMed] [Google Scholar]
- 20.Neuvonen AM, Putkonen M, Översti S, et al. . Vestiges of an ancient border in the contemporary genetic diversity of north-eastern Europe. PLoS One 2015;10:e0130331. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Access to data was regulated by the Finnish law, THL, and Valvira. Permission to conduct this study prohibits disclosing data to third parties without explicit permission from THL and Valvira. Those fulfilling the requirements for viewing confidential data as required by the Finnish law and who receive the permission from THL and Valvira are able to access the data.