

Abstract
Background
This is an updated version of the original Cochrane review published in Issue 3, 2012. That review considered both fibromyalgia and neuropathic pain, but the efficacy of milnacipran for neuropathic pain is now dealt with in a separate review.
Milnacipran is a serotonin‐norepinephrine (noradrenaline) reuptake inhibitor (SNRI) that is licensed for the treatment of fibromyalgia in some countries, including Canada, Russia, and the United States.
Objectives
To assess the analgesic efficacy of milnacipran for pain in fibromyalgia in adults and the adverse events associated with its use in clinical trials.
Search methods
We searched the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE, and EMBASE to 18 May 2015, together with reference lists of retrieved papers and reviews, and two clinical trial registries. For the earlier review, we also contacted the manufacturer.
Selection criteria
We included randomised, double‐blind studies of eight weeks' duration or longer, comparing milnacipran with placebo or another active treatment in fibromyalgia in adults.
Data collection and analysis
We extracted efficacy and adverse event data, and two review authors examined issues of study quality independently.
Main results
We identified one new study with 100 participants for the pooled analysis. We identified two additional reports of a study using an enriched enrolment randomised withdrawal (EERW) design that included participants from earlier randomised controlled trials and an open‐label study. Because this study used the same participants already included in our main analysis, and a different design, we dealt with it separately.
The main analysis included six studies (five from the earlier review; 4238 participants in total), all of which were placebo‐controlled, and used titration to a target dose of milnacipran 100 or 200 mg, with assessment after 8 to 24 weeks of stable treatment. There were no studies with active comparators. Study quality was generally good, although the imputation method used in analyses of the primary outcomes could overestimate treatment effect.
Both doses of milnacipran provided moderate levels of pain relief (at least 30% pain intensity reduction) to about 40% of participants treated, compared to 30% with placebo, giving a number needed to treat for an additional beneficial outcome (NNT) of 6 to 10 (high quality evidence). Using a stricter definition for responder and a more conservative method of analysis gave lower levels of response (while maintaining a 10% difference between milnacipran and placebo) and increased the NNT to 11 (high quality evidence). One EERW study was broadly supportive.
Adverse events were common in both milnacipran (86%) and placebo (78%) groups (high quality evidence), but serious adverse events did not differ between groups (less than 2%) (low quality evidence). Nausea, constipation, and headache were the most common events showing the greatest difference between groups (number needed to treat for an additional harmful outcome (NNH) of 5.7 for nausea, 13 for constipation, and 29 for headache) (moderate quality evidence).
Withdrawals for any reason were more common with milnacipran than placebo, and more common with 200 mg (NNH 9) than 100 mg (NNH 23), compared with placebo. This was largely driven by adverse event withdrawals, where the NNH compared with placebo was 14 for 100 mg and 7.0 for 200 mg (high quality evidence). Withdrawals due to lack of efficacy were less common with milnacipran than placebo but did not differ between doses (number needed to treat to prevent an additional unwanted outcome (NNTp) of 41) (moderate quality evidence).
Authors' conclusions
The evidence available indicates that milnacipran 100 mg or 200 mg is effective for a minority in the treatment of pain due to fibromyalgia, providing moderate levels of pain relief (at least 30%) to about 40% of participants, compared with about 30% with placebo. There were insufficient data to assess substantial levels of pain relief (at least 50%), and the use of last observation carried forward imputation may overestimate drug efficacy. Using stricter criteria for 'responder' and a more conservative method of analysis gave lower response rates (about 26% with milnacipran versus 17% with placebo). Milnacipran was associated with increased adverse events and adverse event withdrawals, which were significantly greater for the higher dose.
Plain language summary
Milnacipran for fibromyalgia in adults
Fibromyalgia is characterised by persistent, widespread pain and tenderness, sleep problems, and fatigue. Common pain‐relieving medicines such as paracetamol and ibuprofen are not usually considered effective. Medicines used to treat epilepsy or depression can be effective in some people with fibromyalgia and other forms of chronic (persistent, long‐lasting) pain where there may be nerve damage. Milnacipran is an antidepressant, and antidepressants are widely recommended for treating fibromyalgia. Milnacipran is licensed to treat fibromyalgia only in some parts of the world, particularly the USA.
This review is an update of one originally published in 2012, which examined how well milnacipran worked in both fibromyalgia and neuropathic pain conditions (pain from damage to nerves or disease affecting the nerves). Here we examine only fibromyalgia. The earlier review showed that milnacipran worked, but only in a small proportion of people with fibromyalgia. This is the same as all other fibromyalgia treatments to date, and for chronic pain conditions generally. We use a definition of 'worked' that involved both a high level of pain relief and the ability to take the tablets over a longer time without side effects being intolerable.
We searched scientific databases for studies that looked at the effects of milnacipran in adults with fibromyalgia who had moderate or severe pain. The treatment had to last at least eight weeks. The evidence is current to May 2015.
We identified six studies that satisfied the inclusion criteria, including one new study for this update. Over 4000 participants were treated with milnacipran 100 or 200 mg, or placebo, for 8 to 24 weeks at the target dose. Overall study quality was good, although the method of analysis for our primary outcomes of pain relief could overestimate treatment effect.
Milnacipran at either dose provided moderate pain relief (at least 30% reduction in pain intensity) to 1 in 10 (10%) more people than did placebo (high quality evidence). This relatively modest effect may be clinically important in this difficult‐to‐treat condition. Adverse events were reported by most participants in all treatment groups, but were more common with milnacipran than placebo (high quality evidence), with nausea (feeling sick) and constipation showing the greatest differences (moderate quality evidence). Serious adverse events were uncommon, fewer than 1 in 50 (2%) participants, and did not differ between treatment groups (low quality evidence). The numbers of participants dropping out of the studies (withdrawals) because of adverse events were also more common with milnacipran than placebo, and were more common with 200 mg than 100 mg (high quality evidence), while withdrawals due to lack of effect were less common with milnacipran, with no difference between doses (moderate quality evidence).
Milnacipran gives good pain relief to some people with fibromyalgia, but only a minority; it will not work for most people.
Summary of findings
for the main comparison.
| Milnacipran compared with placebo for fibromyalgia | ||||||
|
Patient or population: adults with fibromyalgia Settings: community Intervention: milnacipran 100 and 200 mg/day Comparison: placebo | ||||||
| Outcomes | Probable outcome with placebo | Probable outcome with intervention | Risk ratio (95% CI) and NNT or NNH | No of studies and participants | Quality of the evidence (GRADE) | Comments |
| Milnacipran 100 mg/day | ||||||
| At least 50% reduction in pain or equivalent (substantial) | 180 in 1000 | 270 in 1000 | RR 1.6 (1.3 to 2.0) NNT 10 (7.0 to 20) |
2 studies, 1250 participants | Moderate | Only 2 studies, 1 much smaller than other Downgrade because of LOCF analysis |
| At least 30% reduction in pain or equivalent (moderate) | 300 in 1000 | 410 in 1000 | RR 1.4 (1.2 to 1.6) NNT 9.0 (6.5 to 15) |
3 studies, 1925 participants | High | Downgrade because of LOCF analysis, but upgrade as the result is supported by Composite outcome 1 giving a very similar result; this is a BOCF analysis for the outcome plus PGIC much or very much improved |
| Adverse event withdrawals | 120 in 1000 | 190 in 1000 | RR 1.6 (1.3 to 2.0) NNH 14 (10 to 24) |
4 studies, 2379 participants | High | ‐ |
| Serious adverse events | 16 in 1000 | 15 in 1000 | RR 0.90 (0.47 to 1.7) NNH not calculated |
4 studies, 2378 participants |
Low | Few events (36) |
| Death | None reported | |||||
| Milnacipran 200 mg/day | ||||||
| At least 50% reduction in pain or equivalent (substantial) | 140 in 1000 | 280 in 1000 | Not calculated | 1 study, 125 participants | Very low | 1 study, few participants |
| At least 30% reduction in pain or equivalent (moderate) | 290 in 1000 | 390 in 1000 | RR 1.4 (1.2 to 1.5) NNT 10 (7.0 to 18) |
3 studies, 1798 participants | High | Downgrade because of LOCF analysis, but upgrade as the result is supported by composite outcome 1 giving a very similar result; this is a BOCF analysis for the outcome plus PGIC much or very much improved |
| Adverse event withdrawals | 95 in 1000 | 240 in 1000 | RR 2.5 (2.0 to 3.1) NNH 7.0 (5.8 to 8.7) |
4 studies, 2470 participants | High | ‐ |
| Serious adverse events | 21 in 1000 | 19 in 1000 | RR 0.91 (0.52 to 1.6) NNH not calculated |
4 studies, 2463 participants | Low | Few events (49) |
| Death | None reported | |||||
| BOCF: baseline observation carried forward; CI: confidence interval; LOCF: last observation carried forward; NNT: number needed to treat for an additional beneficial outcome; NNH: number needed to treat for an additional harm outcome; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
Background
This is an updated version of the original Cochrane review published in Issue 3, 2012 (Derry 2012). That review considered both fibromyalgia and neuropathic pain, but the efficacy of milnacipran for neuropathic pain is now dealt with in a separate review (Derry 2015).
This review is based on a template for reviews of drugs used to relieve fibromyalgia. The aim is for all reviews to use the same methods, based on new criteria for what constitutes reliable evidence in chronic pain (Moore 2010a; Appendix 1).
Description of the condition
Fibromyalgia has been defined as widespread pain that lasts for longer than three months, with pain on palpation at 11 or more of 18 specified tender points (Wolfe 1990). It is frequently associated with other symptoms such as poor sleep, fatigue, and depression (Wolfe 2014). More recently, a definition of fibromyalgia has been proposed based on symptom severity and the presence of widespread pain, and which does not require palpation of tender points for diagnosis (Wolfe 2010). While some rheumatologists have thought of fibromyalgia as a specific pain disorder, other investigators have characterised it as a bodily distress syndrome or a physical symptom disorder, or somatoform disorder (Wolfe 2014). Our understanding of fibromyalgia is growing. It is a heterogeneous condition in which there is abnormal processing of the sensation of pain. Central nervous system (CNS) dysfunction appears to be the primary pathogenic problem (Schmidt‐Wilcke 2014). It has some features in common with neuropathic pain, and people with these conditions experience similar sensory phenomena (Koroschetz 2011).
Many people with fibromyalgia are significantly disabled, and experience moderate or severe pain for many years. Chronic painful conditions comprised five of the 11 top‐ranking conditions for years lived with disability in 2010 (Vos 2012), and are responsible for considerable loss of quality of life, employment, and increased health costs (Moore 2014a).
Fibromyalgia is common. Numerous studies have investigated prevalence in different settings and countries. The Queiroz 2013 review gives a global mean prevalence of 2.7% (range 0.4% to 9.3%), and a mean in the Americas of 3.1%, in Europe of 2.5%, and in Asia of 1.7%. Fibromyalgia is more common in women, with a female to male ratio of 3:1 (4.2%:1.4%). The change in diagnostic criteria does not appear to have significantly affected estimates of prevalence (Wolfe 2013a). Estimates of prevalence in specific populations vary greatly, but have been reported to be as high as 9% in female textile workers in Turkey and 10% in metalworkers in Brazil (59% in those with repetitive strain injury; Queiroz 2013).
Fibromyalgia pain, like other chronic pain conditions, is known to be difficult to treat effectively, with only a minority of individuals experiencing a clinically relevant benefit from any one intervention. A multidisciplinary approach is now advocated, combining pharmacological interventions with physical or cognitive interventions, or both. Conventional analgesics are usually not effective. Treatment is often by so‐called unconventional analgesics (or pain modulators), such as antidepressants like duloxetine and amitriptyline (Lunn 2014; Moore 2012a; Sultan 2008), or antiepileptics like gabapentin or pregabalin (Moore 2009; Moore 2011a; Wiffen 2013). The proportion of people who achieve worthwhile pain relief (typically at least a 50% reduction in pain intensity; Moore 2013a) is small, generally only 10% to 25% more than with placebo, with numbers needed to treat for an additional beneficial outcome (NNT) usually between 4 and 10 (Kalso 2013; Wiffen 2013). Those who do experience good levels of pain relief may well benefit from substantial reductions in other symptoms, such as fatigue, function, sleep, depression, anxiety, and ability to work, with significant improvement in quality of life (Moore 2010b; Moore 2014a; Straube 2011). Fibromyalgia is not particularly different from other chronic pain in that only a small proportion of trial participants have a good response to treatment (Moore 2013b).
Description of the intervention
Milnacipran (trade names Ixel, Savella) is a serotonin‐norepinephrine (noradrenaline) reuptake inhibitor (SNRI), used to treat depression and fibromyalgia. It is licensed for major depressive disorder in many countries, and for the treatment of fibromyalgia in some countries including Canada, Russia, and the United States, but not as yet in the United Kingdom.
How the intervention might work
5‐hydroxytryptamine (5‐HT or serotonin) and norepinephrine (NE) are involved in the modulation of endogenous analgesic mechanisms via descending inhibitory pain pathways in the brain and the spinal cord (Suzuki 2004). Disinhibition and imbalance of 5‐HT and NE in endogenous pain inhibitory pathways could contribute to persistent pain. An increase in 5‐HT and NE may increase inhibition of painful signals, improving pain relief, but the exact mechanism of action is not fully understood.
Why it is important to do this review
Milnacipran is a recent addition to the pharmacological interventions available in some countries to treat fibromyalgia. It is important to establish its efficacy compared with placebo or other active interventions to understand its place among the available treatment options.
Like the earlier Cochrane review, this update assessed evidence in ways that make both statistical and clinical sense, and used developing criteria for what constitutes reliable evidence in chronic pain (Appendix 1; Moore 2010a). It followed standards set out in the PaPaS Author and Referee Guidance for pain studies of the Cochrane Pain, Palliative and Supportive Care Group (PaPaS 2012).
Objectives
To assess the analgesic efficacy of milnacipran for pain in fibromyalgia in adults and the adverse events associated with its use in clinical trials.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs) with double‐blind assessment of outcomes, and reported after eight weeks of treatment or longer. Full journal publication was required, with the exception of extended abstracts of otherwise unpublished clinical trials. Short abstracts (usually meeting reports) were not included. We excluded non‐randomised studies, studies of experimental pain, case reports, and clinical observations.
Types of participants
We included adults with fibromyalgia diagnosed using the 1990 or 2010 criteria (Wolfe 1990; Wolfe 2010), aged 18 years and above, and with initial pain of at least moderate intensity.
Types of interventions
Milnacipran in any dose, by any route, administered for the relief of fibromyalgia, and compared to placebo, no intervention, or any other active comparator. We excluded studies using milnacipran to treat pain resulting from the use of other drugs.
Types of outcome measures
We expected that a variety of outcome measures would be used in the studies, based on standard subjective scales for pain intensity or pain relief, or both. Acceptable scales for measurement of pain intensity were 100 mm visual analogue scale (VAS) (no pain to worst pain imaginable) or four‐point categorical scale (none, mild, moderate, severe), and for pain relief were 100 mm VAS (no relief to complete relief) or five‐point categorical scale (none, a little, some, a lot, complete, or similar wording). We paid particular attention to Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) definitions for moderate and substantial benefit in chronic pain studies (Dworkin 2008). These are defined as at least 30% pain relief over baseline (moderate), at least 50% pain relief over baseline (substantial), much or very much improved on Patient Global Impression of Change (PGIC) (moderate), and very much improved on PGIC (substantial). These outcomes are different from those set out in an earlier review of antidepressants for fibromyalgia (Saarto 2007), concentrating on dichotomous outcomes where pain responses are not normally distributed.
Primary outcomes
Participant reported pain relief of 30% or greater
Participant reported pain relief of 50% or greater
PGIC much or very much improved
PGIC very much improved
Secondary outcomes
Any pain‐related outcome indicating some improvement
Withdrawals due to lack of efficacy
Participants experiencing any adverse event
Participants experiencing any serious adverse event. Serious adverse events typically include any untoward medical occurrence or effect that at any dose results in death, is life‐threatening, requires hospitalisation or prolongation of existing hospitalisation, results in persistent or significant disability or incapacity, is a congenital anomaly or birth defect, is an 'important medical event' that may jeopardise the person, or may require an intervention to prevent one of the above characteristics or consequences.
Withdrawals due to adverse events
Specific adverse events, particularly somnolence and dizziness
Search methods for identification of studies
Two review authors carried out searches independently; we settled any disagreements or uncertainty by discussion, with a third review author if necessary.
Electronic searches
We searched the following databases.
Cochrane Central Register of Controlled Trials (CENTRAL) (to 18 May 2015).
MEDLINE (via Ovid) (1946 to 18 May 2015).
EMBASE (via Ovid) (1974 to 18 May 2015).
See Appendix 2 for the CENTRAL search strategy, Appendix 3 for the MEDLINE search strategy, and Appendix 4 for the EMBASE search strategy.
There were no language restrictions.
Searching other resources
We searched reference lists of retrieved articles and reviews for any additional studies, and two clinical trial registries (ClinicalTrials.gov (ClinicalTrials.gov) and the World Health Organization (WHO) ICTRP (apps.who.int/trialsearch/)) to identify additional published or unpublished data. For the earlier review, we asked the manufacturer of milnacipran to provide additional unpublished dichotomous outcome data to complement published reports.
Data collection and analysis
Selection of studies
Two review authors read the abstract of each study identified by the search, eliminated studies that clearly did not satisfy the inclusion criteria, and obtained full copies of the remaining studies. The same review authors then independently read these studies to determine eligibility; any disagreements or uncertainty were settled by discussion, with a third review author if necessary. Studies were not anonymised in any way before assessment.
Data extraction and management
Two review authors extracted data using a standard form and agreed data before entry into Review Manager 5 (RevMan 2014), or any other analysis method. Data extracted included information about the pain condition and number of participants treated, drug and dosing regimen, study design (placebo or active control), study duration and follow‐up, analgesic outcome measures and results, withdrawals, and adverse events (participants experiencing any adverse event, or a serious adverse event).
Assessment of risk of bias in included studies
We used the Oxford Quality Score (Jadad 1996) as the basis for inclusion, limiting inclusion to studies that were randomised and double‐blind as a minimum.
Two review authors independently assessed the risk of bias for each study, using the criteria outlined in the 'Risk of bias' tool in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), and adapted from those used by the Cochrane Pregnancy and Childbirth Group. We resolved any disagreements by discussion. We assessed the following for each study.
Random sequence generation (checking for possible selection bias). We assessed the method used to generate the allocation sequence as: low risk of bias (any truly random process such as random number table or computer random number generator); unclear risk of bias (method used to generate sequence not clearly stated). We excluded studies using a non‐random process (for example, odd or even date of birth; hospital or clinic record number).
Allocation concealment (checking for possible selection bias). The method used to conceal allocation to interventions prior to assignment determines whether intervention allocation could have been foreseen in advance of, or during, recruitment, or changed after assignment. We assessed the methods as: low risk of bias (for example, telephone or central randomisation; consecutively numbered sealed opaque envelopes); unclear risk of bias (method not clearly stated). We excluded studies that did not conceal allocation (for example, open list).
Blinding of outcome assessment (checking for possible detection bias). We assessed the methods used to blind study participants and outcome assessors from knowledge of which intervention a participant received. We assessed the methods as: low risk of bias (study stated that it was blinded and described the method used to achieve blinding, for example, identical tablets; matched in appearance and smell); unclear risk of bias (study stated that it was blinded but did not provide an adequate description of how it was achieved). We excluded studies that were not double‐blind.
Incomplete outcome data (checking for possible attrition bias due to the amount, nature, and handling of incomplete outcome data). We assessed the methods used to deal with incomplete data as: low risk of bias (less than 10% of participants did not complete the study or used 'baseline observation carried forward' (BOCF) analysis, or both); unclear risk of bias (used 'last observation carried forward' (LOCF) analysis); high risk of bias (used 'completer' analysis).
Size (checking for possible biases confounded by small size). Small studies have been shown to overestimate treatment effects, probably because the conduct of small studies is more likely to be less rigorous, allowing critical criteria to be compromised (Dechartres 2013; Kjaergard 2001; Nüesch 2010). Small studies with limited data are subject to large chance effects (Moore 1998). Studies were considered to be at low risk of bias if they had 200 participants or more, at unclear risk of bias if they had 50 to 200 participants, and at high risk of bias if they had fewer than 50 participants.
Measures of treatment effect
We used dichotomous data to calculate risk ratio (RR) with 95% confidence intervals (CI) using a fixed‐effect model unless we found significant statistical heterogeneity (see Assessment of heterogeneity). We calculated numbers needed to treat for an additional beneficial outcome (NNTs) as the reciprocal of the absolute risk reduction (McQuay 1998). For unwanted effects, the NNT becomes the number needed to treat for an additional harmful outcome (NNH), and is calculated in the same manner. Where the unwanted effect is less common with treatment than control, we used the term number needed to treat to prevent (NNTp).
We did not use continuous data because it is inappropriate where there is an underlying skewed distribution, as is usually the case with analgesic response.
Unit of analysis issues
We accepted randomisation to individual participant only.
Dealing with missing data
We used intention‐to‐treat (ITT) analysis, where the ITT population consisted of participants who were randomised, took the assigned study medication, and provided at least one post‐baseline assessment. Where possible we assigned missing participants zero improvement.
Assessment of heterogeneity
We dealt with clinical heterogeneity by combining studies that examined similar conditions, while we assessed statistical heterogeneity visually (L'Abbé 1987), and using the I2 statistic.
Assessment of reporting biases
The aim of this review was to use dichotomous data of known utility and of value to patients (Moore 2010b; Moore 2010c; Moore 2010d; Moore 2013a). The review did not depend on what authors of the original studies chose to report or not, though clearly difficulties arose with studies failing to report any dichotomous results. We planned to extract and use continuous data, which probably poorly reflect efficacy and utility, if useful for illustrative purposes only.
We assessed publication bias using a method designed to detect the amount of unpublished data with a null effect required to make any result clinically irrelevant (usually taken to mean an NNT of 10 or higher) (Moore 2008).
Data synthesis
We carried out meta‐analysis using a fixed‐effect model. A random‐effects model for meta‐analysis was also carried out where there was significant clinical heterogeneity and we considered it appropriate to combine studies.
We determined that we would analyse data for each painful condition in three tiers, according to outcome and freedom from known sources of bias.
The first tier used data meeting current best standards, where studies reported the outcome of at least 50% pain intensity reduction over baseline (or its equivalent), without the use of LOCF or other imputation method other than BOCF for drop‐outs, reported an ITT analysis, lasted eight or more weeks, had a parallel‐group design, and had at least 200 participants (preferably at least 400) in the comparison (Moore 2010a; Moore 2012b). We planned to report these top‐tier results first, but in this review outcomes satisfying these criteria were not pre‐specified primary outcomes, so they are reported after the primary outcomes.
The second tier used data from at least 200 participants, but where one or more of the above conditions was not met (for example, reporting at least 30% pain intensity reduction, using LOCF or a completer analysis, or lasting four to eight weeks).
The third tier of evidence used data from fewer than 200 participants, or where there were expected to be significant problems because, for example, of very short duration studies of less than four weeks; where there was major heterogeneity between studies; or where there were shortcomings in allocation concealment, attrition, or incomplete outcome data. For this third tier of evidence, no data synthesis is reasonable, and may be misleading, but an indication of beneficial effects might be possible.
Subgroup analysis and investigation of heterogeneity
We planned a subgroup analysis for dose of milnacipran.
Sensitivity analysis
We planned no sensitivity analyses.
Results
Description of studies
Results of the search
Searches identified 58 potentially relevant studies in CENTRAL, 141 in MEDLINE, and 288 in EMBASE. In addition to the five studies included in the earlier review, we identified 10 additional potentially relevant reports from the searches. After reading the full reports, we included one new study in the main analysis (Bateman 2013), and two separate reports of an enriched enrolment randomised withdrawal (EERW) trial (Clauw 2013). We excluded seven studies (Ang 2013; Arnold 2012; Häuser 2014; Kim 2013; Matthey 2013; Mease 2014; Spera 2012) (Figure 1).
1.
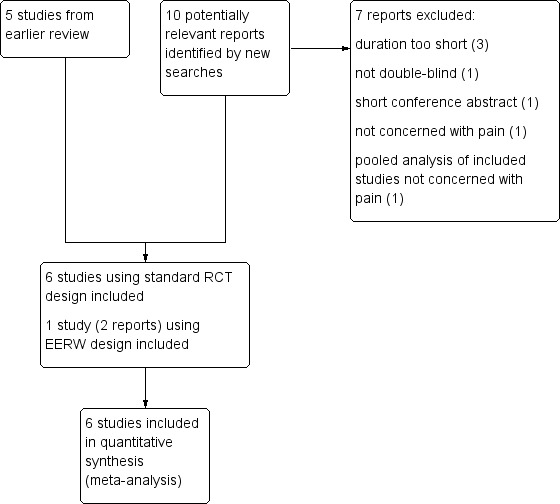
Study flow diagram. EERW: enriched enrolment randomised withdrawal; RCT: randomised controlled trial.
Included studies
Seven studies fulfilled the inclusion criteria. We included five studies from the earlier review, and one new study (100 participants) in the main analysis. We also reported an EERW trial separately (150 participants entered the randomised withdrawal phase). This study included participants who had previously been enrolled in a three‐year, open‐label, flexible‐dosing study of milnacipran for the treatment of fibromyalgia (Arnold 2013), and before that some of the RCTs included in this review.
Most participants were women (92% to 97%) and white (84% to 93%), and the mean age was 47 to 50 years. All except Bateman 2013 had fibromyalgia diagnosed according to American College of Rheumatology (ACR) 1990 criteria (Wolfe 1990) and baseline pain of 60/100 on a VAS, or 10/20 on the Gracely scale. Bateman 2013 did not provide diagnostic criteria and required baseline pain between 40 and 90 out of 100. Full details are in the Characteristics of included studies table.
Arnold 2010: one to four weeks' screening and washout, four to six weeks' flexible dose titration, and 12 weeks of stable dose milnacipran 100 mg/day (50 mg twice daily) or placebo.
Bateman 2013: stable dose of duloxetine 60 mg/day for a minimum of four weeks prior to enrolment, following a two‐week duloxetine 60 mg open‐label period. Direct switch to 10 weeks with milnacipran (100 mg/day with possibility to escalate to 200 mg/day or de‐escalate to 50 to 75 mg/day) or placebo (first week blinded duloxetine 30 mg) (ratio milnacipran:placebo = 4:1).
Branco 2010: similar screening, washout, dose escalation, and stable dose to Arnold 2010, except that the stable target dose was milnacipran 200 mg/day (100 mg twice daily), or placebo. This study also included a nine‐day period of down‐titration, with two‐week follow‐up.
Clauw 2008: similar screening, washout, dose escalation, and stable dose to Arnold 2010, except dose escalation was carried out over three weeks, then 12 weeks at a stable dose of either milnacipran 100 mg/day (50 mg twice daily) or 200 mg/day (100 mg twice daily), or placebo.
Clauw 2013: recruited adults directly from a long‐term, open‐label, flexible‐dose, lead‐in study in which they received milnacipran 50 to 200 mg/day for up to 3.25 years. They had previously received up to 15 months of treatment with milnacipran 100 or 200 mg/day during double‐blind studies, resulting in up to 4.5 years of milnacipran exposure before entering the discontinuation study. Participants taking milnacipran 100 or 200 mg/day and with at least 50% pain reduction in the long term were classified as responders, and were randomised to continuing milnacipran or placebo after a four‐week open run‐in. The study then continued for 12 weeks, with loss of therapeutic response (less than 30% reduction in VAS pain from original baseline) as the primary outcome.
Mease 2009: similar in structure to Arnold 2010, with three weeks of baseline measurement, followed by 24 weeks at a stable dose of milnacipran 100 mg/day (50 mg twice daily) or 200 mg/day (100 mg twice daily), or placebo (ratio 100 mg/day:200 mg/day:placebo = 1:2:1).
Vitton 2004: similar study to Arnold 2010, with two weeks of baseline measurement, four weeks dose titration, and eight weeks stable at a dose of milnacipran 200 mg/day (once daily), milnacipran 200 mg/day (100 mg twice daily), or placebo (ratio once daily:twice daily:placebo = 3:3:2).
A total of 1220 participants received milnacipran 100 mg, 1383 received milnacipran 200 mg, and 1635 received placebo in the standard RCTs, and 226 participants received milnacipran (minimum 100 mg/day) and 111 received placebo in the EERW trial. No study used an active comparator. All studies were carried out over 8 to 24 weeks and all used electronic patient experience diaries (PED) to collect data, except Bateman 2013, which did not give details of the method used to collect data.
Excluded studies
We excluded 10 studies, seven from the updated searches (Ang 2013; Arnold 2012; Branco 2011; Goldenberg 2010; Häuser 2014; Kim 2013; Matthey 2013; Mease 2014; NCT00797797; Spera 2012). Reasons for exclusion are in the Characteristics of excluded studies table.
Risk of bias in included studies
Allocation
All the studies were randomised, but only three adequately described the method used to generate the random sequence (Clauw 2008; Clauw 2013; Vitton 2004). Arnold 2010, Clauw 2008, and Clauw 2013 adequately described the methods used to ensure that allocation of participants to treatment groups was concealed. The remaining four studies did not report the method used (Bateman 2013; Branco 2010; Mease 2009; Vitton 2004).
Blinding
All studies were described as double‐blind, but Mease 2009 and Bateman 2013 did not describe the methods used to ensure that participants and interacting investigators were unable to differentiate between the treatment and control tablets. The other five studies provided adequate information.
Incomplete outcome data
All seven studies used LOCF to input data for participants who withdrew for any reason in analyses of individual pain outcomes. The three studies that reported composite outcomes performed BOCF (any participant who withdrew for any reason was considered a non‐responder) for these analyses (Arnold 2010; Branco 2010; Clauw 2008).
Selective reporting
All seven studies reported the outcomes specified in the methods.
Other potential sources of bias
Bateman 2013 and Vitton 2004 had treatment arms with 51 or fewer participants, making treatment group size an issue. Studies with small group sizes tend to overestimate efficacy (Dechartres 2013; Kjaergard 2001; Nüesch 2010).
Bateman 2013 stated that the participants had the opportunity to escalate to 200 mg/day or de‐escalate to 50 to 75 mg/day. The study did not state if any of the participants used this dose adjustment. For our analyses, we have assumed that all participants were taking 100 mg/day.
See Figure 2 and Figure 3 for a graph and summary of the risk of bias in included studies.
2.
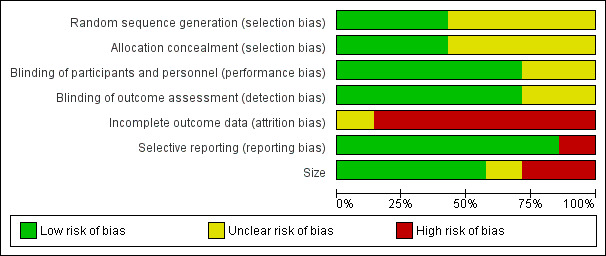
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.
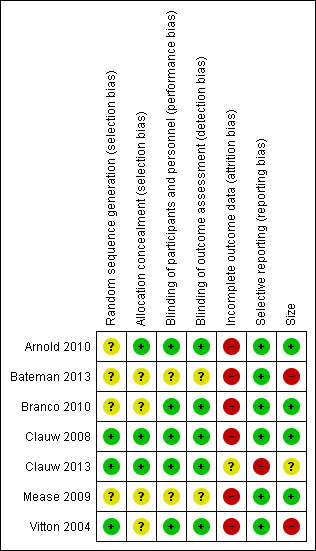
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Effects of interventions
See: Table 1
Details of results in individual studies are in Appendix 5 (efficacy) and Appendix 6 (adverse events and withdrawals).
Efficacy
There were no data for our pre‐specified primary efficacy outcomes that satisfied our criteria for first‐tier evidence because analyses were carried out using LOCF. Some studies also reported composite responder outcomes, which are more difficult to achieve since two or more criteria must be satisfied. Although these were not pre‐specified individual primary efficacy outcomes for this review, we have chosen to report them as they used BOCF analyses (participants who withdrew were considered to be non‐responders and have no pain relief at end of trial), and satisfy our criteria for first‐tier evidence.
Milnacipran 100 mg/day versus placebo
Moderate benefit: second‐tier evidence
Participants experiencing at least 30% improvement from baseline pain intensity, or reporting a PGIC of "much or very much improved" are considered to be experiencing moderate pain relief. Since the same studies reported both outcomes, we used 'at least 30% improvement from baseline' preferentially, but presented both results.
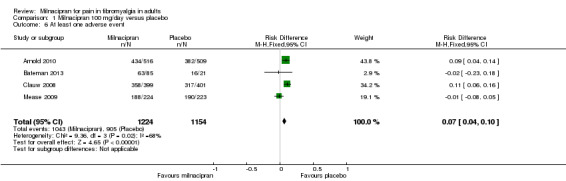
Three studies (1925 participants) contributed data for at least 30% pain relief (Arnold 2010; Bateman 2013; Clauw 2008).
The proportion of participants with at least 30% pain relief with milnacipran 100 mg was 41% (406/994, range 34% to 45%).
The proportion of participants with at least 30% pain relief with placebo was 30% (277/931, range 29% to 31%).
The RR for milnacipran compared with placebo was 1.4 (95% CI 1.2 to 1.6), and the NNT was 9.0 (95% CI 6.5 to 15) for moderate pain relief (Figure 4).
4.
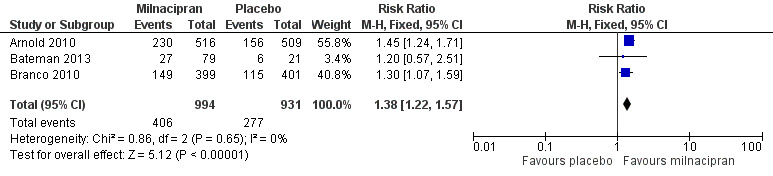
Forest plot of comparison: 1 Milnacipran 100 mg/day versus placebo, outcome: 1.1 At least 30% pain relief.
Using data for participants reporting PGIC gave an RR of 1.5 (1.3 to 1.7), and the NNT was 7.8 (5.9 to 12) for moderate pain relief (Analysis 1.2).
1.2. Analysis.
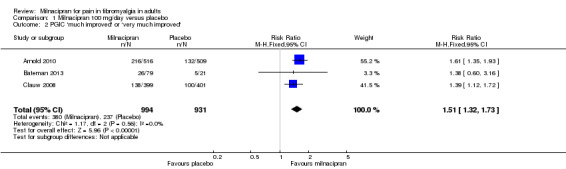
Comparison 1 Milnacipran 100 mg/day versus placebo, Outcome 2 PGIC 'much improved' or 'very much improved'.
Substantial benefit: second‐tier evidence
Two studies (1250 participants) reported at least 50% improvement from baseline pain intensity, which is considered equivalent to substantial benefit (Arnold 2010; Bateman 2013).
The proportion of participants with at least 50% pain relief with milnacipran 100 mg was 27% (190/692, range 27% to 28%).
The proportion of participants with at least 50% pain relief with placebo was 18% (99/558, range 14% to 18%).
The RR for milnacipran compared with placebo was 1.6 (1.3 to 2.0), and the NNT was 10 (6.7 to 20) for substantial pain relief (Analysis 1.3).
1.3. Analysis.
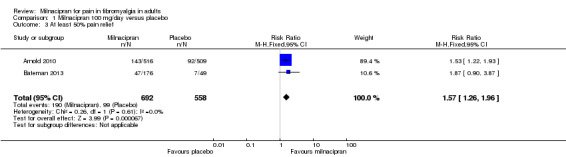
Comparison 1 Milnacipran 100 mg/day versus placebo, Outcome 3 At least 50% pain relief.
Data for at least 30% and at least 50% pain relief and PGIC were reported using LOCF, an imputation method that has been shown to overestimate efficacy in circumstances where there is an imbalance of adverse events withdrawals in test and comparator treatment arms. Some studies also reported composite responder outcomes using BOCF (participants who withdrew were considered to be non‐responders and have no pain relief at end of trial). Composite outcomes are more difficult to achieve, since two or more criteria must be satisfied.
Composite 1: at least 30% pain relief plus PGIC much or very much improved: first‐tier evidence
Three studies (2272 participants) contributed data for this two‐part composite outcome (Arnold 2010; Clauw 2008; Mease 2009).
The proportion of participants achieving Composite 1 with milnacipran 100 mg was 27% (303/1139, range 23% to 29%).
The proportion of participants achieving Composite 1 with placebo was 18% (202/1133, range 18% to 19%).
The RR for milnacipran compared with placebo was 1.5 (1.3 to 1.7), and the NNT was 11 (8.2 to 19) for ≥ 30% pain relief plus PGIC much or very much improved (Analysis 1.4).
1.4. Analysis.
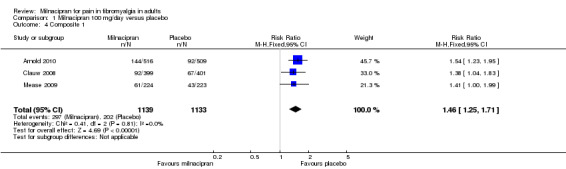
Comparison 1 Milnacipran 100 mg/day versus placebo, Outcome 4 Composite 1.
Composite 2: at least 30% pain relief plus PGIC much or very much improved plus at least a 6‐point improvement in physical function: first‐tier evidence
Three studies (2272 participants) contributed data for the three‐part composite outcome (Arnold 2010; Clauw 2008; Mease 2009).
The proportion of participants achieving Composite 2 with milnacipran 100 mg was 18% (206/1139, range 15% to 20%).
The proportion of participants achieving Composite 2 with placebo was 10% (118/1133, range 9% to 12%).
The RR for milnacipran compared with placebo was 1.7 (1.4 to 2.1), and the NNT was 13 (9.5 to 21) for at least 30% pain relief plus PGIC much or very much improved plus at least a 6‐point improvement in physical function (Analysis 1.5).
1.5. Analysis.
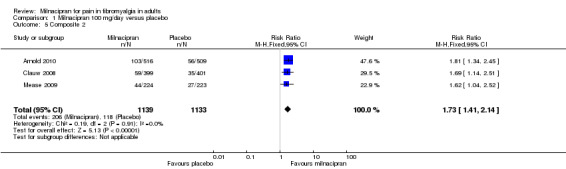
Comparison 1 Milnacipran 100 mg/day versus placebo, Outcome 5 Composite 2.
Milnacipran 200 mg/day versus placebo
Vitton 2004 included two groups of participants receiving milnacipran 200 mg/day, one as a divided dose (100 mg twice daily), and the other as a single dose (200 mg once daily). Since these groups gave numerically very similar responses and individually were too small (51 participants in twice daily group and 46 participants in once daily group) to show a difference reliably, we have combined them for analyses in this review.
Moderate benefit: second‐tier evidence
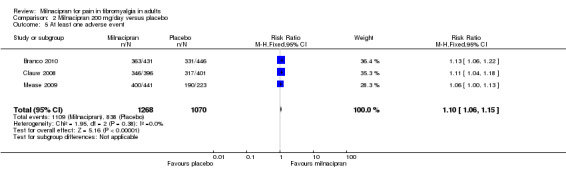
Three studies (1798 participants) contributed data for at least 30% pain relief (Branco 2010; Clauw 2008; Vitton 2004).
The proportion of participants with at least 30% pain relief with milnacipran 200 mg was 39% (361/923, range 35% to 40%).
The proportion of participants with at least 30% pain relief with placebo was 29% (255/875, range 21% to 30%).
The RR for milnacipran compared with placebo was 1.4 (1.2 to 1.5), and the NNT was 10 (7.0 to 18) for moderate pain relief (Analysis 2.1).
2.1. Analysis.
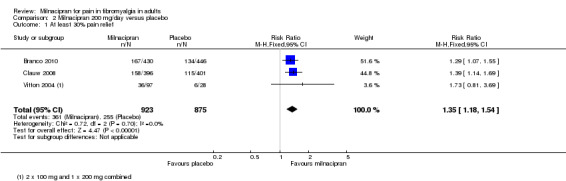
Comparison 2 Milnacipran 200 mg/day versus placebo, Outcome 1 At least 30% pain relief.
Using information from the two studies (1673 participants) with data for participants reporting PGIC gave an RR of 1.6 (1.3 to 1.8), and the NNT was 7.7 (5.8 to 12) for moderate pain relief (Branco 2010; Clauw 2008; Analysis 2.2).
2.2. Analysis.
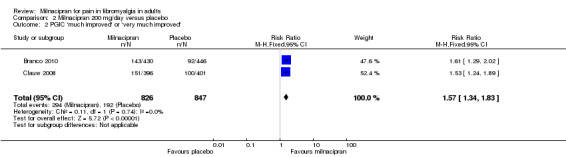
Comparison 2 Milnacipran 200 mg/day versus placebo, Outcome 2 PGIC 'much improved' or 'very much improved'.
Substantial benefit: third‐tier evidence
One study (two small treatment arms) reported at least 50% improvement from baseline pain intensity, which is considered equivalent to substantial benefit (Vitton 2004); 29% (15/51) of participants treated with milnacipran 100 mg/day twice daily, 26% of participants treated with 200 mg once daily, and 14% (4/28) of participants treated with placebo experienced this outcome.
Composite 1: at least 30% pain relief plus PGIC much or very much improved: first‐tier evidence
Three studies (2337 participants) contributed data for the two‐part composite outcome (Branco 2010; Clauw 2008; Mease 2009).
The proportion of participants achieving Composite 1 with milnacipran 200 mg was 25% (320/1267, range 23% to 27%).
The proportion of participants achieving Composite 1 with placebo was 16% (170/1070, range 13% to 19%).
The RR for milnacipran compared with placebo was 1.6 (1.3 to 1.8). The NNT was 11 (7.9 to 16) for at least 30% pain relief plus PGIC much or very much improved (Analysis 2.3).
2.3. Analysis.
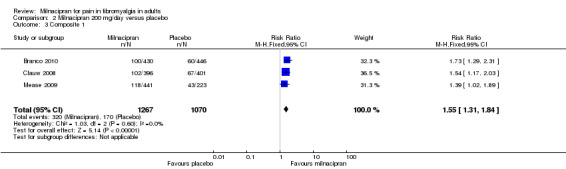
Comparison 2 Milnacipran 200 mg/day versus placebo, Outcome 3 Composite 1.
Composite 2: at least 30% pain relief plus PGIC much or very much improved plus at least a 6‐point improvement in physical function: first‐tier evidence
Two studies (1461 participants) contributed data for the three‐part composite outcome (Clauw 2008; Mease 2009).
The proportion of participants achieving Composite 2 with milnacipran 200 mg was 17% (141/837, range 14% to 19%).
The proportion of participants achieving Composite 2 with placebo was 10% (62/624, range 9% to 12%).
The RR for milnacipran compared with placebo was 1.6 (1.2 to 2.1). The NNT was 14 (9.7 to 29) for at least 30% pain relief plus PGIC much or very much improved plus at least a 6‐point improvement in physical function (Analysis 2.4).
2.4. Analysis.
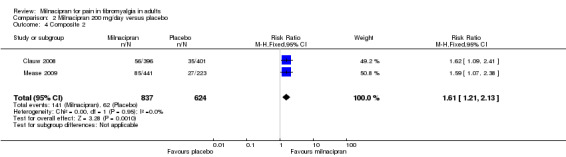
Comparison 2 Milnacipran 200 mg/day versus placebo, Outcome 4 Composite 2.
| Summary of results A: Efficacy outcomes with different doses of milnacipran in fibromyalgia | ||||||
| Outcome ‐ daily dose | Number of | Per cent with outcome | Relative risk (RR (95% CI)) | NNT (95% CI) | ||
| Studies | Participants | Milnacipran | Placebo | |||
| Moderate response: ≥ 30% pain relief | ||||||
| 100 mg | 3 | 1925 | 41 | 30 | 1.4 (1.2 to 1.6) | 9 (6.5 to 15) |
| 200 mg | 3 | 1798 | 39 | 29 | 1.4 (1.2 to 1.5) | 10 (7.0 to 18) |
| Moderate response: PGIC much or very much improved | ||||||
| 100 mg | 3 | 1925 | 38 | 25 | 1.5 (1.3 to 1.7) | 7.8 (5.9 to 12) |
| 200 mg | 2 | 1673 | 36 | 23 | 1.6 (1.3 to 1.8) | 7.7 (5.8 to 12) |
| Substantial response: ≥ 50% pain relief | ||||||
| 100 mg | 2 | 1250 | 27 | 18 | 1.6 (1.3 to 2.0) | 10 (7.0 to 20) |
| 200 mg | 1 | 125 | 28 | 14 | Not calculated | Not calculated |
| Composite outcome 1 | ||||||
| 100 mg | 3 | 2272 | 27 | 18 | 1.5 (1.3 to 1.7) | 11 (8.2 to 19) |
| 200 mg | 3 | 2337 | 25 | 16 | 1.6 (1.3 to 1.8) | 11 (7.9 to 16) |
| Composite outcome 2 | ||||||
| 100 mg | 3 | 2272 | 18 | 10 | 1.7 (1.4 to 2.1) | 13 (9.5 to 21) |
| 200 mg | 2 | 1461 | 17 | 10 | 1.6 (1.2 to 2.1) | 14 (9.7 to 29) |
| Composite 1: ≥ 30% improvement from baseline pain intensity + patient global impression of change (PGIC) much or very much improved Composite 2: ≥ 30% improvement from baseline pain intensity + PGIC much or very much improved + ≥ 6‐point improvement in physical function Moderate and substantial responses analysed using last observation carried forward; composite outcomes analysed using baseline observation carried forward | ||||||
Enriched enrolment randomised withdrawal trial
The principal outcome of loss of therapeutic response occurred in 35% of participants with milnacipran and 64% of participants with placebo(Clauw 2013).
Adverse events
Participants with any adverse event
All the RCTs provided some information on participants experiencing adverse events over the study period, which was collected from spontaneous reports, and clinical observation and evaluation. Vitton 2004 did not provide data suitable for pooled analysis.
Four studies provided data on the number of participants experiencing one or more adverse events with milnacipran 100 mg (2378 participants) and 200 mg (2338 participants) (Arnold 2010; Bateman 2013; Clauw 2008; Mease 2009). Most participants with both milnacipran and placebo reported at least one adverse event over the period, with no difference between milnacipran 200 and 100 mg/day.
For milnacipran 100 mg versus placebo, 85% of participants with active treatment and 78% with placebo had an event, and for milnacipran 200 mg versus placebo, 87% of participants with active treatment and 78% with placebo had an event, giving a RR of 1.1 (1.06 to 1.15) for both doses. The NNH was 15 (10 to 27) for milnacipran 100 mg/day (Analysis 1.6), and 11 (8.2 to 16) for milnacipran 200 mg/day (Analysis 2.5). For milnacipran 100 mg/day, the I2 was 68% using a fixed effect model; using a random‐effects model did not significantly change the estimate.
1.6. Analysis.
Comparison 1 Milnacipran 100 mg/day versus placebo, Outcome 6 At least one adverse event.
2.5. Analysis.
Comparison 2 Milnacipran 200 mg/day versus placebo, Outcome 5 At least one adverse event.
In the EERW study, adverse events in the double‐blind phase were experienced by 47% of participants taking milnacipran, and 58% taking placebo (Clauw 2013).
Serious adverse events
Five of the RCTs reported serious adverse events; 1.5% (18/1224) with milnacipran 100 mg and 1.6% (18/1154) with placebo, and 1.9% (26/1365) with milnacipran 200 mg, and 2.1% (23/1098) with placebo. There were no significant differences between either dose of milnacipran and placebo (milnacipran 100 mg/day: RR 0.90 (0.47 to 1.7) Analysis 1.7; milnacipran 200 mg/day: 0.91 (0.52 to 1.6) Analysis 2.6), or the two doses of milnacipran.
1.7. Analysis.
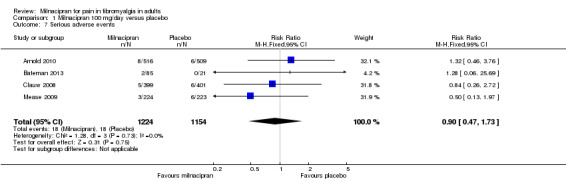
Comparison 1 Milnacipran 100 mg/day versus placebo, Outcome 7 Serious adverse events.
2.6. Analysis.
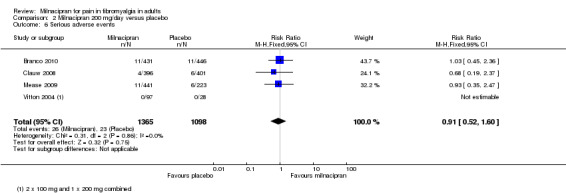
Comparison 2 Milnacipran 200 mg/day versus placebo, Outcome 6 Serious adverse events.
Specific adverse events
Five RCTs provided data for numbers of participants experiencing specific adverse events (Arnold 2010; Bateman 2013; Branco 2010; Clauw 2008; Mease 2009). Rates for each event were very similar for both doses of milnacipran, and are presented below for both doses combined (see 'Summary of results B' table), where there was a significant difference from placebo (Figure 5; Analysis 2.7). Sinusitis, fatigue, diarrhoea, and upper respiratory tract infection were not more common with milnacipran than with placebo. With milnacipran 100 mg/day, the I2 was 66% for nausea and 51% for increased heart rate, and with milnacipran 200 mg/day the I2 was 64% for nausea and 51% for increased heart rate, using a fixed‐effect model; using a random‐effects model did not significantly change the estimate.
5.
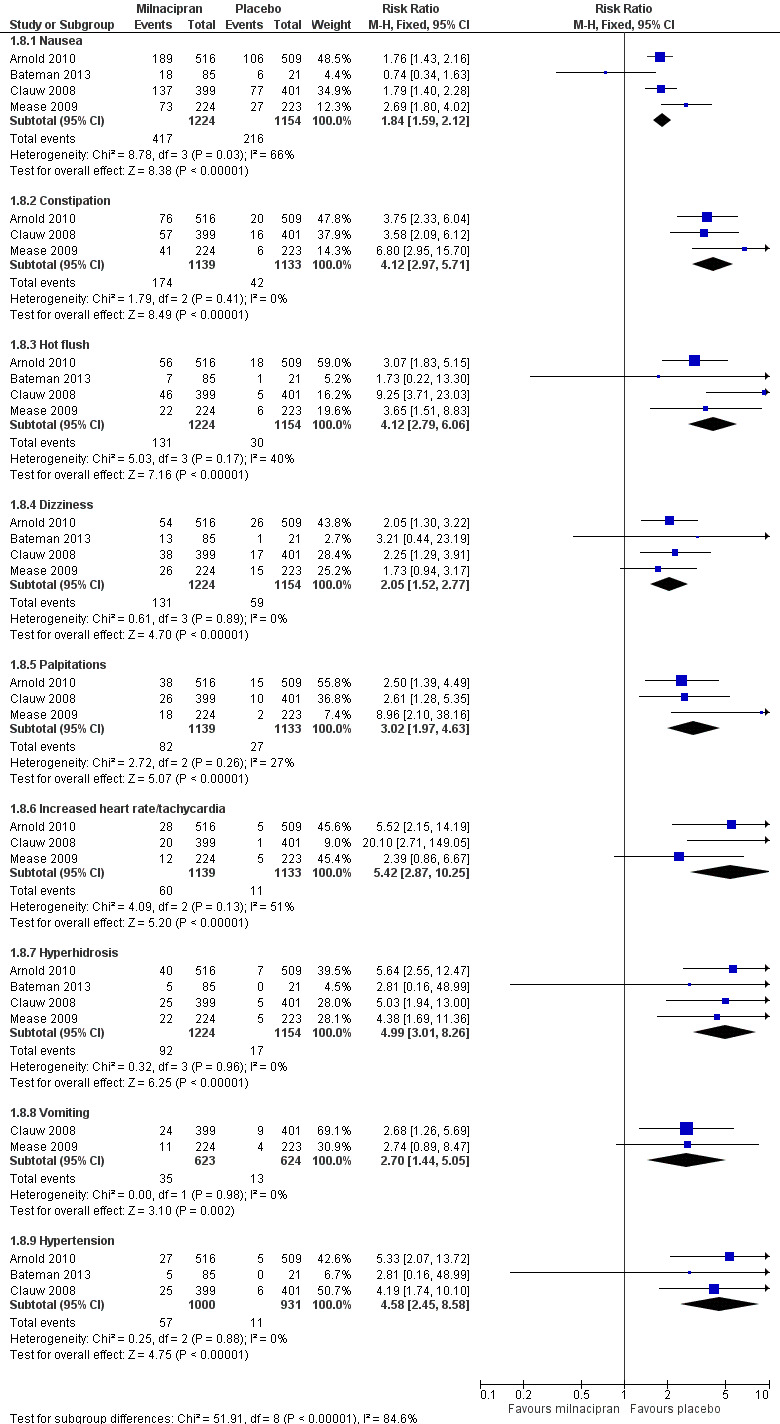
Forest plot of comparison: 1 Milnacipran 100 mg/day versus placebo, outcome: 1.8 Individual adverse events.
2.7. Analysis.
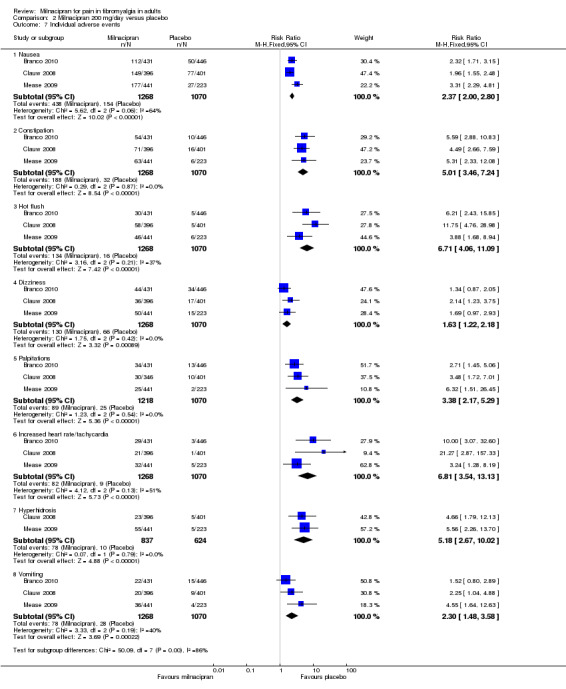
Comparison 2 Milnacipran 200 mg/day versus placebo, Outcome 7 Individual adverse events.
| Summary of results B: Adverse events with different doses of milnacipran in fibromyalgia | ||||||
| Outcome ‐ daily dose | Number of | Per cent with outcome | Risk ratio (RR (95% CI)) | NNH (95% CI) | ||
| Studies | Participants | Milnacipran | Placebo | |||
| Any adverse event | ||||||
| 100 mg | 4 | 2378 | 85 | 78 | 1.1 (1.05 to 1.1) | 15 (10 to 27) |
| 200 mg | 3 | 2338 | 87 | 78 | 1.1 (1.06 to 1.15 | 11 (8.2 to 16) |
| Serious adverse event | ||||||
| 100 mg | 4 | 2378 | 1.5 | 1.6 | 0.90 (0.47 to 1.7) | Not calculated |
| 200 mg | 4 | 2463 | 1.9 | 2.1 | 0.91 (0.52 to 1.6) | Not calculated |
| Specific adverse events ‐ 100 and 200 mg combined | ||||||
| Nausea | 5 | 4092 | 34 | 18 | 2.1 (1.8 to 2.3) | 5.7 (4.9 to 6.6) |
| Constipation | 4 | 3986 | 15 | 3.3 | 4.5 (3.4 to 6.0) | 13 (10 to 18) |
| Hot flush | 5 | 4092 | 11 | 2 | 4.9 (3.4 to 6.9) | 12 (10 to 14) |
| Dizziness | 5 | 4092 | 10 | 6 | 1.8 (1.4 to 2.3) | 21.5 (16 to 33) |
| Palpitations | 4 | 3986 | 7.1 | 2.5 | 3.1 (2.2 to 4.3) | 25 (17 to 42) |
| Tachycardia/increased heart rate | 4 | 3986 | 5.9 | 0.9 | 5.2 (3.2 to 8.6) | 41 (27 to 67) |
| Hyperhidrosis | 4 | 3215 | 8 | 1 | 5.6 (3.6 to 9.2) | 14.8 (12 to 19) |
| Vomiting | 3 | 2961 | 6.0 | 1.8 | 3.4 (2.0 to 5.7) | 38 (21 to 94) |
| Hypertension | 3 | 2327 | 5 | 1 | 4.4 (2.3 to 8.2) | 25.2 (19 to 38) |
Withdrawals
All studies provided data on withdrawals over the study period for all causes, due to lack of efficacy and due to adverse events.
All‐cause withdrawals
For milnacipran 100 mg versus placebo, 35% (425/1225) of participants with active treatment and 30% (349/1154) with placebo withdrew for any reason, giving a RR of 1.1 (1.01 to 1.3). The NNH was 23 (12 to 140) (Analysis 1.9).
1.9. Analysis.
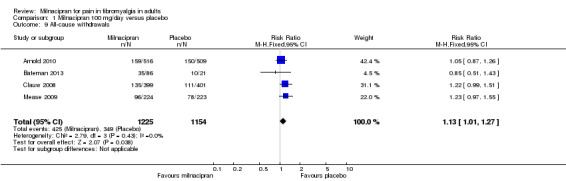
Comparison 1 Milnacipran 100 mg/day versus placebo, Outcome 9 All‐cause withdrawals.
For milnacipran 200 mg versus placebo, 36% (488/1364) of participants with active treatment and 24% (268/1098) with placebo withdrew for any reason, giving a RR of 1.4 (1.2 to 1.6). The NNH was 8.8 (6.7 to 13) (Analysis 2.8).
2.8. Analysis.
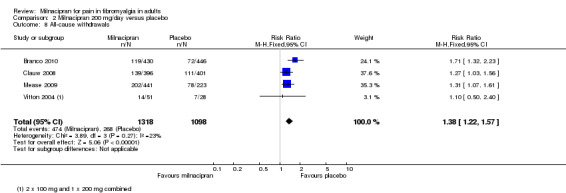
Comparison 2 Milnacipran 200 mg/day versus placebo, Outcome 8 All‐cause withdrawals.
Z = 2.6068, P = 0.009 for difference between doses.
Lack of efficacy withdrawals
For milnacipran 100 mg versus placebo, 7% (86/1225) of participants with active treatment and 9.4% (109/1154) with placebo withdrew due to lack of efficacy, giving a RR of 0.72 (0.55 to 0.94). The NNTp was 41 (22 to 470) (Analysis 1.10).
1.10. Analysis.
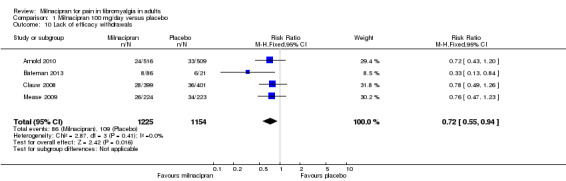
Comparison 1 Milnacipran 100 mg/day versus placebo, Outcome 10 Lack of efficacy withdrawals.
For milnacipran 200 mg versus placebo, 7.2% (98/1364) of participants with active treatment and 10% (106/1098) with placebo withdrew due to lack of efficacy, giving a RR of 0.66 (0.51 to 0.87). The NNTp was 41 (21 to 400) (Analysis 2.9).
2.9. Analysis.
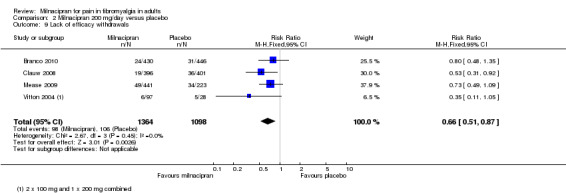
Comparison 2 Milnacipran 200 mg/day versus placebo, Outcome 9 Lack of efficacy withdrawals.
There was no significant difference between the two doses.
Adverse event withdrawals
For milnacipran 100 mg versus placebo, 19% (229/1225) of participants with active treatment and 12% (134/1154) with placebo withdrew due to an adverse event, giving a RR of 1.6 (1.3 to 2.0). The NNH was 14 (10 to 24) (Analysis 1.11).
1.11. Analysis.
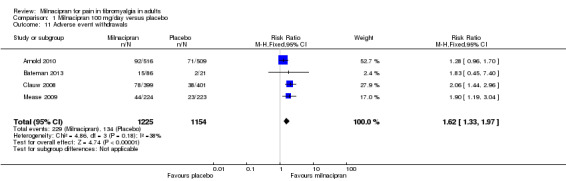
Comparison 1 Milnacipran 100 mg/day versus placebo, Outcome 11 Adverse event withdrawals.
For milnacipran 200 mg versus placebo, 24% (325/1364) of participants with active treatment and 9.5% (104/1098) with placebo withdrew due to adverse events, giving a RR of 2.5 (2.0 to 3.1). The NNH was 7.0 (5.8 to 8.7) (Analysis 2.10).
2.10. Analysis.
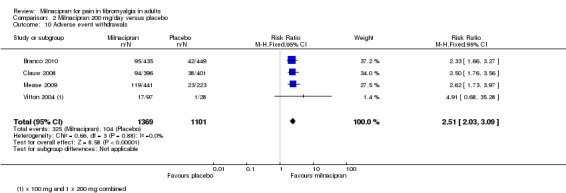
Comparison 2 Milnacipran 200 mg/day versus placebo, Outcome 10 Adverse event withdrawals.
For the difference between doses, Z = 3.5314 and P value = 0.0004.
Death
None of the studies reported any deaths.
Discussion
Summary of main results
Participants in these studies were all treated for fibromyalgia symptoms, including pain, with milnacipran or placebo.
The review included six randomised, double‐blind studies in which over 4000 participants were titrated to a target dose of milnacipran 100 mg, milnacipran 200 mg, or placebo, and assessed following a stable dose period of 8 to 24 weeks.
None of the studies reported first‐tier evidence for our primary outcomes because they used LOCF for analyses. Using second‐tier evidence, a moderate response was experienced by 40% of participants treated with either dose of milnacipran, using either measurement, while response to placebo was about 10% lower (30%) using at least 30% pain relief. NNTs were 6.1 to 10, with no significant difference between doses or outcome measurement (high quality evidence). Two studies reported an outcome equivalent to substantial improvement for milnacipran 100 mg (NNT 10) (moderate quality evidence).
The single EERW study showed an absolute difference between milnacipran and placebo of about 30% in terms of loss of therapeutic response in responders; as only 44% of the original sample were responders, this implies an NNT in the whole sample of about 7 or 8, consistent with the findings in classical placebo controlled studies. Interpretation is difficult, though, as the time between baseline and the study while on open‐label milnacipran was around four years.
Some studies reported two further outcomes. These were composite outcomes, one requiring response to both 30% or greater pain relief and PGIC (Composite 1) and the other requiring response to 30% or greater pain relief, PGIC, and a measure of physical function (Composite 2). These outcomes, while not pre‐specified in the protocol, were analysed because they more closely represent what people with fibromyalgia want from treatment, and because they were calculated using BOCF, without imputation for missing data, which reflects the real world of clinical practice. These outcomes satisfy our criteria for first‐tier evidence. Response rates for both doses of milnacipran were around 25% for Composite 1 and 18% for composite 2, and 8% or 9% lower with placebo, giving NNTs of 11 to 14, with no significant difference between doses or outcome (high quality evidence). The higher (worse) NNTs for the composite outcomes reflect the fact that these are more difficult to achieve than the individual components alone, as well as a potential for overestimation of treatment effect with imputation of efficacy results from participants who have withdrawn from the study (Moore 2010a).
Adverse events were experienced by the majority (about 80%) of participants in all treatment groups, but were slightly more common with milnacipran than placebo, giving NNHs of 11 to 13 (high quality evidence). There were few serious adverse events (< 2%) and they were no more frequent with milnacipran than placebo (low quality evidence). A number of specific adverse events were more common with milnacipran, and in particular nausea and constipation, with NNHs of 7 for nausea and 13 for constipation (moderate quality evidence). The range of adverse events reported was typical of those associated with SNRIs. There was no difference between the doses. It has been suggested that twice daily dosing is better tolerated than once daily, but only one treatment arm in one study used once daily dosing, so we were unable to evaluate this.
Withdrawals for any reason were more common with milnacipran than placebo, and more common with 200 mg/day than 100 mg/day (NNH of 8.8 for 200 mg/day and 23 for 100 mg/day, compared with placebo). This was largely driven by adverse event withdrawals, where the NNH compared with placebo was 7.0 for 200 mg/day and 14 for 100 mg/day (high quality evidence). Withdrawals due to lack of efficacy were more common with milnacipran than placebo but did not differ between doses (NNTp of 45 for 200 mg/day and 41 for 100 mg/day) (moderate quality evidence).
Overall completeness and applicability of evidence
Included studies were not of sufficient duration to determine long‐term use, but extension studies have demonstrated efficacy and tolerability for some participants up to three years (Arnold 2013; Branco 2011; Goldenberg 2010). Studies have not been conducted across all ethnic groups and have excluded participants with other major medical and mental disorders. This may limit applicability in clinical practice.
Quality of the evidence
Studies, with the exception of Vitton 2004 and Bateman 2013, had large sample sizes (more than 200 in each arm), thereby minimising the effects of randomisation by chance and small‐study bias. While these studies reported our preferred dichotomous outcome measures, the individual outcomes were analysed using LOCF as the imputation method for missing data. Where there is an imbalance of withdrawals due to lack of efficacy or adverse events between active and placebo treatment arms, this may lead to an overestimate of efficacy (Moore 2010a). For this reason, we chose also to review data on two composite outcomes where present, since these were presented using BOCF for withdrawals (ie no imputation; withdrawal = non‐responder).
Other aspects of methodological quality were good.
Potential biases in the review process
We used an extensive search strategy, but we can never be certain that some studies have not been identified. We calculated the number of participants who would need to be in trials with zero effect (RR 1.0) needed for the point estimate of the NNT to increase beyond a clinically useful level (Moore 2008). An additional 642 participants treated with milnacipran 100 mg or 360 treated with milnacipran 200 mg would need to be in unidentified studies with zero effect to change the NNT to 12. This is possible, but somewhat unlikely for the 100 mg dose.
Of greater concern is the use of LOCF as the imputation method for the primary outcomes of this review. Recent investigations of the effects of imputation methods in chronic pain trials have shown that LOCF overestimates efficacy outcomes compared with calculations where participants who withdraw from treatment are regarded as not being able to have meaningful pain relief (Moore 2011a). This is particularly the case where there is an imbalance between comparator groups for withdrawals, especially due to adverse events; this may be particularly important for the 200 mg dose, where there was a 12% absolute increase in all‐cause withdrawals for milnacipran over placebo. Where NNTs are relatively large, as here with milnacipran, results using LOCF can overstate the efficacy of the drug.
Agreements and disagreements with other studies or reviews
Bernstein 2013 reviewed the use of milnacipran to treat fibromyalgia, but did not carry out any pooled analyses, concluding that it "provides modest fibromyalgia pain relief and is best used as part of a multidisciplinary treatment approach". Kyle 2010, Recla 2010, and Ormseth 2010 did not carry out any meta‐analyses, but reported that milnacipran was effective and reasonably well tolerated. Häuser 2011 compared pregabalin, duloxetine, and milnacipran (apparently all doses combined), and reported a slightly higher NNT for at least 30% pain relief (19, versus 10 for 200 mg/day in this review), a lower NNTp for lack of efficacy (31, versus 41 for 200 mg/day in this review), and a similar NNH for adverse event withdrawals (7.6, versus 7.0 for 200 mg/day in this review). These differences can be explained by the fact that fewer studies were included. In a more recent review, Hauser et al. carried out a combined analysis for duloxetine and milnacipran, and reported an NNT for at least 30% pain reduction of 11 (95% CI 8 to 14), and an NNT for at least 50% pain reduction of 9 (7 to 11) (Häuser 2013).
Hauser et al also updated their indirect comparisons of duloxetine and milnacipran to incorporate the two most recent studies, and to add a comparison with amitriptyline (Häuser 2011). No data were reported in a way that could be compared with this review.
Two reviews have examined both direct and indirect comparisons of drug treatments in fibromyalgia. One included only a single milnacipran study (Choy 2011), and the other included two studies (Roskell 2011). Neither review had sufficient data to arrive at any definitive conclusions, other than several treatments, including milnacipran, had better results than placebo, although not by a large margin.
Authors' conclusions
Implications for practice.
For people with fibromyalgia
Milnacipran gives good pain relief to some people with fibromyalgia, but only a minority of them; it will not work for most people. This is also the case for other treatments for fibromyalgia, such as duloxetine and pregabalin. People who take milnacipran are likely to experience adverse events, which may be troublesome. Milnacipran is not available to treat fibromyalgia in many countries.
For clinicians
Milnacipran gives really good pain relief to some people with fibromyalgia, but only a minority of them; it will not work for most people. This is also the case for other treatments for fibromyalgia, such as duloxetine and pregabalin. Milnacipran is not licensed for fibromyalgia in many countries. Potential benefit is accompanied by increased numbers experiencing adverse events and stopping their medication.
Since relatively few participants achieve a worthwhile response with milnacipran, it is important to establish stopping rules, so that when someone does not respond within a specified time, they can be switched to an alternative treatment. This will reduce the number of participants exposed to adverse events in the absence of benefit.
For policy‐makers
Since no single treatment is effective in a majority of individuals with fibromyalgia, this relatively small number who benefit may be considered worthwhile, particularly if appropriate stopping rules are in place.
For funders
Milnacipran may be worth considering as a potential treatment, as there are few proven effective treatments.
Implications for research.
General
Because the trials in this review used the last observation carried forward (LOCF) imputation method for study withdrawals, post‐hoc individual participant level analyses using baseline observation carried forward (BOCF) would be appropriate to strengthen the findings, especially if the pain reduction were linked to improved quality of life and function. Future studies should also investigate the relationship between pain relief and other fibromyalgia symptoms using outcomes that are relevant to people with fibromyalgia, such as the proportion of participants whose sleep or fatigue levels return to normal values for the population, or by at least 30% or 50%.
Design
The design of trials is adequate, but reporting of clinically relevant outcomes using appropriate imputation for withdrawal would improve the relevance of the findings for clinical practice.
Measurement (endpoints)
Assessment of fibromyalgia symptoms should be based on dichotomous outcomes of participant reported, proven clinical utility.
Comparison between active treatments
Studies involving other treatments including non‐pharmacological interventions may be valuable in this context. A multi‐component approach reflects current practice.
What's new
| Date | Event | Description |
|---|---|---|
| 22 May 2019 | Review declared as stable | See Published notes |
History
Protocol first published: Issue 1, 2010 Review first published: Issue 3, 2012
| Date | Event | Description |
|---|---|---|
| 3 May 2017 | Review declared as stable | See Published notes. |
| 4 November 2015 | Amended | Minor typo corrected. |
| 18 May 2015 | New citation required but conclusions have not changed | Small amount of additional information did not change or strengthen conclusions. |
| 18 May 2015 | New search has been performed | Updated review examining only fibromyalgia. Split from previous review "Milnacipran for neuropathic pain and fibromyalgia in adults" published in 2012. This update has one new study in the main analysis (Bateman 2013), and two separate reports of an additional enriched enrolment randomised withdrawal (EERW) trial (Clauw 2013). These add 251 new participants. Latest search date 18 May 2015. |
Notes
2017
In May 2017, this review was stabilised following discussion with the authors and editors. Restricted searches identified two new studies (Ahmed 2016; Staud 2015), but we judged that including them would not affect the conclusions of the review. If appropriate, we will update the review if new evidence likely to change the conclusions is published, or if standards change substantially which necessitate major revisions.
Staud R, Lucas YE, Price DD, Robinson ME. Effects of milnacipran on clinical pain and hyperalgesia of patients with fibromyalgia: results of a 6‐week randomized controlled trial. J Pain. 2015 Aug;16(8):750‐9. doi: 10.1016/j.jpain.2015.04.010. (46 participants)
Ahmed M, Aamir R, Jishi Z, Scharf MB. The Effects of Milnacipran on Sleep Disturbance in Fibromyalgia: A Randomized, Double‐Blind, Placebo‐Controlled, Two‐Way Crossover Study. J Clin Sleep Med. 2016 Jan;12(1):79‐86. doi: 10.5664/jcsm.5400. (19 participants)
2019
A restricted search in May 2019 identified one small relevant study (Pickering 2018), but did not identify any potentially relevant studies likely to change the conclusions. Therefore, this review has now been stabilised following discussion with the authors and editors. The review will be re‐assessed for updating in five years. If appropriate, we will update the review before this date if new evidence likely to change the conclusions is published, or if standards change substantially which necessitates major revisions.
Pickering G, Macian N, Delage N, Picard P, Cardot JM, Sickout‐Arondo S, Giron F, Dualé C, Pereira B, Marcaillou F. Milnacipran poorly modulates pain in patients suffering from fibromyalgia: a randomized double‐blind controlled study. Drug Des Devel Ther. 2018 Aug 10;12:2485‐2496. doi: 10.2147/DDDT.S162810. eCollection 2018.
Acknowledgements
Dipender Gill was an author on the original review.
Cochrane Review Group funding acknowledgement: The National Institute for Health Research (NIHR) is the largest single funder of the Cochrane Pain, Palliative and Supportive Care Group. Disclaimer: The views and opinions expressed therein are those of the review authors and do not necessarily reflect those of the NIHR, National Health Service (NHS), or the Department of Health.
We wish thank peer reviewers for their useful comments.
Appendices
Appendix 1. Methodological considerations for chronic pain
There have been several changes in how the efficacy of conventional and unconventional treatments is assessed in chronic painful conditions. The outcomes are now better defined, particularly with new criteria for what constitutes moderate or substantial benefit (Dworkin 2008); older trials may only report participants with 'any improvement'. Newer trials tend to be larger, avoiding problems from the random play of chance. Newer trials also tend to be of longer duration, up to 12 weeks, and longer trials provide a more rigorous and valid assessment of efficacy in chronic conditions. New standards have evolved for assessing efficacy in neuropathic pain, and we are now applying stricter criteria for the inclusion of trials and assessment of outcomes, and are more aware of problems that may affect our overall assessment. To summarise some of the recent insights that must be considered in this new review:
Pain results tend to have a U‐shaped distribution rather than a bell‐shaped distribution. This is true in acute pain (Moore 2011a; Moore 2011b), back pain (Moore 2010d), and arthritis (Moore 2010b), as well as in fibromyalgia (Straube 2010); in all cases average results usually describe the experience of almost no‐one in the trial. Data expressed as averages are potentially misleading, unless they can be proven to be suitable.
As a consequence, we have to depend on dichotomous results (the individual either has or does not have the outcome) usually from pain changes or patient global assessments. The Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) group has helped with their definitions of minimal, moderate, and substantial improvement (Dworkin 2008). In arthritis, trials of less than 12 weeks duration, and especially those shorter than eight weeks, overestimate the effect of treatment (Moore 2010b); the effect is particularly strong for less effective analgesics, and this may also be relevant in neuropathic‐type pain.
The proportion of patients with at least moderate benefit can be small, even with an effective medicine, falling from 60% with an effective medicine in arthritis to 30% in fibromyalgia (Moore 2009; Moore 2010b; Moore 2013b; Moore 2014b; Moore 2014c; Straube 2008; Sultan 2008). One Cochrane review of pregabalin in neuropathic pain and fibromyalgia demonstrated different response rates for different types of chronic pain (higher in diabetic neuropathy and postherpetic neuralgia and lower in central pain and fibromyalgia) (Moore 2009). This indicates that different neuropathic pain conditions should be treated separately from one another, and that pooling should not be done unless there are good reasons for doing so.
Individual patient analyses indicate that patients who get good pain relief (moderate or better) have major benefits in many other outcomes, affecting quality of life in a significant way (Hoffman 2010; Moore 2010c; Moore 2014a).
Imputation methods such as last observation carried forward (LOCF), used when participants withdraw from clinical trials, can overstate drug efficacy especially when adverse event withdrawals with drug are greater than those with placebo (Moore 2012b).
Appendix 2. CENTRAL search strategy
MESH DESCRIPTOR Pain EXPLODE ALL TREES (30104)
MESH DESCRIPTOR Peripheral Nervous System Diseases EXPLODE ALL TREES (2571)
MESH DESCRIPTOR Somatosensory Disorders EXPLODE ALL TREES (705)
MESH DESCRIPTOR Fibromyalgia THIS TERM ONLY (529)
MESH DESCRIPTOR Myofascial Pain Syndromes EXPLODE ALL TREES (339)
MESH DESCRIPTOR Polymyalgia Rheumatica EXPLODE ALL TREES (44)
((pain* or discomfort*) and (central or complex or rheumat* or muscl* or muscul* or myofasci* or nerv* or neuralg* or neuropath*)): TI,AB,KY (18421)
(fibromyalgi* or fibrosti* or FM or FMS): TI,AB,KY (1887)
((neur* or nerv*) and (compress* or damag*)): TI,AB,KY (2028)
#1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 (46698)
milnacipr*:TI,AB,KY (168)
(F‐2207 or Midalcipran or Dalcipran or Ixel or Toledomin or Savella or Milnatsiprana or Iksel):TI,AB,KY (9)
#11 OR #12 (170)
#10 AND #13 (58)
Appendix 3. MEDLINE (via Ovid) search strategy
exp PAIN/ (317821)
exp PERIPHERAL NERVOUS SYSTEM DISEASES/ (119352)
exp SOMATOSENSORY DISORDERS/ (16913)
FIBROMYALGIA/ (6468)
exp MYOFASCIAL PAIN SYNDROMES/ (5832)
POLYMYALGIA RHEUMATICA/ (2185)
((pain* or discomfort*) adj10 (central or complex or rheumat* or muscl* or muscul* or myofasci* or nerv* or neuralg* or neuropath*)).mp. (65028)
(fibromyalgi* or fibrosti* or FM or FMS).mp. (20704)
((neur* or nerv*) adj6 (compress* or damag*)).mp. (49796)
1 or 2 or 3 or 4 or 5 or 6 or 7 or 8 or 9 (498511)
milnacipr*.mp. (507)
(F‐2207 or Midalcipran or Dalcipran or Ixel or Toledomin or Savella or Milnatsiprana or Iksel).mp. (23)
11 or 12 (510)
randomized controlled trial.pt. (394270)
controlled clinical trial.pt. (89400)
randomized.ab. (291237)
placebo.ab. (152068)
drug therapy.fs. (1771119)
randomly.ab. (205257)
trial.ab. (300541)
groups.ab. (1308416)
14 or 15 or 16 or 17 or 18 or 19 or 20 or 21 (3340584)
10 and 13 and 22 (141)
Appendix 4. EMBASE (via Ovid) search strategy
exp PAIN/ (895433)
exp PERIPHERAL NERVOUS SYSTEM DISEASES/ (53098)
exp SOMATOSENSORY DISORDERS/ (68644)
FIBROMYALGIA/ (14202)
exp MYOFASCIAL PAIN SYNDROMES/ (6762)
POLYMYALGIA RHEUMATICA/ (3632)
((pain* or discomfort*) adj10 (central or complex or rheumat* or muscl* or muscul* or myofasci* or nerv* or neuralg* or neuropath*)).mp. (123813)
(fibromyalgi* or fibrosti* or FM or FMS).mp. (32450)
((neur* or nerv*) adj6 (compress* or damag*)).mp. (70482)
1 or 2 or 3 or 4 or 5 or 6 or 7 or 8 or 9 (1057623)
milnacipr*.mp. (2122)
(F‐2207 or Midalcipran or Dalcipran or Ixel or Toledomin or Savella or Milnatsiprana or Iksel).mp. (206)
11 or 12 (2128)
crossover‐procedure/ (42840)
double‐blind procedure/ (122720)
randomized controlled trial/ (373012)
(random* or factorial* or crossover* or cross over* or cross‐over* or placebo* or (doubl* adj blind*) or assign* or allocat*).tw. (1323768)
14 or 15 or 16 or 17 (1404517)
10 and 13 and 18 (288)
Appendix 5. Summary of outcomes: efficacy
| Study | Participant characteristics | Maximum daily dose of milnacipran Titration/fixed | Numbers in trial |
Efficacy Single endpoint |
Efficacy Composite endpoint |
| Arnold 2010 | M 48, F 977 Mean age 49 years 91% white Mean duration of symptoms 10.8 years Baseline PI > 60/100 mm | 1‐ to 4‐week screening and washout
4‐ to 6‐week flexible dose titration
12‐week stable dose
2‐week randomised discontinuation (not considered here)
Target dose 100 mg/day (2 x 50 mg) Treatment duration Max 18 weeks Stable 12 weeks |
N = 1025 Miln = 516 Plac = 509 |
LOCF 24‐h recall PI* ≥ 30% improvement from baseline Miln = 230/516 Plac = 156/509 24‐h recall PI* ≥ 50% improvement from baseline Miln = 143/516 Plac = 92/509 PGIC much improved Miln = 216/516 Plac = 132/509 PCS ≥ 6‐pt improved Miln = 206/516 Plac = 157/509 * time weighted average (AUC) of mean weekly PED 24‐h recall pain scores | BOCF At end of stable dose period 1. ≥ 30% improvement from baseline PI + PGIC much or very much improved Miln = 150/516 Plac = 92/509 2. ≥ 30% improvement from baseline PI + PGIC much or very much improved + ≥ 6‐point improvement in physical function Miln = 103/516 Plac = 56/509 |
| Bateman 2013 | M 8, F 91 Mean age 48.5 years 91% white Baseline PI > 60 mm | 4 weeks' duloxetine 60 mg/day for ≥ 4 weeks prior to enrolment 2 weeks' duloxetine 60 mg 10 weeks randomised Miln (direct switch) 100 mg/day with possibility to escalate to 200 mg/day or de‐escalate to 50‐75 mg/day or 10 weeks' placebo (first week blinded duloxetine 30 mg) 1 week down‐taper period (not considered here) |
N = 100 (ITT population) Miln = 79 Plac = 21 N= 106 (+1) (at randomisation) Miln = 85(+1) Plac = 21 |
End of week 10: PGIC ≤ 2 Miln = 26/79 Plac = 5/21 VAS pain ≥ 30% improvement Miln = 27/79 Plac = 6/21 VAS pain ≥ 40% improvement Miln = 23/79 Plac = 4/21 VAS pain ≥ 50% improvement Miln = 20/79 Plac = 3/21 |
None |
| Branco 2010 | M 58, F 826 Mean age 49 years Mean duration symptoms 9.5 years | 1‐ to 4‐week washout 2‐week training and randomisation 4‐week dose escalation 12‐week stable dose 9‐day down‐titration 2‐week follow‐up Target dose 200 mg/day (2 x 100 mg) Treatment duration Max 16 weeks Stable 12 weeks |
N = 884 Miln = 435 Plac = 449 | LOCF At end of stable dose 24‐h recall PI** ≥ 30% improvement from baseline Miln: 38.6% = 166/430 Plac: 30.0% = 134/446 PGIC much or very much improved Miln: 33.3% = 143/430 Plac: 20.6% = 92/446 FIQ total score mean change (SEM) Miln: ‐14.18 (1.03) Plac: ‐11.18 (0.99) PCS mean change (SEM): Miln: 4.55 (0.36) Plac: 3.57 (0.35) ** daily PI averaged for 2 weeks immediately preceding visit day | BOCF
At end of stable dose period 1. ≥ 30% improvement from baseline PI + PGIC much or very much improved Miln: 100/430 Plac: 60/446 |
| Clauw 2008 | M 45, F 1151 Mean age 50 years 93% white Mean duration of symptoms 10 years Baseline PI > 60/100 mm | 1‐ to 4‐week washout, 2‐week training, randomisation
3‐week dose escalation
12‐week stable dose
Target dose 100 mg/day (2 x 50 mg) or 200 mg/day (2 x 100 mg) Treatment duration Max 15 weeks Stable 12 weeks |
N = 1207 (ITT 1196) Miln 100 mg/day = 401 (399) Miln 200 mg/day = 401 (396) Plac = 405 (401) | LOCF At end of stable dose 24‐h recall PI** ≥ 30% improvement from baseline Miln 100 = 149/399 Miln 200 = 158/396 Plac = 115/401 PGIC ≥ much improved Miln 100 = 138/399 Miln 200 = 151/396 Plac = 100/401 PCS ≥ 6‐point improved Miln 100 = 129/399 Miln 200 = 109/396 Plac = 102/401 ** daily PI averaged for 2 weeks immediately preceding visit day | BOCF At end of stable dose period 1. ≥ 30% improvement from baseline PI + PGIC much or very much improved Miln 100 = 92/399 Miln 200 = 102/396 Plac = 67/401 2. ≥ 30% improvement from baseline PI + PGIC much or very much improved + ≥ 6‐point improvement in physical function Miln 100 = 59/399 Miln 200 = 56/396 Plac = 35/401 |
| Mease 2009 | M 39, F 849 Mean age 49 years 93% white Mean duration of symptoms 5.5 years Baseline PI > 60/100 mm | 1‐4 weeks' screening and washout
2‐week baseline and training, randomisation
3‐week dose escalation
24 weeks stable dose
All medication given as divided doses, 2 x daily Treatment duration Max 27 weeks Stable 24 weeks |
N = 888 Miln 100 mg/day = 224 Miln 200 mg/day = 441 Plac = 223 | Mean data for pain at 15 and 27 weeks ‐ improvements generally similar between 2 doses ≥ 30% and ≥ 50% improvement in PI and PGIC response: % given for observed cases (completers) only | BOCF (modified BOCF) At week 15 (27) 1. ≥ 30% improvement from baseline PI + PGIC much or very much improved Miln 100 = 61/224 (58/224) Miln 200 = 118/441 (113/441) Plac = 43/223 (41/223) At week 15 (27) 2. ≥ 30% improvement from baseline PI + PGIC much or very much improved + ≥ 6‐point improvement in physical function Miln 100 = 44/224 (41/224) Miln 200 = 85/441 (80/441) Plac = 27/223 (29/223) |
| Vitton 2004 | Across groups: F 96% to 98% Mean age 46 to 48 years White 79% to 89% Mean duration of symptoms 3.8 to 4.3 years | 1‐4 weeks' screening and washout
2‐week baseline/training, randomisation
4‐week dose titration
8‐week stable dose Miln given as single or divided dose Treatment duration Max 12 weeks Stable 8 weeks |
N = 125 Miln 2 x 100 mg/day = 51 Miln 1 x 200 mg/day = 46 Plac = 28 | LOCF
Gracely PI ≥ 50%***:
Miln 2 x 100 = 18/51
Miln 1 x 200 = 10/46
Plac = 4/28 VAS PI ≤ 30%***: Miln 2 x 100 = 20/51 Miln 1 x 200 = 16/46 Plac = 6/28 VAS PI ≥ 50%***: Miln 2 x 100 = 15/51 Miln 1 x 200 = 12/46 Plac = 6/28 PGIC "improved" (1‐3) for completers only *** daily PI (Gracely scale) averaged for 2 weeks immediately preceding visit day and compared with 2‐week baseline period |
None |
| AUC: area under the curve; BOCF: baseline observation carried forward; F: female; FIQ: Fibromyalgia Impact Questionnaire; h: hour; ITT: intention‐to‐treat; LOCF: last observation carried forward; M: male; max: maximum; Miln: milnacipran; N: number of participants in study; n: number of participants in treatment arm; PCS: Physical Component Summary; PED: Patient Experience Diary; PGIC: Patient Global Impression of Change; PI: pain intensity; Plac: placebo; SEM: standard error of the mean; VAS: visual analogue scale. | |||||
Appendix 6. Summary of results: adverse events and withdrawals
| Study | Maximum daily dose of milnacipran Titration/fixed | Adverse events (general) | Adverse events (specific) | Withdrawals |
| Arnold 2010 | 1‐ to 4‐week screening and washout
4‐ to 6‐week flexible dose titration
12‐week stable dose
2‐week randomised discontinuation (not considered here)
Target dose 100 mg/day (2 x 50 mg) Treatment duration Max 18 weeks Stable 12 weeks |
Participants with ≥ 1 Miln = 434/516 Plac = 382/509 Most mild to moderate SAE: Miln = 8/516 Plac = 6/509 No deaths | In ≥ 5% Nausea: Miln 189/516, Plac 106/509 Headache: Miln 92/516, Plac 80/509 Constipation: Miln 76/516, Plac 20/509 Hot flush: Miln 56/516, Plac 18/509 Dizziness: Miln 54/516, Plac 26/509 Insomnia: Miln 51/516, Plac 41/509 Hyperhidrosis: Miln 40/516, Plac 7/509 Palpitations: Miln 38/516, Plac 15/509 Fatigue: Miln 31/516, Plac 22/509 |
Miln
All‐cause = 159
AE = 92
LoE = 24
Pt consent = 27 Plac All‐cause = 150 AE = 71 LoE = 33 Participant consent = 21 |
| Bateman 2013 | 4 weeks' duloxetine 60 mg/day for ≥ 4 weeks before enrolment 2 weeks' duloxetine 60 mg 10 weeks' randomised miln (direct switch) 100 mg/day with possibility to escalate to 200 mg/day or de‐escalate to 50‐75 mg/day or 10 weeks' placebo (first week blinded duloxetine 30 mg) 1 week down‐taper period |
Participants with ≥ 1
Miln = 63/85
Plac = 16/21
Most mild to moderate SAE: Miln = 2/85 Plac = 0/21 No deaths |
Nausea: Miln 18/85, Plac 6/21 Diarrhoea: Miln 4/85, Plac 3/21 Irritability: Miln 6/85, Plac 1/21 Fatigue: Miln 3/85, Plac 2/21 Nasopharyngitis: Miln 6/85, Plac 0/21 Blood pressure increased: Miln 5/85, Plac 0/21 Muscle spasms: Miln 0/85, Plac 2/21 Dizziness: Miln 13/85, Plac 1/21 Headache: Miln 10/85, Plac 2/21 Migraine: Miln 1/85, Plac 2/21 Paraesthesia: Miln 1/85, Plac 2/21 Insomnia: Miln 9/85, Plac 2/21 Anxiety: Miln 5/85, Plac 0/21 Hyperhidrosis: Miln 5/85, Plac 0/21 Hot flush: Miln 7/85, Plac 1/21 |
Miln All‐cause = 35/86 AE = 15 LoE = 8 Participant consent = 6 5 lost to follow‐up, 1 did not take any drug Plac All‐cause = 10/21 AE = 2 LoE = 6 Participant consent = 0 2 lost to follow‐up |
| Branco 2010 | 1‐ to 4‐week washout 2‐week training and randomisation 4‐week dose escalation 12‐week stable dose 9‐day down‐titration 2‐week follow‐up Target dose 200 mg/day (2 x 100 mg) Treatment duration Max 16 weeks Stable 12 weeks |
Participants with ≥ 1 Miln: 363/431 Plac: 331/446 Most mild to moderate, and in titration period SAE Miln: 11/431 (14 events) Plac: 11/446 (16 events) No deaths | In ≥ 5% Nausea: Miln 112/431, Plac 50/446 Hyperhidrosis: Miln 102/431, Plac 13/446 Headache: Miln 73/431, Plac 55/446 Constipation: Miln 54/431, Plac 10/446 Dizziness: Miln 44/431, Plac 34/446 Palpitations: Miln 34/431, Plac 13/446 Insomnia: Miln 33/431, Plac 24/446 Nasopharyngitis: Miln 33/431, Plac 33/446 Hot flush: Miln 30/431, Plac 5/446 Tachycardia: Miln 29/431, Plac 3/446 Vomiting: 22/431, Plac 15/446 |
Miln
All‐cause = 119
AE = 95
LoE = 24
Participant consent = 7
Plac
All‐cause = 72
AE = 42
LoE = 31
Pt consent = 7 5 participants with Miln and 3 with Plac did not have evaluable data: problem with 1 study centre (4), did not receive medication (3), missing baseline data (1) |
| Clauw 2008 | 1‐ to 4‐week washout, 2‐week training, randomisation
3‐week dose escalation
12‐week stable dose
Target dose 100 mg/day (2 x 50 mg) or 200 mg/day (2 x 100 mg) Treatment duration Max 15 weeks Stable 12 weeks |
Participants with ≥ 1 Miln 100 = 358/399 Miln 200 = 346/396 Plac = 317/401 Most mild to moderate SAE Miln 100 = 5/399 Miln 200 = 4/396 Plac = 6/401 No deaths | In ≥ 5% active group: Nausea: Miln 100 137/399, Miln 200 149/396, Plac 77/401 Headache: Miln 100 72/399, Miln 200 70/396, Plac 58/401 Constipation: Miln 100 57/399, Miln 200 71/396, Plac 16/401 Insomnia: Miln 100 42/399, Miln 200 62/396, Plac 42/401 Hot flush: Miln 100 46/399, Miln 200 58/396, Plac 5/401 Dizziness: Miln 100 38/399, Miln 200 36/396, Plac 17/401 Palpitations: Miln 100 26/399, Miln 200 30/396, Plac 10/401 Fatigue: Miln 100 25/399, Miln 200 28/396, Plac 22/401 Sinusitis: Miln 100 22/399, Miln 200 26/396, Plac 18/401 Hyperhidrosis: Miln 100 25/399, Miln 200 23/396, Plac 5/401 Hypertension: Miln 100 25/399, Miln 200 15/396, Plac 6/401 Vomiting: Miln 100 24/399, Miln 200 20/396, Plac 9/401 Diarrhoea: Miln 100 22/399, Miln 200 13/396, Plac 24/401 Increased heart rate: Miln 100 20/399, Miln 200 21/396, Plac 1/401 Migraine: Miln 100 20/399, Miln 200 20/396, Plac 9/401 Depression: Miln 100 14/399, Miln 200 12/396, Plac 24/401 |
Miln 100
All‐cause = 135
AE = 78
LoE = 28
Participant consent = 14
Miln 200
All‐cause = 139
AE = 94
LoE = 19
Participant consent = 15
Placebo
All‐cause = 111
AE = 38
LoE = 36
Participant consent = 20 2 participants with Miln 100, 5 with Miln 200, and 4 with Plac excluded from all analyses: problem with 1 study centre (9), did not receive medication (1), enrolled at 2 centres (1) |
| Mease 2009 | 1‐4 weeks' screening and washout
2‐week baseline and training, randomisation
3‐week dose escalation
24 weeks' stable dose
All medication given as divided doses, 2 x daily Treatment duration Max 27 weeks Stable 24 weeks |
Participants with ≥ 1: Miln 100 = 188/224 Miln 200 = 400/441 Plac = 190/223 Most mild or moderate SAE: Miln 100 = 3/224 Miln 200 = 11/441 Plac = 6/223 | In ≥ 5%: Nausea: Miln 100 73/224, Miln 200 177/441, Plac 47/223 Headache: Miln 100 35/224, Miln 200 78/441, Plac 26/223 Constipation: Miln 100 41/224, Miln 200 63/441, Plac 6/223 Hyperhidrosis: Miln 100 22/224, Miln 200 55/441, Plac 5/223 Dizziness: Miln 100 26/224, Miln 200 50/441, Plac 15/223 Hot flush: Miln 100 22/224, Miln 200 46/441, Plac 6/223 Insomnia: Miln 100 24/224, Miln 200 41/441, Plac 15/223 Vomiting: Miln 100 11/224, Miln 200 36/441, Plac 4/223 Sinusitis: Miln 100 11/224, Miln 200 32/441, Plac 18/223 Increased heart rate: Miln 100 12/224, Miln 200 32/441, Plac 5/223 Dry mouth: Miln 100 13/224, Miln 200 31/441, Plac 6/223 URTI: Miln 100 20/224, Miln 200 30/441, Plac 16/223 Palpitations: Miln 100 18/224, Miln 200 25/441, Plac 2/223 Diarrhoea: Miln 100 10/224, Miln 200 23/441, Plac 16/223 |
Miln 100 All‐cause = 96/224 AE = 44/224 LoE = 26/224 Miln 200 All‐cause = 202/441 AE = 119/441 LoE = 49/441 Plac All‐cause = 78/223 AE = 23/223 LoE = 34/223 |
| Vitton 2004 | 1‐4 weeks‐ screening and washout
2‐week baseline/training, randomisation
4‐week dose titration
8‐week stable dose Miln given as single or divided dose Treatment duration Max 12 weeks Stable 8 weeks |
No data for participants with ≥ 1 AE No SAE Most AE of mild or moderate intensity | No data | Miln 2 x 100
All‐cause = 14/51
AE = 7/51
LoE = 3/51 Miln 1 x 200 All‐cause = 14/46 AE = 10/46 LoE = 3/46 Plac All‐cause = 7/28 AE = 1/28 LoE = 5/28 |
| AE: adverse event; LoE: lack of efficacy; max: maximum; Miln: milnacipran; Plac: placebo; SAE: serious adverse event; URTI: upper respiratory tract infection | ||||
Data and analyses
Comparison 1. Milnacipran 100 mg/day versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 At least 30% pain relief | 3 | 1925 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.38 [1.22, 1.57] |
| 2 PGIC 'much improved' or 'very much improved' | 3 | 1925 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.51 [1.32, 1.73] |
| 3 At least 50% pain relief | 2 | 1250 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.57 [1.26, 1.96] |
| 4 Composite 1 | 3 | 2272 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.46 [1.25, 1.71] |
| 5 Composite 2 | 3 | 2272 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.73 [1.41, 2.14] |
| 6 At least one adverse event | 4 | 2378 | Risk Difference (M‐H, Fixed, 95% CI) | 0.07 [0.04, 0.10] |
| 7 Serious adverse events | 4 | 2378 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.47, 1.73] |
| 8 Individual adverse events | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 Nausea | 4 | 2378 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.84 [1.59, 2.12] |
| 8.2 Constipation | 3 | 2272 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.12 [2.97, 5.71] |
| 8.3 Hot flush | 4 | 2378 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.12 [2.79, 6.06] |
| 8.4 Dizziness | 4 | 2378 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.05 [1.52, 2.77] |
| 8.5 Palpitations | 3 | 2272 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.02 [1.97, 4.63] |
| 8.6 Increased heart rate/tachycardia | 3 | 2272 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.42 [2.87, 10.25] |
| 8.7 Hyperhidrosis | 4 | 2378 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.99 [3.01, 8.26] |
| 8.8 Vomiting | 2 | 1247 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.70 [1.44, 5.05] |
| 8.9 Hypertension | 3 | 1931 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.58 [2.45, 8.58] |
| 9 All‐cause withdrawals | 4 | 2379 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [1.01, 1.27] |
| 10 Lack of efficacy withdrawals | 4 | 2379 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.72 [0.55, 0.94] |
| 11 Adverse event withdrawals | 4 | 2379 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.62 [1.33, 1.97] |
1.1. Analysis.
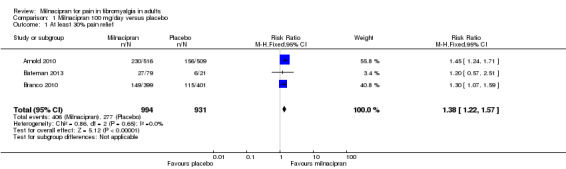
Comparison 1 Milnacipran 100 mg/day versus placebo, Outcome 1 At least 30% pain relief.
1.8. Analysis.
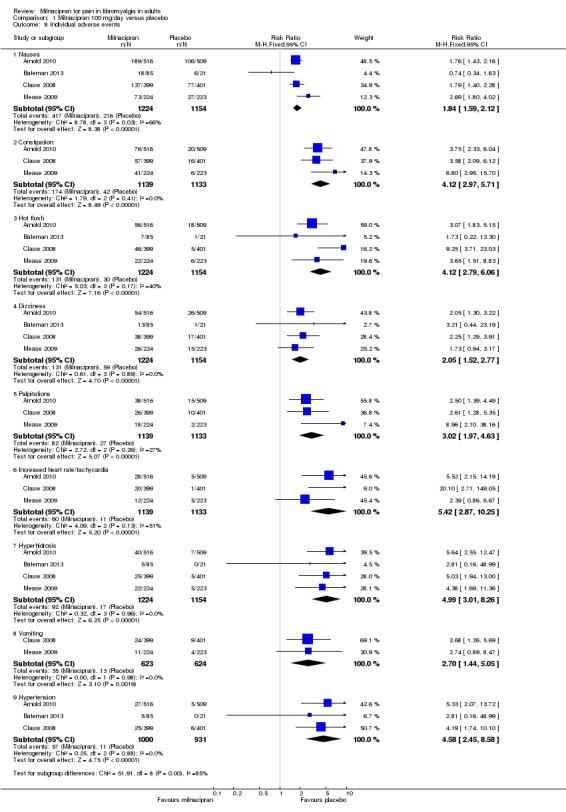
Comparison 1 Milnacipran 100 mg/day versus placebo, Outcome 8 Individual adverse events.
Comparison 2. Milnacipran 200 mg/day versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 At least 30% pain relief | 3 | 1798 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.35 [1.18, 1.54] |
| 2 PGIC 'much improved' or 'very much improved' | 2 | 1673 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.57 [1.34, 1.83] |
| 3 Composite 1 | 3 | 2337 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.55 [1.31, 1.84] |
| 4 Composite 2 | 2 | 1461 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.61 [1.21, 2.13] |
| 5 At least one adverse event | 3 | 2338 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [1.06, 1.15] |
| 6 Serious adverse events | 4 | 2463 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.52, 1.60] |
| 7 Individual adverse events | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 Nausea | 3 | 2338 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.37 [2.00, 2.80] |
| 7.2 Constipation | 3 | 2338 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.01 [3.46, 7.24] |
| 7.3 Hot flush | 3 | 2338 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.71 [4.06, 11.09] |
| 7.4 Dizziness | 3 | 2338 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.63 [1.22, 2.18] |
| 7.5 Palpitations | 3 | 2288 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.38 [2.17, 5.29] |
| 7.6 Increased heart rate/tachycardia | 3 | 2338 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.81 [3.54, 13.13] |
| 7.7 Hyperhidrosis | 2 | 1461 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.18 [2.67, 10.02] |
| 7.8 Vomiting | 3 | 2338 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.30 [1.48, 3.58] |
| 8 All‐cause withdrawals | 4 | 2416 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.38 [1.22, 1.57] |
| 9 Lack of efficacy withdrawals | 4 | 2462 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.66 [0.51, 0.87] |
| 10 Adverse event withdrawals | 4 | 2470 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.51 [2.03, 3.09] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Arnold 2010.
| Methods | Prospective, multicentre, randomised, double‐blind, placebo‐controlled trial with parallel groups. Participants recruited from outpatient clinical/research centres in the USA and Canada 1‐ to 4‐week screening and washout (all FM therapy stopped), 4‐ to 6‐week flexible dose titration, 12‐week stable dose of milnacipran 100 mg/day (50 mg twice daily). Participants unable to tolerate 100 mg/day were discontinued from study Data collected using electronic PED; pain improvement based on time weighted average of mean weekly 24‐h recall pain scores |
|
| Participants | Inclusion: age 18 to 70 years, ACR criteria for FM; physical function (FIQ) ≥ 4, and BDI > 25 at screening, mean PI ≥ 40 and ≤ 90/100 mm over 14‐day baseline period Excluded people with various medical and psychiatric conditions/risk factors, and previous exposure to milnacipran; women not using adequate contraception N = 1025 Mean age ˜ 49 years, M:F 48:977, 91% white, mean duration of symptoms ˜ 10.8 years, baseline pain > 60/100 mm |
|
| Interventions | Milnacipran 100 mg/day, n = 516 Placebo, n = 509 Permitted analgesics: paracetamol, aspirin, NSAIDs Short‐term rescue medication up to week 4: tramadol, hydrocodone |
|
| Outcomes | PI using 100 mm VAS: at least 30% and 50% improvement from baseline PGIC using 7‐point scale: much or very much improved Composite pain scores: 2‐part BOCF composite responder criteria: 24‐h and weekly recall pain scores using 100 mm VAS ≥ 30% improvement and PGIC score 'much' or 'very much' improved on 7‐point scale 3‐part BOCF composite responder criteria: 24‐h and weekly recall pain scores using 100 mm VAS ≥ 30% improvement, PGIC score 'much' or 'very much' improved on 7‐point scale, and SF‐36 PCS ≥ 6‐point improvement Adverse events Withdrawals |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomization assignments generated in blocks of four" |
| Allocation concealment (selection bias) | Low risk | "Assignments to treatment groups was conducted centrally (i.e. at the study level) using an interactive voice response system" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "identical‐appearing capsules were used by all patients during all phases of the study" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Clinical staff, investigators, patients, and the study sponsor were blinded to treatment allocation" |
| Incomplete outcome data (attrition bias) All outcomes | High risk | LOCF in analysis of individual outcomes, but BOCF in analysis of composite outcomes |
| Selective reporting (reporting bias) | Low risk | All relevant outcomes in methods were reported in some way, although not necessarily as our preferred outcome |
| Size | Low risk | Both groups > 200 participants |
Bateman 2013.
| Methods | Multicentre, randomised, double‐blind, placebo‐controlled switch study. Participants recruited from multiple centres in the United States All participants used stable dosage of duloxetine 60 mg/day for ≥ 4 weeks before enrolment, followed by open‐label duloxetine 60 mg for 2 weeks. Participants with PI ≥ 40/100 then randomised to double‐blind treatment for 10 weeks with milnacipran (direct switch) or placebo (with 1 week blinded taper of duloxetine 30 mg/day) 1 week double‐blind down‐taper period at end of study |
|
| Participants | Inclusion: FM (diagnostic criteria not given) with inadequate response to duloxetine 60 mg/day for ≥ 4 weeks, age 18 to 70 years, VAS 1‐week pain recall score ≥ 40 mm and ≤ 90 mm Excluded: people with various medical and psychiatric conditions or risk factors; women not using adequate contraception; concomitant medication except paracetamol, aspirin, and NSAIDs N = 100 Mean age ˜ 49 years, M:F 8:92, 91% white, mean baseline pain > 60/100 mm |
|
| Interventions | Milnacipran 100 to 200 mg/day with option to reduce to 50 mg/day or 75 mg/day during the first 2 weeks if not tolerated, n = 79 Placebo, n = 21 |
|
| Outcomes | PGIC using 7‐point scale: much or very much improved PI using 100 mm VAS: change from baseline |
|
| Notes | Oxford Quality Score: R = 1, DB = 1, W = 1. Total = 3 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of sequence generation not described |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Method to maintain blinding not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Method to maintain blinding not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | LOCF in individual outcome analysis |
| Selective reporting (reporting bias) | Low risk | No problems detected |
| Size | High risk | < 50 participants in each treatment arm |
Branco 2010.
| Methods | Prospective, multicentre, randomised, double‐blind, placebo‐controlled trial with parallel groups. Participants recruited from outpatient centres in Europe 1‐ to 4‐week screening and washout (all FM therapy stopped), 4‐week dose escalation, 12‐week stable dose with target milnacipran 200 mg/day (100 mg twice daily), 9‐day down‐titration, 2‐week follow‐up Data collected using electronic PED: daily PI averaged for 2 weeks immediately preceding visit day Adverse event data collected by spontaneous reporting, non‐leading questions, and clinical evaluation |
|
| Participants | Inclusion: age 18 to 70 years, met ACR criteria for FM; physical function (FIQ) ≥ 3, BDI > 25 at screening; mean PI ≥ 40 and ≥ 90 over 14‐day baseline period Excluded: people with various medical and psychiatric conditions or risk factors, considered unlikely to comply with treatment; women not using adequate contraception or pregnant N = 884 Mean age ˜ 49 years, M:F 58:826, mean duration of symptoms ˜ 9.5 years |
|
| Interventions | Milnacipran 200 mg/day, n = 435 Placebo, n = 449 |
|
| Outcomes | PI using 100 mm VAS: 30% improvement from baseline PGIC using 7‐point scale: much or very much improved Composite pain score: 2‐measure BOCF composite responder criteria: 24‐h morning recall pain scores ≥ 30% improvement using 100 mm VAS, PGIC score of 'very much' or 'much' improved Adverse events Withdrawals |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of sequence generation not described |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "In patients receiving placebo, twice‐daily sham dosing was used to maintain blinding" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "In patients receiving placebo, twice‐daily sham dosing was used to maintain blinding" |
| Incomplete outcome data (attrition bias) All outcomes | High risk | LOCF in analysis of individual outcomes, but BOCF in analysis of composite outcomes |
| Selective reporting (reporting bias) | Low risk | All relevant outcomes in methods were reported in some way, although not necessarily as our preferred outcome |
| Size | Low risk | Both groups > 200 participants |
Clauw 2008.
| Methods | Prospective, multicentre, randomised, double‐blind, placebo‐controlled trial with parallel groups. Participants recruited from outpatient centres in United States. 1‐ to 4‐week washout (all FM therapy stopped), 3‐week dose escalation, 12‐week stable dose with 100 mg/day (50 mg twice daily) or 200 mg/day (100 mg twice daily) milnacipran Data collected using electronic PED: daily PI averaged for 2 weeks immediately preceding visit day Adverse event data collected by spontaneous reporting and clinical evaluation |
|
| Participants | Inclusion: age 18 to 70 years, met ACR criteria for FM, physical function (FIQ) ≥ 4 and BDI > 25 at screening, baseline PI ≥ 40/100 Excluded: various medical and psychiatric conditions/risk factors, interfering medication over the last 30 days, women not using adequate contraception or pregnant N = 1151 Mean age ˜50 years, M:F 45:1151, ˜ 93% white, mean duration of symptoms ˜ 10 years, baseline pain > 60/100 mm |
|
| Interventions | Milnacipran 100 mg/day, n = 401 (396 for analysis) Milnacipran 200 mg/day, n = 410 (399 for analyses) Placebo, n = 405 (401 for analysis) |
|
| Outcomes | PI using 100 mm VAS: 30% improvement from baseline PGIC using 7‐point scale: much or very much improved Composite pain scores: 2‐measure BOCF composite responder criteria: 24‐h and weekly recall pain scores using 100 mm VAS ≥ 30% improvement and PGIC score 'much' or 'very much' improved on 7‐point scale 3‐measure BOCF composite responder criteria: 24‐h and weekly recall pain scores using 100 mm VAS ≥ 30% improvement, PGIC score 'much' or 'very much' improved on 7‐point scale, and SF‐36 PCS ≥ 6‐point improvement Adverse events Withdrawals |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1, Total = 5/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomization lists for each site were generated by a computer program" |
| Allocation concealment (selection bias) | Low risk | "randomization assignments were made via an interactive voice response system" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Milnacipran and placebo capsules were visually identical" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Clinical staff, investigators, patients, and the study sponsor were blinded to treatment allocation" |
| Incomplete outcome data (attrition bias) All outcomes | High risk | LOCF in analysis of individual outcomes, but BOCF in analysis of composite outcomes |
| Selective reporting (reporting bias) | Low risk | All relevant outcomes in methods were reported in some way, although not necessarily as our preferred outcome |
| Size | Low risk | All groups > 200 participants |
Clauw 2013.
| Methods | Double‐blind randomisation to placebo or continuing on milnacipran 50 to 200 mg ‐ the established daily dose for individual participants. Participants recruited from 58 centres in the United States There was a 4‐week maintenance phase, a 12‐week randomised withdrawal phase, and 1 week of tapering |
|
| Participants | Adults meeting the 1990 ACR criteria for FM entered directly from a long‐term, open‐label, flexible‐dose, lead‐in study in which they received milnacipran 50 to 200 mg/day for up to 3.25 years. They had previously received up to 15 months of treatment with milnacipran 100 or 200 mg/day during double‐blind studies resulting in up to 4.5 years of milnacipran exposure before entering the discontinuation study. Participants had to be classified as responders (≥ 50% pain improvement after long‐term treatment) and be receiving a minimum of milnacipran 100 mg/day |
|
| Interventions | Milnacipran 100 or 200 mg/day, n = 100 Placebo, n = 51 |
|
| Outcomes | Loss of therapeutic response, defined as time from randomisation to the first double‐blind study visit in which a participant had < 30% reduction in VAS pain from pre‐milnacipran exposure or worsening of FM requiring alternative treatment, as judged by the study's principal investigator PGIC Quality of life (SF‐36) |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1. Total = 5. Note, though, that this score is not designed for EERW trials | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generation implied |
| Allocation concealment (selection bias) | Low risk | Remote allocation using "interactive voice and/or web response system" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Medication "sealed and coded to maintain the double‐blinding" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Medication "sealed and coded to maintain the double‐blinding" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | LOCF imputation for missing data |
| Selective reporting (reporting bias) | High risk | Participants who did not experience loss of therapeutic response or withdrew for other reasons were censored in the primary efficacy analysis |
| Size | Unclear risk | 50 to 200 participants per treatment arm |
Mease 2009.
| Methods | Prospective, multicentre, randomised, double‐blind, placebo‐controlled trial with parallel groups. Participants recruited from 59 outpatient clinical/research centres in the United States Electronic PED used to collect data; 1‐ to 4‐week screening and washout (all FM therapy stopped), 3‐week baseline measurement and PED training, 24‐week stable dose with placebo or milnacipran 100 mg/day (50 mg twice daily) or milnacipran 200 mg/day (100 mg twice daily) (ratio milnacipran 100 mg/day:milnacipran 200 mg:placebo = 1:2:1) Adverse events collected from spontaneous reports, clinical observation, and clinical evaluation |
|
| Participants | Inclusion: age 18 to 70 years, met ACR criteria for FM, physical function (FIQ) ≥ 4, BDI > 25, and PI > 50/100 mm Exclusion: people with various medical and psychiatric conditions/risk factors; women not using adequate contraception N = 888 Mean age ˜ 49 years, M:F 39:849, ˜ 93% white, mean duration of symptoms ˜ 5.5 years, baseline PI > 60/100 at screening Analgesics prohibited, except for paracetamol, aspirin, stable doses of NSAIDs, and hydrocortisone |
|
| Interventions | Milnacipran 100 mg/day, n = 224 Milnacipran 200 mg/day, n = 441 Placebo, n = 223 |
|
| Outcomes | Composite pain scores: 2‐measure BOCF composite responder criteria: 24‐h and 2‐week mean recall pain scores using 100 mm VAS ≥ 30% improvement and PGIC score 'much' or 'very much' improved on 7‐point scale 3‐measure BOCF composite responder criteria: 24‐h and weekly recall pain scores using 100 mm VAS ≥ 30% improvement, PGIC score 'much' or 'very much' improved on 7‐point scale, and SF‐36 PCS ≥ 6‐point improvement Adverse events Withdrawals |
|
| Notes | Oxford Quality Score: R = 1, DB = 1, W = 1. Total = 3/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of sequence generation not described |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Method to maintain blinding not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Method to maintain blinding not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | LOCF in individual outcome analysis |
| Selective reporting (reporting bias) | Low risk | All relevant outcomes in methods were reported in some way, although not necessarily as our preferred outcome |
| Size | Low risk | All groups > 200 participants |
Vitton 2004.
| Methods | Prospective, multicentre, randomised, double‐blind, placebo‐controlled trial with parallel groups. Participants recruited from outpatient centres in US with experience in treating FM Electronic PED used to collect data; 1‐ to 4‐week screening and washout (all FM therapy stopped), 2‐week baseline measurement and PED training, 4‐week dose titration, 8‐week stable dose with milnacipran 200 mg/day (once daily), milnacipran 200 mg/day (100 mg twice daily), or placebo (ratio once daily:twice daily:placebo = 3:3:2) Adverse events collected from spontaneous reports, clinical observation, and clinical evaluation |
|
| Participants | Inclusion: age 18 to 70 years, met ACR criteria for FM; baseline PI ≥ 10/20 (Gracely log‐scale) Exclusion: people with various medical and psychiatric conditions/risk factors, women not using adequate contraception N = 125 participants Mean age 46 to 48 years, 96% to 98% female, 79% to 89% white, mean duration of symptoms 3.8 to 4.3 years Analgesics prohibited, except for stable doses of paracetamol, aspirin, and NSAIDs |
|
| Interventions | Milnacipran 200 mg/day (once daily), n = 46 Milnacipran 200 mg/day (twice daily), n = 51 Placebo, n = 28 |
|
| Outcomes | Pain (Short‐form McGill Pain Questionnaire, VAS, Gracely and Kwilosz anchored logarithmic scale) PGIC at end of study (completers analysis only) Adverse events Withdrawals |
|
| Notes | Oxford Quality Score: R = 2 (from Gendreau 2005), DB = 2 (from Gendreau 2005), W = 1. Total = 5/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomization was performed by an independent contract research organization that generated randomisation assignments" (from Gendreau 2005 ‐ see Vitton 2004) |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Blinding was rigorously maintained, as all patients took capsules morning and evening that were visually identical" (from Gendreau 2005 ‐ see Vitton 2004) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Patients and investigators remained blinded to patients' treatment allocation" (from Gendreau 2005 ‐ see Vitton 2004) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | LOCF for all analyses |
| Selective reporting (reporting bias) | Low risk | All relevant outcomes in methods were reported in some way, although not necessarily as our preferred outcome |
| Size | High risk | All groups ≤ 51 |
ACR: American College of Rheumatology; BDI: Beck Depression Inventory, BOCF: baseline observation carried forward; DB: double‐blinding; EERW: enriched enrolment randomised withdrawal; F: female; FIQ: fibromyalgia impact questionnaire; FM: fibromyalgia; h: hour; LOCF: last observation carried forward; M: male; N: number of participants in study; n: number of participants in treatment arm; NSAID: non‐steroidal anti‐inflammatory drug; PCS: physical component summary; PED: patient experience diary; PGIC: patient global impression of change; PI: pain intensity; R: randomisation; SF‐36: 36‐item short form; VAS: visual analogue scale; W: withdrawals.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Ang 2013 | Less than 8 weeks of treatment phase |
| Arnold 2012 | Study of effect on body weight not pain |
| Branco 2011 | Participants from Branco 2010 re‐randomised for extension study |
| Goldenberg 2010 | No placebo control (trial followed a 6‐month lead‐in study, where placebo controls were re‐randomised to treatment with milnacipran) |
| Häuser 2014 | Systematic review |
| Kim 2013 | Less than 8 weeks of treatment phase |
| Matthey 2013 | Less than 8 weeks of treatment phase |
| Mease 2014 | Pooled analysis of three fibromyalgia trials |
| NCT00797797 | Open‐label study |
| Spera 2012 | Short conference abstract, not full publication |
Differences between protocol and review
For the earlier review, we included analyses of two composite efficacy outcomes that were not pre‐specified in the original protocol. We chose to do this because they more closely reflect what is desirable in clinical practice (what patients with fibromyalgia want), and because they were calculated using baseline observation carried forward, without imputation for missing data, which reflects the situation in clinical practice (if you cannot take the medication you cannot get any benefit from it).
For this update in 2015, we made minor changes to the 'Background' after peer review comments. We also made minor changes in methods, principally upgrading the risk of bias assessment, and carrying out analysis according to three tiers of evidence, to distinguish the strength of evidence, in line with other reviews of interventions for chronic painful conditions.
Contributions of authors
RAM and SD wrote the protocol.
For the earlier review, DG and TP carried out searches and assessed studies for inclusion. SD, DG, and TP extracted data and carried out analyses. RAM acted as arbitrator. All review authors were involved in writing the review.
For this update, MC and PW carried out the searches and selected studies. MC, SD, and PW extracted data and carried out analyses. RAM acted as arbitrator. All review authors were involved in writing the review.
MC will be responsible for updating the review.
Sources of support
Internal sources
-
Oxford Pain Relief Trust, UK.
General institutional support
External sources
-
The National Institute for Health Research (NIHR), UK.
NIHR Cochrane Programme Grant: 13/89/29 ‐ Addressing the unmet need of chronic pain: providing the evidence for treatments of pain
Declarations of interest
MC has no conflicts relating to this review or any similar product.
SD has no conflicts relating to this review or any similar product.
TP has no conflicts relating to this review or any similar product.
RAM has no conflicts relating to this review or any similar product.
PW has no conflicts relating to this review or any similar product.
We are funded by the National Institute for Health Research (NIHR) for work on a series of reviews informing the unmet need of chronic pain and providing the evidence for treatments of pain.
Stable (no update expected for reasons given in 'What's new')
References
References to studies included in this review
Arnold 2010 {published data only}
- Arnold LM, Gendreau RM, Palmer RH, Gendreau JF, Wang Y. Efficacy and safety of milnacipran 100mg/day in patients with fibromyalgia. Arthritis and Rheumatism 2010;62(9):2745‐56. [DOI: 10.1002/art.27559] [DOI] [PubMed] [Google Scholar]
- Arnold LM, Palmer RH, Gendreau RM, Chen W. Relationships among pain, depressed mood, and global status in fibromyalgia patients: post hoc analyses of a randomized, placebo‐controlled trial of milnacipran. Psychosomatics 2012;53(4):371‐9. [DOI: 10.1016/j.psym.2012.02.005] [DOI] [PubMed] [Google Scholar]
- Saxe PA, Arnold LM, Palmer RH, Gendreau RM, Chen W. Short‐term (2‐week) effects of discontinuing milnacipran in patients with fibromyalgia. Current Medical Research and Opinion 2012;28(5):815‐21. [DOI: 10.1185/03007995.2012.677418] [DOI] [PubMed] [Google Scholar]
Bateman 2013 {published data only}
- Study of milnacipran in patients with inadequate response to duloxetine for the treatment of fibromyalgia, 2011. www.clinicaltrials.gov/ct2/show/study/NCT01077375 (accessed 22 September 2015). [CTG: NCT01077375]
- Bateman L, Palmer RH, Trugman JM, Lin Y. Results of switching to milnacipran in fibromyalgia patients with an inadequate response to duloxetine: a phase IV pilot study. Journal of Pain Research 2013;6:311‐8. [DOI: 10.2147/JPR.S43395] [DOI] [PMC free article] [PubMed] [Google Scholar]
Branco 2010 {published data only}
- Branco JC, Cherin P, Montagne A, Bouroubi A. Longterm therapeutic response to milnacipran treatment for fibromyalgia. A European 1‐year extension study following a 3‐month study. Journal of Rheumatology 2011;38(7):1403‐12. [DOI: 10.3899/jrheum.101025] [DOI] [PubMed] [Google Scholar]
- Branco JC, Zachrisson O, Perrot S, Mainguy Y. A European multicenter randomized double‐blind placebo‐controlled monotherapy clinical trial of milnacipran in treatment of fibromyalgia. Journal of Rheumatology 2010;37(4):851‐9. [DOI: 10.3899/jrheum.090884] [DOI] [PubMed] [Google Scholar]
Clauw 2008 {published data only}
- Clauw DJ, Mease P, Palmer RH, Gendreau RM, Wang Y. Milnacipran for the treatment of fibromyalgia in adults: a 15‐week, multicenter, randomized, double‐blind, placebo‐controlled, multiple‐dose clinical trial. Clinical Therapeutics 2008;30(11):1988‐2004. [DOI: 10.1016/j.clinthera.2008.11.009] [DOI] [PubMed] [Google Scholar]
Clauw 2013 {published data only}
- Clauw DJ, Mease PJ, Palmer RH, Trugman JM, Wang Y. Continuing efficacy of milnacipran following long‐term treatment in fibromyalgia: a randomized trial. Arthritis Research and Therapy 2013;15(4):R88. [DOI: 10.1186/ar4268] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mease PJ, Clauw DJ, Trugman JM, Palmer RH, Wang Y. Efficacy of long‐term milnacipran treatment in patients meeting different thresholds of clinically relevant pain relief: subgroup analysis of a randomized, double‐blind, placebo‐controlled withdrawal study. Journal of Pain Research 2014;7:679‐87. [DOI: 10.2147/JPR.S70200] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mease 2009 {published data only}
- Mease PJ, Clauw DJ, Gendreau RM, Rao SG, Kranzler J, Chen W, et al. The efficacy and safety or milnacipran for treatment of fibromyalgia. A randomized, double‐blind, placebo‐controlled trial. Journal of Rheumatology 2009;36(2):398‐409. [DOI: 10.3899/jrheum.080734] [DOI] [PubMed] [Google Scholar]
Vitton 2004 {published data only}
- Gendreau RM, Thorn MD, Gendreau JF, Kranzler JD, Ribeiro S, Gracely RH, et al. Efficacy of milnacipran in patients with fibromyalgia. Journal of Rheumatology 2005;32:1975‐85. [PUBMED: 16206355] [PubMed] [Google Scholar]
- Vitton O, Gendreau M, Gendreau J, Kranzler J, Rao SG. A double‐blind placebo‐controlled trial of milnacipran in the treatment of fibromyalgia. Human Psychopharmacology 2004;19:S27‐35. [DOI: 10.1002/hup.622] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Ang 2013 {published data only}
- Ang DC, Jensen MP, Steiner JL, Hilligoss J, Gracely RH, Saha C. Combining cognitive‐behavioral therapy and milnacipran for fibromyalgia: a feasibility randomized‐controlled trial. Clinical Journal of Pain 2013;29(9):747‐54. [DOI: 10.1097/AJP.0b013e31827a784e] [DOI] [PMC free article] [PubMed] [Google Scholar]
Arnold 2012 {published data only}
- Arnold LM, Palmer RH, Hufford MR, Chen W. Effect of milnacipran on body weight in patients with fibromyalgia. International Journal of General Medicine 2012;5:879‐87. [DOI: 10.2147/IJGM.S36444] [DOI] [PMC free article] [PubMed] [Google Scholar]
Branco 2011 {published data only}
- Branco JC, Cherin P, Montagne A, Bouroubi A, on behalf of the Multinational Coordinator Study Group. Longterm therapeutic response to milnacipran treatment for fibromyalgia. A European 1‐year extension study following a 3‐month study. Journal of Rheumatology 2011;38(7):1403‐12. [DOI: 10.3899/jrheum.101025] [DOI] [PubMed] [Google Scholar]
Goldenberg 2010 {published data only}
- Goldenberg DL, Clauw DJ, Palmer RH, Mease P, Chen W, Gendreau RM. Durability of therapeutic response to milnacipran treatment for fibromyalgia. Results of a randomized, double‐blind, monotherapy 6‐month extension study. Pain Medicine 2010;11(2):180‐94. [DOI: 10.1111/j.1526-4637.2009.00755.x] [DOI] [PubMed] [Google Scholar]
Häuser 2014 {published data only}
- Häuser W, Sarzi‐Puttini P, Töle TR, Wolfe F. Placebo and nocebo responses in randomised controlled trials of drugs applying for approval for fibromyalgia syndrome treatment: systematic review and meta‐analysis. Clinical and Experimental Rheumatology 2014;30(6 (Suppl 74)):78‐87. [PUBMED: 23137770] [PubMed] [Google Scholar]
Kim 2013 {published data only}
- Kim JL, Rele S, Marks DM, Masand PS, Yerramsetty P, Millet RA, et al. Effects of milnacipran on neurocognition, pain, and fatigue in fibromyalgia: a 13‐week, randomized, placebo‐controlled, crossover trial. Primary Care Companion for CNS Disorders 2013;15(6):PCC.13m01555. [DOI: 10.4088/PCC.13m01555] [DOI] [PMC free article] [PubMed] [Google Scholar]
Matthey 2013 {published data only}
- Matthey A, Cedraschi C, Piguet V, Besson M, Chabert J, Daali Y, et al. Dual reuptake inhibitor milnacipran and spinal pain pathways in fibromyalgia patients: a randomized, double‐blind, placebo‐controlled trial. Pain Physician 2013;16(5):E553‐62. [PUBMED: 24077206] [PubMed] [Google Scholar]
Mease 2014 {published data only}
- Mease PJ, Palmer RH, Wang Y. Effects of milnacipran on the multidimensional aspects of fatigue and the relationship of fatigue to pain and function: pooled analysis of 3 fibromyalgia trials. Journal of Clinical Rheumatology 2014;20(4):195‐202. [DOI: 10.1097/RHU.0000000000000103] [DOI] [PubMed] [Google Scholar]
NCT00797797 {unpublished data only}
- A multicenter, randomized, open‐label, controlled study to evaluate the safety, tolerability, and efficacy of milnacipran when added to pregabalin in the treatment of fibromyalgia, 2011. clinicaltrials.gov/ct2/show/NCT00797797 (accessed 12 August 2011).
Spera 2012 {published data only}
- Spera A, Clauw DJ, Arnold LM, Ma Y. Long‐term efficacy and tolerability of milnacipran for the management of fibromyalgia: results from an open‐label, flexible‐dosing study followed by a randomized, double‐blind, placebo‐controlled discontinuation study. Journal of General Internal Medicine. Conference: 35th Annual Meeting of the Society of General Internal Medicine, SGIM. 2012.
Additional references
Arnold 2013
- Arnold LM, Palmer RH, Ma Y. A 3‐year, open‐label, flexible‐dosing study of milnacipran for the treatment of fibromyalgia. Clinical Journal of Pain 2013;29(12):1021‐8. [DOI: 10.1097/AJP.0b013e31828440ab] [DOI] [PubMed] [Google Scholar]
Bernstein 2013
- Bernstein CD, Albrecht KL, Marcus DA. Milnacipran for fibromyalgia: a useful addition to the treatment armamentarium. Expert Opinion on Pharmacotherapy 2013;14(7):905‐16. [DOI: 10.1517/14656566.2013.779670] [DOI] [PubMed] [Google Scholar]
Choy 2011
- Choy E, Marshall D, Gabriel ZL, Mitchell SA, Gylee E, Dakin HA. A systematic review and mixed treatment comparison of the efficacy of pharmacological treatments for fibromyalgia. Seminars in Arthritis and Rheumatism 2011;41(3):335‐45.e6. [DOI: 10.1016/j.semarthrit.2011.06.003] [DOI] [PubMed] [Google Scholar]
Dechartres 2013
- Dechartres A, Trinquart L, Boutron I, Ravaud P. Influence of trial sample size on treatment effect estimates: meta‐epidemiological study. BMJ 2013;346:f2304. [DOI: 10.1136/bmj.f2304] [DOI] [PMC free article] [PubMed] [Google Scholar]
Derry 2015
- Derry S, Phillips T, Moore RA, Wiffen PJ. Milnacipran for neuropathic pain in adults. Cochrane Database of Systematic Reviews 2015, Issue 7. [DOI: 10.1002/14651858.CD011789] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dworkin 2008
- Dworkin RH, Turk DC, Wyrwich KW, Beaton D, Cleeland CS, Farrar JT, et al. Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. Journal of Pain 2008;9(2):105‐21. [DOI: 10.1016/j.jpain.2007.09.005] [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S. Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Altman DG, Sterne JAC (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hoffman 2010
- Hoffman DL, Sadosky A, Dukes EM, Alvir J. How do changes in pain severity levels correspond to changes in health status and function in patients with painful diabetic peripheral neuropathy?. Pain 2010;149(2):194‐201. [DOI: 10.1016/j.pain.2009.09.017] [DOI] [PubMed] [Google Scholar]
Häuser 2011
- Häuser W, Petzke F, Üçeyler N, Sommer C. Comparative efficacy and acceptability of amitriptyline, duloxetine and milnacipran in fibromyalgia syndrome: a systematic review with meta‐analysis. Rheumatology (Oxford) 2011;50(3):532‐43. [DOI: 10.1093/rheumatology/keq354] [DOI] [PubMed] [Google Scholar]
Häuser 2013
- Häuser W, Urrútia G, Tort S, Uçeyler N, Walitt B. Serotonin and noradrenaline reuptake inhibitors (SNRIs) for fibromyalgia syndrome. Cochrane Database of Systematic Reviews 2013, Issue 1. [DOI: 10.1002/14651858.CD010292] [DOI] [PubMed] [Google Scholar]
Jadad 1996
- Jadad AR, Moore RA, Carroll D, Jenkinson C, Reynolds DJ, Gavaghan DJ, et al. Assessing the quality of reports of randomized clinical trials: is blinding necessary?. Controlled Clinical Trials 1996;17(1):1‐12. [DOI: ] [DOI] [PubMed] [Google Scholar]
Kalso 2013
- Kalso E, Aldington DJ, Moore RA. Drugs for neuropathic pain. BMJ 2013;347:f7339. [DOI: 10.1136/bmj.f7339] [DOI] [PubMed] [Google Scholar]
Kjaergard 2001
- Kjaergard LL, Villumsen J, Gluud C. Reported methodologic quality and discrepancies between large and small randomized trials in meta‐analyses. Annals of Internal Medicine 2001;135(11):982‐9. [DOI: 10.7326/0003-4819-135-11-200112040-00010] [DOI] [PubMed] [Google Scholar]
Koroschetz 2011
- Koroschetz J, Rehm SE, Gockel U, Brosz M, Freynhagen R, Tölle TR, et al. Fibromyalgia and neuropathic pain ‐ differences and similarities. A comparison of 3057 patients with diabetic painful neuropathy and fibromyalgia. BMC Neurology 2011;11:55. [DOI: 10.1186/1471-2377-11-55] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kyle 2010
- Kyle JA, Dugan BD, Testerman KK. Milnacipran for treatment of fibromyalgia. Annals of Pharmacotherapy 2010;44(9):1422‐9. [DOI: 10.1345/aph.1P218] [DOI] [PubMed] [Google Scholar]
L'Abbé 1987
- L'Abbé KA, Detsky AS, O'Rourke K. Meta‐analysis in clinical research. Annals of Internal Medicine 1987;107:224‐33. [DOI: 10.7326/0003-4819-107-2-224] [DOI] [PubMed] [Google Scholar]
Lunn 2014
- Lunn MP, Hughes RA, Wiffen PJ. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database of Systematic Reviews 2014, Issue 1. [DOI: 10.1002/14651858.CD007115.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
McQuay 1998
- McQuay H, Moore R. An Evidence‐based Resource for Pain Relief. Oxford: Oxford University Press, 1998. [ISBN: 0‐19‐263048‐2] [Google Scholar]
Moore 1998
- Moore RA, Gavaghan D, Tramèr MR, Collins SL, McQuay HJ. Size is everything ‐ large amounts of information are needed to overcome random effects in estimating direction and magnitude of treatment effects. Pain 1998;78(3):209‐16. [DOI: 10.1016/S0304-3959(98)00140-7] [DOI] [PubMed] [Google Scholar]
Moore 2008
- Moore RA, Barden J, Derry S, McQuay HJ. Managing potential publication bias. In: McQuay HJ, Kalso E, Moore RA editor(s). Systematic Reviews in Pain Research: Methodology Refined. Seattle: IASP Press, 2008:15‐24. [ISBN: 978‐0‐931092‐69‐5] [Google Scholar]
Moore 2009
- Moore RA, Straube S, Wiffen PJ, Derry S, McQuay HJ. Pregabalin for acute and chronic pain in adults. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD007076] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2010a
- Moore RA, Eccleston C, Derry S, Wiffen P, Bell RF, Straube S, et al. ACTINPAIN Writing Group of the IASP Special Interest Group on Systematic Reviews in Pain Relief, Cochrane Pain, Palliative and Supportive Care Systematic Review Group Editors. "Evidence" in chronic pain‐establishing best practice in the reporting of systematic reviews. Pain 2010;150(3):386‐9. [DOI: 10.1016/j.pain.2010.05.011] [DOI] [PubMed] [Google Scholar]
Moore 2010b
- Moore RA, Moore OA, Derry S, Peloso PM, Gammaitoni AR, Wang H. Responder analysis for pain relief and numbers needed to treat in a meta‐analysis of etoricoxib osteoarthritis trials: bridging a gap between clinical trials and clinical practice. Annals of the Rheumatic Diseases 2010;69(2):374‐9. [DOI: 10.1136/ard.2009.107805] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2010c
- Moore RA, Straube S, Paine J, Phillips CJ, Derry S, McQuay HJ. Fibromyalgia: moderate and substantial pain intensity reduction predicts improvement in other outcomes and substantial quality of life gain. Pain 2010;149(3):260‐4. [DOI: 10.1016/j.pain.2010.02.039] [DOI] [PubMed] [Google Scholar]
Moore 2010d
- Moore RA, Derry S, McQuay HJ, Straube S, Aldington D, Wiffen P, et al. Clinical effectiveness: an approach to clinical trial design more relevant to clinical practice, acknowledging the importance of individual differences. Pain 2010;149(2):173‐6. [DOI: 10.1016/j.pain.2009.08.007] [DOI] [PubMed] [Google Scholar]
Moore 2011a
- Moore RA, Straube S, Paine J, Derry S, McQuay HJ. Minimum efficacy criteria for comparisons between treatments using individual patient meta‐analysis of acute pain trials: examples of etoricoxib, paracetamol, ibuprofen, and ibuprofen/paracetamol combinations after third molar extraction. Pain 2011;152(5):982‐9. [DOI: 10.1016/j.pain.2010.11.030] [DOI] [PubMed] [Google Scholar]
Moore 2011b
- Moore RA, Mhuircheartaigh RJ, Derry S, McQuay HJ. Mean analgesic consumption is inappropriate for testing analgesic efficacy in post‐operative pain: analysis and alternative suggestion. European Journal of Anaesthesiology 2011;28(6):427‐32. [DOI: 10.1097/EJA.0b013e328343c569] [DOI] [PubMed] [Google Scholar]
Moore 2012a
- Moore RA, Derry S, Aldington D, Cole P, Wiffen PJ. Amitriptyline for neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews 2012, Issue 12. [DOI: 10.1002/14651858.CD008242.pub2] [DOI] [PubMed] [Google Scholar]
Moore 2012b
- Moore RA, Straube S, Eccleston C, Derry S, Aldington D, Wiffen P, et al. Estimate at your peril: imputation methods for patient withdrawal can bias efficacy outcomes in chronic pain trials using responder analyses. Pain 2012;153(2):265‐8. [DOI: 10.1016/j.pain.2011.10.00] [DOI] [PubMed] [Google Scholar]
Moore 2013a
- Moore RA, Straube S, Aldington D. Pain measures and cut‐offs ‐ 'no worse than mild pain' as a simple, universal outcome. Anaesthesia 2013;68(4):400‐12. [DOI: 10.1111/anae.12148] [DOI] [PubMed] [Google Scholar]
Moore 2013b
- Moore A, Derry S, Eccleston C, Kalso E. Expect analgesic failure; pursue analgesic success. BMJ 2013;346:f2690. [DOI: 10.1136/bmj.f2690] [DOI] [PubMed] [Google Scholar]
Moore 2014a
- Moore RA, Derry S, Taylor RS, Straube S, Phillips CJ. The costs and consequences of adequately managed chronic non‐cancer pain and chronic neuropathic pain. Pain Practice 2014;14(1):79‐94. [DOI: 10.1111/papr.12050] [DOI] [PubMed] [Google Scholar]
Moore 2014b
- Moore RA, Wiffen PJ, Derry S, Toelle T, Rice AS. Gabapentin for chronic neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews 2014, Issue 4. [DOI: 10.1002/14651858.CD007938.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2014c
- Moore RA, Cai N, Skljarevski V, Tölle TR. Duloxetine use in chronic painful conditions ‐ individual patient data responder analysis. European Journal of Pain 2014;18(1):67‐75. [DOI: 10.1002/j.1532-2149.2013.00341.x] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nüesch 2010
- Nüesch E, Trelle S, Reichenbach S, Rutjes AW, Tschannen B, Altman DG, et al. Small study effects in meta‐analyses of osteoarthritis trials: meta‐epidemiological study. BMJ 2010;341:c3515. [DOI: 10.1136/bmj.c3515] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ormseth 2010
- Ormseth MJ, Eyler AE, Hammonds CL, Boomershine CS. Milnacipran for the management of fibromyalgia syndrome. Journal of Pain Research 2010;1(3):15‐24. [DOI: 10.2147/JPR.S7883] [DOI] [PMC free article] [PubMed] [Google Scholar]
PaPaS 2012
- PaPaS author and referee guidance. papas.cochrane.org/papas‐documents (accessed 11 August 2015).
Queiroz 2013
- Queiroz LP. Worldwide epidemiology of fibromyalgia. Current Pain and Headache Reports 2013;17(8):356. [DOI: 10.1007/s11916-013-0356-5] [DOI] [PubMed] [Google Scholar]
Recla 2010
- Recla JM. New and emerging therapeutic agents for the treatment of fibromyalgia: an update. Journal of Pain Research 2010;22(3):89‐103. [DOI: 10.2147/JPR.S6792] [DOI] [PMC free article] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Roskell 2011
- Roskell NS, Beard SM, Zhao Y, Le TK. A meta‐analysis of pain response in the treatment of fibromyalgia. Pain Practice 2011;11(6):516‐27. [DOI: 10.1111/j.1533-2500.2010.00441.x] [DOI] [PubMed] [Google Scholar]
Saarto 2007
- Saarto T, Wiffen PJ. Antidepressants for neuropathic pain. Cochrane Database of Systematic Reviews 2007, Issue 4. [DOI: 10.1002/14651858.CD005454.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schmidt‐Wilcke 2014
- Schmidt‐Wilcke T, Ichesco E, Hampson JP, Kairys A, Peltier S, Harte S, et al. Resting state connectivity correlates with drug and placebo response in fibromyalgia patients. Neuroimage Clinical 2014;6:252‐61. [DOI: 10.1016/j.nicl.2014.09.007] [DOI] [PMC free article] [PubMed] [Google Scholar]
Straube 2008
- Straube S, Derry S, McQuay HJ, Moore RA. Enriched enrollment: definition and effects of enrichment and dose in trials of pregabalin and gabapentin in neuropathic pain. A systematic review. British Journal Clinical Pharmacology 2008;66(2):266‐75. [DOI: 10.1111/j.1365-2125.2008.03200.x] [DOI] [PMC free article] [PubMed] [Google Scholar]
Straube 2010
- Straube S, Derry S, Moore RA, Paine J, McQuay HJ. Pregabalin in fibromyalgia ‐ responder analysis from individual patient data. BMC Musculoskeletal Disorders 2010;11:150. [DOI: 10.1186/1471-2474-11-150] [DOI] [PMC free article] [PubMed] [Google Scholar]
Straube 2011
- Straube S, Moore RA, Paine J, Derry S, Phillips CJ, Hallier E, et al. Interference with work in fibromyalgia: effect of treatment with pregabalin and relation to pain response. BMC Musculoskeletal Disorders 2011;12:125. [DOI: 10.1186/1471-2474-12-125] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sultan 2008
- Sultan A, Gaskell H, Derry S, Moore RA. Duloxetine for painful diabetic neuropathy and fibromyalgia pain: systematic review of randomised trials. BMC Neurology 2008;8:29. [DOI: 10.1186/1471-2377-8-29] [DOI] [PMC free article] [PubMed] [Google Scholar]
Suzuki 2004
- Suzuki R, Rygh LJ, Dickenson AH. Bad news from the brain: descending 5‐HT pathways that control spinal pain processing. Trends in the Pharmacological Sciences 2004;2512:613‐7. [DOI: 10.1016/j.tips.2004.10.002] [DOI] [PubMed] [Google Scholar]
Vos 2012
- Vos T, Flaxman AD, Naghavi M, Lozano R, Michaud C, Ezzati M, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012;380(9859):2163‐96. [DOI: 10.1016/S0140-6736(12)61729-2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wiffen 2013
- Wiffen PJ, Derry S, Moore RA, Aldington D, Cole P, Rice ASC, et al. Antiepileptic drugs for neuropathic pain and fibromyalgia ‐ an overview of Cochrane reviews. Cochrane Database of Systematic Reviews 2013, Issue 11. [DOI: 10.1002/14651858.CD010567.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wolfe 1990
- Wolfe F, Smythe HA, Yunus MB, Bennett RM, Bombardier C, Goldenberg DL, et al. The American College of Rheumatology 1990 Criteria for the Classification of Fibromyalgia. Report of the Multicenter Criteria Committee. Arthritis and Rheumatism 1990;33:160‐72. [PUBMED: 2306288] [DOI] [PubMed] [Google Scholar]
Wolfe 2010
- Wolfe F, Clauw DJ, Fitzcharles MA, Goldenberg DL, Katz RS, Mease P, et al. The American College of Rheumatology preliminary diagnostic criteria for fibromyalgia and measurement of symptom severity. Arthritis Care and Research 2010;62(5):600‐10. [DOI: 10.1002/acr.20140] [DOI] [PubMed] [Google Scholar]
Wolfe 2013a
- Wolfe F, Brähler E, Hinz A, Häuser W. Fibromyalgia prevalence, somatic symptom reporting, and the dimensionality of polysymptomatic distress: results from a survey of the general population. Arthritis Care and Research 2013;645(5):777‐85. [DOI: 10.1002/acr.21931] [DOI] [PubMed] [Google Scholar]
Wolfe 2014
- Wolfe F, Walitt BT, Häuser W. What is fibromyalgia, how is it diagnosed and what does it really mean?. Arthritis Care and Research 2014;66(7):969‐71. [DOI: 10.1002/acr.22207] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Derry 2012
- Derry S, Gill D, Phillips T, Moore RA. Milnacipran for neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews 2012, Issue 3. [DOI: 10.1002/14651858.CD008244.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]