

Abstract
Historically, soy protein and extracts have been used extensively in foods due to their high protein and mineral content. More recently, soy protein has received attention for a variety of its potential health benefits, including enhanced skin regeneration. It has been reported that soy protein possesses bioactive molecules similar to extracellular matrix (ECM) proteins and estrogen. In wound healing, oral and topical soy has been heralded as a safe and cost-effective alternative to animal protein and endogenous estrogen. However, engineering soy protein-based fibrous dressings, whilst recapitulating ECM microenvironment and maintaining a moist environment, remains a challenge. Here, we describe the development of an entirely plant-based nanofibrous dressing comprised of cellulose acetate (CA) and soy protein hydrolysate (SPH) using rotary jet spinning. The spun nanofibers successfully mimic physicochemical properties of the native skin ECM and exhibit high water-retaining capability. In vitro, CA/SPH nanofibers promote fibroblast proliferation, migration, infiltration, and integrin β1 expression. In vivo, CA/SPH scaffolds accelerate re-epithelialization and epidermal thinning as well as reduce scar formation and collagen anisotropy in a similar fashion to other fibrous scaffolds, but without the use of animal proteins or synthetic polymers. These results affirm the potential of CA/SPH nanofibers as a novel wound dressing.
Keywords: Fibers, Composite Materials, Biomedical Applications, Tissue Engineering
Graphical Abstract
A plant-based biomimetic cellulose/soy protein nanofibrous wound dressing is fabricated using rotary jet spinning. Its physicochemical properties mimic native extracellular matrix in skin and exhibit high water-retaining capability for enhanced wound healing. The plant hybrid wound dressing accelerates in vitro dermal fibroblast proliferation, spreading, and migration. Furthermore, it promotes re-epithelialization and reduces epidermal thickness and scar area in mouse model.

1. Introduction
Soy protein is a dietary protein extracted from the soy beans, which have received considerable attention in the last couple of decades for their potential health benefits. Epidemiological and clinical studies supporting this claim ultimately enabled US Food and Drug Administration (FDA) approval in 1999 of soy protein for protective effects on coronary heart disease.[1] Alternatively, soy protein has also been explored more recently as a “green” and renewable substitute for petroleum- or animal-derived polymers in biomedical applications.[2] It was found that soy protein has bioactive peptides similar to extracellular matrix (ECM) proteins, present in human tissues.[3] These ECM-mimetic peptides can promote cell adhesion, proliferation, and migration critical for supporting tissue regeneration.[3–4] Furthermore, soy protein carries phytoestrogens that act as a structural and functional analogue to the female sex hormone estrogen,[5] which affects the regulation and development of various organs by binding to estrogen receptors (ERs).[6] Binding of estrogen to ERs forms dimers that perform as co-activators to stimulate transcription of target gene expressions in various regions.[7] Interestingly, soy phytoestrogens preferentially bind to an ER that has been shown to have positive effects on age-related diseases, including delayed wound healing.[8] In cutaneous wound healing, soy protein has attracted increased attention as a safe and cost-effective alternative to animal protein and endogenous estrogen.[3, 8c, 9] Previous studies have shown that cryptic peptides in soy protein improved wound healing by increasing dermal ECM synthesis and stimulating re-epithelialization.[3, 9b, 9c] Soy phytoestrogens have demonstrated to accelerate the healing process via ER-mediated signaling pathways.[8b, 8c, 10] They also possess anti-bacterial,[2b, 11] anti-inflammatory,[12] and anti-oxidant properties[13] that support and enhance wound healing. Moreover, clinical trials have reported that oral intake of soy (both protein and phytoestrogens) accelerates skin regeneration in aged women and burn patients.[13b, 14]
Nanofibrous scaffolds have emerged as a promising approach to develop wound dressings,[15] as they can replicate the fibrous dermal ECM microenvironment that provides structural support for wound healing and functional cues for directing tissue regeneration.[15–16] Biodegradable synthetic polymers such as polycaprolactone (PCL) have been widely used to produce nanofibers due to their versatile spinning capabilities.[15–16] Yet, they remain poorly-suited building blocks for developing wound dressings as they are much stiffer than natural skin.[17] Furthermore, many of them are hydrophobic, limiting their ability to keep wounds hydrated.[17] Synthetic polymers also lack cell binding domains and therefore cannot enhance cellular attachment or functionality.[18] Nanofibers spun from animal-sourced ECM proteins, such as gelatin and collagen in combination with synthetic polymers, have been previously reported in literature to contain bioactive molecules which support healing.[15b, 17b] Whilst adding ECM proteins to a nanofibrous scaffold enhances its biological and mechanical properties, ECM proteins are costly and susceptible to common liabilities of animal-derived products: immunogenicity, antigenicity, disease transmission, and pathogen contamination.[2b, 19] Furthermore, the utilization of collagen alone, the most common ECM protein used in wound dressings, has been shown to cause extensive wound contraction and scarring.[20]
Because of its pro-regenerative traits, soy protein-based nanofiber wound dressings have recently been developed as an alternative to the animal-derived ECM protein nanofibers.[2a, 9c, 21] By mimicking the fibrous dermal ECM microenvironment, they can provide potent structural and functional cues for directing tissue regeneration.[20, 22] However, current methods for engineering soy protein nanofibers require the use of synthetic polymers as carriers, due to the low molecular weight of soy protein that inhibits the production of nanofibers alone.[2a, 21a, 23] As described above, synthetic polymers are not ideal for developing wound dressings as they possess physicochemical properties different from the native skin.[17] Soy protein hydrogels necessitate additional crosslinking agents that can be toxic and can alter the original structure of soy peptides.[4, 21b] As such, the development and validation of effective soy protein-based nanofiber scaffolds for wound healing applications remain an essential challenge.
In this study, we report the fabrication of plant hybrid cellulose acetate (CA) / soy protein hydrolysate (SPH) nanofibers for wound healing applications. We hypothesized that CA/SPH nanofibers could recapitulate the dermal ECM microenvironment and maintain a moist environment while delivering soy protein to potentiate skin regeneration. Cellulose acetate was selected as a co-spinning polymer because it readily dissolves in various solvents and self-assembles into nanofibers, enabling recapitulation of the native ECM fibrous structure and high water retention ability.[24] It is also abundant and exhibits low immunogenicity to humans because of its non-animal origins.[2b, 9a, 24a, 24c] Dermal ECM-mimetic CA and SPH nanofibers were manufactured via rotary jet spinning (RJS) system that utilizes centrifugal forces to extrude fibers in the nanometer range. We optimized physicochemical properties of the spun nanofibers by functionalizing the CA nanofibers with SPH. The RJS-spun CA/SPH nanofibers have higher production rate and better control of fiber morphology without an additional modification or high-voltage electric fields in the system, when compared to the existing electro-spun soy-based nanofibers.[25] Lastly, in vitro and in vivo functionalities of our dressings were tested by investigating dermal fibroblast behaviors and then further assessing wound closure rate and skin regeneration in an excisional wound splinting mice model, respectively. In comparison with the current fibrous scaffolds, the CA/SPH nanofibers have a healing ability similar to or better than other fibrous dressings, but our scaffolds are free of animal-derived proteins or synthetic polymers that are suboptimal. Our results underscored the potential of such soy-based nanofiber scaffolds as potent and cost-effective alternative to existing pro-regenerative strategies.
2. Results and Discussion
2.1. Fabrication of Plant Hybrid Nanofibers
Plant-based hybrid nanofibers were fabricated by co-spinning CA and SPH in hexafluoroisopropanol (HFIP) using a RJS system, which produces defect-free nanofibers under centrifugally induced shear forces (Figure 1).[25–26] CA was chosen to supplement the low molecular weight of soy protein, and SPH was chosen as the soy protein source. SPH is a mixture of amino acids, peptides, phytoestrogens, and soy derivatives obtained by hydrolyzing soy protein isolate to minimize inorganic ions and to maximize protein content.[27] Continuous CA and CA/SPH nanofibers were spun at a centimeter scale by extruding polymer solution from a rotating reservoir (Figure 1a–b).
Figure 1.
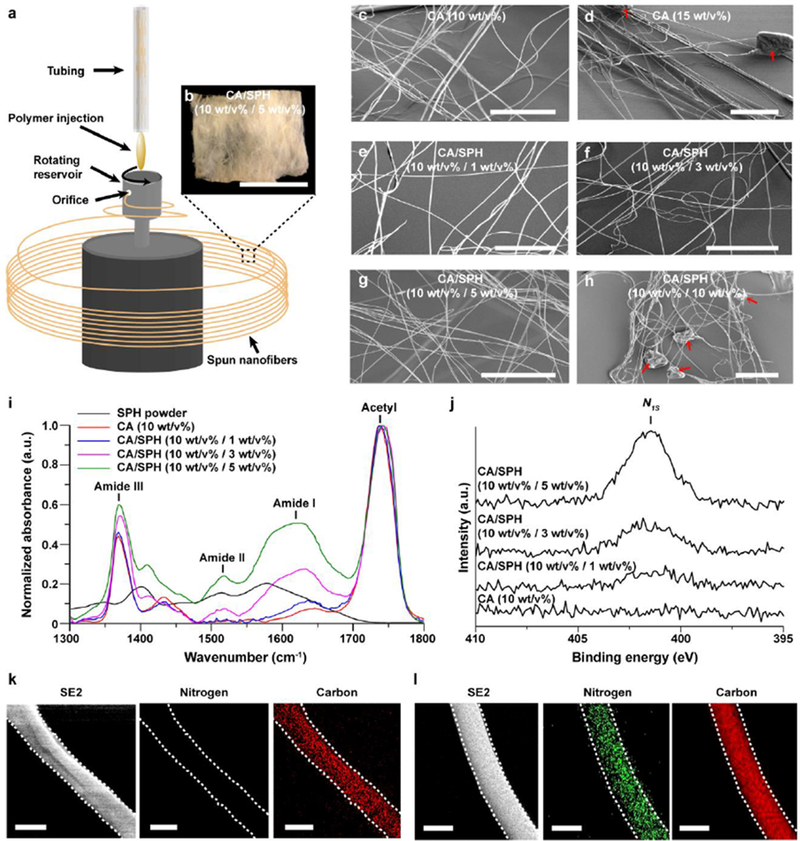
Fabrication and chemical composition of plant hybrid nanofiber. (a) Schematic for nanofiber fabrication by RJS. (b) Bright field image of CA/SPH (10:5) nanofiber scaffolds. Scales are 5mm. (c–h) SEM images of CA and CA/SPH. The red arrows indicate beading. Scales are 50 μm. (i) FT-IR spectrum of different CA/SPH nanofibers and SPH powder. (j) High resolution XPS spectra of N1s for different CA/SPH nanofibers. (k–l) Elemental analysis by Energy-dispersive X-ray spectroscopy (EDS) for nitrogen (NK) and carbon (CK) together with corresponding secondary electron (SE2) images in (k) CA (10 wt/v%) and (l) CA/SPH (10 wt/v% / 5 wt/v%) nanofibers. The white dots indicate the shape of nanofibers. Scales are 500 nm.
For the RJS system, the spinnability and beading of CA and SPH nanofibers were significantly influenced by their polymer concentrations (w/v%). Table 1 shows that SPH alone could not be spun into nanofibers because its molecular weight is too low. The short chains of SPH molecules cannot overlap and entangle, suggesting that SPH would require a co-spinning polymer with longer chains.[23b, 25a] Experimentation with fixed rotation and injection speeds showed that adding 10 w/v% of CA to various concentrations of SPH (1, 3, 5 w/v%) resulted in continuous nanofiber formation without beading (Table 1, Figure S1, and Figure 1c–d). A higher concentration of SPH (10 w/v%) in contrast showed beading in fibers (Table 1 and Figure 1e–h). Moving forward, 10 w/v% of CA was therefore selected as the carrier polymer for SPH. The developed continuous nanofibers had an intercalated nanofibrous structure that resembles the native extracellular matrix. This morphological similarity supports cell–fiber interactions that promote wound healing.[20, 22]
Table 1.
Spinnability of CA and SPH in HFIP
| Material | Morphology | Corresponding image | |
|---|---|---|---|
| Carrier polymer (w/v%) | Soy protein (w/v%) | ||
| CA (5) | None | No fiber | N/A |
| CA (10) | None | Continuous fibers | Figure 1c and S1a |
| CA (15) | None | Continuous fibers with beads | Figure 1d and S1b |
| CA (10) | SPH (1) | Continuous fibers | Figure 1e and S1c |
| CA (10) | SPH (3) | Continuous fibers | Figure 1f and S1d |
| CA (10) | SPH (5) | Continuous fibers | Figure 1g and S1e |
| CA (10) | SPH (10) | Continuous fibers with beads | Figure 1h and S1f |
| None | SPH (10) | No fiber | N/A |
2.2. Chemical Structure of Plant Hybrid Nanofibers
To ensure a uniform structure, elements must be homogenously dispersed at the nanofiber surface.[28] ATR-FTIR spectroscopy was performed to determine the relative amounts of proteins in the spun nanofibers. In the FTIR spectrum, amide I peaks (1600–1700 cm−1) are representative of the secondary structure of amino acids in SPH, and acetyl peaks (1700–1800 cm−1) are representative of C=O stretching of acetyl groups in CA (Figure 1i). After subtracting background intensity from CA in the amide I peak, the peak area-to-peak area ratios (amide I peak over acetyl peak) were linearly related to the amounts of SPH (Figure S2a), showing that SPH can be added into fibers in an amount up to 5 w/v% without causing the loss of soy protein molecules.
XPS was performed to confirm the elemental composition of the nanofiber surfaces. The nitrogen content gradually increased as the concentration of SPH increased (Figure 1j and S2b), confirming that SPH was incorporated into CA nanofibers. High resolution analysis of the C1s peaks additionally confirmed the increasing protein content on the nanofiber surface. This peak was deconvoluted, into four peaks corresponding to the following chemical bonds: C–C, C–O, O–C–O/N–C=O, and O–C=O (Figure S2c). Increasing SPH content thus led to relatively higher concentrations of C–C and O–C–O/N–C=O bonds (Figure S2c and Table S1). More amino acids and phytoestrogens in higher concentration of SPH ascribe to the increase of C–C and O–C–O/N–C=O bonds. These results demonstrated that SPH successfully integrated with CA.
To analyze the distribution of CA and SPH in individual fibers, EDS was performed to obtain an elemental mapping of nitrogen and carbon atoms (Figure 1k–l). Carbon mapping showed uniform distribution of carbon atoms on the spun nanofibers, matching the corresponding secondary electron (SE2) images. Nitrogen atoms appeared exclusively on CA/SPH nanofibers owing to the presence of SPH and were homogeneously distributed throughout individual fibers. This confirms and concludes that spinning CA at 10 w/v% and SPH at 5 w/v% improved fiber spinnability and yielded fibers with high concentrations of uniformly distributed protein. In the following studies, CA (10 w/v%) and CA/SPH (10 w/v% / 5 w/v%) nanofibers were selected as pure CA nanofibers and CA/SPH nanofibers, respectively.
2.3. Characterization of Mechanical Properties and Surface Chemistry
The physico-mechanical properties of nanofibers—fiber diameter, pore diameter, and stiffness—influence wound healing. Recent studies have shown that fiber diameter (200–400 nm) and scaffold pore diameter (6–20 μm), similar to the native ECM, enhance adhesion, proliferation, and infiltration of human dermal fibroblasts, while minimizing bacterial infiltration.[29] Fiber stiffness has also been shown to affect cell behavior.[30] To encourage assembly of new ECM, the stiffness of wound dressing materials should mimic the stiffness of the native ECM microenvironment (5–600 kPa),[31] however the stiffness of common synthetic polymer nanofiber scaffolds is usually one to several orders of magnitude higher.[17]
It was observed that fiber diameter ranges from 300.30 ± 0.76 nm in CA nanofibers and to 396.66 ± 0.90 nm in CA/SPH nanofibers (Figure 2a–b). In contrast, PCL nanofibers showed thicker fiber diameter (644.04 ± 5.20 nm) than CA-based nanofibers. Pore diameter ranges from 6.63 ± 0.14 μm in CA scaffolds to 6.13 ± 0.17 μm in CA/SPH scaffolds, while for PCL scaffold pore size decreased to 3.82 ± 0.38 μm. Next, scaffold thickness can be controlled by spinning a different amount of polymer solution (Figure S3a–c). Figure S3d showed that our system is able to produce fiber scaffolds with thickness ranging from a couple hundred micrometers to several millimeters. However, scaffold thickness does not significantly change pore diameters of nanofiber scaffolds (Figure S3e). The stiffness of the CA and the CA/SPH nanofibers was between 100 and 600 kPa in the longitudinal and transverse directions respectively (see Figure 2c and Table S2). On the other hand, the stiffness of the PCL nanofibers was in a MPa range, much stiffer when compared to native skin or CA-based nanofibers. These results suggest that fiber and pore diameter of both CA and CA/SPH nanofibers are well suited to support growth and migration of human dermal fibroblasts and that their stiffness resembles that of human skin ECM.[31]
Figure 2.
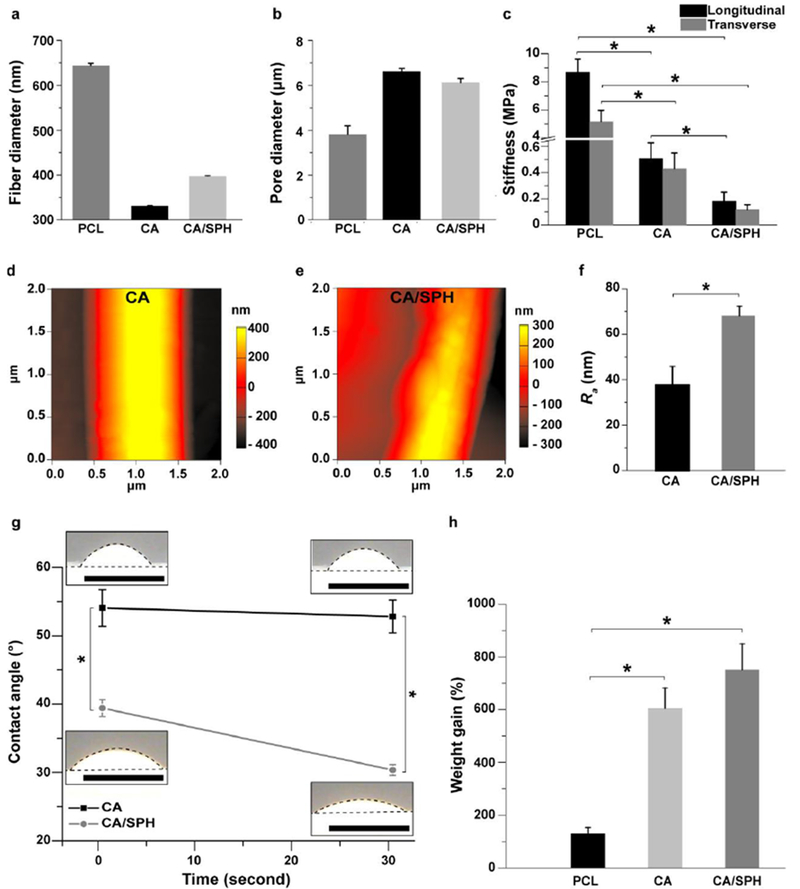
Characterization of mechanical properties and surface chemistry. (a–b) Fiber diameter and pore diameter analysis for PCL (6 wt/v%), CA (10 wt/v%), and CA/SPH (10 wt/v% / 5 wt/v%) nanofiber scaffolds. Bars represent standard error, n=10 from 3 productions. (c) Stiffness measurement for PCL (6 wt/v%), CA (10 wt/v%), and CA/SPH (10 wt/v% / 5 wt/v%) nanofibers in the wet state on the longitudinal and transverse directions. Bars represent standard error, n=5 from 3 productions, * indicates p < 0.05. (d–f)AFM images of (d) CA (10 wt/v%) and (e) CA/SPH (10 wt/v% / 5 wt/v%) nanofibers with (f) roughness (Ra) of nanofibers (n=3, FOV=3 from 3 productions). (g) Bright field images of water droplets on CA (10 wt/v%) and CA/SPH (10 wt/v% / 5 wt/v%) cast films with contact angle analysis (n=3 from 3 productions). Dots delimit water droplet and film. Scales are 5 mm. (h) In vitro water absorption measurements by weight gain (n=6 from 3 productions). Bars represent standard error, * indicates p < 0.05.
Next, we investigated the fiber surface roughness that affects cellular behaviors at both nano- and micro-scales since cells sense and react differently on various micro-topographies.[32] Current studies have reported that rough surfaces enhance cell adhesion, migration, and growth by triggering expression of integrin receptors and production of growth factors and ECM proteins.[32] To estimate the effect of the addition of SPH on the surface roughness of CA nanofibers, the average deviation (Ra) of the surface roughness was calculated from atomic force microscopy images (Figure 2d–e). Figure 2f showed that the Ra value for the CA/SPH nanofibers (68.19 ± 4.13 nm) was significantly higher than that of the CA nanofibers (38.06 ± 7.98 nm). Several factors may account for the effect of SPH on fiber roughness: the distribution of proteins throughout the surface and inside the nanofibers (Figure 1i–l), the aggregation of different materials within the nanofibers, and the short peptides that SPH carries.
The incorporation of SPH introduces polar moieties such as hydroxyl, amino, and carboxylic groups into the fibers. This increases the hydrophilicity as well as improves cell attachment by providing cell-binding functional groups.[33], [23b] High hydrophilicity and water retaining properties are vital for removing wound exudates and providing a moist environment for cell growth.[16, 33] To evaluate the chemical composition influence on the hydrophilicity of the materials, we performed contact angle measurement of uniform cast films (Figure 2g). The contact angles were significantly reduced by raising the ratio of SPH in the films, indicative of increased hydrophilicity. A similar trend was seen for fibrous samples, though rapid diffusion of water into the samples was seen for all samples (Figure S4a-e). The increased hydrophilicity was reflected by an increased water absorption capacity (Figure 2h). When CA was used as a backbone in nanofibers, their water-absorbing capabilities were significantly greater than that of hydrophobic PCL nanofibers which are frequently used as a backbone polymer to spin nanofiber scaffolds. The addition of SPH to the CA backbone further increased the water uptake.
An ideal nanofibrous scaffolds should be highly biodegradable so that it is gradually replaced by natural tissues during wound healing.[34] Figure S4f shows that over a 15-day period CA/SPH nanofibers lost significantly more mass than CA or PCL nanofibers due to hydrolysis of soy proteins. The rate of soy protein hydrolysis within the hybrid nanofibers resulted in the degradation, which correlates with the rate of protein breakdown.[2a, 34b] The lower mechanical strength and higher surface wettability of the hybrid nanofibers also contributed to their rate of degradation.[2a, 34b] In addition, the release kinetics of soy protein from CA/SPH nanofiber scaffolds resulted in a burst release of soy protein within 24 hours due to the fast hydrolysis of soy protein and high hydrophilicity (Figure S4g). After the initial burst release, a sustained soy release over 2 weeks was observed. The two phases of in vitro release (the initial burst and the sustained release over a long period) are typical release profiles of nanofiber-loaded molecules.[35] Therefore, a dressing made from plant-based hybrid nanofibers could provide structural cues until wound healing is completed and be naturally replaced by native tissue.
2.4. In Vitro Fibroblast Study
We hypothesized that the addition of SPH into CA nanofibers could promote wound healing-relevant cellular activity of human dermal fibroblasts (HNDF) via the presence of bioactive molecules, increased roughness, and enhanced water-retaining capabilities. In an effort to test this hypothesis, we began by analyzing several indicative markers for wound closure and tissue regeneration, including proliferation, surface coverage, migration, and infiltration of HNDFs (Figure 3, S5, and S6).[36] The behaviors of dermal fibroblasts were tested in vitro because they are a critical skin cell type that remodels the dermal ECM, communicates with other skin cells (such as keratinocytes), and thus regulates dermal function.[36a, 37] Cytotoxicity tests of the nanofiber scaffolds were likewise conducted as a standard pre-clinical experiment.[38] PCL nanofibers were used as a reference since it is one of the most common biocompatible and biodegradable synthetic polymers in nanofiber fabrication for biomedical applications.[28]
Figure 3.
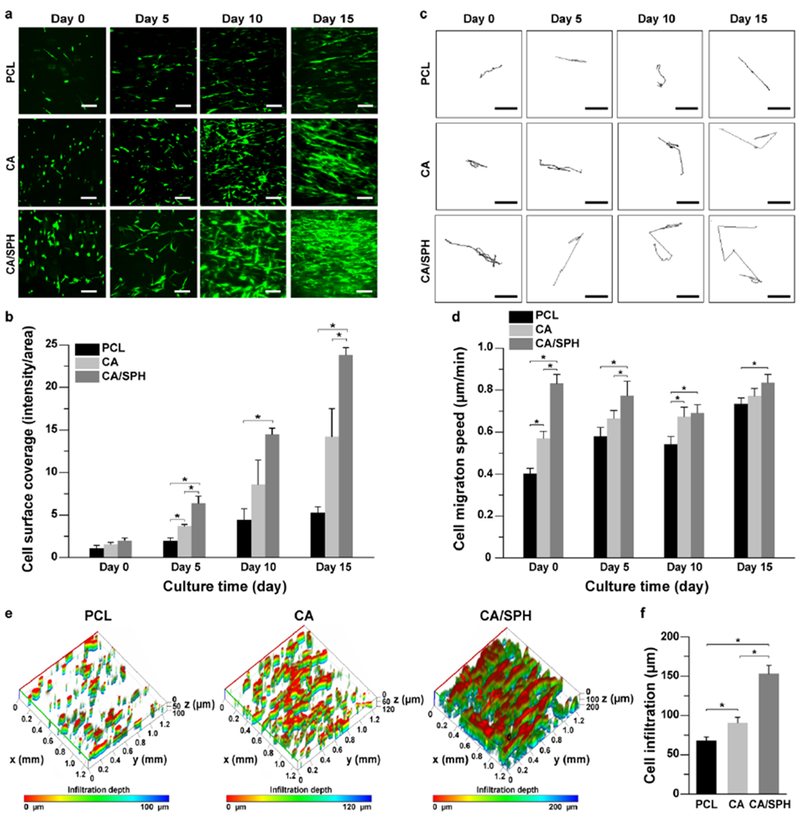
In vitro fibroblast coverage, migration, and infiltration. (a–b) (a) Representative confocal microscopy images of GFP-expressing human neonatal dermal fibroblasts (HNDF) on nanofibers with (b) analysis of surface area covered by cells at day 0, 5, 10, and 15. Scales are 50 μm. Bars represent standard error, n=5, FOV=5 from 3 productions, * indicates p < 0.05. (c–d) (c) Representative binary images of tracking a single cell on nanofibers at day 0, 5, 10, and 15 for calculating (d) migration speed of HNDF. Scales are 50 μm. Bars represent standard error, n=5, FOV=5 from 3 productions, * indicates p < 0.05. (e-f) (e) Representative 3D-recontructed confocal microscopy images of HNDF on nanofibers after 15 days of cell culture with (f) quantitative analysis of cell infiltration depth. Bars represent standard error, n=5 for PCL and n=8 for CA and CA/SPH, FOV=3 from 3 productions, * indicates p < 0.05.
Immunostaining analysis with the Ki-67 antibody – a marker specific to proliferative nuclei[39] – showed that CA/SPH nanofibers induced higher cell proliferation than PCL or CA nanofibers (Figure S5a–b). Nanofiber cytotoxicity was calculated by using a common LDH assay.[25b, 38] Both CA and CA/SPH nanofiber scaffolds exhibited low cytotoxicity, with similar values to PCL nanofibers (Figure S5c). Furthermore, we observed that the cell surface coverage on the CA/SPH nanofibers was significantly higher than on the PCL and CA nanofibers after 5 days in culture (Figure 3a–b). The CA nanofibers showed greater cell coverage at day 5 and day 15 versus the PCL nanofibers. HNDFs migrated faster on CA-based nanofibers than on PCL nanofibers (Figure 3c–d), whilst the addition of bioactive SPH into CA nanofibers resulted in increased cell migration compared to pure CA nanofibers. These results reflect the preferential properties of dermal ECM-mimetic CA-based nanofibers (fiber diameter, pore diameter, and stiffness as shown in Figure 2), and underscore the suboptimal properties of PCL. In addition, soy protein has been reported to trigger the expression of extracellular signal-regulated kinase (ERK), transforming growth factor (TGF β1), and integrin β1 that promote cell migration.[2a, 9b] In an effort to assess cell infiltration, cells were seeded on the surface of nanofiber scaffolds. Cells adhered to nanofibers and started to grow. At day 0, there is no significant difference in cell infiltration between different nanofibers (Figure S6). After 15 days of cell culture, CA-based nanofibers showed an increase in cell infiltration depth compared to PCL nanofibers (Figure 3e–f and S6) which was again further increased by functionalizing CA nanofibers with SPH. As CA-based nanofiber scaffolds have higher pore diameters than PCL nanofibers (Figure 2b), cells infiltrate faster on CA-based nanofibers.[29b] However, there is no significant difference in pore diameters between CA and CA/SPH nanofiber scaffolds, suggesting that the existence of SPH promoted cell migration (Figure 3c–d) and thus cells on CA/SPH nanofibers penetrated faster than CA nanofibers.
Next, immunocytochemical and western blot analysis for integrin β1 were performed to understand the effect of SPH on cell growth and migration. The integrin β1 is ECM protein receptors which regulates the behavior of ECM proteins and cells.[40] It also enables crosstalk with other growth factors and plays a crucial role in tissue repair.[41] During wound healing, dermal fibroblasts migrate to the wound site and express integrin β1 to mature the developing matrix.[41] It has been found that decreased expression of integrin β1 reduces the ability of fibroblasts and keratinocytes to migrate, lay down a collagen matrix, and ultimately enable wound closure.[40] After 15 days of cell culture, immunocytochemical (Figure 4a) and western blot (Figure 4b–c) analysis indicated that the integrin β1 expression was significantly increased on CA/SPH nanofibers, compared to CA nanofibers. These results indicate that soy protein in our scaffolds can trigger the expression of integrin β1 that in turn accelerates the cell migration and the production of new ECM proteins for wound closure. The increased integrin β1 expression by functionalizing CA nanofibers with SPH is in line with previously published work that reported that soy protein peptides up-regulated the expression of integrin β1 in fibroblasts.[2a]
Figure 4.
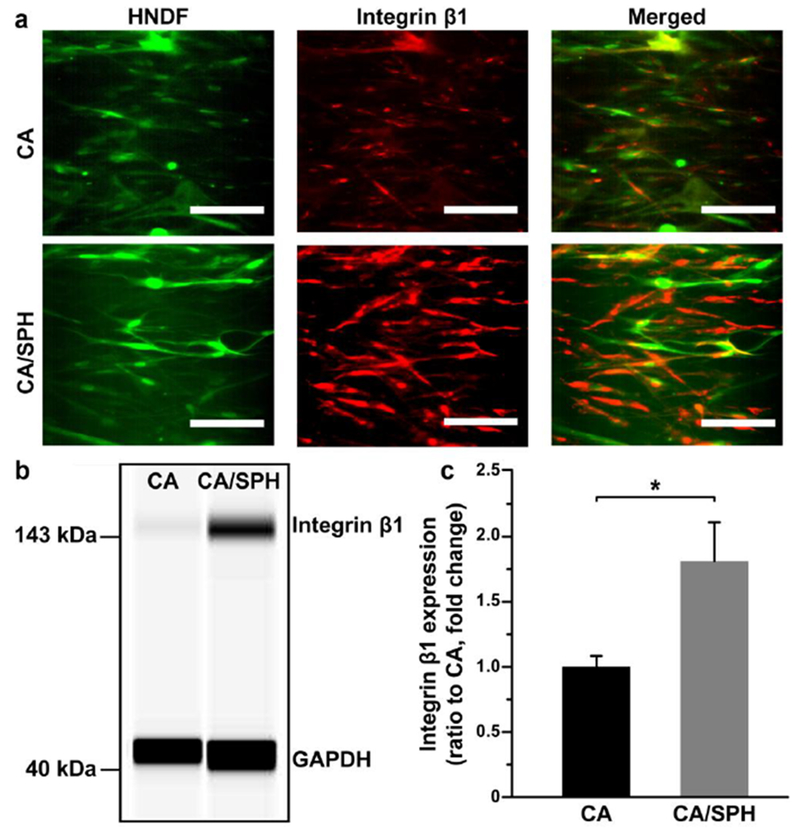
In vitro integrin β1 expression by fibroblast. (a) Representative immunostaining and (b) Western blotting images for integrin β1 with (c) quantitative analysis from Western blotting. Scales are 100 μm. Bars represent standard error, n=6 for CA and n=7 for CA/SPH from 3 productions, * indicates p < 0.05.
In summary, our in vitro fibroblast study demonstrated that CA nanofibers supported stronger cell growth, proliferation, migration, and infiltration than PCL nanofibers. These enhanced cellular activities occurred because CA provides a soft and hydrophilic backbone similar to that of a collagen matrix found in native dermal tissue. Further functionalization of CA nanofibers with SPH accelerated proliferation, growth, migration, infiltration, and integrin β1 expression of HNDFs. Accordingly, it can be extrapolated that CA/SPH nanofibers should possess the ability to provide structural and biological cues to promote wound healing in vivo.
2.5. In Vivo Wound Healing Study in a Rodent Model
To investigate the potency of CA/SPH in vivo, we tested our nanofiber scaffolds on a mouse excisional wound splinting model.[42] Wound contraction was inhibited by suturing a silicon splint to the peripheral edge of the wound in an effort to study the healing process via re-epithelialization and thus improving recapitulation of the wound healing process of humans (Figure 5a and S7).[43] Nanofiber scaffolds were held in place with a Tegaderm™ film. The control group wounds received no nanofiber treatment and were only covered with the Tegaderm™ film. It was observed that CA/SPH nanofibers significantly accelerated wound closure (Figure 5b-c). On Day 7 after surgery, CA nanofibers showed 42% faster wound closure than our control. The addition of SPH in the CA nanofibers further accelerated wound closure by 21% and showed an overall 72% increase when compared to the non-treated control. After 14 days, the wounds treated with CA/SPH nanofibers were fully closed (Figure 5b-c). Moreover, the wound closure potentiated by CA/SPH nanofibers significantly higher than both the control and CA nanofibers.
Figure 5.
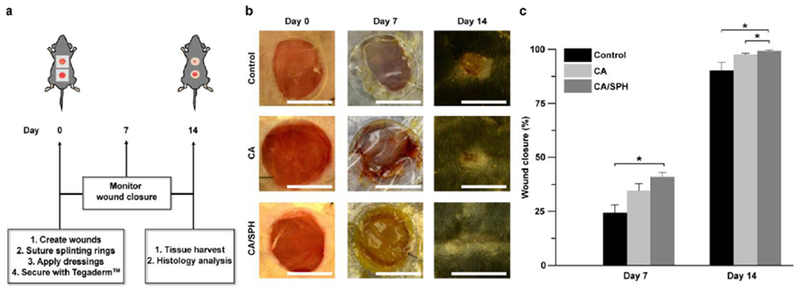
In vivo wound healing study with a mouse excisional wound splinting model. All wounds were covered with the Tegaderm™ film. Control wounds received no nanofibers. (a) Schematic representation of in vivo wound healing experiment. (b) Representative images of wounds from the study groups: untreated control, treated with CA nanofibers, and treated with CA/SPH nanofibers at Day 0, 7, and 14. Scales are 5 mm. (c) Analysis of wound closure. Fiber wound dressings were prepared from 3 productions for each condition. Bars represent standard error, n=4 wounds and 3 mice for control, n=5 wounds and 3 mice for CA and CA/SPH. * indicates p < 0.05.
In an effort to further assess the regenerative capacity of our treatment conditions, histological analysis of healed tissues was performed at day 14 post surgery (Figure 6a). Restoration of the dermal and epidermal layers are key parameters for evaluating wound healing and tissue regeneration.[42, 44] It is commonly analyzed by quantifying the epithelial gap, epithelial thickness, and scar size.[42, 44] H&E staining confirmed our previous macroscopic observation that CA/SPH nanofiber-treated wounds were re-epithelialized at day14 post-surgery. However, wounds from the control and CA nanofiber-treated groups remained open, resulting in epithelial gaps a few hundred micrometers in diameter after 14 days of treatment (Figure 6b). In addition, the control or CA nanofibers-treated wounds exhibited significantly thicker epidermises than CA/SPH nanofibers-treated wounds, indicating slower regeneration of the epidermis (Figure 6c). It should nonetheless be noted that the epidermal thicknesses of CA/SPH nanofibers-treated wounds were still higher than that of healthy tissues (Figure 6c and S8). We also measured the scar size using a quantitative scar index (Figure 6d),[44] which found that CA/SPH nanofibers significantly reduced the scar size compared to the control or CA nanofibers after 14 days treatment. Lastly, the alignment of the newly synthesized collagen in the dermis was calculated (Figure 6e). The dermal collagen was significantly less aligned in CA/SPH nanofiber-treated wounds than control or CA nanofiber-treated wounds. However, the alignment of CA/SPH nanofiber–treated wounds was still higher than that of healthy tissues that possess typically basket-woven fiber organization. In line with our in vitro results, the in vivo data supported our hypothesis that both a nanofibrous architecture and bioactive soy protein accelerated wound closure and supported regeneration of the dermal and epidermal layers. These observation also corroborate previously published results in which ECM-mimetic peptide[9c, 13b] and phytoestrogens[8c, 45] in soy protein promoted re-epithelialization and dermal tissue regeneration. When comparing our results to other reports of pro-regenerative fibrous scaffolds that consist of natural materials (such as ECM proteins, silk, and chitosan),[46] similar trends can be established for both wound closure rate and tissue regeneration (re-epithelialization, epidermal thinning, collagen alignment, and scar formation). Yet our scaffolds present a significant advantage gained by their inherent plant-based origin as they are free of animal-derived materials or synthetic carrier polymers that raise manufacturing cost, immunogenicity, and ethical concerns.
Figure 6.
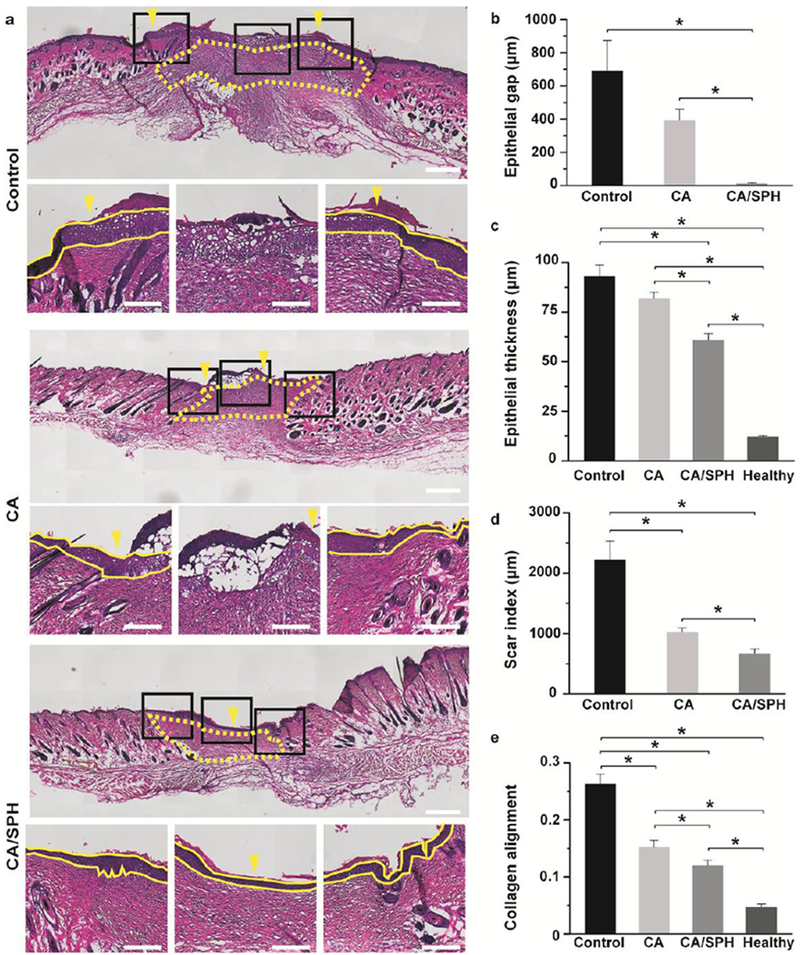
Histology analysis. (a) H&E staining of wounds after 14 day post-surgery (untreated control, treated with CA nanofibers, and treated with CA/SPH nanofibers). In the top panel, the yellow arrows indicate the edge of the epidermal layer and the yellow dots outline the scar area. The yellow lines in the bottom panels of the zoom-in images delimit the epidermal layer in the skin tissue. Scales in the top and bottom panels are 500 μm and 200 μm, respectively. (b–e) Quantitative analysis of b) epithelial gap, c) epithelial thickness, d) scar index, and e) collagen alignment from H&E staining images. Fiber wound dressings were prepared from 3 productions for each condition Bars represent standard error, n=3 wounds and 3 mice for control, n=4 wounds and 3 mice for CA and CA/SPH nanofibers, n=5 wounds and 5 mice for healthy tissue, at least 3 sections per wound, * indicates p < 0.05.
3. Conclusions
To the best of our knowledge, the present study reports the first fabrication and optimization of CA/SPH nanofibers produced using an RJS system. CA and SPH molecules were homogeneously distributed along the nanofibers for equal functionality at the fiber surface. Using CA as a co-spinning polymer enabled recapitulation of fiber morphology, fiber diameter, pore diameter, and stiffness of the native ECM thus creating optimal conditions for dermal fibroblasts to thrive. Functionalization of CA nanofibers with SPH enhanced surface roughness, hydrophilicity, and water absorption capacity. The in vitro study indicated that CA/SPH nanofibers increased proliferation, growth, migration, and infiltration of fibroblasts and exhibited low cytotoxicity, compared to both PCL and CA nanofibers. The addition of SPH into CA nanofibers further up-regulated the expression of integrin β1, which has been attributed to enhanced cell migration and tissue regeneration. Finally, the in vivo mouse study revealed that CA/SPH nanofibers accelerated wound closure and tissue regeneration in comparison to CA nanofibers or the non-treated control. Altogether, these findings confirmed the utility of CA/SPH nanofibers for enhanced wound healing. We envision that our one-step, cost-effective, and regenerative scaffolds comprised of plant-based materials will be the next generation of regenerative dressings to push the envelope of nanofiber technology and the wound care market.
4. Experimental Section
Materials:
PCL (Mn 70,000-90,000; Sigma-Aldrich), CA (Mn 50,000; Sigma-Aldrich), SPH (Amisoy™; Sigma-Aldrich), and Hexafluoroisopropanol (HFIP, Oakwood Chemical) were used as received.
Fiber Fabrication by Rotary Jet Spinnings:
Nanofibers were spun by using rotary jet spinning (RJS) system as described in previous studies.[25–26] Briefly, CA and CA/SPH with different compositions and concentrations (weight per volume percent, wt/v %) were dissolved in HFIP and stirred for overnight. As a reference group, PCL (6 wt/v %) was also dissolved in HFIP. After mixing, solutions were flowed to the rotating reservoir through polyfluoroalkoxy alkane tubing (Saint-Gobain) at 2 mL/min by using an automatic syringe pump (Harvard Apparatus). Then, the solutions (10 mL in total) were sprayed from the reservoir at at 60,000 rpm for 5 min, elongating polymers into nanofibers and evaporating HFIP rapidly in the air from the orifice (diameter of 360 μm). The spun nanofibers were dried overnight in a desiccator to fully remove excess solvent. For cell culture, the spun nanofibers were collected on coverslips and sterilized overnight under UV-light.
Scanning Electron Microscopy (SEM):
Fiber samples were imaged by using a field emission scanning electron microscopy (FESEM, Carl Zeiss). The fiber samples were mounted on sample stubs, sputter-coated with 5 nm thickness of Pt/PD (Denton Vacuum), and imaged by using FESEM.
Characterization of Chemical Compositions:
Attenuated Total Reflectance-Fourier Transform Infrared spectroscopy (ATR-FTIR, Bruker) was used to obtain FT-IR spectra of nanofibers over 600–4000 cm−1 at a resolution of 2 cm−1 with 16 scans. The samples were mounted on sample stage and contacted with ATR-crystal for measurement. The FT-IR spectrum of the dried samples were measured and normalized from 0 to 1. For Gaussian curve fitting and area analysis, OriginPro 8.6 software (Origin Lab Corporation) was used. For statistical analysis, n=3 from 3 productions for each condition. X-ray photoelectron spectrometer (XPS, K-Alpha XPS system, Thermo Scientific) was used to further evaluate fiber surface composition. Fibrous test samples were prepared on silicon wafer substrates. Survey and high resolution elemental spectra were obtained using monochromatized aluminum Kα radiation (pass energy 200 eV). An argon flood gun was applied to offset sample charging. Peak detection and high resolution C1s peaks were deconvoluted using Lorentzian/Gaussian product mix (30% L) functions. For statistical analysis, n=3 from 3 productions for each condition. Energy-dispersive X-ray spectroscopy (EDS) in FESEM was used to investigate elemental mapping of nitrogen (NK near 0.392 eV) and carbon (CK near 0.277 eV) atoms, together with corresponding type II secondary electron (SE2) images. The fiber sample was also sputter-coated with Pd/Pt on sample stub and imaged by using EDS.
Characterization of Fiber diameter, Pore Diameter, and Fiber Thickness:
Fiber diameter, pore diameter, and fiber thickness were analyzed by using SEM images of the nanofibers and ImageJ (NIH) with the plug-in DiameterJ.[47] For fiber thickness analysis, nanofiber scaffolds were prepared from different injection volume (10, 30, and 60 mL in total) and the cross-sectioned scaffolds were imaged and analyzed. DiameterJ was used to determine fiber and pore diameters by using algorithm as described in previous study.[47] Here, the pore diameters refer to the pores of the fibrous scaffolds (between fibers). For statistical analysis, n=10 from 3 productions for each condition.
Biaxial Tensile Test for Stiffness Measurement:
The stiffness in the wet state was determined by using biaxial tensile tester (CellScale). The spun fiber scaffolds were loaded by using clamps to hold the samples and immersed in phosphate buffered saline (PBS, ThermoFisher Scientific) at 37°C. Sample was loaded equibiaxially at a strain rate of 5% per second to 20% strain. A built-in software (CellScale) was used to record force/displacement measurements and images at 15 Hz. By using these measurements and the thickness of the samples, stress-strain curves were then produced. Stiffness was determined by calculating the slope of the stress-strain curves. For statistical analysis, n=5 from 3 productions for each condition.
Atomic Force Microscopy (AFM) for Roughness Measurement:
Roughness (average deviation, Ra) was calculated by using built-in software in atomic force microscopy (AFM, MFP-3D™, Asylum). The fiber samples were mounted on sample stage and imaged with tapping mode.[48] For statistical analysis, n=3 (field of view (FOV)=3) from 3 productions for each condition.
Contact Angle and Water Absorption Measurements:
The cast film samples were prepared on coverslips using spin coater (at 2000 rpm for 1 min). The nanofiber samples were directly spun onto coverslips. A camera was used to record water droplet formation on the surfaces of the substrates. Contact angle was calculated by using ImageJ with the plug-in Drop Shape Analysis.[49] For statistical analysis, n=3 from 3 productions for each condition. Water absorbency was measured as % mass gain like a standard method reported before.[50] First, dry weight of the samples was recorded. The samples were immersed in PBS for 24 h at 37°C. The excess PBS on the wet samples was removed by placing it on a paper towel. Then, weight of the water-absorbing samples was measured. The water absorption ability was defined as described below;
| (1) |
where A is the water absorption ability (%), W1 is the weight before wet, and W2 is the weight after wet. For statistical analysis, n=3 from 3 productions for each condition.
Biodegradation Measurement.
In vitro biodegradation was measured as % mass loss as detailed in previous studies.[2a, 34b, 51] The initial weight of the scaffold was measured, after which the samples were immersed in PBS at 37°C and 5% CO2. At day 5, 10, and 15, the samples were washed three times with fresh PBS and dried in an oven at 60°C overnight. After complete dehydration, the weight of the dried samples was measured. The in vitro biodegradation was defined as follows:
| (2) |
where D is the in vitro biodegradation (%), W1 is the initial weight, and W3 is the final weight after degradation. For statistical analysis, n=3 from 3 productions for each condition.
Soy Protein Release Kinetics.
In vitro release profile of soy protein from the nanofibers was measured as % loss of amide I peaks. The samples were immersed in PBS at 37°C and 5% CO2. At day 0, 3, 5, 7, and 15, the samples were washed three times with fresh PBS and freeze-dried. The FT-IR spectrum of the dried samples were measured and normalized from 0 to 1. The relative areas of amide I peaks were analyzed from the normalized spectrum to calculate the % release of soy protein from the scaffolds. For statistical analysis, n=3 from 3 productions for each condition.
Cell Culture:
Green fluorescent protein (GFP)-expressing human neonatal dermal fibroblasts (HNDFs, Angio-Proteomie) were properly treated as described in protocol from the manufacturer (Angio-Proteomie) for cell culture. Briefly, HNDFs were delivered at passage 3 in a frozen vial and stored in a liquid nitrogen tank before use. Cells were subcultured to passage 7 with Dulbecco’s modified eagle medium (DMEM, ThermoFisher Scientific) containing Fetal Bovine Serum (FBS, 5%) and antibiotics (penicillin-streptomycin, ThermoFisher Scientific, 1%) in a T25 flask at 37°C incubator with 5% CO2 and 21% O2. Once the cells reach passage 7, trypsin/ ethylenediaminetetraacetic acid solution (trypsin/EDTA, Lonza, 2 mL) was added to the T25 flask. Seeding density was fixed at 30,000 cells per sample. Cell media was changed every 2 days before imaging and fixation.
Analysis of Growth, Migration, and Infiltration of Dermal Fibroblasts:
GFP-expressing HDNFs on the fibers were imaged by using confocal microscopy (Zeiss LSM 5 LIVE) at 37 °C in a temperature controlled chamber. 4-(2-hydroxyethly)-1-piperazineethanesulfonic acid (HEPES, ThermoFisher Scientific, 2.5 %) buffer was added to the media during imaging in an effort to keep the pH constant. For cellular growth study, the intensity of GFP-expressing HNDF per area was calculated from the confocal images by using ImageJ. For cellular migration study, the migration of GFP-expressing cells on fibers was tracked (1 frame/10 min for at least 40 frames). Once all images were collected, ImageJ plug-in StackReg was used to correct the center of each image.[52] For statistical analysis, n=5 (field of view (FOV)=5) from 3 productions for each condition. Migration of each cell was analyzed by using the plug-in Mtrack2 in ImageJ.[53] The Mtrack2 calculates the total distance each cell has migrated. Migration speed of cells was calculated by dividing the total distance by total imaging time. For statistical analysis, n=5 (field of view (FOV)=5) from 3 productions for each condition. In cellular infiltration study, z-stack confocal images of GFP-expressing cells on fibers were captured at 15 days of cell culture. The cell infiltration depth from the z-stack images was calculated using the z-axis profile function in ImageJ as previously reported.[26d] The cross-sectional view (in yz plane) of cells was processed from ImageJ by using the orthogonal view function. For statistical analysis, n=5 for PCL and n=8 for CA and CA/SPH nanofibers (field of view (FOV) =3) from 3 productions for each condition.
Cytotoxicity Measurement:
In vitro cytotoxicity of cells on the fibers was measured by using lactate dehydrogenase (LDH) cytotoxicity assay (Promega) as described previously.[25b, 38] Briefly, HNDFs were cultured on nanofibers for 15 days and successively incubated with reaction solution and stop solution (1 M acetic acid) from the assay kit. A commercial plate reader was used to measure absorbance at 490 nm. The % cytotoxicity was defined as follows;
| (3) |
where S is the readout from the sample, C is the readout from the control (medium only without cell), and M is the readout from maximum LDH release.[25b, 38] For statistical analysis, n=17 in triplicate from 3 productions for each condition. For box plot in Figure S3, the box range is 25–75%, the whisker range is 10–90% using OriginPro 8.6 software.
Immunocytochemical Analysis:
After 15 days of culture, HNDFs grown on nanofibers were fixed in paraformaldehyde (PFA, 4%) and Triton-X (0.05%) for 10 min. Following fixation, samples were incubated with primary antibody (rabbit polyclonal anti-Ki67 with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) for proliferation study or rabbit monoclonal anti-integrin β1 antibody, Abcam) and with secondary antibody (goat anti-rabbit IgG (H+L) secondary antibody with Alexa fluor® 546, Invitrogen) during 1 h at room temperature for both primary and secondary antibody incubation. Following immunostaining, samples were mounted on glass slides by using Prolong Gold anti-fade agent (Invitrogen) and imaged on the confocal microscopy. Cell proliferation was calculated by dividing the number of Ki-67 positive cells by the number of DAPI-positive cells. For statistical analysis, n=5 for PCL and n=6 for CA and CA/SPH (field of view (FOV)=25) from 3 productions for each condition.
Western Blot Analysis:
HNDFs were cultured on nanofibers for 15 days and were lysed at 4 °C using radioimmunoprecipitation assay (RIPA) lysis buffer (SLBG8489, Sigma) with Complete Mini (11836153001, Roche Diagnostic) and Halt-Protease and Phosphotase Inhibitor (1861281, ThermoFisher Scientific). A capillary-based Wes Simple Western (ProteinSimple) was used to detect and quantify the expression of integrin β1 in cell lysates following the manufacturer’s protocol. In brief, each capillary loaded 5 μg of sample lysates and separated proteins by size. The samples were incubated with primary antibodies for Integrin β1 and Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as a loading control (ab52971 and ab9485 respectively, ABCAM). Target proteins were labeled with secondary antibodies and chemiluminescent reagents provided by the manufacturer (ProteinSimple). Signals were detected and quantified using CompassSoftware (ProteinSimple). Expression of integrin β1 was normalized to GAPDH loading control and compared across sample conditions. For statistical analysis, n=6 for CA and n=7 for CA/SPH from 3 productions for each condition.
Mouse Excisional Wound Splinting Model:
All mouse wound healing experiments were performed using IACUC approved protocols (Protocol ID 11-11). Based on the previous publications,[43] the mouse excisional splinting model was carried out in order to analyze cutaneous wound closure in murine skin by excluding wound contraction. Briefly, splinting rings were prepared by cutting 8 mm holes in a 0.5 mm-thick silicon sheet (Grace Bio-Labs) using a sterile biopsy punch (Integra® Miltex®). The prepared rings were washed and sterilized by ethanol (70 % vol/vol), and then were air-dried in a sterile culture hood before surgery. C57BL/6 male mice (Charles River Laboratories, 52 days old) were anesthetized with isofurane through the duration of procedure. Once anesthesia was confirmed by a toe pinch test, the dorsal side of mice was shaved using electric and manual razor. After hair removal, the skin was cleaned with betadine (Santa Cruz Biotechnology) and ethanol (70 % vol/vol). The full-thickness excisional wounds were created on the midline by punching through the skin with a 6-mm-diameter sterile biopsy punch. The punched tissues were used for histological analysis of healthy skin (Day 0). An instant-bonding adhesive (Krazy glue) was put on one side of a splint. The splints were fixed into place around the wound with instant bonding adhesive followed by suturing with nylon suture (Ethicon). Nanofiber wound dressings were applied to the wound and covered with Tegaderm™ (Nexcare™) patches to keep the scaffolds in place and the surgical area clean. Control wounds received no nanofibers and were covered with Tegaderm™ patches only. Tegaderm™ is a clinical standard wound dressing.[43] The mice were monitored daily. Before tissue harvest on Day 7 and 14, mice were sacrificed via IACUC approved methods.
In Vivo Wound Closure Analysis:
Wound area was photographed with a digital camera on Day 0, 7, and 14. The wound area was manually quantified using ImageJ. Wound closure was defined as described below;[43a]
| (4) |
where O is the area of the original wound and A is the area of the wound at a given time point. Fiber wound dressings were prepared from 3 productions for each condition. For statistical analysis, n=4 wounds and 3 mice for control, n=5 wounds and 3 mice for CA and CA/SPH.
Histological analysis:
Histological analysis was preformed based on previously published methods.[43a] Tissues were harvested from Day 0 and 14 and fixed with PFA (4%) at 4 °C overnight. The fixed tissue was washed using PBS five times for 30 min each. The tissue was incubated with sucrose (Sigma, 20% and 40% wt/vol) in PBS at room temperature for 2 h each. Then, the tissue was embedded in O.C.T. compound (Electron Microscopy Science) with cryomold (Tissue-Tek®). The frozen wound tissues were sectioned with 10 μm thickness, stained with hematoxylin and eosin (H&E), and imaged by slide scanner (Olympus VS120). Re-epithelialization was analyzed by manually calculating distance among the newly synthesized epithelial layers from H&E staining tissue sections (marked as yellow arrows in Figure 5a).[42] Epithelial thickness was also manually measured using ImageJ. Scar index was quantified by using a previously published method.[44] Briefly, scar area (yellow dotted area in Figure 5a) and dermal thickness were manually measured using ImageJ. Then, scar index was defined as described below;[44]
| (5) |
where S is the scar area (μm2) and D is the average dermal thickness (μm). Dermal collagen alignment in the wounds was calculated by using OrientationJ in ImageJ as previously published.[54] The OrientationJ computes the coherency that is between 0 (isotropic) and 1 (anisotropic). Fiber wound dressings were prepared from 3 productions for each condition. For statistical analysis, n=3 wounds and 3 mice for control, n=4 wounds and 3 mice for CA and CA/SPH nanofibers, n=5 wounds and 5 mice for healthy tissue, at least 3 sections per wound.
Statistical Analysis:
All data is displayed as mean ± standard error of mean (SEM). One-way analysis of variance (ANOVA) with the post-hoc Tukey’s test in OriginPro 8.6 software was used for statistical comparisons. Statistical significance was determined at * p < 0.05.
Supplementary Material
Acknowledgements
This work was funded by the Wyss Institute for Biologically Inspired Engineering at Harvard University. Research reported in this publication was supported by the Harvard University Materials Research Science and Engineering Center (MRSEC), NSF Award number DMR-1420570. Seungkuk Ahn received Fulbright Scholarship (Fulbright Foreign Student Program for 2014-2015) from the United States Department of State to accomplish this research. This work was also in part conducted at Harvard Center for Nanoscale Systems (CNS), which is a member of the National Nanotechnology Infrastructure Network (NNIN), supported by the National Science Foundation under NSF award no. 1541959. We thank Dana-Farber/Harvard Cancer Center in Boston, MA, for the use of the Rodent Histopathology Core, which provided tissue sectioning, embedding, and H&E staining services for histology analysis. Dana-Farber/Harvard Cancer Center is supported in part by a NCI Cancer Center Support Grant # NIH 5 P30 CA06516. We acknowledge the Neurobiology Department and the Neurobiology Imaging Facility for consultation and instrument availability that supported this work. This facility is supported in part by the Neural Imaging Center as part of an NINDS P30 Core Center grant #NS072030. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors appreciate the graphic works provided by Grant Gonzalez.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
References
- [1].a) Sacks FM, Lichtenstein A, Van Horn L, Harris W, Kris-Etherton P, Winston M, Circulation 2006, 113, 1034; [DOI] [PubMed] [Google Scholar]; b) Potter SM, Baum JA, Teng H, Stillman RJ, Shay NF, Erdman J, Am. J. Clin. Nutr 1998, 68, 1375S. [DOI] [PubMed] [Google Scholar]
- [2].a) Lin L, Perets A, Har-el Y. e., Varma D, Li M, Lazarovici P, Woerdeman DL, Lelkes PI, J. Tissue. Eng. Regen. Med 2013, 7, 994; [DOI] [PubMed] [Google Scholar]; b) Tansaz S, Boccaccini AR, J. Biomed. Mater. Res., Part A 2016, 104, 553. [DOI] [PubMed] [Google Scholar]
- [3].Tokudome Y, Nakamura K, Kage M, Todo H, Sugibayashi K, Hashimoto F, Int. J. Food Sci. Nutr 2012, 63, 689. [DOI] [PubMed] [Google Scholar]
- [4].a) Chien KB, Makridakis E, Shah RN, Tissue Eng., Part C 2012, 19, 417; [DOI] [PubMed] [Google Scholar]; b) Chien KB, Shah RN, Acta Biomater 2012, 8, 694. [DOI] [PubMed] [Google Scholar]
- [5].a) Setchell KD, J. Am. Coll. Nutr 2001, 20, 354S; [DOI] [PubMed] [Google Scholar]; b) Cederroth CR, Nef S, Mol. Cell. Endocrinol 2009, 304, 30. [DOI] [PubMed] [Google Scholar]
- [6].Ascenzi P, Bocedi A, Marino M, Mol. Aspects Med 2006, 27, 299. [DOI] [PubMed] [Google Scholar]
- [7].Mendelsohn ME, Karas RH, Engl N. J. Med 1999, 340, 1801. [DOI] [PubMed] [Google Scholar]
- [8].a) Hall G, Phillips TJ, J. Am. Acad. Dermatol 2005, 53, 555; [DOI] [PubMed] [Google Scholar]; b) Ashcroft GS, Dodsworth J, Van Boxtel E, Tarnuzzer R, Horan MA, Schultz GS, Ferguson M, Nat. Med 1997, 3, 1209; [DOI] [PubMed] [Google Scholar]; c) Emmerson E, Campbell L, Ashcroft GS, Hardman MJ, Mol. Cell. Endocrinol 2010, 321, 184. [DOI] [PubMed] [Google Scholar]
- [9].a) Santin M, Ambrosio L, Expert Rev. Med. Devices 2008, 5, 349; [DOI] [PubMed] [Google Scholar]; b) Lee J, Roh K-B, Kim S-C, Lee J, Park D, J. Nutr. Biochem 2012, 23, 1341; [DOI] [PubMed] [Google Scholar]; c) Gerstenhaber JA, Brodsky R, Huneke RB, Lelkes PI, Wound Medicine 2014, 5, 9. [Google Scholar]
- [10].a) Sirotkin AV, Harrath AH, Eur. J. Pharmacol 2014, 741, 230; [DOI] [PubMed] [Google Scholar]; b) Hsia SY, Hsiao YH, Li WT, Hsieh JF, Sci. Rep 2016, 6, 35718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Hong H, Landauer MR, Foriska MA, Ledney GD, J. Basic Microbiol 2006, 46, 329; [DOI] [PubMed] [Google Scholar]; b) Ben Arfa A, Preziosi-Belloy L, Chalier P, Gontard N, J. Agric. Food Chem 2007, 55, 2155. [DOI] [PubMed] [Google Scholar]
- [12].Chacko BK, Chandler RT, Mundhekar A, Khoo N, Pruitt HM, Kucik DF, Parks DA, Kevil CG, Barnes S, Patel RP, Am. J. Physiol. Heart Circ. Physiol 2005, 289, H908. [DOI] [PubMed] [Google Scholar]
- [13].a) Peñta-Ramos E, Xiong Y, J. Food Sci 2002, 67, 2952; [Google Scholar]; b) Babajafari S Akhlaghi M, Mazloomi SM, Ayaz M, Noorafshan A, Jafari P, Hojhabrimanesh A, Burns 2017. [DOI] [PubMed] [Google Scholar]
- [14].a) Jenkins G, Wainwright L, Holland R, Barrett K, Casey J, Int. J. Cosmet. Sci 2014, 36, 22; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Izumi T, Saito M, Obata A, Arii M, Yamaguchi H, Matsuyama A, J. Nutr. Sci. Vitaminol 2007, 53, 57. [DOI] [PubMed] [Google Scholar]
- [15].a) Rieger KA, Birch NP, Schiffman JD, J. Mater. Chem. B 2013, 1, 4531; [DOI] [PubMed] [Google Scholar]; b) Hassiba AJ, El Zowalaty ME, Nasrallah GK, Webster TJ, Luyt AS, Abdullah AM, Elzatahry AA, Nanomedicine 2016. [DOI] [PubMed] [Google Scholar]
- [16].Boateng JS, Matthews KH, Stevens HN, Eccleston GM, J. Pharm. Sci 2008, 97, 2892. [DOI] [PubMed] [Google Scholar]
- [17].a) Kumbar S, James R, Nukavarapu S, Laurencin C, Biomed. Mater 2008, 3, 034002; [DOI] [PubMed] [Google Scholar]; b) Sell SA, Wolfe PS, Garg K, McCool JM, Rodriguez IA, Bowlin GL, Polymers 2010, 2, 522. [Google Scholar]
- [18].Place ES, George JH, Williams CK, Stevens MM, Chem. Soc. Rev 2009, 38, 1139. [DOI] [PubMed] [Google Scholar]
- [19].a) Peng YY, Glattauer V, Ramshaw JA, Werkmeister JA, J. Biomed. Mater. Res., Part A 2010, 93, 1235; [DOI] [PubMed] [Google Scholar]; b) Liu W, Burdick JA, van Osch GJ, Tissue Eng., Part A 2013, 19, 1489. [DOI] [PubMed] [Google Scholar]
- [20].Zhong S, Zhang Y, Lim C, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol 2010, 2, 510. [DOI] [PubMed] [Google Scholar]
- [21].a) Thirugnanaselvam M, Gobi N, Karthick SA, Fibers Polym 2013, 14, 965 [Google Scholar]; b) Curt S, Subirade M, Rouabhia M, Tissue Eng., Part A 2008, 15, 1223. [DOI] [PubMed] [Google Scholar]
- [22].Li WJ, Laurencin CT, Caterson EJ, Tuan RS, Ko FK, J. Biomed. Mater. Res., Part A 2002, 60, 613. [DOI] [PubMed] [Google Scholar]
- [23].a) Sinha-Ray S, Zhang Y, Yarin A, Davis S, Pourdeyhimi B, Biomacromolecules 2011, 12, 2357; [DOI] [PubMed] [Google Scholar]; b) Xu X, Jiang L, Zhou Z, Wu X, Wang Y, ACS Appl. Mater. Interfaces 2012, 4, 4331; [DOI] [PubMed] [Google Scholar]; c) Sett S, Lee MW, Weith M, Pourdeyhimi B, Yarin A, J. Mater. Chem. B 2015, 3, 2147. [DOI] [PubMed] [Google Scholar]
- [24].a) Lee KY, Jeong L, Kang YO, Lee SJ, Park WH, Adv. Drug Delivery Rev 2009, 61, 1020; [DOI] [PubMed] [Google Scholar]; b) Miao J, Pangule RC, Paskaleva EE, Hwang EE, Kane RS, Linhardt RJ, Dordick JS, Biomaterials 2011, 32, 9557; [DOI] [PubMed] [Google Scholar]; c) Rodríguez K, Gatenholm P, Renneckar S, Cellulose 2012, 19, 1583; [Google Scholar]; d) Huang R, Li W, Lv X, Lei Z, Bian Y, Deng H, Wang H, Li J, Li X, Biomaterials 2015, 53, 58. [DOI] [PubMed] [Google Scholar]
- [25].a) Badrossamay MR, McIlwee HA, Goss JA, Parker KK, Nano Lett 2010, 10, 2257; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Badrossamay MR, Balachandran K, Capulli AK, Golecki HM, Agarwal A, Goss JA, Kim H, Shin K, Parker KK, Biomaterials 2014, 35, 3188. [DOI] [PubMed] [Google Scholar]
- [26].a) Mellado P, McIlwee HA, Badrossamay MR, Goss JA, Mahadevan L, Parker KK, Appl. Phys. Lett 2011, 99, 203107; [Google Scholar]; b) Golecki HM, Yuan H, Glavin C, Potter B, Badrossamay MR, Goss JA, Phillips MD, Parker KK, Langmuir 2014, 30, 13369; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gonzalez GM, MacQueen LA, Lind JU, Fitzgibbons SA, Chantre CO, Huggler I, Golecki HM, Goss JA, Parker KK, Macromol. Mater. Eng 2017, 302; [Google Scholar]; d) Capulli AK, Emmert MY, Pasqualini FS, Kehl D, Caliskan E, Lind JU, Sheehy SP, Park SJ, Ahn S, Weber B, Biomaterials 2017, 133, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Achouri A, Zhang W, Shiying X, Food Res. Int 1998, 31, 617. [Google Scholar]
- [28].Pham QP, Sharma U, Mikos AG, Tissue Eng 2006, 12, 1197. [DOI] [PubMed] [Google Scholar]
- [29].a) Hodgkinson T, Yuan X-F Bayat A, J. Tissue Eng 2014, 5, 2041731414551661; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lowery JL, Datta N, Rutledge GC, Biomaterials 2010, 31, 491; [DOI] [PubMed] [Google Scholar]; c) Huang Z-M, Zhang Y-Z Kotaki M, Ramakrishna S, Compos. Sci. Technol 2003, 63, 2223. [Google Scholar]
- [30].a) Discher DE, Janmey P, Wang Y.-l., Science 2005, 310, 1139; [DOI] [PubMed] [Google Scholar]; b) Wells RG, Hepatology 2008, 47, 1394. [DOI] [PubMed] [Google Scholar]
- [31].a) Agache P, Monneur C, Leveque J, De Rigal J, Arch. Dermatol. Res 1980, 269, 221; [DOI] [PubMed] [Google Scholar]; b) Liang X, Boppart SA, IEEE Trans. Biomed. Eng 2010, 57, 953; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kuwazuru O, Saothong J, Yoshikawa N, Med. Eng. Phys 2008, 30, 516; [DOI] [PubMed] [Google Scholar]; d) Pailler-Mattei C, Bec S, Zahouani H, Med. Eng. Phys 2008, 30, 599; [DOI] [PubMed] [Google Scholar]; e) Pan J.-f., Liu N.-h., Sun H, Xu F, PLoS One 2014, 9, e112885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].a) Chung T-W, Liu D-Z, Wang S-Y, Wang S-S, Biomaterials 2003, 24, 4655; [DOI] [PubMed] [Google Scholar]; b) Xu C, Yang F, Wang S, Ramakrishna S, J. Biomed. Mater. Res., Part A 2004, 71, 154. [DOI] [PubMed] [Google Scholar]
- [33].Jin G, Prabhakaran MP, Kai D, Annamalai SK, Arunachalam KD, Ramakrishna S, Biomaterials 2013, 34, 724. [DOI] [PubMed] [Google Scholar]
- [34].a) Hutmacher DW, Biomaterials 2000, 21, 2529; [DOI] [PubMed] [Google Scholar]; b) Xue J, He M, Liu H, Niu Y Crawford A, Coates PD, Chen D, Shi R, Zhang L, Biomaterials 2014, 35, 9395. [DOI] [PubMed] [Google Scholar]
- [35].Hu J, Prabhakaran MP, Tian L, Ding X, Ramakrishna S, RSC Adv 2015, 5, 100256. [Google Scholar]
- [36].a) Eckes B, Zigrino P, Kessler D, Holtkötter O, Shephard P, Mauch C, Krieg T, Matrix Biol 2000, 19, 325; [DOI] [PubMed] [Google Scholar]; b) Gurtner GC, Werner S, Barrandon Y, Longaker MT, Nature 2008, 453, 314; [DOI] [PubMed] [Google Scholar]; c) Ekaputra AK, Prestwich GD, Cool SM, Hutmacher DW, Biomaterials 2011, 32, 8108; [DOI] [PubMed] [Google Scholar]; d) Jiang J, Li Z, Wang H, Wang Y, Carlson MA, Teusink MJ, MacEwan MR, Gu L, Xie J, Adv. Healthcare Mater 2016, 5, 2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].a) Grinnell F, Trends Cell Biol 2000, 10, 362; [DOI] [PubMed] [Google Scholar]; b) Sorrell JM, Caplan AI, J. Cell Sci 2004, 117, 667; [DOI] [PubMed] [Google Scholar]; c) Sriram G, Bigliardi PL, Bigliardi-Qi M, Eur. J. Cell Biol 2015, 94, 483. [DOI] [PubMed] [Google Scholar]
- [38].Chan FK-M, Moriwaki K, De Rosa MJ, Methods Mol. Biol 2013, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Scholzen T, Gerdes J, J. Cell. Physiol 2000, 182, 311. [DOI] [PubMed] [Google Scholar]
- [40].a) Liu S, Shi-Wen X, Blumbach K, Eastwood M, Denton CP, Eckes B, Krieg T, Abraham DJ, Leask A, J. Cell Sci 2010, 123, 3674; [DOI] [PubMed] [Google Scholar]; b) Huang C, Fu X, Liu J, Qi Y, Li S, Wang H, Biomaterials 2012, 33, 1791; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Grose R, Hutter C, Bloch W, Thorey I, Watt FM, Fässler R, Brakebusch C, Werner S, Development 2002, 129, 2303; [DOI] [PubMed] [Google Scholar]; d) Li J, Chen J, Kirsner R, Clin. Dermatol 2007, 25, 9. [DOI] [PubMed] [Google Scholar]
- [41].Margadant C, Sonnenberg A, EMBO Rep 2010, 11, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Galiano RD, Michaels V, Dobryansky M, Levine JP, Gurtner GC, Wound Repair Regen 2004, 12, 485. [DOI] [PubMed] [Google Scholar]
- [43].a) Wang X, Ge J, Tredget EE, Wu Y, Nat. Protoc 2013, 8, 302; [DOI] [PubMed] [Google Scholar]; b) Griffin DR, Weaver WM, Scumpia PO, Di Carlo D, Segura T, Nat. Mater 2015, 14, 737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Khorasani H, Zheng Z, Nguyen C, Zara J, Zhang X, Wang J, Ting K, Soo C, Am. J. Pathol 2011, 178, 621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].a) Marini H, Polito F, Altavilla D, Irrera N, Minutoli L, Calo M, Adamo E, Vaccaro M, Squadrito F, Bitto A, Br. J. Pharmacol 2010, 160, 1185; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Park E, Lee SM, Jung I-K, Lim Y, Kim J-H, Biochem. Biophys. Res. Commun 2011, 410, 514; [DOI] [PubMed] [Google Scholar]; c) Tie L, An Y, Han J, Xiao Y, Xiaokaiti Y, Fan S, Liu S, Chen AF, Li X, J. Nutr. Biochem 2013, 24, 88. [DOI] [PubMed] [Google Scholar]
- [46].a) Ma K, Liao S, He L, Lu J, Ramakrishna S, Chan CK, Tissue Eng., Part A 2011, 17, 1413; [DOI] [PubMed] [Google Scholar]; b) Gil ES, Panilaitis B, Bellas E, Kaplan DL, Adv. Healthcare Mater 2013, 2, 206; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Anjum F, Agabalyan NA, Sparks HD, Rosin NL, Kallos MS, Biernaskie J, Sci. Rep 2017, 7, 10291; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Levengood SL, Erickson AE, Chang FC, Zhang M, J. Mater. Chem. B 2017, 5, 1822; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Xu HL, Chen PP, ZhuGe DL, Zhu QY, Jin BH, Shen BX, Xiao J, Zhao YZ, Adv. Healthcare Mater 2017, 6, 1700344. [DOI] [PubMed] [Google Scholar]
- [47].Hotaling NA, Bharti K, Kriel H, Simon CG, Biomaterials 2015, 61, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Deravi LF, Su T, Paten JA, Ruberti JW, Bertoldi K, Parker KK, Nano Lett 2012, 12, 5587. [DOI] [PubMed] [Google Scholar]
- [49].Stalder A, Kulik G, Sage D, Barbieri L, Hoffmann P, Colloids Surf., A 2006, 286, 92. [Google Scholar]
- [50].Liu X, Lin T, Gao Y, Xu Z, Huang C, Yao G, Jiang L, Tang Y, Wang X, J. Biomed. Mater. Res., Part B 2012, 100, 1556. [DOI] [PubMed] [Google Scholar]
- [51].Lin J, Li C, Zhao Y, Hu J, Zhang L-M, ACS Appl. Mater. Interfaces 2012, 4, 1050. [DOI] [PubMed] [Google Scholar]
- [52].Thevenaz P, Ruttimann UE, Unser M, IEEE Trans. Image Process 1998, 7, 27. [DOI] [PubMed] [Google Scholar]
- [53].Klopfenstein DR, Vale RD, Mol. Biol. Cell 2004, 15, 3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Rezakhaniha R, Agianniotis A, Schrauwen JTC, Griffa A, Sage D, Bouten C. v., Van de Vosse F, Unser M, Stergiopulos N, Biomech. Model. Mechanobiol 2012, 11, 461. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.