

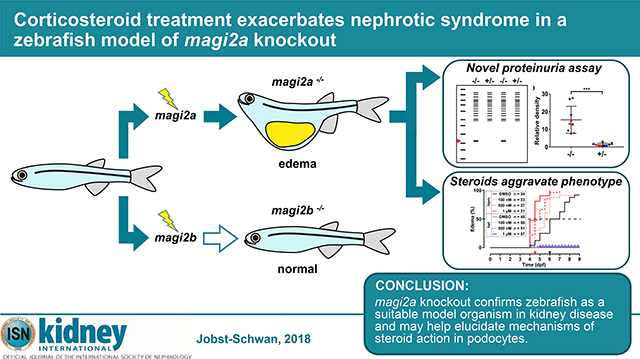
Abstract
Recently, recessive mutations of MAGI2 were identified as a cause of steroid resistant nephrotic syndrome (SRNS) in humans and mice. To further delineate the pathogenesis of MAGI2 loss-of-function, we generated stable knockout lines for the two zebrafish orthologues magi2a and magi2b by CRISPR/Cas9. We also developed a novel assay for the direct detection of proteinuria in zebrafish independent of transgenic background. Whereas knockout of magi2b did not yield a nephrotic syndrome phenotype, magi2a−/− larvae developed ascites, periorbital edema, and proteinuria, as indicated by increased excretion of low molecular weight protein. Electron microscopy demonstrated extensive podocyte foot process effacement. As in human SRNS, we observed genotype/ phenotype correlation, with edema onset occurring earlier in zebrafish with truncating alleles (5–6 days post fertilization) versus hypomorphic alleles (19–20 days post fertilization). Paradoxically, corticosteroid treatment exacerbated the phenotype, with earlier onset of edema. In contrast, treatment with cyclosporine A or tacrolimus had no significant effect. Although RhoA signaling has been implicated as a downstream mediator of MAGI2 activity, targeting of the RhoA pathway did not modify the nephrotic syndrome phenotype. In the first CRISPR/Cas9 zebrafish knockout model of SRNS, we found that corticosteroids may have a paradoxical effect in the setting of specific genetic mutations.
Keywords: kidney development, proteinuria, podocyte, chronic kidney disease, focal segmental glomerulosclerosis
Graphical Abstract
INTRODUCTION
Nephrotic syndrome (NS) is the second most frequent cause of chronic kidney disease in the first 3 decades of life, requiring dialysis or transplantation for survival.1 Based on its response to steroid treatment, steroid sensitive NS (SSNS, ~80% of cases) is distinguished from steroid resistant NS (SRNS, ~20% of cases).
Recently, recessive mutations in the MAGI2 gene have been identified as a cause of SRNS in human patients.6, 7 MAGI2 is a member of the membrane-associated guanylate kinase family of scaffolding proteins, important in neurologic functioning.8, 9 MAGI proteins are involved in tethering of cell surface receptors to the cytoskeleton. In podocyte foot processes, MAGI2 belongs to the nephrin multi-protein complex, where it interacts with nephrin, spectrins and alpha-actinin to build up the glomerular slit diaphragm.10 Moreover, MAGI2 has been associated with actin cytoskeleton regulation in podocytes via RhoA control and signaling.6, 7, 11, 12
Magi2-deleted mice display podocyte foot process effacement and severe proteinuria with early death from end-stage renal failure.16–18 We previously discovered recessive MAGI2 mutations as causing SRNS in humans, and replicated that regulation of RhoA activity omits pathogenesis.6 To further characterize disease mechanisms of SRNS in MAGI2 loss-of-function, we generated stable CRISPR/Cas9-mediated zebrafish knockout lines (KO). Similar to all teleosts, the zebrafish genome features gene duplications,30, 31and we targeted both zebrafish orthologues, magi2a and magi2b, that are annotated for the human MAGI2 gene. Expression of magi2 without specific differentiation between magi2a and magi2b in pronephric zebrafish glomeruli has been shown previously by whole mount in situ hybridization (WISH), and morpholino knockdown (KD) of magi2 altered pronephros morphology.11 We show that loss-of-function of magi2a, but not magi2b, recapitulates human NS in zebrafish. Additionally, by drug-treatment studies we find that steroid treatment paradoxically exacerbates rather than mitigates the nephrotic phenotype. Taken together, we present the first genomic disease model for SRNS in zebrafish by magi2a KO, and provide the first evidence for an ambivalent effect of steroids on podocyte pathology.
RESULTS
KO of magi2a, but not magi2b causes nephrotic syndrome and reduced survival in an allele dependent manner
A nephrotic phenotype in zebrafish has been described as periorbital edema, ascites or a combination of both in the presence of proteinuria.26, 32, 33 Since two zebrafish orthologues for human MAGI2 exist, magi2a and magi2b (Ensembl Genome Browser, Suppl. Fig S1 A-C), we generated CRISPR/Cas9-mediated KO larvae for magi2a and magi2b to determine the relevant zebrafish orthologue for human MAGI2 in podocyte function.
Larvae were observed for onset of edema and survival until 21 days post fertilization (dpf). Neither magi2b+/− nor magi2b−/− larvae developed periorbital edema or ascites until 21 dpf (Suppl. Fig. S2 A-D). Additionally, survival after 21 days was not impaired (Suppl. Fig. S2 E and F). Consistent with these results and in contrast to magi2a (Suppl. Fig. S3 A-C), WISH of magi2b did not show any glomerular expression at 4 dpf and 6 dpf (Suppl. Fig. S3 D-F). Both findings suggest that magi2b is dispensable in glomerular development.
Four different magi2a hypothetical null or hypomorphic alleles were created (Suppl. Table S1, S2 and Suppl. Fig. S4). Immunoblotting showed a strong reduction of a band at 140 kDa compatible with Magi2a in magi2a−/− larvae compared to magi2a+/+ or magi2a+/− controls (Suppl. Fig. S1 E-G). However, the peptide derived from human MAGI2 that was used for the generation of the antibody shows high similarity with the according amino acid sequence of not only zebrafish Magi2a, but also Magi2b and other paralogues, resulting in unspecific bands (Suppl. Fig. S1 H).
We performed qPCR at 6 dpf for the three magi2a alleles (magi2acl604, magi2acl605, magi2acl607) that would potentially undergo nonsense-mediated decay (NMD) but did not observe a reduction in magi2a transcript levels for neither of the alleles (Suppl. Fig. S5 A-C). In contrast, the allele magi2acl607 showed a significant increase compared to wildtype.
WISH in magi2a−/− larvae (magi2acl604, Suppl. Fig. S5 E) revealed a pronephric expression pattern unchanged from magi2a+/− control (Suppl. Fig. S5 F). From these findings, we conclude that NMD does not play a role in magi2a pathogenesis in our zebrafish model.
Macroscopically, magi2a−/− larvae, but not heterozygous magi2a+/− controls, developed a distinct edema phenotype with ascites and periorbital edema (Fig. 1 A-B).
Figure 1. Mutations in magi2a cause a distinct edema phenotype with proteinuria in zebrafish.

(A) No ascites (arrow) was observed in magi2a+/− control larvae by 21 dpf.
(B) In contrast, magi2a−/− zebrafish larvae presented with an edema phenotype of ascites (arrow) and periorbital edema with median onset between 5 and 21 dpf, depending on allele. Scale bars, 1 mm.
(C) At 6 dpf, larvae from magi2a c.69_71delinsGCTA, p.Pro24Leufs*76 were selected by phenotype and housed individually in 60 μl fish water for 24 h. Individual water samples were preserved, and the larvae were genotyped individually. 6 samples from larvae with the same genotype were pooled, concentrated by acetone precipitation, and 3 pooled samples each for magi2a−/− and magi2a+/− were run on SDS-PAGE. Fish water only (FW) served as negative control. The gel was stained by ultrasensitive DBF-staining that detects 0.025 ng protein per band. The bands at ~22 and ~24 kDa were observed only in magi2a−/− samples. Note that the bands >40 kDa that appear in all lanes including negative control are most likely derived from skin cells or normal bacterial colonization of the environment as they are present in the negative control (FW).
(D) Densitometry was performed for the low molecular weight band at ~22 kDa and normalized by the negative control and defined as relative density. Results for the relative density from 3 independent experiments are shown together, each individual experiment in a different color. magi2a−/− larvae show a significantly higher excretion of low molecular protein than magi2a+/− larvae (P<0.0001, two-tailed unpaired t-test).
magi2a−/− larvae exhibited proteinuria as determined by ultra-sensitive DBF-staining showing significantly higher excretion of low molecular weight protein (~22 kDa) by magi2a−/− larvae compared to magi2a+/− larvae (Fig. 1 C-D).
Kaplan-Meier plots for edema onset showed an early edema phenotype in larvae homozygous for two hypothetical magi2a null alleles (Suppl. Fig. S4 A-B) c.69_71delinsGCTA, p.Pro24Leufs*76, (magi2acl604, Fig. 2 A) and c.54_71delinsA, p.Val19Glufs*75, (magi2acl605, Fig. 2 B) with median onset of edema at 7.5 and 6 dpf, respectively. In contrast, the hypomorphic alleles (Suppl. Fig. S4 C, E) c.64_70delinsG, p.Arg22_Pro24delinsAla (magi2acl606, Fig. 2 C), and c.61_73delinsGGG, p.Ser2_Met218del (magi2acl607, Fig. 2 D) caused median onset of edema at 19 and 20 dpf, respectively. Additionally, survival was impaired for both null allele larvae that presented with an early onset edema with survival rates of 30–53% at 21 dpf. In contrast, the two hypomorphic alleles show survival rates of 86–92% at 21 dpf (Fig 2 E-H), consistent with late edema onset. These results demonstrate an allele specific effect on onset of phenotype and survival.
Figure 2. Edema phenotype and reduced survival caused by mutations in magi2a are allele dependent.

Specific alleles are indicated above panels. Numbers of fish with each genotype within each clutch are indicated for wildtype (wt), heterozygous (het) and homozygous (hom) (color coded), and were compatible with Mendelian ratios. Zebrafish larvae were monitored daily for 21 consecutive days, separated when presenting with edema, and observed further, until determination of the magi2a genotype at 21 dpf.
(A-D) Kaplan-Meier plot for onset of edema for magi2a−/− zebrafish larvae. Larvae carrying either one of the null alleles c.69_71delinsGCTA, p.Pro24Leufs*76 (A) or magi2a c.54_71delinsA, p.Val19Glufs*75 (B) show an early edema phenotype with 50% of edema at 7.5 dpf and 6 dpf respectively (red arrow heads on x-axis). In contrast, for two hypomorphic alleles c.64_70delinsG, p.Arg22_Pro24delinsAla (C) and c.61_73delinsGGG, p.Ser2_Met218del (D), 50% of the larvae develop edema only at 19 dpf and 20 dpf, respectively (red arrow heads on x-axis). Note that onset of the edema phenotype was allele dependent, null vs. hypomorphic.
(E-H) Kaplan-Meier plots for survival of magi2a−/− larvae. Larvae carrying either one of the null alleles showed reduced survival (median survival of 14 dpf (E) and 51% survival at the end of the experiment (F), red arrow heads on x-axis). In contrast, survival was only slightly impaired for larvae carrying the hypomorphic alleles. Therefore, a median survival point could not be determined during the observation period. At 21 dpf, 92% of larvae survived for c.64_70delinsG, p.Arg22_Pro24delinsAla (G) compared to 86% for c.61_73delinsGGG, p.Ser2_Met218del (H). Note that alleles resulting in early onset 741 edema correlate with a reduced survival of magi2a−/− larvae at 21 dpf. Numbers of 742 included larvae and color coding are indicated.
Loss of magi2a results in podocyte foot process effacement and lack of glomerular fusion
To examine the histological correlates of the observed edema phenotypes, magi2a+/− and magi2a−/− larvae were processed for light and transmission electron microscopy (TEM) at 21 dpf for the allele magi2acl604. TEM revealed normal podocyte morphology with rhythmic arrangements of distinct podocyte foot processes in magi2a+/− control larvae (Fig. 3 A, C). In contrast, magi2a−/− larvae showed long segments of foot process effacement (Fig. 3 B, D) and capillary dilation (Fig. 3 D, asterisks). H&E in paraffin sections and toluidine blue staining in resin sections showed a morphologically normal glomerulus in magi2a+/− larvae (Fig. 3 E, E’) compared to an increased size of the glomerulus and fusion defects of pronephric glomeruli in magi2a−/− (Fig. 3 F, F’). Quantitation of glomerular surface area and number of nuclei showed an increased glomerular surface area and reduced cell count for magi2a−/−, suggesting either hypertrophy or expansion of the glomerular capillaries rather than hyperproliferation of cells (Fig. 3 G,H).Thus, TEM and light microscopy in magi2a−/− showed abnormal morphology of podocytes and glomeruli, indicating that loss of magi2a severely affects podocyte and glomerular structure and function.
Figure 3. Loss of magi2a results in podocyte foot process effacement and increased sectional glomerular surface area.

Pronephric glomeruli of magi2a+/− and magi2a−/− (c.70_72delinsGTCC, p.Pro24Valfs*76) larvae were analyzed for morphology by light and transmission electron microscopy (TEM) at 21 dpf. Normally, zebrafish larvae develop a bilateral pair of pronephric glomeruli that fuse in the midline by 2 dpf with fully functional slit diaphragms at 4 dpf 20, 67
(A-B) Evidence of podocyte foot process effacement in magi2−/− larvae. High magnification reveals normal podocyte morphology with rhythmic arrangements of distinct podocyte foot processes in magi2a+/− (A, arrows). Note the long-segment podocyte foot process effacement in magi2a−/− larvae (B, arrows). Arrowheads mark the fenestrated endothelium in both E and F. Scale bars, 500 nm.
(C-D) Low magnification in TEM shows normal glomerular morphology in magi2a+/− larvae (C) whereas magi2a−/− larva exhibit capillary dilatation (D, asterisks). Scale bars, 10 μM.
(E-F) Light microscopy shows increased glomerular surface and fusion defects of the bilateral glomeruli in magi2a−/− larvae. H&E in paraffin sections (E) and toluidine blue in resin sections (E’) staining show normal glomerular morphology in representative glomeruli of magi2a+/− larvae, whereas magi2a−/− clutch mates exhibit increased glomerular surface and fusion defects of the bilateral glomeruli in (F, H&E in paraffin sections; F’, toluidine blue in resin sections). Note the dark pigment that is part of the normal larval anatomy. Scale bars, 100 μM.
(G) Quantitation of glomerular surface area of magi2a+/− and magi2a−/− larval glomeruli, show an increase in surface area for magi2a−/− glomeruli (n=11) compared to glomeruli of magi2a+/− larvae (n=8) (P = 0.0002).
(H) Quantitation of nuclei per 1,000 μm2 glomerular surface, for magi2a+/− and magi2a−/− larvae. Number of nuclei per surface area of glomeruli of magi2a−/− larvae is significantly lower compared to glomeruli of magi2a+/− larvae (n = 11 for magi2a−/− and n = 8 for magi2a+/−). P = 0.0002, represented as ***.
We conclude that KO of magi2a results in edema, impaired survival, foot process effacement of the podocytes and proteinuria. Our model thus recapitulates phenotypes and morphological changes of the podocyte similar to human NS patients.6, 7 We therefore used our magi2a−/− zebrafish model to evaluate the effect of drugs that are commonly used in human NS (Dexamethasone, Prednisolone, Cyclosporine A and Tacrolimus) for their effect on edema of magi2a−/− mutant fish (Fig. 4, Suppl. Fig. S6–S8).
Figure 4. Steroid treatment exacerbates the edema phenotype in magi2a−/− larvae.

At 48 hpf embryos were mechanically dechorionated and treated with different concentrations of dexamethasone, prednisolone, tacrolimus, RhoA activator II, lysophosphatidic acid, ROCK inhibitor or vehicle control (0.1% DMSO/water/PBS). Larvae were monitored (twice) daily for nine consecutive days (eight days for tacrolimus). Larvae were genotyped individually by Sanger sequencing at the end of the experiment. The graphs show Kaplan-Meier curves for onset of edema with magi2a−/− groups in red (vehicle control in black), magi2a+/− groups in blue (vehicle control in brown). The drug concentrations are depicted by lines from dotted over dashed to solid in order of increasing concentrations. Drug concentrations and numbers of larvae per treatment group are indicated in the legends. Statistical analysis was performed by Log-rank (Mantel-Cox) test. Age of median onset of edema is indicated as black (control) and red (highest treatment dose) arrow heads on the x-axis.
(A) The observed phenotype in magi2a−/− larvae at 9 dpf was ascites (arrows) and periorbital edema, defined as a clear, fluid filled space around the pigmented epithelium of the eye, arrowheads. The c.70_72delinsGTCC, p.Pro24Valfs*76 allele was used for all experiments displayed in this figure. Scale bar, 0.5 mm.
(B) Increasing doses of dexamethasone cause a significantly earlier onset of the edema phenotype in magi2a−/− larvae (median onset 5.5 dpf [100nM]), 5 dpf [500nM], 4.5 dpf [1 μM]) compared to DMSO control (6 dpf), P<0.0001.
(C) Increasing doses of prednisolone cause a significantly earlier onset of the edema phenotype in magi2a−/− (median onset 4 dpf for all concentrations) compared to DMSO control (median onset 5 dpf), P<0.0001.
(D) Increasing doses of tacrolimus do not cause a significantly earlier onset of the edema phenotype in magi2a−/− (median onset 5 dpf [200 nM], 4.75 dpf [500 nM], 5 dpf [1 μM]) compared to DMSO control (median onset 4.5 dpf), P=0.391. Note that 10% of magi2a+/− larvae in the highest concentration develop edema as well.
(E) Increasing doses of RhoA activator II do not cause a significantly earlier onset of the edema phenotype in magi2a−/− (median onset 5 dpf for all concentrations), compared to water control (median onset 5 dpf), (P= 0.8516). The curves of control and treatment groups for heterozygotes coincide without any appearance of edema, and therefore overlap in one single line.
(F) Increasing doses of lysophosphatidic acid do not cause a significantly earlier onset of the edema phenotype in magi2a−/− (median onset 5 dpf for all concentrations, but 7dpf for 1 μM) compared to PBS control (median onset 5 dpf), (P=0.4634).
(G) Increasing doses of ROCK inhibitor Y-27632 do not cause a significantly earlier onset of the edema phenotype in magi2a−/− (median onset 4.5 dpf for all concentrations) compared to PBS control (median onset 5 dpf), (P=0.5933).
Steroids, but not Calcineurin inhibitors exacerbate the edema phenotype in magi2a−/− larvae.
For all treatment experiments, clutches from magi2a+/− parents were treated from 2 dpf to 9 dpf with increasing drug doses. Treatment was initiated at 2 dpf to avoid drug effects on structural brain development.34 All larvae were genotyped at the end of each experiment. Genotypes observed for all experiments were compatible with Mendelian distribution. None of the drugs showed a significant influence on the edema phenotype of magi2a+/− negative controls (Fig. 4).
Application of increasing doses of Dexamethasone exacerbated, rather than mitigated the edema phenotype in magi2a−/− larvae (Fig. 4 B, Suppl. Fig. S6). For DMSO control median onset of the edema was at 6 dpf, and for Dexamethasone at 5.5 dpf for 100 nM, 5 dpf for 500 nM, and 4.5 dpf for 1 μM. Also, treatment with 10 μM, 50 μM, or 100 μM prednisolone significantly expedited edema onset to 4 dpf compared to 5 dpf for DMSO control (Fig. 4 C and Suppl. Fig. S7). Both, Tacrolimus and Cyclosporine A did not significantly influence edema onset (Fig. 4 D, and Suppl. Fig. S8). These results show that treatment with glucocorticosteroids paradoxically accelerates the development of a NS phenotype in magi2a−/− mutant fish.
Pharmacological targeting of RhoA activation does not modify the edema phenotype in magi2a−/− larvae.
We showed previously that KD of MAGI2 protein in HEK293 cells causes a reduction in RhoA activation whereas MAGI2 overexpression increases active RhoA.6
We therefore hypothesized that increasing RhoA activity would ameliorate the phenotype in magi2a−/− zebrafish larvae, whereas reduction of RhoA activity would exacerbate the phenotype.
We aimed to asses RhoA levels in magi2a KO larvae by a GLISA assay as described before in cell lines.6 However, the assay did not show any signal for the control larvae that was different from background control, most likely due to lack of cross reactivity of the kit antibody to zebrafish RhoA.
To increase RhoA activity, larvae were treated with the direct RhoA activator II (Fig. 4 E). Concentrations were chosen based on previous usage in zebrafish.35 Neither of the three concentrations applied (1.25 μg/ml, 2.5 μg/ml, 5 μg/ml) changed the course of edema development significantly compared to control (P=0.8516). Similar results were obtained for the indirect RhoA activator lysophosphatidic acid (Fig. 4 F) without effect for treatment with 10nM, 100nM, 1μM or 10uM (P=0.4925).
We then aimed to reduce RhoA activity to exacerbate the edema phenotype. The ROCK inhibitor Y-27632 acts downstream of RhoA, and has been employed previously to inhibit RhoA effects on actin rearrangements, mimic reduction of active RhoA.36 Yet, treatment with Y-27632 at 10 μM or 100 μM (Fig. 4 G) did not have any significant effect on edema development (P=0.5933). Taken together, modulation of RhoA activity does not affect the progression of the edema phenotype in the magi2a−/− zebrafish model.
DISCUSSION
Here, we established stable KO lines for magi2a as the first monogenic disease model for SRNS in zebrafish. We show that KO of magi2a results in allele-dependent onset of characteristic edema phenotypes with ascites and periorbital edema, resulting in impaired survival. magi2a KO larvae exhibit proteinuria as shown by a novel proteinuria assay in zebrafish larvae. The microscopic kidney phenotype is characterized by increased glomerular size, and total podocyte foot process effacement, partially recapitulating the mouse and human NS phenotypes.7, 16–18 Surprisingly, treatment of magi2a KO larvae with steroids paradoxically exacerbates the NS phenotype. Treatment with calcineurin inhibitors and drugs that modulate RhoA activity did not alter the severity of the phenotype.
KO of magi2b does not cause a noticeable pronephric phenotype, confirming that magi2a is likely the only functional zebrafish orthologue of human MAGI2 in the pronephros. These findings are consistent with MO experiments targeting magi2a as published previously,11 and are supported by the lack of pronephric magi2b expression (Suppl. Fig. S3 D-F). Both genes are co-expressed in the brain, but neither magi2a KO nor magi2b KO leads to a significant neuronal phenotype, suggesting a mutual compensation in the brain.
Different mutations in magi2a lead to different latency to onset of disease. As predicted, early truncating mutations cause a more severe and earlier phenotype as an inframe deletion (Fig. 2) as has been described for monogenic disease genes in humans.37 magi2a−/− null alleles resulting in early termination of proteins (magi2acl604, magi2acl605; Suppl. Fig. S4 A-B) with a transcriptional stop in Exon 2, developed an early edema phenotype, accompanied with impaired survival. In contrast, larvae carrying the hypomorphic alleles magi2acl607 and magi2acl606 showed delayed edema onset with slightly impaired survival until the end of the observation period (Fig. 2).
The allele magi2acl607 leads to an early stop gain in Exon 1. New stop codons in any exon from exon 2 to the second to last exon would typically lead to NMD, whereas newly introduced stop codons in exon 1 may escape NMD and initiate an alternative translation initiation site (TIS) at a canonical or even a non-canonical start codon.38, 39 For magi2acl607, three different prediction programs predicted an alternative TIS at the ATG 642–644 with the protein change p.Ser2_Met218del.40–42 This results in an N-terminally truncated protein (Suppl. Fig. S4 D), bearing the PDZ0 and the GK domain. The milder course of disease in magi2acl607 is comparable to the in-frame mutation magi2acl606and therefore supports the hypothesis of an N-terminally truncated protein. Additionally, qPCR for magi2acl607 showed a significant increase of magi2a transcript in homozygous larvae compared to wildtype which may be a compensatory upregulation of a partially functional protein.43
In contrast, the alleles magi2acl604 and magi2acl605 would be subject to NMD and can be considered as null alleles (Suppl. Fig. S4 A-B).
However, qPCR for magi2a for these two magi2a alleles, and WISH of magi2−/− larvae revealed normal mRNA expression for either of the alleles (Suppl. Fig. S5), suggesting translational repression rather than NMD. Escape from NMD with translational repression has been described for truncating mutations where the newly introduced stop is not located within the first or last exon presenting with normal or elevated transcript levels, but absence of any protein.44 Despite the lack of reduced expression in the magi2a−/− larvae (Suppl. Fig. S1 and S5), the clear genotype phenotype correlation for all magi2a alleles (Fig. 2) strongly supports our interpretation.
Moreover, both non-truncating mutations suggest an important role of the PDZ0 domain for magi2a function in the zebrafish podocyte (Suppl. Fig. S1). However, in contrast to the remaining structural domains of MAGI2, no specific binding proteins have been described for the affected PDZ0 (Suppl. Fig S9).45
Proteinuria is a symptom of nephrotic syndrome. Direct proteinuria detection has been established in transgenic zebrafish that express vitamin D binding protein-GFP in the serum with quantitation by GFP-ELISA in water of pooled zebrafish larvae.26, 28, 33 Our magi2a KO models were generated in this line, but we were not able to detect a significant GFP excretion. Other indirect methods (retinal plexus fluorescence intensity, intravascular fluorescence) of proteinuria quantification are published based on this transgenic line.46 However they are highly depending on the (variable) expression of the transgene and lack a direct proof of protein excretion.46 Here, we present a novel proteinuria assay in zebrafish by DBF-staining in SDS-PAGE gels that is independent of a transgenic background (Fig. 1 C).47 Surprisingly, the differential band at 25 kDa that distinguishes between homozygous KO and heterozygous controls represents a low molecular weight protein, which stands in contrast to glomerular high molecular weight proteinuria in humans and mice.48 This fact might be related to the different physiological requirements in an aqueous environment, but the mechanisms remain unclear to date.
Several studies highlighted the importance of these small GTPases, including RhoA, Rac1 and Cdc42 for podocyte physiology, and imply a dysregulation of these being associated with the pathogenesis of monogenic SRNS.33, 51–53
Our previous findings identified mutations in the genes MAGI2, TNS2, DLC1 in patient with NS and showed interaction of the gene products in a pathway that regulates RhoA activity with MAGI2 acting upstream of the other proteins.6 However, pharmacological modulation of the RhoA pathway does not affect the course of disease in our zebrafish model. Findings from other groups implicate a role of dendrin in the pathogenesis of MAGI2 KO.16 Human mutations in MAGI2 were found to cause more severe phenotype with SRNS,7 whereas mutations in TNS2 and DLC1 caused a less severe phenotype with partially steroid sensitive NS.6 Taken together, these finding potentially implicate MAGI2 in different regulatory pathways where mitigation of the defect in one of the pathways may not be sufficient to rescue a severe phenotype.
The response to the initial steroid treatment of NS patients determines the disease entity of SSNS or SRNS.54, 55 As indicated above, patients with MAGI2 mutations do not respond to treatment attempts with steroids. However, we showed that patients with TNS2 and DLC1 mutations, which are both downstream of MAGI2 in the RhoA pathway, do respond to steroids.6 Therefore, we hypothesized that steroids might positively influence disease outcome in magi2a KO zebrafish. Surprisingly, we observed the opposite effect; steroid treatment of magi2a KO larvae paradoxically exacerbated the phenotype, by inducing earlier onset of edema (Fig. 4). Applied concentrations were based on previous applications in zebrafish,34, 56–58 and do not induce an obvious phenotypic effect in heterozygous and wild type clutch mates, suggesting a steroid specific effect. This assumption is supported by the fact that application of calcineurin inhibitors, another class of drugs commonly applied in NS treatment, did not affect the onset of edema.
Since MAGI2 binds β-catenin at adherence junctions,59, 60 it has been suggested that MAGI2 could modulate Wnt/β-catenin signaling.61 Pharmacologic activation of β-catenin induced albuminuria in wild-type mice but not in β-catenin-knockout littermates.62 As shown in Con8 rat mammary epithelial tumor cells, dexamethasone prevents β-catenin phosphorylation, and recruits it from the nucleus to adherence junctions.63 Therefore, in the absence of Magi2a in our KO fish, β-catenin is most likely not bound at the adherence junctions upon steroid treatment, and instead, accumulates in the nucleus and aggravates the podocyte dysfunction.62, 64
METHODS
A complete and detailed description of the methods is enclosed in Suppl. Material S1.
Zebrafish experiments were performed in Danio rerio, Tg(l-fabp::VDBP-EGFP) mi1000Tg. All national and institutional guidelines for the care and use of laboratory animals were followed. The zebrafish experiments were approved by the Boston Children’s Hospital (BCH) Institutional Animal Care and Use Committee (IACUC). Embryos were generated by timed breeding for all experiments.
Generation of zebrafish mutant lines by CRISPR/Cas9
Single guide RNA (sgRNA) targets were selected using the CHOPCHOP online tool v1 (Suppl. Table S1) and generated by in vitro transcription as described before.65 Mosaic F0 zebrafish were generated by coinjection of sgRNA and Cas9 protein in onecell stage zebrafish embryos, raised and outcrossed against wildtype fish to assure germline transmission and establish a stable line.
Edema and survival analysis
Larvae were transferred to rotifer feeding solution at 5 dpf. The dishes were monitored daily until 22 dpf. Edema phenotype contained whole body edema with ascites and periorbital edema (Fig. 1A-B, Fig 3A) and scoring of the edema phenotype was performed in dorsal view of the larvae. The endpoint for survival was reached when minimal residual cardiac activity without visible blood flow in the tail vein was observed. Genotypes were confirmed by Sanger sequencing.
Proteinuria assay
Larvae were sorted for edema phenotype at 6 dpf, and housed individually in 60 μl fish water for 24 h. Water samples were snap frozen and the larvae were genotyped. 6 water samples per genotype were pooled and concentrated by acetone precipitation. Resulting pellets were reconstituted 1× SDS-sample buffer, and SDS-PAGE was performed on 1mm Bis-Tris gradient gels (4–12%) according to standard procedures. Negative protein stain by 4′,5′-Dibromofluorescein (DBF) was performed as described before,47 and the gel was imaged immediately.
Immunoblotting
Larvae were sorted for edema phenotype at 6 dpf. The tail was clipped for DNA extraction. Remaining tissue was pooled according to phenotype, and pooled protein was extracted. Sanger sequencing confirmed genotypes with only magi2a−/− larvae in the edema pools. Only pools without homozygous larvae were used as controls. SDS-PAGE and transfer to nitrocellulose membranes was performed according to standard procedures. After blocking with SuperBlock T20 (TBS) (Thermo Fisher, Waltham, MA) membranes were stained with biotinylated polyclonal rabbit MAGI2 antibody (ARP61404_P050, Aviva Systems Biology, San Diego, CA) over night at 4°C followed by incubation with streptavidin-HRP complex (DY998, R&D systems, Minneapolis, MN). For loading control, membranes were stained with monoclonal mouse GAPDH antibody (sc47724, Santa Cruz Biotechnology, Dallas, TX) over night at 4°C, followed by incubation with HRP-tagged donkey anti-mouse antibody (sc-2314, Santa Cruz Biotechnology, Dallas, TX).
Densitometry
Densitometry for DBF staining and immunoblotting was performed in Image Lab 4.1 (Biorad, Hercules, CA).
qPCR
RNA from whole individual zebrafish larvae at 6 dpf was extracted using the RNeasy Micro kit (Qiagen, Germantown, MD), and larvae were genotyped. RNA from 7 larvae was pooled according to genotype. cDNA was generated using the ProtoScript II First Strand cDNA Synthesis Kit (NEB, Ipswich, MA). qPCR was performed with iTaq Universal SYBR Green Supermix (Biorad, Hercules, CA) on a Step One Plus real-time PCR system (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions with primers spanning the exon-exon junction of exon 4 and 5 (forward primer: CAAGTCCGTCAGCAACATGG, reverse primer: TAGGGGCATCTGTGGAGGTC), and normalized by gapdh expression (pre-tested primers, assay ID: qDreCED0021000, Biorad).
Whole mount in situ hybridization (WISH)
WISH was performed at 4 and 6 dpf following a WISH standard protocol using an anti-Dig Ab-dilution of 1:10:000.66 Antisense and sense probes for magi2a and magi2b were amplified from zebrafish poly-A larval cDNA with primers containing a T7 (antisense probe) or SP6 (sense probe) promoter. Antisense (T7) and sense probe (SP6) were transcribed using the DIG RNA Labeling Kit (SP6/T7) (Roche, Westborough, MA). Primers for magi2a were designed as described previously.11 Probes for magi2b were designed by using following forward (GAGATTTAGGTGACACTATAGCATACGTGACAACCTCTACC) and reverse (GAGTAATACGACTCACTATAGGGGGTTCCTCTCTGAATGAC) primer.
Drug treatment experiments
Embryos were dechorionated mechanically at 48 hpf and distributed randomly in treatment groups. Drugs or vehicle control were added to the fish water at 48 hpf and changed every other day. Larvae were monitored twice a day until 9 dpf (8 dpf for Tacrolimus and Cyclosporine), and genotyped by Sanger sequencing.
Dexamethasone (Sigma, St. Louis, MO), Prednisolone (Medisca, Plattsburgh, NY), Tacrolimus (Medisca, Plattsburgh, NY) and Cyclosporine A (Medisca, Plattsburgh, NY) were dissolved in DMSO. Fish water of treatment groups and vehicle control contained 0.001% DMSO each. ROCK inhibitor Y-27632 (STEMCELL Technologies, Cambridge, MA) and lysophosphatidic acid (LPA; Santa Cruz Biotechnology, Dallas, TX) were dissolved in phosphate buffered saline (PBS). Fish water of treatment groups and vehicle control contained 0.1% PBS each. RHO Activator II (Cytoskeleton, Denver, CO) was dissolved in water and treatment groups were compared to untreated controls.
Imaging
Larval imaging was performed on a Leica M205 FA stereoscope with a Leica DFC 300 FX camera (Leica Microsystems, Wetzlar, Germany) at 5 and 21 dpf.
Electron microscopy
Zebrafish larvae were fixed in 5.0% glutaraldehyde 2.5% paraformaldehyde and 0.03% picric acid in 0.1 M sodium cacodylate buffer (pH 7.4) for 24h at 4°C, and embedded in resin for EM according to standard procedures. Ultrathin sections (80 nm) were cut on a Reichert Ultracut-S microtome, stained with lead citrate and examined in a JEOL 1200EX Transmission electron microscope with an AMT 2k CCD camera.
Light microscopy and staining
Zebrafish larvae were fixed in 4% paraformaldehyde in PBS for 24h at 4°C. Samples were decalcified in 0.5 mM EDTA for 3 days at room temperature and embedded in paraffin according to standard procedures. Transverse 8 μm sections were obtained on a Leica RM2255 microtome (Leica Microsystems, Wetzlar, Germany). Resin embedded samples were processed as indicated above, but 0.5 μm sections were obtained. H&E and toluidine blue staining were performed according to standard procedures. Imaging was performed on a Nikon Eclipse Ni compound microscope with a Nikon DS-Fi2 camera (Nikon Instruments, Melville, NY).
Statistical analysis
Statistical analysis was performed using Graph Pad Prism® (version 7.00; GraphPad Software, Inc, La Jolla, CA). Significance was calculated using unpaired t-test (two-tailed, standard confidence interval of 95%) to perform proteinuria assay and determine areas of glomerular surface and nuclei in μm2. Significance was calculated using unpaired one-way ANOVA with multiple comparisons (standard confidence interval of 95%) for qPCR. Post hoc analysis was performed according to Tukey. Kaplan-Meier-blots for onset of edema and survival were analyzed by Log-rank (Mantel-Cox) test (standard confidence interval of 95%).
Supplementary Material
Supplemental Figure S1: Zebrafish orthologues of human MAGI2.
Supplemental Figure S7: Replicates of Prednisolone treatment of magi2a−/− (c.69_71delinsGCTA, p.Pro24Leufs*76) larvae.
Supplemental Figure S8: Calcineurin inhibitor treatment of magi2a−/− (c.69_71delinsGCTA, p.Pro24Leufs*76) larvae does not alter the edema phenotype.
Supplemental Figure S9: Domain specific binding of known MAGI2 interactors.
Supplemental Material S1: Complete methods.
Supplemental Table S1: Oligonucleotides used for targeting magi2a and magi2b.
Supplemental Table S2: magi2a and magi2b allele annotations.
Supplemental Figure S2: Biallelic truncating mutations in magi2b do not cause an edema phenotype or impaired survival.
Supplemental Figure S3: Whole mount in situ hybridization for magi2a and magi2b.
Supplemental Figure S4: Primary structure of the zebrafish Magi2a proteins (peptides) that result from the specific null vs. hypomorphic alleles introduced by CRISPR/Cas9.
Supplemental Figure S5: Nonsense mediated decay is not involved in the pathogenesis of magi2a KO.
Supplemental Figure S6: Replicates of Dexamethasone treatment of magi2a−/− (c.70_72delinsGTCC, p.Pro24Valfs*76) larvae.
TRANSLATIONAL STATEMENT.
Steroids are the initial standard therapy for nephrotic syndrome in humans with the therapeutic effect differentiating between steroid sensitive and steroid resistant nephrotic syndrome. However, in this magi2a knock-out zebrafish model steroids exacerbate the phenotype, suggesting that under the circumstances of a specific monogenic pathology steroid treatment may have paradoxical effects in exacerbating rather than improving disease. Thus, further studies may conclude that rapid genetic testing before initiation of steroid therapy in nephrotic syndrome could be beneficial.
ACKNOWLEDGMENTS
This research was supported by grants from the National Institutes of Health to F.H. (DK076683) and the Isabella Julian Forrest Fund. F.H. is the William E. Harmon Professor. T.J.S. is supported by a grant of the Deutsche Forschungsgemeinschaft (Jo 1324/1–1). A.M. is supported by the National Institutes of Health funded Training Grant T32DK007726–31A1, the Harvard Stem Cell Institute Kidney Group Inter-laboratory Post-doctoral Fellowship, and the ASN Ben J. Lipps Research Fellowship Program. EW is supported by the Leopoldina Fellowship Program, German National Academy of Sciences Leopoldina (LPDS 2015–07). The authors thank Johannes Wedel for advice regarding optimization of the DBF staining protocol, and Louis Trakimas from the Electron Microscope Core Facility, Harvard Medical School, for excellent EM services.
Footnotes
No other authors have competing financial interests.
DISCLOSURE
F.H. is a cofounder and SAC member and holds stock in Goldfinch-Bio.
ACCESSION NUMBERS
D. rerio magi2a cDNA (NM_001327853), D. rerio magi2b cDNA (ENSDART00000110879.3).
ZFIN LINE DESIGNATION
magi2a c.69_71delinsGCTA, p.Pro24Leufs*76, (magi2acl604); magi2a c.54_71delinsA, p.Val19Glufs*75, (magi2acl605); magi2a c.64_70delinsG, p.Arg22_Pro24delinsAla (magi2acl606); magi2a c.61_73delinsGGG, p.Ser2_Met218del (magi2acl607)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Sadowski CE, Lovric S, Ashraf S, et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol 2015; 26: 1279–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lovric S, Ashraf S, Tan W, et al. Genetic testing in steroid-resistant nephrotic syndrome: when and how? Nephrol Dial Transplant 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vivante A, Hildebrandt F. Exploring the genetic basis of early-onset chronic kidney disease. Nature reviews Nephrology 2016; 12: 133–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Warejko JK, Tan W, Daga A, et al. Whole Exome Sequencing of Patients with Steroid-Resistant Nephrotic Syndrome. Clin J Am Soc Nephrol 2018; 13: 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tan W, Lovric S, Ashraf S, et al. Analysis of 24 genes reveals a monogenic cause in 11.1% of cases with steroid-resistant nephrotic syndrome at a single center. Pediatr Nephrol 2018; 33: 305–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ashraf S, Kudo H, Rao J, et al. Mutations in six nephrosis genes delineate a pathogenic pathway amenable to treatment. Nature communications 2018; 9: 1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bierzynska A, Soderquest K, Dean P, et al. MAGI2 Mutations Cause Congenital Nephrotic Syndrome. J Am Soc Nephrol 2017; 28: 1614–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirao K, Hata Y, Ide N, et al. A novel multiple PDZ domain-containing molecule interacting with N-methyl-D-aspartate receptors and neuronal cell adhesion proteins. The Journal of biological chemistry 1998; 273: 21105–21110. [DOI] [PubMed] [Google Scholar]
- 9.Shoji H, Tsuchida K, Kishi H, et al. Identification and characterization of a PDZ protein that interacts with activin type II receptors. The Journal of biological chemistry 2000; 275: 5485–5492. [DOI] [PubMed] [Google Scholar]
- 10.Lehtonen S, Ryan JJ, Kudlicka K, et al. Cell junction-associated proteins IQGAP1, MAGI-2, CASK, spectrins, and alpha-actinin are components of the nephrin multiprotein complex. Proceedings of the National Academy of Sciences of the United States of America 2005; 102: 9814–9819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong L, Pietsch S, Tan Z, et al. Integration of Cistromic and Transcriptomic Analyses Identifies Nphs2, Mafb, and Magi2 as Wilms’ Tumor 1 Target Genes in Podocyte Differentiation and Maintenance. J Am Soc Nephrol 2015; 26: 2118–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang L, Ellis MJ, Gomez JA, et al. Mechanisms of the proteinuria induced by Rho GTPases. Kidney Int 2012; 81: 1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim YH, Goyal M, Kurnit D, et al. Podocyte depletion and glomerulosclerosis have a direct relationship in the PAN-treated rat. Kidney Int 2001; 60: 957–968. [DOI] [PubMed] [Google Scholar]
- 14.Wharram BL, Goyal M, Wiggins JE, et al. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol 2005; 16: 2941–2952. [DOI] [PubMed] [Google Scholar]
- 15.Roselli S, Heidet L, Sich M, et al. Early glomerular filtration defect and severe renal disease in podocin-deficient mice. Molecular and cellular biology 2004; 24: 550–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shirata N, Ihara KI, Yamamoto-Nonaka K, et al. Glomerulosclerosis Induced by Deficiency of Membrane-Associated Guanylate Kinase Inverted 2 in Kidney Podocytes. J Am Soc Nephrol 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ihara K, Asanuma K, Fukuda T, et al. MAGI-2 is critical for the formation and maintenance of the glomerular filtration barrier in mouse kidney. Am J Pathol 2014; 184: 2699–2708. [DOI] [PubMed] [Google Scholar]
- 18.Balbas MD, Burgess MR, Murali R, et al. MAGI-2 scaffold protein is critical for kidney barrier function. Proceedings of the National Academy of Sciences of the United States of America 2014; 111: 14876–14881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zon LI, Peterson RT. In vivo drug discovery in the zebrafish. Nat Rev Drug Discov 2005; 4: 35–44. [DOI] [PubMed] [Google Scholar]
- 20.Kramer-Zucker AG, Wiessner S, Jensen AM, et al. Organization of the pronephric filtration apparatus in zebrafish requires Nephrin, Podocin and the FERM domain protein Mosaic eyes. Dev Biol 2005; 285: 316–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lieschke GJ, Currie PD. Animal models of human disease: zebrafish swim into view. Nature reviews Genetics 2007; 8: 353–367. [DOI] [PubMed] [Google Scholar]
- 22.Hentschel DM, Mengel M, Boehme L, et al. Rapid screening of glomerular slit diaphragm integrity in larval zebrafish. American journal of physiology Renal physiology 2007; 293: F1746–1750. [DOI] [PubMed] [Google Scholar]
- 23.Ebarasi L, Oddsson A, Hultenby K, et al. Zebrafish: a model system for the study of vertebrate renal development, function, and pathophysiology. Current opinion in nephrology and hypertension 2011; 20: 416–424. [DOI] [PubMed] [Google Scholar]
- 24.Drummond IA, Davidson AJ. Zebrafish kidney development. Methods Cell Biol 2010; 100: 233–260. [DOI] [PubMed] [Google Scholar]
- 25.Zhou W, Otto EA, Cluckey A, et al. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat Genet 2012; 44: 910–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou W, Hildebrandt F. Inducible podocyte injury and proteinuria in transgenic zebrafish. J Am Soc Nephrol 2012; 23: 1039–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou W, Hildebrandt F. Molecular cloning and expression of phospholipase C epsilon 1 in zebrafish. Gene Expr Patterns 2009; 9: 282–288. [DOI] [PubMed] [Google Scholar]
- 28.Gee HY, Zhang F, Ashraf S, et al. KANK deficiency leads to podocyte dysfunction and nephrotic syndrome. J Clin Invest 2015; 125: 2375–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gee HY, Ashraf S, Wan X, et al. Mutations in EMP2 cause childhood-onset nephrotic syndrome. Am J Hum Genet 2014; 94: 884–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Howe K, Clark MD, Torroja CF, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013; 496: 498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inoue J, Sato Y, Sinclair R, et al. Rapid genome reshaping by multiple-gene loss after whole-genome duplication in teleost fish suggested by mathematical modeling. Proceedings of the National Academy of Sciences of the United States of America 2015; 112: 14918–14923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hinkes B, Wiggins RC, Gbadegesin R, et al. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet 2006; 38: 1397–1405. [DOI] [PubMed] [Google Scholar]
- 33.Gee HY, Saisawat P, Ashraf S, et al. ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. J Clin Invest 2013; 123: 3243–3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clift DE, Thorn RJ, Passarelli EA, et al. Effects of embryonic cyclosporine exposures on brain development and behavior. Behav Brain Res 2015; 282: 117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamm MJ, Kirchmaier BC, Herzog W. Sema3d controls collective endothelial cell migration by distinct mechanisms via Nrp1 and PlxnD1. The Journal of cell biology 2016; 215: 415–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gu C, Lee HW, Garborcauskas G, et al. Dynamin Autonomously Regulates Podocyte Focal Adhesion Maturation. J Am Soc Nephrol 2017; 28: 446–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chaki M, Hoefele J, Allen SJ, et al. Genotype-phenotype correlation in 440 patients with NPHP-related ciliopathies. Kidney Int 2011; 80: 1239–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pereira FJC, Teixeira A, Kong J, et al. Resistance of mRNAs with AUG-proximal nonsense mutations to nonsense-mediated decay reflects variables of mRNA structure and translational activity. Nucleic acids research 2015; 43: 6528–6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neu-Yilik G, Amthor B, Gehring NH, et al. Mechanism of escape from nonsense-mediated mRNA decay of human beta-globin transcripts with nonsense mutations in the first exon. RNA (New York, NY) 2011; 17: 843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu H, Han H, Li J, et al. DNAFSMiner: a web-based software toolbox to recognize two types of functional sites in DNA sequences. Bioinformatics 2005; 21: 671–673. [DOI] [PubMed] [Google Scholar]
- 41.Nishikawa T, Ota T, Isogai T. Prediction whether a human cDNA sequence contains initiation codon by combining statistical information and similarity with protein sequences. Bioinformatics 2000; 16: 960–967. [DOI] [PubMed] [Google Scholar]
- 42.Pedersen AG, Nielsen H. Neural network prediction of translation initiation sites in eukaryotes: perspectives for EST and genome analysis. Proceedings International Conference on Intelligent Systems for Molecular Biology 1997; 5: 226–233. [PubMed] [Google Scholar]
- 43.El-Brolosy MA, Stainier DYR. Genetic compensation: A phenomenon in search of mechanisms. PLoS Genetics 2017; 13: e1006780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.You KT, Li LS, Kim NG, et al. Selective translational repression of truncated proteins from frameshift mutation-derived mRNAs in tumors. PLoS Biol 2007; 5: e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nagashima S, Kodaka M, Iwasa H, et al. MAGI2/S-SCAM outside brain. J Biochem 2015; 157: 177–184. [DOI] [PubMed] [Google Scholar]
- 46.Hanke N, King BL, Vaske B, et al. A Fluorescence-Based Assay for Proteinuria Screening in Larval Zebrafish (Danio rerio). Zebrafish 2015; 12: 372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu D, Wang Y, Zhang S, et al. An ultrasensitive stain for negative protein detection in SDS-PAGE via 4’,5’-Dibromofluorescein. Journal of proteomics 2017; 165: 21–25. [DOI] [PubMed] [Google Scholar]
- 48.D’Amico G, Bazzi C. Pathophysiology of proteinuria. Kidney Int 2003; 63: 809–825. [DOI] [PubMed] [Google Scholar]
- 49.Nobes CD, Hall A. Rho, rac and cdc42 GTPases: regulators of actin structures, cell adhesion and motility. Biochem Soc Trans 1995; 23: 456–459. [DOI] [PubMed] [Google Scholar]
- 50.Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: conserved structure and molecular mechanism. Nature 1991; 349: 117–127. [DOI] [PubMed] [Google Scholar]
- 51.Gee HY, Sadowski CE, Aggarwal PK, et al. FAT1 mutations cause a glomerulotubular nephropathy. Nature communications 2016; 7: 10822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blattner SM, Hodgin JB, Nishio M, et al. Divergent functions of the Rho GTPases Rac1 and Cdc42 in podocyte injury. Kidney Int 2013; 84: 920–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu L, Jiang R, Aoudjit L, et al. Activation of RhoA in podocytes induces focal segmental glomerulosclerosis. J Am Soc Nephrol 2011; 22: 1621–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hildebrandt F Genetic kidney diseases. Lancet 2010; 375: 1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Benoit G, Machuca E, Antignac C. Hereditary nephrotic syndrome: a systematic approach for genetic testing and a review of associated podocyte gene mutations. Pediatr Nephrol 2010; 25: 1621–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chatzopoulou A, Roy U, Meijer AH, et al. Transcriptional and metabolic effects of glucocorticoid receptor alpha and beta signaling in zebrafish. Endocrinology 2015; 156: 1757–1769. [DOI] [PubMed] [Google Scholar]
- 57.Pasqualetti S, Congiu T, Banfi G, et al. Alendronate rescued osteoporotic phenotype in a model of glucocorticoid-induced osteoporosis in adult zebrafish scale. International journal of experimental pathology 2015; 96: 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wan X, Chen Z, Choi WI, et al. Loss of Epithelial Membrane Protein 2 Aggravates Podocyte Injury via Upregulation of Caveolin-1. Journal of the American Society of Nephrology : JASN 2016; 27: 1066–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kawajiri A, Itoh N, Fukata M, et al. Identification of a novel beta-catenininteracting protein. Biochemical and biophysical research communications 2000; 273: 712–717. [DOI] [PubMed] [Google Scholar]
- 60.Nishimura W, Yao I, Iida J, et al. Interaction of synaptic scaffolding molecule and Beta -catenin. J Neurosci 2002; 22: 757–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Empitu MA, Kadariswantiningsih IN, Aizawa M, et al. MAGI-2 and scaffold proteins in glomerulopathy. American Journal of Physiology-Renal Physiology 2018; 315: F1336–F1344. [DOI] [PubMed] [Google Scholar]
- 62.Dai C, Stolz DB, Kiss LP, et al. Wnt/beta-catenin signaling promotes podocyte dysfunction and albuminuria. J Am Soc Nephrol 2009; 20: 1997–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guan Y, Rubenstein NM, Failor KL, et al. Glucocorticoids control beta-catenin protein expression and localization through distinct pathways that can be uncoupled by disruption of signaling events required for tight junction formation in rat mammary epithelial tumor cells. Mol Endocrinol 2004; 18: 214–227. [DOI] [PubMed] [Google Scholar]
- 64.Wang D, Dai C, Li Y, et al. Canonical Wnt/β-catenin signaling mediates transforming growth factor-β1-driven podocyte injury and proteinuria. Kidney Int 2011; 80: 1159–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jobst-Schwan T, Schmidt JM, Schneider R, et al. Acute multi-sgRNA knockdown of KEOPS complex genes reproduces the microcephaly phenotype of the stable knockout zebrafish model. PloS one 2018; 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thisse C, Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nature protocols 2008; 3: 59–69. [DOI] [PubMed] [Google Scholar]
- 67.Ichimura K, Bubenshchikova E, Powell R, et al. A comparative analysis of glomerulus development in the pronephros of medaka and zebrafish. PloS one 2012; 7: e45286. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1: Zebrafish orthologues of human MAGI2.
Supplemental Figure S7: Replicates of Prednisolone treatment of magi2a−/− (c.69_71delinsGCTA, p.Pro24Leufs*76) larvae.
Supplemental Figure S8: Calcineurin inhibitor treatment of magi2a−/− (c.69_71delinsGCTA, p.Pro24Leufs*76) larvae does not alter the edema phenotype.
Supplemental Figure S9: Domain specific binding of known MAGI2 interactors.
Supplemental Material S1: Complete methods.
Supplemental Table S1: Oligonucleotides used for targeting magi2a and magi2b.
Supplemental Table S2: magi2a and magi2b allele annotations.
Supplemental Figure S2: Biallelic truncating mutations in magi2b do not cause an edema phenotype or impaired survival.
Supplemental Figure S3: Whole mount in situ hybridization for magi2a and magi2b.
Supplemental Figure S4: Primary structure of the zebrafish Magi2a proteins (peptides) that result from the specific null vs. hypomorphic alleles introduced by CRISPR/Cas9.
Supplemental Figure S5: Nonsense mediated decay is not involved in the pathogenesis of magi2a KO.
Supplemental Figure S6: Replicates of Dexamethasone treatment of magi2a−/− (c.70_72delinsGTCC, p.Pro24Valfs*76) larvae.